
The threshold of pentylenetetrazole-induced convulsive seizures, but not that of
9
This article appeared in a journal published by Elsevier. The attached copy is furnished to the author for internal non-commercial research and education use, including for instruction at the authors institution and sharing with colleagues. Other uses, including reproduction and distribution, or selling or licensing copies, or posting to personal, institutional or third party websites are prohibited. In most cases authors are permitted to post their version of the article (e.g. in Word or Tex form) to their personal website or institutional repository. Authors requiring further information regarding Elsevier’s archiving and manuscript policies are encouraged to visit: http://www.elsevier.com/authorsrights
Transcript of The threshold of pentylenetetrazole-induced convulsive seizures, but not that of

This article appeared in a journal published by Elsevier. The attachedcopy is furnished to the author for internal non-commercial researchand education use, including for instruction at the authors institution
and sharing with colleagues.
Other uses, including reproduction and distribution, or selling orlicensing copies, or posting to personal, institutional or third party
websites are prohibited.
In most cases authors are permitted to post their version of thearticle (e.g. in Word or Tex form) to their personal website orinstitutional repository. Authors requiring further information
regarding Elsevier’s archiving and manuscript policies areencouraged to visit:
http://www.elsevier.com/authorsrights

Author's personal copy
The threshold of pentylenetetrazole-induced convulsive seizures, but not that ofnonconvulsive seizures, is controlled by the nitric oxide levels in murine brains
Masatomo Watanabe a, Asuka Miyai a,1, Sonoko Danjo b,c, Yu Nakamura c, Kouichi Itoh a,b,⁎a Laboratory of Molecular and Cellular Neuroscience, Kagawa School of Pharmaceutical Sciences, Tokushima Bunri University, Sanuki, Kagawa 769-2193, Japanb Laboratory for Brain Science, Kagawa School of Pharmaceutical Sciences, Tokushima Bunri University, Sanuki, Kagawa 769-2193, Japanc Department of Neuropsychiatry, School of Medicine, Kagawa University, Kita, Kagawa 761-0793, Japan
a b s t r a c ta r t i c l e i n f o
Article history:Received 6 September 2012Revised 8 February 2013Accepted 28 February 2013Available online 7 March 2013
Keywords:PentylenetetrazoleNitric oxideNeuronal nitric oxide synthaseElectron paramagnetic resonanceGeneralized seizureAntiepileptic drugs
Alterations in the NO pathway play an important role in the development of convulsive seizures via theglutamatergic and GABAergic systems in acute pentylenetetrazole (PTZ) seizure animals. We previouslyreported that the background NO levels under physiological conditions negatively regulate convulsiveseizures, while excess NO levels under pathologic conditions positively regulate PTZ-induced convulsiveseizures. In this study, the NO content in various brain regions after a single dose injection of PTZ was quan-titatively and directly measured using the ex vivo X-band electron paramagnetic resonance method with anNO-trapping agent. Experimental data demonstrated the effects of NO on the convulsive seizure threshold: a1.5-fold increase in the NO level in all brain regions compared to that observed in the control state showedproconvulsive properties without any involvement with nonconvulsive seizures. The distribution of the back-ground NO content in the normal animals was higher in the temporal region of the cerebral cortex, includingthe amygdala, than in the hippocampus, cerebellum and other regions of the cerebral cortex. However, thelevels of NO after the occurrence of acute PTZ-induced convulsive seizures significantly increased by morethan 50% in all brain regions, thus suggesting that the NO levels in all brain regions contribute toPTZ-induced convulsions as a seizure threshold. In a pharmacological study, the inhibitor of neuronal NOsynthase and antagonists of ionotropic glutamate receptors prevented PTZ-induced convulsions and excessiveNO generation. In addition, therapeutic drugs, such as valproate and ethosuximide used to treat generalizedseizures not only inhibited the increase in NO generation induced by PTZ, but also prevented both convulsiveand nonconvulsive seizures caused by PTZ. We herein provide novel insight into the involvement of NO inPTZ-seizure susceptibility at the whole-animal level.
© 2013 Elsevier Inc. All rights reserved.
Introduction
Approximately 50 million people suffer from epilepsy worldwide,and the disease is one of the most widespread neurological disorders(Chang and Lowenstein, 2003; World Health Organization, 2009). Ep-ileptic seizures have been effectively prevented with antiepilepticdrugs (AEDs) since phenobarbital was created a century ago(Shorvon, 2009). Pentylenetetrazole (PTZ) and maximal electroshockseizure tests are widely used as the gold standard to evaluate the ef-fectiveness of AEDs. Although the anticonvulsive properties of mostAEDs were discovered using gold standard tests with simple andeasy procedures, the underlying mechanisms are only partially un-derstood and thus remain elusive. In this study, we focused on theacute seizures induced by single doses of PTZ as a chemical convul-sant. This animal model is well known for studying primary general-ized epilepsy (Löscher et al., 1991); however, the pathophysiologicalmechanisms of PTZ remain unknown. Generally, PTZ-induced convul-sive seizures are caused by the enhancement of glutamatergic neuro-nal activity by antagonism of γ-aminobutyric acidA receptors(GABAAR) in the brain (Psarropoulou et al., 1994; Rocha et al.,
Experimental Neurology 247 (2013) 645–652
Abbreviations: 3Br7NI, 3-bromo-7-nitroindazole; AEDs, antiepileptic drugs; AMPA,DL-alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; AMPAR, AMPA receptor;CGP39551, (E)-(±)-2-amino-4-methyl-5-phospho no-3-pentenoic acid ethyl ester;CBZ, carbamazepine; CP465022, 3-(2-Chlorophenyl)-2-[2-[6-[(diethylamino) methyl]-2-pyridinyl]ethenyl]-6-fluoro-4(3H)-quinazolinone hydrochloride; DMSO, dimethyl sulfoxide;DETC, N,N-diethyldithiocarbamate Na; EPR, electron paramagnetic resonance; ESM,ethosuximide; GABAAR, gamma-aminobutyric acid A receptor; L-NNA, Nω-nitro-L-ar-ginine; LTG, lamotrigine; MK-801, (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]-cyclohepten-5,10-imine maleate; NBQX, 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7 -sulfonamide; NMDA, N-methyl-D-aspartate; NMDAR, NMDA receptor;nNOS, neuronal nitric oxide synthase; NO, nitric oxide; PHT, phenytoin; PTZ,pentylenetetrazole; SMTC, S-methyl L-thiocitrulline; VPA, sodium valproate.⁎ Corresponding author at: Laboratory for Brain Science and Laboratory of Molecular
and Cellular Neuroscience, Kagawa School of Pharmaceutical Sciences, TokushimaBunri University, Sanuki, Kagawa 769-2193, Japan. Fax: +81 87 894 0181.
