
Advanced glycation end products induce moesin phosphorylation in murine retinal endothelium
10
Research Report Advanced glycation end products induce moesin phosphorylation in murine brain endothelium Qiaoqin Li, Hongxia Liu, Jing Du, Bo Chen, Qiang Li, Xiaohua Guo, Xuliang Huang, Qiaobing Huang ⁎ Department of Pathophysiology, Key Lab for Shock and Microcirculation Research, Southern Medical University, 1023 Shatai Road, Tonghe, Guangzhou 510515, PR China ARTICLE INFO ABSTRACT Article history: Accepted 10 December 2010 Available online 16 December 10 Advanced glycation end products (AGEs) have been found to play an important role in the development of diabetes, and AGE levels are correlated with the severity of diabetic complications. We have demonstrated that moesin, a protein linker between actin filaments and the plasma membrane, undergoes phosphorylation of its threonine 558 residue by AGE stimulation in human dermal microvascular endothelial cells through activation of p38 and Rho kinase (ROCK) pathways. In this study, we observed in situ whether AGEs caused phosphorylation of vascular endothelial cells in the brains of AGE-stimulated mice. The animals were injected with AGE-modified mouse serum albumin (AGE-MSA) for 7 consecutive days. Immunohistochemistry was conducted to assess the phosphorylation of moesin in brain vessels. The level of moesin protein phosphorylation was also assessed in cerebral microvessels by western blotting. The effects of p38 and ROCK activation were determined by application of a p38 inhibitor (SB203580) and a ROCK inhibitor (Y27632) at 30 min before each AGE administration. The results showed specific expression of moesin in murine brain vascular endothelial cells. AGE treatment induced a significant increase of threonine 558 phosphorylation in moesin, while inhibition of p38 and ROCK remarkably attenuated the phosphorylation of moesin. The level of moesin protein phosphorylation was also increased in cerebral microvessels, along with an increased permeability of the blood–brain barrier, while inhibition of the p38 and ROCK attenuated these responses. These results demonstrate that AGEs cause the phosphorylation of moesin in murine brain microvascular endothelial cells, with p38 and ROCK being involved in this process. © 2010 Elsevier B.V. All rights reserved. Keywords: Advanced glycation end product Blood–brain barrier Endothelial cell ERM (ezrin/radixin/moesin) 1. Introduction Diabetes mellitus is a major risk factor for cerebrovascular disease and the most common diabetic cerebrovascular complications are ischemic stroke and cerebral atrophy. Ischemic stroke is due to accelerated macrovascular athero- sclerosis (Sonnen et al., 2009), while cerebral atrophy is known to result from microvascular lesions (Messier et al., 2004). The resulting alterations of cognitive function are a diabetes- related disability. Endothelial dysfunction and damage are believed to play an important role in the pathogenesis of these diabetic macrovascular and microvascular complications (Bakker et al., 2009; Huber, 2008). Disruption of the blood– brain barrier and an increase of brain vascular permeability BRAIN RESEARCH 1373 (2011) 1 – 10 ⁎ Corresponding author. Fax: +86 20 61648299. E-mail address: [email protected] (Q. Huang). 0006-8993/$ – see front matter © 2010 Elsevier B.V. All rights reserved. doi:10.1016/j.brainres.2010.12.032 available at www.sciencedirect.com www.elsevier.com/locate/brainres
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Advanced glycation end products induce moesin phosphorylation in murine retinal endothelium

B R A I N R E S E A R C H 1 3 7 3 ( 2 0 1 1 ) 1 – 1 0
0d
ava i l ab l e a t www.sc i enced i r ec t . com
www.e l sev i e r . com/ loca te /b ra i n res
Research Report
Advanced glycation end products induce moesinphosphorylation in murine brain endothelium
Qiaoqin Li, Hongxia Liu, Jing Du, Bo Chen, Qiang Li, Xiaohua Guo,Xuliang Huang, Qiaobing Huang⁎
Department of Pathophysiology, Key Lab for Shock and Microcirculation Research, Southern Medical University, 1023 Shatai Road, Tonghe,Guangzhou 510515, PR China
A R T I C L E I N F O
⁎ Corresponding author. Fax: +86 20 61648299E-mail address: [email protected]
006-8993/$ – see front matter © 2010 Elsevieoi:10.1016/j.brainres.2010.12.032
A B S T R A C T
Article history:Accepted 10 December 2010Available online 16 December 10
Advanced glycation end products (AGEs) have been found to play an important role in thedevelopment of diabetes, and AGE levels are correlated with the severity of diabeticcomplications. We have demonstrated that moesin, a protein linker between actin filamentsand the plasma membrane, undergoes phosphorylation of its threonine 558 residue by AGEstimulation in human dermal microvascular endothelial cells through activation of p38 andRho kinase (ROCK) pathways. In this study, we observed in situ whether AGEs causedphosphorylation of vascular endothelial cells in the brains of AGE-stimulated mice. Theanimals were injectedwith AGE-modifiedmouse serum albumin (AGE-MSA) for 7 consecutivedays. Immunohistochemistrywas conducted to assess the phosphorylationofmoesin in brainvessels. The level of moesin protein phosphorylation was also assessed in cerebralmicrovessels by western blotting. The effects of p38 and ROCK activation were determinedbyapplicationof ap38 inhibitor (SB203580) andaROCK inhibitor (Y27632) at 30minbefore eachAGE administration. The results showed specific expression of moesin in murine brainvascular endothelial cells. AGE treatment induced a significant increase of threonine 558phosphorylation in moesin, while inhibition of p38 and ROCK remarkably attenuated thephosphorylationofmoesin. The level ofmoesinproteinphosphorylationwas also increased incerebral microvessels, along with an increased permeability of the blood–brain barrier, whileinhibition of the p38 and ROCK attenuated these responses. These results demonstrate thatAGEs cause the phosphorylation of moesin in murine brain microvascular endothelial cells,with p38 and ROCK being involved in this process.
© 2010 Elsevier B.V. All rights reserved.
