Impact of testosterone on cardiac L-type calcium channels and Ca 2+ sparks: Acute actions antagonize...
11
Cell Calcium 41 (2007) 467–477 Impact of testosterone on cardiac L-type calcium channels and Ca 2+ sparks: Acute actions antagonize chronic effects Fikret Er a , Guido Michels a , Mathias C. Brandt a , Ismail Khan a , Hannelore Haase b , Michael Eicks a , Michael Lindner a , Uta C. Hoppe a,c,∗ a Department of Internal Medicine III, University of Cologne, Kerpener Str. 62, 50937 Cologne, Germany b Max Delbruck Center for Molecular Medicine (MDC), Berlin c Center for Molecular Medicine Cologne (CMMC), University of Cologne, Germany Received 24 July 2006; received in revised form 29 August 2006; accepted 8 September 2006 Available online 7 November 2006 Abstract While androgens generally have been associated with an increased cardiovascular risk, recent studies indicate potential beneficial acute effects of testosterone. However, detailed evaluation of chronic and acute actions of testosterone on the function of cardiac I Ca,L and intracellular Ca 2+ handling is limited. To clarify this situation we performed whole-cell and single-channel analysis of I Ca,L , recordings of Ca 2+ sparks, mea- surements of contractility and quantitative real-time RT-PCR in rat cardiomyocytes following testosterone pretreatment and acute testosterone application. Pretreatment with testosterone 100 nM for 24–30 h increased whole-cell I Ca,L from 3.8 ± 0.8 pA/pF (n = 10) to 10.1 ± 0.31 pA/pF (n = 9) at +10 mV (p < 0.001). Increase of I Ca,L density was caused by both, increased expression levels of the alpha 1C subunit of L-type calcium channel and a pronounced increment of the single-channel activity (availability 81.8 ± 3.15% versus 37.1 ± 7.01%; open probability 12.8 ± 3.09% versus 1.0 ± 0.62%, p < 0.01). Moreover, testosterone pretreatment significantly increased the frequency of Ca 2+ sparks and improved myocytes contractility without altering SR Ca 2+ load. All chronic effects could be inhibited by flutamide. In contrast acute testos- terone administration significantly reduced I Ca,L density. Indeed, on the single-channel level acute testosterone application completely reversed the chronic testosterone-mediated effects, and antagonized the chronic testosterone effects on Ca 2+ spark frequency, which was unaffected by flutamide. Thus, testosterone pretreatment activates I Ca,L via nuclear receptor-mediated pathways, while testosterone acutely blocks I Ca,L in a direct manner. Thus, testosterone chronically affects the basal level of intracellular Ca 2+ handling, which in addition rapidly may be modulated by acute changes of hormone levels. © 2006 Elsevier Ltd. All rights reserved. Keywords: Ion channels; Hormones; Ca channel; Ca sparks; Electrophysiology; Myocytes 1. Introduction Epidemiological and clinical evidence indicates that gender and sex hormone status influence cardiovascular physiology and pathophysiology. Even when corrections are made for sex-dependent differences in risk factors, cardiovascular mortality and the incidence of coronary heart ∗ Corresponding author at: Department of Internal Medicine III, University of Cologne, Kerpener Str. 62, 50937 Cologne, Germany. Tel.: +49 221 4786465; fax: +49 221 4787929. E-mail address: [email protected] (U.C. Hoppe). disease is approximately twice as prevalent in middle-aged men as it is in premenopausal women of similar age [1]. These findings led many to conclude that estrogens attenuate and, conversely, testosterone exacerbates the incidence and severity of cardiovascular disease. However, recent clinical studies have failed to support the concept that estrogens are beneficial to the cardiovascular system, while the effects of testosterone remain poorly understood [2,3]. Androgens have been associated with an increased cardiovascular risk by adversely affecting the plasma lipid and lipoprotein profile, thrombosis and suspected proatherogenic effects [1,4,5]. However, beside these genetic, mainly testosterone 0143-4160/$ – see front matter © 2006 Elsevier Ltd. All rights reserved. doi:10.1016/j.ceca.2006.09.003
Transcript of Impact of testosterone on cardiac L-type calcium channels and Ca 2+ sparks: Acute actions antagonize...

A
eCsa(c1ittfldb©
K
1
gpac
oT
0d
Cell Calcium 41 (2007) 467–477
Impact of testosterone on cardiac L-type calcium channels and Ca2+
sparks: Acute actions antagonize chronic effects
Fikret Er a, Guido Michels a, Mathias C. Brandt a, Ismail Khan a, Hannelore Haase b,Michael Eicks a, Michael Lindner a, Uta C. Hoppe a,c,∗
a Department of Internal Medicine III, University of Cologne, Kerpener Str. 62, 50937 Cologne, Germanyb Max Delbruck Center for Molecular Medicine (MDC), Berlin
c Center for Molecular Medicine Cologne (CMMC), University of Cologne, Germany
Received 24 July 2006; received in revised form 29 August 2006; accepted 8 September 2006Available online 7 November 2006
bstract
While androgens generally have been associated with an increased cardiovascular risk, recent studies indicate potential beneficial acuteffects of testosterone. However, detailed evaluation of chronic and acute actions of testosterone on the function of cardiac ICa,L and intracellulara2+ handling is limited. To clarify this situation we performed whole-cell and single-channel analysis of ICa,L, recordings of Ca2+ sparks, mea-
urements of contractility and quantitative real-time RT-PCR in rat cardiomyocytes following testosterone pretreatment and acute testosteronepplication. Pretreatment with testosterone 100 nM for 24–30 h increased whole-cell ICa,L from 3.8 ± 0.8 pA/pF (n = 10) to 10.1 ± 0.31 pA/pFn = 9) at +10 mV (p < 0.001). Increase of ICa,L density was caused by both, increased expression levels of the alpha 1C subunit of L-typealcium channel and a pronounced increment of the single-channel activity (availability 81.8 ± 3.15% versus 37.1 ± 7.01%; open probability2.8 ± 3.09% versus 1.0 ± 0.62%, p < 0.01). Moreover, testosterone pretreatment significantly increased the frequency of Ca2+ sparks andmproved myocytes contractility without altering SR Ca2+ load. All chronic effects could be inhibited by flutamide. In contrast acute testos-erone administration significantly reduced ICa,L density. Indeed, on the single-channel level acute testosterone application completely reversedhe chronic testosterone-mediated effects, and antagonized the chronic testosterone effects on Ca2+ spark frequency, which was unaffected by
utamide. Thus, testosterone pretreatment activates ICa,L via nuclear receptor-mediated pathways, while testosterone acutely blocks ICa,L in airect manner. Thus, testosterone chronically affects the basal level of intracellular Ca2+ handling, which in addition rapidly may be modulatedy acute changes of hormone levels.2006 Elsevier Ltd. All rights reserved.
ogy; M
dmTa
eywords: Ion channels; Hormones; Ca channel; Ca sparks; Electrophysiol
. Introduction
Epidemiological and clinical evidence indicates thatender and sex hormone status influence cardiovascular
hysiology and pathophysiology. Even when correctionsre made for sex-dependent differences in risk factors,ardiovascular mortality and the incidence of coronary heart∗ Corresponding author at: Department of Internal Medicine III, Universityf Cologne, Kerpener Str. 62, 50937 Cologne, Germany.el.: +49 221 4786465; fax: +49 221 4787929.