E-mail addresses: [email protected] (M. Watanabe),[email protected] (A. Miyai), [email protected] (S. Danjo),[email protected] (Y. Nakamura), [email protected] (K. Itoh).
1 Present address: Department of Pharmacy, National Hospital Organization KochiNational Hospital, Kochi 780-8077, Japan.
0014-4886/$ – see front matter © 2013 Elsevier Inc. All rights reserved.http://dx.doi.org/10.1016/j.expneurol.2013.02.019
Contents lists available at SciVerse ScienceDirect
Experimental Neurology
j ourna l homepage: www.e lsev ie r .com/ locate /yexnr

Author's personal copy
1996). Neuron-derived nitric oxide (NO) synthesized from L-arginineby neuronal NO synthase (nNOS) in the brain is considered to be botha neurotransmitter and a neuromodulator, and the involvement ofNO in epileptic disorders has been demonstrated in experimentsusing systemic injections of NOS inhibitors (Banach et al., 2011;Snyder and Bredt., 1991). We previously reported a relationship be-tween NO-nNOS machinery and PTZ-induced convulsion susceptibil-ity. NO generation triggers motor seizures such as clonic and tonic–clonic convulsions. NO was generated by nNOS through the activationof N-methyl-D-aspartate receptors (NMDAR) in brain of PTZ-inducedseizure models. Therefore, we showed that PTZ-induced convulsiveseizures occur following NO generation through the activation ofnNOS induced by hyperexcitability of glutamatergic neurons (Itoh etal., 2004; Kaneko et al., 2002). On the other hand, high doses ofnNOS inhibitors have been reported to increase seizure severity in re-sponse to subconvulsive doses of PTZ, and PTZ-induced convulsionsusceptibility is facilitated in mice lacking the nNOS gene comparedto that observed in wild type mice, thus suggesting that the motorseizure threshold is regulated by the background NO levels (Itohand Watanabe, 2009). Therefore, NO may be a neuromodulator thatexhibits dual proconvulsive and anticonvulsive properties under cer-tain circumstances, and the therapeutic modulation of the NO levelsto treat epilepsy may be complicated by adaptive responses to thistherapy.
In the current study, the NO content in the brain at the time ofPTZ-induced convulsive seizures was quantitatively and directly mea-sured using the ex vivo X-band electron paramagnetic resonance(EPR) method with an NO-trapping agent due to the method's highresolution and ability to specifically detect only NO (Itoh et al.,2004; Kaneko et al., 2002). We determined whether the backgroundlevels of NO in various brain regions are regulated by activation ofnNOS induced by excitability of glutamatergic neurons using a phar-macological study and nNOS gene-deficient mice. Next, we investi-gated the relationship between PTZ-induced generalized convulsiveseizures and upregulation of NO generation in various brain regions.This study indicated that the threshold of PTZ-induced convulsive sei-zures is regulated by certain NO levels in murine brains. In addition,valproate and ethosuximide, chosen treatments for generalized con-vulsive seizures, inhibited the PTZ-induced high levels of NO and con-vulsive seizures.
Experimental procedures
Experimental animals
The protocols for all animal experiments were approved byTokushima Bunri University Animal Care Committees according tothe National Institutes of Health (USA) Animal Care and Use Protocol.All efforts were made to minimize the number of animals used andtheir suffering. Ninety male ICR mice, nine to 14 weeks old, were pur-chased from Japan SLC (Shizuoka, Japan). Mice homozygous for nNOSdeficiency (nNOS−/−, B6.129SNOS1tm1Plh/J) were generated byintercrossing heterozygote nNOS+/− mice and genotyped using PCR,as previously described (Itoh and Watanabe, 2009). All mice weremaintained with laboratory chow and water ad libitum on a 12-hrlight/dark cycle.
Administration of NOS inhibitors, NMDAR antagonists, AMPARantagonists and AEDs
(5S,10R) - (+)-5 -methyl - 10, 11 -dihydro -5H-dibenzo[ a, d ] -cyclohepten-5,10-imine maleate (MK-801), (E)-(±)-2-amino-4-methyl-5 -phospho no -3 -pentenoic acid ethyl ester (CGP39551),3 - (2-Chlorophenyl) -2-[2-[6-[(diethylamino)methyl]-2-pyridinyl]ethenyl]-6-fluoro-4(3H)-quinazolinone hydrochloride (CP465022) and2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide
(NBQX)were obtained from Tocris Biosciences (Ellisville, MO, USA). Thedoses of MK-801, CGP39551, CP465022 and NBQX used in this studywere 0.05, 10, 3 and 30 mg/kg, respectively (Itoh and Watanabe, 2009;Löscher and Hönack, 1991; Löscher et al., 1993; Menniti et al., 2003).Nω-nitro-L-arginine (L-NNA, in saline; Sigma-Aldrich Corp.) and3-bromo-7-nitroindazole (3Br7NI, in dimethyl sulfoxide (DMSO);Calbiochem, San Diego, CA, USA) were used as nNOS inhibitors (Hara etal., 1996; Kaneko et al., 2002). All inhibitors dissolved in saline were ad-ministered intraperitoneally (i.p.) 30 min before treatment with PTZ. AllAEDs, sodium valproate (VPA, Wako Pure Chem. Ltd., Oosaka, Japan),ethosuximide (ESM, Sigma-Aldrich Corp.), carbamazepine (CBZ,Wako Pure Chem. Ltd.), phenytoin (PHT, Dainippon Sumitomo PharmaCo., Ltd., Oosaka, Japan) and lamotrigine (LTG, Sigma-Aldrich Corp.)were dissolved in 0.5% (w/v) carboxymethyl cellulose 400 solution(Wako Pure Chem. Ltd.). Each AEDwas orally administered at an injec-tion volume of 0.05 ml/10 g 60 min before PTZ injection. The doses ofeach AED were selected based on the findings of previous reports(Mandhane et al., 2007; Watanabe et al., 2010).
