
Angiotensin II‐induced increase in myocardial distensibility and its modulation by the endocardial...
10
Exp Physiol 94.6 pp 665–674 665 Experimental Physiology – Research Paper AngiotensinII-induced increase in myocardial distensibility and its modulation by the endocardial endothelium in the rabbit heart Paulo Castro-Chaves, Ricardo Fontes-Carvalho, Mariana Pintalhao, Pedro Pimentel-Nunes and Adelino F. Leite-Moreira Department of Physiology, Faculty of Medicine, University of Porto, Porto, Portugal As recently demonstrated, angiotensin II (Ang II) induces an increase in myocardial distensibility. Although endothelin-1 and the endocardial endothelium (EE) also modulate myocardial diastolic properties, their interaction with Ang II at this level has not yet been investigated. Increasing concentrations of Ang II (from 10 −8 to 10 −5 m) were studied in rabbit right papillary muscles in the following conditions: (1) baseline; (2) after selective removal of EE with Triton X-100; and (3) with intact EE in presence of a non-selective endothelin receptor antagonist (PD-145065), a selective endothelin type A receptor antagonist (BQ-123), an inhibitor of nitric oxide synthesis (N G -nitro-l-arginine (l-NA) or an inhibitor of the NAD(P)H oxidase (apocynin). At baseline, Ang II induced a concentration-dependent positive inotropic effect and an increase in passive muscle length (L) up to 1.020 ± 0.004L/L max . After restoring muscle length to maximal physiological length (L max ), passive tension decreased by 46.1 ± 4.0%. When the EE was removed, the effect on myocardial distensibility was abolished. With intact EE in presence of PD-145065, BQ-123 or l-NA, the effects of Ang II on myocardial distensibility were attenuated, with a maximal increase in passive muscle length of 1.0087 ± 0.0012, 1.0068 ± 0.0022 and 1.0066 ± 0.0020L/L max and a decrease in resting tension of 22.6 ± 3.6, 16.1 ± 6.0 and 20.4 ± 5.6%, respectively. In the presence of apocynin, the effect on myocardial distensibility was abolished. In conclusion, the Ang II-dependent acute increase in myocardial distensibility is abolished by the selective removal of the EE and attenuated in the presence of endothelin-1 receptor antagonists, an inhibitor of nitric oxide synthesis or an inhibitor of NAD(P)H oxidase. (Received 17 December 2008; accepted after revision 17 March 2009; first published online 3 April 2009) Corresponding author A. F. Leite-Moreira: Department of Physiology, Faculty of Medicine, University of Porto, Alameda Prof. Hernˆ ani Monteiro, 4200-319 Porto, Portugal. Email: [email protected] The octapeptide angiotensin II (Ang II) is the final product of the classical cascade of the renin–angiotensin system and is its main effector. Once synthesized, this peptide can act on two main G protein-coupled receptor subtypes, AT 1 and AT 2 (de Gasparo et al. 2000; Kim & Iwao, 2000). Classically, Ang II has been considered to have endocrine actions and a central role in blood pressure regulation, aldosterone release and sodium homeostasis. This view has been expanded in recent years, and the concept has emerged of a local renin–angiotensin system with dominant paracrine, autocrine and even intracrine effects (Touyz & Schiffrin, 2000; Paul et al. 2006). These local systems were first described in the kidney (Kimbrough et al. 1977; Levens et al. 1981, 1983) and later in the heart (Dzau & Re, 1987; Re, 1987), vasculature (Re et al. 1982; Dzau, 1984; Kifor & Dzau, 1987) and adrenal gland (Mulrow & Franco-Saenz, 1996). Conclusive evidence now exists that all components of the renin– angiotensin system necessary for the biosynthesis of Ang II are present in the heart and that Ang II is synthesized in this tissue (Carey & Siragy, 2003). In the heart, the local renin–angiotensin system regulates several aspects of both acute and chronic myocardial function (Kim et al. 1995; Wollert & Drexler, 1999; de Gasparo et al. 2000). Some of these effects depend, at least in part, on the interaction of this system with other autocrine/paracrine mediators, such as endothelin-1 (Ito et al. 1993; Moreau et al. 1997; Herizi et al. 1998; Rossi et al. 1999; Castro-Chaves et al. C 2009 The Authors. Journal compilation C 2009 The Physiological Society DOI: 10.1113/expphysiol.2008.046458
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Angiotensin II‐induced increase in myocardial distensibility and its modulation by the endocardial...

Exp Physiol 94.6 pp 665–674 665
Experimental Physiology – Research Paper
AngiotensinII-induced increase in myocardial distensibilityand its modulation by the endocardial endothelium in therabbit heart
Paulo Castro-Chaves, Ricardo Fontes-Carvalho, Mariana Pintalhao, Pedro Pimentel-Nunesand Adelino F. Leite-Moreira
Department of Physiology, Faculty of Medicine, University of Porto, Porto, Portugal
As recently demonstrated, angiotensin II (Ang II) induces an increase in myocardialdistensibility. Although endothelin-1 and the endocardial endothelium (EE) also modulatemyocardial diastolic properties, their interaction with Ang II at this level has not yet beeninvestigated. Increasing concentrations of Ang II (from 10−8 to 10−5
m) were studied in rabbitright papillary muscles in the following conditions: (1) baseline; (2) after selective removal ofEE with Triton X-100; and (3) with intact EE in presence of a non-selective endothelin receptorantagonist (PD-145065), a selective endothelin type A receptor antagonist (BQ-123), an inhibitorof nitric oxide synthesis (N G-nitro-l-arginine (l-NA) or an inhibitor of the NAD(P)H oxidase(apocynin). At baseline, Ang II induced a concentration-dependent positive inotropic effectand an increase in passive muscle length (L) up to 1.020 ± 0.004L/Lmax. After restoring musclelength to maximal physiological length (Lmax), passive tension decreased by 46.1 ± 4.0%. Whenthe EE was removed, the effect on myocardial distensibility was abolished. With intact EE inpresence of PD-145065, BQ-123 or l-NA, the effects of Ang II on myocardial distensibility wereattenuated, with a maximal increase in passive muscle length of 1.0087 ± 0.0012, 1.0068 ± 0.0022and 1.0066 ± 0.0020L/Lmax and a decrease in resting tension of 22.6 ± 3.6, 16.1 ± 6.0 and20.4 ± 5.6%, respectively. In the presence of apocynin, the effect on myocardial distensibilitywas abolished. In conclusion, the Ang II-dependent acute increase in myocardial distensibilityis abolished by the selective removal of the EE and attenuated in the presence of endothelin-1receptor antagonists, an inhibitor of nitric oxide synthesis or an inhibitor of NAD(P)H oxidase.
