
Block by calcium of ATP-activated channels in pheochromocytoma cells
Upload
independentCategory
view
3download
0
A R T I C L E S
118 VOLUME 7 | NUMBER 2 | FEBRUARY 2004 NATURE NEUROSCIENCE
1Department of Physiology & Biophysics, Cellular and Molecular Neurobiology Research Group, University of Calgary, 3330 Hospital Drive NW, Calgary, Alberta T2N 4N1, Canada. 2Département de Physiologie, LGF CNRS UPR2580, Montpellier 34396, France. Correspondence should be addressed to G.W.Z. ([email protected]).
Published online 18 January 2004; doi:10.1038/nn1180
Agonist-independent modulation of N-type calciumchannels by ORL1 receptorsAaron M Beedle1, John E McRory1, Olivier Poirot2, Clinton J Doering1, Christophe Altier1, Christian Barrere2,Jawed Hamid1, Joel Nargeot2, Emmanuel Bourinet2 & Gerald W Zamponi1
We have investigated modulation of voltage-gated calcium channels by nociceptin (ORL1) receptors. In rat DRG neurons and in tsA-201 cells, nociceptin mediated a pronounced inhibition of N-type calcium channels, whereas other calcium channelsubtypes were unaffected. In tsA-201 cells, expression of N-type channels with human ORL1 resulted in a voltage-dependent G-protein inhibition of the channel that occurred in the absence of nociceptin, the ORL1 receptor agonist. Consistent with thisobservation, native N-type channels of small nociceptive dorsal root ganglion (DRG) neurons also had tonic inhibition by G proteins. Biochemical characterization showed the existence of an N-type calcium channel–ORL1 receptor signaling complex,which efficiently exposes N-type channels to constitutive ORL1 receptor activity. Calcium channel activity is thus regulated bychanges in ORL1 receptor expression, which provides a possible molecular mechanism for the development of tolerance to opioidreceptor agonists.
The transmission of nociceptive signals is modulated by activation of avariety of G protein–coupled receptors, including opioid receptorsand opioid receptor–like (ORL1) receptors. Opioid receptor agonistsinhibit pain neurotransmission by activating postsynaptic K+ currentsand by inhibiting presynaptic calcium channels through Gβγ sub-units1. Activation of ORL1 receptors by intrathecal injection of thepeptide nociceptin (or orphanin FQ)2,3 diminishes pain sensation andspinal neurotransmission4–6. At the cellular level, opioid and ORL1receptors target overlapping effector systems, including voltage-gatedcalcium channels7,8. Whereas opioid receptors seem to couple pre-dominantly to N-type and P/Q-type channels, nociceptin reportedlyinhibits L- and R-type calcium channels in some neuronal prepara-tions and mediates a potent inhibition of T-type channels in rat DRGneurons7,9–11. This contrasts with nociceptin modulation in mousetrigeminal ganglion neurons, which is specific for high-voltage acti-vated (HVA) channels12. Here we show that the ORL1 receptor doesnot regulate native or transiently expressed T-type channels. Instead,ORL1 receptor activation predominantly inhibited N-type channelactivity. In addition, ORL1 receptors and N-type channels formed aphysical signaling complex that results in an agonist-independent,receptor concentration–dependent inhibition of the channels inexpression systems and in DRG neurons. Our data raise the possibilitythat the activity of N-type calcium channels, and consequently trans-mission of pain signals, may be regulated by ORL1 receptor density.
RESULTSWe first investigated the inhibition of calcium currents by ORL1receptors in acutely dissociated DRG neurons by whole-cell voltage
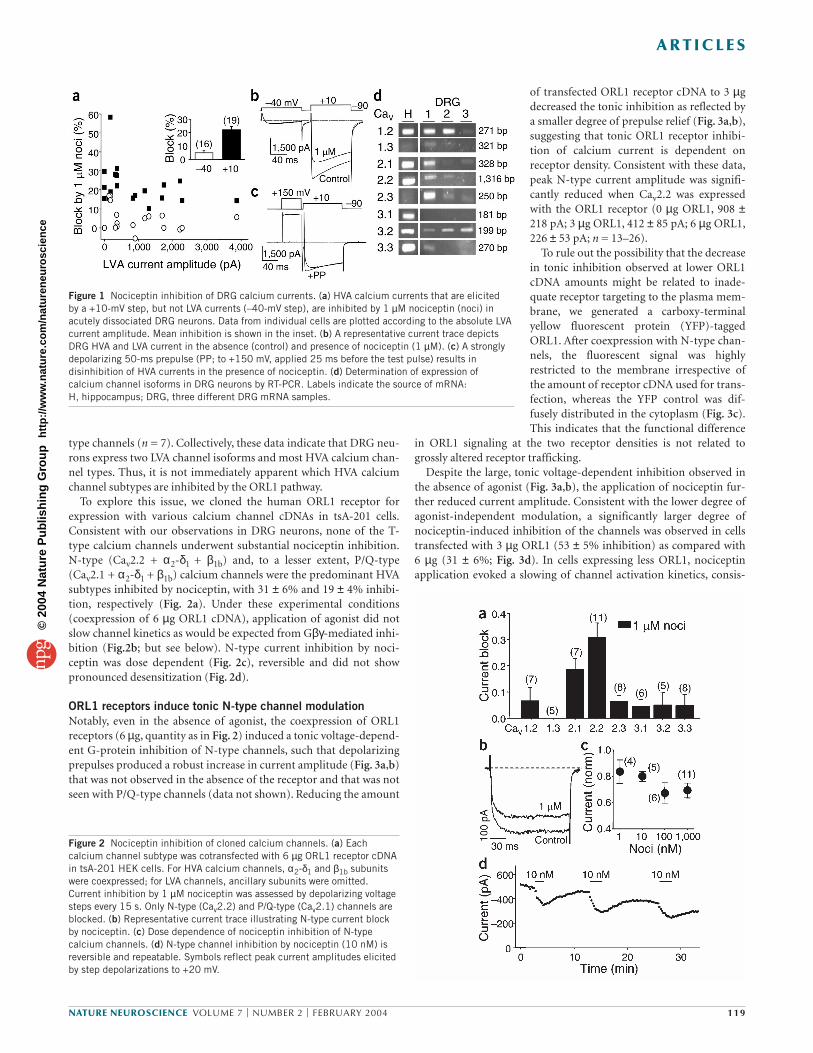
clamp. Robust HVA current (0.5 to >5 nA) was measured in all cellsexamined, whereas low-voltage activated (LVA) current ranged frombeing absent to nearly 3 nA. Application of 1 µM nociceptin inhibitedHVA current but had little effect on LVA current (Fig. 1a,b). Thistrend was consistent across a number of cells, which had an averageHVA current block of 22 ± 2%, whereas LVA current was virtuallyunchanged (mean LVA inhibition, 5 ± 2%; Fig. 1a). Nociceptin inhibi-tion of LVA calcium channels in DRG neurons may be increased incells expressing LVA current that is greater than 1 nA11. In our experi-ments, little if any LVA channel inhibition was apparent for currentsas large as 2 to 4 nA (Fig. 1a). The inhibition of HVA currents was par-tially reversed by application of a strong depolarizing prepulse (Fig. 1c), consistent with Gβγ-mediated voltage-dependent inhibi-tion13. Yet, the prepulse did not fully reverse the effects of nociceptin(Fig. 1c), perhaps indicating the presence of an additional voltage-independent mechanism, such as the activation of a cytoplasmic mes-senger cascade.
