
Effects of energy restriction on thyroid hormone metabolism in chickens
Upload
khangminh22Category
view
0download
0
A Novel Cell Type-Specific Mechanism for ThyroidHormone-Dependent Negative Regulation of theHuman Type 1 Deiodinase Gene
SUNG-WOO KIM, SEONG-JUNE HONG, KYUNG MIN KIM, SUNG-CHUL HO, EDWARD C. SO,JOHN W. HARNEY, AND P. REED LARSEN
Thyroid Section, Division of Endocrinology, Diabetes and Hypertension, Department of Medicine,Brigham and Women’s Hospital, Harvard Institute of Medicine, Boston, Massachusetts 02115
We have identified a cell type-specific, negativethyroid hormone-responsive element in the humantype 1 iodothyronine deiodinase (hdio1) gene. Thisfragment, termed a JEG response element, boundtightly to a JEG-cell nuclear protein [JEG cell-specific transcription factor (JTF)] also present inplacenta but not in COS-7, HeLa, or human embry-onic kidney-293 cells. In JEG-3 cells, three copiesof the JEG response element conferred a morethan 40-fold transcriptional stimulation to the het-erologous rat GH promoter which was further in-creased 2-fold by apo-thyroid hormone receptor(TR) and reduced 3-fold by T3. Dimethyl sulfide
footprinting showed overlapping contact sites forthe high- affinity interaction of JTF and low-affinitybinding of TR-retinoid X receptor. Expression ofthe same construct was unaffected by TR or T3 inCOS cells, indicating JTF was required for negativeregulation by T3-TR. Mutations of the critical thy-roid hormone responsive element binding P boxamino acids EG to GS in TR�1 or TR�2 eliminatedthe apo-TR and T3-TR effects. These studies iden-tify a novel mechanism for cell type-specific,promoter-independent negative regulation by T3.(Molecular Endocrinology 18: 2924–2936, 2004)
THYROID HORMONE PLAYS important roles ingrowth, development, differentiation, metamor-
phosis, metabolic regulation, and thermogenesis. Thebiologically active hormone, T3, is produced from theprohormone T4 by the type 1 and 2 iodothyroninedeiodinases (D1 and D2), which remove an iodine fromthe outer ring. D1 is abundantly expressed in liver,kidney, and thyroid in all species studied (1). The hu-man, rat, and mouse dio1 genes are positively regu-lated by T3 in the liver and kidney (1). Two thyroidhormone-response elements (TREs) have been iden-tified within 700 bp of the human dio1 transcriptionstart site (TSS) that confer the positive response of thisgene to T3 (2, 3). Positive transcriptional regulation byT3 requires thyroid hormone receptor (TR)-DNA bind-ing either as a homodimer or as a heterodimer with aretinoid X receptor (RXR). Binding of T3 changes theconformation of the TR causing a dissociation of core-pressors such as the nuclear receptor corepressor
(NCoR), the silencing mediator for retinoid and TRs,and histone deacetylases (HDACs) but recruiting co-activators such as cAMP response element bindingprotein (CREB)-binding protein/p300 and the CREB-binding protein-associated factor, which contain his-tone acetyl transferase activities (4–13). Subsequentacetylation of histone molecules is thought to increaseaccessibility of chromatin to RNA polymerase II (14).
The negative regulation of gene expression by thy-roid hormone is common, but despite considerableefforts, no single mechanism that explains this pro-cess has been elucidated. There are several modelsfor negative regulation by T3 (14). Most frequentlystudied have been those genes that are part of thefeedback-regulatory loop controlling thyroid function,e.g. the genes encoding the TSH� and � subunits andTRH (15–17). A direct TR-DNA-binding model pro-poses that binding of T3 to TR at so called “negativeTREs” on target genes is required for negative regu-lation by T3. For example, the mutation of a TRE half-site sequence near the TSS of the TSH� gene abol-ishes hTR� binding or the use of a hTR� with amutation in the zinc finger region of the receptor elim-inates the T3-dependent repression of this gene (18–20). As a mechanism for the negative regulation bythese DNA sequences, some have shown recruitmentof HDACs to the TR-T3 complex (21). Other systems inwhich direct TR binding is required for negative regu-lation are in genes containing a binding site for theCCCTC-binding factor (CTCF). This is present in sev-eral negatively regulated genes including the lysozymesilencer F1, genomic DNA segment 144, and mamma-
Abbreviations: APP, amyloid precursor protein; CAT,chloramphenicol acetyltransferase; DMS, dimethyl sulfide;FBS, fetal bovine serum; 5�-FR, 5�-flanking region; HDAC,histone deacetylase; hdio1, human type 1 iodothyroninedeiodinase gene; HEK, human embryonic kidney; NCoR, nu-clear receptor corepressor; NE, nuclear extract; JRE, JTF-responsive element; JTF, JEG cell-specific transcription fac-tor; RXR, retinoid X receptor; TR, thyroid hormone receptor;TRE, thyroid responsive element; TSS, transcription start site;WT, wild-type.
Molecular Endocrinology is published monthly by TheEndocrine Society (http://www.endo-society.org), theforemost professional society serving the endocrinecommunity.
0888-8809/04/$15.00/0 Molecular Endocrinology 18(12):2924–2936Printed in U.S.A. Copyright © 2004 by The Endocrine Society
doi: 10.1210/me.2004-0255
2924
Dow
nloaded from https://academ
ic.oup.com/m
end/article/18/12/2924/2741793 by guest on 13 January 2022

lian c-myc (22–24). Another mechanism requiring TR-DNA binding occurs in genes with TR binding sites thatoverlap those for transcriptional enhancers. Examplesare the human epidermal growth factor receptor and the�-amyloid precursor protein (APP) genes (25, 26).
Other proposals are that unoccupied TR enhancesexpression of negatively regulated genes by bindingcorepressors-HDAC complexes facilitating their ex-pression in a indirect manner. Such a “squelching”mechanism has been reported for the TSH� subunitgene (17, 27). Yet another model proposes that TRinteracts with DNA indirectly by protein-protein inter-action such as in the jun-fos heterodimers of activatorprotein 1 sites (14, 28).
In the course of studies of hdio1 transcriptional reg-ulation, we were surprised to find that in the JEG-3choriocarcinoma cell line, an approximately 5-kb 5�-flanking region (FR) chloramphenicol acetyltransferase(CAT) construct was negatively regulated by T3 whencoexpressed with either TR� or TR� in transient trans-fection studies whereas it is positively regulated inCOS-7 and HEK-293 cells. This suggested that therewere specific properties of the JEG cell that facilitatedT3-dependent negative regulation. In this work, wehave analyzed this cell type-specific mechanism fornegative regulation by T3. Negative regulation of hdio1in these cells requires TR-DNA binding but possiblynot to the hdio1 gene. We have identified a nuclearprotein in JEG-3 cells which binds with high affinity toa site in the 5�-FR of the human dio1 gene, markedlyenhancing its transcription. In transient expressionstudies in JEG-3, but not in COS-7 cells, dio1 geneexpression is further enhanced by coexpressing TRand expression is markedly reduced by addition of T3.
There is no evidence for binding of TR-RXR to the JEGcell protein-DNA complex in vivo in COS-7 cells al-though it does bind with low affinity in vitro. Nonethe-less, a mutation in the TR that blocks its capacity tobind to a TRE eliminates negative regulation. Such anexample of negative regulation by T3 has not beendescribed previously, suggesting yet another mecha-nism for thyroid hormone action.