(Received 17 December 2008; accepted after revision 17 March 2009; first published online 3 April 2009)Corresponding author A. F. Leite-Moreira: Department of Physiology, Faculty of Medicine, University of Porto, AlamedaProf. Hernani Monteiro, 4200-319 Porto, Portugal. Email: [email protected]
The octapeptide angiotensin II (Ang II) is the final productof the classical cascade of the renin–angiotensin systemand is its main effector. Once synthesized, this peptide canact on two main G protein-coupled receptor subtypes, AT1
and AT2 (de Gasparo et al. 2000; Kim & Iwao, 2000).Classically, Ang II has been considered to have
endocrine actions and a central role in blood pressureregulation, aldosterone release and sodium homeostasis.This view has been expanded in recent years, and theconcept has emerged of a local renin–angiotensin systemwith dominant paracrine, autocrine and even intracrineeffects (Touyz & Schiffrin, 2000; Paul et al. 2006).These local systems were first described in the kidney(Kimbrough et al. 1977; Levens et al. 1981, 1983) and
later in the heart (Dzau & Re, 1987; Re, 1987), vasculature(Re et al. 1982; Dzau, 1984; Kifor & Dzau, 1987) andadrenal gland (Mulrow & Franco-Saenz, 1996). Conclusiveevidence now exists that all components of the renin–angiotensin system necessary for the biosynthesis of Ang IIare present in the heart and that Ang II is synthesized inthis tissue (Carey & Siragy, 2003). In the heart, the localrenin–angiotensin system regulates several aspects of bothacute and chronic myocardial function (Kim et al. 1995;Wollert & Drexler, 1999; de Gasparo et al. 2000). Someof these effects depend, at least in part, on the interactionof this system with other autocrine/paracrine mediators,such as endothelin-1 (Ito et al. 1993; Moreau et al. 1997;Herizi et al. 1998; Rossi et al. 1999; Castro-Chaves et al.
C© 2009 The Authors. Journal compilation C© 2009 The Physiological Society DOI: 10.1113/expphysiol.2008.046458

666 P. Castro-Chaves and others Exp Physiol 94.6 pp 665–674
2006). The predominant physiological role of the cardiacrenin–angiotensin system appears to be the maintenanceof an appropriate cellular milieu, by the balancing ofstimuli, induction and inhibition of cell growth andproliferation, as well as mediating adaptive responsesto myocardial stress after myocardial stretch (Paul et al.2006). Therefore, chronic AT1 receptor stimulation bylocally or systemically produced Ang II eventually leadsto ventricular remodelling (Weber et al. 1995), cardiachypertrophy (Schluter & Wenzel, 2008) and, ultimately,to the deterioration of systolic and diastolic functions(McElmurray et al. 1999).
These chronic effects have, for a long time, beenconsidered the main mechanisms through which Ang IIand other neurohumoral agents may influence myocardialdiastolic properties (Kass et al. 2004). However, recentstudies suggest that diastolic stiffness may also be acutelymodulated by neurohumoral factors, such as nitric oxide(Shah et al. 1994; Heymes et al. 1999), endothelin-1 (Leite-Moreira et al. 2003; Bras-Silva & Leite-Moreira, 2006) andAng II (Leite-Moreira et al. 2006). In fact, endothelin-1 acutely decreases myocardial stiffness in conditions ofcardiac overload (Leite-Moreira et al. 2003). Althoughmediated by endothelin type A (ETA) receptor stimulation(Leite-Moreira et al. 2003), this effect requires an intactendocardial endothelium (EE) and active endothelialendothelin type B1 (ETB1) receptors (Bras-Silva & Leite-Moreira, 2006) and is dependent on nitric oxide andprostaglandin release (Bras-Silva et al. 2008). As recentlydemonstrated by our group, Ang II can also acutelymodulate diastolic properties by significantly decreasingmyocardial stiffness. This effect requires AT1 receptoractivation and is mediated by protein kinase C and Na+–H+ exchanger (Leite-Moreira et al. 2006).
In the heart, there may be a significant interactionbetween these neurohumoral systems at the endotheliallevel. Cardiac endothelial cells, like all other endothelialcells, express and release a variety of autocrineand paracrine agents, which directly influence cardiacmetabolism, growth, contractile performance andrhythmicity (Brutsaert, 2003). In the normal adult heart,cardiac endothelial cells produce nitric oxide, endothelin-1 and prostacyclin and transform angiotensin I intoactive angiotensin II (Brutsaert, 2003). Also at theendothelial level, Ang II, through AT1 receptor activation,increases superoxide production via NAD(P)H oxidasestimulation, and this may imbalance the modulatoryinfluence of nitric oxide on angiotensin II effects (Justet al. 2007; Toda et al. 2007). It is therefore reasonableto hypothesize that the endocardium may play animportant role in the modulation of the acute effect ofangiotensin II in myocardial distensibility. In this study,we test this hypothesis and investigate possible underlyingmechanisms, namely the interaction with other mediators,such as endothelin-1 and nitric oxide.
Methods
The investigation conformed to the Guide for the Care andUse of Laboratory Animals published by the US NationalInstitutes of Health (NIH publication no. 85-23, revised1996).
Experimental preparation
Male New Zealand White rabbits (Oryctolagus cuniculus;2.0–3.0 kg; 11 weeks old; n = 32) were anaesthetizedwith intravenous pentobarbitone sodium (25 mg kg−1).A left thoracotomy was performed, and beatinghearts were quickly excised and immersed in amodified Krebs–Ringer solution [composition (mM):98 NaCl, 4.7 KCl, 2.4 MgSO4.7H2O, 1.2 KH2PO4,4.5 glucose, 1.8 CaCl2.2H2O, 17 NaHCO3, 15 C3H3NaO3,5 CH3COONa, and 0.02 atenolol] at 35◦C, withcardioplegic 2,3-butanedione monoxime (3%) and 5%newborn calf serum. Atenolol was used to preventβ-adrenoceptor-mediated effects. The solution was inequilibrium with 95% O2 and 5% CO2, maintainingthe pH between 7.38 and 7.42. After dissection,papillary muscles (n = 64; length 3.3 ± 1.8 mm; weight2.7 ± 0.2 mg; and preload 4.4 ± 0.1 mN) were verticallymounted in a 10 ml Plexiglass organ bath containing theaforementioned solution. The lower muscular end wasfixed in a phosphorbronze clip and the upper tendinousend was attached to an electromagnetic length–tensiontransducer (University of Antwerp, Belgium). Preloadwas initially set between 3 and 4 mN according to muscledimensions. The preparations were stimulated at 0.6 Hzwith a voltage of 10% above threshold (typically 3–6 mV)by rectangular pulses of 5 ms duration through twoplatinum electrodes arranged longitudinally alongside theentire muscle. Twenty minutes later, the bathing solutionwas replaced with a corresponding solution without2,3-butanedione monoxime and 1 h later replaced witha corresponding one without calf serum. During thenext 2 h, the muscles were stabilized. Finally, the muscleswere stretched to a muscle length at which active forcedevelopment was maximal. This length (in mm) is knownas maximal physiological length (Lmax). The muscleswere again left to stabilize for approximately 30 mins,and afterwards the experimental protocols were initiated.At the end of the experiment, the muscles were lightlyblotted and then weighed. Muscle cross-sectional areawas calculated by dividing the weight of the muscle by itslength at Lmax. A cylindrical shape and a specific gravityof 1.0 were assumed (Brutsaert et al. 1988; Gillebert &Raes, 1994). Muscle tension was then expressed as forcenormalized per cross-sectional area (in mN mm−2).