To determine which calcium channel subtypes contributed to thewhole-cell LVA and HVA currents, we carried out PCR after reversetranscription of RNA (RT-PCR) using whole DRG mRNA as a sub-strate. Cav1.2 (L-type channel), Cav2.1 (P/Q-type channel), Cav2.2(N-type channel), Cav2.3 (R-type channel), Cav3.2 (T-type channel),Cav3.3 (T-type channel) and Cav1.3 (L-type channel) transcripts weredetected in DRG neurons (Fig. 1d), but no Cav3.1 T-type calciumchannel transcript was present. Consistent with these data, applica-tion of the calcium channel blockers nifedipine, ω-conotoxin GVIAand ω-agatoxin IVA showed that the total HVA current comprised 16 ± 5% L-type, 63 ± 12% N-type, 17 ± 11% P/Q-type and 4 ± 3% R-
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
uren
euro
scie
nce
A R T I C L E S
NATURE NEUROSCIENCE VOLUME 7 | NUMBER 2 | FEBRUARY 2004 119
type channels (n = 7). Collectively, these data indicate that DRG neu-rons express two LVA channel isoforms and most HVA calcium chan-nel types. Thus, it is not immediately apparent which HVA calciumchannel subtypes are inhibited by the ORL1 pathway.
To explore this issue, we cloned the human ORL1 receptor forexpression with various calcium channel cDNAs in tsA-201 cells.Consistent with our observations in DRG neurons, none of the T-type calcium channels underwent substantial nociceptin inhibition.N-type (Cav2.2 + α2-δ1 + β1b) and, to a lesser extent, P/Q-type(Cav2.1 + α2-δ1 + β1b) calcium channels were the predominant HVAsubtypes inhibited by nociceptin, with 31 ± 6% and 19 ± 4% inhibi-tion, respectively (Fig. 2a). Under these experimental conditions(coexpression of 6 µg ORL1 cDNA), application of agonist did notslow channel kinetics as would be expected from Gβγ-mediated inhi-bition (Fig.2b; but see below). N-type current inhibition by noci-ceptin was dose dependent (Fig. 2c), reversible and did not showpronounced desensitization (Fig. 2d).
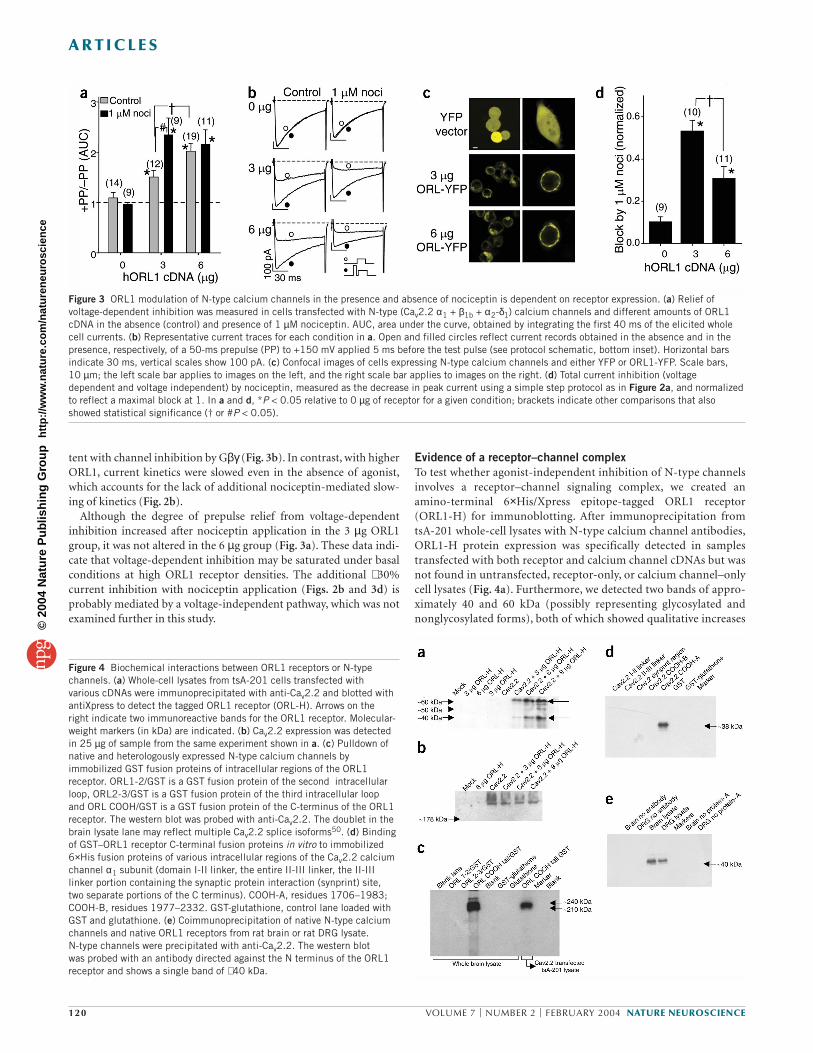
ORL1 receptors induce tonic N-type channel modulationNotably, even in the absence of agonist, the coexpression of ORL1receptors (6 µg, quantity as in Fig. 2) induced a tonic voltage-depend-ent G-protein inhibition of N-type channels, such that depolarizingprepulses produced a robust increase in current amplitude (Fig. 3a,b)that was not observed in the absence of the receptor and that was notseen with P/Q-type channels (data not shown). Reducing the amount
of transfected ORL1 receptor cDNA to 3 µgdecreased the tonic inhibition as reflected bya smaller degree of prepulse relief (Fig. 3a,b),suggesting that tonic ORL1 receptor inhibi-tion of calcium current is dependent onreceptor density. Consistent with these data,peak N-type current amplitude was signifi-cantly reduced when Cav2.2 was expressedwith the ORL1 receptor (0 µg ORL1, 908 ±218 pA; 3 µg ORL1, 412 ± 85 pA; 6 µg ORL1,226 ± 53 pA; n = 13–26).
To rule out the possibility that the decreasein tonic inhibition observed at lower ORL1cDNA amounts might be related to inade-quate receptor targeting to the plasma mem-brane, we generated a carboxy-terminalyellow fluorescent protein (YFP)-taggedORL1. After coexpression with N-type chan-nels, the fluorescent signal was highlyrestricted to the membrane irrespective ofthe amount of receptor cDNA used for trans-fection, whereas the YFP control was dif-fusely distributed in the cytoplasm (Fig. 3c).This indicates that the functional difference
in ORL1 signaling at the two receptor densities is not related togrossly altered receptor trafficking.
Despite the large, tonic voltage-dependent inhibition observed inthe absence of agonist (Fig. 3a,b), the application of nociceptin fur-ther reduced current amplitude. Consistent with the lower degree ofagonist-independent modulation, a significantly larger degree ofnociceptin-induced inhibition of the channels was observed in cellstransfected with 3 µg ORL1 (53 ± 5% inhibition) as compared with 6 µg (31 ± 6%; Fig. 3d). In cells expressing less ORL1, nociceptinapplication evoked a slowing of channel activation kinetics, consis-
Figure 1 Nociceptin inhibition of DRG calcium currents. (a) HVA calcium currents that are elicited by a +10-mV step, but not LVA currents (–40-mV step), are inhibited by 1 µM nociceptin (noci) inacutely dissociated DRG neurons. Data from individual cells are plotted according to the absolute LVAcurrent amplitude. Mean inhibition is shown in the inset. (b) A representative current trace depictsDRG HVA and LVA current in the absence (control) and presence of nociceptin (1 µM). (c) A stronglydepolarizing 50-ms prepulse (PP; to +150 mV, applied 25 ms before the test pulse) results indisinhibition of HVA currents in the presence of nociceptin. (d) Determination of expression ofcalcium channel isoforms in DRG neurons by RT-PCR. Labels indicate the source of mRNA: H, hippocampus; DRG, three different DRG mRNA samples.