RESULTS
TR-T3-Dependent, Promoter-IndependentNegative Regulation Is Directed by Sequencesfrom the 5-kb hdio1 Promoter in JEG-3 But NotCOS-7, HEK-293, or HepG2 Cells
Analyses of hdio1 5�-FR-reporter constructs showedthat when sequences of 3.5 kb or greater were in-cluded in the construct, a significant negative regula-tion of the hdio1 gene was conferred by T3 in thepresence of coexpressed TR in JEG-3 cells (Fig. 1).This was further confirmed by showing a similar effecton a heterologous promoter-CAT [137-bp rat GH (rGH)CAT] plasmid using TR�1. Functional studies of this5�-FR suggested that the critical sequences were in a100-bp fragment located between �3.4 and �3.3 kbrelative to the TSS (Fig. 1). The negative regulation ofhdio1 by T3 was TR dependent as well as cell typespecific. In the absence of TR, the 5-kb hdio1 con-struct was not significantly affected by T3 (Table 1).However, in the presence of TR� (or TR�, data notshown) in JEG-3 cells, T3 caused an approximately7-fold decrease in CAT expression in the JEG-3 cells
Fig. 1. Localization of hdio1 Sequences Conferring a Negative Response to T3-TR in JEG-3 CellsA, Effect of T3 on the transient expression of hdio1 5�-FR-CAT of various lengths. A mouse TR�1 (mTR�1) expressing vector
and TKhGH as a transfection efficiency control were cotransfected, and CAT expression was normalized to hGH expression.Results shown are the mean CAT/hGH � SD ratios of the constructs. B, Effects of the sequences of the hdio1 fragment conferringa negative response to T3 on expression of the heterologous 137-bp rGH promoter in JEG-3 cells. CAT expression wasnormalized to hGH expression as described. Values are mean � SD.
Kim et al. • Negative Regulation of the hdio1 Gene in JEG-3 Cells Mol Endocrinol, December 2004, 18(12):2924–2936 2925D
ownloaded from
https://academic.oup.com
/mend/article/18/12/2924/2741793 by guest on 13 January 2022

but a 3-fold increase in COS-7 cells (Table 1) as well asin HEK-293 and HepG2 cells (data not shown). Takentogether, these results indicated that there were spe-cific sequences between approximately �3.4 and�3.3 kb that confer T3-dependent negative regulationto either the hdio1 or rGH promoters in JEG-3, but notin three other cell lines.
JEG-3 Cell Nuclear Protein Binding Sequences inthe hdio1 Promoter
To determine whether this negative regulation couldbe explained by an hdio1 binding protein in JEG cells,we performed DNAse I footprinting of sequences be-tween �3.4 and �3.3 kb. We also exposed thesesequences to either bacterially expressed chickenTR� and human RXR� (cTR�/hRXR�) or a nuclearextract (NE) of JEG cells transiently expressing TRexposed or not exposed to T3 (Fig. 2). All three com-plexes protected sequences between �3367 and�3390 with no effect of the coexpression of TR on theJEG cell extract pattern with or without T3 (Fig. 2).DNAse I hypersensitivity sites were present at �3369with the NEs and about 3368–67 with TR-RXR, sug-gesting a protein-DNA interaction and increasedDNAse accessibility at this position. Protection by TR-RXR and NE plus or minus T3 was protein concentra-tion dependent, and there was no obvious effect of T3
on the NE binding.These results indicated that there were binding sites
for a nuclear protein and/or TR-RXR complexes lo-cated in this functionally active region of the hdio1promoter. This was confirmed by EMSAs using the33-bp fragment corresponding to the footprinted re-gion (�3393 to �3359) and either bacterially ex-pressed TR-RXR or NEs from cells transfected with orwithout rat TR�1 (Fig. 3). The mobility of the TR�-RXR�-bound DNA was slightly greater than that ofNEs from JEG cells expressing or not expressing TR.Neither the TR�1 cotransfection nor exposure to 50nm T3 changed the mobility or amount of the NE DNAcomplex. Because the stimulation of hdio1 appearedto be JEG-3 cell specific, we compared JEG NE withthat from COS-7, HeLa, and HEK-293 cells. Little, if any,
retardation of the hdio1 sequences (the slowest migrat-ing band) occurred with these nuclear proteins (Fig. 3B).Together with the transient expression studies and thefootprinting results, these data suggested that a JEG cellnuclear protein conferred the apo-TR and T3-TR effectson hdio1 gene expression in these cells.
The Nuclear Protein in JEG-3 Cells Is anEnhancer of hdio1 Expression
Given the specific binding of the functionally activehdio1 sequences by JEG nuclear protein, we com-
Table 1. Cell Type-Specific Negative Regulation of 5-kb hdio1-CAT by Thyroid Hormone
CAT/hGH Ratio
�TR�1 �TR�1
�T3 �T3 �T3/�T3 �T3 �T3 �T3/�T3
JEG-3 0.12 � 0.03a 0.09 � 0.01 0.71a 0.81 � 0.06a 0.12 � 0.01 0.15a
COS-7 0.37 � 0.01b 0.36 � 0.03 0.97a 0.21 � 0.02b 0.57 � 0.03 2.7a
The 5-kb 5�FR hdio1-CAT reporter construct with or without a vector expressing mouse TR�1 was transiently transfected intoJEG-3 or COS-7 cells, which were subsequently exposed to 0 or 50 nM T3 for 24 h. As an internal control, a TK-human GH (hGH)expression vector was cotransfected. CAT expression was normalized to hGH. hGH production was not affected by theseadditions. Values are mean � SD from four independent experiments. P values were determined by ANOVA with Tukey’s test.a P � 0.001.b P � 0.01.
Fig. 2. Identification of JEG Cell Nuclear Protein-BindingSites in the Functionally Active Fragment of hdio1 5� FR UsingDNAse I Protection
A, A 32P-labeled 54-bp hdio1 fragment (�3431 to �3378)was incubated with various concentrations of bacterially ex-pressed cTR�1-hRXR� or JEG cell NE isolated from JEGcells transfected with mTR�1 cultured in the presence orabsence of T3. Unprotected DNA was partially digested byDNAse I (see Materials and Methods) and compared withnaked DNA under the same conditions (lane 1). The symbolsindicate the quantity of protein added.
2926 Mol Endocrinol, December 2004, 18(12):2924–2936 Kim et al. • Negative Regulation of the hdio1 Gene in JEG-3 CellsD
ownloaded from
https://academic.oup.com
/mend/article/18/12/2924/2741793 by guest on 13 January 2022

pared the effects of this 33-bp footprinted fragment ona heterologous rGH CAT promoter in the absence andpresence of TR and T3. Expression of the native rGHpromoter in JEG-3 cells was more than 40-fold en-hanced by three copies of the 33-bp hdio1 gene se-quences (Fig. 4). That expression was doubled bycoexpression of TR� but transient expression in thepresence of TR was reduced about 3-fold by T3. In
COS-7 cells, there was no effect of the dio1 se-quences on expression of the rGH-CAT construct. TRand T3 were also without effect. These results estab-lish that the 33-bp fragment is a binding site for apotent transcriptional enhancer protein present in JEGcell nuclei. For convenience, we will refer to this celltype-specific protein as the JEG transcription factor(JTF) and the sequences to which it binds in the hdio1
Fig. 3. EMSA Using the 33-bp Footprinted DNA FragmentA, Interaction with bacterially expressed cTR�-hRXR� (lane 1) or NE isolated from JEG cells transfected (lanes 2 and 3) or not
transfected (lanes 4 and 5) with hTR�1 expression vector in the presence and absence of 50 nM T3. B, Cell type-specificexpression of the JEG DNA-binding protein. NEs from JEG-3, COS-7, HeLa, and HEK293 cells were incubated with a 32P-labeled33-bp fragment. A 1-ng probe was reacted with 0.16/0.05 �g of bacterially expressed cTR�-hRXR� or 1.2 �g of NE protein forEMSA.
Fig. 4. The JEG Cells Nuclear Protein Acts as a Transcriptional Enhancer by Interacting with Sequences from the hdio1 Gene5�-FR 3X hdio1-rGHCAT and rGHCAT plasmids (33 bp) were transfected with or without mTR�1 expression plasmid in the
absence or presence of 50 nM T3 into JEG-3 and COS-7 cells (see scale difference). CAT expression was normalized to hGHexpression as described.
Kim et al. • Negative Regulation of the hdio1 Gene in JEG-3 Cells Mol Endocrinol, December 2004, 18(12):2924–2936 2927D
ownloaded from
https://academic.oup.com
/mend/article/18/12/2924/2741793 by guest on 13 January 2022
promoter as the JTF-response element or JRE. Theseresults also suggested that the APO-TR and T3-TReffects were not due to direct interaction of TR-RXRwith the JRE because they do not occur in COS cells.