Acute PTZ-induced seizures protocol
The animals were placed in a plastic chamber (15 × 15 × 30 cm)and their behavior was observed before and after PTZ administration.After the animals displayed a resting posture, they were injected i.p.with varying doses of PTZ (Sigma-Aldrich, Corp., St. Louis, MO,USA). The mice were acutely treated with subconvulsive doses (20and 40 mg/kg) and convulsive doses (60 mg/kg) of PTZ. The controlmice received 0.2 ml/10 g saline injections. After each PTZ injection,the convulsive behaviors of the mice were observed for 10 min andresultant convulsions were classified and scored according to thecriteria of a previous report (Itoh et al., 2004; Itoh and Watanabe,2009) as follows: 0: normal; 1: immobilization; 2: facial, vibrissaland forelimb clonus (short myoclonic jerk); 3: myoclonic jerkingconsisted of a whole body jerk with or without irregular, bilateralforelimb movements; 4: generalized clonic seizures (GCS) with kan-garoo posture; 5: generalized tonic–clonic seizures (GTCS) with lossof posture tone. In this study, “immobilization” and “myoclonicjerking”were given scores ranging from 1 to 3 as a nonconvulsive sei-zure and “severe GCS and GTCS” were assigned scores of 4 and 5 asconvulsive seizures, respectively.
Measurement of NO content with ex vivo X-band EPR
The NO content in brain tissue was measured using ex vivoX-band EPR, as previously described (Itoh et al., 2004; Kaneko etal., 2002). Briefly, mice were injected i.p. with 500 mg/kg of N,N-diethyldithiocarbamate Na (DETC, Wako Pure Chem. Ltd.) andsubsequently injected subcutaneously (s.c.) with an iron-citratecomplex (50 mg/kg·FeSO4·7H2O + 250 mg/kg·sodium citrate)30 min prior to PTZ injection. After undergoing PTZ injection, micethat had also received the NO-trapping agents (DETC andiron-citrate complex) were observed for 10 min and scored for be-havioral changes. The mice were then killed, their brains were ex-cised and each cortical region (frontal, temporal, parietal andoccipital), the hippocampus and the cerebellum were immediatelydissected and stored in liquid N2 until EPRmeasurement. The excisedsections of brain tissue were weighed and subsequently measured inglass capillaries (Microcaps 100, Drummond Scientific Co., Broomall,PA, USA) at ambient temperature on an Elexsys E500 X-band EPRspectrometer (Bruker BioSpin K.K., Kanagawa, Japan). The typicalspectrometer conditions were: microwave power = 20 mW;micro-wave frequency = 9.84 GHz; modulation frequency = 100 kHz;modulation amplitude = 2.0 G; receiver gain = 60 dB; responsetime = 20.48 mS; number of points = 1024; sweep time = 40 s;number of scans = 10; central field = 3450 G; sweep width = 100 G.The concentration of trapped NO was determined using double
646 M. Watanabe et al. / Experimental Neurology 247 (2013) 645–652
Author's personal copy
integration from the area between the third peak top and its −50 Gcompared with a standard curve. The standard curve was linear be-tween 12 and 400 pmol of (DETC)2-Fe(II)-NO complex prepared from(DETC)2-Fe(II) and a NO donor SNAP (S-nitroso N-acetylpenicilamine,DOJINDO Laboratories, Kumamoto, Japan) in DMSO.
Determination of the rate of NO generation in brain homogenate
The initial rate of NO generation inmurine brain homogenate wasdetermined using a hemoglobin assay (Hevel and Marletta, 1994), aspreviously described (Itoh and Watanabe, 2009). Briefly, the reac-tion was initiated by the addition of NADPH and L-arginine to thereaction mixture containing cerebral homogenate (150 μg), oxyhe-moglobin and AEDs at 0.5–2 mM. The concentrations of the AEDswere chosen according to the effective therapeutic concentrations:50 to 100 μg/ml for VPA and 40 to 100 μg/ml for ESM. After incuba-tion was performed for 20 min at 37 °C, the reaction mixture wascentrifuged at 20,000 ×g for 10 min and then the absorbance of thesupernatant containing hemoglobin was monitored. The increase inabsorbance at 401–421 nm induced by the conversion of oxyhe-moglobin to methemoglobin was used to quantitate the reactionusing an extinction coefficient of 77 mM−1 cm−1 between 401 and421 nm.
Statistical analysis
All data are expressed as the mean ± SEM. The differences be-tween the mean values for each group of NO content and behavioralscores were analyzed using the one-way ANOVA followed byDunnett's test and the Kruskal–Wallis test followed by Steel's test, re-spectively. The interaction effect between the dose of PTZ and func-tional brain regions was analyzed using two-way ANOVA followedby Scheffe's test. A P-value less than 0.05 was considered to be statis-tically significant.