E-mail address: [email protected] (U.C. Hoppe).
ssbohbp[
143-4160/$ – see front matter © 2006 Elsevier Ltd. All rights reserved.oi:10.1016/j.ceca.2006.09.003
yocytes
isease is approximately twice as prevalent in middle-ageden as it is in premenopausal women of similar age [1].hese findings led many to conclude that estrogens attenuatend, conversely, testosterone exacerbates the incidence andeverity of cardiovascular disease. However, recent clinicaltudies have failed to support the concept that estrogens areeneficial to the cardiovascular system, while the effectsf testosterone remain poorly understood [2,3]. Androgens
ave been associated with an increased cardiovascular risky adversely affecting the plasma lipid and lipoproteinrofile, thrombosis and suspected proatherogenic effects1,4,5]. However, beside these genetic, mainly testosterone
4 lcium 4
riais
oa[amHtooadfpMt[tsetiasfacc
2
UI1
2
SdpetS3a2tm
2
2c
mi27tpa1D6
2c
ct−mTtHpwB1m1uta>sOcp(aaIocSsocri
68 F. Er et al. / Cell Ca
eceptor-mediated pathways recent experimental datandicated another, genomic independent way of testosteronection on the cardiovascular system, i.e. short-term admin-stration of testosterone acutely induces vasodilation in theystemic, coronary and pulmonary vascular beds [6,7].
While previous studies mainly examined androgen effectsn the vascular system, we recently demonstrated also ancute direct effect of testosterone on cardiac myocytes8]: testosterone directly and acutely protected cardiocytesgainst ischemic injury by opening KATP channels of the inneritochondrial membrane in isolated cells and mitoplasts.owever, before drawing any clinical consequences from
hese initial findings of potential beneficial androgen actionsn the myocardium further detailed experimental evaluationsf acute and chronic testosterone effects on cardiac functionnd arrhythmogenesis are still warranted. In the heart, car-iac L-type calcium channels (ICa,L) play an important roleor the intracellular calcium homeostasis, for cardiac actionotential duration and thus the induction of arrhythmias.oreover, alterations of ICa,L are believed to be involved in
he development and progression of structural heart diseases9,10]. While chronic testosterone exposure has been showno increase L-type calcium channel expression in coronarymooth muscle cells [11] and myocardium [12,13] detailedvaluation of chronic and acute effects of testosterone onhe function of cardiac ICa,L and intracellular Ca2+ handlings limited. To clarify this situation we performed whole-cellnd single-channel analysis of ICa,L, recordings of calciumparks and measurements of contractility in cardiomyocytesollowing testosterone pretreatment and acute testosteronepplication. Interestingly, we obtained distinct acute versushronic testosterone effects on ICa,L whole-cell and single-hannel properties and calcium handling.
. Methods
The investigation conforms to the Guide for the Care andse of Laboratory Animals published by the US National
nstitutes of Health (NIH Publication No. 85-23, revised996).
.1. Myocyte isolation
Single adult ventricular cells were isolated from femaleprague–Dawley rats (weight, 200–250 g) by enzymaticigestion, as previously described [8]. Dissociated cells werelated for primary culture on laminin-coated (Sigma) cov-rslips. A standard trypsin dissociation method was usedo prepare ventricular myocytes of 1–2-day-old neonatalprague–Dawley rats [14]. For patch-clamp experiments,–5-day-old monolayer cultures were dispersed by trypsin,
nd re-plated at a low density to study isolated cells within–8 h. Eighteen to 24 h before patch-clamp experimentshe culture medium was exchanged against a serum-freeedium.
orbl
1 (2007) 467–477
.2. Electrophysiology
.2.1. Patch-clamp recordings in the whole-cellonfiguration
Whole-cell Ca2+ currents were recorded using standardicroelectrode patch-clamp techniques [15,16]. The record-
ng bath solution was composed of (mM): TEA-Cl 136, CaCl2, MgCl2 2, HEPES 25 and glucose 20, pH was adjusted to.4 with TEA-OH. The corresponding pipette solution con-ained (mM): CsCl 130, EGTA 10, Mg-ATP 2, HEPES 25,H was adjusted to 7.2 with CsOH. Currents were recordednd digitized with an Axopatch 200B amplifier and Digidata200 interface (Axon Instruments) by using custom software.ata were not corrected for the liquid junction potential of.7 mV.
.2.2. Patch-clamp recordings of single channels in theell-attached configuration
Single calcium channels were recorded in cell-attachedonfiguration of the patch-clamp technique (depolarizingest pulses of 150 ms duration at 1.67 Hz, holding potential
80 mV, test potential +20 mV). Single-channel measure-ents and analysis were done as previously reported [8,9,17].he external solution for single-channel experiments con-
ained (mM): potassium glutamate 120, KCl 25, MgCl2 2,EPES 10, EGTA 2, CaCl2 1, Na-ATP 1 and dextrose 10,H was adjusted to 7.4 with KOH. This high-K+ solutionas used to achieve a resting membrane potential of zero.orosilicate pipettes (5–8 M�) were filled with (mM) BaCl210 and HEPES 10, pH of 7.4 was adjusted with tetraethylam-onium hydroxide. An Axopatch 200B amplifier, Digidata
200 interface (Axon Instruments) and custom software weresed for pulse generation, data acquisition (10 kHz) and fil-ering (2 kHz, −3 dB, 4-pole Bessel filter). Experiments werenalysed whenever the channel activity persisted for at least120 sweeps. Linear leak and capacity currents were digitallyubtracted using the average currents of non-active sweeps.penings and closures were identified by the half-height
riterion. The open probability (defined as the relative occu-ancy of the open state during active sweeps), the availabilityfraction of sweeps containing at least one channel opening)nd Ipeak (the peak ensemble average current, obtained visu-lly) were calculated from single- and multichannel patches.n the latter case, they were corrected for n, the numberf channels in the patch. n was defined as the maximumurrent amplitude observed, divided by the unitary current.ingle-channel amplitudes were determined by direct mea-urements of fully resolved openings or as the maximumf Gaussian fits on amplitude histograms. Peak current wasorrected by division through n. The availability was cor-ected by the square root method: (1 − availabilitycorrected)s the nth root of (1 − availabilityuncorrected). The corrected
pen probability was calculated on the basis of the cor-ected number of active sweeps, i.e. total open time dividedy (n × availabilitycorrected × number of test pulses × pulseength). Closed-time and first-latency analyses were carried
lcium 4
oamta
2a
cillt3ssEtesottbwcmtegTa
2
pmmmtimcp4>(woOaap
esflgtob
itifrv
2
cswRRffDcfACS(cAAQi2aaomsr
2
awcb
F. Er et al. / Cell Ca
ut only in one-channel patches. Time constants of open-nd closed-time histograms (τopen, τclosed) were obtained byaximum likelihood estimation (PStat software). The inac-
ivation (%) was determined by the term of the ensembleverage current (1 − (Iminimal/Ipeak)).
.3. Confocal quantification of myocyte contractilitynd calcium transient recordings
Images were obtained with a Leica TCS SP2 confo-al laser-scanning microscope equipped with a DM IRE2nverted fluorescence microscope and argon/helium–neonasers at room temperature 21–22 ◦C. Myocytes wereoaded with 10 �M fluorescent Ca2+ indicator Fluo-3 (ace-oxymethylester, Molecular Probes, Eugene, OR, USA) by0 min incubation at 37 ◦C. Contraction and calcium tran-ients of cardiomyocytes were induced by electrical fieldtimulation at 0.5 Hz with 2 ms square wave bipolar pulses.ach experiment was preceded by a run-in phase with con-
inuous field stimulation at 0.5 Hz. Fluo-3 fluorescence wasxcited with the 488 nm line of an argon laser and emis-ion was recorded at >515 nm. The degree of contractilityf cardiomyocytes was defined as the difference betweenhe lengths of fully diastolic relaxation (rel) and fully sys-olic contraction (cont). The difference (cont − rel) betweenoth values was calculated using the confocal analysis soft-are Leica TCS SL. The fractional shortening (fs) was cal-
ulated using the equation fs = [(cont − rel) × 100]/rel. Theaximum fluorescence of Ca2+-transients was calculated as
he peak fluorescence intensity immediately following thelectrical stimulation impulse (�F) divided by the back-round fluorescence intensity value before stimulation (F0).herefore, the fluorescence of Ca2+-transients is expresseds �F/F0.