Specific Identification of Nuclear Protein or TR-RXR Binding Sites Using Dimethyl Sulfide (DMS)Protection and Mutational Analyses
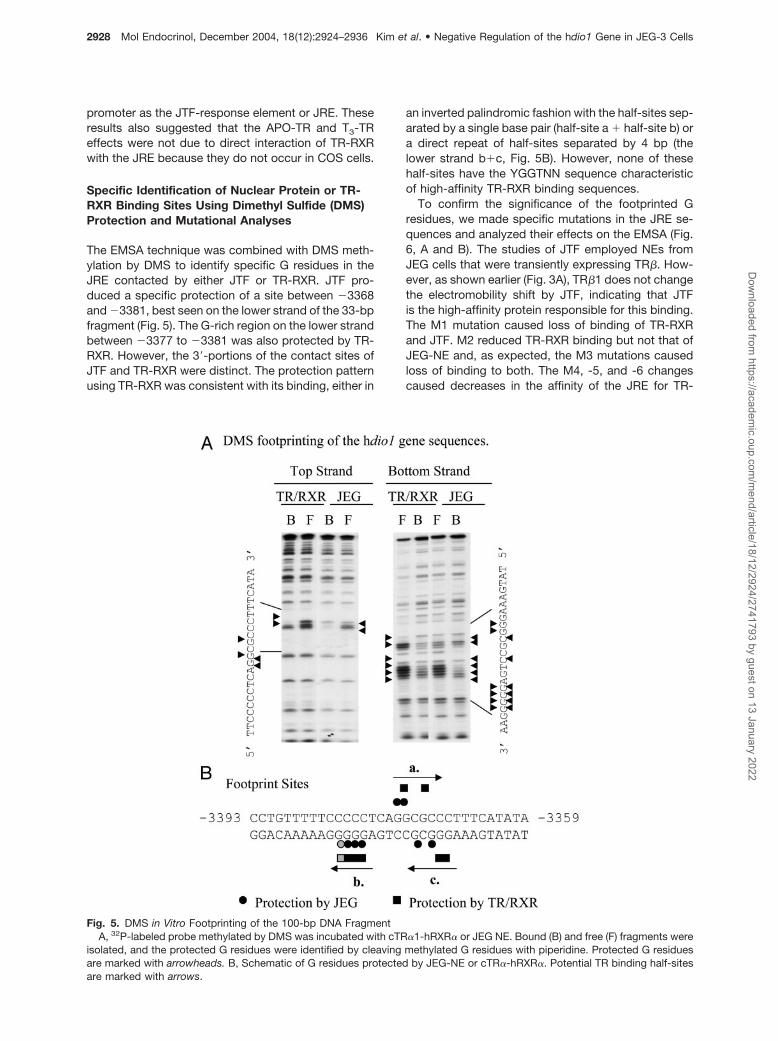
The EMSA technique was combined with DMS meth-ylation by DMS to identify specific G residues in theJRE contacted by either JTF or TR-RXR. JTF pro-duced a specific protection of a site between �3368and �3381, best seen on the lower strand of the 33-bpfragment (Fig. 5). The G-rich region on the lower strandbetween �3377 to �3381 was also protected by TR-RXR. However, the 3�-portions of the contact sites ofJTF and TR-RXR were distinct. The protection patternusing TR-RXR was consistent with its binding, either in
an inverted palindromic fashion with the half-sites sep-arated by a single base pair (half-site a � half-site b) ora direct repeat of half-sites separated by 4 bp (thelower strand b�c, Fig. 5B). However, none of thesehalf-sites have the YGGTNN sequence characteristicof high-affinity TR-RXR binding sequences.
To confirm the significance of the footprinted Gresidues, we made specific mutations in the JRE se-quences and analyzed their effects on the EMSA (Fig.6, A and B). The studies of JTF employed NEs fromJEG cells that were transiently expressing TR�. How-ever, as shown earlier (Fig. 3A), TR�1 does not changethe electromobility shift by JTF, indicating that JTFis the high-affinity protein responsible for this binding.The M1 mutation caused loss of binding of TR-RXRand JTF. M2 reduced TR-RXR binding but not that ofJEG-NE and, as expected, the M3 mutations causedloss of binding to both. The M4, -5, and -6 changescaused decreases in the affinity of the JRE for TR-
Fig. 5. DMS in Vitro Footprinting of the 100-bp DNA FragmentA, 32P-labeled probe methylated by DMS was incubated with cTR�1-hRXR� or JEG NE. Bound (B) and free (F) fragments were
isolated, and the protected G residues were identified by cleaving methylated G residues with piperidine. Protected G residuesare marked with arrowheads. B, Schematic of G residues protected by JEG-NE or cTR�-hRXR�. Potential TR binding half-sitesare marked with arrows.
2928 Mol Endocrinol, December 2004, 18(12):2924–2936 Kim et al. • Negative Regulation of the hdio1 Gene in JEG-3 CellsD
ownloaded from
https://academic.oup.com
/mend/article/18/12/2924/2741793 by guest on 13 January 2022

RXR, but there was still some association. Despite theM4 mutations, JTF still bound to the JRE, but thisbinding was lost for the M5 mutation and reduced forthe single nucleotide mutation in M6. These resultsconfirmed that the “b site” on the lower strand (Fig. 5B)was critical for JEG-NE and TR/RXR binding. The foot-printed GG residues on the top strand are also criticalfor JTF binding but less important for that of TR-RXR.The role of the “a” and “c” sites for TR-RXR bindingwas ambiguous. The results of these studies sug-gested that the binding of JTF was much stronger andmore specific than that of TR-RXR and confirmed thatcotransfection of TR�1 did not affect the specificity ofthis binding.
Because JEG-3 cells are a human choriocarcinomacell line, we compared the binding of NE from humanplacenta to that obtained with JEG cell NE with bothwild-type (WT) JRE and fragments containing M1, -2,and -3 mutations (Fig. 6C). The quantity of JTFretarded was much higher for JEG-3 cells than forhuman placental NE using comparable amounts ofprotein. However, the pattern of retardation of theWT and mutant JREs was similar with JEG andplacental NEs suggesting the presence of a similarnuclear protein presumably in the trophoblastspresent in that organ.
TR-TR or TR-RXR Does Not Inhibit JTF Bindingto the JRE
Because of the overlap of the binding sequences forJTF and TR-RXR, we examined the possibility of in-teraction or interference with JTF-JRE binding by TRor TR-RXR and also for binding of TR by JTF (Fig. 7).There was potent self-displacement of labeled JREbinding by the unlabeled WT sequences with a neardisappearance of labeled JRE binding by JEG NE withas little as a 10-fold increase in unlabeled JRE. Theinhibition of binding, however, was not complete, evenwith a 1000-fold excess of JRE, suggesting eithersome degree of nonspecific binding or a subset oflow-affinity, high-capacity binding sites. Importantly,neither the chicken lysozyme F2 TRE nor the malicenzyme TRE interfered with the JTF-JRE interactionnor did random DNA at a 1000-fold excess (Fig. 7A).Furthermore, JEG NE bound only weakly to the F2chicken lysozyme silencer TRE and did not retard themalic enzyme TRE (Fig. 7B). These and the aboveresults suggested that JTF bound sites in JRE whichcould also bind TR-RXR but that the JTF affinity wasmuch higher.
We therefore tested whether TR�, without or withRXR�, could perturb or compete with JTF-JRE inter-actions. Neither low nor high quantities of TR� inter-
Fig. 6. Analysis of Mutations Designed to Block JTF or cTR�-hRXR� BindingA, Mutation sites within the JRE sequence. 1–3 bases were mutated as denoted in M1–M6. B, EMSA of WT and mutated
32P-labeled 33-bp fragments with bacterially expressed cTR�1/hRXR� or JEG NE (see Materials and Methods). C, Comparisonof the binding pattern of NEs from JEG-3 cells and freshly obtained human placenta. Probe (1 ng) was reacted with 0.16/0.05 �gcTR�1/hRXR�, 1.2 �g JEG-protein, and placenta.
Kim et al. • Negative Regulation of the hdio1 Gene in JEG-3 Cells Mol Endocrinol, December 2004, 18(12):2924–2936 2929D
ownloaded from
https://academic.oup.com
/mend/article/18/12/2924/2741793 by guest on 13 January 2022

fered with JTF binding to either the WT or the M2-JRE.This mutation was specifically designed to interferewith TR-RXR binding but not with that of JTF (Fig. 7C).Despite this, high concentrations of TR led to theretardation of the WT JRE and M2 by putative TR�homodimers. There was TR-RXR heterodimer bindingto the JRE (Fig. 7C) with much higher affinity than forTR homodimers. Despite the binding of significantquantities of JRE by either TR homodimer or TR-RXRheterodimer, the JTF-JRE complex was unchanged inamount and position. This indicates that the binding ofeither TR homodimer or heterodimer is weak and doesnot interfere with JTF binding.
