
The insulin receptor: a new anticancer target for peroxisome proliferator-activated receptor- ...
11
The insulin receptor: a new anticancer target for peroxisome proliferator-activated receptor-g (PPARg) and thiazolidinedione- PPARg agonists V Costa, D Foti, F Paonessa, E Chiefari, L Palaia, G Brunetti 1 , E Gulletta, A Fusco 2 and A Brunetti Dipartimento di Medicina Sperimentale e Clinica ‘G. Salvatore’, Universita ` di Catanzaro ‘Magna Græcia’, 88100 Catanzaro, Italy 1 Dipartimento di Scienze Biomolecolari e Biotecnologie, Universita ` di Milano, 20133 Milano, Italy 2 Dipartimento di Biologia e Patologia Cellulare e Molecolare c/o Istituto di Endocrinologia ed Oncologia Sperimentale del Consiglio Nazionale delle Ricerche, Universita ` di Napoli Federico II, e NOGEC-CEINGE, Biotecnologie Avanzate, 80131 Napoli, Italy (Correspondence should be addressed to A Brunetti; Email: [email protected]) V Costa and D Foti contributed equally to this work Abstract The peroxisome proliferator-activated receptor-g (PPARg) is a member of the nuclear hormone receptor superfamily. Ligand activation of PPARg is associated with differentiation and inhibition of proliferation in the normal and malignant cells. Herein, we studied the effects of PPARg and the PPARg agonists thiazolidinediones (TZDs) on the insulin receptor (IR), a cell membrane tyrosine kinase receptor protein, whose role is of paramount importance in mediating the metabolic and growth-promoting effects of the peptide hormone insulin. Overexpression of the PPARg1 in human hepatocellular (HepG2) cells was associated with decreased IR gene transcription and protein expression levels, and these reductions were more evident in the presence of TZDs. Since no PPARg response elements were identified on the IR promoter, we postulated that PPARg adversely affects the IR gene transcription by perturbing the assembly and stability of the transcriptionally active multiprotein-DNA complex identified previously, which includes the high-mobility group A1 protein, the ubiquitously expressed transcription factor (Sp1), the CAAT enhancer-binding protein (C/EBPb), and, in some cell lines, the developmentally regulated activator protein-2 (AP-2) transcription factor. Using glutathione S-transferase pull-down assays combined with electrophoretic mobility shift assay and chromatin immunoprecipitation, we demonstrated that by interacting with Sp1, C/EBPb, and AP-2, PPARg can prevent Sp1/AP-2 protein–protein association and inhibit binding of Sp1 and C/EBPb to DNA, thus reducing IR gene transcription. Our results demonstrate that IR is a new target gene of PPARg, and support a potential use of TZDs as anti-proliferative agents in selected neoplastic tissues overexpressing IRs. Endocrine-Related Cancer (2008) 15 325–335 Introduction The peroxisome proliferator-activated receptor-g (PPARg) is a member of a subfamily of nuclear receptors involved in the control of several aspects of lipid metabolism, including fatty acid transport, uptake by the cells, intracellular binding and activation, as well as catabolism and storage (Desvergne & Wahli 1999, Kliewer et al. 2001, Berger & Moller 2002). It is highly expressed in adipose cells and its induction precedes the activation of most adipose-specific genes (Rosen et al. 1999, Keller et al. 1993a,b). Although PPARg is mainly expressed in the adipose tissue, its expression also occurs in many normal cells Endocrine-Related Cancer (2008) 15 325–335 Endocrine-Related Cancer (2008) 15 325–335 1351–0088/08/015–325 q 2008 Society for Endocrinology Printed in Great Britain DOI: 10.1677/ERC-07-0226 Online version via http://www.endocrinology-journals.org
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of The insulin receptor: a new anticancer target for peroxisome proliferator-activated receptor- ...