Keywords:Advanced glycation end productBlood–brain barrierEndothelial cellERM (ezrin/radixin/moesin)
1. Introduction
Diabetes mellitus is a major risk factor for cerebrovasculardisease and the most common diabetic cerebrovascularcomplications are ischemic stroke and cerebral atrophy.Ischemic stroke is due to accelerated macrovascular athero-sclerosis (Sonnen et al., 2009), while cerebral atrophy is known
.(Q. Huang).
r B.V. All rights reserved.
to result from microvascular lesions (Messier et al., 2004). Theresulting alterations of cognitive function are a diabetes-related disability. Endothelial dysfunction and damage arebelieved to play an important role in the pathogenesis of thesediabetic macrovascular and microvascular complications(Bakker et al., 2009; Huber, 2008). Disruption of the blood–brain barrier and an increase of brain vascular permeability

2 B R A I N R E S E A R C H 1 3 7 3 ( 2 0 1 1 ) 1 – 1 0
are early changes during the development of diabetes in bothanimal models (Huber et al., 2006) and clinical observation(Hovsepyan et al., 2004). However, the pathological featuresand molecular pathogenesis of cerebrovascular complicationsrelated to diabetes are not fully understood.
Hyperglycemia and its related pathological alterations leadto the onset of vascular complications (Geraldes et al., 2009).There isgrowingevidence thatadvancedglycationendproducts(AGEs) produced by non-enzymatic reactions between reducingsugars and amino reactive groups of proteins and lipids areespecially relevant to the processes of macrovascular andmicrovascular damages (Takeuchi and Yamagishi, 2009). It hasbeen noticed that microvascular endothelial cells in the brainare more susceptible to AGE-induced inflammatory damagethan aortic endothelial cells (Niiya et al., 2006). By binding to aspecific receptor known as RAGE, AGEs trigger oxidative stressand the activation of the mitogen-activated protein kinases(MAPKs) and RhoA kinase (ROCK) signaling pathways inendothelial cells, leading to up-regulation of pro-inflammatorymolecules anddisruption of endothelial barrier function (Forbeset al., 2003; Guo et al., 2005). The activation of MAPK and ROCKsignaling strengthens the construction of F-actin and opensinter-endothelial junctions (Mehta andMalik, 2006). It has beennoted in numerous studies that the ezrin/radixin/moesin (ERM)family of plasma membrane–actin linking proteins plays animportant role in mediating the response to kinase signals andF-actin (Niggli and Rossy, 2008). Moesin is regarded as the mostimportant player in this endothelial response since it is the ERMdominantly expressed by endothelial cells (Berryman et al.,1993), aswell as cerebrovascular endothelial cells (Hayashi et al.,1999; Johnson et al., 2002). Our previous study demonstratedthatmoesin is phosphorylated at threonine residue 558 byAGE-induced signaling, and this change plays an important role inmodulating the endothelial cytoskeleton and barrier function inhuman microvascular endothelial cells (HMVECs) (Guo et al.,2009). We have already detected an increase of murine retinalmicrovascular permeability in AGE-stimulated mice (unpub-lished data). The purpose of the present studywas to assess thechanges of blood–brain barrier function after AGE stimulationand to explore the role of threonine phosphorylation inmoesinin this response. The involvement of MAPK and ROCK in AGE-induced phosphorylation of moesin in brain microvascularendothelial cells was also assessed in this study.
2. Results
2.1. Effect of AGE-MSA on immunohistochemical stainingof moesin and threonine 558-phosphorylated moesin in themurine brain
Cellular expression of moesin was immunohistochemicallyidentified with an antibody against moesin or an antibodyagainst 558 threonine-phosphorylated moesin. Immunohisto-chemistry of brain tissue showed thatmoesinwas strongly andpredominately expressed in murine brain microvascular endo-thelial cells (Fig. 1A, arrow). There was almost no phosphory-lated moesin in control brain endothelial cells. Administrationof AGE-MSA did not alter total moesin expression, but itsignificantly strengthened the staining of moesin phosphory-
latedat the558 threonine residue incerebrovascularendothelialcells (Fig. 1A, hollow arrow). Pretreatment with a p38 MAPKinhibitor (SB203580) or a ROCK inhibitor (Y27632) had noinfluence on total moesin expression, but markedly lessenedAGE-MSA-induced moesin phosphorylation in the cerebrovas-cular endothelium (Fig. 1A). Immunofluorescence of murinebrains revealed similar results, with very strong and specificexpression of moesin in brain endothelial cells. AGE-MSA-induced phosphorylation of moesin was obviously detectedwith a fluorescent-labeled secondary antibody (Fig. 2A), whileinhibition of the p38 MAPK or ROCK pathways attenuatedphosphorylation of moesin at the 558 threonine residue.Quantitative analysis after immunohistochemical and immu-nofluorescent staining further confirmed these observations(Figs. 1B and 2B).
2.2. Effect of AGE-MSA on the expression of moesin andthreonine 558-phosphorylated moesin in murine brainmicrovessels
Murine brain microvessels were isolated by dissection, and theexpression of moesin and the amount of phosphorylatedmoesin in microvascular tissues were investigated. Sincemoesin is specifically expressed by vascular endothelial cells,the amount of moesin or phosphorylated moesin reflects thepattern of expression by cerebrovascular endothelial cells. Theresults demonstrated that expression of total moesin was notaltered by either stimulation with AGE-MSA alone or bypretreating mice with SB203580 or Y27632 before AGE-MSAadministration. The amount of 558 threonine-phosphorylatedmoesin in AGE-MSA-treatedmice showed a significant increase(Fig. 3A, B), with quantitative analysis demonstrating that thedensity ratio of phospho-moesin to total moesin was increasedremarkably (p<0.05 compared with control) (Fig. 3C, D). Theseresults suggested that AGE-MSA treatment leads to phosphor-ylation ofmoesin inmurine cerebral microvascular endothelialcells.
Prior administration of SB203580 or Y27632 attenuated theAGE-MSA-induced phosphorylation of moesin, with obviousdecrease of the density of phospho-moesin/moesin for 10.11%or 26.47% respectively (p<0.05 compared with AGE-MSA treat-ment). However, inhibition of p38 or ROCK alone could nottotally abolish AGE-MSA-induced phosphorylation of moesin,while the density ratio of phospho-moesin to total moesin wasstill higher than control (p<0.05 compared with control) (Fig. 3).These results indicated that both p38 and ROCK MAPK areinvolved in AGE-induced threonine phosphorylation ofmoesin.The inhibitory effect of SB203580 on p38 MAPK activation orY27632 on ROCK activation was also confirmed by westernblotting. The data showed that the activation of p38 MAPK orROCK by AGE-MSA was significantly attenuated for 29.29% or15.99%, respectively, by prior administration of SB203580(Fig. 4A, C) or Y27632 (Fig. 4B, D) in AGE-MSA-treated mice.