.4. Ca2+ sparks recording
Ca2+ sparks were recorded in bath solution at room tem-erature using a Leica TCS SP2 confocal laser-scanningicroscope equipped with a DM IRE2 inverted fluorescenceicroscope (as above) [18]. Before spark measurementsyocytes were loaded with 10 �M fluorescent Ca2+ indica-
or Fluo-3 (acetoxymethylester, Molecular Probes) by 30 minncubation at 37 ◦C. Experiments were carried out in restingyocytes under steady-state conditions after loading the sar-
oplasmic reticulum with Ca2+ through field stimulation (10ulses at 1 Hz). Fluo-3 fluorescence was excited with the88 nm line of an argon laser and emission was recorded at515 nm. Every recording sequence consisted of eight imagescontinuous recording) with a dimension of 169 �m scanidth and 2237 ms scan duration. The total scan duration ofne sequence was 17.896 s. The images were analysed using
rigin 5.0 (Microcal Software Inc., Northampton, MA, USA)nd IDL 6.3 software (Research Systems, Boulder, CO, USA)s previously described [18,19]. The calcium spark detectionrogram used was based on an algorithm developed by Cheng
ct6p
1 (2007) 467–477 469
t al. [20]. The images were filtered and normalized beforetarting the spark detection algorithm. Only spark islets withuorescence above a threshold given by the normalized back-round fluorescence added with the product of at least 3.2imes its standard deviation were accepted. The calculationf spark duration, width, rise and decay time was performedased on these spark islets.
Sarcoplasmic reticulum (SR) Ca2+ content was estimatedn some cells by rapid caffeine (10 mM) application to releasehe SR Ca2+ content. During this procedure the cell wasmaged in the line-scan mode along the longitudinal lineor 15 s, which allowed recording both caffeine-evoked Ca2+
elease and contraction (estimated by cell shortening) as pre-iously reported [18,21].
.5. RNA isolation and quantitative real-time PCR
Total RNA was extracted from neonatal and adultardiomyocytes 20 and 8 h after application of the testubstance using the RNeasy kit, and first strand cDNAas synthesized by reverse transcription of 2 �g of totalNA from different treatment groups using the Omniscripteverse Transcription Kit (Qiagen) according to the manu-
acture’s instructions. Quantitative real-time PCR was per-ormed on a LightCyclerTM 1.5 instrument using FastStartNA MasterPlus SYBR Green I (Roche Molecular Bio-
hemical). Gene-specific primers for rat Cav1.2 were theollowing—forward-primer: 5′-AGC AAC TTC CCT CAGCG TTT G-3′; backward-primer: 5′-CGG CAC ATG TAGTG TCA TTG-3′ (GenBank accession No. M67515) [22].uccinate dehydrogenase complex, subunit A, flavoproteinSDHA) expression of each sample was used as endogenousontrol—forward-primer: 5′-TGG GAA CAA GAG GGCTC TG-3′; backward-primer: 5′-CCA CCA CTG CAT CAATT CAT G-3′ (GenBank accession No. NM 130428) [23].uantitative real-time PCR was carried out under the follow-
ng cycling conditions: stage 1, 95 ◦C for 10 min (rep 1); stage, 95 ◦C for 10 s, 60 ◦C for 5 s and 72 ◦C for 10 s with signalcquisition at 81 ◦C for 1 s (determined from melting curvenalysis) (rep 40); stage 3, 65 ◦C for 10 s (rep 1). Analysisf the PCR curves was performed with the second derivateaximum method of the LightCycler software [24,25]. All
ample measurements were repeated at least three times andesults are given as mean ± S.E.M.
.6. Materials and statistical analysis
In some experiments dihydrotestosterone (testosterone)nd/or flutamide (Sigma–Aldrich Chemie GmbH, Munich)ere added to the bath solutions or culture medium, as indi-
ated. Testosterone and flutamide were dissolved in ethanolefore they were added to experimental solutions. The final
oncentration of ethanol was <0.01% (maximal concentra-ion 6 nM). In all control experiments vehicle, i.e. ethanolnM was added (if not otherwise indicated). Pooled data areresented as mean ± S.E.M. Comparisons between groups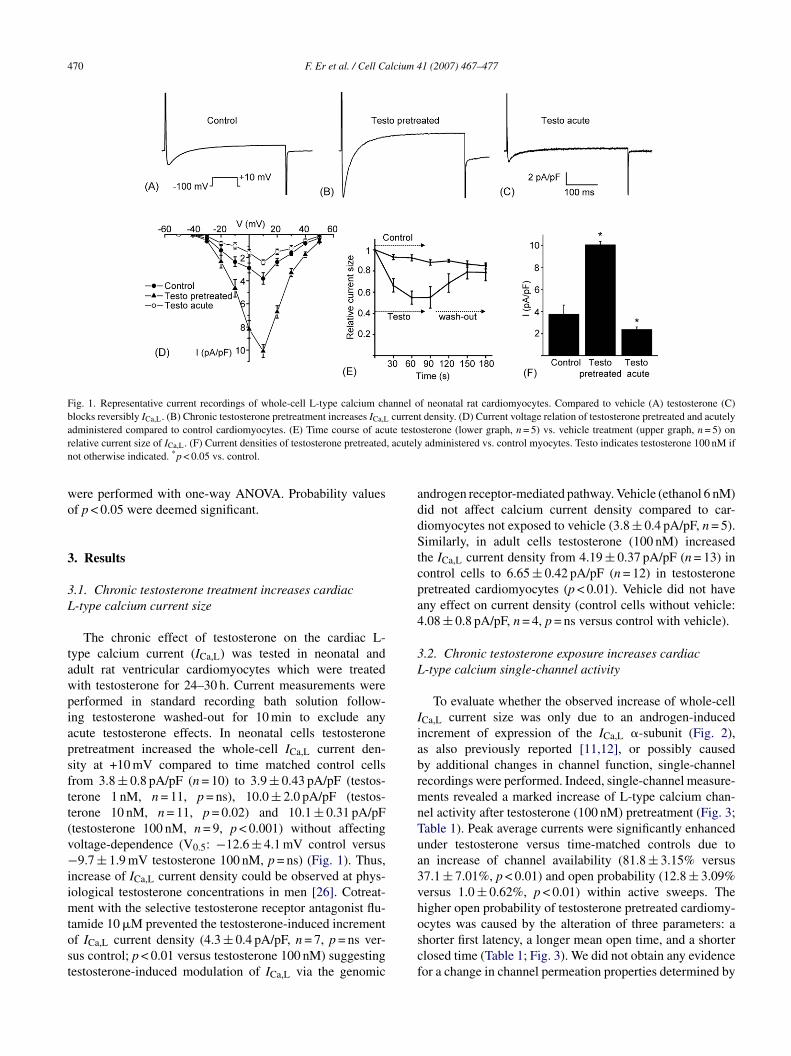
470 F. Er et al. / Cell Calcium 41 (2007) 467–477
Fig. 1. Representative current recordings of whole-cell L-type calcium channel of neonatal rat cardiomyocytes. Compared to vehicle (A) testosterone (C)blocks reversibly ICa,L. (B) Chronic testosterone pretreatment increases ICa,L current density. (D) Current voltage relation of testosterone pretreated and acutelya te testor , acutelyn
wo
3
3L
tawpiapsftt(v−iimtost
addStcpa4
3L
IiabrmnTua3vh
dministered compared to control cardiomyocytes. (E) Time course of acuelative current size of ICa,L. (F) Current densities of testosterone pretreatedot otherwise indicated. *p < 0.05 vs. control.
ere performed with one-way ANOVA. Probability valuesf p < 0.05 were deemed significant.