Results
Background levels of NO in the brain are produced by nNOS activation
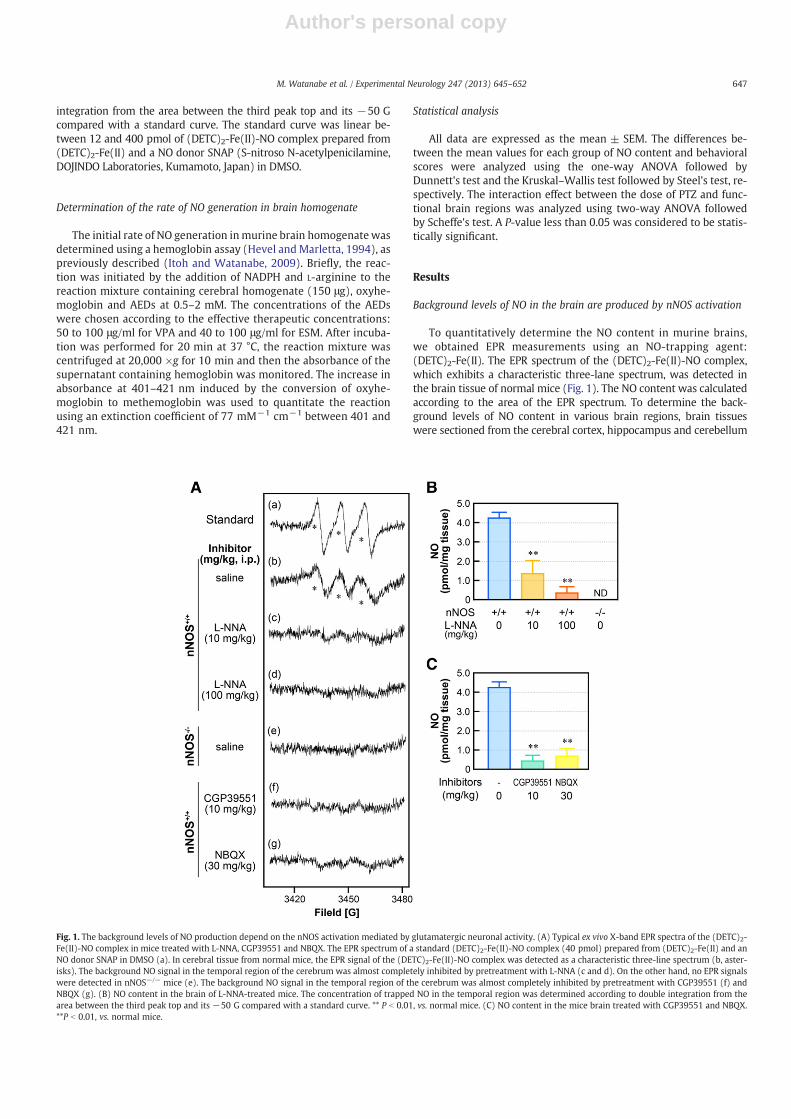
To quantitatively determine the NO content in murine brains,we obtained EPR measurements using an NO-trapping agent:(DETC)2-Fe(II). The EPR spectrum of the (DETC)2-Fe(II)-NO complex,which exhibits a characteristic three-lane spectrum, was detected inthe brain tissue of normal mice (Fig. 1). The NO content was calculatedaccording to the area of the EPR spectrum. To determine the back-ground levels of NO content in various brain regions, brain tissueswere sectioned from the cerebral cortex, hippocampus and cerebellum
Fig. 1. The background levels of NO production depend on the nNOS activation mediated by glutamatergic neuronal activity. (A) Typical ex vivo X-band EPR spectra of the (DETC)2-Fe(II)-NO complex in mice treated with L-NNA, CGP39551 and NBQX. The EPR spectrum of a standard (DETC)2-Fe(II)-NO complex (40 pmol) prepared from (DETC)2-Fe(II) and anNO donor SNAP in DMSO (a). In cerebral tissue from normal mice, the EPR signal of the (DETC)2-Fe(II)-NO complex was detected as a characteristic three-line spectrum (b, aster-isks). The background NO signal in the temporal region of the cerebrum was almost completely inhibited by pretreatment with L-NNA (c and d). On the other hand, no EPR signalswere detected in nNOS−/− mice (e). The background NO signal in the temporal region of the cerebrum was almost completely inhibited by pretreatment with CGP39551 (f) andNBQX (g). (B) NO content in the brain of L-NNA-treated mice. The concentration of trapped NO in the temporal region was determined according to double integration from thearea between the third peak top and its −50 G compared with a standard curve. ** P b 0.01, vs. normal mice. (C) NO content in the mice brain treated with CGP39551 and NBQX.**P b 0.01, vs. normal mice.
647M. Watanabe et al. / Experimental Neurology 247 (2013) 645–652

Author's personal copy
and the cerebral cortex was compartmentalized to its functionalorganization (frontal, temporal, parietal and occipital regions)according to previous reports (Dorr et al., 2007; Paxinos andFranklin, 2001; Supplemental Fig. 1). The background levels ofNO in the four cortical regions, the hippocampus and the cerebel-lum were 2.77 ± 0.17, 4.19 ± 0.28, 2.63 ± 0.22, 3.03 ± 0.25,3.22 ± 0.23 and 2.29 ± 0.15 pmol/mg, respectively (Table 1). TheNO content in various brain regions was dose-dependently de-creased by the nNOS specific inhibitor L-NNA and no EPR signalswere detected in the brain tissue from nNOS−/− mice (Figs. 1A, B).Therefore, the background levels of NO in all brain regions were de-termined by nNOS activation, not eNOS or iNOS activation under thedetection limit of this assay.
Background NO generation is inhibited by NMDAR and AMPARantagonists
To elucidate whether the background NO levels are modulated byglutamatergic transmission via NMDAR and AMPAR, the mice weretreated with specific competitive NMDAR and AMPAR antagonists(CGP39551 and NBQX, respectively). The background levels of NO invarious brain regions were significantly decreased by treatmentwith CGP39551 and NBQX (Figs. 1A, C, Supplemental Table 1).These results indicate that the background NO generation is con-trolled by the activation of nNOS by glutamatergic neuronal activitythrough NMDAR and AMPAR.