Experimental protocol
Effects of increasing concentrations of Ang II (10−8, 10−7,10−6 and 10−5 M) on myocardial contraction, relaxation
C© 2009 The Authors. Journal compilation C© 2009 The Physiological Society
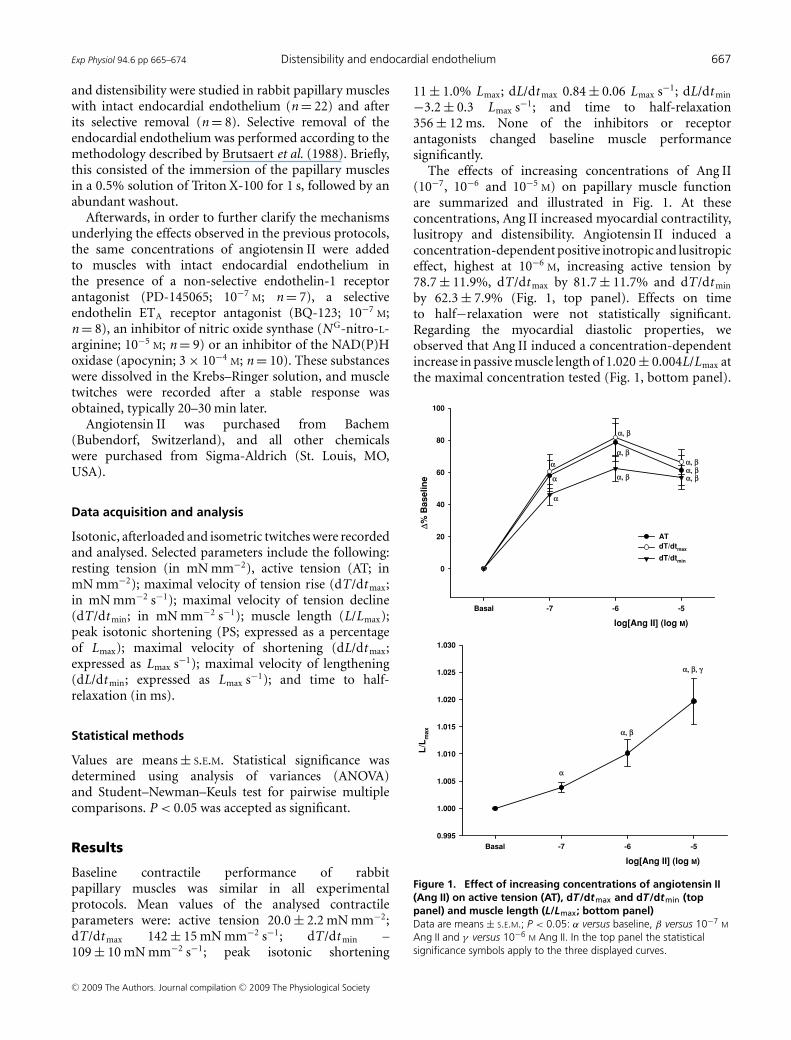
Exp Physiol 94.6 pp 665–674 Distensibility and endocardial endothelium 667
and distensibility were studied in rabbit papillary muscleswith intact endocardial endothelium (n = 22) and afterits selective removal (n = 8). Selective removal of theendocardial endothelium was performed according to themethodology described by Brutsaert et al. (1988). Briefly,this consisted of the immersion of the papillary musclesin a 0.5% solution of Triton X-100 for 1 s, followed by anabundant washout.
Afterwards, in order to further clarify the mechanismsunderlying the effects observed in the previous protocols,the same concentrations of angiotensin II were addedto muscles with intact endocardial endothelium inthe presence of a non-selective endothelin-1 receptorantagonist (PD-145065; 10−7 M; n = 7), a selectiveendothelin ETA receptor antagonist (BQ-123; 10−7 M;n = 8), an inhibitor of nitric oxide synthase (N G-nitro-L-arginine; 10−5 M; n = 9) or an inhibitor of the NAD(P)Hoxidase (apocynin; 3 × 10−4 M; n = 10). These substanceswere dissolved in the Krebs–Ringer solution, and muscletwitches were recorded after a stable response wasobtained, typically 20–30 min later.
Angiotensin II was purchased from Bachem(Bubendorf, Switzerland), and all other chemicalswere purchased from Sigma-Aldrich (St. Louis, MO,USA).
Data acquisition and analysis
Isotonic, afterloaded and isometric twitches were recordedand analysed. Selected parameters include the following:resting tension (in mN mm−2), active tension (AT; inmN mm−2); maximal velocity of tension rise (dT/dt max;in mN mm−2 s−1); maximal velocity of tension decline(dT/dt min; in mN mm−2 s−1); muscle length (L/Lmax);peak isotonic shortening (PS; expressed as a percentageof Lmax); maximal velocity of shortening (dL/dt max;expressed as Lmax s−1); maximal velocity of lengthening(dL/dt min; expressed as Lmax s−1); and time to half-relaxation (in ms).
Statistical methods
Values are means ± S.E.M. Statistical significance wasdetermined using analysis of variances (ANOVA)and Student–Newman–Keuls test for pairwise multiplecomparisons. P < 0.05 was accepted as significant.
Results
Baseline contractile performance of rabbitpapillary muscles was similar in all experimentalprotocols. Mean values of the analysed contractileparameters were: active tension 20.0 ± 2.2 mN mm−2;dT/dt max 142 ± 15 mN mm−2 s−1; dT/dt min –109 ± 10 mN mm−2 s−1; peak isotonic shortening
11 ± 1.0% Lmax; dL/dt max 0.84 ± 0.06 Lmax s−1; dL/dt min
−3.2 ± 0.3 Lmax s−1; and time to half-relaxation356 ± 12 ms. None of the inhibitors or receptorantagonists changed baseline muscle performancesignificantly.
The effects of increasing concentrations of Ang II(10−7, 10−6 and 10−5 M) on papillary muscle functionare summarized and illustrated in Fig. 1. At theseconcentrations, Ang II increased myocardial contractility,lusitropy and distensibility. Angiotensin II induced aconcentration-dependent positive inotropic and lusitropiceffect, highest at 10−6 M, increasing active tension by78.7 ± 11.9%, dT/dt max by 81.7 ± 11.7% and dT/dt min
by 62.3 ± 7.9% (Fig. 1, top panel). Effects on timeto half−relaxation were not statistically significant.Regarding the myocardial diastolic properties, weobserved that Ang II induced a concentration-dependentincrease in passive muscle length of 1.020 ± 0.004L/Lmax atthe maximal concentration tested (Fig. 1, bottom panel).
log[Ang II] (log M)
Basal -7 -6 -5
Δ% B
aselin
e
0
20
40
60
80
100
AT
dT/dtmax
dT/dtmin
α
α
α
α, β
α, β
α, β
α, βα, βα, β
log[Ang II] (log M)
Basal -7 -6 -5
L/L
max
0.995
1.000
1.005
1.010
1.015
1.020
1.025
1.030
α
α, β
α, β, γ
Figure 1. Effect of increasing concentrations of angiotensin II(Ang II) on active tension (AT), dT/dtmax and dT/dtmin (toppanel) and muscle length (L/Lmax; bottom panel)Data are means ± S.E.M.; P < 0.05: α versus baseline, β versus 10−7 M
Ang II and γ versus 10−6 M Ang II. In the top panel the statisticalsignificance symbols apply to the three displayed curves.