2.3. Effect of AGE-MSA on blood–brain barrier function
The Evansblue leakage assay revealed that AGE-MSA treatmentled to impairment of cerebral microvascular endothelial barrierfunction. The amount of Evans blue (EB) dye extravasationincreased from 0.809±0.045 μg/g in control mice to 2.218±

Fig. 1 – Immunohistochemistry of total moesin expression and the level of phosphorylated (p-moesin) in murine brains. Micewere treated with 10 mg/kg of AGE-MSA per day for seven consecutive days. The same dose of native MSA was given as acontrol. SB203580 (1 mg/kg) or Y27632 (5 mg/kg) was injected at 30 min before every dose of AGE-MSA in the relevant groups.Moesin expression (arrow) and the level of threonine 558 phosphorylation (hollow arrow) were detected byimmunohistochemistry (A) using anti-moesin or anti-T558 phospho-moesin antibody as the primary antibodies.Representative results are shown from 6 independent experiments. The gray scale data on total and phosphorylatedmoesin inbrain vessels was quantified and represented as arbitrary units (B) (n=6, mean±SD). *P<0.05.
3B R A I N R E S E A R C H 1 3 7 3 ( 2 0 1 1 ) 1 – 1 0
0.109 μg/g in AGE-MSA-treated mice (p<0.05, Fig. 5A). Fluores-cent images of frozen sections also demonstratedmuch greaterEvans blue dye leakage from the vessels and spreading of thedye into the brain tissue in the AGE-MSA-treated group than inthecontrol group (Fig. 5B). AdministrationofSB203580orY27632before each dose of AGE-MSA significantly attenuated the AGE-induced increase of brain microvascular permeability, withEvans blue leakage declining from 2.218±0.109 to 0.871±0.038 μg/g or 0.873±0.052 μg/g, respectively (p<0.05, Fig. 5A).Observation of frozen sections also revealed much weakerfluorescent staining in the SB203580- or Y27632-treated groupscompared with the control group (Fig. 5B).
3. Discussion
The present study demonstrated that AGEs induced thephosphorylation of moesin at threonine residue 558 inmurinebrain microvascular endothelial cells, and that phosphoryla-tion of moesin was accompanied by an increase of cerebralmicrovascular permeability. Inhibition of p38MAPK and ROCKnot only attenuated this AGE-evoked moesin phosphoryla-tion, but also preserved blood–brain barrier function, indicat-ing involvement of the p38 MAPK and ROCK pathways in thisAGE-induced cerebral microvascular response. These findings

Fig. 2 – Fluorescence images of total and phosphorylatedmoesin (p-moesin) expression in murine brains. Mice were treated asmentioned above. Representative results are shown from 6 independent experiments. The fluorescence intensity of total andphosphorylated moesin in brain vessels was quantified and represented as arbitrary units (B) (n=6, mean±SD). *P<0.05.
4 B R A I N R E S E A R C H 1 3 7 3 ( 2 0 1 1 ) 1 – 1 0
have shown for the first time that phosphorylation of moesinat threonine residue 558 plays an important role in cerebro-vascular functional changes triggered by AGEs.
The AGE-MSAmousemodel used in this studywas adaptedfrom Moore et al. (2003). Glycation of macromolecules in vivois a slow and complicated process that leads to the glycation ofvarious proteins, lipids, collagen, and other macromolecules.This process is highly diverse, making it difficult to evaluateand control the experimental conditions. Although theinjection method and the amount of glycated and in vitroMSA used in this study could not perfectly mimic the changesdue to glycation (AGE formation) that occurred in vivo,administration of exogenous AGE-MSA did at least reflectthe influence of glycated albumin on morphological changesand organ function.
In contrast to the other ERM proteins (radixin and ezrin),moesin is strongly expressed in the endothelium of bloodvessels (Schwartz-Albiez et al., 1995). Several authors havereported that moesin is specifically expressed by the vascularendothelial cells in the brain. For example, only endothelialcells were stained very intensely in the mouse cerebrum by aratmAb specifically targeting the phosphorylated threonine atthe COOH terminus of each ERM protein (T567 of ezrin, T564 ofradixin, and T558 of moesin) (Hayashi et al., 1999). It was alsoreported that moesin was most abundantly expressed in thecerebral vascular endothelium and was not detectable incortical neurons during human brain development from20 weeks of gestation until adulthood (Johnson et al., 2002).Furthermore, predominant expression of moesin in theendothelium was confirmed by showing that moesin was

Fig. 3 – Effect of AGE-MSA on moesin phosphorylation in murine brain microvessels and the role of p38MAPK and ROCKactivation. Mice were treated as mentioned above. Brain microvessels were isolated by dissection and were digested. Proteinswere collected through filtration. Total cellular proteins were analyzed by Western blotting to detect moesin andphosphorylatedmoesin (p-moesin). Results shown are representativeWestern blots (A and B) and densitometry data (C and D).n=3 independent experiments. *P<0.05.
5B R A I N R E S E A R C H 1 3 7 3 ( 2 0 1 1 ) 1 – 1 0
only weakly detected in neuroblasts of the rostral migratorystream inmice (Persson et al., 2010). The results of the presentstudy are consistent with those previous reports on moesinexpression in cerebral endothelial cells. Although Sanchez et
Fig. 4 – Effects of inhibitors (SB203580 or Y27632) on p38 and ROCBrain microvessels were isolated by dissection and were digesteproteins were analyzed by Western blotting to detect total p38 oResults shown are representative Western blots (A and B) and de*P<0.05.
al.'s recent studies have also emphasized the importance ofmoesin and its phosphorylation in neural synapse growth(Sanchez and Simoncini, 2010; Sanchez et al., 2009), but other'sand our data have indicated that the dominantly expressed
K phosphorylation. Mice were treated as mentioned above.d. Proteins were collected through filtration. Total cellularr ROCK and phosphorylated p38 (p-p38) or ROCK (p-RCOK).nsitometry data (C and D). n=3 independent experiments.