Endocrine-Related Cancer (2008) 15 325–335
The insulin receptor: a new anticancertarget for peroxisome proliferator-activatedreceptor-g (PPARg) and thiazolidinedione-PPARg agonists
V Costa, D Foti, F Paonessa, E Chiefari, L Palaia, G Brunetti1,E Gulletta, A Fusco2 and A Brunetti
Dipartimento di Medicina Sperimentale e Clinica ‘G. Salvatore’, Universita di Catanzaro ‘Magna Græcia’, 88100 Catanzaro, Italy1Dipartimento di Scienze Biomolecolari e Biotecnologie, Universita di Milano, 20133 Milano, Italy2Dipartimento di Biologia e Patologia Cellulare e Molecolare c/o Istituto di Endocrinologia ed Oncologia Sperimentale del Consiglio
Nazionale delle Ricerche, Universita di Napoli Federico II, e NOGEC-CEINGE, Biotecnologie Avanzate, 80131 Napoli, Italy
(Correspondence should be addressed to A Brunetti; Email: [email protected])
V Costa and D Foti contributed equally to this work
Abstract
The peroxisome proliferator-activated receptor-g (PPARg) is a member of the nuclear hormonereceptor superfamily. Ligand activation of PPARg is associated with differentiation andinhibition of proliferation in the normal and malignant cells. Herein, we studied the effects ofPPARg and the PPARg agonists thiazolidinediones (TZDs) on the insulin receptor (IR), a cellmembrane tyrosine kinase receptor protein, whose role is of paramount importance inmediating the metabolic and growth-promoting effects of the peptide hormone insulin.Overexpression of the PPARg1 in human hepatocellular (HepG2) cells was associated withdecreased IR gene transcription and protein expression levels, and these reductions were moreevident in the presence of TZDs. Since no PPARg response elements were identified on the IRpromoter, we postulated that PPARg adversely affects the IR gene transcription by perturbingthe assembly and stability of the transcriptionally active multiprotein-DNA complex identifiedpreviously, which includes the high-mobility group A1 protein, the ubiquitously expressedtranscription factor (Sp1), the CAAT enhancer-binding protein (C/EBPb), and, in some celllines, the developmentally regulated activator protein-2 (AP-2) transcription factor. Usingglutathione S-transferase pull-down assays combined with electrophoretic mobility shift assayand chromatin immunoprecipitation, we demonstrated that by interacting with Sp1, C/EBPb,and AP-2, PPARg can prevent Sp1/AP-2 protein–protein association and inhibit binding ofSp1 and C/EBPb to DNA, thus reducing IR gene transcription. Our results demonstrate that IRis a new target gene of PPARg, and support a potential use of TZDs as anti-proliferativeagents in selected neoplastic tissues overexpressing IRs.
Endocrine-Related Cancer (2008) 15 325–335
Introduction
The peroxisome proliferator-activated receptor-g(PPARg) is a member of a subfamily of nuclear
receptors involved in the control of several aspects of
lipid metabolism, including fatty acid transport, uptake
by the cells, intracellular binding and activation, as
Endocrine-Related Cancer (2008) 15 325–335
1351–0088/08/015–325 q 2008 Society for Endocrinology Printed in Great
well as catabolism and storage (Desvergne & Wahli
1999, Kliewer et al. 2001, Berger & Moller 2002). It is
highly expressed in adipose cells and its induction
precedes the activation of most adipose-specific genes
(Rosen et al. 1999, Keller et al. 1993a,b). Although
PPARg is mainly expressed in the adipose tissue,
its expression also occurs in many normal cells
Britain
DOI: 10.1677/ERC-07-0226
Online version via http://www.endocrinology-journals.org

V Costa, D Foti et al.: Effects of PPARg and TZD-PPARg on IR expression
(Vidal-Puig et al. 1997), and in several cancer cell lines
and tissues, including breast, prostate, colon, lung, and
B-lineage cells (Padilla et al. 2000, Allred & Kilgore
2005, Nunez et al. 2005). Besides a fundamental role in
adipogenesis, PPARg plays a critical role in placental,
cardiac, and embryonic development (Barak et al.
1999, Maloney & Rees 2005) and influences the
production of cytokines, growth factor release, and cell
cycle progression leading to differentiation-inducing,
anti-inflammatory, and anti-proliferative effects
(Kersten et al. 2000, Tan et al. 2005, Chou et al.
2007). The PPARg generally exerts its biological
function as heterodimer with the retinoid X receptor,
which binds to a specific cis-acting sequence on DNA,
a peroxisome proliferator response element, initiating
gene transcription (Desvergne & Wahli 1999, Tan
et al. 2005). The PPARg activators identified so far
include some inflammatory activators, like a prosta-
glandin J2 metabolite (15-deoxy-D12,14-PGJ2), and
oxidized LDL particles (9-and 13-HODE; Desvergne
& Wahli 1999, Kersten et al. 2000), and the synthetic
thiazolidinedione (TZD) compounds (Lehmann et al.
1995). The TZDs are a new class of insulin-sensitizing
drugs widely used in the treatment of human type 2
diabetes mellitus, including rosiglitazone, pioglita-
zone, and ciglitazone (Lehmann et al. 1995, Saltiel &
Olefsky 1996). However, despite the positive role of
TZDs in the control of glucose homeostasis, increasing
evidences support a potential use of these drugs as anti-
proliferative agents.