C© 2009 The Authors. Journal compilation C© 2009 The Physiological Society

668 P. Castro-Chaves and others Exp Physiol 94.6 pp 665–674
At the end of the experiment, correction of muscle lengthto its initial value resulted in a 46.1 ± 4.0% decrease ofresting tension, without changing the other contractileparameters. Therefore, these results indicate an increase inmuscle distensibility or, conversely, a decrease in musclestiffness.
After selective removal of the endocardial endothelium,the addition of the same concentrations of Ang II alsoinduced a positive inotropic effect. Maximal effects wereobserved at 10−5 M, which increased active tension by71.8 ± 19%, dT/dt max by 60.4 ± 17% and dT/dt min by52.3 ± 16%, without changing time to half-relaxation(Fig. 2, top panel). However, concerning the diastolicproperties, the effect of Ang II on muscle length afterdamage of the endocardial endothelium was abolished,corresponding to a non-significant decrease in passivetension of 2.86 ± 8.1% (Fig. 2, bottom panel).
Basal -7 -6 -5
Δ% B
aselin
e
0
20
40
60
80
100
AT
dT/dtmax
dT/dtmin
α
α
α
α, β
α, β
α, β
Basal -7 -6 -5
L/L
ma
x
0.995
1.000
1.005
1.010
1.015
1.020
1.025
1.030
Ang II with intact EE
Ang II after EE removal
α
α, β
α, β, γ
∗ ∗∗
log[Ang II] (log M)
log[Ang II] (log M)
Figure 2. Effect of increasing concentrations of Ang II on AT,dT/dtmax and dT/dtmin (top panel) and L/Lmax (bottom panel) inmuscles after the selective removal of the endocardialendothelium (EE)Data are means ± S.E.M. P < 0.05: α versus baseline, β versus 10−7 M
Ang II, γ versus 10−6 M Ang II and ∗ versus with intact EE.
The effects of the same concentrations of Ang IIin the presence of a non-selective endothelin receptorantagonist (PD-145065), a selective endothelin ETA
receptor antagonist (BQ-123), an inhibitor of nitric oxidesynthesis (L-NA) or an inhibitor of NAD(P)H oxidase(apocynin) are illustrated in Figures 3, 4 and 5. Figure 5summarizes the effects of Ang II (10−5 M) on restingmuscle tension at baseline and in the various experimentalconditions. This effect was assessed at the end of theexperiment by correcting muscle length to its initial value.
In the presence of the non-selective endothelinreceptor antagonist PD-145065, the Ang II-inducedpositive inotropic effect was significantly attenuated. At10−5 M, Ang II increased active tension by 44 ± 11%,dT/dt max by 51 ± 10% and dT/dt min by 41 ± 18%(Fig. 3). Regarding the diastolic properties, the effecton muscle length was significantly attenuated. Therewas an increase of only 1.0087 ± 0.0012L/Lmax at theconcentration of 10−5 M (Fig. 4). This corresponded toa decrease in passive tension of 22.6 ± 3.6% at the sameconcentration of 10−5 M (Fig. 5). A similar effect wasobserved in the presence of the selective endothelin ETA
receptor antagonist BQ-123. As shown in Figs 3, 4 and5, in this protocol, the maximal concentration of Ang II(10−5 M) increased active tension by 40 ± 8%, dT/dt max
by 38 ± 8%, dT/dt min by 45 ± 10% and muscle length to1.0068 ± 0.0022L/Lmax, which corresponded to a decreasein passive tension of only 16.1 ± 6.0%.
Afterwards, we tested the effects of Ang II in the presenceof an inhibitor of nitric oxide synthesis (N G-nitro-L-arginine). With regard to the contractile parameters, therewas an increase, again maximal at 10−5 M, in active tensionof 54 ± 8%, dT/dt max 57 ± 9% and dT/dt min 55 ± 6.0%(Fig. 3). Concerning the diastolic properties, Ang II inthe presence of N G-nitro-L-arginine increased musclelength up to 1.0066 ± 0.0020L/Lmax (Fig. 4) and decreasedpassive tension by only 20.4 ± 5.6% (Fig. 5), at the highestconcentration tested.
Finally, we observed that the addition of Ang II in thepresence of apocynin, an inhibitor of NADP(H) oxidase,abolished the effect on myocardial distensibility. Therewas only a non-significant increase in muscle lengthof 1.0036 ± 0.0020L/Lmax and a non-significant decreasein passive tension of only 10.9 ± 6.0% (Figs 4 and 5).Regarding contractile properties, the effect of Ang II inthe presence of apocynin was significantly blunted. Weobserved an increase of active tension of only 13.9 ± 5.6%,dT/dt max 13.2 ± 7.4% and dT/dt min 19.6 ± 8.0% at themaximal concentration of Ang II (Fig. 3).
Although this description of the results has focusedon the effects on parameters derived from isometriccontractions (AT, dT/dt max and dT/dt min), similarobservations were made on parameters derived fromisotonic contractions (PS, dL/dt max and dL/dt min), as canbe seen in Table 1.
C© 2009 The Authors. Journal compilation C© 2009 The Physiological Society

Exp Physiol 94.6 pp 665–674 Distensibility and endocardial endothelium 669
basal -7 -6 -5
Ac
tive
te
ns
ion
(Δ%
baselin
e)
0
20
40
60
80
100Ang II basal
Ang II with BQ-123
Ang II with PD-145065
Ang II with L-NA
Ang II with APO
α
α, β
α, β
α
α, β
α, β, γ
α
α, β
α, β, γ
αα
α
basal -7 -6 -5
0
20
40
60
80
100Ang II basal
Ang II with BQ-123
Ang II with PD-145065
Ang II with L-NA
Ang II with APO
dT
/dtm
ax
ma
x( Δ
% b
ase
lin
e)
α
α, β
α, β
α
α
α
α, β
α, β, γ
αα, β
basal -7 -6 -5
0
20
40
60
80
Ang II basal
Ang II with BQ-123
Ang II with PD-145065
Ang II with L-NA
Ang II with APO
dT
/dt m
in (
Δ% b
as
eli
ne
)
α
α, β
α, β
α
α
α, β
α, β, γ
α
α, β
α, β, γ
α
log[Ang II] (log M)
log[Ang II] (log M)
log[Ang II] (log M)
Figure 3. Effect of increasing concentrations of Ang II on AT,dT/dtmax and dT/dtmin in baseline conditions and in thepresence of PD-145065, BQ-123, NG-nitro-L-arginine (L-NA) andapocynin (APO)Data are means ± S.E.m. P < 0.05: α versus baseline, β versus 10−7 M
Ang II and γ versus 10−6 M Ang II.
basal -7 -6 -5
0.995
1.000
1.005
1.010
1.015
1.020
1.025
1.030
Ang II basal
Ang II with BQ-123
Ang II with PD-145065
Ang II with L-NA
Ang II with APO
L/L
ma
x
α
α, β
α, β, γ
α, βα, β, ∗
α
α, ∗
αα, β
α, β, γ, ∗
∗
log[Ang II] (log M)
Figure 4. Effect of increasing concentrations of Ang II on L/Lmaxin baseline conditions and in the presence of PD-145065,BQ-123, L-NA and APOData are means ± S.E.m. P < 0.05: α versus baseline, β versus 10−7 M
Ang II, γ versus 10−6 M Ang II and ∗ versus Ang II basal.