PTZ-induced convulsive seizures require 1.5-fold increases in the back-ground NO levels
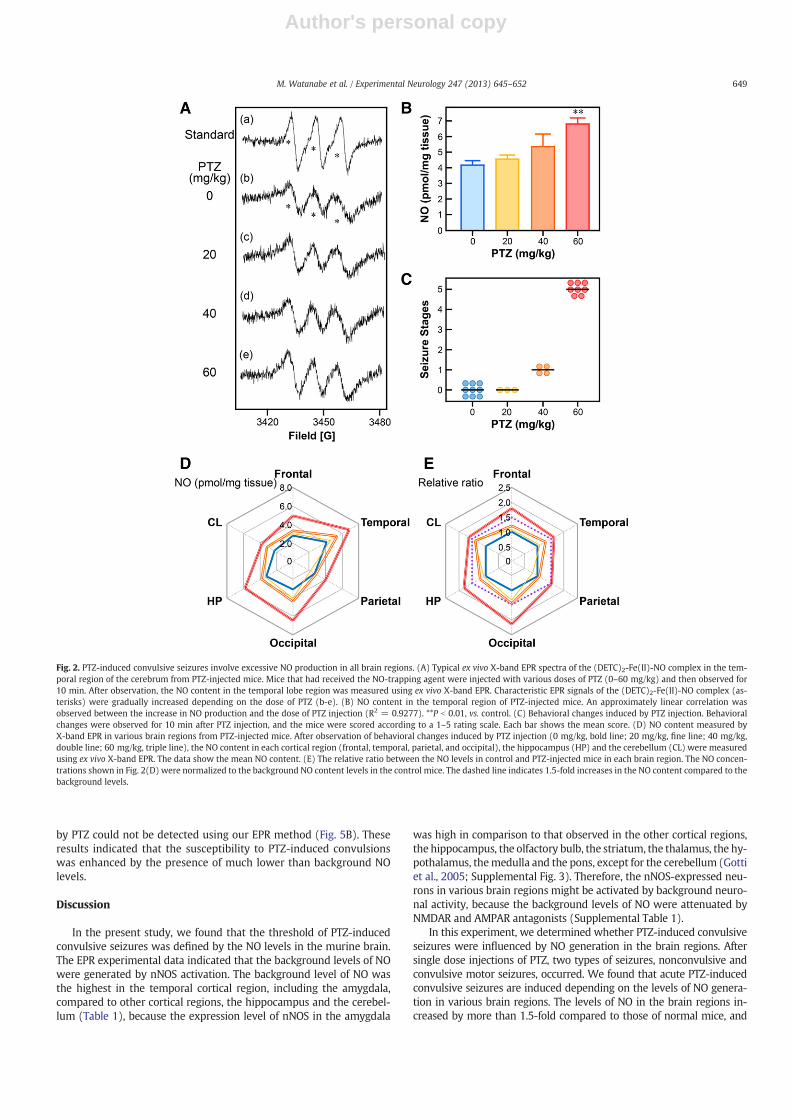
Increased NO generation dependent on the dose of PTZ (20–60 mg/kg) was observed in various regions measured in this study(Fig. 2). Although the levels of NO were dose-dependently in-creased by PTZ (Fig. 2B), the severity of the convulsions inducedby PTZ was not correlated with the dose of PTZ (Fig. 2C). Interest-ingly, the relative ratio between the NO levels in control andPTZ-injected mice was increased significantly by more than1.5-fold in all brain regions following PTZ (60 mg/kg)-inducedGCS and GTCS (Fig. 2E), although the NO contents showed no signif-icant statistical interactions between the dose of PTZ and the brainregions (two-way ANOVA, F (15, 114) = 1.3146, P = 0.2048).These results suggest that at least 1.5-fold increases in the back-ground levels of the NO content in various brain regions are neces-sary to provoke convulsive seizures by PTZ. Therefore, the thresholdof convulsive seizures induced by PTZ is regulated by the NO levelsin brain regions.
Increased NO levels and convulsive seizures induced by PTZ areprevented by nNOS inhibitor and NMDAR and AMPAR antagonists
PTZ-induced GCS and GTCS were pharmacologically prevented bya nNOS inhibitor (3Br7NI), NMDAR antagonists (CGP39551, MK801)and AMPAR antagonists (NBQX, CP465022) (Fig. 3A). The preven-tive effects of these agents were not significantly different. The
PTZ-induced severe motor convulsions apparently disappeared, al-though the nonconvulsive seizures, including forelimb jerking, werenot attenuated. The levels of NO in the pharmacologically treated micerecovered to the basal levels from more than 150% increases inducedby PTZ (Fig. 3B, Supplemental Table 2). These results indicate thatthe levels of NO produced by nNOS, which is activated by thehyperexcitability of glutamatergic neurons, in all brain regions mightthus exceed the initiation threshold of severe motor convulsions suchas GCS and GTCS but not four limb jerking.
VPA inhibits excessive NO generation induced by the activation ofglutamatergic transmission
To determine the relationship between the anti-seizure effects ofAEDs and the NO levels in acute PTZ-induced seizures, treatmentwith VPA and ESM for generalized seizures and CBZ, PHT and LTGfor partial seizures was administered before giving single injectionsof PTZ. Treatment with VPA and ESM inhibited both severe convulsiveand nonconvulsive seizures (Fig. 4A), and recovered the basal NOlevels from the excessive NO generation induced by PTZ injection inthe all brain regions (Fig. 4D, Supplemental Table 3). On the otherhand, treatment with CBZ, PHT and LTG for partial seizures did notsuppress GCS and GTCS (Fig. 4A); however, it prolonged the duration(Fig. 4B), but not the latency (Fig. 4C), of convulsions induced by PTZ.The levels of NO in the various brain regions remained at the highlevels induced by PTZ in the CBZ-, PHT- and LTG-pretreated mice(Fig. 4D, Supplemental Table 3). Overall, it was elucidated that theanti-seizure effects of AEDs involve the inhibition of the NO levelthat is the threshold for GCS and GTCS. To evaluate whether VPA in-fluences the nNOS activity and the expression of nNOS, the nNOS cat-alytic activity was examined in vitro and an immunoblot analysis wasperformed using anti-nNOS antibodies in murine brains, respectively.VPA and ESM did not inhibit the nNOS catalytic activity in vitro(Table 2), and the expression levels of nNOS in the brain regions didnot change following treatment with VPA (Supplemental Fig. 2).These results indicate that the downregulation of the NO levels in-duced by VPA and ESM is not influenced by the direct inhibition ofnNOS.
Convulsive seizures were induced by PTZ under much lower than back-ground NO levels
NO not only positively, but also negatively, regulates the onset ofPTZ-induced convulsions (Itoh and Watanabe, 2009). The nNOS−/−
mice exhibited GCS and GTCS induced by a subconvulsive dose(40 mg/kg) of PTZ (Fig. 5A). NO was not detected in any brain regionsin the mice (Fig. 5B). These results indicated that the depletion of NOgenerated by nNOS facilitated the susceptibility to PTZ-induced con-vulsions. To confirm the susceptibility to PTZ-induced convulsions,the nNOS+/+ mice was treated with a nNOS-specific inhibitor(L-NNA) 30 min before injection with a subconvulsive dose of PTZ.As shown in Fig. 5A, GCS and GTCS were induced by PTZ in thenNOS+/+ mice pretreated with a high dose (100 mg/kg) of L-NNA.In the L-NNA-pretreated nNOS+/+ mice, the changes in NO induced
Table 1Effects of PTZ on the NO content in various brain regions.
PTZ (mg/kg) n Cerebral cortex Hippocampus Cerebellum
Frontal Temporal Parietal Occipital
0 9 2.77 ± 0.17 4.19 ± 0.28 2.63 ± 0.22 3.03 ± 0.25 3.22 ± 0.23 2.29 ± 0.1520 3 2.99 ± 0.08 4.55 ± 0.27 2.08 ± 0.27 4.01 ± 0.34 3.41 ± 0.30 3.19 ± 0.2240 4 3.32 ± 0.09 5.38 ± 0.81 2.79 ± 0.47 4.39 ± 0.95 3.84 ± 0.51 3.15 ± 0.1160 7 4.97 ± 0.40a 6.83 ± 0.40a 3.99 ± 0.28a 6.46 ± 0.60a 5.86 ± 0.36a 3.79 ± 0.39a
All values are expressed as the mean ± SEM of the NO content (pmol/mg tissue). Statistical significance was assessed using the one-way ANOVA followed by Dunnett's test.a P b 0.05 for PTZ-injected mice vs. control mice.