. Results
.1. Chronic testosterone treatment increases cardiac-type calcium current size
The chronic effect of testosterone on the cardiac L-ype calcium current (ICa,L) was tested in neonatal anddult rat ventricular cardiomyocytes which were treatedith testosterone for 24–30 h. Current measurements wereerformed in standard recording bath solution follow-ng testosterone washed-out for 10 min to exclude anycute testosterone effects. In neonatal cells testosteroneretreatment increased the whole-cell ICa,L current den-ity at +10 mV compared to time matched control cellsrom 3.8 ± 0.8 pA/pF (n = 10) to 3.9 ± 0.43 pA/pF (testos-erone 1 nM, n = 11, p = ns), 10.0 ± 2.0 pA/pF (testos-erone 10 nM, n = 11, p = 0.02) and 10.1 ± 0.31 pA/pFtestosterone 100 nM, n = 9, p < 0.001) without affectingoltage-dependence (V0.5: −12.6 ± 4.1 mV control versus9.7 ± 1.9 mV testosterone 100 nM, p = ns) (Fig. 1). Thus,
ncrease of ICa,L current density could be observed at phys-ological testosterone concentrations in men [26]. Cotreat-
ent with the selective testosterone receptor antagonist flu-
amide 10 �M prevented the testosterone-induced incrementf ICa,L current density (4.3 ± 0.4 pA/pF, n = 7, p = ns ver-us control; p < 0.01 versus testosterone 100 nM) suggestingestosterone-induced modulation of ICa,L via the genomicoscf
sterone (lower graph, n = 5) vs. vehicle treatment (upper graph, n = 5) onadministered vs. control myocytes. Testo indicates testosterone 100 nM if
ndrogen receptor-mediated pathway. Vehicle (ethanol 6 nM)id not affect calcium current density compared to car-iomyocytes not exposed to vehicle (3.8 ± 0.4 pA/pF, n = 5).imilarly, in adult cells testosterone (100 nM) increased
he ICa,L current density from 4.19 ± 0.37 pA/pF (n = 13) inontrol cells to 6.65 ± 0.42 pA/pF (n = 12) in testosteroneretreated cardiomyocytes (p < 0.01). Vehicle did not haveny effect on current density (control cells without vehicle:.08 ± 0.8 pA/pF, n = 4, p = ns versus control with vehicle).
.2. Chronic testosterone exposure increases cardiac-type calcium single-channel activity
To evaluate whether the observed increase of whole-cellCa,L current size was only due to an androgen-inducedncrement of expression of the ICa,L �-subunit (Fig. 2),s also previously reported [11,12], or possibly causedy additional changes in channel function, single-channelecordings were performed. Indeed, single-channel measure-ents revealed a marked increase of L-type calcium chan-
el activity after testosterone (100 nM) pretreatment (Fig. 3;able 1). Peak average currents were significantly enhancednder testosterone versus time-matched controls due ton increase of channel availability (81.8 ± 3.15% versus7.1 ± 7.01%, p < 0.01) and open probability (12.8 ± 3.09%ersus 1.0 ± 0.62%, p < 0.01) within active sweeps. Theigher open probability of testosterone pretreated cardiomy-
cytes was caused by the alteration of three parameters: ahorter first latency, a longer mean open time, and a shorterlosed time (Table 1; Fig. 3). We did not obtain any evidenceor a change in channel permeation properties determined by
F. Er et al. / Cell Calcium 41 (2007) 467–477 471
Table 1Gating parameters of single L-type calcium channel recordings: chronic testosterone (100 nM) effect
Parameter Control n Testosterone pretreated n Testosterone + flutamide pretreated n
Open probability (%) 1.0 ± 0.62 8 12.8 ± 3.09* 12 1.32 ± 0.33** 6Availability (%) 37.1 ± 7.01 8 81.8 ± 3.15* 12 36.2 ± 4.85** 6Mean open time (ms) 0.34 ± 0.07 8 0.53 ± 0.04 12 0.26 ± 0.04 6τopen (ms) 0.44 ± 0.05 4 0.55 ± 0.05 12 0.47 ± 0.04 5Mean closed time (ms) 13.3 ± 4.51 5 3.06 ± 0.62* 8 10.6 ± 2.34** 5τclosed, fast (ms) 0.82 ± 0.14 5 0.52 ± 0.04* 8 0.82 ± 0.04** 5τclosed, slow (ms) 19.4 ± 1.0 5 9.84 ± 1.24* 8 20.6 ± 1.03** 5Mean first latency (ms) 55.4 ± 7.28 5 20.6 ± 2.15* 8 41.3 ± 4.17** 5Amplitude (pA) −0.72 ± 0.06 5 −0.67 ± 0.02 11 −0.70 ± 0.01 5Inactivation (%) 41.6 ± 4.28 8 32.7 ± 3.0 12 38.8 ± 4.62 6Ipeak (fA) 14 ± 6 8 119 ± 27* 12 12 ± 3** 6
Unitary single-channel amplitude, from amplitude histogram; τopen, time constant of the open-time histogram; τclosed, fast, time constant of the fast component ofthe closed-time histogram; τclosed, slow, time constant of the slow component of the closed time histogram. Ipeak was measured from ensemble average currents.For closed time and latency analysis, only experiments containing just one detected
* p < 0.05 vs. control group.** p < 0.05 vs. pretreated group.
Fig. 2. Chronic testosterone treatment increases expression of the L-type cal-cium channel Cav1.2 in cardiomyocytes. Quantitative real-time PCR resultsof Cav1.2 mRNA levels normalized to SDHA are summarized as bar graphs(log-corrected). Adult (A) and neonatal rat cardiomyocytes (B) were cul-tured for 8 and 20 h, respectively, under control conditions with or withoutvehicle, in the presence of testosterone (100 nM), flutamide (10 �M) ortestosterone + flutamide. Testosterone increased the expression of Cav1.2,while flutamide abolished the testosterone-induced effect. *p < 0.05 vs. con-trol.
tctmi(
3c
cdTtalaFC2vin0eAccp
3f
ico
open level were used for calculation.
he size of the single-channel amplitude (Table 1) and single-hannel conductance (control: 15.7 ± 0.90 pS, n = 5; testos-erone pretreatment: 16.0 ± 1.12 pS, n = 3, p = ns). Cotreat-
ent with flutamide abolished the chronic testosterone-nduced alterations of single calcium channel functionTable 1).