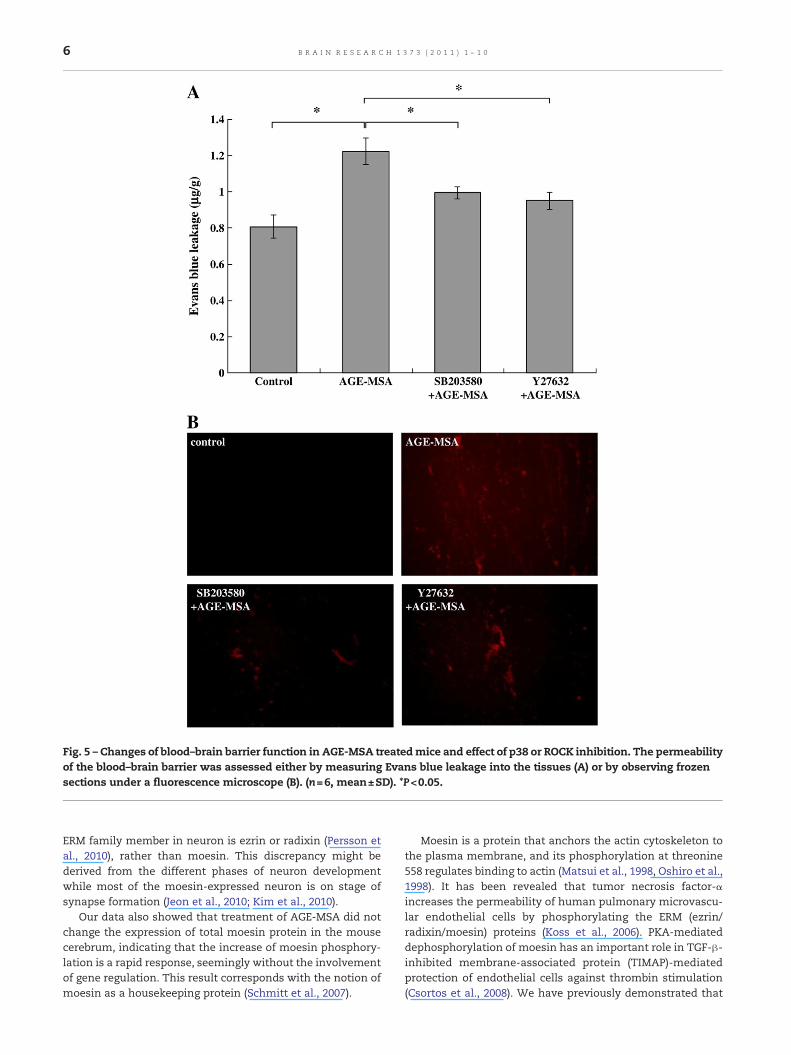
Fig. 5 – Changes of blood–brain barrier function in AGE-MSA treatedmice and effect of p38 or ROCK inhibition. The permeabilityof the blood–brain barrier was assessed either by measuring Evans blue leakage into the tissues (A) or by observing frozensections under a fluorescence microscope (B). (n=6, mean±SD). *P<0.05.
6 B R A I N R E S E A R C H 1 3 7 3 ( 2 0 1 1 ) 1 – 1 0
ERM family member in neuron is ezrin or radixin (Persson etal., 2010), rather than moesin. This discrepancy might bederived from the different phases of neuron developmentwhile most of the moesin-expressed neuron is on stage ofsynapse formation (Jeon et al., 2010; Kim et al., 2010).
Our data also showed that treatment of AGE-MSA did notchange the expression of total moesin protein in the mousecerebrum, indicating that the increase of moesin phosphory-lation is a rapid response, seemingly without the involvementof gene regulation. This result corresponds with the notion ofmoesin as a housekeeping protein (Schmitt et al., 2007).
Moesin is a protein that anchors the actin cytoskeleton tothe plasma membrane, and its phosphorylation at threonine558 regulates binding to actin (Matsui et al., 1998, Oshiro et al.,1998). It has been revealed that tumor necrosis factor-αincreases the permeability of human pulmonary microvascu-lar endothelial cells by phosphorylating the ERM (ezrin/radixin/moesin) proteins (Koss et al., 2006). PKA-mediateddephosphorylation of moesin has an important role in TGF-β-inhibited membrane-associated protein (TIMAP)-mediatedprotection of endothelial cells against thrombin stimulation(Csortos et al., 2008). We have previously demonstrated that

7B R A I N R E S E A R C H 1 3 7 3 ( 2 0 1 1 ) 1 – 1 0
AGEs cause a significant increase of phosphorylatedmoesin incultured HMVECs (Guo et al., 2009). The present study is thefirst to demonstrate that AGEs induce the phosphorylation ofmoesin in mouse cerebrovascular endothelial cells and thatthis change was accompanied by a remarkable increase ofblood–brain barrier permeability. Although we did not obtaindirect evidence of a regulatory relation between phosphory-lation of moesin and cerebrovascular permeability, treatmentwith moesin siRNA suppressed the AGE-stimulated redistri-bution of F-actin and increase of albumin flux in HMVECmonolayers in our previous study (Guo et al., 2009), making itreasonable to postulate that moesin phosphorylation plays arole in AGE-MSA-induced disruption of the blood–brainbarrier. It would be ideal if we could suppress moesinexpression by siRNA in an animal model. Indubitably, as animportant protein regulating endothelial motility, moesinplays some other critical roles in vascular development, suchas vascular lumen formation (Strilić et al., 2009; Wang et al.,2010), or even in endothelial senescence (Lee et al., 2010).