Figure 2 Nociceptin inhibition of cloned calcium channels. (a) Eachcalcium channel subtype was cotransfected with 6 µg ORL1 receptor cDNAin tsA-201 HEK cells. For HVA calcium channels, α2-δ1 and β1b subunitswere coexpressed; for LVA channels, ancillary subunits were omitted.Current inhibition by 1 µM nociceptin was assessed by depolarizing voltagesteps every 15 s. Only N-type (Cav2.2) and P/Q-type (Cav2.1) channels areblocked. (b) Representative current trace illustrating N-type current blockby nociceptin. (c) Dose dependence of nociceptin inhibition of N-typecalcium channels. (d) N-type channel inhibition by nociceptin (10 nM) isreversible and repeatable. Symbols reflect peak current amplitudes elicitedby step depolarizations to +20 mV.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
uren
euro
scie
nce
A R T I C L E S
120 VOLUME 7 | NUMBER 2 | FEBRUARY 2004 NATURE NEUROSCIENCE
tent with channel inhibition by Gβγ(Fig. 3b). In contrast, with higherORL1, current kinetics were slowed even in the absence of agonist,which accounts for the lack of additional nociceptin-mediated slow-ing of kinetics (Fig. 2b).
Although the degree of prepulse relief from voltage-dependentinhibition increased after nociceptin application in the 3 µg ORL1group, it was not altered in the 6 µg group (Fig. 3a). These data indi-cate that voltage-dependent inhibition may be saturated under basalconditions at high ORL1 receptor densities. The additional ∼ 30%current inhibition with nociceptin application (Figs. 2b and 3d) isprobably mediated by a voltage-independent pathway, which was notexamined further in this study.
Evidence of a receptor–channel complexTo test whether agonist-independent inhibition of N-type channelsinvolves a receptor–channel signaling complex, we created anamino-terminal 6×His/Xpress epitope-tagged ORL1 receptor(ORL1-H) for immunoblotting. After immunoprecipitation fromtsA-201 whole-cell lysates with N-type calcium channel antibodies,ORL1-H protein expression was specifically detected in samplestransfected with both receptor and calcium channel cDNAs but wasnot found in untransfected, receptor-only, or calcium channel–onlycell lysates (Fig. 4a). Furthermore, we detected two bands of appro-ximately 40 and 60 kDa (possibly representing glycosylated andnonglycosylated forms), both of which showed qualitative increases
Figure 3 ORL1 modulation of N-type calcium channels in the presence and absence of nociceptin is dependent on receptor expression. (a) Relief ofvoltage-dependent inhibition was measured in cells transfected with N-type (Cav2.2 α1 + β1b + α2-δ1) calcium channels and different amounts of ORL1cDNA in the absence (control) and presence of 1 µM nociceptin. AUC, area under the curve, obtained by integrating the first 40 ms of the elicited wholecell currents. (b) Representative current traces for each condition in a. Open and filled circles reflect current records obtained in the absence and in thepresence, respectively, of a 50-ms prepulse (PP) to +150 mV applied 5 ms before the test pulse (see protocol schematic, bottom inset). Horizontal barsindicate 30 ms, vertical scales show 100 pA. (c) Confocal images of cells expressing N-type calcium channels and either YFP or ORL1-YFP. Scale bars, 10 µm; the left scale bar applies to images on the left, and the right scale bar applies to images on the right. (d) Total current inhibition (voltagedependent and voltage independent) by nociceptin, measured as the decrease in peak current using a simple step protocol as in Figure 2a, and normalizedto reflect a maximal block at 1. In a and d, *P < 0.05 relative to 0 µg of receptor for a given condition; brackets indicate other comparisons that alsoshowed statistical significance († or #P < 0.05).
Figure 4 Biochemical interactions between ORL1 receptors or N-typechannels. (a) Whole-cell lysates from tsA-201 cells transfected with various cDNAs were immunoprecipitated with anti-Cav2.2 and blotted withantiXpress to detect the tagged ORL1 receptor (ORL-H). Arrows on the right indicate two immunoreactive bands for the ORL1 receptor. Molecular-weight markers (in kDa) are indicated. (b) Cav2.2 expression was detectedin 25 µg of sample from the same experiment shown in a. (c) Pulldown ofnative and heterologously expressed N-type calcium channels byimmobilized GST fusion proteins of intracellular regions of the ORL1receptor. ORL1-2/GST is a GST fusion protein of the second intracellularloop, ORL2-3/GST is a GST fusion protein of the third intracellular loop and ORL COOH/GST is a GST fusion protein of the C-terminus of the ORL1receptor. The western blot was probed with anti-Cav2.2. The doublet in thebrain lysate lane may reflect multiple Cav2.2 splice isoforms50. (d) Bindingof GST–ORL1 receptor C-terminal fusion proteins in vitro to immobilized6×His fusion proteins of various intracellular regions of the Cav2.2 calciumchannel α1 subunit (domain I-II linker, the entire II-III linker, the II-IIIlinker portion containing the synaptic protein interaction (synprint) site, two separate portions of the C terminus). COOH-A, residues 1706–1983;COOH-B, residues 1977–2332. GST-glutathione, control lane loaded withGST and glutathione. (e) Coimmunoprecipitation of native N-type calciumchannels and native ORL1 receptors from rat brain or rat DRG lysate. N-type channels were precipitated with anti-Cav2.2. The western blot was probed with an antibody directed against the N terminus of the ORL1receptor and shows a single band of ∼ 40 kDa.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
uren
euro
scie
nce

A R T I C L E S
NATURE NEUROSCIENCE VOLUME 7 | NUMBER 2 | FEBRUARY 2004 121
in protein expression associated with increased quantities of trans-fected DNA. Western blot analysis (Fig. 4b) detected similaramounts of Cav2.2 protein in all cells transfected with the N-typechannel irrespective of ORL1 receptor density, indicating that dif-ferences in receptor amounts (Fig. 4a) are not due to variation in theexpression of the channel protein. Thus, the expression of N-typecalcium channel/ORL1-H complexes is regulated by the amount ofORL1 receptor cDNA used for transfection and, consequently,ORL1 mRNA levels.
To examine the biochemical basis for the formation of this signal-ing complex, we generated GST and 6×His fusion proteins of themajor intracellular regions of the ORL1 receptor and the Cav2.2 cal-cium channel α1 subunit, respectively. The C-terminal region, butnot the second or third intracellular loops, of the ORL1 receptorefficiently pulled down native N-type calcium channels from whole-brain extract, and from Cav2.2-transfected tsA-201 cell lysate (Fig. 4c). Moreover, the ORL1 receptor C terminus interacted invitro with the proximal C-terminal region of the Cav2.2 calciumchannel α1 subunit but not with the domain I–II linker, the domainII–III linker or the distal C-terminal region (Fig. 4d). Collectively,these data indicate that the formation of the N-type channel–ORL1receptor signaling complex is due to a physical interaction betweenthe C termini of these two proteins. To rule out the possibility thatthese observations might be due to an expression system artifact, we
created an antibody against the N-terminalregion of the ORL1 receptor for detection ofnative ORL1 receptors. Robust levels ofORL1 protein were detected after immuno-precipitation of N-type channels from ratbrain and rat DRG lysate (Fig. 4e), indicat-ing that complexes between ORL1 receptorsand N-type calcium channels are indeed aphysiological feature of neurons. Takentogether, our data indicate that N-type cal-cium channels and ORL1 receptors form aphysical signaling complex that may resultin the tonic inhibition of channel activity byG proteins.