.3. Chronic testosterone treatment enhancesontractility of cardiomyocytes
To analyse the long-term effect of testosterone onardiac contractility and calcium transients, adult car-iomyocytes were treated with testosterone for 24–30 h.he fractional shortening and the peak Ca2+-transients of
estosterone (10 nM) treated myocytes (9.76 ± 1.86%, n = 8,nd 3.04 ± 0.17, n = 14, respectively) were significantlyarger compared to control cells (3.69 ± 0.64%, n = 11,nd 2.36 ± 0.22, n = 11, respectively, p = 0.003 and 0.002;ig. 4). Vehicle did not have any effect on contractility anda2+-transients (control without vehicle 4.15 ± 0.91% and.29 ± 0.14, respectively, n = 16, p = ns versus control withehicle). Flutamide 10 �M prevented these testosterone-nduced changes (4.20 ± 0.44%, n = 5, and 2.41 ± 0.14,= 14, respectively, p = ns versus control; p = 0.04 and.008 versus testosterone), indicating a positive inotropicffect of testosterone mediated by testosterone receptors.cute testosterone application to testosterone pretreated
ardiomyocytes inhibited the chronically induced increase ofontractility (4.48 ± 0.31, n = 6, p < 0.01 versus testosteroneretreatment alone).
.4. Chronic testosterone treatment increases therequency of Ca2+ sparks
Since the observed chronic testosterone-mediatedncrease of L-type calcium current might affect intracellularalcium handling, we recorded intracellular calcium releasef adult rat ventriculocytes following pretreatment with

472 F. Er et al. / Cell Calcium 41 (2007) 467–477
Fig. 3. Effect of testosterone (100 nM) on the L-type calcium channel in cell-attached patches, test potential +20 mV, holding potential −80 mV. Left: sequentialunitary current records before (control); middle: after 24–30 h treatment with testosterone (pretreated); right: acute testosterone effect of pretreated cells (acuteeffect). Bottom: ensemble-average currents calculated from sweeps before (left: control 240 sweeps), after 2-day testosterone-therapy (middle: pretreated 300s bars inr
tadsnpicvtrcmrrtc(ipTvtcaot1
owcrt
w(ptctdvopTiwc
3L
cuc1dt32Aab
weeps) and after drug addition to bath (right: acute effect 180 sweeps). Scaleespectively.
estosterone (10 nM) for 24–30 h. Line-scan images andverage data of Ca2+ sparks of a sequence of 8 recordingsemonstrated significantly more spontaneously arising Ca2+
parks in testosterone pretreated myocytes (62.70 ± 10.22,= 23) compared to control cells (15.17 ± 2.90, n = 23,< 0.001) (Fig. 5). The testosterone-induced increase
n the frequency of Ca2+ sparks could be prevented byotreatment with flutamide (12.60 ± 2.91, n = 21, p < 0.001ersus testosterone; p = ns versus control) (Fig. 5). Onhe line-scan image Ca2+ sparks occurred at an averageate of 125.72 ± 7.70 s−1 in testosterone pretreated cellsompared to 83.92 ± 7.38 and 69.85 ± 6.28 s−1 in controlyocytes and in cells exposed to testosterone + flutamide,
espectively. Within the entire recording sequences, theseates remained constant. The mean time to peak Ca2+ (riseime) was significantly delayed in testosterone pretreatedardiomyocytes (32.50 ± 2.67 ms) compared to controls18.87 ± 1.37 ms, p < 0.001). Cotreatment with flutamidenhibited this testosterone-induced delay (18.00 ± 1.79 ms,= ns versus control, p < 0.001 versus testosterone).estosterone slowed the decay time (93.22 ± 5.92 ms)ersus controls (65.04 ± 6.34 ms, p < 0.001) and the testos-erone + flutamide group (51.85 ± 5.82 ms, p = ns versusontrol; p < 0.001 versus testosterone). The Ca2+ sparkmplitude (as indicated by the ratio of peak fluorescencever background fluorescence) was unaffected by testos-erone (1.52 ± 0.03 controls; 1.67 ± 0.04 testosterone;.46 ± 0.04 testosterone + flutamide, p = ns; Fig. 5E).
Since SR Ca2+ content may contribute to the probabilityf spontaneous Ca2+ sparks occurring in resting myocytes,
e estimated SR Ca2+ content by rapidly applying 10 mMaffeine [21,27]. However, the amplitude of the fluorescenceatio (peak of the fluorescence signal after caffeine applica-ion normalized to the signal before caffeine application) [21]
ptIs
dicate 50 ms and 1 pA (unitary current traces) or 30 fA (ensemble averages),
as not significantly different in testosterone treated cells3.0 ± 0.5, n = 10) compared to controls (2.6 ± 0.2, n = 10,= ns). The absence of any change in SR load after testos-
erone exposure was further confirmed by the analysis ofaffeine-induced cell contraction. Myocytes shortening athe maximum caffeine-evoked twitch was not significantlyifferent in the absence (cell length 115.79 ± 4.08 �m praeersus 103.42 ± 3.14 �m post-caffeine, n = 10) or presencef testosterone pretreatment (cell length 113.50 ± 9.78 �mrae versus 98.92 ± 8.69 �m post-caffeine, n = 10, p = ns).hese results demonstrate a significant chronic testosterone-
nduced increase in the frequency of spontaneous Ca2+ sparksithout alterations of SR Ca2+ content, which effectively
ould be abolished by flutamide.
.5. Acute testosterone exposure decreases whole-cell-type calcium current
To test the acute effect of testosterone on L-type cal-ium channels, ICa,L was recorded in whole-cell config-ration of cardiomyocytes consecutively superfused withontrol solution, control solution + testosterone 1, 10 and00 nM and after wash-out. The initial current densityecreased upon acute testosterone exposure from 3.82 ± 0.33o 3.74 ± 0.32 pA/pF (testosterone 1 nM, p = ns, n = 5),.3 ± 0.34 pA/pF (testosterone 10 nM, p = ns, n = 5) and.4 ± 0.2 pA/pF (testosterone 100 nM, p < 0.01, n = 5; Fig. 1).fter testosterone wash-out the current density increased
gain, demonstrating the reversibility of ICa,L inhibitiony testosterone (3.5 ± 0.20 pA/pF, p = ns versus baseline,
= 0.03 versus testosterone 100 nM) (Fig. 1E). Acute testos-erone application did not affect the voltage-dependence ofCa,L (V0.5: −10.5 ± 2.2 mV testosterone 100 nM, p = ns ver-us control) (Fig. 1D).

F. Er et al. / Cell Calcium 41 (2007) 467–477 473
Fig. 4. Impact of testosterone on contractility and Ca2+-release in adult rat cardiomyocytes. (A) Fractional shortening during field stimulation of myocytespretreated with 100 nM testosterone (black column), testosterone and 10 �M flutamide (dark grey column) and vehicle (light grey column) for 24–30 h,compared to controls without vehicle (white column). Fractional shortening in testosterone pretreated cells is significantly increased compared to no vehicle,testo + flutamide and vehicle (p = 0.003, 0.004 and 0.007, respectively). (B–F) Confocal line-scan recordings of Ca2+-transients elicited by field stimulationof Fluo-3 loaded cardiomyocytes. The different groups were prepared as (A, above). (B) Peak fluorescence intensities of Ca2+-transients calculated as �F/F0
and averaged for all groups of experiments. In myocytes pretreated with 100 nM testosterone, Ca2+-transients are significantly increased compared to controlswithout vehicle (p = 0.002, n = 14 vs. n = 16), to testosterone + flutamide (p = 0.008, n = 15) and to vehicle alone (p = 0.022, n = 11). Representative line-scanrecordings during repetitive electrical field-stimulation of cardiomyocytes under control conditions without vehicle (C), pretreated with 100 nM testosterone(D), 100 nM testosterone and 10 �M flutamide (E) and vehicle (F). The upper part shows original line-scan images during electric stimulation. Two typicalc k square
3c
tic
btc
onsecutive impulses were taken from the original series, indicated by blacxpressed as �F/F0, fitted into the same timescale.