Like other inflammatory mediators, AGEs bind to variousspecific receptors, including the receptor for AGEs (RAGE), AGEreceptor 1–3 (AGER1–3) and several other AGE-related receptors(Goldin et al., 2006). Activation of those receptors exerts acomplex influenceon thedevelopmentofdiabetic retinaldiseasevia multiple signal transduction pathways (Pachydaki et al.,2006), including the kinases that phosphorylatemoesin and ERMproteins. Our previous report has revealed in cellular level thatAGEs exerted its effects by binding to RAGE, whilst an anti-RAGEIgGblocked thephosphorylationofmoesinand thesubsequentlyelevation of endothelial monolayer permeability. Several down-stream pathways of RAGE have been unveiled, including ROCKandMAPK. In a recent study, it was revealed that RAGE-deficientApoE-null mice displayed reduced ROCK activity (Bu et al., 2010).Tanikawa et al. (2009) also indicated that AGEs induce vascularinflammatory response through RAGE/p38 MAPK.
It has been demonstrated by several experiments that Rho-ROCK is a typical upstream pathway for the phosphorylationof moesin (Matsui et al., 1998). p38 MAPK and protein kinase Chave also been found to phosphorylate moesin and other ERMproteins (Koss et al., 2006). Koss et al. reported that 20-μmolSB203580 prevented TNF-α-induced activation of p38 in ECs,followed by complete abolition of the phosphorylation ofmoesin and ERM. Inhibition of p38 or ERM protein expressioncompletely prevents TNF-α-induced reorganization of F-actinand paracellular gap formation, and also increases the flux ofalbumin and dextran across EC membranes. We havepreviously demonstrated that RhoA, ROCK, and p38 MAPKare phosphorylated by AGEs and that suppression of ROCK orp38 activation with inhibitors (Y27632 or SB203580) not onlyblocked phosphorylation of moesin but also attenuated theAGE-triggered hyperpermeability response of HMVECs (Guo etal., 2009; Wang et al., 2009). Our previous study has also ruledout the possible influences of other MAPK sub-family on thisAGE-induced response, whilst the extracellular signal regu-lated kinase (ERK) or the c-JunN-terminal kinase (JNK) was notactivated during AGE-treated period and the inhibition of ERKor JNK with PD98059 or SP600125 did not alter AGE-evokedmoesin phosphorylation in cultured endothelium.
In thepresent study, thep38MAPK inhibitor SB20358 and theROCK inhibitor Y276320 not only decreased the relative
phosphorylation of p38 or ROCK, but also significantly sup-pressed thephosphorylationofmoesin in the cerebral vessels ofAGE-treated mice (Figs. 4 and 5). These findings indicate thatactivation of p38 and ROCK mediates the phosphorylation ofmoesin in AGE-treated mice. While the inhibition of ROCK isonly 15.99% (Fig. 4B), the moesin phosphorylation is 26.47%(Fig. 3B).We consider this discrepancy resulting from the notionthat ROCK is the typical upstream kinase to phosphorylatemoesin and the inhibition of ROCK has major impact onphosphorylation of moesin. Down-regulation of p38 or ROCKwas accompanied by a marked decrease of Evans blue leakagefrom brain microvessels (Fig. 5), indicating involvement of thep38 and ROCK pathways in AGE-triggered blood–brain barrierdysfunction. Although we have not yet shown a directrelationship of moesin phosphorylation to the morphologicaland functional changes of cerebral endothelial cells, ourprevious study of a cellular model directly demonstrated theinvolvement of moesin in regulating endothelial cell F-actinformation and monolayer permeability by knockdown ofmoesin expression with siRNA (Guo et al., 2009). Down-regulation of moesin expression by siRNA treatment preventedAGE-induced changes of the F-actin distribution in the cyto-skeleton andan increase of permeability inHMVECs. Itwouldbeeven more convincing to carry out an experiment in a moesinknockoutmousemodel, and we intended to do so in the future.
There have been several studies showing that there is across talk between the Rho/ROCK and p38 MAPK pathways,and most of the reports have demonstrated that ROCK is theupstream regulator of p38 MAPK activation (Bogatcheva et al.,2007; Marinissen et al., 2001). We showed in a previous studythat there was an additive attenuated effect on endothelialmonolayer permeability when the activities of ROCK and p38MAPK were both inhibited (Guo et al., 2009). The further studyin cultured cell level in our lab has shown that the inhibition ofROCK did weaken the phosphorylation of p38, while inhibitionof p38 affected ROCK activation as well (data not shown). A lotmore evidence is needed to clearly clarify the complicatedrelationship between Rho-ROCK and p38 MAPK in these AGE-induced EC responses.
Except for ROCK and p38 MAPK, some other signalingpathways might be involved in threonine 558 phosphorylationof moesin, while PKC (Koss et al., 2006) was involved in TNF-αinduced moesin phosphorylation. PKC (Kim et al., 2010), PI3K/Akt, Rac1 (Jeon et al., 2010), and Leucine-rich repeat kinase 2(LRRK2) (Parisiadou et al., 2009) were mentioned to phosphor-ylate moesin in developing neuronal cells. As an importantprotein in regulating cellular shape, motility and development,it is predictable that there will be more signaling moleculesunveiled inmodulating thephosphorylationof thisERMprotein.
4. Experimental procedures
4.1. Materials
Mouse serum albumin (MSA, fraction V), D-glucose, and Evansblue were purchased from Sigma (St. Louis, MO, USA). Theantibody specifically recognizing phospho-moesin (T558) wasobtained from Santa Cruz Biotechnology (CA, USA). Antibodiesrecognizing total moesin, total MAPK, and phospho-p38 MAPK

8 B R A I N R E S E A R C H 1 3 7 3 ( 2 0 1 1 ) 1 – 1 0
were purchased from CST (USA). Antibody against ROCK I wasobtained from Chemicon (USA) and antibody against phospho-ROCK I was fromUpstate (USA). The p38 inhibitor SB203580 andtheRhokinase inhibitorY27632wereacquired fromCalbiochem(San Diego, CA, USA). Other chemicals were purchased fromSigma (St. Louis, MO, USA), unless otherwise indicated. Immu-nohistochemical detectionkitswere fromDako (Ely, UK) and thefluorescein isothiocyanate (FITC)-labeled secondary antibody(FITC-labeled goat anti-rabbit IgG) was from Beyotime Instituteof Biotechnology Co. Ltd. (Jiangsu, China).
