
Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by...
12
05.10.15 14:02 Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A Page 1 of 12 http://hyper.ahajournals.org/content/30/6/1598.full + Hypertension Hypertension hyper.ahajournals.org Hypertension. 1997; 1997; 30: 1598-1605 1598-1605 doi: 10.1161/01.HYP.30.6.1598 doi: 10.1161/01.HYP.30.6.1598 Articles Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A Johann Bauersachs Johann Bauersachs, , Ingrid Fleming Ingrid Fleming, , Dimitri Scholz Dimitri Scholz, , Rüdiger Popp Rüdiger Popp, Rudi Busse Rudi Busse Author Affiliations Correspondence to Dr Johann Bauersachs, Institut für Kardiovaskuläre Physiologie, Zentrum der Physiologie, Klinikum der J.W. Goethe-Universität, Theodor-Stern-Kai 7, D-60590 Frankfurt am Main, Germany. E-mail [email protected] Abstract Abstract The subcellular localization of the enzymes synthesizing endothelium- derived vasodilator autacoids has been proposed to play a role in determining the ability of endothelial cells to enhance autacoid production in response to stimulation. We therefore investigated the effects of brefeldin A–induced disruption of the Golgi apparatus and Golgi-plasma membrane trafficking on the production of nitric oxide (NO), prostacyclin, and the endothelium-derived hyperpolarizing factor (EDHF) by native and cultured endothelial cells. In porcine coronary artery segments, brefeldin A (35 μmol/L, 90 minutes) did not affect relaxations to sodium nitroprusside or the K channel opener cromakalim but elicited a rightward shift in the concentration-response curve to bradykinin without altering the maximum vasodilator response (R max ). Brefeldin A failed to attenuate the bradykinin-induced, NO-mediated relaxation under depolarizing conditions but inhibited the bradykinin response under conditions of combined cyclooxygenase/NO synthase blockade, suggesting that this agent selectively interferes with the production of EDHF. Indeed, incubation of porcine coronary arteries with brefeldin A, which did not affect the bradykinin-induced accumulation of either cyclic GMP or 6-keto-prostaglandin F 1α , markedly and reversibly attenuated the EDHF-mediated hyperpolarization of detector smooth muscle cells in a patch-clamp bioassay system. The microtubule destabilizer nocodazole also affected both the EC 50 and R max to bradykinin in porcine coronary arteries. Since EDHF is thought to be a cytochrome P450–derived metabolite of arachidonic acid and both brefeldin A and nocodazole are known to interfere with the targeting of cytochrome P450 from the Golgi apparatus to the plasma membrane, it is conceivable that brefeldin A inhibits EDHF formation by preventing the targeting of the EDHF-synthesizing enzymes to the plasma membrane. Key Words: Key Words: endothelium-derived hyperpolarizing factor nitric oxide cytochrome P450 brefeldin A microtubule prostacyclin endothelial cells Endothelial cells synthesize and release the vasodilator autacoids NO and prostacyclin as well as the so-called EDHF (for review, see Reference 1 ). While the intracellular site of synthesis of the more unstable autacoids (NO and EDHF) is likely to have important implications on their sphere of influence and thus biological activity, the cellular distribution of the respective generating enzymes and the relationship between localization and enzymatic activity are unclear. + 1
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by...

05.10.15 14:02Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A
Page 1 of 12http://hyper.ahajournals.org/content/30/6/1598.full
++
HypertensionHypertensionhyper.ahajournals.org
Hypertension. 1997; 1997; 30: 1598-16051598-1605doi: 10.1161/01.HYP.30.6.1598doi: 10.1161/01.HYP.30.6.1598
ArticlesEndothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, IsReversibly Inhibited by Brefeldin A
Johann BauersachsJohann Bauersachs, , Ingrid FlemingIngrid Fleming, , Dimitri ScholzDimitri Scholz, , Rüdiger PoppRüdiger Popp,,Rudi BusseRudi Busse
Author Affiliations
Correspondence to Dr Johann Bauersachs, Institut für Kardiovaskuläre Physiologie,Zentrum der Physiologie, Klinikum der J.W. Goethe-Universität, Theodor-Stern-Kai7, D-60590 Frankfurt am Main, Germany. E-mail [email protected]
Abstract
Abstract The subcellular localization of the enzymes synthesizing endothelium-derived vasodilator autacoids has been proposed to play a role in determining theability of endothelial cells to enhance autacoid production in response tostimulation. We therefore investigated the effects of brefeldin A–induceddisruption of the Golgi apparatus and Golgi-plasma membrane trafficking on theproduction of nitric oxide (NO), prostacyclin, and the endothelium-derivedhyperpolarizing factor (EDHF) by native and cultured endothelial cells. In porcinecoronary artery segments, brefeldin A (35 µmol/L, 90 minutes) did not affectrelaxations to sodium nitroprusside or the K channel opener cromakalim butelicited a rightward shift in the concentration-response curve to bradykininwithout altering the maximum vasodilator response (Rmax). Brefeldin A failed toattenuate the bradykinin-induced, NO-mediated relaxation under depolarizingconditions but inhibited the bradykinin response under conditions of combinedcyclooxygenase/NO synthase blockade, suggesting that this agent selectivelyinterferes with the production of EDHF. Indeed, incubation of porcine coronaryarteries with brefeldin A, which did not affect the bradykinin-inducedaccumulation of either cyclic GMP or 6-keto-prostaglandin F1α, markedly andreversibly attenuated the EDHF-mediated hyperpolarization of detector smoothmuscle cells in a patch-clamp bioassay system. The microtubule destabilizernocodazole also affected both the EC50 and Rmax to bradykinin in porcinecoronary arteries. Since EDHF is thought to be a cytochrome P450–derivedmetabolite of arachidonic acid and both brefeldin A and nocodazole are known tointerfere with the targeting of cytochrome P450 from the Golgi apparatus to theplasma membrane, it is conceivable that brefeldin A inhibits EDHF formation bypreventing the targeting of the EDHF-synthesizing enzymes to the plasmamembrane.
Key Words:Key Words:endothelium-derived hyperpolarizing factornitric oxidecytochrome P450brefeldin Amicrotubuleprostacyclinendothelial cells
Endothelial cells synthesize and release the vasodilator autacoids NO andprostacyclin as well as the so-called EDHF (for review, see Reference 1 ). Whilethe intracellular site of synthesis of the more unstable autacoids (NO and EDHF) islikely to have important implications on their sphere of influence and thusbiological activity, the cellular distribution of the respective generating enzymesand the relationship between localization and enzymatic activity are unclear.