.6. Testosterone acutely blocks L-type calcium singlehannels
Given these distinct acute versus chronic effects of testos-erone on whole-cell ICa,L we further characterized the block-ng effect of testosterone in additional cell-attached singlealcium channel recordings. Given our observation of higher
epIc
es. The lower part shows the corresponding Fluo-3 fluorescence intensity
asal L-type calcium channel activity following chronicestosterone treatment, we decided to evaluate whether thehronic testosterone action could be antagonized by the acute
ffect. Testosterone 100 nM acutely changed the rapid gatingroperties indicating an antagonistic effect on ICa,L current.ndeed, on the single-channel level acute testosterone appli-ation completely reversed the chronic testosterone-mediated
474 F. Er et al. / Cell Calcium 41 (2007) 467–477
Fig. 5. Impact of testosterone (10 nM) on Ca2+ sparks. (A–C) Line-scan images with spontaneously arising Ca2+ sparks recorded in three representativecardiomyocytes. Testosterone (B) significantly increased the frequency of Ca2+ sparks compared to controls (A), which could be inhibited by flutamide (C).Each set of images shows the raw line-scan image on the left and the normalized, median filtered data on the right. Line plots (bottom illustrations) derivedf red arroA rone pret ot affect version
eops
rom line-scan images at the locations along the scan line indicated by theverage numbers of Ca2+ sparks in a sequence of eight recordings. Testoste
o control and testosterone + flutamide treated cells. (E) Testosterone does nhe references to color in this figure legend, the reader is referred to the web
ffects (Fig. 3; Table 2): we observed a pronounced reductionf the open probability (15.1 ± 4.54% versus 2.3 ± 0.88%,< 0.01 versus testosterone pretreated cells, p = ns ver-
us controls), the peak current from ensemble averages
(pmp
ws from each scan demonstrate the fluorescence as a function of time. (D)treated myocytes demonstrate markedly higher Ca2+ spark rates compared
t the Ca2+ spark amplitude compared to control cells. (For interpretation ofof this article.)
168 ± 44 fA versus 28 ± 8 fA, p < 0.01 versus testosteroneretreated cells; p = ns versus controls) and an increase of theean first latency (19.2 ± 3.45 ms versus 36.2 ± 3.87 ms,< 0.01 versus testosterone pretreated cells; p = ns versus

F. Er et al. / Cell Calcium 41 (2007) 467–477 475
Table 2Gating parameters of single L-type calcium channel recordings: acute testosterone (100 nM) effect of pretreated cells
Parameter Control (pretreated) n Testosteroneacute
n Control wash-out(pretreated)
n Testosterone +flutamide acute
n
Open probability (%) 15.1 ± 4.54 6 2.3 ± 0.88* 6 10.5 ± 4.38 6 2.1 ± 0.48* 6Availability (%) 81.0 ± 3.24 6 42.3 ± 9.90* 6 82.7 ± 5.74 6 39.1 ± 8.60* 6Mean open time (ms) 0.54 ± 0.05 6 0.43 ± 0.04 6 0.52 ± 0.07 6 0.45 ± 0.04 6τopen (ms) 0.62 ± 0.03 6 0.48 ± 0.07 6 0.48 ± 0.08 6 0.48 ± 0.05 6Mean closed time (ms) 2.92 ± 1.04 5 5.22 ± 1.16 5 3.21 ± 0.78 3 4.72 ± 0.56 3τclosed, fast (ms) 0.47 ± 0.05 5 0.92 ± 0.28 5 0.60 ± 0.07 3 0.61 ± 0.11 3τclosed, slow (ms) 11.6 ± 1.44 5 12.6 ± 2.78 5 6.85 ± 0.62 3 7.19 ± 1.03 3Mean first latency (ms) 19.2 ± 3.45 5 36.2 ± 3.87* 5 22.0 ± 2.75 3 35.7 ± 2.26* 3Amplitude (pA) −0.63 ± 0.02 6 −0.71 ± 0.02 6 −0.72 ± 0.02 5 −0.70 ± 0.02 5Inactivation (%) 26.0 ± 4.48 6 51.0 ± 7.64* 6 39.3 ± 1.36 6 49.3 ± 4.22* 6Ipeak (fA) 168 ± 44 6 28 ± 8* 6 69 ± 18 6 28 ± 7* 6
Unitary single-channel amplitude, from amplitude histogram; τopen, time constant of the open-time histogram; τclosed, fast, time constant of the fast component ofthe closed-time histogram; τclosed, slow, time constant of the slow component of the closed time histogram. Ipeak was measured from ensemble average currents.F etected
caap(Stcn021c
b(t
3f
i(tcd6acdtmpsam
4
ccscbm
sCttL[Cocldmsaaeccn
iot
or closed time and latency analysis, only experiments containing just one d* p < 0.05 vs. control.
ontrols). Moreover, testosterone acutely reduced thevailability from 81.0 ± 3.24 to 42.3 ± 10.0% supporting andditional slow blocking effect (p < 0.01 versus testosteroneretreated cells; p = ns versus controls). A very rapid blocka reduction of single-channel amplitude) was not obtained.ingle-channel conductance remained unchanged upon acute
estosterone exposure (16.7 ± 0.83 pS, n = 4, p = ns versusontrol). Acute testosterone application (100 nM) also sig-ificantly reduced the open probability (2.38 ± 1.42% versus.39 ± 0.15%, n = 3), the availability (57.4 ± 4.54% versus5.2 ± 6.32%, n = 3) and the peak current (31 ± 6 fA versus0 ± 3 fA, n = 3) of non-pretreated cells (all p < 0.05 versusontrol).
Flutamide 10 �M did not prevent the testosterone-inducedlock of ICa,L in the whole-cell or cell-attached configurationTable 2), indicating a receptor-independent acute action ofestosterone.
.7. Effect of acute testosterone application on therequency of Ca2+ sparks
While acute testosterone exposure (120–300 s) of freshlysolated cardiocytes did not significantly affect Ca2+ sparksfrequency of Ca2+ sparks: controls 4.9 ± 0.97 s−1 versusestosterone 3.2 ± 0.33 s−1, n = 24, p = ns; amplitude:ontrols 1.78 ± 0.02 versus 1.80 ± 0.05, p = ns; sparkuration: controls 64.21 ± 3.14 ms versus testosterone2.95 ± 6.38 ms, p = ns), acute testosterone applicationbolished the chronic effect of testosterone on intra-ellular calcium release: the number of calcium sparksecreased significantly to 14.75 ± 2.86 when testos-erone was acutely administered to testosterone pretreated
yocytes (n = 12, p < 0.01 versus testosterone pretreatment,
= ns versus control). The calcium spark amplitude, thepark duration, rise and decay times were unchanged bycute testosterone application to testosterone pretreatedyocytes.
qtc(
open level were used for calculation.
. Discussion
In the present study, we identified distinct acute versushronic actions of testosterone on cardiac L-type calciumhannel function, contractility and frequency of calciumparks. While long-term androgen exposure increased ICa,Lurrent size via a genomic pathway, testosterone acutelylocked ICa,L directly in an androgen receptor-independentanner.Recent data demonstrated an upregulation of the expres-
ion of the L-type calcium channel pore forming subunitav1.2 by testosterone in cardiomyocytes [13,28]. Gonadec-
omy of male rats decreased Cav1.2 mRNA, while testos-erone treatment of castrated rats resulted in a tenfold higher-type calcium channel mRNA level compared to controls
13,28]. We could confirm a receptor-mediated elevation ofav1.2 expression upon chronic testosterone treatment. Thus,ur observed increase of whole-cell ICa,L current densityould readily be explained by an androgen-induced upregu-ation of Cav1.2 expression upon long-term exposure of car-iomyocytes to testosterone. However, we obtained a secondechanism underlying the change of whole-cell ICa,L current
ize. Interestingly, chronic testosterone application addition-lly enhanced ICa,L single-channel activity by increasing thevailability and open probability though we might have over-stimated this effect since an increase of the real number ofhannels in each patch cannot absolutely be exclude despiteorrection of the availability and open probability for theumber of channels using BayK 8644 in most recordings.