648 M. Watanabe et al. / Experimental Neurology 247 (2013) 645–652
Author's personal copy
by PTZ could not be detected using our EPR method (Fig. 5B). Theseresults indicated that the susceptibility to PTZ-induced convulsionswas enhanced by the presence of much lower than background NOlevels.
Discussion
In the present study, we found that the threshold of PTZ-inducedconvulsive seizures was defined by the NO levels in the murine brain.The EPR experimental data indicated that the background levels of NOwere generated by nNOS activation. The background level of NO wasthe highest in the temporal cortical region, including the amygdala,compared to other cortical regions, the hippocampus and the cerebel-lum (Table 1), because the expression level of nNOS in the amygdala
was high in comparison to that observed in the other cortical regions,the hippocampus, the olfactory bulb, the striatum, the thalamus, the hy-pothalamus, themedulla and the pons, except for the cerebellum (Gottiet al., 2005; Supplemental Fig. 3). Therefore, the nNOS-expressed neu-rons in various brain regions might be activated by background neuro-nal activity, because the background levels of NO were attenuated byNMDAR and AMPAR antagonists (Supplemental Table 1).
In this experiment, we determined whether PTZ-induced convulsiveseizures were influenced by NO generation in the brain regions. Aftersingle dose injections of PTZ, two types of seizures, nonconvulsive andconvulsive motor seizures, occurred. We found that acute PTZ-inducedconvulsive seizures are induced depending on the levels of NO genera-tion in various brain regions. The levels of NO in the brain regions in-creased by more than 1.5-fold compared to those of normal mice, and
Fig. 2. PTZ-induced convulsive seizures involve excessive NO production in all brain regions. (A) Typical ex vivo X-band EPR spectra of the (DETC)2-Fe(II)-NO complex in the tem-poral region of the cerebrum from PTZ-injected mice. Mice that had received the NO-trapping agent were injected with various doses of PTZ (0–60 mg/kg) and then observed for10 min. After observation, the NO content in the temporal lobe region was measured using ex vivo X-band EPR. Characteristic EPR signals of the (DETC)2-Fe(II)-NO complex (as-terisks) were gradually increased depending on the dose of PTZ (b-e). (B) NO content in the temporal region of PTZ-injected mice. An approximately linear correlation wasobserved between the increase in NO production and the dose of PTZ injection (R2 = 0.9277). **P b 0.01, vs. control. (C) Behavioral changes induced by PTZ injection. Behavioralchanges were observed for 10 min after PTZ injection, and the mice were scored according to a 1–5 rating scale. Each bar shows the mean score. (D) NO content measured byX-band EPR in various brain regions from PTZ-injected mice. After observation of behavioral changes induced by PTZ injection (0 mg/kg, bold line; 20 mg/kg, fine line; 40 mg/kg,double line; 60 mg/kg, triple line), the NO content in each cortical region (frontal, temporal, parietal, and occipital), the hippocampus (HP) and the cerebellum (CL) were measuredusing ex vivo X-band EPR. The data show the mean NO content. (E) The relative ratio between the NO levels in control and PTZ-injected mice in each brain region. The NO concen-trations shown in Fig. 2(D) were normalized to the background NO content levels in the control mice. The dashed line indicates 1.5-fold increases in the NO content compared to thebackground levels.
649M. Watanabe et al. / Experimental Neurology 247 (2013) 645–652

Author's personal copy
their increases that occurred following PTZ injection initiated the GCS,which consisted of GCS involving the entire body, and acute GTCSconsisted of GCS followed by tonic phases. Each of the PTZ-sensitivebrain areas is a potential candidate contributing to the genesis of gener-alized convulsive seizures. Therefore, the seizure paradigm used in thisstudy was the assessment of the GCS threshold induced by the NOlevels.
The increased NO production induced by PTZ recovered to thebackground levels following treatment with NMDAR and AMPAR an-tagonists and the nNOS inhibitor (Fig. 3B). These reagents exhibitedanticonvulsant effects against severe convulsive seizures consistingof GCS and GTCS; however, myoclonic jerking was not inhibited(Fig. 3A). Generally, the order of increasing PTZ-induced seizure se-verity began with absence seizures, including facial myoclonus andvibrissal myoclonic jerking as nonconvulsive seizures, and progressedto GCS and GTCS. These results indicate that the genesis of convulsiveseizures induced by PTZ requires more than a 1.5-fold increase in thebackground levels of NO generated by normal glutamatergic activity.The possible genesis mechanisms underlying the development ofnonconvulsive seizures such as absence seizures induced by PTZ re-main obscure. This evidence suggests that the development of gener-alized convulsive seizures, but not nonconvulsive seizures, involvesthe NO biosynthetic pathways via glutamatergic and GABAergicactivities.
VPA is a conventional second-generation AED that exhibits abroad spectrum of antiepileptic activities (Löscher, 1993; Rogawskiand Löscher, 2004) and is the first choice for treatment of generalizedseizures in Japan (Societas Neurologica Japonica, 2010). VPA has beenshown to be rapidly altered by acute PTZ episodes of generalized sei-zures in a well-known generalized seizure model (Löscher et al.,1991). In this study, the elevated NO levels induced by PTZ recovered
to the background levels following treatment with VPA, andPTZ-induced convulsive and nonconvulsive seizures were completelyprevented by pretreatment with VPA (Figs. 4A, D). These results sug-gest that PTZ-induced nonconvulsive seizures are inhibited regardlessof the NO levels in the brain. Therefore, the genesis of nonconvulsiveseizures as absence seizures does not involve the NO pathway, and in-stead involves other mechanisms. To address this question, a pharma-cological study was conducted using ESM. ESM specifically blocksT-type calcium (Ca) channels (Khosravani and Zamponi, 2006;Rogawski and Löscher, 2004), and patients with generalized seizures,especially absence seizures, are treated with ESM as a second choice
Fig. 3. Enhanced NO content mediated by glutamatergic neuronal excitation in all brainregions is the threshold for induction of GCS and GTCS in PTZ-injected mice. (A) Effectsof 3Br7NI, CGP39551, MK801, NBQX and CP465022 on PTZ-induced convulsive seizures.Each inhibitor was injected 30 min before PTZ injection. The bars indicate the meanscore. *P b 0.05, vs. control. (B) Effects of 3Br7NI, CGP39551, MK801, NBQX andCP465022 on excessive NO production in PTZ-injectedmurine brains. After observing be-havioral changes induced by PTZ injection, the NO content in the temporal region wasmeasured using ex vivo X-band EPR. The NO concentrations in the inhibitor-treated mu-rine brains were normalized to the background NO content levels of normal mice.**P b 0.01, vs. PTZ-injected mice.