+
1

05.10.15 14:02Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A
Page 2 of 12http://hyper.ahajournals.org/content/30/6/1598.full
There are conflicting reports concerning the major intracellular site of NOformation and the impact of endothelial NO synthase (NOS III) localization on thegeneration of bioactive NO. Histochemical studies suggested that NOS III ispredominantly situated in the Golgi apparatus, and association with thisorganelle is reportedly necessary for the enzyme to respond to intracellularsignals and for efficient NO synthesis. However, more recent studies haveindicated that functionally active NOS III is concentrated in caveolin-richmembrane domains, although NOS III, and the caveola marker proteincaveolin, may cycle from the Golgi apparatus to caveolae.
Compelling evidence exists for the release of a diffusible endothelium-derivedautacoid that induces relaxation by hyperpolarizing the underlying smooth musclecell layers. However, since the chemical identity of this EDHF is still elusive,no information is available regarding the intracellular localization of the “EDHFsynthase.” Pharmacological studies have, however, suggested that the EDHFgenerated by the coronary and femoral endothelium may be anarachidonic acid metabolite generated by cytochrome P450–dependent enzymes,which are normally present in the endoplasmic reticulum.
The aim of the present study was to determine the role of the Golgi apparatus andthe endoplasmic reticulum in the synthesis of all three vasodilator autacoids, ie,NO, prostacyclin, and EDHF, by endothelial cells. We therefore investigated theeffects of brefeldin A, which causes reversible disassembly of the Golgi apparatusand swelling of the endoplasmic reticulum, on autacoid formation in nativeporcine coronary artery and cultured human endothelial cells. Since brefeldin Aalso affects microtubule-dependent transport processes, we further studied theeffect of interfering with microtubule polymerization and depolymerization onendothelial autacoid production.
Methods
Materials
Bradykinin was from Bachem Biochemica GmbH; HEPES and L-NNA were fromServa. Taxol was obtained from Calbiochem, and nocodazol from Fluka Chemika.Diclofenac (Voltaren injection solution) was from CIBA-Geigy, and U46619 (9,11-dideoxy-11α,9 α-epoxymethano-prostaglandin F2α) was provided by Upjohn.The specific NOS III antibody was purchased from Transduction Laboratories.Brefeldin A, cromakalim, phenylephrine, ACh, isobutyl-methyl-xanthine (IBMX),SNP, and all other chemicals were purchased from Sigma Chemical.
Cell Culture
Human umbilical vein or porcine aortic endothelial cells were seeded onfibronectin-coated culture dishes containing medium M-119 (GIBCO Berlin) and20% heat-inactivated fetal calf serum (Vitromex) supplemented with penicillin (50U/mL), streptomycin (50 µg/mL), L-glutamine (1 mmol/L), glutathione (5 mg/mL),and L(+)-ascorbic acid (5 mg/mL). Rat aortic smooth muscle cells were isolatedand cultured as described. Confluent cultures of smooth muscle cells werepassaged by using trypsin-EDTA (1 mmol/L), and experiments were performedusing cells between passages 12 and 16.
Organ Bath Studies
Porcine coronary artery or rabbit aortic rings (3 mm in length) were mountedbetween force transducers (Scaime) and a rigid support for measurement ofisometric force and incubated in organ baths containing warmed (37°C),oxygenated (95% O2, 5% CO2) Krebs-Henseleit solution (pH 7.4) of the followingcomposition (mmol/L): NaCl 118.4; NaHCO3 25.0; KCl 4.7; CaCl2 1.6; KH2PO4 1.2;MgSO4 1.2; and glucose 11.1.
Porcine Coronary Arteries
Passive tension was adjusted over a 60-minute equilibration period to 5 g;thereafter, the segments were repeatedly exposed to KCl (70 mmol/L) until stableconstrictions were obtained. After washing, 10 g of tension (≈70% of the maximalconstriction) was applied by constriction using U46619 (0.1 to 0.3 µmol/L). Theintegrity of the endothelium was assessed using the endothelium-dependentvasodilator bradykinin (1 µmol/L), and vessels exhibiting <70% relaxation were
2 3
4
5 67
8 9 10
10 11 12 13 14 15
16
17
10

05.10.15 14:02Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A
Page 3 of 12http://hyper.ahajournals.org/content/30/6/1598.full
discarded.
Rabbit Aortae
Passive tension was adjusted over a 30-minute equilibration period to 2 g;thereafter, the segments were repeatedly exposed to phenylephrine (1 µmol/L)until stable constrictions were obtained (≈70% of the maximal constriction). Theintegrity of the endothelium was assessed using ACh (1 µmol/L), and vesselsexhibiting <70% relaxation were discarded.
All vessels used were preconstricted to the same tension using the appropriateagonist before the addition of the vasodilator (bradykinin or ACh). As the relaxantresponses observed were transient, the dose-response curve represents the peakresponse observed to cumulative applications of the agonists.
Detection of EDHF Release
The release of EDHF was detected by recording changes in the membranepotential of cultured rat aortic smooth muscle cells exposed to the effluate of aperfused porcine coronary artery. Briefly, a segment of porcine coronary arterywas cannulated at both ends, mounted in an organ chamber, and perfusedintraluminally (1 mL/min) with PSS of the following composition (mmol/L): NaCl140; KCl 4.7; MgCl2 1; CaCl2 1.3; glucose 5; HEPES 10 (pH 7.4, 37°C) containingthe NOS inhibitor L-NNA (100 µmol/L) and the cyclooxygenase inhibitordiclofenac (1 µmol/L). Effluate from the segment superfused cultured smoothmuscle cells, the membrane potential of which was recorded by the slow whole-cell configuration of the patch-clamp technique using nystatin (100 µg/mL) in thepipette as described. The cell membrane potentials, measured in the currentclamp mode, were recorded continuously. Only detector smooth muscle cells thathad a stable resting membrane potential for more than 2 minutes and exhibitedno further change in the input resistance were used in the bioassay system.
Detection of Prostacyclin Release
In a separate series of experiments, the concentration of 6-keto PGF1α, the stablehydrolysis product of prostacyclin, was measured by a specific radioimmunoassayeither in the effluate of perfused (0.5 mL/min) porcine coronary arteries (collectedfor 3 minutes before and after application of bradykinin, 0.1 µmol/L) or in thesupernatant of cultured human endothelial cells under resting conditions andafter stimulation with bradykinin (10 nmol/L, 5 minutes).