Discussion
The present study shows that, in multicellular cardiacmuscle preparations, the effect of Ang II on myocardialdiastolic properties is modulated by the endocardialendothelium. This experimental model is particularlyadvantageous because it allows a full assessment ofboth systolic (contraction and relaxation) and diastolicproperties (resting tension), neither of which canbe ignored for a full evaluation of cardiac muscleperformance.
Angiotensin II has a role in the acute modulationof myocardial diastolic properties. In fact, a previousstudy showed that, in this animal species, Ang IIinduces a concentration-dependent acute increase in
Ang II Ang II -EE Ang II+PD Ang II+BQ Ang II+L-NA Ang II+APO
Re
sti
ng
Ten
sio
n (
Δ% B
as
eli
ne
)
-80
-60
-40
-20
0
20
40
60
α
β
αβαβαβ
β
Figure 5. Effects of Ang II (10−5 M) on resting tension atbaseline (Ang II), after selective removal of the endocardialendothelium (−EE) and in the presence of PD-145065 (PD),BQ-123 (BQ), NG-nitro-L-arginine (L-NA) and apocynin (APO)Data are means ± S.E.M. P < 0.05: α versus baseline and β versusAng II alone.
C© 2009 The Authors. Journal compilation C© 2009 The Physiological Society

670 P. Castro-Chaves and others Exp Physiol 94.6 pp 665–674
Table 1. Effects of 10−6 M Ang II on isotonic contractile parameters at baseline, after selective removal of the endocardialendothelium (−EE) and in the presence of PD-145065, BQ-123, NG-nitro-L-arginine (L-NA) and apocynin
Ang II + PD- Ang II + BQ- Ang II +Ang II (�% Ang II −EE (D% 145065 (�% 123 (�% Ang II + L-NA Apocynin (�%
Parameter baseline) baseline) baseline) baseline) (�% baseline) baseline)
PS 75.8 ± 13∗ 38.5 ± 6.7∗ 31.9 ± 8.1∗ 26.3 ± 6.5∗ 32.2 ± 4.9∗ 4.8 ± 1.4dL/dtmax 65.6 ± 11∗ 37.6 ± 13∗ 33.1 ± 9.6∗ 18.5 ± 6.4∗ 25.1 ± 5.1∗ 4.2 ± 1.9dL/dtmin 109 ± 29∗ 54.6 ± 19∗ 11.2 ± 7.2 39.3 ± 12∗ 41.3 ± 9.1∗ 4.7 ± 1.5
Represented parameters: peak isotonic shortening (PS), maximal velocity of shortening (dL/dtmax) and maximal velocityof lengthening (dL/dtmin). The results are presented as percentage of variation from baseline (�% baseline). Data aremeans ± S.E.M.; ∗P < 0.05 versus baseline.
myocardial resting length (Leite-Moreira et al. 2006). Thisincrease in muscle length corresponded to a decrease inresting tension, a rightward and downward shift of thepassive length–tension relation and thus to a decreasein myocardial stiffness. This effect was dependent onangiotensin AT1 receptor stimulation and was modulatedby protein kinase C and Na+–H+ exchanger (Leite-Moreira et al. 2006).
However, the role of endocardial endothelium in theacute actions of Ang II on myocardial diastolic propertieswas not clearly established. It is known that the endocardialendothelium is a key regulator of myocardial function,since it is an interface between the ventricular lumen,subendothelial layer and myocytes and releases a numberof vasoactive substances with paracrine and autocrineactions. In the normal adult heart, cardiac endothelialcells produce: nitric oxide, by the action of constitutivenitric oxide synthase (ecNOS or NOSIII); endothelin-1after conversion of pre-pro-endothelin to pro-endothelinand into endothelin-1 through the endothelin-convertingenzyme; eicosanoids; prostacyclin; and active Ang IIfrom angiotensin I (Brutsaert, 2003). Angiotensin II isproduced locally in the heart, and most of the effectsof Ang II on cardiac growth and contractile performanceare now thought to result from locally produced ratherthan circulating Ang II (Dzau, 1988). Angiotensin IIis synthesized locally through angiotensin-convertingenzyme or chymase-dependent pathways, both of whichare expressed in coronary vascular endothelium and incardiac endocardial endothelium (Dzau, 1988; Dostal &Baker, 1999). Both angiotensin AT1 and AT2 receptors areexpressed in the vascular and cardiac endothelium, wherethey can be involved in activation of different pathways,such as production of free radical oxygen species, nitricoxide, prostacyclin and endothelin-1 (Toda et al. 2007).
In the present study, we aimed to clarify the roleof endocardial endothelium in the acute modulation ofmyocardial diastolic properties. We observed that theeffects of Ang II on myocardial distensibility depend onthe functional integrity of the endocardial endothelium. Infact, after selective removal of this layer, the effects of Ang IIon muscle length and passive tension were abolished.
These results are particularly interesting because theyinvolve this layer as a critical regulator of both acutecontractile and diastolic myocardial properties. They alsosuggest that the effects on myocardial contractility anddistensibility may be mediated by distinct subcellularmechanisms. In fact, in what concerns myocardialcontractility, endocardial endothelium removal induces adownward and rightward deviation of the Ang II dose–response curve, while the effect on distensibility wascompletely abolished after selective removal of this layer.As represented in Fig. 1, this dissociation was alreadypresent in the baseline protocol, since we observed thatfor active tension, dT/dt max and dT/dt min the maximaleffect was observed at an Ang II concentration of 10−6 M,whereas for muscle length the values continued to increaseup to the concentration of 10−5 M. It is possible that, inthese conditions, a decline of the curve might be observedat higher concentrations. However, higher concentrationswere not used because they would be to far from thephysiological range and therefore would lack physiologicalrelevance.
In the following protocols, we tried to clarify some of thepathways involved in this endothelium-dependent effect.