The Functional Activity of JRE Correlates withJTF Binding
As previously demonstrated, the 33-bp hdio1 frag-ment conferred an increase in promoter expression tothe basal rGH promoter (Fig. 4). Accordingly, wetested the relevance of the JTF-JRE physical interac-tion for its enhancer activity by inserting three copiesof WT or mutated JREs 5� to the rGH-CAT construct(Fig. 8). The WT, M2, and M4 JREs, each of whichbinds JTF, markedly increase expression from the rGHpromoter in the absence of TR. This expression wasincreased by TR, and, in addition, T3 decreased rGH-CAT expression to less than its basal level. Neither theM1, M3, or M5 JRE constructs show any enhance-ment of basal 137rGH CAT expression in the absenceor presence of TR. The effect of M6, a JRE that con-tains a single G nucleotide mutation which decreasesJTF binding, is markedly reduced. The loss of en-hancer function of the mutated JRE fragments corre-lates precisely with the loss of JTF binding but not with
their RXR-TR binding affinity (Fig. 6). The M2 JRE,which has a reduced TR-RXR affinity, confers a WTincrease in rGH-CAT activity with APO-TR, which wasmarkedly reduced by T3. This suggests that despitethe binding of TR-RXR to the JRE, such an interactionis unlikely to explain its in vivo effects.
That TR-DNA interactions are required for theAPO-TR effects is suggested in Fig. 8B. Coexpressionof mTR�1-GS71, which contains mutations (EG to GS)in the P box of the first zinc finger that eliminate TREbinding and T3-dependent positive TRE regulation(Ref. 19 and data not shown), does not confer in-creased expression to the WT JRE (Fig. 8B). We alsoobtained similar results using this same mutation inTR�2 (GS125) kindly provided by Dr. F. Wondisford(19).
The effect of WT APO-TR to increase transient ex-pression of negatively regulated genes has been at-tributed to HDAC binding to TR-DNA-corepressorcomplexes. This effect was increased by expression ofNCoR (17, 27). We found no effect of transient expres-sion of NCoR on the APO-TR effect (Fig. 8B), suggest-ing that either the NCoR concentration was alreadymaximal or that another mechanism was operative.
Isolation of JTF Candidate Proteins Using DNA-Affinity Chromatography
Using the high-affinity DNA-binding characteristics ofJTF, we performed JRE-affinity chromatography topurify JTF from JEG-3 cells. DNA affinity columnswere generated by coupling three copies of WT or M1mutant (M1) JREs to Sepharose or magnetic beads asdescribed in Materials and Methods. In the first step,JEG NE was subjected to two-step affinity chroma-
Fig. 7. The JTF-JRE Interaction Is Not Perturbed by TR-RXR or by TRE BindingA, The EMSAs were performed with 32P-labeled JRE (1 ng) and JEG NE (0.4 �g). Competition was with various amounts of
unlabeled JRE, 27 bp lysozyme silencer F2-TRE (0.1 or 1 �g), and 28 bp rat malic enzyme (ME) TRE (0.1 or 1 �g) or random DNA(RD) (1 �g). B, EMSA using 32P-labeled WT JRE (1 ng), F2-TRE (1 ng), ME-TRE (1 ng), and JEG-NE (0.4 �g). C, Absence of effectsof cTR�-hRXR� on the interaction of JTF (from 0.4 �g JEG-NE) with WT or M2-mutant 32P-labeled JRE.
2930 Mol Endocrinol, December 2004, 18(12):2924–2936 Kim et al. • Negative Regulation of the hdio1 Gene in JEG-3 CellsD
ownloaded from
https://academic.oup.com
/mend/article/18/12/2924/2741793 by guest on 13 January 2022

tography purification procedure using M1- followed byWT-JRE-Sepharose columns. We found that non-JTFDNA-binding proteins were eluted from the M1-JRE-Sepharose column at lower ionic strength. Eluatescontaining JTF activity were pooled and repurified us-ing the same approach. A pool of JTF containing elu-ates from the second WT-JRE-magnetic bead affinitychromatography was fractionated to more than 100kDa, 30 to approximately 100 kDa, and less than 30-kDa proteins by ultrafiltration through Centricon YM30and 100 filters (Fig. 9A). EMSA with JRE showed vir-tually all activity was in the 30 to approximately 100-kDa fraction (Fig. 9A). Finally, the JTF-containing frac-tion was purified by Mono-S anion-exchangechromatography using 0.1–1 M NaCl in hypotonicbuffer. The fractions were analyzed by EMSA andSDS-PAGE (Fig. 9, B and C). Silver staining showedthe enrichment of five proteins in fractions containingJTF activity. When these bands were sequenced bymass spectrometry, they were identified as Ku86 (86kDa), Ku70 (70 kDa), Ku86 variant (69 kDa), andHistone1B (two bands at 30 to �40 kDa). These pro-teins are ubiquitously expressed and bind to double-stranded DNA termini. We obtained Ku70 and Ku80expression vectors and transfected COS-7 cells to-gether with WT JRE-rGHCAT. These proteins had noeffect on expression of these constructs, indicatingthey are not JTF.
DISCUSSION
The present studies reveal a complex mechanism forthe surprising observation that a gene which is posi-
tively regulated in vivo in liver cells can be negativelyregulated in a cell type-specific fashion in vitro. Theresults satisfy rigorous criteria for the presence of a“negative” TRE in the hdio1 5�-FR. There was bothstimulation of expression by APO-TR of JRE-contain-ing sequences and a 3- to 6-fold down-regulation ofexpression when T3 is added (Table 1). The negativeregulation can be transferred to a heterologous (rGH)promoter by the JRE sequences (Figs. 1, 4, and 8).Nonetheless, this negative regulation does not occurin COS-7 cells in which there is 3-fold increase inhdio1 expression with T3 as was initially demonstratedwith a 2.5-kb 5�-FR construct in the original studies ofthis promoter in HEK-293 cells (Table 1) (2). This indi-cates the negative regulation is cell type specific. Theprominent effects of APO-TR and T3 on the 5-kb dio1construct led to an initial hypothesis that TR bound tohdio1 sequences at approximately �3.4, perhaps inconcert with a protein expressed in JEG-3, but not inCOS-7, cells. Further studies, however, showed a dif-ferent explanation was more likely. Footprinting withTR-RXR and NEs from JEG cells confirmed colocal-ization of binding sites for TR-RXR and a JEG nuclearprotein that was not expressed in COS-7, HEK-293, orHeLa cells (Fig. 3). The region footprinted in the func-tionally active JRE sequences was GC rich (Fig. 2), andDMS footprinting showed that the binding sites forTR-RXR and JTF had some overlapping and somedistinct G residue contact sites (Fig. 4).
Given the direct and specific binding of a JEG-3nuclear protein, we compared effects of the se-quences protected by the JEG protein (�3393 to�3359, Figs. 3 and 4) on expression of rGH CAT inJEG-3 and COS-7 cells in the absence and presence
Fig. 8. Functional Effects Conferred by WT and Mutant JRE from hdio1 on the 137-bp rGHCAT Vector in JEG-3 CellsComparison of the expression of rGHCAT containing three copies of WT or mutant 33-bp JRE fragments inserted 5� to the rGH
promoter-CAT plasmid. A, Transient transfection assays with WT and mutations M1–6 (see Fig. 6). TR and hGH expressionvectors were cotransfected with or without 50 nM T3 or a mouse TR�1 expression plasmid. CAT expression was normalized tohGH expression as described. B, Disruption of TR�1-TRE binding by introduction of mutations in the P box GS71 containsmutations converting EG at positions 71 and 72 to GS. Empty vector, mTR�1, mutant GS71, or mTR�1�NCoR expression vectorswere cotransfected with WT 33-bp JRE-rGH-CAT plasmid into JEG-3 cells.