Mechanisms of ORL1 receptor inhibitionIncubating cells with pertussis toxin (PTX)reduced agonist-independent inhibition,suggesting the involvement of Gα i/o sub-units (Fig. 5a). Subsequent application ofnociceptin did not result in any additional voltage-dependent inhibition, as no increasein prepulse relief could be observed (Fig. 5b). After cotransfection with the C-terminal fragment of the β-adrenergicreceptor kinase (βARK-ct), a Gβγ sink14,agonist-independent inhibition was signifi-cantly attenuated, albeit not completelyabolished. This indicates that although Gβγ-mediated signaling is involved in the tonicvoltage-dependent regulation, βARK-ctmay have incomplete access to all Gβγ sub-units in the signaling complex. After noci-ceptin application, the degree ofvoltage-dependent modulation did not dif-fer significantly from that in control cells(Fig. 5b), suggesting that βARK-ct was inef-
fective against agonist-induced voltage-dependent inhibition.Many G protein–coupled receptors, including the ORL1 receptor,
contain a highly conserved threonine residue in the second intracellu-lar loop. In 5-HT1A receptors, mutagenesis of this threonine uncou-ples receptor–Gβγ signaling with only minimal effects on Gαactivity15–17. A similar mutation to alanine in ORL1 receptors abol-ished both agonist-independent and agonist-dependent voltage-dependent inhibition of the channels without affecting membranetargeting (Fig. 5a–c), indicating that the second intracellular loop ofthe receptor may be a key determinant of the observed effects. Hence,Thr163 seems to be essential for ORL1 signaling to N-type channels,possibly by uncoupling receptor–G protein interactions.
We also examined the effects of PTX, βARK-ct and the alaninemutation on the voltage-independent component of ORL1 receptorsignaling (Fig. 5d). Treatment with PTX abolished nociceptin-mediated inhibition of channel activity irrespective of ORL1 receptorexpression. In contrast, when the voltage-dependent pathway was sat-urated (6 µg ORL1 cDNA), βARK-ct did not affect the additionalvoltage-independent nociceptin-mediated inhibition of the channel.These data indicate that voltage-independent inhibition is probablymediated by a Gαi/o pathway rather than by Gβγ. Nociceptin-mediated voltage-independent regulation of the channel was abol-ished in the T163A mutant, suggesting that Thr 163 is a key determi-nant of all ORL1 receptor signaling to N-type channels.
Figure 5 Determinants of ORL1 receptor modulation of N-type calcium channels expressed in tsA-201 cells. Relief from (a) agonist-independent and (b) nociceptin (1 µM)-induced voltage-dependentinhibition in the presence of wild-type ORL1 (black), wild-type ORL1 with overnight PTX treatment(500 ng/ml medium; white), wild-type ORL1 cotransfected with βARK-ct (blue) or mutant T163A-ORL1 (red). Representative current traces are also shown. (c) Confocal images of the YFP-taggedT163A-ORL1 mutant demonstrating appropriate expression and membrane targeting. Scale bar, 10µm. (d) Current block by 1 µM nociceptin measured using the standard step protocol. The coloring ofthe bars corresponds to that in a. Data were recorded and plotted and statistical significance isindicated as in Figure 3.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
uren
euro
scie
nce
A R T I C L E S
122 VOLUME 7 | NUMBER 2 | FEBRUARY 2004 NATURE NEUROSCIENCE
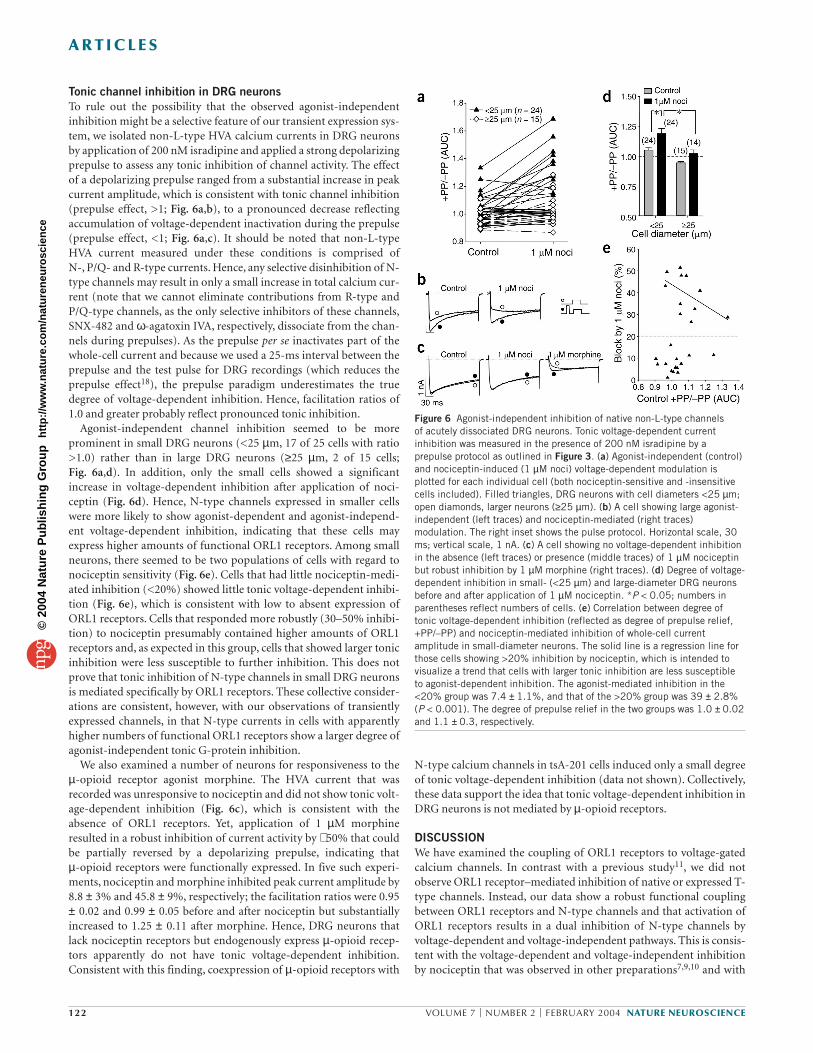
Tonic channel inhibition in DRG neuronsTo rule out the possibility that the observed agonist-independentinhibition might be a selective feature of our transient expression sys-tem, we isolated non-L-type HVA calcium currents in DRG neuronsby application of 200 nM isradipine and applied a strong depolarizingprepulse to assess any tonic inhibition of channel activity. The effectof a depolarizing prepulse ranged from a substantial increase in peakcurrent amplitude, which is consistent with tonic channel inhibition(prepulse effect, >1; Fig. 6a,b), to a pronounced decrease reflectingaccumulation of voltage-dependent inactivation during the prepulse(prepulse effect, <1; Fig. 6a,c). It should be noted that non-L-typeHVA current measured under these conditions is comprised ofN-, P/Q- and R-type currents. Hence, any selective disinhibition of N-type channels may result in only a small increase in total calcium cur-rent (note that we cannot eliminate contributions from R-type andP/Q-type channels, as the only selective inhibitors of these channels,SNX-482 and ω-agatoxin IVA, respectively, dissociate from the chan-nels during prepulses). As the prepulse per se inactivates part of thewhole-cell current and because we used a 25-ms interval between theprepulse and the test pulse for DRG recordings (which reduces theprepulse effect18), the prepulse paradigm underestimates the truedegree of voltage-dependent inhibition. Hence, facilitation ratios of1.0 and greater probably reflect pronounced tonic inhibition.
Agonist-independent channel inhibition seemed to be moreprominent in small DRG neurons (<25 µm, 17 of 25 cells with ratio>1.0) rather than in large DRG neurons (≥25 µm, 2 of 15 cells;Fig. 6a,d). In addition, only the small cells showed a significantincrease in voltage-dependent inhibition after application of noci-ceptin (Fig. 6d). Hence, N-type channels expressed in smaller cellswere more likely to show agonist-dependent and agonist-independ-ent voltage-dependent inhibition, indicating that these cells mayexpress higher amounts of functional ORL1 receptors. Among smallneurons, there seemed to be two populations of cells with regard tonociceptin sensitivity (Fig. 6e). Cells that had little nociceptin-medi-ated inhibition (<20%) showed little tonic voltage-dependent inhibi-tion (Fig. 6e), which is consistent with low to absent expression ofORL1 receptors. Cells that responded more robustly (30–50% inhibi-tion) to nociceptin presumably contained higher amounts of ORL1receptors and, as expected in this group, cells that showed larger tonicinhibition were less susceptible to further inhibition. This does notprove that tonic inhibition of N-type channels in small DRG neuronsis mediated specifically by ORL1 receptors. These collective consider-ations are consistent, however, with our observations of transientlyexpressed channels, in that N-type currents in cells with apparentlyhigher numbers of functional ORL1 receptors show a larger degree ofagonist-independent tonic G-protein inhibition.