Endothelin-1 is synthesized in the endocardialendothelium, cardiac muscle and coronary endothelium.This local synthesis suggests that this mediator canexert both paracrine and autocrine actions in the heart(Mebazaa et al. 1993; Evans et al. 1994; McClellanet al. 1994). Furthermore, the effect of endothelin-1on cardiac function seems to be modulated by theendocardial endothelium (Leite-Moreira & Bras-Silva,2004). This endocardial endothelium dependence isalso present when we analyse specifically the effect ofendothelin-1 on acute myocardial diastolic properties.As previously demonstrated, endothelin-1 induces anincrease in myocardial distensibility, mediated by ETA
receptors, that requires an intact endocardial endotheliumand active endothelial ETB1 receptors (Bras-Silva & Leite-Moreira, 2006). Endothelin-1 and Ang II share multiplepoints of interaction, and there is a reciprocal regulationof their synthesis and release (Imai et al. 1992; Ito et al.1993; Rossi et al. 1999). These two neurohumoral systems
C© 2009 The Authors. Journal compilation C© 2009 The Physiological Society

Exp Physiol 94.6 pp 665–674 Distensibility and endocardial endothelium 671
are also interrelated at a functional level. Chronically, theeffects of Ang II in myocardial remodelling, hypertrophyand fibrosis are dependent on endothelin-1 synthesis andrelease in several animal models and in humans (Ito et al.1993; Moreau et al. 1997; Serneri et al. 2001). Acutely,the effects of Ang II on vascular tone and myocardialcontractility have also been shown to be partly mediatedby endothelin-1 (Herizi et al. 1998; Riggleman et al. 2001;Castro-Chaves et al. 2006). The present study introducesa novel point of interaction between these two systems.In fact, regarding the acute myocardial properties, weobserved in the present study that endothelin-1 is notonly important for the modulation of the inotropic effect,but also modulates the acute increase in myocardialdistensibility induced by Ang II.
However, there are other endocardial factors, besidesendothelin-1, that may be involved in the Ang II-induced increase in myocardial distensibility. In fact, asstated previously, the endocardium also synthesizes otherbioactive substances that may be partly responsible forthe experimental observations. In order to define theseunderlying mechanisms better, we tested the effects ofAng II on myocardial distensibility after the inhibition ofnitric oxide synthase or NAD(P)H oxidase.
Nitric oxide seems to exert a dual action on myocardialcontractile performance (Shah & MacCarthy, 2000).In the lower concentration range, corresponding toendogenously generated nitric oxide through endothelialconstitutive nitric oxide synthase, it exerts a slightpositive inotropic action, contributing to maintenanceof basal contractile performance of the heart inphysiological conditions (Kojda et al. 1996; Mohanet al. 1996; Sarkar et al. 2000). At higher nitric oxidelevels, a negative response in terms of peak contractileperformance is consistently observed (Vila-Petroff et al.1999; Layland et al. 2002). Besides these effects oncontractility, nitric oxide induces an earlier onset ofventricular relaxation, an increase in left ventricularend-diastolic volume, a fall in left ventricular end-diastolic pressure and a downward shift in the diastolicleft ventricular pressure–volume relation (Paulus et al.1994). These effects favour ventricular relaxation, rapidearly filling, diastolic compliance and diastolic coronaryperfusion, which may be particularly beneficial as acompensatory mechanism against ventricular overloadingor in conditions of maladaptive hypertrophy and/ortachycardia. Angiotensin II is able, through AT1 or AT2
receptor activation at the endothelial level, to increasenitric oxide synthesis by enhancing the expression andactivity of nitric oxide synthase (Ikeda et al. 1995; Seyediet al. 1995; Gomez Llambi et al. 1996; Pueyo et al. 1998;Brede et al. 2003; Kurisu et al. 2003; Suzuki et al. 2006).We have previously observed that the effect of Ang II onmyocardial distensibility is dependent on AT1 receptoractivation and is not present when AT2 receptors are
selectively stimulated (Castro-Chaves et al. 2006, 2008).Thus, it seems reasonable to speculate that the attenuationof myocardial distensibility observed when Ang II wasadded in the presence of L-NA depends on stimulationof nitric oxide synthase by endothelial AT1 receptors.Since L-NA is a non-specific inhibitor of nitric oxidesynthase, it is not possible to identify clearly which ofthe isoforms is specifically involved, although it it appearsto inhibit constitutive NOS isoforms preferentially overinducible nitric oxide synthase (Southan & Szabo, 1996).It is also known that endothelial AT1 receptors may rapidlyphosphorylate the Ser1179 residue in constitutive nitricoxide synthase and thus enhance nitric oxide production(Suzuki et al. 2006).
Angiotensin II, through endothelial AT1 receptorstimulation, is able to increase NAD(P)H oxidase activityand thus endothelial synthesis of free radical oxygenspecies in several vascular beds and in the heart (Todaet al. 2007). In fact, endothelial dysfunction in responseto long-term Ang II treatment has been shown to besecondary to increased superoxide production withinthe endothelium, the media and/or the adventitial layer,and NAD(P)H oxidase was identified as a predominantsuperoxide source (Mollnau et al. 2002). Interestingly,some of the cardiac effects of endothelin-1 also may bemediated by NAD(P) oxidase activation, thus reinforcingthe possible functional interaction between Ang II andendothelin-1 (De Keulenaer et al. 1995). In the presentstudy, the addition of Ang II in the presence of apocyninsignificantly attenuated the effect on both myocardialcontractility and distensibility. Thus, this pathway alsoseems to be involved in the observed effects.
It is therefore reasonable to hypothesize that theendocardial endothelium plays an important rolein the acute interaction between the local renin–angiotensin system, nitric oxide, radical oxygen speciesand the endothelin system in what concerns myocardialdistensibility. It is interesting to note that although theeffect of Ang II on myocardial stiffness seems to bedependent on endothelin-1 release, the effects of bothagents have some differences between each other. Infact, unlike what is observed with Ang II, increasedmyocardial distensibility in response to endothelin-1 isobserved only in acutely loaded cardiac muscles and iscompletely abolished by Na+–H+ exchanger inhibition(Leite-Moreira et al. 2003, 2006; Ladeiras-Lopes et al.2009). The interaction between these multiple pathwaysmay be rather complex. In fact, many of the agents releasedby the endocardial endothelium modulate the actions ofthe other endothelium-derived agents on the same targetcell, actions that may be mutually additive, synergisticor inhibitory. They may even result in novel effects, notseen with either agents alone, which may be determinedby the milieu in which the interactions occur. It maytherefore be simplistic to try to define their properties
C© 2009 The Authors. Journal compilation C© 2009 The Physiological Society

672 P. Castro-Chaves and others Exp Physiol 94.6 pp 665–674
independently from one another (Brutsaert, 2003). Theobservation that the different antagonists only partlyinhibit the effect of Ang II may reflect the interactionbetween different neurohumoral pathways in the acutemodulation of myocardial distensibility.
In conclusion, the acute effect of Ang II on myocardialstiffness is dependent on the functional integrity of theendocardial endothelium and is modulated by nitric oxide,by endothelin-1 synthesis and release and by NAD(P)Hoxidase activity. This acute decrease of myocardial restingtension may be an important adaptive mechanism in anacute overload context, allowing the ventricle to reachhigher filling volumes at lower filling pressures. Theseresults add to our knowledge, since they reinforce previousevidence in favour of an active control of diastolic stiffnessby neurohumoral agents.
References
Bras-Silva C & Leite-Moreira AF (2006). Obligatory role of theendocardial endothelium in the increase of myocardialdistensibility induced by endothelin-1. Exp Biol Med(Maywood) 231, 876–881.