Kim et al. • Negative Regulation of the hdio1 Gene in JEG-3 Cells Mol Endocrinol, December 2004, 18(12):2924–2936 2931D
ownloaded from
https://academic.oup.com
/mend/article/18/12/2924/2741793 by guest on 13 January 2022

of cotransfected TR. Remarkably, these sequencescaused a more than 40-fold increase in expression inJEG-3 cells but had no effect in COS-7 cells (Fig. 4).This indicated that the JEG nuclear protein acted as apotent, cell type-specific enhancer (JTF) when itbound to the JRE sequences. Of further interest wasthe observation that the effect of the APO-TR to furtherincrease JTF-rGHCAT expression and the effect of T3
to reduce that expression below the levels present inthe absence of TR occurred only in JEG-3 cells (Fig. 4).
Because of this important result, we directed ourattention toward understanding how APO-TR couldsynergize with JTF to enhance hdio1 expression andwhy T3 inhibited its action. Despite the fact that foot-printing and EMSA results showed that TR-TR or TR-RXR could bind to the JRE in vitro (Figs. 2–4), coex-pression of TR with or without T3 does not change theEMSA pattern of JRE binding (Fig. 3). Furthermore, theinteraction of labeled JRE with JTF is readily blockedby unlabeled JRE but not by the TREs such as the F2or malic enzyme elements (Fig. 7, A and B). This indi-cated that JTF does not bind to authentic TREs withhigh affinity. Furthermore, the binding of high concen-trations of TR or RXR-TR to the JRE did not influencethe JTF-JRE EMSA either qualitatively or quantitatively(Fig. 7C). These results indicated that JTF had a much
higher affinity for JRE than TR homo- or heterodimercomplexes and that the TR-RXR footprint (Fig. 2) prob-ably resulted from the use of high concentrations ofbacterially expressed TR and RXR in these experi-ments. Further studies confirmed that WT, M2, and M4JRE sequences, all of which bind JTF with high affinity(Fig. 6), confer the same TR-dependent enhancementin the absence of T3 and suppression of heterologouspromoter expression in the presence of T3 eventhough the affinity of TR-RXR for M2 and M4 is sig-nificantly less than for the intact JRE. Further criticalevidence that TR-RXR does not directly affect JRE-driven promoters is that TR, with or without T3, has noeffect on expression of the 3XJRE rGH CAT in COS-7cells (Fig. 4). If TR did bind to the JRE, we would haveanticipated repression with APO-TR and stimulationby T3 analogous to the effects of TR-T3 on the 5-kbhdio1 5�FR-CAT construct (Table 1). This is furtherevidence that the TR-RXR footprint reflects an in vitrophenomenon that does not occur in vivo. It is compat-ible with the sequences of the RXR-TR half-sites of theJRE that are not optimal for RXR-TR binding.
Because these in vivo results provided no evidenceof direct or indirect TR-JRE interaction to explain theAPO-TR effect on a promoter containing JRE, weasked whether DNA binding was required for the effectof TR. Surprisingly, coexpression of either a non-DNA-binding mutant TR� or TR�2 eliminated both theAPO-TR effect and the negative regulation by T3 inJEG cells as was shown previously for the TRH gene(20). It is analogous to more recent results with theTSH� and � subunit negative regulation that is elimi-nated both in transient expression assays and that ofTSH� in vivo with the same mutation knocked into theTR�1 gene (19). These mutated receptors do not con-fer T3 regulation to genes containing positive TREs(19).
A recent report shows that a similar E to G mutationat position 71 of TR�1 receptor does not abrogate theincreased expression of the �-APP gene but doeseliminate the repression of that gene by T3 (25). Theseauthors proposed that the positive effects on tran-scription of the APO-TR on the negatively regulatedAPP gene promoter occurred via partitioning core-pressors and associated HDACs to non-DNA-boundRXR-TR complex, thus permitting its indirect activa-tion. Coexpression of NCoR was reported to enhancethis effect consistent with this hypothesis. This is avariation of the squelching model hypothesized fornegative T3 regulation by Tagami et al. (17, 27). How-ever, because DNA binding is required for repressionof the APP gene, it implies the necessity for TR-DNAcontact for the suppressive effect of T3. In the case ofthe APP gene, the authors postulate the necessity ofweak DNA binding of RXR-TR, which interferes withSp1 binding to sequences in close proximity to a neg-ative TRE. A similar interference by TR with Sp1 bind-ing has been proposed as the explanation for T3-TRsuppression of the human epidermal growth factor
Fig. 9. Purification of JTF from JEG-3 NEJTF was partially purified with JEG NE obtained from 109
cells through a two-step affinity chromatography using M1mutant and WT JRE-Sepharose and magnetic beads as de-scribed in Materials and Methods. A, The partially purified JTFwas fractionated into three molecular sizes, which were morethan 100 kDa, 30–100 kDa, and less than 30-kDa proteins asdescribed in Materials and Methods. B, The 30- to 100-kDafraction containing most of the JTF was further resolved byMono-S anion-exchange chromatography. LD, Loading; FT,flow through; E1–9, eluted fractions during 0.3 M-1 M NaClgradient. JTF-enriched fractions were identified by EMSA. C,Proteins of the eluted fractions were visualized by silver stain-ing after SDS-PAGE.
2932 Mol Endocrinol, December 2004, 18(12):2924–2936 Kim et al. • Negative Regulation of the hdio1 Gene in JEG-3 CellsD
ownloaded from
https://academic.oup.com
/mend/article/18/12/2924/2741793 by guest on 13 January 2022

receptor expression and for attenuation of the positiveT3 response to TRE2 of the hdio1 promoter (3, 26).
In addition to Sp1 and JTF, there are a number ofexamples in which endogenous transcription factorsare required for T3-dependent negative regulation ofgene expression. These include CTCF, the CCTC-binding factor, which is involved in negative regulationof the rat F1 sequence of the S-2.4 lysozyme tran-scriptional silencer, the rat TRE-containing genomicDNA element 144, the human c-myc gene enhancer,and perhaps even the APP gene promoter mentionedearlier (23, 29, 30). In these examples, TR-bindingsequences are located near, although not adjacent to,CTCF binding sites, and there is no evidence of directbinding of TR or RXR to CTCF such as occurs withT3-TR interference with AP1 activation (29, 31). How-ever, many TRE half-sites in these genes are the high-affinity PuGGTNN sequences, unlike the JRE whichhas low affinity for RXR-TR (Fig. 7) (23). In some ofthese examples, APO-TR alone as well as TR-T3 mayrepress gene function rather than increase its expres-sion as does the JTF-JRE-APO-TR combination (22)or the negative TRE in the TSH� and TRH genes (18,19, 22, 23).
The mechanism for negative regulation by T3 in thepresent study is not yet clear. The EMSA studies showthat TR-RXR can only bind to the JRE when present athigh concentrations (Fig. 7C), and the transient ex-pression studies show no effect of mutations that de-crease RXR-TR affinity on the APO-TR or T3 effect aslong as JTF binding is preserved (Figs. 6 and 8). Fur-thermore, there is no effect of transient TR expressionon the EMSA in the JEG-NE (Fig. 3) or on the 3XJRE-rGHCAT expression in COS-7 cells. The squelchingmodel does not explain these observations becauseelimination of the DNA binding blocks TR effects.These P box substitutions have been shown to pre-serve corepressor binding and transactivation proper-ties of the TR�2 (19). This is supported by the absenceof an effect of coexpressed NCoR on the APO-TRtranscriptional enhancement. The enhancement ofTR-JRE binding by an identified protein expressed inJEG, but not COS cells, cannot be ruled out althoughwe have no direct evidence of this. The fact that RXR�is more highly expressed in JEG than in COS cells, andthat RXR-TR binds to JRE with high affinity (Fig. 7C)could contribute to such a possibility, especially in thepresence of T3, which enhances the stability ofRXR-TR complexes (32–34). Because the TR-bindingsequences overlap those for JTF (Fig. 5) and even ahigh concentration of RXR-TR does not interfere withJTF binding (Fig. 7C), it seems unlikely that JTF-JREinteraction could facilitate RXR-TR binding to JRE.Another possibility is that RXR-TR complexes arebinding with high affinity to unidentified TREs in theJEG cell genome, thus partitioning corepressors orcoactivators away from the JTF-JRE complexes in theabsence and presence of T3, respectively. This wouldimply that DNA-bound RXR-TR complexes are muchmore effective than the soluble RXR-TR complexes
thought to be involved in the squelching model (14,17). Many of these possible explanations can be betteraddressed once JTF is cloned.