Fig. 4. Effects of AEDs on PTZ-induced convulsive seizures. (A) Anticonvulsive effects ofAEDs on PTZ-induced convulsive seizures. VPA, ESM, CBZ, PHT and LTG in 0.5% (w/v)CMC were injected orally 60 min before PTZ injection. Each bar shows the meanscore. *P b 0.05, vs. control. (B) Effects of AEDs on seizure duration of GCS and GTCSprovoked by PTZ injection. *P b 0.05, vs.without AEDs. (C) Effects of AEDs on the laten-cy of GCS and GTCS provoked by PTZ injection. (D) The relative ratio of NO productionin PTZ-injected mice treated with AEDs. The NO concentrations in the AED-treated mu-rine brains were normalized to the background NO content levels of normal mice.*P b 0.05, vs. PTZ-injected mice.
650 M. Watanabe et al. / Experimental Neurology 247 (2013) 645–652

Author's personal copy
in Japan (Societas Neurologica Japonica, 2010). ESM completelyinhibited PTZ-induced convulsive and nonconvulsive seizures as ef-fectively as VPA (Fig. 4A), and the levels of NO were suppressed to ap-proximately 60% (2.44 pmol/mg tissue) of the background NO levelsfollowing treatment with ESM (Fig. 4D, Supplemental Table 3).These results suggested that at least one possible explanation forthe genesis of nonconvulsive seizures induced by PTZ is that the sei-zures are triggered by Ca2+ influx via T-type Ca channels. However,a relationship between the PTZ-induced seizures and the NO levelslower than the background NO levels cannot be ruled out.
The anti-seizure effects of AEDs involve inhibition of the NO levelthat is the threshold for convulsive seizures; however, it is not clearwhether AED-induced seizure aggravations involve further increasesin the NO levels. It is generally known that treatment with CBZ andPHT, which are effective for partial seizures, cause seizure aggravationin patients with generalized epilepsy (Somerville, 2009). In the ex-periments using an acute PTZ-induced generalized seizure model,pretreatment with CBZ, PHT and LTG significantly prolonged the du-ration of severe convulsive seizures by two-fold (Fig. 4B); however,the latency was not affected (Fig. 4C). Additionally, the increasedNO levels induced by PTZ in each brain region were not further in-creased (Fig. 4D). These findings suggest that AED-induced seizureaggravations might not involve the NO pathway. Therefore, the re-sults suggest that the NO levels contribute to the grade of seizurescore rather than seizure duration or latency.
Our previous report suggested that the NO of background levelsserved as an endogenous inhibitory mechanism that allowed neuronsto inhibit aberrant electrical activity (Itoh and Watanabe, 2009). Asshown in Fig. 5, in the nNOS−/− mice and L-NNA-pretreatednNOS+/+ mice, the changes in the nNOS-derived NO production byPTZ could not be detected using our EPR method, and these animalsdeveloped GCS and GTCS. These results suggested that one of the par-adoxical functions of NO, such as a negative feedback mechanism,was involved in the PTZ-induced GCS and GTCS. In this study, wefound that two different NO levels, one more than 1.5-fold the normallevel, and the value much lower than background NO levels, wereboth involved in the induction of GCS and GTCS.
In conclusion, investigating the negative and positive feedbackmechanisms involved in theNOpathway is necessary in order to under-stand the causes of abnormal brain activity in patients with epilepsy.The tissue NO levels around 0.6 pmol/mg have the potential to nega-tively regulate PTZ-induced seizure susceptibility. On the other hand,the nonconvulsive seizures induced by PTZ are not involved in NO gen-eration caused by glutamatergic neuronal activation. Therefore, the cur-rent study indicates that increases of 1.5-fold in the NO levels arerequired to trigger convulsive seizures, but not nonconvulsive seizures,induced by PTZ.We provide novel insight into the involvement of NO inPTZ-seizure susceptibility at thewhole-animal level. Therefore, control-ling the levels of NO synthetized by nNOS in the brain will serve as animportant target for the development of AEDs in the future.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.expneurol.2013.02.019.
Acknowledgments
We are grateful to Dr. Syoji Ueki (Kagawa School of PharmaceuticalSciences, Tokushima Bunri University) for providing valuable technicaladvice on the EPR measurements. This work was supported byGrants-in-Aid for Scientific Research (C) (to M. W., Y. N. and K. I.)from the JSPS and was financially supported in part by TokushimaBunri University.
References
Banach, M., Piskorska, B., Czuczwar, S.J., Borowicz, K.K., 2011. Nitric oxide, epilepticseizures, and action of antiepileptic drugs. CNS Neurol. Disord. Drug Targets 10,808–819.
Chang, B.S., Lowenstein, D.H., 2003. Epilepsy. N. Engl. J. Med. 349, 1257–1266.Dorr, A., Sled, J.G., Kabani, N., 2007. Three-dimensional cerebral vasculature of the CBA
mouse brain: a magnetic resonance imaging and micro computed tomographystudy. NeuroImage 35, 1409–1423.
Gotti, S., Sica, M., Viglietti-Panzica, C., Panzica, G., 2005. Distribution of nitric oxidesynthase immunoreactivity in the mouse brain. Microsc. Res. Tech. 68, 13–35.
Hara, S., Kuriiwa, F., Iwata, N., Mukai, T., Kano, S., Endo, T., 1996. Distinct effects of Nomega-nitro-L-arginine on seizures induced by several drugs in mice. Pharmacol.Biochem. Behav. 53, 673–677.
Hevel, J.M., Marletta, M.A., 1994. Nitric-oxide synthase assays. Methods Enzymol. 233,250–258.