Determination of Cyclic GMP Concentration
The concentration of cyclic GMP in confluent cultured human endothelial cellsunder resting conditions and after stimulation with bradykinin (10 nmol/L, 5minutes) in the presence of the phosphodiesterase inhibitor IBMX (0.1 mmol/L)was determined with a specific radioimmunoassay.
Isolation of Caveolin-Rich Membrane Domains and Determination of NOS IIIActivity
Freshly isolated porcine aortae were slit longitudinally, mounted in an openchamber, washed twice in HEPES-modified Tyrode solution (mmol/L: NaCl 132,KCl 4, CaCl2 1, MgCl2 0.5, HEPES 9.5, and glucose 5), and the exposed endotheliallayer was incubated at 37°C in the presence and absence of agonists as indicatedin “Results.” Thereafter, the incubation was stopped by exchanging the incubationmedium with ice-cold HEPES buffer, and the cells were harvested by scraping.Thereafter, the cytosolic cell fraction and caveolin-rich membrane domains wereprepared by detergent-free sequential centrifugation as described, and thepresence of NOS III in each fraction was determined by Western blotting afterseparation of proteins by SDS-PAGE. NOS activity in aliquots (4 µg protein) of thecytosolic and caveolae cell fractions was determined by assessing the L-NNA (1mmol/L)–sensitive conversion of L-[ H]arginine to L-[ H]citrulline as describedpreviously.
Assay of Cytochrome P450 Activity
Cytochrome P450–dependent metabolic activity was assayed as the dealkylation of7-ethoxyresorufin in confluent porcine aortic endothelial cells cultivated in theabsence and presence of β-naphthoflavone (3 µmol/L, 48 hours).
10 18
19
3 320
10

05.10.15 14:02Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A
Page 4 of 12http://hyper.ahajournals.org/content/30/6/1598.full
View larger version:In this page In a new windowDownload as PowerPoint Slide
View this table:In this window In a new window
Immunofluorescence Experiments
Samples (cells or vessels) were fixed using 4% formaldehyde in phosphate-buffered saline for 30 minutes, permeabilized in 0.05% Triton X-100 for 10minutes followed by blocking in 100 mmol/L glycine for 10 minutes. Thereafter,samples were incubated in a reaction buffer consisting of 0.01% Triton X-100,0.1% carboxylated bovine serum albumin (AURION, Netherlands) in phosphate-buffered saline, pH 7.6. The fixed samples were incubated for 1 hour with amouse monoclonal antibody directed against the NOS III (3 µg/mL, TransductionLaboratories) in a 50-mmol/L TRIS solution (pH 7.5) containing NaCl 200 mmol/L,Triton X-100 0.2%, bovine serum albumin 3%, and horse serum 10%. Sampleswere then incubated with a biotinylated donkey anti-mouse antibody for 1 hourfollowed by streptavidin-Cy2 (Biotrend). After nuclear staining with 7-amino-actinomycin D (Molecular Probes), the preparations were mounted in Mowiol(Hoechst) and examined using a confocal microscope (Leica TCS).
Statistics
Relaxant responses are given as percentage relaxation relative to preconstrictionlevels. All data in the figures and in the text are expressed as mean±SEM of nexperiments using segments from different animals. Statistical analysis wasperformed by one-way ANOVA followed by a Bonferroni t test or by the Mann-Whitney test for unpaired data where appropriate, with values of P<.05 beingconsidered statistically significant.
Results
Effects of Brefeldin A on Endothelium-Dependent and -IndependentRelaxations
In porcine coronary artery segments preconstricted with U46619 (0.1 to 0.3µmol/L), brefeldin A (35 µmol/L, 90 minutes) elicited a rightward shift in theconcentration-response curve to bradykinin (Fig 1⇓, Table 1⇓) but did not alterthe maximum vasodilator response (Rmax). The effect of brefeldin A was selectivefor endothelium-dependent vasodilation because brefeldin A treatment did notaffect relaxations induced by SNP (Table 1⇓) or the K channel opener cromakalim(0.1, 1 µmol/L; Rmax, control 89±9% versus 98±2% after brefeldin A treatment).
Figure 1.Figure 1.
Effects of brefeldin A on thebradykinin (BK)-inducedrelaxations in porcine coronaryarteries preconstricted withU46619 (0.1 to 0.3 µmol/L).Original tracings (A) andstatistical summary (B) ofexperiments performed in theabsence (○) and presence (•) ofbrefeldin A (35 µmol/L, 90minutes). Results are expressedas the mean±SEM of 7 separateexperiments. *P<.05, **P<.01 vs
control.
Table 1.Table 1.
EC50 Values and MaximumRelaxation (Rmax) forEndothelium-DependentRelaxation to Bradykinin and Endothelium-Independent Relaxation toSNP in Porcine Coronary Arteries in the Absence or Presence of BrefeldinA
Effect of Brefeldin A on NO Production
In porcine coronary artery segments preconstricted with depolarizing
+
05.10.15 14:02Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A
Page 5 of 12http://hyper.ahajournals.org/content/30/6/1598.full