We also examined a number of neurons for responsiveness to theµ-opioid receptor agonist morphine. The HVA current that wasrecorded was unresponsive to nociceptin and did not show tonic volt-age-dependent inhibition (Fig. 6c), which is consistent with theabsence of ORL1 receptors. Yet, application of 1 µM morphineresulted in a robust inhibition of current activity by ∼ 50% that couldbe partially reversed by a depolarizing prepulse, indicating that µ-opioid receptors were functionally expressed. In five such experi-ments, nociceptin and morphine inhibited peak current amplitude by8.8 ± 3% and 45.8 ± 9%, respectively; the facilitation ratios were 0.95± 0.02 and 0.99 ± 0.05 before and after nociceptin but substantiallyincreased to 1.25 ± 0.11 after morphine. Hence, DRG neurons thatlack nociceptin receptors but endogenously express µ-opioid recep-tors apparently do not have tonic voltage-dependent inhibition.Consistent with this finding, coexpression of µ-opioid receptors with
N-type calcium channels in tsA-201 cells induced only a small degreeof tonic voltage-dependent inhibition (data not shown). Collectively,these data support the idea that tonic voltage-dependent inhibition inDRG neurons is not mediated by µ-opioid receptors.
DISCUSSIONWe have examined the coupling of ORL1 receptors to voltage-gatedcalcium channels. In contrast with a previous study11, we did notobserve ORL1 receptor–mediated inhibition of native or expressed T-type channels. Instead, our data show a robust functional couplingbetween ORL1 receptors and N-type channels and that activation ofORL1 receptors results in a dual inhibition of N-type channels byvoltage-dependent and voltage-independent pathways. This is consis-tent with the voltage-dependent and voltage-independent inhibitionby nociceptin that was observed in other preparations7,9,10 and with
Figure 6 Agonist-independent inhibition of native non-L-type channels of acutely dissociated DRG neurons. Tonic voltage-dependent currentinhibition was measured in the presence of 200 nM isradipine by aprepulse protocol as outlined in Figure 3. (a) Agonist-independent (control)and nociceptin-induced (1 µM noci) voltage-dependent modulation isplotted for each individual cell (both nociceptin-sensitive and -insensitivecells included). Filled triangles, DRG neurons with cell diameters <25 µm;open diamonds, larger neurons (≥25 µm). (b) A cell showing large agonist-independent (left traces) and nociceptin-mediated (right traces)modulation. The right inset shows the pulse protocol. Horizontal scale, 30ms; vertical scale, 1 nA. (c) A cell showing no voltage-dependent inhibitionin the absence (left traces) or presence (middle traces) of 1 µM nociceptinbut robust inhibition by 1 µM morphine (right traces). (d) Degree of voltage-dependent inhibition in small- (<25 µm) and large-diameter DRG neuronsbefore and after application of 1 µM nociceptin. *P < 0.05; numbers inparentheses reflect numbers of cells. (e) Correlation between degree oftonic voltage-dependent inhibition (reflected as degree of prepulse relief,+PP/–PP) and nociceptin-mediated inhibition of whole-cell currentamplitude in small-diameter neurons. The solid line is a regression line forthose cells showing >20% inhibition by nociceptin, which is intended tovisualize a trend that cells with larger tonic inhibition are less susceptibleto agonist-dependent inhibition. The agonist-mediated inhibition in the<20% group was 7.4 ± 1.1%, and that of the >20% group was 39 ± 2.8%(P < 0.001). The degree of prepulse relief in the two groups was 1.0 ± 0.02and 1.1 ± 0.3, respectively.
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
uren
euro
scie
nce

A R T I C L E S
NATURE NEUROSCIENCE VOLUME 7 | NUMBER 2 | FEBRUARY 2004 123
inhibition of N-type currents by norepinephrine, GABAB and mus-carinic receptors19–22. The voltage-independent pathway may involvekinase activity22,23 but will need to be characterized in more detail. Incontrast, it is widely accepted that voltage-dependent inhibition of N-type channels is mediated by direct interactions with Gβγ sub-units23–27. The sensitivity of the tonic voltage-dependent inhibitionto PTX indicates that this Gβγ-mediated inhibition also requires theactivation of Gαi/o subunits. Thus, these data indicate that ORL1receptors may have a low level of constitutive activity in the absence ofagonist, as reported for numerous other types of G protein–coupledreceptors28.
The observation that ORL1 receptors and N-type channels form asignaling complex is consistent with an emerging view of macromol-ecular receptor–effector complexes that mediate the specificity ofreceptor signaling29,30 and suggests the following scenario. At highreceptor densities, a large fraction of channels are present in a com-plex with receptors. Within each complex, Gβγ molecules that disso-ciate from Gα subunits as a result of low amounts of constitutivereceptor activity effectively interact with N-type channels because oftheir spatial proximity. This would result in tonic inhibition of mostchannels, thus precluding any additional voltage-dependent inhibi-tion after agonist binding. This is consistent with the observation thatno further agonist-induced voltage-dependent inhibition could beobserved after coexpression of 6 µg ORL1 receptor cDNA. At lowreceptor density, most channels would not be complexed with ORL1receptors and thus would be unaffected by low amounts of constitu-tive receptor activity. Agonist application would sufficiently increasereceptor activity to inhibit noncomplexed channels. The observationthat the µ-opioid receptor did not show substantial agonist-independent inhibition of N-type channels in expression systems andin DRG neurons may reflect an absence of constitutive µ-opioidreceptor activity, or perhaps an inability to bind to N-type channels.The latter would be consistent with the notion that the C-terminalregion of the ORL1 receptor bears only little sequence homology toother members of the opioid receptor family.
Our model also accommodates our observations with PTX andβARK-ct. Because constitutive activity still involves G-proteinturnover, the application of PTX prevents the dissociation of Gβγsub-units, thus blocking agonist-independent inhibition. The coexpressionof βARK-ct directly interferes with Gβγsignaling, thus attenuating thevoltage-dependent inhibition. The finding that voltage-dependentinhibition was not completely blocked by βARK-ct may arise fromincomplete access to Gβγsubunits in the receptor–channel complex. Itis more difficult to assess to what extent voltage-independent inhibitionis affected by increased receptor density, because we have no way ofdetermining whether voltage-independent inhibition is already presentin the absence of agonist. Under conditions where voltage-dependentinhibition is saturated, however, application of nociceptin still medi-ated additional voltage-independent inhibition, consistent with oursuggestion that voltage-independent and voltage-dependent inhibitioninvolve separate mechanisms.
The role of Thr163 of the ORL1 receptor remains enigmatic. Theobservation that voltage-dependent inhibition was abolished in theT163A mutant is consistent with 5-HT receptor experiments in whichGβγ modulation of N-type channels was lost with mutation of thecorresponding threonine16,17. Although mutant 5-HT receptor con-tinues to partially couple to Gα subunits, agonist-dependent receptorsignaling was lost with T163A-ORL, indicating that the mutation maypreclude Gα activation.