The peptide hormone insulin is a major regulator of
glucose homeostasis and cell growth. The first step in
insulin action is the binding of the hormone to the
insulin receptor (IR), a phylogenetically ancient
receptor protein embedded in the plasma membrane
of insulin target cells. The IR belongs to the tyrosine
kinase growth factors receptor superfamily and
consists of two identical extracellular a-subunits that
bind insulin, and two transmembrane b-subunits withintrinsic tyrosine kinase activity (Ullrich et al. 1985,
Goldfine 1987, White & Kahn 1994). When insulin
binds to the IR, the receptor is first activated by
tyrosine autophosphorylation and then the IR tyrosine
kinase phosphorylates various effector molecules, like
the insulin receptor substrate-1 (IRS-1), leading to
hormone action (Ullrich et al. 1985, Goldfine 1987,
White & Kahn 1994). In target cells, the IR has been
shown to be under the regulation of hormones,
metabolites, and differentiation (Brunetti et al. 1989,
Mamula et al. 1990). To understand the molecular
mechanisms controlling IR gene regulation, many
groups including our own group have previously
identified and characterized the 5 0-flanking region of
326
the IR gene (Araki et al. 1987, Seino et al. 1989,
Brunetti et al. 1993). Later, we have provided evidence
that transcriptional activation of the human IR
promoter requires the assembly of a transcriptionally
active multiprotein-DNA complex which includes, in
addition to the architectural factor high-mobility group
A1 (HMGA1), the transcription factors Sp1 and CAAT
enhancer-binding protein (C/EBPb). Functional integ-rity of this nucleoprotein complex is required for full
transactivation of the IR gene by Sp1 and C/EBPb in
cells readily expressing IRs (Brunetti et al. 2001, Foti
et al. 2003, 2005). Recently, we published data
showing that the nuclear transcription factor activator
protein-2 (AP-2), by interacting with Sp1 and
HMGA1, may stabilize this multiprotein complex at
the level of the IR promoter, leading to IR over-
expression in breast cancer cells (Paonessa et al. 2006).
In this paper, we demonstrate for the first time that
PPARg and its ligands inhibit IR gene transcription.
We show that PPARg physically interacts with Sp1,
AP-2, and C/EBPb in vitro, and this protein–protein
interaction significantly reduces the transactivation
of the IR gene by Sp1, C/EBPb, and AP-2 in vivo, in
the absence of a consensus sequence for PPARg on the
IR promoter.
Materials and methods
Cells and protein extracts
HepG2 human hepatoma cells, 3T3-L1 mouse fibro-
blasts, andMCF-7 human breast cancer cells (American
Type Culture Collection, Manassas, VA, USA) were
maintained in DMEM (GIBCO-BRL) supplemented
with 10% fetal bovine serum. The 3T3-L1 fibroblasts
were differentiated into 3T3-L1 adipocytes as described
elsewhere (Foti et al. 2003). Nuclear and cytoplasmic
extracts were prepared from cultured cells as previously
described (Brunetti et al. 2001). For each nuclear
extract, an equal number of nuclei were homogenized,
and the final protein concentration in the extracts
was determined by the modified Bradford method
(Bio-Rad Laboratories).
RT-PCR, immunoprecipitation, and western
blot analysis
Total cellular RNA was extracted from cells using
the RNAqueous-4PCR kit and subjected to DNase
treatment (Ambion, Austin, TX, USA). The cDNA was
synthesized from total RNA with the RETROscript
first strand synthesis kit (Ambion) and used for
PCR amplification. The PCR products were electro-
phoretically resolved on 2% agarose gel, visualized by
www.endocrinology-journals.org

Endocrine-Related Cancer (2008) 15 325–335
ethidium bromide staining, and quantified by densito-
metry. The values obtained were then normalized to
those of the tubulin gene. Western blot analyses of IR,
PPARg, HMGA1, Sp1, C/EBPb, and AP-2 were done
on total cellular lysates or nuclear extracts as
previously described (Brunetti et al. 2001, Foti et al.
2003, 2005). For immunoprecipitation studies with IR
antibody, aliquotes of HepG2 cytoplasmic extracts
were incubated for 12 h with rotation at 4 8C with 10 mlIR antibody-coupled protein A beads. Covalent
coupling of antibody was performed as previously
described (Paonessa et al. 2006). Beads were recovered
by gentle centrifugation and washed thrice with 500 mlNETN wash buffer (0.1% NP40, 150 mM NaCl, 1 mM
EDTA, 50 mM Tris–HCl (pH 8.0)) for 5 min. Protein
was removed from the beads by boiling in sample
buffer for 5 min and analyzed by SDS-PAGE and
immunoblotting. Antibodies used for these studies
were as follows: anti-HMGA1 (Brunetti et al. 2001),
anti-PPARg1 (H-100), anti-Sp1 (PEP 2), anti-C/EBPb(C-19), anti-AP-2a (H-79), and anti-IRb (C-19; Santa
Cruz Biotechnology, Santa Cruz, CA, USA).