To date, our attempts to identify JTF have beenunsuccessful. Our initial approach was to take advan-tage of DNA-affinity columns using inactive JRE se-quences such as M1 coupled to Sepharose to clearthe NE of irrelevant DNA binding proteins. This wasfollowed by chromatography with WT JRE-Sepharoseconjugates to isolate JTF. We were encouraged by theresults using this approach because the elution of JREbinding activity occurred at a significantly higher mo-larity than did that to the M1-Sepharose conjugates.This allowed partial purification and determination ofan approximate molecular mass range of 30–100 kDafor JTF protein (Fig. 9). However, the purified activefraction still contained four to five visible bands onsilver-stained gels. Mass spectrometry of these pro-teins showed they were Ku proteins and one histonesubunit (Fig. 9). Both direct testing of a Ku-expressingconstructs as well as the fact that the Ku proteins areubiquitously expressed DNA-binding proteins indicatethat these are not JTF (35–39). A different approachwill be required to identify this protein despite its highDNA binding affinity.
The negative regulation of the hdio1 gene by T3-TRand the presence of JTF-like activity in placenta (Fig.6), suggests that D1 activity might be expressed in thecyto- or syncytiotrophoblasts of the human placenta.If thyroid hormone levels were reduced, this could leadto a paradoxical increase in expression of D1. Undernormal circumstances, D1 is not expressed in humanplacenta even though there is high expression of type3 deiodinase and low expression of the type 2 deio-dinase (40, 41), indicating that all of the elementsrequired for selenoprotein synthesis are present inthese cells.
A computer search showed that exact copies of thecore JRE sequences �3382 CCCCCTCAGGCGC�3369 are present in 22 mammalian and 27 othereukaryotic genes. In addition, the same sequenceswere identified in human DNA in several chromosomes(nos. 1, 6, 9, 11, 13, 19, and 22). This suggests that JTFcould be present and regulate JRE-containing genesin other cells. Confirmation of this will require success-ful cloning of this protein.
In summary, we have demonstrated promoter-inde-pendent, TR-T3-dependent negative regulation of aspecific gene in which the negative TRE may not bindTR in vivo but can in vitro. The negative effect requiresan as yet unidentified JEG cell transcription factor,JTF. The fact that TR-DNA binding is required for theeffect but that the JRE does not interact with TR inother cells suggests that binding of transiently ex-pressed APO-TRs to other unidentified TREs in thesecells could be important given the high potency of JTFas a transcription factor. There are a number of otherpossibilities for this effect, which will require furtherstudies once the identity of JTF is known. Whateverthe mechanism, the present results indicate that it
Kim et al. • Negative Regulation of the hdio1 Gene in JEG-3 Cells Mol Endocrinol, December 2004, 18(12):2924–2936 2933D
ownloaded from
https://academic.oup.com
/mend/article/18/12/2924/2741793 by guest on 13 January 2022

does not fit any of the currently proposed models forT3-dependent negative regulation.
MATERIALS AND METHODS
Cell Culture
JEG-3, COS-7, HeLa, and HEK-293 cells were maintained inHam’s F10 medium containing 14 mM sodium bicarbonate(pH 7.4) and 2 mM L-glutamine supplemented with 10% fetalbovine serum (FBS, Life Technologies, Gaithersburg, MD)until cells were grown to about 70–80% confluence (42). Forthe stimulation with T3, the culture medium was removedfrom cells, the cells were rinsed with PBS once, and Ham’sF10 medium containing charcoal-stripped 10% FBS wasadded. The medium was changed after 1 d, and the incuba-tion continued for another 2 d. T3 (50 nM) was diluted in Ham’sF10 medium�charcoal-stripped 10% FBS and added to cellsfor the indicated times.
Plasmid Constructions and Mutagenesis
Standard techniques were used for all plasmid constructions(43). 5�FR genomic fragments (5, 3.7, 3.5, 3.3, 2.5 and 0.8 kb)were amplified by PCR and cloned into BamHI site of plasmidpOCAT2, a promoter insertion chloramphenicol acetyltrans-ferase (CAT) expression vector (44). These were designated–5 kb, �3.7 kb, �3.5 kb, �3.3 kb, �2.5 kb, and �0.8 kb 5�FR hdio1-CAT. To insert hdio1 fragment to the heterologouspromoter-CAT, 137 bp basal rGH promoter-pOCAT plasmid,PCR-amplified 200-bp, 100-bp, and 33-bp DNA fragmentsfrom �3.5 kb 5�FR hdio1-CAT were cloned into the BamHIsite of the 137-bp rGH promoter-pOCAT plasmid (137 bprGH-CAT) (45), which were designated 200 bp, 100 bp, and33 bp hdio1-137 bp rGH-CAT. Point mutations of core se-quences in the 33 bp JRE were generated by synthesizingtop-strand and bottom-strand oligos and hybridizing two oli-gos to make double-stranded fragments. Three copies of33-bp WT or mutated sequences (M1–M6) were inserted into137-bp rGHCAT. To generate a mTRa1 mutant, GS71, twobases at 614 (A to G) converting amino acid 71 from E to Gand at 616 converting amino acid from G to S from TSS ofmTRa1 cDNA-CDM expression vector were mutated by usingQuikChange Site-Directed Mutagenesis kit (Stratagene, LaJolla, CA) according to manufacturer’s protocol. All con-structs were confirmed by sequencing and restriction diges-tion mapping. GS125 containing the same mutation in hTRb2was kindly provided in a pSG5 expression vector by Dr. F.Wondisford (19).
Transient Transfections and CAT Assay
Transfections were performed as previously described (42,46) with FuGENE 6 Transfection Reagent (Roche ClinicalLaboratories, Indianapolis, IN) in JEG-3, COS-7, HeLa, andHEK293 cells. Each plate was transfected with 2 �g CATreporter plasmids and 0.5 �g TKGH, which constitutivelyexpresses human GH (hGH) as an transfection efficiencycontrol. CDM8 vector (0.5 �g) expressing mouse TRa1 (WT ormutant) or a pSG5 vector expressing hTR�2 was cotrans-fected. As a control, CDM8 and pSG5 empty vectors wereused. After transfection, cells were incubated in 60-mm cul-ture dishes in the absence of thyroid hormone for 2 d andthen exposed to 50 nm T3 for 24 h at 37 C. CAT activity wasdetermined by a phase extraction procedure with minor mod-ifications as described previously (47). CAT expression wasnormalized to hGH expression in the absence and presenceof T3 to calculate T3 responsiveness. Transfection data arethe means � SD of a minimum of triplicate samples.
Isolation of NE
NE was isolated according to a standard protocol (43).Briefly, JEG-3, COS-7, HeLa, and 293-HEK cells were cul-tured in twenty 100-mm dishes in the presence or absence of50 nM T3. Cells were harvested into 50-ml Conical tubes,rinsed with cold PBS and cold hypotonic solution (10 mM
HEPES, pH 7.9; 1.5 mM MgCl2; 10 mM KCl) with proteaseinhibitors. Cells were then dispersed in 3 volumes of hypo-tonic solution containing protease inhibitors, incubated on icefor 10 min, and then homogenized. Nuclei were isolated byremoving the cytoplasmic fraction after centrifugation at1850 � g for 15 min at 4C. NE was isolated from nuclei in 300mM KCl by incubating for 30 min at 4 C and centrifuging at25,000 � g for 30 min. NE from placenta tissue was isolatedin the same way except for a minor modification at the ho-mogenization step. In this case, 1 g placenta tissue wasminced into small pieces, which were incubated in hypotonicbuffer on ice for 10 min and then homogenized for NEpreparation.
EMSA
EMSA was performed as described previously (3). Double-strand oligonucleotides of WT and mutant JREs were radio-labeled with [32P]dCTP (New England Nuclear, Boston, MA)by fill-in reaction using Klenow DNA polymerase and gelpurified. Chicken TR�1 and human RXR� were over-expressed in Escherichia coli and purified as described pre-viously (48). NEs were prepared from JEG-3, COS-7, HeLa,and HEK293 mammalian cells as described above. Radiola-beled probes were reacted with bacterially synthesizedcTR�, cTR��hRXR� or NEs and were resolved on a nonde-naturing PAGE.