View larger version:In this page In a new windowDownload as PowerPoint Slide
View larger version:In this page In a new windowDownload as PowerPoint Slide
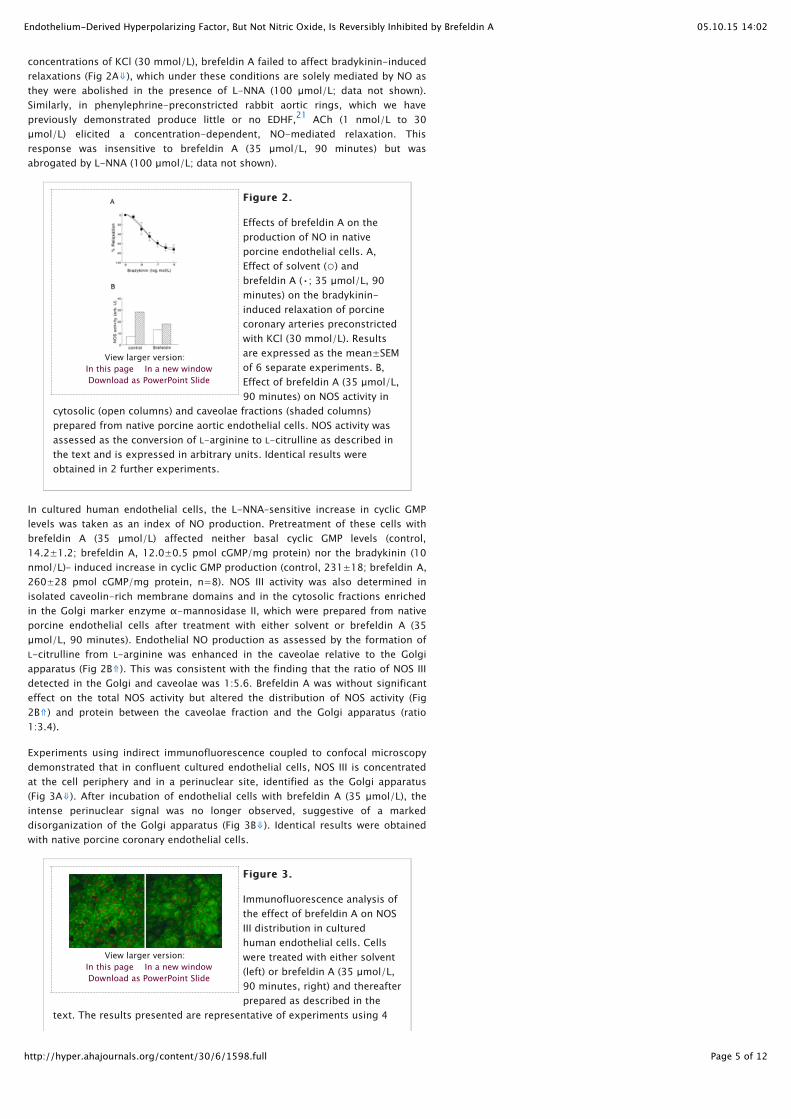
concentrations of KCl (30 mmol/L), brefeldin A failed to affect bradykinin-inducedrelaxations (Fig 2A⇓), which under these conditions are solely mediated by NO asthey were abolished in the presence of L-NNA (100 µmol/L; data not shown).Similarly, in phenylephrine-preconstricted rabbit aortic rings, which we havepreviously demonstrated produce little or no EDHF, ACh (1 nmol/L to 30µmol/L) elicited a concentration-dependent, NO-mediated relaxation. Thisresponse was insensitive to brefeldin A (35 µmol/L, 90 minutes) but wasabrogated by L-NNA (100 µmol/L; data not shown).
Figure 2.Figure 2.
Effects of brefeldin A on theproduction of NO in nativeporcine endothelial cells. A,Effect of solvent (○) andbrefeldin A (•; 35 µmol/L, 90minutes) on the bradykinin-induced relaxation of porcinecoronary arteries preconstrictedwith KCl (30 mmol/L). Resultsare expressed as the mean±SEMof 6 separate experiments. B,Effect of brefeldin A (35 µmol/L,90 minutes) on NOS activity in
cytosolic (open columns) and caveolae fractions (shaded columns)prepared from native porcine aortic endothelial cells. NOS activity wasassessed as the conversion of L-arginine to L-citrulline as described inthe text and is expressed in arbitrary units. Identical results wereobtained in 2 further experiments.
In cultured human endothelial cells, the L-NNA–sensitive increase in cyclic GMPlevels was taken as an index of NO production. Pretreatment of these cells withbrefeldin A (35 µmol/L) affected neither basal cyclic GMP levels (control,14.2±1.2; brefeldin A, 12.0±0.5 pmol cGMP/mg protein) nor the bradykinin (10nmol/L)– induced increase in cyclic GMP production (control, 231±18; brefeldin A,260±28 pmol cGMP/mg protein, n=8). NOS III activity was also determined inisolated caveolin-rich membrane domains and in the cytosolic fractions enrichedin the Golgi marker enzyme α-mannosidase II, which were prepared from nativeporcine endothelial cells after treatment with either solvent or brefeldin A (35µmol/L, 90 minutes). Endothelial NO production as assessed by the formation ofL-citrulline from L-arginine was enhanced in the caveolae relative to the Golgiapparatus (Fig 2B⇑). This was consistent with the finding that the ratio of NOS IIIdetected in the Golgi and caveolae was 1:5.6. Brefeldin A was without significanteffect on the total NOS activity but altered the distribution of NOS activity (Fig2B⇑) and protein between the caveolae fraction and the Golgi apparatus (ratio1:3.4).
Experiments using indirect immunofluorescence coupled to confocal microscopydemonstrated that in confluent cultured endothelial cells, NOS III is concentratedat the cell periphery and in a perinuclear site, identified as the Golgi apparatus(Fig 3A⇓). After incubation of endothelial cells with brefeldin A (35 µmol/L), theintense perinuclear signal was no longer observed, suggestive of a markeddisorganization of the Golgi apparatus (Fig 3B⇓). Identical results were obtainedwith native porcine coronary endothelial cells.
Figure 3.Figure 3.
Immunofluorescence analysis ofthe effect of brefeldin A on NOSIII distribution in culturedhuman endothelial cells. Cellswere treated with either solvent(left) or brefeldin A (35 µmol/L,90 minutes, right) and thereafterprepared as described in the
text. The results presented are representative of experiments using 4
21

05.10.15 14:02Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A
Page 6 of 12http://hyper.ahajournals.org/content/30/6/1598.full
View larger version:In this page In a new windowDownload as PowerPoint Slide
different cell batches.
Effect of Brefeldin A on Prostacyclin Release
In porcine coronary arteries, prostacyclin does not contribute appreciably toagonist-induced endothelium-dependent relaxations as confirmed by the lack ofeffect of diclofenac on bradykinin-induced relaxations (data not shown).Therefore, formation of prostacyclin was assessed as the accumulation of thestable derivative 6-keto-PGF1α in the effluate from porcine coronary arterysegments. The application of bradykinin (0.1 µmol/L) elicited an increase in therelease of 6-keto-PGF1α from 1437±190 to 2479±296 pg 6-keto-PGF1α/mL(n=8). Pretreatment with brefeldin A (35 µmol/L) affected neither the basal(1564±203 pg 6-keto-PGF1α/mL) nor the bradykinin-induced increase in 6-keto-PGF1α (2291±278 pg 6-keto-PGF1α/mL). Brefeldin A also failed to affect thebradykinin-induced increase in 6-keto-PGF1α in cultured human endothelial cells(control, 72.5±17.5 versus 1925±213 pg 6-keto-PGF1α/mg protein; brefeldin A,87.5±25.0 versus 2275±174 pg 6-keto-PGF1α/mg protein, in the absence andpresence of bradykinin, respectively).