4.2. Preparation of AGE-MSA
AGE-MSA was prepared according to the protocol of Hou et al.(2001). Briefly,mouse serumalbumin (3.5 mg/ml)was incubatedin PBS (pH 7.4) with 100 mmol/l of D-glucose for 10 weeks at37 °C in a sterile environment. Control albumin was incubatedunder the same conditionswithout glucose. The solutions werethen extensively dialyzed against PBS and concentrated with aMillipore 15ml ultrafiltration column. Endotoxin was removedby using Pierce Endotoxin Removing Gel. AGE-specific fluores-cencewasdetermined by ratio spectrofluorometry. Itwas foundthat AGE-MSA contained 72.50 arbitrary units of AGEs per gramof protein, while native albumin only contained 0.85 arbitraryunits of AGEs per gram of protein.
4.3. Animals and anesthesia
The female C57/BL6 mice used in this experiment wereobtained from the Laboratory Animal Center of SouthernChina. The research protocol was approved by the AnimalCare Committee of the Southern Medical University of Chinaand animal studies were performed according to NationalInstitute of Health guidelines. Mice were anesthetized with anintramuscular injection of 13.3% ethyl carbamate plus 0.5%chloralose (0.65 ml/kg) before all surgical manipulations.
4.4. AGE-MSA treatment of mice
Twenty-fourmice (10–12 weeks old) were randomly assigned tofour groups of 6 animals. According to the protocol of Moore etal. (2003), control and AGE-treated mice were injected intraper-itoneally (i.p.) with either native mouse serum albumin (MSA:10mg/kg) or AGE-MSA (10 mg/kg) on a daily basis for sevenconsecutive days. SB203580 (1 mg/kg) or Y27632 (5 mg/kg) wasinjected intraperitoneally (i.p.) at 30 min before every dose ofAGE-MSA in p38 or ROCK inhibition group, respectively.
4.5. Immunohistochemical and immunofluorescenceobservations of moesin and phosphorylated moesin in the brain
After anesthesia, the animals were perfused with saline fromthe inferior vena cava to the left ventricle for 2 min and thenwith 10%neutral buffered formalin for 1 min to achieve primaryfixation. The brains were removed and the pia mater wasdiscarded. Then each brain was kept in 10% neutral bufferedformalin for at least 24 h at room temperature. Next, the brainswere embedded in paraffin and 20-μm sections were cut on asliding microtome for immunohistochemistry. All immuno-staining was carried out by standard protocols using anti-
moesin or anti-phospho-moesin as the primary antibodies anda biotin-free horseradish peroxidase (HRP)-labeled antibody asthe secondary antibody. TheHRP complexwas identifiedwith3,3′-diaminobenzidine (EnVisionplusdetection system,Dako Ltd,Ely, UK). For immunofluorescence, the brain sections wereprepared and treated as described above, except that thesections were incubated with an FITC-labeled secondaryantibody. The images were observed with a Nikon ECLIPSE 80imicroscope and the arbitrary unit for gray scale of each set ofslices was quantitatively analyzed by using a Leica Q500MCimage analysis program (Leica Instruments Ltd., Germany).
4.6. Quantification and observation of blood–brain barrierfunction
To clarify the functional impact of AGE-MSA on blood–brain-barrier permeability, Evans blue (EB) dye extravasation withfluorescein was applied according to Uuama's protocol(Uuama et al., 1988). Briefly, mice were anesthetized and EBwas injected through the tail vein over 10 s at a dosage of45 mg/kg and the distribution of the dye was confirmed byimmediate visualization of a blue color. After the dye hadcirculated for 60 min, the chest cavity was opened, and salinewas perfused via the left ventricle at 110 mmHg, while theright atrium was opened to flush out all the blood. Then 1%paraformaldehyde was diluted in 0.05 mol/l citric acid buffer(pH 3.5) at 37 °C and was perfused via the left ventricle toremove EB inside the vessels (Brokaw and McDonald, 1988).The brainwas removed and one hemisphere wasweighed andthen homogenized with 2 ml of 50 trichloroacetic acid (TCA) at4 °C and 15,000 rpm for 20 min. The supernatant was dilutedin 100% ethanol at 1:3 volume and then fluorescence wasdetected by using a fluorescence spectrophotometer. Theamount of extravasated Evans blue was determined from astandard curve established with the dye from 31.25 to 2000 ng/ml and was expressed as micrograms of Evans blue per gramof brain tissue (μg/g). To directly observe dye leakage frombrain vessels, the other half of the brainwas used to cut frozencoronal sections at a thickness of 8 μm. EBwas observed undera fluorescence microscope with excitation at 450–470 nm.
4.7. Western blot analysis
Brain microvascular tissues were isolated and collectedaccording to the method of Roux (Wu et al., 2003). After2 min of saline perfusion, anesthetized animals were sacri-ficed. The brain was removed and the pia mater and whitematter were discarded on ice. The remaining brain tissue,mainly with ectocinerea and vessel-rich arachnoid mem-brane, was cut to small pieces and gently milled in 6 ml of PBS(pH 7.2) at 4 °C. The milled tissue was centrifuged at 4 °C andthe resuspended pellet was incubated with 0.1 g/l of type IIcollagenase in PBS at 37 °C with shaking for 60 min. Thedigested product was centrifuged at 1500 rpm for 10 min. Thesediment,mainly withmicrovessel, was extracted by lysis andsonication in lysis buffer with an anti-protease and anti-phosphatase cocktail. Samples were subjected to SDS-PAGE,and the proteins were transferred to polyvinylidene difluoridemembranes. Blots were blocked and incubated with therelevant primary antibody (1:1000) overnight at 4 °C on a

9B R A I N R E S E A R C H 1 3 7 3 ( 2 0 1 1 ) 1 – 1 0
rocker. After three washes with TPBS for 5 min each, the blotswere incubated with an HRP-conjugated species-specificsecondary antibody (1:1000) (Dako Ltd, Ely, UK) for 1 h atroom temperature. After three washes with TPBS for 5 mineach, protein bands were visualized by chemiluminescence.
4.8. Statistics
Results are presented as themean±SD. Data were analyzed byusing one-way ANOVA followed by post hoc comparisons.P<0.05 was considered significant.
Acknowledgments
This work was supported by General Program from NaturalScience Foundation of China (30771028, 30971201); Program forChangjiangScholarsand InnovativeResearchTeaminUniversity(PCSIRT) (No. IRT0731); and National Key Foundation for BasicScience Research of China (G2005CB522601).