In Vitro DNAse I Footprinting
The DNAse I digestion reaction was performed according tostandard protocol with minor modifications (43). Briefly, 32P-labeled 54-bp DNA fragment (�3405 to ��3352) was pre-pared by PCR using 32P-labeled 5�-end primer at �3405(5�-GAATATATGCTTTTCCTGTTTT) and unlabeled 3�-endprimer at �3352 (5�-TGAAAGCTATATGAAAGGGC). Gel-pu-rified probe was reacted with cTR�, cTR��hRXR� or NEs inEMSA reaction condition as described above, which werethen exposed to 20 U of DNAse I (Worthington BiochemicalCorp., Freehold, NJ) for 2 min at room temperature. Thereaction was stopped by addition of stop solution and DNAprecipitated in dry-ice ethanol. Protection sites were ana-lyzed by denatured sequencing gel electrophoresis.
Methylation Interference Assay
DMS methylation interference assay was performed using astandard protocol as described previously (2).
Purification
Three copies of 33-bp WT or mutant (M1) JRE sequenceswere amplified from WT and M1 JRE-rGHCAT plasmids byPCR using biotinylated specific primers. The amplified DNAwere conjugated to streptavidin-Sepharose (Sigma ChemicalCo., St. Louis, MO) or streptavidin-magnetic beads (DynAl,Great Neck, NY) according to the manufacturer’s protocol.JEG NEs from 109 JEG-3 cells were diluted to 0.1 M NaCl inhypotonic buffer, which was partially purified by M1 mutantand WT JRE-Sepharose affinity chromatography. M1 mutantJRE-Sepharose column was first used to remove nonspecifi-cally binding proteins during the affinity chromatography.JEG NE was passed through M! JRE-Sepharose column,
2934 Mol Endocrinol, December 2004, 18(12):2924–2936 Kim et al. • Negative Regulation of the hdio1 Gene in JEG-3 CellsD
ownloaded from
https://academic.oup.com
/mend/article/18/12/2924/2741793 by guest on 13 January 2022

which were subsequently loaded into WT JRE-Sepharosecolumn. Nonspecific weak binding proteins were eluated by0.3 M NaCl whereas specific binding proteins by 0.3–1 M NaClstep-gradient. JTF-enriched fractions were identified byEMSA using 32P-labeled JRE. JTF- enriched fractions werepooled and purified again using M1-mutant and WT JREmagnetic bead affinity chromatography. Once more, nontar-get DNA-binding proteins in the partially purified eluates wereremoved by incubating the partially purified proteins with M1mutant coupled to magnetic beads. Then JTF was bound toWT JRE-magnetic beads by incubating 1 h at 4C with gentleshaking. JTF was again eluted by 0.3–1M NaCl step gradient.JTF fractions were pooled and used to fractionate protein intodifferent sizes, more than100 kDa, 30–100 kDa, and less than30 kDa by ultrafiltration using Centricon YM-30 and -100(Millipore Corp., Bedford, MA). The JTF-containing fraction(30–100 kDa) was finally purified by Mono-S anion-exchangechromatography and resolved using 0.1–1 M NaCl salt gra-dient. The most active fractions were identified by EMSA, andpurified proteins were visualized by SDS-PAGE and silverstaining.
Miscellaneous
All chemicals were of reagent grade and obtained from com-mercial sources. All molecular biological manipulations wereperformed using standard techniques (10). Human tissueswere obtained under an Institutional Review Board (IRB)-approved protocol.
Acknowledgments
We thank Dr. Ann Marie Zavacki for a gift of TR� and-�-expressing bacterial clones, Dr. Fredric E. Wondisford fora TR�2-expressing plasmid, Dr. Herbert Samuels for a chickTRa1 bacterial expression, and Dr. Michael G. Rosenfeld forNCoR expression vectors.
Received June 24, 2004. Accepted August 9, 2004.Address all correspondence and requests for reprints to:
S.-W. Kim, Thyroid Section, Division of Endocrinology, Dia-betes and Hypertension, Department of Medicine, Brighamand Women’s Hospital, Harvard Institute of Medicine, 77Avenue Louis Pasteur, Boston, Massachusetts 02115. E-mail: [email protected].
This work was supported by National Institutes of Health(NIH) Grant DK 44128, NIH KO1 Grant DK02867, and NIHTraining Grant DK07529.
REFERENCES
1. Bianco AC, Salvatore D, Gereben B, Berry MJ, Larsen PR2002 Biochemistry, cellular and molecular biology andphysiological roles of the iodothyronine selenodeiodi-nases. Endocr Rev 23:38–89
2. Toyoda N, Zavacki AM, Maia AL, Harney JW, Larsen PR1995 A novel retinoid X receptor-independent thyroidhormone response element is present in the human type1 deiodinase gene. Mol Cell Biol 15:5100–5112
3. Zhang C, Kim S, Harney JW, Larsen PR 1998 Furthercharacterization of thyroid hormone response elementsin the human type 1 iodothyronine deiodinase gene. En-docrinology 139:1156–1163
4. Horlein AJ, Naar AM, Heinzel T, Torchia J, Gloss B,Kurokawa R, Ryan A, Kamei Y, Soderstrom M, Glass CK1995 Ligand-independent repression by the thyroid hor-
mone receptor mediated by a nuclear receptor co-repressor. Nature 377:397–404
5. Heinzel T, Lavinsky RM, Mullen TM, Soderstrom M, La-herty CD, Torchia J, Yang WM, Brard G, Ngo SD, DavieJR, Seto E, Eisenman RN, Rose DW, Glass CK, Rosen-feld MG 1997 A complex containing N-CoR, mSin3 andhistone deacetylase mediates transcriptional repression.Nature 387:43–48
6. Chen JD, Evans RM 1995 A transcriptional co-repressorthat interacts with nuclear hormone receptors. Nature377:454–457
7. Kao HY, Downes M, Ordentlich P, Evans RM 2000 Iso-lation of a novel histone deacetylase reveals that class Iand class II deacetylases promote SMRT-mediated re-pression. Genes Dev 14:55–66
8. Nagy L, Kao HY, Chakravarti D, Lin RJ, Hassig CA, AyerDA, Schrieber SL, Evans RM 1997 Nuclear receptor re-pression mediated by a complex containing SMRT,mSin3A, and histone deacetylase. Cell 89:373–380
9. Huang EY, Zhang J, Miska EA, Guenther MG, KouzaridesT, Lazar MA 2000 Nuclear receptor corepressors partnerwith class II histone deacetylases in a Sin3-independentrepression pathway. Genes Dev 14:45–54
10. Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Na-katani Y 1996 The transcriptional coactivators p300 andCBP are histone acetyltransferases. Cell 87:953–959
11. Korzus E, Torchia J, Rose DW, Xu L, Kurokawa R, McIn-erney EM, Mullen TM, Glass CK, Rosenfeld MG 1998Transcription factor-specific requirements for coactiva-tors and their acetyltransferase functions. Science 279:703–707
12. Blanco JC, Minucci S, Lu J, Yang XJ, Walker KK, Chen H,Evans RM, Nakatani Y, Ozato K 1998 The histone acety-lase PCAF is a nuclear receptor coactivator. Genes Dev12:1638–1651
13. Hassig CA, Tong JK, Fleischer TC, Owa T, Grable PG,Ayer DE, Schreiber SL 1998 A role for histone deacety-lase activity in HDAC1-mediated transcriptional repres-sion. Proc Natl Acad Sci USA 95:3519–3524
14. Lazar MA 2003 Thyroid hormone action: a binding con-tract. J Clin Invest 112:497–499
15. Wondisford FE, Radovick S, Moates JM, Usala SJ, Wein-traub BD 1988 Isolation and characterization of the hu-man thyrotropin �-subunit gene: differences in genestructure and promoter function from murine species.J Biol Chem 263:12538–12542
16. Carr FE, Kaseem LL, Wong NCW 1992 Thryoid hormoneinhibits thyrotropin gene expression via a position-inde-pendent negative L-triiodothyronine-responsive element.J Biol Chem 267:18689–18694