GST pull-down assay
The 35S-labeled HA-tagged HMGA1, Sp1, C/EBPb,and AP-2 (Foti et al. 2003, Paonessa et al. 2006) were
synthesized in vitro using the TNT-T7 quick-coupled
transcription/translation system (Promega). The gluta-
thione S-transferase (GST) fusion protein expression
vectors pGEX4T-3-human PPARg2 (kindly provided
by H Nishizawa and T Funahashi, University of Osaka;
Nishizawa et al. 2002) and the pGEX-2TK control
vector (Amersham Pharmacia Biotech) were trans-
formed into the BL21(D3) strain of Escherichia coli
(Stratagene, La Jolla, CA, USA), expanded in suspen-
sion culture, and induced for 2 h with 0.5 mM
isopropyl-D-thiogalactopyranoside (Sigma). Bacteria
were pelleted, sonicated in ice-cold PBS lysis buffer
containing 1% NP-40, 10% glycerol, 1 mM dithio-
threitol (DTT), 1 mM phenylmethylsulphonyl fluoride
(PMSF), 10 mg pepstatinA, leupeptin, and aprotinin/ml,
and 0.4 mg lysozyme/ml and centrifuged. The resultant
supernatant was then added to 300 ml glutathione-
agarose beads, mixed on a rotating wheel at 4 8C for 2 h
and centrifuged. The bound GST-fused proteins in the
pellet were washed five times with lysis buffer and
resuspended in 300 ml binding buffer (50 mM NaCl,
20 mM Tris–HCl (pH 8.0), 0.05% NP-40, 0.25% BSA,
1 mM PMSF, 1 mM DTT). The bound protein was
quantitated with the Coomassie protein assay reagent
(Pierce Co., Rockford, IL, USA), and 0.5 mg of each
GST-protein bound to glutathione-agarose beads was
www.endocrinology-journals.org
incubated with 7 ml in vitro-translated 35S-labeled
protein in 150 ml binding buffer at 4 8C for 2 h.
Reactions were terminated by centrifugation, the
precipitate was washed three times with protein-
binding buffer and subjected to a 10% PAGE
(SDS-10% PAGE), and proteins were visualized by
autoradiography.
Plasmids, transfections, and electrophoretic
mobility shift assay (EMSA)
Recombinant chloramphenicol acetyltransferase
(CAT) plasmids (pIR-CAT, pCAT-C2 and pCAT-E3)
containing different fragments of the human IR gene
promoter have been described previously (Brunetti
et al. 1993, Foti et al. 2003, Paonessa et al. 2006). The
human PPARg1 expression vector was obtained using
standard techniques after PCR amplification and
cloning of PPARg full-length cDNA into the
expression vector pcDNA3. The pC-hu/AP-2aexpression vector was kindly provided by K Fujimori
(Osaka Bioscience Institute). The IR-CAT reporter
plasmids, together with effector vectors for PPARgand/or AP-2, in the absence or presence of the PPARgagonist rosiglitazone (a kind gift of Glaxo Smith Kline)
or ciglitazone (Vinci-Biochem Alexis, Vinci, Italy)
were transiently transfected into cultured cells by the
calcium phosphate precipitation method, and CAT
activity was assayed 48 h later, as previously described
(Brunetti et al. 1993). The pSV-b-galactosidase controlvector served as an internal control of transfection
efficiency, together with measurements of protein
expression levels (Brunetti et al. 2001). For EMSA,
the C2 sequence (300 bp) of the human IR gene
promoter was generated by PCR amplification, using
the recombinant plasmid pCAT-C2 (Brunetti et al.
1993). The 32P-labeled C2 was used in gel shift assays
as previously described (Brunetti et al. 2001).
Chromatin immunoprecipitation (ChIP)
ChIP assay was done as described previously
(Foti et al. 2005), using HepG2 cells transfected with
the AP-2a expression vector and untransfected MCF-7
cells. Formaldehyde-fixed DNA–protein complex was
immunoprecipitated with anti-HMGA1, anti-Sp1, anti-
C/EBPb, or anti-AP-2a antibody. Primers for the IR
sequence (Paonessa et al. 2006) were used for PCR
amplification of immunoprecipitated DNA (30 cycles)
using PCR ready-to-go beads (Amersham Pharmacia
Biotech). The PCR products were electrophoretically
resolved on 1.5% agarose gel and visualized by
ethidium bromide staining.
327

V Costa, D Foti et al.: Effects of PPARg and TZD-PPARg on IR expression
Statistical analysis
All experiments were performed at least three times.
The mean and S.E.M. were calculated for each set of
results and the significance of the difference between
values was assessed by the Student’s t-test. For all
analyses, P!0.05 was considered significant.
Results
PPARg and TZDs reduce IR protein and mRNA
expression in HepG2 cells
To see whether PPARg had any effect on IR
expression, we first measured the IR protein content
in HepG2 cells, before and after forced expression of
exogenous PPARg1. The HepG2 cells were consideredideally suited for studying the effects of PPARg on IR
expression since IRs are relatively abundant in this cell
line, while endogenous PPARg is barely detectable.
Using immunoprecipitation and western blot analyses,
we showed that IR protein content was considerably
reduced in cells overexpressing PPARg (w50% less
than control cells), and this reduction was more
pronounced in PPARg1-overexpressing cells exposed
to 10K5 M TZD (rosiglitazone; Fig. 1). It is notable
that rosiglitazone alone also reduced IR protein
content, and it is likely that this occurred, at least
in part, by stimulating endogenous PPARg (Tontonoz
et al. 1994). As measured by RT-PCR, the abundance
of IR mRNA transcripts in HepG2 cells closely
Figure 1 Inhibition of IR protein and mRNA levels by PPARgand TZDs. (A) Total cellular extracts from untransfected(control) or HepG2 cells transfected with PPARg expressionvector, in the absence or presence of TZD (rosiglitazone,10K7 M), were resolved by SDS-PAGE, and the IR proteincontent was measured by immunoprecipitation and the westernblot (IP/WB) with an anti-IRb antibody. Tubulin was measuredas an internal control. (B) In parallel experiments, IR and tubulinmRNA levels were quantified by RT-PCR.