Bras-Silva C, Monteiro-Sousa D, Duarte AJ, Guerra M,Fontes-Sousa AP, Moura C, Areias JC & Leite Moreira AF(2008). Nitric oxide and prostaglandins – important playersin endothelin-1 induced myocardial distensibility. PhysiolRes 57, 165–174.
Brede M, Roell W, Ritter O, Wiesmann F, Jahns R, Haase A,Fleischmann BK & Hein L (2003). Cardiac hypertrophy isassociated with decreased eNOS expression in angiotensinAT2 receptor-deficient mice. Hypertension 42, 1177–1182.
Brutsaert DL (2003). Cardiac endothelial-myocardial signaling:its role in cardiac growth, contractile performance, andrhythmicity. Physiol Rev 83, 59–115.
Brutsaert DL, Meulemans AL, Sipido KR & Sys SU (1988).Effects of damaging the endocardial surface on themechanical performance of isolated cardiac muscle. Circ Res62, 358–366.
Carey RM & Siragy HM (2003). Newly recognized componentsof the renin-angiotensin system: potential roles incardiovascular and renal regulation. Endocr Rev 24, 261–271.
Castro-Chaves P, Roncon-Albuquerque R Jr & Leite-MoreiraAF (2006). Endothelin ETA receptors and endotheliumpartially mediate the positive inotropic and lusitropic effectsof angiotensin II. Eur J Pharmacol 544, 91–96.
Castro-Chaves P, Soares S, Fontes-Carvalho R & Leite-MoreiraAF (2008). Negative inotropic effect of selective AT2 receptorstimulation and its modulation by the endocardialendothelium. Eur J Pharmacol 578, 261–269.
de Gasparo M, Catt KJ, Inagami T, Wright JW & Unger T(2000). International union of pharmacology. XXIII. Theangiotensin II receptors. Pharmacol Rev 52, 415–472.
De Keulenaer GW, Andries LJ, Sys SU & Brutsaert DL (1995).Endothelin-mediated positive inotropic effect induced byreactive oxygen species in isolated cardiac muscle. Circ Res76, 878–884.
Dostal DE & Baker KM (1999). The cardiac renin-angiotensinsystem: conceptual, or a regulator of cardiac function? CircRes 85, 643–650.
Dzau VJ (1984). Vascular wall renin-angiotensin pathway incontrol of the circulation. A hypothesis. Am J Med 77, 31–36.
Dzau VJ (1988). Circulating versus local renin-angiotensinsystem in cardiovascular homeostasis. Circulation 77, I4–13.
Dzau VJ & Re RN (1987). Evidence for the existence of renin inthe heart. Circulation 75, I134–I136.
Evans HG, Lewis MJ & Shah AM (1994). Modulation ofmyocardial relaxation by basal release of endothelin fromendocardial endothelium. Cardiovasc Res 28, 1694–1699.
Gillebert TC & Raes DF (1994). Preload, length-tensionrelation, and isometric relaxation in cardiac muscle. Am JPhysiol Heart Circ Physiol 267, H1872–H1879.
Gomez Llambi H, Manni F, La Padula P, Carretero OA &Taquini CM (1996). Myocardial contractility is modulatedby angiotensin II via nitric oxide. Hypertension 27, 704–708.
Herizi A, Jover B, Bouriquet N & Mimran A (1998). Preventionof the cardiovascular and renal effects of angiotensin II byendothelin blockade. Hypertension 31, 10–14.
Heymes C, Vanderheyden M, Bronzwaer JG, Shah AM &Paulus WJ (1999). Endomyocardial nitric oxide synthase andleft ventricular preload reserve in dilated cardiomyopathy.Circulation 99, 3009–3016.
Ikeda U, Maeda Y, Kawahara Y, Yokoyama M & Shimada K(1995). Angiotensin II augments cytokine-stimulated nitricoxide synthesis in rat cardiac myocytes. Circulation 92,2683–2689.
Imai T, Hirata Y, Emori T, Yanagisawa M, Masaki T & MarumoF (1992). Induction of endothelin-1 gene by angiotensin andvasopressin in endothelial cells. Hypertension 19, 753–757.
Ito H, Hirata Y, Adachi S, Tanaka M, Tsujino M, Koike A,Nogami A, Murumo F & Hiroe M (1993). endothelin-1 is anautocrine/paracrine factor in the mechanism ofangiotensin II-induced hypertrophy in cultured ratcardiomyocytes. J Clin Invest 92, 398–403.
Just A, Olson AJ, Whitten CL & Arendshorst WJ (2007).Superoxide mediates acute renal vasoconstriction producedby angiotensin II and catecholamines by a mechanismindependent of nitric oxide. Am J Physiol Heart Circ Physiol292, H83–H92.
Kass DA, Bronzwaer JG & Paulus WJ (2004). Whatmechanisms underlie diastolic dysfunction in heart failure?Circ Res 94, 1533–1542.
Kifor I & Dzau VJ (1987). Endothelial renin-angiotensinpathway: evidence for intracellular synthesis and secretion ofangiotensins. Circ Res 60, 422–428.
Kim S & Iwao H (2000). Molecular and cellular mechanisms ofangiotensin II-mediated cardiovascular and renal diseases.Pharmacol Rev 52, 11–34.
Kim S, Ohta K, Hamaguchi A, Yukimura T, Miura K & Iwao H(1995). Angiotensin II induces cardiac phenotypicmodulation and remodeling in vivo in rats. Hypertension 25,1252–1259.
Kimbrough HM Jr, Vaughan ED Jr, Carey RM & Ayers CR(1977). Effect of intrarenal angiotensin II blockade on renalfunction in conscious dogs. Circ Res 40, 174–178.
C© 2009 The Authors. Journal compilation C© 2009 The Physiological Society

Exp Physiol 94.6 pp 665–674 Distensibility and endocardial endothelium 673
Kojda G, Kottenberg K, Nix P, Schluter KD, Piper HM & NoackE (1996). Low increase in cGMP induced by organic nitratesand nitrovasodilators improves contractile response of ratventricular myocytes. Circ Res 78, 91–101.
Kurisu S, Ozono R, Oshima T, Kambe M, Ishida T, Sugino H,Matsuura H, Chayama K, Teranishi Y, Iba O, Amano K &Matsubara H (2003). Cardiac angiotensin II type 2 receptoractivates the kinin/NO system and inhibits fibrosis.Hypertension 41, 99–107.
Ladeiras-Lopes R, Ferreira-Martins J & Leite-Moreira AF(2009). Acute neurohumoral modulation of diastolicfunction. Peptides 30, 419–425.
Layland J, Li JM & Shah AM (2002). Role of cyclicGMP-dependent protein kinase in the contractile responseto exogenous nitric oxide in rat cardiac myocytes. J Physiol540, 457–467.
Leite-Moreira AF & Bras-Silva C (2004). Inotropic effects ofETB receptor stimulation and their modulation byendocardial endothelium, NO, and prostaglandins. Am JPhysiol Heart Circ Physiol 287, H1194–H1199.