R E F E R E N C E S
Bakker, W., Eringa, E.C., Sipkema, P., van Hinsbergh, V.W., 2009.Cell endothelial dysfunction and diabetes: roles ofhyperglycemia, impaired insulin signaling and obesity. TissueRes. 335, 165–189.
Berryman, M., Franck, Z., Bretscher, A., 1993. Ezrin is concentratedin the apical microvilli of a wide variety of epithelial cellswhereas moesin is found primarily in endothelial cells. J. CellSci. 105, 1025–1043.
Bogatcheva, N.V., Adyshev, D., Mambetsariev, B., Moldobaeva, N.,Verin, A.D., 2007. Involvement of microtubules, p38, and Rhokinases pathway in 2-methoxyestra-diol-induced lungvascular barrier dysfunction. Am. J. Physiol. Lung Cell. Mol.Physiol. 292, L487–L499.
Brokaw, J.J., McDonald, D.M., 1988. Neurally mediated increase invascular permeability in the rat trachea: onset, duration, andtachyphylaxis. Exp. Lung Res. 14, 757–767.
Bu, D.X., Rai, V., Shen, X., Rosario, R., Lu, Y., D'Agati, V., Yan, S.F.,Friedman, R.A., Nuglozeh, E., Schmidt, A.M., 2010. Activation ofthe ROCK1 branch of the transforming growth factor-betapathway contributes to RAGE-dependent acceleration ofatherosclerosis in diabetic ApoE-null mice. Circ. Res. 106,1040–1051.
Csortos, C., Czikora, I., Bogatcheva, N.V., Adyshev, D.M., Poirier, C.,Olah, G., Verin, A.D., 2008. TIMAP is a positive regulator ofpulmonary endothelial barrier function. Am. J. Physiol.: LungCell Mol. Physiol. 295, L440–L450.
Forbes, J.M., Cooper, M.E., Oldfield, M.D., Thomas, M.C., 2003. Roleof advanced glycation end products in diabetic nephropathy.J. Am. Soc. Nephrol. 14, S254–S258.
Geraldes, P., Hiraoka-Yamamoto, J., Matsumoto, M., Clermont, A.,Leitges, M., Marette, A., Aiello, L.P., Kern, T.S., King, G.L., 2009.Activation of PKC-delta and SHP-1 by hyperglycemia causesvascular cell apoptosis and diabetic retinopathy. Nat. Med. 15,1298–1306.
Goldin, A., Beckman, J.A., Schmidt, A.M., Creager, M.A., 2006.Advanced glycation end products, sparking the developmentof diabetic vascular injury. Circulation 114, 597–605.
Guo, X., Wang, L., Chen, B., Li, Q., Wang, J., Zhao, M., Wu, W., Zhu,P., Huang, X., Huang, Q., 2009. ERM protein moesin isphosphorylated by advanced glycation end products and
modulates endothelial permeability. Am. J. Physiol.: Heart Circ.Physiol. 297, H238–H246.
Guo, X.H., Huang, Q.B., Chen, B., Wang, Sh.Y., Hou, F.F., Fu, N.,2005. Mechanism of advanced glycation end products-inducedhyperpermeability in endothelial cells. Sheng Li Xue Bao 57,205–210.
Hayashi, K., Yonemura, S., Matsui, T., Tsukita, S., 1999.Immunofluorescence detection of ezrin/radixin/moesin(ERM) proteins with their carboxyl-terminal threoninephosphorylated in cultured cells and tissues. J. Cell Sci. 112,1149–1158.
Hou, F.F., Miyata, T., Boyce, J., Yuan, Q., Chertow, G.M., Kay, J.,Schmidt, A.M., Owen,W.F., 2001. Beta(2)-Microglobulinmodifiedwith advanced glycation end products delays monocyteapoptosis. Kidney Int. 59, 990–1002.
Hovsepyan, M.R., Haas, M.J., Boyajyan, A.S., Guevorkyan, A.A.,Mamikonyan, A.A., Myers, S.E., Mooradian, A.D., 2004.Astrocytic and neuronal biochemical markers in the sera ofsubjects with diabetes mellitus. Neurosci. Lett. 369, 224–227.
Huber, J.D., 2008. Diabetes, cognitive function, and the blood–brainbarrier. Curr. Pharm. Des. 14, 1594–1600.
Huber, J.D., VanGilder, R.L., Houser, K.A., 2006.Streptozotocin-induced diabetes progressively increasesblood–brain barrier permeability in specific brain regions inrats. Am. J. Physiol.: Heart Circ. Physiol. 291, H2660–H2668.
Jeon, S., Park, J.K., Bae, C.D., Park, J., 2010. NGF-induced moesinphosphorylation is mediated by the PI3K, Rac1 and Akt andrequired for neurite formation in PC12 cells. J. Neurochem Int.56, 810–818.
Johnson, M.W., Miyata, H., Vinters, H.V., 2002. Ezrin and moesinexpression within the developing human cerebrum andtuberous sclerosis-associated cortical tubers. ActaNeuropathol. 104, 188–196.
Kim, H.S., Bae, C.D., Park, J., 2010. Glutamate receptor-mediatedphosphorylation of ezrin/radixin/moesin proteins isimplicated in filopodial protrusion of primary culturedhippocampal neuronal cells. J. Neurochem. 113, 1565–1576.
Koss, M., Pfeiffer II, G.R., Wang, Y., Thomas, S.T., Yerukhimovich,M., Gaarde, W.A., Doerschuk, C.M., Wang, Q., 2006. Ezrin/radixin/moesin proteins are phosphorylated by TNF-alpha andmodulate permeability increases in human pulmonarymicrovascular endothelial cells. J. Immunol. 176, 1218–1227.
Lee, J.H., Yoo, J.H., Oh, S.H., Lee, K.Y., Lee, K.H., 2010. Knockdown ofmoesin expression accelerates cellular senescence of humandermal microvascular endothelial cells. Yonsei Med. J. 51,438–447.
Marinissen, M.J., Chiariello, M., Gutkind, J.S., 2001. Regulation ofgene expression by the small GTPase Rho through the ERK6(p38 gamma) MAP kinase pathway. Genes Dev. 15, 535–553.