328
correlated with IR protein content in every condition
analyzed, indicating that changes at the protein level
reflected differences in IR mRNA (Fig. 1).
PPARg and TZDs reduce IR gene transcription
in cultured cells
In order to reveal whether PPARg and its agonists
inhibited the transcriptional activation of IR gene, we
performed reporter gene analysis using the pIR-CAT
recombinant vector that contains the entire 50-flanking
region of the human IR gene (Foti et al. 2003). Forced
expression of PPARg1 in HepG2 cells transfected with
pIR-CAT determined a 40–50% reduction in CAT
activity that was similar to the reduction observed in
cells exposed to rosiglitazone alone, and resulted more
marked in PPARg1-overexpressing cells simultaneously
treated with TZD rosiglitazone (Fig. 2A). The possibility
that PPARg could theoretically decrease IR gene
transcription by reducing the intracellular expression of
HMGA1, Sp1 or C/EBPb was excluded by western blot
analysis of nuclear extracts showing that no changeswere
observed in the endogenous levels of these proteins under
these experimental conditions (Fig. 2A). The hypothesis
that PPARg and its agonists could negatively interfere
with the crosstalk between these various transcription
factors in the context of the IR gene was substantiated
further by transiently cotransfecting the AP-2-null cell
lineHepG2with thepIR-CATpromoter, togetherwith an
expression vector encoding the wild-type AP-2a. Asshown in Fig. 2A, overexpressionofAP-2 inHepG2 cells
efficiently induced IR gene transcription. However, this
effect was totally abolished by PPARg and TZDs that
reverted gene transcription to levels similar to those
observed in the absence of AP-2 (Fig. 2A). These
findings were confirmed in PPARg overexpressing
MCF-7 cells, a cell line naturally expressing only
detectable levels of PPARg (Tontonoz et al. 1994), and
3T3-L1 adipocytes producing relatively high amounts of
endogenous PPARg (Mueller et al. 1998). As shown in
Fig. 2B and C, exposure of both cell lines to either
rosiglitazone and/or ciglitazone reduced IR gene
transcription in a dose-dependent manner.
We have previously shown that transcriptional
activation of the human IR gene by HMGA1, Sp1,
and C/EBPb requires the assembly and cooperation
among these various nuclear factors at the levels of two
AT-rich sequences of the IR gene promoter, C2 and E3
(K674 to K874 and K1662 to K1818 bp upstream
of the IR ATG codon respectively), which have a
significant ability to drive transcription when intro-
duced into mammalian cells (Brunetti et al. 1993,
2001, Foti et al. 2003). As demonstrated before, the
www.endocrinology-journals.org

Figure 2 Functional significance of PPARg and TZDs for IR gene transcription. (A) HepG2, (B) MCF-7, and (C) 3T3-L1 cells weretransfected with a CAT reporter plasmid containing the IR promoter element (pCAT-IR, 2 mg), in the absence or presence of PPARg(2 mg) and/or AP-2 (1 mg) expression vector, with or without treatment with rosiglitazone or ciglitazone at the indicated molarconcentrations. For each cell line, data represent the meansGS.E.M. for three separate experiments; values are expressed relative tothe CAT activity obtained in transfections with the pCAT-IR alone that is assigned an arbitrary value of 1. *P!0.05 versus cellstransfected with the pCAT-IR alone; #P!0.01 versus cells transfected with the pCAT-IR plus AP-2 effector vector. In someexperiments (D), HepG2 cells were transfected as above, using the CAT reporter vector containing the proximal C2 (pCAT-C2) ordistal E3 (pCAT-E3) element of the IR gene promoter. *P!0.05 versus cells transfected with the pCAT-C2 alone. White bars, mock(no DNA); black bars, pCAT-basic (vector without an insert). Shown are representative western blots of PPARg, Sp1, C/EBPb, andHMGA1 from nuclear extracts of transfected HepG2 cells.
Endocrine-Related Cancer (2008) 15 325–335
lack of any of these factors, or a scarcity of one or
more, may severely perturb IR gene transcription in
cells and tissues readily expressing IR (Brunetti et al.
2001, Foti et al. 2003, 2005). When the recombinant
plasmid pCAT-C2 was transfected into HepG2 cells
(and other cell lines), PPARg, and TZDs, either alone
or in combination, inhibited pCAT-C2 activity to the
same extent than they did in HepG2 cells transfected
with the full-length pCAT-IR promoter (Fig. 2D),
indicating that interference of PPARg and agonists
with IR gene-transcription machinery occurs at the
level of the proximal promoter region, C2. This
conclusion was supported by the observation that in
experiments using the pCAT-E3 reporter vector,
containing the more distant E3 sequence of the IR
www.endocrinology-journals.org
gene, no effects on CAT activity were observed in cells
exposed to PPARg and/or TZDs (Fig. 2D).