Inhibition of EDHF Release by Brefeldin A
In the combined presence of diclofenac (1 µmol/L) and L-NNA (100 µmol/L),conditions under which endothelium-dependent relaxation is mediated entirely byEDHF, brefeldin A induced a pronounced rightward shift in the concentration-response curve to bradykinin as well as a marked attenuation of Rmax in porcinecoronary arteries. The inhibitory effect of brefeldin A on EDHF production wasreversible, since washout (3 hours) resulted in an almost complete restoration ofthe bradykinin-induced EDHF-mediated relaxation (Fig 4C⇓).
Figure 4.Figure 4.
Effects of brefeldin A on thebradykinin (BK)-inducedrelaxation of porcine coronaryarteries preconstricted withU46619 (0.1 to 0.3 µmol/L) inthe combined presence ofdiclofenac (1 µmol/L) and L-NNA (100 µmol/L). A, Originaltracings showing bradykinin-induced relaxation in theabsence and presence ofbrefeldin A. B, Statisticalsummary of experiments
performed in the absence (○) and presence (•) of brefeldin A (35µmol/L, 90 minutes). C, Statistical summary of experiments performedeither in the absence of brefeldin A (○) or following its washout (•).Results are expressed as the mean±SEM of 5 to 7 separateexperiments. **P<.01 vs control.
The effects of brefeldin A on EDHF production were further analyzed in a patch-clamp setup for the detection of EDHF. In this system, the effects of brefeldin A onEDHF release from the donor endothelium were investigated separately fromeffects on the membrane potential and EDHF-induced hyperpolarization of thedetector cells.
Stimulation of the donor porcine coronary arteries with bradykinin (0.1 µmol/L) inthe presence of both diclofenac and L-NNA led to the hyperpolarization (14±2mV, n=6) of detector smooth muscle cells situated downstream from the donor.After pretreatment of the donor segments with brefeldin A (35 µmol/L), thebradykinin-induced, EDHF-mediated hyperpolarization of detector smooth musclecells was significantly reduced (Fig 5⇓). Removal of brefeldin A from the donorperfusate (3 hours) resulted in a substantial restoration of the smooth muscle cellhyperpolarization elicited by effluate from bradykinin-stimulated arterialsegments (Fig 5⇓). The effect of brefeldin A could be attributed to inhibition ofthe release of EDHF from the donor artery because the direct application of

05.10.15 14:02Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A
Page 7 of 12http://hyper.ahajournals.org/content/30/6/1598.full
View larger version:In this page In a new windowDownload as PowerPoint Slide
View larger version:In this page In a new windowDownload as PowerPoint Slide
brefeldin A (35 µmol/L, 90 minutes) to smooth muscle cells failed to affect eitherthe resting membrane potential or the hyperpolarization elicited by the perfusatefrom bradykinin-stimulated donor arteries (data not shown).
Figure 5.Figure 5.
Effect of brefeldin A on thehyperpolarization elicited by theEDHF in cultured rat aorticsmooth muscle cells. Themembrane potential wasrecorded (patch clamp, currentclamp mode) in detector cellsexposed to the effluate of aluminally perfused porcinecoronary artery segmentpretreated with L-NNA (100µmol/L) and diclofenac (1µmol/L). A, Original tracings
showing the bradykinin (100 nmol/L)–induced hyperpolarization in theabsence and presence of brefeldin A (BfA; 35 µmol/L, 90 minutes) andafter its washout (3 hours). B, Summarized data showing thebradykinin-induced, EDHF-mediated hyperpolarization of vascularsmooth muscle cells in the absence (open column), presence (shadedcolumn), and after washout (hatched column) of brefeldin A. Results areexpressed as the mean±SEM of 6 separate experiments. *P<.05 vscontrol.
Figure 6.Figure 6.
Effect of nocodazol onbradykinin-induced relaxationof porcine coronary arteriespreconstricted with U46619 (0.1to 0.3 µmol/mL). Experimentswere performed in the absence(○) and presence (•) ofnocodazol (30 µmol/L, 90
minutes) and in the absence (A) or presence (B) of both diclofenac (1µmol/L) and L-NNA (100 µmol/L). Results are expressed as the mean±SEM of 8 separate experiments.
Brefeldin A (35 µmol/L) was without effect on basal P450 oxygenase activity asassessed in either solvent or β-naphthoflavone-treated porcine aortic endothelialcells (control, 4.5±0.5 versus brefeldin A; 4.7±0.4 nmol resorufin · L · minin solvent-treated cells and control, 68±8 versus brefeldin A, 71±7 nmolresorufin · L · min in β-naphthoflavone–treated cells; n=12).
Effects of Nocodazol and Taxol on Agonist-Induced Relaxations in PorcineCoronary Arteries
The effects of interfering with microtubule polymerization and depolymerizationon endothelial autacoid production were investigated using nocodazole andtaxol, which have been previously shown to interfere with microtubuleorganization. Treatment of porcine coronary arteries with nocodazole (30µmol/L, 90 minutes) failed to affect the mainly NO-mediated relaxation of arteriesin response to bradykinin (Table 2⇓). A distinct attenuation of both the EC50 andRmax to bradykinin was observed when the effects of nocodazole wereinvestigated in rings pretreated with L-NNA; however, this effect just failed toattain statistical significance. Endothelium-independent relaxation to SNP was notaffected by nocodazole (Table 2⇓). Taxol (10 µmol/L, 120 minutes), whichreportedly stabilizes microtubules, was without effect on the bradykinin-inducedproduction of either NO or EDHF (Table 2⇓).
Table 2.Table 2.
−1 −1
−1 −1
22 2324

05.10.15 14:02Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A
Page 8 of 12http://hyper.ahajournals.org/content/30/6/1598.full
View this table:In this window In a new window
EC50 Values and MaximumRelaxation (Rmax) forEndothelium-DependentRelaxation to Bradykinin andEndothelium-Independent Relaxation to SNP in Porcine CoronaryArteries in the Absence or Presence of Nocodazol or Taxol
Discussion
In the present study, we demonstrated that in both native and cultured endothelialcells the disruption of the Golgi apparatus by brefeldin A does not alter NO orprostacyclin formation but leads to a pronounced inhibition of EDHF production.Brefeldin A is therefore the first inhibitor described to date that selectively andreversibly interferes with the generation of EDHF in endothelial cells.