Leite-Moreira AF, Bras-Silva C, Pedrosa CA & Rocha-Sousa AA(2003). ET−1 increases distensibility of acutely loadedmyocardium: a novel ETA and Na+/H+ exchanger-mediatedeffect. Am J Physiol Heart Circ Physiol 284, H1332–H1339.
Leite-Moreira AF, Castro-Chaves P, Pimentel-Nunes P,Lima-Carneiro A, Guerra MS, Soares JB & Ferreira-Martins J(2006). Angiotensin II acutely decreases myocardial stiffness:a novel AT1, PKC and Na+/H+ exchanger-mediated effect.Br J Pharmacol 147, 690–697.
Levens NR, Freedlender AE, Peach MJ & Carey RM (1983).Control of renal function by intrarenal angiotensin II.Endocrinology 112, 43–49.
Levens NR, Peach MJ & Carey RM (1981). Role of theintrarenal renin-angiotensin system in the control of renalfunction. Circ Res 48, 157–167.
McClellan G, Weisberg A, Rose D & Winegrad S (1994).Endothelial cell storage and release of endothelin as acardioregulatory mechanism. Circ Res 75, 85–96.
McElmurray JH 3rd, Mukherjee R, New RB, Sampson AC, KingMK, Hendrick JW, Goldberg A, Peterson TJ, Hallak H, ZileMR & Spinale FG (1999). Angiotensin-converting enzymeand matrix metalloproteinase inhibition with developingheart failure: comparative effects on left ventricularfunction and geometry. J Pharmacol Exp Ther 291,799–811.
Mebazaa A, Mayoux E, Maeda K, Martin LD, Lakatta EG,Robotham JL & Shah AM (1993). Paracrine effects ofendocardial endothelial cells on myocyte contractionmediated via endothelin. Am J Physiol Heart Circ Physiol 265,H1841–H1846.
Mohan P, Brutsaert DL, Paulus WJ & Sys SU (1996).Myocardial contractile response to nitric oxide and cGMP.Circulation 93, 1223–1229.
Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M,Li H, Bodenschatz M, August M, Kleschyov AL, TsilimingasN, Walter U, Forstermann U, Meinertz T, Griendling K &Munzel T (2002). Effects of angiotensin II infusion on theexpression and function of NAD(P)H oxidase andcomponents of nitric oxide/cGMP signaling. Circ Res 90,E58–E65.
Moreau P, d’Uscio LV, Shaw S, Takase H, Barton M & LuscherTF (1997). Angiotensin II increases tissue endothelin andinduces vascular hypertrophy: reversal by ETA-receptorantagonist. Circulation 96, 1593–1597.
Mulrow PJ & Franco-Saenz R (1996). The adrenalrenin-angiotensin system: a local hormonal regulator ofaldosterone production. J Hypertens 14, 173–176.
Paul M, Poyan Mehr A & Kreutz R (2006). Physiology of localrenin-angiotensin systems. Physiol Rev 86, 747–803.
Paulus WJ, Vantrimpont PJ & Shah AM (1994). Acute effects ofnitric oxide on left ventricular relaxation and diastolicdistensibility in humans. Assessment by bicoronary sodiumnitroprusside infusion. Circulation 89, 2070–2078.
Pueyo ME, Arnal JF, Rami J & Michel JB (1998). Angiotensin IIstimulates the production of NO and peroxynitrite inendothelial cells. Am J Physiol Cell Physiol 274, C214–C220.
Re R (1987). The myocardial intracellular renin-angiotensinsystem. Am J Cardiol 59, 56A–58A.
Re R, Fallon JT, Dzau V, Ouay SC & Haber E (1982). Reninsynthesis by canine aortic smooth muscle cells in culture.Life Sci 30, 99–106.
Riggleman A, Harvey J & Baylis C (2001). Endothelin mediatessome of the renal actions of acutely administeredangiotensin II. Hypertension 38, 105–109.
Rossi GP, Sacchetto A, Cesari M & Pessina AC (1999).Interactions between endothelin-1 and therenin–angiotensin–aldosterone system. Cardiovasc Res 43,300–307.
Sarkar D, Vallance P, Amirmansour C & Harding SE (2000).Positive inotropic effects of NO donors in isolatedguinea-pig and human cardiomyocytes independent of NOspecies and cyclic nucleotides. Cardiovasc Res 48, 430–439.
Schluter KD & Wenzel S (2008). Angiotensin II: a hormoneinvolved in and contributing to pro-hypertrophic cardiacnetworks and target of anti-hypertrophic cross-talks.Pharmacol Ther 119, 311–325.
Serneri GG, Boddi M, Cecioni I, Vanni S, Coppo M, Papa ML,Bandinelli B, Bertolozzi I, Polidori G, Toscano T, MaccheriniM & Modesti PA (2001). Cardiac angiotensin II formation inthe clinical course of heart failure and its relationship withleft ventricular function. Circ Res 88, 961–968.
Seyedi N, Xu X, Nasjletti A & Hintze TH (1995). Coronarykinin generation mediates nitric oxide release afterangiotensin receptor stimulation. Hypertension 26, 164–170.
Shah AM & MacCarthy PA (2000). Paracrine and autocrineeffects of nitric oxide on myocardial function. PharmacolTher 86, 49–86.
Shah AM, Spurgeon HA, Sollott SJ, Talo A & Lakatta EG(1994). 8−Bromo-cGMP reduces the myofilament responseto Ca2+ in intact cardiac myocytes. Circ Res 74, 970–978.
Southan GJ & Szabo C (1996). Selective pharmacologicalinhibition of distinct nitric oxide synthase isoforms. BiochemPharmacol 51, 383–394.
Suzuki H, Eguchi K, Ohtsu H, Higuchi S, Dhobale S, FrankGD, Motley ED & Eguchi S (2006). Activation of endothelialnitric oxide synthase by the angiotensin II type 1 receptor.Endocrinology 147, 5914–5920.
Toda N, Ayajiki K & Okamura T (2007). Interaction ofendothelial nitric oxide and angiotensin in the circulation.Pharmacol Rev 59, 54–87.
C© 2009 The Authors. Journal compilation C© 2009 The Physiological Society

674 P. Castro-Chaves and others Exp Physiol 94.6 pp 665–674
Touyz RM & Schiffrin EL (2000). Signal transductionmechanisms mediating the physiological andpathophysiological actions of angiotensin II in vascularsmooth muscle cells. Pharmacol Rev 52, 639–672.
Vila-Petroff MG, Younes A, Egan J, Lakatta EG & Sollott SJ(1999). Activation of distinct cAMP-dependent andcGMP-dependent pathways by nitric oxide in cardiacmyocytes. Circ Res 84, 1020–1031.
Weber KT, Sun Y & Campbell SE (1995). Structuralremodelling of the heart by fibrous tissue: role of circulatinghormones and locally produced peptides. Eur Heart J16(Suppl N), 12–18.
Wollert KC & Drexler H (1999). The renin–angiotensin systemand experimental heart failure. Cardiovasc Res 43,838–849.
C© 2009 The Authors. Journal compilation C© 2009 The Physiological Society



