Our observations of tonic voltage-dependent inhibition of HVAcurrents in rat DRG neurons contrast with findings in mouse trigem-
inal ganglion neurons12. In rat stellate ganglion neurons, which areresponsive to nociceptin but not to agonists of other opioid receptors,N-type channels do seem to have tonic, agonist-independent voltage-dependent inhibition (V. Ruiz-Velasco, personal communication).Therefore, tonic voltage-dependent inhibition of N-type channels byORL1 receptors may be dependent on tissue preparation or perhapsspecies.
As N-type channels are important in neurotransmitter release,their modulation profoundly influences the sensitivity of synapticcircuits to incoming stimuli. A number of studies demonstrate inhibi-tion of excitatory and/or inhibitory neurotransmission by nociceptinin the rat spinal cord and other brain structures31–34. Within thisframework, agonist-independent inhibition of N-type currents couldbe of multiple significance. High amounts of ORL1 receptor mRNA,but little nociceptin binding, are found within DRGs, indicating thatORL1 receptor protein may be localized to cellular processes outsidethe ganglion, which is in agreement with the notion that nociceptinmodulates transmission of centrally projecting small nociceptiveneurons12,35–37. Thus, higher ORL1 receptor density in a subpopula-tion of DRG neurons could mediate long-term synaptic depression ofpain sensation by tonic inhibition of calcium channel current and/ora dampening of sensitivity to neuromodulators like nociceptin. Thisis consistent with findings showing that ORL1–/– mice have greatersynaptic potentiation38. Second, it has been reported that ORL1receptor gene expression is increased after sciatic nerve ligation39, andan upregulation of nociceptin-binding sites in the spinal cord isobserved after peripheral inflammation or morphine tolerance40,41. Itis conceivable that agonist-independent receptor modulation of N-type channels could contribute to the development of tolerance inresponse to endogenous and/or exogenous receptor agonists by pre-venting agonist-induced regulation of N-type channel activity42,43.
In summary, our data offer evidence for a signaling complex ofORL1 receptors and N-type channels that results in a receptor den-sity–dependent, agonist-independent inhibition of these channels.This could provide a mechanism by which variation of ORL1 receptorexpression in response to pharmacological and/or physiological stim-uli may contribute to the regulation of nociceptive signaling.
METHODSDRG neuron preparation and mRNA extraction. Lumbar DRGs with attachedroots were dissected from adult Sprague-Dawley rats and were collected inNeurobasal A medium (Gibco BRL) with 10% heat-inactivated horse serum.DRGs were treated with 0.25% collagenase (Boehringer Mannheim) for two 90-min incubations, washed twice in PBS, treated with 0.25% trypsin for 30 min(Gibco BRL), washed three times in Neurobasal A/10% horse serum and takenup in 2 ml of Neurobasal A/B27 with 200 mM glutamine, 50 µg/ml DNAse andsoybean trypsin inhibitor (Sigma). Single-cell suspensions were obtained by sixto eight passages through a reduced fire-polished Pasteur pipette tip. Cells wereplated on dishes coated with 500 µg/ml polyornithine/5 µg/ml laminin. Patch-clamp recordings were carried out at room temperature 6–24 h after plating.
Adult rat DRGs (L4 and L5) and whole hippocampus were frozen in liquidnitrogen before RNA extraction (SNAP purification system; Invitrogen), andcDNA was synthesized from 1 µg of RNA. Additional reactions were done inthe absence of reverse transcriptase (not shown). PCR was carried out withTaq polymerase (Invitrogen) using 35 cycles of amplification (94 °C, 30 s;55 °C, 1 min; 72° C, 1.5 min). HVA primers have been reported previously44;LVA primers with amplicon sizes of 200–300 bp were as follows: rCaV3.1-F,GCTCACACCGTCGTCTGTC; rCaV3.1-R, CCAGAGGGGAGTGGGTTAGA;rCaV3.2-F, GCTGGCGTAAGAAGGTGGAT; rCaV3.2-R, CAACCTGTTGT-CACCAGCAC; rCaV3.3-F, CTGGTACCATGGGTACTGCC; rCaV3.3-R,CTGACGCAGCTACCTCCAC.
cDNA constructs. Rat α1A/Cav2.1, α1B/Cav2.2, α1C/Cav1.2, α1E/Cav2.3, α2-δ1
and β1b were donated by T. Snutch (University of British Columbia), rat
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
uren
euro
scie
nce

A R T I C L E S
124 VOLUME 7 | NUMBER 2 | FEBRUARY 2004 NATURE NEUROSCIENCE
α1D/Cav1.3 was a gift from J. Striessnig (University of Innsbruck) and humanα1H/Cav3.2 was provided by E. Perez-Reyes (University of Virginia). Humanα1G/Cav3.1, α1I/Cav3.3, βARK-ct pIRES and 6×His synprint constructs weredescribed previously45–48.
The ORL1 receptor was cloned from human cerebellum cDNA (Clontech)by PCR of two separate fragments. Both constructs were inserted into pGEM TEasy (Promega), sequenced (ABI Prism), joined by ligation and inserted intoPMT2SX. A mutant Thr-163-Ala receptor (T163A-ORL) was created byQuikChange Mutagenesis (Stratagene). C-terminal YFP-tagged ORL1 andT163A-ORL1 constructs were generated by in-frame insertion into pEYFP-N1(Clontech). For biochemical analyses, an ORL1 clone with a shortened 5′untranslated region was subcloned into pcDNA3.1/HisB (Invitrogen) to createan ORL1 receptor that was tagged at its N terminus with 6×His/Xpress Epitope(ORL1-H). Intracellular regions of ORL1 were PCR amplified and insertedinto pGEX4.1 (Stratagene). Intracellular loops of the Cav2.2 channel wereamplified by PCR and subcloned into pTrcHis (Invitrogen).
Cell culture and transient transfection. Culturing and transfection of tsA-201cells was carried out as described previously45. Green fluorescent protein (1.5 µg pEGFP; Clontech) was included as a marker in experiments lacking analternative fluorescent construct. For PTX experiments, cells were incubated inPTX (Sigma) at 500 ng/ml medium for 12–20 h before recordings were made.
Imaging and biochemistry. ORL1-YFP, T163A-YFP and YFP vectors weretransfected with the N-type channel, as described above. Cells were transferredinto 20 mM barium external solution, and YFP was imaged using a fluores-cence confocal microscope (Olympus) with Fluoview 3.3 software.
Immunoblot detection was carried out using transfected or mock-trans-fected tsA-201 cell lysates as follows. Plated HEK cells were washed twice withPBS and lysed in 0.5 ml of Triton X-100 lysis buffer: 300 mM NaCl; 50 mMTrisHCl, pH 7.5; 0.5% Triton X-100; 0.01 ml protease inhibitor cocktail(Sigma)/ml buffer. Cell lysate was iced for 30 min and microcentrifuged(10,000g). Total protein in the supernatant was quantified. Cell lysate (100 µg)was incubated overnight with Cav2.2 rabbit polyclonal IgG (anti-Cav2.2;Calbiochem). Precleared protein A–Sepharose 4 fast flow (AmershamBiosciences) was incubated with samples for 1 h, and slurries were washedwith buffer B (10 mM TrisHCl,pH 7.5; 0.2% NP40; 2 mM EDTA; 0.15 MNaCl) and with buffer C (10 mM TrisHCl, pH 7.5; 0.2% NP40; 2 mM EDTA;0.5 M NaCl). The immunoprecipitate (in 10 mM TrisHCl, pH 7.5) was pre-pared in sample buffer with 0.1 M DTT, separated by SDS–PAGE elec-trophoresis and transferred to Hybond ECL nitrocellulose membrane forimmunoblot analysis. Membranes were blocked in PBS/0.1% Tween 20 (PBS-T) with 5% milk prior to overnight incubation with 1:8,000 AntiXpress anti-body (Invitrogen). Membranes were washed, incubated with 1:1,000 sheepanti-mouse Ig for 30 min (Amersham Biosciences) and washed again beforesignal detection using ECL+ (Amersham Biosciences). Cell lysates (25 µg)were electrophoresed for standard western blotting with anti-Cav2.2 (1:200)and donkey anti-rabbit Ig (1:1,000; Amersham Biosciences).