Two intracellular pools of NOS III protein, the Golgi apparatus and caveolae, havebeen described in native and cultured endothelial cells. However, contradictoryreports exist regarding the relative contribution of these intracellular sites to theproduction of bioactive NO. Indeed, while functionally active NOS III isconcentrated in caveolae prepared from native and cultured endothelial cells, brefeldin A–induced disruption of the Golgi apparatus has been reported toabrogate NO production in cerebrovascular endothelial cells. In the presentstudy, brefeldin A failed to affect NO production in either native or culturedendothelial cells despite complete loss of the immunofluorescent perinuclear NOSIII signal.
Biochemical analysis revealed that NOS III protein was up to six times moreconcentrated in caveolae than in the Golgi-containing cytosolic fraction, a findingthat is reflected by the differences in NOS activity detected in the two subcellularfractions. Although brefeldin A did not attenuate overall NOS activity, it did inducea distinct decrease in caveolar and an increase in cytosolic NO formation. Thisbrefeldin A–induced change in NOS activity was accompanied by a shift in thedistribution of NOS III between these two cellular compartments. Although underdepolarizing conditions brefeldin A did not attenuate NO-mediated relaxation, wecannot exclude the possibility that the shift in the concentration-response curveto bradykinin under control conditions may be partially attributable to the effectsof brefeldin A on NOS III compartmentalization. Moreover, since brefeldin Ainhibits endoplasmic reticulum/Golgi trafficking, our results suggest that there isa rapid turnover of the caveolae-associated NOS III and that a continual exchangeof NOS III takes place between the Golgi apparatus and caveolae in nativeendothelial cells.
The lack of effect of brefeldin A on prostacyclin formation in porcine coronary andcultured human endothelial cells indicates that disorganization of the Golgiapparatus and endoplasmic reticulum has, as expected, no effect on theenzymatic activity of either cyclooxygenase I or phospholipase A2, both of whichare cytosolic enzymes.
The reversible inhibition of the bradykinin-induced, NO/prostacyclin–independentvasodilation of porcine coronary arteries suggests that brefeldin A selectivelyaffects the EDHF synthesis/signaling pathway. EDHF-induced hyperpolarization isbrought about by an increase in the K conductance of vascular smooth musclecells via activation of Ca -dependent K (K Ca) channels and is abolished byselective K Ca channel inhibitors. The transient nature of the EDHF-induced hyperpolarization observed in patch-clamp bioassay is also evident inintact vascular segments where the hyperpolarization of smooth muscle usuallyprecedes and is more transient than the accompanying relaxation induced byendothelium-dependent vasodilators.
Since neither the resting membrane potential nor the hyperpolarization inducedby EDHF released from an untreated segment were affected by prolongedincubation of detector cells with brefeldin A, the inhibitory effect of brefeldin A onthe EDHF response appears unrelated to effects on the EDHF-mediated activationof K Ca channels. Moreover, in our patch-clamp setup, an attenuated EDHF-induced hyperpolarization of detector vascular smooth muscle cells was observedonly after incubation of the donor artery with brefeldin A. Additional studies usinga variety of different cell types have also failed to observe a direct inhibitory effect
1 2
5 6
3 4 55 6
25
+2+ + +
+ 9 10 12 14
26 27 28
+
+ −1 2+ 29 30 31

05.10.15 14:02Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A
Page 9 of 12http://hyper.ahajournals.org/content/30/6/1598.full
of brefeldin A on K , Cl , and Ca channel activity.
It is possible only to speculate on the mechanism by which brefeldin A inhibitsEDHF synthesis, since the chemical identity of the EDHF produced by the coronaryendothelium has not been fully elucidated. Experimental evidence, however,suggests that this factor may be a cytochrome P450–derived metabolite ofarachidonic acid, such as an epoxyeicosatrienoic acid. Moreover, inductionof cytochrome P450 enzymes using β-naphthoflavone has been shown to enhancethe release of EDHF from native rat mesenteric as well as cultured porcine andhuman endothelial cells. Recently, a specific cannabinoid receptor (CB1)antagonist has been shown to attenuate EDHF-induced relaxation of isolated ratmesenteric vessels, whereas anandamide, an endogenous cannabinoid, elicited“EDHF-like” relaxations. These results do not necessarily imply that twocandidates for EDHF exist. Indeed, anandamide can be enzymatically synthesizedfrom, and may well prove to be a carrier of, arachidonic acid, the putativeprecursor of EDHF. Moreover, anandamide can be metabolized by cytochromeP450, and the induction of P450 enzymes results in the increased formation ofseveral anandamide metabolites. The mechanism by which brefeldin Aattenuates EDHF formation, however, does not appear to be related to a decreasedavailability of arachidonic acid, since the lack of effect of brefeldin A onprostacyclin formation suggests that the liberation of arachidonic acid is notinhibited. Nor does brefeldin A treatment result in the global inhibition ofcytochrome P450–dependent monooxygenase activity, since this agent did notaffect the ability of endothelial cells to dealkylate the P450 substrate 7-ethoxyresorufin. Rather, the effects of brefeldin A on the trafficking of proteinsbetween the endoplasmic reticulum, Golgi apparatus, and the plasma membranemay be responsible for the observed inhibition of EDHF release. Althoughcytochrome P450 is synthesized on polyribosomes bound to the endoplasmicreticulum and is cotranslationally inserted into the endoplasmic reticulummembrane, there is an extensive flow of vesicles to the Golgi apparatus as wellas a microtubule-dependent transport from the Golgi to the plasma membrane.Indeed, the trafficking of P4502B between the Golgi apparatus and the plasmamembrane has been demonstrated in cultured rat hepatocytes. In the latterstudy, moreover, incubation of hepatocytes with brefeldin A for up to 2 hoursessentially suppressed the expression and activity of P4502B on the plasmamembrane without inducing a measurable decrease in total cellular P450activity. That a similar phenomenon is responsible for the effects of brefeldin Aon EDHF production is supported by the observation that the microtubule inhibitornocodazole also attenuated EDHF-mediated relaxations in the present study. Thefinding that the inhibitory effect of nocodazole was not as marked as that inducedby brefeldin A may also be explained by the fact that this agent was only half aseffective as brefeldin A in reducing the expression of P450 at the plasmamembrane. Corroboration of this hypothesis requires experiments usingantibodies raised against the cytochrome P450 implicated in the generation ofEDHF and must therefore wait until a suitable candidate has been proposed fromthe multitude of isoforms described to date.
In summary, our data indicate that acute disruption of the Golgi apparatus andinhibition of protein transport to the plasma membrane by brefeldin A have littleeffect on NO or prostacyclin production but almost completely inhibits thesynthesis of EDHF by endothelial cells. Thus, brefeldin A, which inhibits a decisivestep in the EDHF-forming pathway (probably by preventing the targeting of thenecessary enzymatic machinery to the plasma membrane), will be an invaluabletool for the identification of this elusive factor.