PPARg physically interacts with Sp1, C/EBPb,
and AP-2, in vitro, in the absence of DNA
In our studies, no peroxisome proliferator response
elements (PPRE) have been identified on the promoter
region of the IR gene. A similar observation has been
previously provided by us for the transcription factor
AP-2, for which DNA-binding activity was undetectable
with the IR gene promoter, and transactivation of the IR
gene by AP-2 occurred indirectly through physical and
functional cooperation with HMGA1 and Sp1 (Paonessa
et al. 2006). Therefore, in an attempt to identify the
biochemical mechanisms underlying the negative effects
329
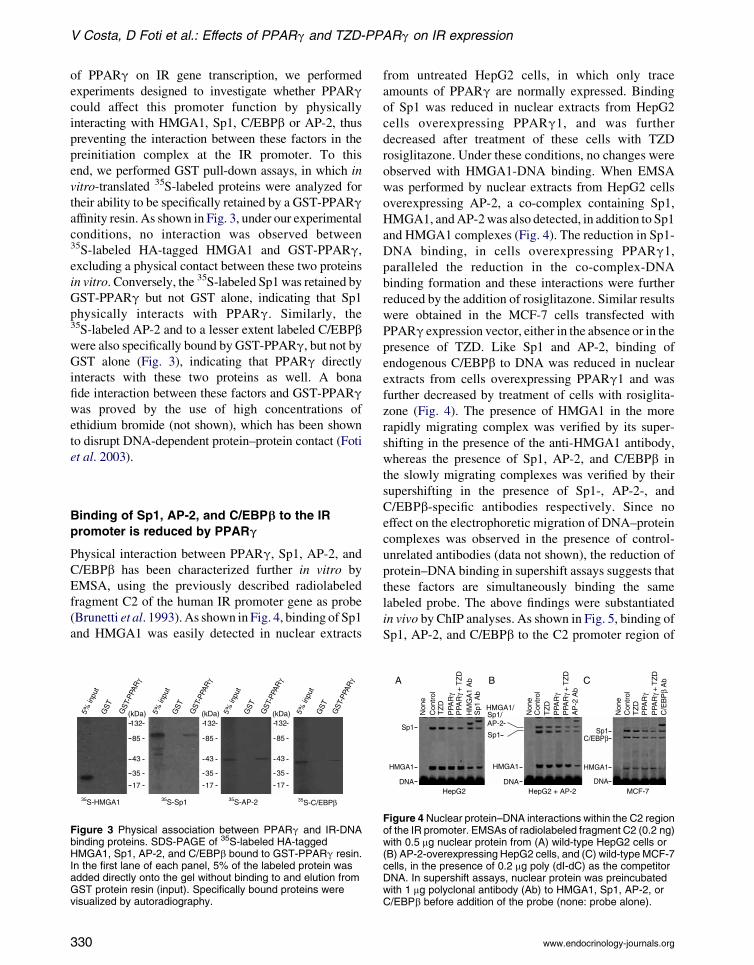
V Costa, D Foti et al.: Effects of PPARg and TZD-PPARg on IR expression
of PPARg on IR gene transcription, we performed
experiments designed to investigate whether PPARgcould affect this promoter function by physically
interacting with HMGA1, Sp1, C/EBPb or AP-2, thus
preventing the interaction between these factors in the
preinitiation complex at the IR promoter. To this
end, we performed GST pull-down assays, in which in
vitro-translated 35S-labeled proteins were analyzed for
their ability to be specifically retained by a GST-PPARgaffinity resin. As shown in Fig. 3, under our experimental
conditions, no interaction was observed between35S-labeled HA-tagged HMGA1 and GST-PPARg,excluding a physical contact between these two proteins
in vitro. Conversely, the 35S-labeled Sp1was retained by
GST-PPARg but not GST alone, indicating that Sp1
physically interacts with PPARg. Similarly, the35S-labeled AP-2 and to a lesser extent labeled C/EBPbwere also specifically bound by GST-PPARg, but not byGST alone (Fig. 3), indicating that PPARg directly
interacts with these two proteins as well. A bona
fide interaction between these factors and GST-PPARgwas proved by the use of high concentrations of
ethidium bromide (not shown), which has been shown
to disrupt DNA-dependent protein–protein contact (Foti
et al. 2003).
Binding of Sp1, AP-2, and C/EBPb to the IR
promoter is reduced by PPARg
Physical interaction between PPARg, Sp1, AP-2, andC/EBPb has been characterized further in vitro by
EMSA, using the previously described radiolabeled
fragment C2 of the human IR promoter gene as probe
(Brunetti et al. 1993).As shown in Fig. 4, binding of Sp1
and HMGA1 was easily detected in nuclear extracts
Figure 3 Physical association between PPARg and IR-DNAbinding proteins. SDS-PAGE of 35S-labeled HA-taggedHMGA1, Sp1, AP-2, and C/EBPb bound to GST-PPARg resin.In the first lane of each panel, 5% of the labeled protein wasadded directly onto the gel without binding to and elution fromGST protein resin (input). Specifically bound proteins werevisualized by autoradiography.