For pulldown experiments, proteins were expressed in BL-21 bacteria.Whole Cav2.2 channel pulldowns were carried out using intracellular regionsof the ORL receptor, generated by PCR and expressed in a glutathione S-transferase (GST) vector (pGex4.1). GST fusion peptides were bound to glu-tathione Sepharose. Then either whole rat brain lysate or a lysate from tsA-201cells transfected with Cav2.2 was added. The sample was washed and elec-trophoresed by SDS–PAGE. Channel peptide was detected by western blottingwith anti-Cav2.2, according to the above protocols. The GST-ORL C-terminalpeptide was bound to glutathione followed by the addition of the differentintracellular regions of the calcium channels tagged with 6×His/Xpress. Thesamples were washed, electrophoresed through a 12% SDS–polyacrylamidegel and detected with an anti-Xpress antibody.
ORL/Cav2.2 coimmunoprecipitations from native tissue were carried out asdescribed previously49, using an ORL1-specific antibody that we generated.Briefly, a unique region (20 amino acid residues) from the rat ORL N terminuswas synthesized, linked to Mariculture keyhole limpet hemocyanin (PierceBiotech) and injected into rabbits for antibody production.
Electrophysiology and data analysis. The external recording solution forDRGs contained 1 mM CaCl2, 10 mM HEPES, 160 mM TEACl and 10 mMglucose, pH 7.4 (with 200 nM isradipine for Fig. 6 only). For tsA-201 cells, it
had 20 mM BaCl2, 1 mM MgCl2, 10 mM HEPES, 40 mM TEACl, 10 mM glu-cose, 65 mM CsCl (HVA) or 2 mM BaCl2, 1 mM MgCl2, 10 mM HEPES,40 mM TEACl, 10 mM glucose and 105 mM CsCl (LVA). Borosilicate glasspipettes (2–4 MΩ) were filled with internal solution containing 110 mM CsCl,3 mM MgCl2, 10 mM EGTA, 10 mM HEPES, 3 mM MgATP and 0.6 mM GTP,pH 7.2 (for DRGs) or 108 mM CsMeSO4, 4 mM MgCl2, 9 mM EGTA, 9 mMHEPES, 2.6 mM MgATP and 0.6 mM LiGTP, pH to 7.2 (for tsA-201 cells).Nociceptin (Calbiochem) was prepared daily in external solution from 1 mMfrozen stocks and was applied to cells by microperfusion. Currents wereelicited from a holding potential of –90 mV (DRGs) or –100 mV (tsA-201 cells). Voltage-dependent inhibition was assessed by integrating cur-rents during the first 40 ms of a +20-mV test pulse with and without priorapplication of a 50-ms prepulse to +150 mV. Data were plotted as mean ±s.e.m.; statistical difference (P < 0.05) was determined using paired t-tests orone-way analysis of variance.
ACKNOWLEDGMENTSThis work was supported by an operating grant to G.W.Z. from the CanadianInstitutes of Health Research (CIHR). G.W.Z. is a CIHR Investigator and anAlberta Heritage Foundation for Medical Research (AHFMR) senior scholar.A.M.B. held studentship awards from AHFMR and the Natural Sciences andEngineering Research Council. C.J.D. is the recipient of an AHFMR studentship,C.A. holds postdoctoral fellowships from the AHFMR and the Heart and StrokeFoundation of Canada. E.B. is supported by the IUD foundation and the PICSprogram of the CNRS.
COMPETING INTERESTS STATEMENTThe authors declare that they have no competing financial interests.
Received 24 November; accepted 18 December 2003Published online at http://www.nature.com/natureneuroscience/
1. Beedle, A.M. & Zamponi, G.W. Modulation of high voltage–activated calcium chan-nels by G protein–coupled receptors. in Calcium Channel Pharmacology (ed.,McDonough, S.) (Kluwer Academic/Plenum Publishing, New York, 2003).
2. Bunzow, J.R. et al. Molecular cloning and tissue distribution of a putative memberof the rat opioid receptor gene family that is not a mu, delta or kappa opioid recep-tor type. FEBS Lett. 347, 284–288 (1994).
3. Bertorelli, R. et al. Nociceptin and the ORL-1 ligand [Phe1psi (CH2-NH)Gly2]noci-ceptin(1-13)NH2 exert anti-opioid effects in the Freund’s adjuvant-induced arthriticrat model of chronic pain. Br. J. Pharmacol. 128, 1252–1258 (1999).
4. Ahmadi, S., Liebel, J.T. & Zeilhofer, H.U. The role of the ORL1 receptor in the mod-ulation of spinal neurotransmission by nociceptin/orphanin FQ and nocistatin. Eur.J. Pharmacol. 412, 39–44 (2001).
5. Mollereau, C., Moisand, C., Butour, J.L., Parmentier, M. & Meunier, J.C.Replacement of Gln280 by His in TM6 of the human ORL1 receptor increases affin-ity but reduces intrinsic activity of opioids. FEBS Lett. 395, 17–21 (1996).
6. Xu, X., Hao, J.W. & Wiesenfeld-Hallin, Z. Nociceptin of antinociceptin: potent spinalantinociceptive effect of orphanin FQ/nociceptin in the rat. Neuroreport 7,2092–2094 (1996).
7. Knoflach, F., Reinscheid, R.K., Civelli, O. & Kemp, J.A. Modulation of voltage-gatedcalcium channels by orphanin FQ in freshly dissociated hippocampal neurons. J. Neurosci. 16, 6657–6664 (1996).
8. Connor, M. & Christie, M.J. Modulation of Ca2+ channel currents of acutely dissoci-ated rat periaqueductal grey neurons. J. Physiol. 15, 47–58 (1998).
9. Larsson, K.P., Olsen, U. B. & Hansen, A.J. Nociceptin is a potent inhibitor of N-typeCa2+ channels in rat sympathetic ganglion neurons. Neurosci. Lett. 296, 121–124(2000).
10. Morikawa, H. et al. Nociceptin receptor-mediated Ca2+ channel inhibition and itsdesensitization in NG108-15 cells. Eur. J. Pharmacol. 351, 247–252 (1998).
11. Abdulla, F.A. & Smith, P.A. Nociceptin inhibits T-type Ca2+ channel current in ratsensory neurons by a G-protein-independent mechanism. J. Neurosci. 17,8721–8728 (1997).
12. Borgland, S.L., Connor, M. & Christie, M.J. Nociceptin inhibits calcium channel cur-rents in a subpopulation of small nociceptive trigeminal ganglion neurons in mouse.J. Physiol. 536, 35–47 (2001).
13. Zamponi, G.W. Determinants of G protein inhibition of presynaptic calcium chan-nels. Cell. Biochem. Biophys. 34, 79–94 (2001).
14. Koch, W.J., Hawes, B.E., Inglese, J., Luttrell, L.M. & Lefkowitz, R.J. Cellular expres-sion of the carboxyl terminus of a G protein-coupled receptor kinase attenuates Gbeta gamma-mediated signalling. J. Biol. Chem. 269, 6193–6197 (1994).
15. Albert, P.R., Morris, S.J., Ghahremani, M.H., Storring, J.M. & Lembo, P.M. A puta-tive alpha-helical G beta gamma-coupling domain in the second intracellular loop ofthe 5-HT1A receptor. Ann. NY Acad. Sci. 861, 146–161 (1998).
16. Lembo, P.M., Ghahremani, M.H., Morris, S.J. & Albert, P.R. A conserved threonineresidue in the second intracellular loop of the 5–hydroxytryptamine 1A receptordirects signalling specificity. Mol. Pharmacol. 52, 164–171 (1997).