Acknowledgments
This study was supported by the Deutsche Forschungsgemeinschaft (Bu 436/6-1)and the Commission of the European Communities (BMH4-CT96-0979). Theauthors are indebted to Isabel Winter, Andreas Schäfer and Michaela Stächele forexpert technical assistance.
Received March 3, 1997.Revision received March 19, 1997.Accepted June 6, 1997.
References
1. Fleming I, Bauersachs J, Busse R. Paracrine functions of the coronary vascular
+ −1 2+ 29 30 31
13 14 32
10 28
33
34
35
3637
38
38
38
39

05.10.15 14:02Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A
Page 10 of 12http://hyper.ahajournals.org/content/30/6/1598.full
endothelium. Mol Cell Biochem.. 1996;157:137-145. MedlineMedline
2. Morin AM, Stanboli A. Nitric oxide synthase localisation in culturedendothelial cells of cerebrovascular origin: cytochemistry. J Neurosci Res..1993;36:272-279. CrossRefCrossRef MedlineMedline
3. O’Brien AJ, Young HM, Povey JM, Furness JB. Nitric oxide synthase is localizedpredominantly in the Golgi apparatus and cytoplasmic vesicles of vascularendothelial cells. Histochemistry.. 1995;103:221-225. CrossRefCrossRef MedlineMedline
4. Sessa WC, García-Cardena G, Liu J, Keh A, Pollock JS, Bradley J, Thiru S,Braverman IM, Desai KM. The Golgi association of endothelial nitric oxidesynthase is necessary for the efficient synthesis of nitric oxide. J Biol Chem..1995;270:17641-17644. AbstractAbstract//FREE FREE Full TextFull Text
5. Shaul PW, Smart EJ, Robinson LJ, German Z, Yuhanna IS, Ying YS, AndersonRGW, Michel T. Acylation targets endothelial nitric oxide synthase toplasmalemmal caveolae. J Biol Chem.. 1996;271:6518-6522.
AbstractAbstract//FREE FREE Full TextFull Text
6. García-Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitricoxide synthase to endothelial caveolae via palmitoylation: implications fornitric oxide signaling. Proc Natl Acad Sci U S A.. 1996;93:6448-6453.
AbstractAbstract//FREE FREE Full TextFull Text
7. Smart EJ, Ying Y-S, Conrad PA, Anderson RGW. Caveolin moves from caveolaeto the Golgi apparatus in response to cholesterol oxidation. J Cell Biol..1994;127:1185-1197. AbstractAbstract//FREE FREE Full TextFull Text
8. Feletou M, Vanhoutte PM. Endothelium-dependent hyperpolarisation ofcanine coronary smooth muscle. Br J Pharmacol.. 1988;93:515-524.
CrossRefCrossRef MedlineMedline
9. Chen G, Yamamoto Y, Miwa K, Suzuki H. Hyperpolarization of arterial smoothmuscle by endothelial humoral substances. Am J Physiol.. 1991;260:H1888–H1892. AbstractAbstract//FREE FREE Full TextFull Text
10. Popp R, Bauersachs J, Hecker M, Fleming I, Busse R. A transferable, β-naphthoflavone-inducible, hyperpolarizing factor is synthesized by nativeand cultured porcine coronary endothelial cells. J Physiol (Lond).1996;497.3:699-709.
11. Komori K, Vanhoutte PM. Endothelium-derived hyperpolarizing factor. BloodVessels. 1990;27:238-245. MedlineMedline
12. Fulton D, McGiff JC, Quilley J. Role of K channels in the vasodilator responseto bradykinin in the rat heart. Br J Pharmacol.. 1994;113:954-958. MedlineMedline
13. Bauersachs J, Hecker M, Busse R. Display of the characteristics ofendothelium-derived hyperpolarizing factor by a cytochrome P450-derivedarachidonic acid metabolite in the coronary microcirculation. Br J Pharmacol..1994;113:1548-1553. CrossRefCrossRef MedlineMedline
14. Hecker M, Bara AT, Bauersachs J, Busse R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonicacid metabolite in mammals. J Physiol (Lond).. 1994;481:407-414. MedlineMedline
15. Rubanyi GM, Vanhoutte PM. Nature of endothelium-derived relaxing factor:are there two relaxing mediators? Circ Res. 1987;61(suppl II):II-61-II-67.
16. Nelson DR, Strobel HW. On the membrane topology of vertebrate cytochromeP-450 proteins. J Biol Chem.. 1988;263:6038-6050.
AbstractAbstract//FREE FREE Full TextFull Text
17. Doms RW, Russ G, Yewdell JW. Brefeldin A redistributes resident and itinerantGolgi proteins to the endoplasmatic reticulum. J Cell Biol.. 1989;109:61-72.
AbstractAbstract//FREE FREE Full TextFull Text
18. Bauersachs J, Popp R, Hecker M, Sauer E, Fleming I, Busse R. Nitric oxideattenuates the release of endothelium-derived hyperpolarizing factor.Circulation. 1996;94:3314-3347.
19. Smart EJ, Ying YS, Mineo C, Anderson RGW. A detergent-free method forpurifying caveolae membrane from tissue culture cells. Proc Natl Acad Sci U SA.. 1995;92:10104-10108. AbstractAbstract//FREE FREE Full TextFull Text
20. Fleming I, Hecker M, Busse R. Intracellular alkalinization induced bybradykinin sustains activation of the constitutive nitric oxide synthase inendothelial cells. Circ Res.. 1994;74:1220-1226. AbstractAbstract//FREE FREE Full TextFull Text
21. Galle J, Bauersachs J, Bassenge E, Busse R. Arterial size determines theenhancement of contractile responses after suppression of endothelium-derived relaxing factor formation. Pflugers Arch.. 1993;422:564-569.
CrossRefCrossRef MedlineMedline
22. Lee JC, Field DJ, Lee LYY. Effects of nocodazole on structures of calf braintubulin. Biochemistry. 1980;19:6209-6215. CrossRefCrossRef MedlineMedline
23. Schiff PB, Fant J, Horwitz SB. Promotion of microtubule assembly in vitro bytaxol. Nature. 1979;277:665-667. CrossRefCrossRef MedlineMedline
24. Hutcheson IR, Griffith TM. Mechanotransduction through the endothelialcytoskeleton: mediation of flow- but not agonist-induced EDRF release. Br J
+

05.10.15 14:02Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A
Page 11 of 12http://hyper.ahajournals.org/content/30/6/1598.full
Pharmacol.. 1996;118:720-726. CrossRefCrossRef MedlineMedline
25. Stanboli A, Morin AM. Nitric oxide synthase in cerebrovascular endothelialcells is inhibited by brefeldin A. Neurosci Lett.. 1994;171:209-212.