330
from untreated HepG2 cells, in which only trace
amounts of PPARg are normally expressed. Binding
of Sp1 was reduced in nuclear extracts from HepG2
cells overexpressing PPARg1, and was further
decreased after treatment of these cells with TZD
rosiglitazone. Under these conditions, no changes were
observed with HMGA1-DNA binding. When EMSA
was performed by nuclear extracts from HepG2 cells
overexpressing AP-2, a co-complex containing Sp1,
HMGA1, andAP-2was also detected, in addition to Sp1
and HMGA1 complexes (Fig. 4). The reduction in Sp1-
DNA binding, in cells overexpressing PPARg1,paralleled the reduction in the co-complex-DNA
binding formation and these interactions were further
reduced by the addition of rosiglitazone. Similar results
were obtained in the MCF-7 cells transfected with
PPARg expression vector, either in the absence or in the
presence of TZD. Like Sp1 and AP-2, binding of
endogenous C/EBPb to DNA was reduced in nuclear
extracts from cells overexpressing PPARg1 and was
further decreased by treatment of cells with rosiglita-
zone (Fig. 4). The presence of HMGA1 in the more
rapidly migrating complex was verified by its super-
shifting in the presence of the anti-HMGA1 antibody,
whereas the presence of Sp1, AP-2, and C/EBPb in
the slowly migrating complexes was verified by their
supershifting in the presence of Sp1-, AP-2-, and
C/EBPb-specific antibodies respectively. Since no
effect on the electrophoretic migration of DNA–protein
complexes was observed in the presence of control-
unrelated antibodies (data not shown), the reduction of
protein–DNA binding in supershift assays suggests that
these factors are simultaneously binding the same
labeled probe. The above findings were substantiated
in vivo by ChIP analyses. As shown in Fig. 5, binding of
Sp1, AP-2, and C/EBPb to the C2 promoter region of
Figure 4 Nuclear protein–DNA interactions within the C2 regionof the IR promoter. EMSAs of radiolabeled fragment C2 (0.2 ng)with 0.5 mg nuclear protein from (A) wild-type HepG2 cells or(B) AP-2-overexpressing HepG2 cells, and (C) wild-type MCF-7cells, in the presence of 0.2 mg poly (dI-dC) as the competitorDNA. In supershift assays, nuclear protein was preincubatedwith 1 mg polyclonal antibody (Ab) to HMGA1, Sp1, AP-2, orC/EBPb before addition of the probe (none: probe alone).
www.endocrinology-journals.org

Figure 5 ChIP assay. ChIP of the IR promoter gene in AP-2a-producing HepG2 cells and MCF-7 cells both induced tooverexpress PPARg, either in the absence or in the presence ofTZD (rosiglitazone). ChIP was done using antibodies (Ab)against either HMGA1, Sp1, AP-2, or C/EBPb.
Endocrine-Related Cancer (2008) 15 325–335
the IR gene was considerably attenuated in the HepG2
and MCF-7 cells overexpressing PPARg, either in the
absence or presence of the TZD rosiglitazone. Thus,
these data suggest that overexpression and/or
activation of PPARg adversely affects IR gene
transcription in the absence of PPRE on the IR gene
promoter. We propose that, by causing a displacement
of Sp1,AP-2, andC/EBPb, PPARgmay play significant
molecular roles in the transcriptional activities of these
factors in the context of the IR gene, both in physiology
and pathology (Fig. 6).
Discussion
The TZDs are insulin-sensitizing drugs that improve
insulin sensitivity in insulin-resistant states such as type
2 diabetes mellitus and obesity. These drugs are
high-affinity ligands for the nuclear receptor PPARg,which regulate transcription of target genes involved in
the homeostasis of nutrients. In addition to the effects on
lipid and glucose metabolism, many evidences have
shown that PPARg and its ligands play an important
role in modulating many processes, including cell
proliferation and differentiation, as well as inflam-
mation, angiogenesis, and immune system function.
The IR is critical in the insulin-mediated effects on
cell metabolism and cell growth. Various studies have
shown that IRs are increased in most human breast
cancers, and both ligand-dependent malignant
www.endocrinology-journals.org
transformation and increased cell growth occur in
cultured breast cells overexpressing the IR (Osborne
et al. 1978, Milazzo et al. 1992, Paonessa et al. 2006).
Overexpression of functional IRs has also been
involved in thyroid carcinogenesis (Farid et al.