This increased calcium influx was associated with a pos-tive inotropic effect in testosterone pretreated cardiomy-cytes. Moreover, exposure of ventricular cells to testos-erone for 24–30 h induced a marked increase of the fre-
uency of spontaneously arising calcium sparks comparedo time-matched controls. Calcium sparks are the visualizedorrelate of Ca2+ release through the ryanodine receptorsRyR) of the sarcoplasmic reticulum (SR). Previously no
4 lcium 4
ecrstcltacpgiLgfmaasiootp
nNgibssoaomItaawoSmotlnnTf
pAS
awmsgoi[tpeo
ebgoidubssaigarume
A
aDM
R
76 F. Er et al. / Cell Ca
ffect of testosterone on the expression of various intra-ellular calcium regulatory proteins (i.e. phospholamban,yanodine receptor type 2, SERCA 2a) has been demon-trated [29]. In resting myocytes two main factors appearo contribute to the probability of spontaneous Ca2+ sparks:ytosolic Ca2+ and SR Ca2+ content [21,27]. While SR Ca2+
oad remained unchanged upon testosterone exposure, testos-erone treatment significantly increased ICa,L current densitynd single-channel activity. The probability of L-type Ca2+
hannel opening is low in myocytes at rest, but remains pro-ortional to channel density [30]. Moreover, the couplingain between ICa,L and RyRs is very high at negative rest-ng membrane potentials and Ca2+-influx through a single-type Ca2+ channel correlates with the probability of trig-ering a calcium spark [31]. Therefore, the higher Ca2+ sparkrequency in our experiments is consistent with testosterone-ediated increase of ICa,L current density and single-channel
ctivity [21]. Though we did not exclude any direct alter-tions of RyR function by testosterone, no change of Ca2+
park amplitude occurred upon androgen exposure suggest-ng that testosterone did not affect the functional propertiesf ryanodine receptors [32]. All observed chronic effectsf testosterone could be abolished by cotreatment with flu-amide indicating a genomic testosterone receptor-mediatedathway.
In addition, to chronic hormone actions evidence for rapid,on-genomic effects of androgens are increasing [6,8,33].on-genomic testosterone actions can be distinguished fromenomic effects by a more rapid onset (seconds to minutes),nsensitivity to inhibition of RNA and protein synthesis, andy the fact that observed effects may not be blocked by clas-ical testosterone-receptor antagonists [34]. In the presenttudy, testosterone, given as a bolus, led to an acute inhibitionf the L-type single calcium channel. Moreover, testosteronecutely suppressed contractility and reduced the frequencyf spontaneous Ca2+ sparks of testosterone pretreated cellsost likely secondarily to the acute testosterone-induced
Ca,L blockade. The interaction of testosterone with the L-ype calcium channel seemed to rely on two mechanisms: (i)rapid mechanism with a reduction of the open probability
nd the ensemble average current, and (ii) a slow mechanismith a reduction of the channel availability, while no effectn current amplitude (very rapid mechanism) was detectable.ince no hormone was present in the pipette, and even smallolecules do not diffuse through the seal area between the rim
f the pipette and membrane surface it is likely, that testos-erone, a highly lipophilic molecule might act through theipid phase of the membrane on the L-type calcium chan-el. Due to its very poor water solubility testosterone isot expected to enter the channel pore from the cytoplasm.herefore, the hormone might rather bind at or near the pore-
orming Cav1.2 subunit similar to dihydropyridine derivates.
Recently, we demonstrated testosterone-induced cellrotection via direct activation of cardiac mitochondrialTP-dependent potassium channels by testosterone [8].imilar to the androgen-mediated action on mitoKATP,
1 (2007) 467–477
cute testosterone-induced inhibition of ICa,L occurredithin minutes via a non-genomic mechanism, possiblyediated by testosterone membrane receptors. Very recently,
uppression of ICa,L whole-cell current has been reported inuinea-pig myocytes, which in contrast to our acute effectsn ICa,L single-channel activity and Ca2+ sparks could benhibited by the testosterone receptor antagonist nilutamide35]. While it is generally believed, that conventional nuclearestosterone receptor antagonists have hardly any affinity toutative testosterone membrane receptors (with nilutamidexhibiting even lower affinity than flutamide) [34], thebserved differences might be species specific.
Interestingly, in our experiments the chronic testosteroneffect on L-type single-channels was completely antagonisedy the acute testosterone application. Our observations sug-est a balance between acute and chronic testosterone actionn ICa,L and intracellular Ca2+ handling. Testosterone chron-cally seems to be involved in setting the basal ICa,L currentensity, while acute changes of hormone levels rapidly mod-late L-type single calcium channel activity. Any shift of thisalance will not only influence cardiac contractility and pos-ibly arrhythmogenesis [35]. In addition, it has been demon-trated that even minor changes in [Ca2+]i are sufficient toffect transcription of various proteins [21,36]. Thus, elevatedntracellular calcium levels likely contribute to cardiac patho-enesis during chronic androgen abuse, while acute androgenpplication might additionally modulate cardiac inotropy andepolarization. Further in vivo studies are warranted to eval-ate a possible clinical relevance of androgen-mediated ICa,Lodulation during physiological variability of hormone lev-
ls and androgen replacement therapy.
cknowledgments
We thank Nadine Henn and Iris Berg for skillful technicalssistance. This study was supported by a grant from theeutsche Forschungsgemeinschaft (Ho 2146/3-1), and by thearga und Walter Boll-Stiftung.
eferences
[1] F.C. Wu, A. von Eckardstein, Androgens and coronary artery disease,Endocr. Rev. 24 (2003) 183–217.
[2] D. Grady, D. Herrington, V. Bittner, et al., Cardiovascular diseaseoutcomes during 6.8 years of hormone therapy: Heart and Estro-gen/progestin Replacement Study follow-up (HERS II), JAMA 288(2002) 49–57.
[3] J.E. Rossouw, G.L. Anderson, R.L. Prentice, et al., Risks and benefitsof estrogen plus progestin in healthy postmenopausal women: principalresults From the Women’s Health Initiative randomized controlled trial,JAMA 288 (2002) 321–333.
[4] M.R. Adams, J.K. Williams, J.R. Kaplan, Effects of androgens on coro-
nary artery atherosclerosis and atherosclerosis-related impairment ofvascular responsiveness, Arterioscler. Thromb. Vasc. Biol. 15 (1995)562–570.[5] R.B. Melchert, A.A. Welder, Cardiovascular effects of androgenic-anabolic steroids, Med. Sci. Sports Exerc. 27 (1995) 1252–1262.

lcium 4
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
F. Er et al. / Cell Ca
[6] K.M. English, R.D. Jones, T.H. Jones, A.H. Morice, K.S. Channer,Testosterone acts as a coronary vasodilator by a calcium antagonisticaction, J. Endocrinol. Invest. 25 (2002) 455–458.