CrossRefCrossRef MedlineMedline
26. Komori K, Lorenz RR, Vanhoutte PM. Nitric oxide, ACh, and electrical andmechanical properties of canine arterial smooth muscle. Am J Physiol..1988;255:143-150.
27. Nakashima M, Mombouli J-V, Taylor AA, Vanhoutte PM. Endothelium-dependent hyperpolarization caused by bradykinin in human coronaryarteries. J Clin Invest.. 1993;92:2867-2871.
28. Chen G, Cheung DW. Modulation of endothelium-dependenthyperpolarization and relaxation to acetylcholine in rat mesenteric artery bycytochrome P450 enzyme activity. Circ Res.. 1996;79:827-833.
AbstractAbstract//FREE FREE Full TextFull Text
29. Passafaro M, Rosa P, Sala C, Clementi F, Sher E. N-type Ca channels arepresent in secretory granules and are transiently translocated to the plasmamembrane during regulated exocytosis. J Biol Chem.. 1996;271:30096-30104. AbstractAbstract//FREE FREE Full TextFull Text
30. Gregory RB, Barritt GJ. Store-activated Ca inflow in Xenopus laevis oocytes:inhibition by primaquine and evaluation of the role of membrane fusion.Biochem J.. 1996;319:755-760.
31. Hug T, Koslowsky T, Ecke D, Greger R, Kunzelmann K. Actin-dependentactivation of ion conductances in bronchial epithelial cells. Pflugers Arch..1995;429:682-690. CrossRefCrossRef MedlineMedline
32. Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification ofepoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors.Circ Res.. 1996;78:415-423. AbstractAbstract//FREE FREE Full TextFull Text
33. Randall MD, Alexander SPH, Bennett T, Boyd EA, Fry JR, Gardiner SM, KempPA, McCulloch AI, Kandall DA. An endogenous cannabinoid as anendothelium-derived vasorelaxant. Biochem Biophys Res Commun..1996;229:114-120. CrossRefCrossRef MedlineMedline
34. Di Marzo V, Fontana A. Anandamide, an endogenous cannabinomimeticeicosanoid: ‘killing two birds with one stone.’ Prostaglandins Leukot EssentFatty Acids. 1995;53:1-11. CrossRefCrossRef MedlineMedline
35. Bornheim LM, Kim KY, Chen B, Correia MA. Microsomal cytochrome P450-mediated liver and brain metabolism. Biochem Pharmacol.. 1995;50:677-686. CrossRefCrossRef MedlineMedline
36. Sakaguchi M, Mihara K, Sato R. Signal recognition particle is required forcotranslational insertion of cytochrome P450 into microsomal membranes.Proc Natl Acad Sci U S A.. 1984;81:3361-3364. AbstractAbstract//FREE FREE Full TextFull Text
37. Pryer NK, Wuestehube LJ, Schekman R. Vesicle-mediated protein sorting.Annu Rev Biochem.. 1992;61:471-516. CrossRefCrossRef MedlineMedline
38. Robin M-A, Maratrat M, Loeper J, Durand-Schneider A-M, Tinel M, Ballet F,Beaune P, Feldmann G, Pessayre D. Cytochrome P4502B follows a vesicularroute to the plasma membrane in cultured rat hepatocytes.Gastroenterology. 1995;208:1110-1123. CrossRefCrossRef
39. Nelson DR, Kamatki T, Waxman DJ, Guengerich FP, Estabrook RW, FeyereisenR, Gonzalez FJ, Coon MJ, Gunsalus IC, Gotoh O, Okuda K, Nebert DW. TheP450 superfamily: update on sequences, gene mapping, accession numbers,early trivial names of enzymes, and nomenclature. DNA Cell Biol..1993;12:1-51. CrossRefCrossRef MedlineMedline
Articles citing this article
Targeting of Endothelial Nitric-oxide Synthase to theTargeting of Endothelial Nitric-oxide Synthase to theCytoplasmic Face of the Golgi Complex or Plasma MembraneCytoplasmic Face of the Golgi Complex or Plasma MembraneRegulates Akt- Versus Calcium-dependent Mechanisms forRegulates Akt- Versus Calcium-dependent Mechanisms forNitric Oxide ReleaseNitric Oxide Release
J Biol Chem. 2004;279:30349-30357,AbstractAbstract Full TextFull Text PDFPDF
Molecular mechanisms involved in the regulation of theMolecular mechanisms involved in the regulation of theendothelial nitric oxide synthaseendothelial nitric oxide synthase
Am. J. Physiol. Regul. Integr. Comp. Physiol.. 2003;284:R1-R12,AbstractAbstract Full TextFull Text PDFPDF
Trauma induced by nontraumatic coronary devices and itsTrauma induced by nontraumatic coronary devices and itsimpact on vascular reactivity and morphologyimpact on vascular reactivity and morphology
Am. J. Physiol. Heart Circ. Physiol.. 2002;283:H2356-H2362,
2+
2+

05.10.15 14:02Endothelium-Derived Hyperpolarizing Factor, But Not Nitric Oxide, Is Reversibly Inhibited by Brefeldin A
Page 12 of 12http://hyper.ahajournals.org/content/30/6/1598.full
AbstractAbstract Full TextFull Text PDFPDF
Cytochrome P450 2C expression and EDHF-mediated relaxationCytochrome P450 2C expression and EDHF-mediated relaxationin porcine coronary arteries is increased by cortisolin porcine coronary arteries is increased by cortisol
Cardiovasc Res. 2002;54:669-675,AbstractAbstract Full TextFull Text PDFPDF
Signal transduction of eNOS activationSignal transduction of eNOS activationCardiovasc Res. 1999;43:532-541,AbstractAbstract Full TextFull Text PDFPDF
Afferent Arteriolar Vasodilation to the Sulfonimide Analog ofAfferent Arteriolar Vasodilation to the Sulfonimide Analog of11,12-Epoxyeicosatrienoic Acid Involves Protein Kinase A11,12-Epoxyeicosatrienoic Acid Involves Protein Kinase A
Hypertension. 1999;33:408-413,AbstractAbstract Full TextFull Text PDFPDF




