Fig. 5. The background NO level negatively regulates the susceptibility to PTZ-inducedconvulsive seizures. (A) The facilitatory effects of treatment with L-NNA in nNOS+/+
mice on the PTZ-induced convulsive seizures. Male nNOS−/− and +/+ mice wereinjected with PTZ at subconvulsive doses of 40 mg/kg. The mice were pretreatedwith L-NNA by i.p. injection 30 min before PTZ (40 mg/kg) injection. Each bar showsthe mean score. #P b 0.05, vs. PTZ (40 mg/kg)-injected nNOS+/+ mice. (B) The NO con-tent in PTZ (40 mg/kg)-injected mice pretreated with L-NNA. The gray dotted lineshows that the NO levels were below the limit of detection by EPR. *P b 0.05, vs. controlnNOS+/+ mice. #P b 0.05, vs. PTZ (40 mg/kg)-injected nNOS+/+ mice.
Table 2Effects of AEDs on the nNOS catalytic activity.
Inhibitors Relative activity(pmol/min/mg)
(Saline) – 22.73 ± 1.48SMTC 1 μM 4.65 ± 1.27a
VPA 0.5 mM 23.48 ± 0.93VPA 1 mM 23.27 ± 0.54VPA 2 mM 23.70 ± 0.72ESM 1 mM 20.67 ± 0.54ESM 2 mM 20.24 ± 0.85(DMSO) – 13.31 ± 1.50CBZ 2 mM 11.15 ± 0.91
All values are expressed as the mean ± SEM of the total NOS activity in the cerebralhomogenates. Statistical significance was assessed using the one-way ANOVA followedby Dunnett's test.
a P b 0.05 vs. control.
651M. Watanabe et al. / Experimental Neurology 247 (2013) 645–652

Author's personal copy
Itoh, K., Watanabe, M., 2009. Paradoxical facilitation of pentylenetetrazole-induced con-vulsion susceptibility in mice lacking neuronal nitric oxide synthase. Neuroscience159, 735–743.
Itoh, K., Watanabe, M., Yoshikawa, K., Kanaho, Y., Berliner, L.J., Fujii, H., 2004. Magnetic res-onance and biochemical studies during pentylenetetrazole-kindling development: therelationship between nitric oxide, neuronal nitric oxide synthase and seizures. Neuro-science 129, 757–766.
Kaneko, K., Itoh, K., Berliner, L.J., Miyasaka, K., Fujii, H., 2002. Consequences of nitric oxidegeneration in epileptic-seizure rodent models as studied by in vivo EPR. Magn.Reson. Med. 48, 1051–1056.
Khosravani, H., Zamponi, G.W., 2006. Voltage-gated calcium channels and idiopathicgeneralized epilepsies. Physiol. Rev. 86, 941–966.
Löscher, W., 1993. In vivo administration of valproate reduces the nerve terminal (syn-aptosomal) activity of GABA aminotransferase in discrete brain areas of rats.Neurosci. Lett. 160, 177–180.
Löscher, W., Hönack, D., 1991. Anticonvulsant and behavioral effects of two novel competi-tive N-methyl-D-aspartic acid receptor antagonists, CGP 37849 and CGP 39551, in thekindlingmodel of epilepsy. ComparisonwithMK-801 and carbamazepine. J. Pharmacol.Exp. Ther. 256, 432–440.
Löscher, W., Hönack, D., Fassbender, C.P., Nolting, B., 1991. The role of technical, biolog-ical and pharmacological factors in the laboratory evaluation of anticonvulsantdrugs. III. Pentylenetetrazole seizure models. Epilepsy Res. 8, 171–189.
Löscher, W., Rundfeldt, C., Hönack, D., 1993. Low doses of NMDA receptor antagonistssynergistically increase the anticonvulsant effect of the AMPA receptor antagonistNBQX in the kindling model of epilepsy. Eur. J. Neurosci. 5, 1545–1550.
Mandhane, S.N., Aavula, K., Rajamannar, T., 2007. Timed pentylenetetrazol infusiontest: a comparative analysis with s.c. PTZ\ and MES models of anticonvulsantscreening in mice. Seizure 16, 636–644.
Menniti, F.S., Buchan, A.M., Chenard, B.L., Critchett, D.J., Ganong, A.H., Guanowsky, V.,Seymour, P.A., Welch, W.M., 2003. CP-465,022, a selective noncompetitive AMPAreceptor antagonist, blocks AMPA receptors but is not neuroprotective in vivo.Stroke 34, 171–176.
Paxinos, G., Franklin, K.B.J., 2001. The Mouse Brain in Stereotaxic Coordinates, secondedition. Academic Press, San Diego.
Psarropoulou, C., Matsokis, N., Angelatou, F., Kostopoulos, G., 1994. Pentylenetet-razol-induced seizures decrease gamma-aminobutyric acid-mediated recur-rent inhibition and enhance adenosine-mediated depression. Epilepsia 35,12–19.
Rocha, L., Ackermann, R.F., Engel Jr., J., 1996. Chronic and single administration of pen-tylenetetrazol modifies benzodiazepine receptor-binding: an autoradiographicstudy. Epilepsy Res. 24, 65–72.
Rogawski, M.A., Löscher, W., 2004. The neurobiology of antiepileptic drugs. Nat. Rev.Neurosci. 5, 553–564.
Shorvon, S.D., 2009. Drug treatment of epilepsy in the century of the ILAE: the first50 years, 1909–1958. Epilepsia 50 (Suppl. 3), 69–92.
Snyder, S.H., Bredt, D.S., 1991. Nitric oxide as a neuronal messenger. Trends Pharmacol. Sci.12, 125–128.
Societas Neurologica Japonica, 2010. Guideline for Epilepsy Diagnose and Treatment inJapan. Igaku-Shoin Ltd., Tokyo.
Somerville, E.R., 2009. Some treatments cause seizure aggravation in idiopathic epilep-sies (especially absence epilepsy). Epilepsia 50, 31–36.
Watanabe, Y., Takechi, K., Fujiwara, A., Kamei, C., 2010. Effects of antiepileptics onbehavioral and electroencephalographic seizure induced by pentetrazol inmice. J. Pharmacol. Sci. 112, 282–289.
World Health Organization (WHO), 2009. Epilepsy Fact sheet N°999. http://www.who.int/mediacentre/factsheets/fs999/en/index.html.
652 M. Watanabe et al. / Experimental Neurology 247 (2013) 645–652




