17. Wu, X., Kushwaha, N., Albert, P.R. & Penington, N.J. A critical protein kinase C
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
uren
euro
scie
nce

A R T I C L E S
NATURE NEUROSCIENCE VOLUME 7 | NUMBER 2 | FEBRUARY 2004 125
phosphorylation site on the 5-HT(1A) receptor controlling coupling to N-type calcium channels. J. Physiol. 538, 41–51 (2002).
18. Arnot, M.I., Stotz, S.C., Jarvis, S.E. & Zamponi, G.W. Differential modulation of N-type 1B and P/Q-type 1A calcium channels by different G protein subunit isoforms.J. Physiol. 527, 203–212 (2000).
19. Beech, D.J., Bernheim, L. & Hille, B. Pertussis toxin and voltage dependence dis-tinguish multiple pathways modulating calcium channels of rat sympthetic neurons.Neuron 8, 90–106 (1992).
20. Luebke, J.I. & Dunlap, K.I. Sensory neuron N-type calcium currents are inhibited byboth voltage-dependent and -independent mechnaisms. Pflugers Arch. 428,499–507 (1994).
21. Kammermeier, P.J., Ruiz-Valesco, V. & Ikeda, S.R. A voltage-independent calciumcurrent inhibitory pathway activated by muscarinic agonists in rat sympathetic neu-rons requires both Gαq/11 and Gβγ. J. Neurosci. 20, 5623–5629 (2000).
22. Schiff, M.L. et al. Tyrsoine-kinase-dependent recruitment of RGS12 to the N-typecalcium channel. Nature 408, 723–727 (2000).
23. Diverse-Pieluissi, M., Goldsmith, P.K.& Dunlap, K. Transmitter-mediated inhibitionof N-type calcium channels in sensory neurons involves multiple GTP-binding pro-teins and subunits. Neuron 14, 191–200 (1995).
24. De Waard, M. et al. Direct binding of G-protein betagamma complex to voltage-dependent calcium channels. Nature 385, 446–450 (1997).
25. Herlitze, S. et al. Modulation of Ca2+ channels by G protein βγ subunits. Nature380, 258–262 (1996).
26. Ikeda, S.R. Voltage-dependent modulation of N-type calcium channels by G-proteinβγ subunits. Nature 380, 255–258 (1996).
27. Zamponi, G.W., Bourinet, E., Nelson, D., Nargeot, J. & Snutch, T.P. Crosstalkbetween G proteins and protein kinase C mediated by the calcium channel alpha1subunit. Nature 385, 442–446 (1997).
28. Seifert, R. & Wenzel-Seifert, K. Constitutive activity of G protein-coupled receptors:cause of disease and common property of wild-type receptors. NauynSchmiedebergs Arch. Pharmacol. 366, 381–416 (2002).
29. Davare, M.A. et al. A beta2 adrenergic receptor signalling complex assembled withthe Ca2+ channel Cav1.2. Science 293, 98–101 (2001).
30. Lavine, N. et al. G protein-coupled receptors form stable complexes with inwardlyrectifying potassium channels and adenylyl cyclase. J. Biol. Chem. 277,46010–46019 (2002).
31. Emmerson, P.J. & Miller, R.J. Pre- and postsynaptic actions of opioid and orphanopioid agonists in the rat arcuate nucleus and ventromedial hypothalamus in vitro. J. Physiol. 517, 431–445 (1999).
32. Meis, S. & Pape, H.C. Control of glutamate and GABA release bynociceptin/orphanin FQ in the rat lateral amygdala. J. Physiol. 532, 701–712(2001).
33. Tallent, M.K., Madamba, S.G. & Siggins, G.R. Nociceptin reduces epileptiformevents in CA3 hippocampus via presynaptic and postsynaptic mechanisms. J. Neurosci. 21, 6940–6948 (2001).
34. Venkatesan, P., Wang, J., Evans, C., Irnaten, M. & Mendelowitz, D. Nociceptininhibits gamma-aminobutyric acidergic inputs to cardiac parasympathetic neurons
in the nucleus ambiguus. J. Pharmacol. Exp. Ther. 300, 78–82 (2002).35. Monteillet-Agius, G., Fein, J., Anton, B. & Evans, C.J. ORL-1 and mu opioid recep-
tor antisera label different fibers in areas involved in pain processing. J. Comp.Neurol. 399, 373–383 (1998).
36. Neal, C.R. et al. Localization of orphanin FQ (nociceptin) peptide and messengerRNA in the central nervous system of the rat. J. Comp. Neurol. 406, 503–547(1999).
37. Wick, M.J. et al. Isolation of a novel cDNA encoding a putative membrane receptorwith high homology to the cloned mu, delta, and kappa opioid receptors. Brain Res.Mol. Brain Res. 27, 37–44 (1994).
38. Manabe, T. et al. Facilitation of long-term potentiation and memory in mice lackingnociceptin receptors. Nature 394, 577–581 (1998).
39. Briscini, L., Corradini, L., Ongini, E. & Bertorelli, R. Up-regulation of ORL-1 recep-tors in spinal tissue of allodynic rats after sciatic nerve injury. Eur. J. Pharmacol.447, 59–65 (2002).
40. Gouarderes, C., Tafani, J.A., Meunier, J., Jhamandas, K. & Zajac, J. Nociceptinreceptors in the rat spinal cord during morphine tolerance. Brain Res. 838, 85–94(1999).
41. Jia, Y., Linden, D.R., Serie, J.R. & Seybold, V. S. Nociceptin/orphanin FQ bindingincreases in superficial laminae of the rat spinal cord during persistent peripheralinflammation. Neurosci. Lett. 250, 21–24 (1998).
42. Kest, B. et al. Morphine tolerance and dependence in nociceptin/orphanin FQ trans-genic knock-out mice. Neurosci. 104, 217–222 (2001).
43. Ueda, H., Inoue, M., Takeshima, H. & Iwasawa, Y. Enhanced spinal nociceptinreceptor expression develops morphine tolerance and dependence. J. Neurosci. 20,7640–7647 (2000).
44. Latour, I., Hamid, J., Beedle, A.M., Zamponi, G.W. & Macvicar, B.A. Expression ofvoltage-gated Ca2+ channel subtypes in cultured astrocytes. Glia 41, 347–353(2003).
45. Beedle, A.M., Hamid, J. & Zamponi, G.W. Inhibition of transiently expressed low-and high-voltage-activated calcium channels by trivalent metal cations. J. Membr.Biol. 187, 225–238 (2002).
46. Monteil, A. et al. Specific properties of T-type calcium channels generated by thehuman alpha 1I subunit. J. Biol. Chem. 275, 16530–16535 (2000).
47. Magga, J.M., Jarvis, S.E., Arnot, M.I., Zamponi, G.W. & Braun, J.E. Cysteine stringprotein regulates G protein modulation of N-type calcium channels. Neuron 28,195–204 (2000).
48. Jarvis, S.E., Magga, J.M., Beedle, A.M., Braun, J.E. & Zamponi, G.W. G proteinmodulation of N-type calcium channels is facilitated by physical interactionsbetween syntaxin 1A and Gbetagamma. J. Biol. Chem. 275, 6388–6394 (2000).
49. Ouardouz, M. et al. Depolarization-induced Ca2+ release in ischemic spinal cordwhite matter involves L-type Ca2+ channel activation of ryanodine receptors. Neuron40, 56–63 (2003).
50. Kaneko, S. et al. Identification and characterization of novel human Cav2.2(alpha1B) calcium channel variants lacking the synaptic protein interaction site. J. Neurosci. 22, 82–92 (2002).
©20
04 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
uren
euro
scie
nce
Copyright © 2022 FDOKUMEN




