Matsui, T., Maeda, M., Doi, Y., Yonemura, S., Amano, M., Kaibuchi,K., Tsukita, S., Tsukita, S., 1998. Rho-kinase phosphorylatesCOOH-terminal threonines of ezrin/radixin/moesin (ERM)proteins and regulates their head-to-tail association. J. CellBiol. 140, 647–657.
Mehta, D., Malik, A.B., 2006. Signaling mechanisms regulatingendothelial permeability. Physiol. Rev. 86, 279–367.
Messier, C., Awad, N., Gagnon, M., 2004. The relationships betweenatherosclerosis, heart disease, type 2 diabetes and dementia.Neurol. Res. 26, 567–572.
Moore, T.C., Moore, J.E., Kaji, Y., 2003. The role of advancedglycation end products in retinal microvascular leukostasis.Invest. Ophth. Vis. Sci. 4, 4457–4464.
Niggli, V., Rossy, J.R., 2008. Ezrin/radixin/moesin, versatilecontrollers of signaling molecules and of the cortical skeleton.Int. J. Biochem. Cell Biol. 40, 344–349.
Niiya, Y., Abumiya, T., Shichinohe, H., Kuroda, S., Kikuchi, S., Ieko,M., Yamagishi, S., Takeuchi, M., Sato, T., Iwasaki, Y., 2006.Susceptibility of brain microvascular endothelial cells toadvanced glycation end products-induced tissue factor

10 B R A I N R E S E A R C H 1 3 7 3 ( 2 0 1 1 ) 1 – 1 0
upregulation is associated with intracellular reactive oxygenspecies. Brain Res. 1108, 179–187.
Oshiro, N., Fukata, Y., Kaibuchi, K., 1998. Phosphorylation ofmoesin by rho-associated kinase (Rho-kinase) plays a crucialrole in the formation of microvilli-like structures. J. Biol. Chem.273, 34663–34666.
Pachydaki, S.I., Tari, S.R., Lee, S.E., Ma, W., Tseng, J.J., Sosunov,A.A., Cataldergirmen, G., Scarmeas, N., Caspersen, C., Chang,S., Schiff, W.M., Schmidt, A.M., Barile, G.R., 2006. Upregulationof RAGE and its ligands in proliferative retinal disease. Exp.Eye Res. 82, 807–815.
Parisiadou, L., Xie, C., Cho, H.J., Lin, X., Gu, X.L., Long, C.X.,Lobbestael, E., Baekelandt, V., Taymans, J.M., Sun, L.,Cai, H., 2009. Phosphorylation of ezrin/radixin/moesinproteins by LRRK2 promotes the rearrangement of actincytoskeleton in neuronal morphogenesis. J. Neurosci. 29,13971–13980.
Persson, A., Lindwall, C., Curtis, M.A., Kuhn, H.G., 2010. Expressionof ezrin radixin moesin proteins in the adult subventricularzone and the rostral migratory stream. Neuroscience 167,312–322.
Sanchez, A.M., Flamini, M.I., Fu, X.D., Mannella, P., Giretti, M.S.,Goglia, L., Genazzani, A.R., Simoncini, T., 2009. Rapid signalingof estrogen to WAVE1 and moesin controls neuronal spineformation via the actin cytoskeleton. Mol. Endocrinol. 23,1193–1202.
Sanchez, A.M., Simoncini, T., 2010. Extra-nuclear signaling ofERalpha to the actin cytoskeleton in the central nervoussystem. Steroids 75, 528–532.
Schmitt, T.L., Martignoni, M.E., Bachmann, J., Fechtner, K., Friess,H., Kinscherf, R., Hildebrandt, W., 2007. Activity of theAkt-dependent anabolic and catabolic pathways in muscleand liver samples in cancer-related cachexia. Mol. Med. 85,647–654.
Schwartz-Albiez, R., Merling, A., Spring, H., Möller, P., Koretz, K.,1995. Differential expression of the microspike-associatedprotein moesin in human tissues. Eur. J. Cell Biol. 67, 189–198.
Sonnen, J.A., Larson, E.B., Brickell, K., Crane, P.K., Woltjer, R.,Montine, T.J., Craft, S., 2009. Different patterns of cerebralinjury in dementia with or without diabetes. Arch. Neurol. 66,315–322.
Strilić, B., Kucera, T., Eglinger, J., Hughes, M.R., McNagny, K.M.,Tsukita, S., Dejana, E., Ferrara, N., Lammert, E., 2009. Themolecular basis of vascular lumen formation in the developingmouse aorta. Dev. Cell 17, 505–515.
Takeuchi, M., Yamagishi, S., 2009. Involvement of toxic AGEs(TAGE) in the pathogenesis of diabetic vascular complicationsand Alzheimer's disease. J. Alzheimers Dis. 16, 845–858.
Tanikawa, T., Okada, Y., Tanikawa, R., Tanaka, Y., 2009. Advancedglycation end products induce calcification of vascular smoothmuscle cells through RAGE/p38 MAPK. J. Vasc. Res. 46, 572–580.
Uuama, O., Okamura, N., Yanse, M., Narita, M., Kawabata, K.,Sugita, M., 1988. Quantitative evaluation of vascularpermeability in the gerbil brain after transient ischemia usingEvans blue fluorescence. J. Cereb. Blood FlowMetab. 8, 282–284.
Wang, J.P., Guo, X.H.,Wang, L.J., Li, Q., Chen, B.,Wu,W.,Huang, X.L.,Huang, Q.B., 2009. Effects of Rho/ROCK signal pathway onAGEs-induced morphological and functional changes inhuman dermal microvascular endothelial cells. Sheng Li XueBao 61, 132–138.
Wang, Y., Kaiser, M.S., Larson, J.D., Nasevicius, A., Clark, K.J.,Wadman, S.A., Roberg-Perez, S.E., Ekker, S.C., Hackett, P.B.,McGrail, M., Essner, J.J., 2010. Moesin1 and Ve-cadherin arerequired in endothelial cells during in vivo tubulogenesis.Development 137, 3119–3128.
Wu, Z., Hofman, F.M., Zlokovic, B.V., 2003. A simple method forisolation and characterization of mouse brain microvascularendothelial cells. J. Neurosci. Methods 130, 53–63.




