17. Tagami T, Park Y, Jameson JL 1999 Mechanisms thatmediate negative regulation of the thyroid-stimulatinghormone � gene by the thyroid hormone receptor. J BiolChem 274:22345–22353
18. Shibusawa N, Hashimoto K, Nikrodhanond AA, LibermanMC, Applebury ML, Liao XH, Robbins JT, Refetoff S,Cohen RN, Wondisford FE 2003 Thyroid hormone actionin the absence of thyroid hormone receptor DNA-bindingin vivo. J Clin Invest 112:588–597
19. Shibusawa N, Hollenberg AN, Wondisford FE 2003 Thy-roid hormone receptor DNA binding is required for bothpositive and negative gene regulation. J Biol Chem 278:732–738
20. Takeda T, Nagasawa T, Miyamoto T, Hashizume K, De-Groot LJ 1997 The function of retinoid X receptors onnegative thyroid hormone response elements. Mol CellEndocrinol 128:85–96
21. Sasaki S, Lesoon-Wood LA, Dey A, Myers FA, ThorneAW, Crane-Robinson C, Bonifer C, Filippova GN, Lo-banenkov V, Renkawitz R 1999 Ligand-induced recruit-ment of a histone deacetylase in the negative-feedbackregulation of the thyrotropin � gene. EMBO J 18:5389–5398
Kim et al. • Negative Regulation of the hdio1 Gene in JEG-3 Cells Mol Endocrinol, December 2004, 18(12):2924–2936 2935D
ownloaded from
https://academic.oup.com
/mend/article/18/12/2924/2741793 by guest on 13 January 2022

22. Lutz M, Baniahmad A, Renkawitz R 2000 Modulation ofthyroid hormone receptor silencing function by co-repressors and a synergizing transcription factor. Bio-chem Soc Trans 28:386–389
23. Lutz M, Burke LJ, LeFevre P, Myers FA, Thorne AW,Crane-Robinson C, Bonifer C, Filippova GN, LobanenkovV, Renkawitz R 2003 Thyroid hormone-regulated en-hancer blocking: cooperation of CTCF and thyroid hor-mone receptor. EMBO J 22:1579–1587
24. Perez-Juste G, Garcia-Silva S, Aranda A 2000 An ele-ment in the region responsible for premature terminationof transcription mediates repression of c-myc gene ex-pression by thyroid hormone in neuroblastoma cells.J Biol Chem 275:1307–1314
25. Villa A, Santiago J, Belandia B, Pascual A 2004 A re-sponse unit in the first exon of the �-amyloid precursorprotein gene containing thyroid hormone receptor andSp1 binding sites mediates negative regulation by 3,5,3�-triiodothyronine. Mol Endocrinol 18:863–873
26. Xu J, Thompson KL, Shephard LB, Hudson LG, Gill GN1993 T3 receptor suppression of Sp1-dependent tran-scription from epidermal growth factor receptor pro-moter via overlapping DNA-binding sites. J Biol Chem268:16065–16073
27. Tagami T, Madison LD, Nagaya T, Jameson JL 1997Nuclear receptor corepressors activate rather than sup-press basal transcription of genes that are negativelyregulated by thyroid hormone. Mol Cell Biol 17:2642–2648
28. Lopez G, Schaufele F, Webb P, Holloway JM, Baxter JD,Kushner PJ 1993 Positive and negative modulation ofJun action by thyroid hormone receptor at a unique AP1site. Mol Cell Biol 13:3042–3049
29. Awad TA, Bigler J, Ulmer JE, Hu YJ, Moore JM, Lutz M,Neiman PE, Collins SJ, Renkawitz R, Lobanenkov VV,Filippova GN 1999 Negative transcriptional regulationmediated by thyroid hormone response element 144 re-quires binding of the multivalent factor CTCF to a noveltarget DNA sequence. J Biol Chem 274:27092–27098
30. Bigler J, Eisenman RN 1994 Isolation of a thyroidhormone-responsive gene by immunoprecipitation ofthyroid hormone receptor-DNA complexes. Mol Cell Biol14:7621–7632
31. Zhang XK, Wills KN, Husmann M, Hermann T, Pfahl M1991 Novel pathway for thyroid hormone receptor actionthrough interaction with jun and fos oncogene activities.Mol Cell Biol 11:6016–6025
32. Hsu J-H, Zavacki AM, Harney JW, Brent GA 1995 Reti-noid-X-receptor (RXR) differentially augments thyroidhormone response in cell lines as a function of the re-sponse element and endogenous RXR content. Endocri-nology 136:421–430
33. Zhang J, Zamir I, Lazar MA 1997 Differential recognitionof liganded and unliganded thyroid hormone receptor byretinoid X receptor regulates transcriptional repression.Mol Cell Biol 17:6887–6897
34. Collingwood TN, Butler A, Tone Y, Clifton-Bligh RJ,Parker MG, Chatterjee VK 1997 Thyroid hormone-medi-
ated enhancement of heterodimer formation betweenthyroid hormone receptor � and retinoid X receptor.J Biol Chem 272:13060–13065
35. Chan DW, Ye R, Veillette CJ, Lees-Miller SP 1999 DNA-dependent protein kinase phosphorylation sites in Ku70/80 heterodimer. Biochemistry 38:1819–1828
36. Mimori T, Hardin JA, Steitz JA 1986 Characterization ofthe DNA-binding protein antigen Ku recognized by au-toantibodies from patients with rheumatic disorders.J Biol Chem 261:2274–2278
37. Blier PR, Griffith AJ, Craft J, Hardin JA 1993 Binding ofKu protein to DNA. Measurement of affinity for ends anddemonstration of binding to nicks. J Biol Chem 268:7594–7601
38. Falzon M, Fewell JW, Kuff EL 1993 EBP-80, a transcrip-tion factor closely resembling the human autoantigen Ku,recognizes single- to double-strand transitions in DNA.J Biol Chem 268:10546–10552
39. Cary RB, Peterson SR, Wang J, Bear DG, Bradbury EM,Chen DJ 1997 DNA looping by Ku and the DNA-depen-dent protein kinase. Proc Natl Acad Sci USA 94:4267–4272
40. Huang SA, Dorfman DM, Genest DR, Salvatore D, LarsenPR 2003 Type 3 iodothyronine deiodinase is highly ex-pressed in the human uteroplacental unit and in fetalepithelium. J Clin Endocrinol Metab 88:1384–1388
41. Kaplan MM, Shaw EA 1984 Type II iodothyronine 5�-deiodination by human and rat placenta in vitro. J ClinEndocrinol Metab 59:253–257
42. Kim SW, Ahn IM, Larsen PR 1996 In vivo genomic foot-printing of thyroid hormone-responsive genes in pituitarytumor cell lines. Mol Cell Biol 16:4465–4477
43. Ausubel FM, Brent R, Kingston RE, Moore DD, SeidmanJG, Smith JA, Struhl K 1992 Ligation-mediated PCR forgenomic sequencing and footprinting. In: Current proto-cols in molecular biology. New York: Wiley and Sons,Inc.; 15.5.1–15.6.10
44. Prost E, Moore DD 1986 CAT vectors for analysis ofeukaryotic promoters and enhancers. Gene 45:107–111
45. Brent GA, Larsen PR, Harney JW, Koenig RJ, Moore DD1989 Functional characterization of the rat growth hor-mone promoter elements required for induction by thy-roid hormone with and without a co-transfected � typethyroid hormone receptor. J Biol Chem 264:178–182
46. Bartha T, Kim SW, Salvatore D, Gereben B, Tu HM,Harney JW, Rudas P, Larsen PR 2000 Characterizationof the 5�-flanking and 5�-untranslated regions of the cy-clic adenosine 3�,5�-monophosphate-responsive humantype 2 iodothyronine deiodinase gene. Endocrinology141:229–237
47. Seed B, Sheen J-Y 1988 A simple phase-extraction as-say for chloramphenicol acetyltransferase activity. Gene67:271–277
48. Zavacki AM, Zhang CY, Harney JW, Larsen PR 1996Enhancement of thyroid hormone receptor isoformspecificity by insertion of a distant half-site into a thy-roid hormone response element. Endocrinology 137:1438–1446
Molecular Endocrinology is published monthly by The Endocrine Society (http://www.endo-society.org), the foremostprofessional society serving the endocrine community.
2936 Mol Endocrinol, December 2004, 18(12):2924–2936 Kim et al. • Negative Regulation of the hdio1 Gene in JEG-3 CellsD
ownloaded from
https://academic.oup.com
/mend/article/18/12/2924/2741793 by guest on 13 January 2022
Copyright © 2022 FDOKUMEN




