[7] J.L. Scragg, R.D. Jones, K.S. Channer, T.H. Jones, C. Peers, Testos-terone is a potent inhibitor of L-type Ca(2+) channels, Biochem. Bio-phys. Res. Commun. 318 (2004) 503–506.
[8] F. Er, G. Michels, N. Gassanov, F. Rivero, U.C. Hoppe, Testosteroneinduces cytoprotection by activating ATP-sensitive K+ channels inthe cardiac mitochondrial inner membrane, Circulation 110 (2004)3100–3107.
[9] F. Schroder, R. Handrock, D.J. Beuckelmann, et al., Increased availabil-ity and open probability of single L-type calcium channels from fail-ing compared with nonfailing human ventricle, Circulation 98 (1998)969–976.
10] G. Klein, F. Schroder, D. Vogler, et al., Increased open probability ofsingle cardiac L-type calcium channels in patients with chronic atrialfibrillation role of phosphatase 2A, Cardiovasc. Res. 59 (2003) 37–45.
11] D.K. Bowles, K.K. Maddali, V.K. Ganjam, L.J. Rubin, D.L. Tharp,J.R. Turk, C.L. Heaps, Endogenous testosterone increases L-type Ca2+
channel expression in porcine coronary smooth muscle, Am. J. Physiol.Heart Circ. Physiol. 287 (2004) H2091–H2098.
12] K.L. Golden, J.D. Marsh, Y. Jiang, Testosterone regulates mRNA levelsof calcium regulatory proteins in cardiac myocytes, Horm. Metab. Res.36 (2004) 197–202.
13] K.L. Golden, J.D. Marsh, Y. Jiang, T. Brown, J. Moulden, Gonadectomyof adult male rats reduces contractility of isolated cardiac myocytes,Am. J. Physiol. Endocrinol. Metab. 285 (2003) E449–E453.
14] F. Er, R. Larbig, A. Ludwig, M. Biel, F. Hofmann, D.J. Beuckel-mann, U.C. Hoppe, Dominant-negative suppression of HCN chan-nels markedly reduces the native pacemaker current I(f) and under-mines spontaneous beating of neonatal cardiomyocytes, Circulation107 (2003) 485–489.
15] U.C. Hoppe, E. Marban, D.C. Johns, Distinct gene-specific mechanismsof arrhythmia revealed by cardiac gene transfer of two long QT diseasegenes, HERG and KCNE1, Proc. Natl. Acad. Sci. U.S.A. 98 (2001)5335–5340.
16] U.C. Hoppe, E. Marban, D.C. Johns, Molecular dissection of cardiacrepolarization by in vivo Kv4.3 gene transfer, J. Clin. Invest. 105 (2000)1077–1084.
17] G. Michels, F. Er, I.F. Khan, M. Sudkamp, S. Herzig, U.C. Hoppe,Single-channel properties support a potential contribution of HCNchannels and if to cardiac arrhythmias, Circulation 111 (2005) 399–404.
18] N. Gassanov, M.C. Brandt, G. Michels, M. Lindner, F. Er, U.C. Hoppe,Angiotensin II-induced changes of calcium sparks and ionic currentsin human atrial myocytes: potential role for early remodeling in atrialfibrillation, Cell Calcium 39 (2006) 175–186.
19] M. Lindner, M.C. Brandt, H. Sauer, J. Hescheler, T. Bohle, D.J. Beuck-elmann, Calcium sparks in human ventricular cardiomyocytes frompatients with terminal heart failure, Cell Calcium 31 (2002) 175–
182.20] H. Cheng, L.S. Song, N. Shirokova, A. Gonzalez, E.G. Lakatta, E.Rios, M.D. Stern, Amplitude distribution of calcium sparks in confo-cal images: theory and studies with an automatic detection method,Biophys. J. 76 (1999) 606–617.
[
1 (2007) 467–477 477
21] J.P. Benitah, E. Perrier, A.M. Gomez, G. Vassort, Effects of aldosteroneon transient outward K+ current density in rat ventricular myocytes, J.Physiol. 537 (2001) 151–160.
22] D.J. Martin, D. McClelland, M.B. Herd, et al., Gabapentin-mediatedinhibition of voltage-activated Ca2+ channel currents in cultured sen-sory neurones is dependent on culture conditions and channel subunitexpression, Neuropharmacology 42 (2002) 353–366.
23] M. Fischer, M. Skowron, F. Berthold, Reliable transcript quantificationby real-time reverse transcriptase-polymerase chain reaction in primaryneuroblastoma using normalization to averaged expression levels of thecontrol genes HPRT1 and SDHA, J. Mol. Diagn. 7 (2005) 89–96.
24] M. Riessland, L. Brichta, E. Hahnen, B. Wirth, The benzamide M344,a novel histone deacetylase inhibitor, significantly increases SMN2RNA/protein levels in spinal muscular atrophy cells, Hum. Genet.(2006).
25] S.H. Chu, K. Sutherland, J. Beck, J. Kowalski, P. Goldspink, D. Schw-ertz, Sex differences in expression of calcium-handling proteins andbeta-adrenergic receptors in rat heart ventricle, Life Sci. 76 (2005)2735–2749.
26] Harrisons’s, Principles of Internal Medicine, 13th ed., Isselbacher,Braunwald, Wilson, Martin, Fauci, Kasper, 1994, p. 2009 (Chapter339).
27] H. Satoh, L.A. Blatter, D.M. Bers, Effects of [Ca2+]i, SR Ca2+ load, andrest on Ca2+ spark frequency in ventricular myocytes, Am. J. Physiol.272 (1997) H657–H668.
28] K.L. Golden, J.D. Marsh, Y. Jiang, Castration reduces mRNA levels forcalcium regulatory proteins in rat heart, Endocrine 19 (2002) 339–344.
29] F. Callies, H. Stromer, R.H. Schwinger, et al., Administration oftestosterone is associated with a reduced susceptibility to myocardialischemia, Endocrinology 144 (2003) 4478–4483.
30] A. Cavalie, D. Pelzer, W. Trautwein, Fast and slow gating behaviourof single calcium channels in cardiac cells. Relation to activation andinactivation of calcium-channel current, Pflugers Arch. 406 (1986)241–258.
31] L.F. Santana, H. Cheng, A.M. Gomez, M.B. Cannell, W.J. Lederer,Relation between the sarcolemmal Ca2+ current and Ca2+ sparks andlocal control theories for cardiac excitation-contraction coupling, Circ.Res. 78 (1996) 166–171.
32] X. Zhu, B.A. Altschafl, R.J. Hajjar, H.H. Valdivia, U. Schmidt, AlteredCa2+ sparks and gating properties of ryanodine receptors in aging car-diomyocytes, Cell Calcium 37 (2005) 583–591.
33] J. Hall, R.D. Jones, T.H. Jones, K.S. Channer, C. Peers, Selective inhi-bition of L-type Ca2+ channels in A7r5 cells by physiological levels oftestosterone, Endocrinology 147 (2006) 2675–2680.
34] A.M. Braun, P. Thomas, Biochemical characterization of a membraneandrogen receptor in the ovary of the atlantic croaker, Biol. Reprod. 71(2004) 146–155.
35] C. Bai, J. Kurokawa, M. Tamagawa, H. Nakaya, T. Furukawa, Nontran-scriptional regulation of cardiac repolarization currents by testosterone,
Circulation 112 (2005) 1701–1710.36] L. Cartin, K.M. Lounsbury, M.T. Nelson, Coupling of Ca(2+) to CREBactivation and gene expression in intact cerebral arteries from mouse:roles of ryanodine receptors and voltage-dependent Ca(2+) channels,Circ. Res. 86 (2000) 760–767.