1994). The IR can exert its oncogenic potential in
malignant cells via abnormal stimulation of multiple
cellular signaling cascades, enhancing growth factor-
dependent proliferation and/or by directly affecting
cell metabolism. Both breast and thyroid neoplastic
cells express PPARg, and PPARg agonists have been
shown to inhibit proliferation in these and other cell
systems (Tontonoz et al. 1994, Martelli et al. 2002,
Grommes et al. 2004). In this light, we undertook to
investigate whether IR expression could be affected by
PPARg. Our study shows that IR gene transcription
and receptor protein content were reduced in cells with
forced PPARg1 overexpression, or TZD-induced
PPARg activation. Although these results apparently
run contrary to what might have been predicted based
on the known insulin-sensitizing effects of TZD,
seeming to exclude the possibility that TZDs may act
as insulin sensitizers through the IR, they are
compatible with the pleiotropic effects of PPARg. Inthis regard, the IR may be considered a new target gene
that accounts for the anti-mitogenic response to
PPARg and its agonists, and this is the first description
of a tyrosine kinase receptor involved in PPARg-induced anti-proliferative mechanisms. To unravel the
molecular basis underlying the decrease in IR gene
expression produced by PPARg, we performed
protein–protein and DNA–protein interaction studies
together with ChIP analysis, combined with transient
transcription assays in the living cells expressing
variable amounts of the IRs. Since no PPRE have
been detected within the IR gene promoter and no
reduction in the expression of HMGA1, Sp1, or
C/EBPb was observed in cells after PPARg or TZD
stimulation, we propose that PPARg acts as a negative
regulator of IR transcription by adversely affecting
binding of Sp1, C/EBPb, or AP-2 to the IR gene. In
other PPARg-responsive genes that carry a PPRE
consensus in their sequence, a functional cooperation
between Sp1 and PPARg has been described (Krey
et al. 1995). On the other hand, functional cooperation
between C/EBPb (and other members of the C/EBP
family of proteins) and PPARg has been reported in
the nutrient signaling system during fetal development
(Maloney & Rees 2005), regulation of vascular
inflammation (Takata et al. 2002), and inhibition of
adhesive interaction between multiple myeloma
and bone marrow stromal cells (Wang et al. 2007).
Herein, we show that PPARg physically interacts with
331

Figure 6 Proposed model for the action of PPARg in the context of the IR gene promoter (C2 region). (A and B) By binding tosequence-specific sites located in the C2 regulatory region of the IR gene, HMGA1, Sp1, and C/EBPb exert a positive control overthe rates of transcription. The PPARg can displace Sp1 and C/EBPb from their binding sites, decreasing the rate of transcription.(C and D) AP-2 overexpression accounts for increased IR expression in both breast cancer cell lines and breast cancer tissues, inwhich HMGA1 acts by facilitating and stabilizing interactions between AP-2 and Sp1 in the preinitiation complex at the IR promoter(Paonessa et al. 2006). By binding to AP-2 and Sp1, PPARg may attenuate the stimulatory role of AP-2 in IR gene expression inbreast carcinogenesis.
V Costa, D Foti et al.: Effects of PPARg and TZD-PPARg on IR expression
Sp1, C/EBPb, and AP-2 reducing the IR gene
transcription in the absence of PPARg DNA-binding
sites on the IR gene. We suggest that, in the absence of
PPRE in the context of the IR promoter, this nuclear
receptor may produce its adverse effects on IR gene
transcription by interacting physically with these
factors, thus reducing their availability to the basic
transcription machinery of the IR gene. With a similar
mechanism involving Sp1/PPARg protein interaction,
PPARg has been shown to exert an anti-proliferative
role by suppressing transcription of the thromboxane
receptor, the cyclin-dependent kinase inhibitor p21,
and the fibronectin genes (Sugawara et al. 2002, Hong
et al. 2004, Han et al. 2005). The molecular
mechanism herein described is therefore in agreement
with the increasing repertoire of ‘non-canonical’
PPARg target genes that now encompasses non-
PPREs containing genes (Tan et al. 2005).
Many potential target genes of PPARg have been
already reported, including bcl-2, b-catenin, and the
PTEN tumor suppressor gene (Elstner et al. 1998, Patel
et al. 2001, Mulholland et al. 2005). However, how
PPARg and its agonists may induce their anti-
proliferative effects is not fully understood yet.
Recently, non-genomic crosstalks between PPARgand cytoplasmic proteins, like extracellular signal-
regulated kinase (ERK) 1/2 and MAPK kinases, have
been reported in cancer cells and functional importance
has been given to the subcellular localization of PPARg
332
(Burgermeister & Seger 2007, Papageorgiou et al.
2007). However, the last three decades of medical
research examining the molecular pathogenesis of
cancers have provided compelling evidence for the
universal disruption of the cell cycle in human tumors,
and recent studies have demonstrated a critical interface
between hormonal signaling and the cell cycle
(Hilakivi-Clarke et al. 2004). In this context, mitogens
like insulin, via the IR, may promote the progression
through the G1 phase by inducing competence of the
cyclin D1/cyclin-dependant kinase 4 (CDK4) complex.
It has been previously demonstrated that the PPARgagonists inhibit cyclin D1 (Wang et al. 2001). Our
findings support the conclusion that PPARg and TZDs
may interfere with the hormonal control of the cell
cycle, at least in part, through the inhibition of the IR.
Over the last decade, PPARg has emerged as an
important drug target in type 2 diabetes mellitus
(Savkur & Miller 2006), and TZDs are widely used for
treatment of diabetic patients. However, conflicting
results on the pro-carcinogenic and anti-tumorigenic
effects of TZDs in humans with diabetes can be found
in the literature. For instance, whereas a population-
based report showed that TZDs were associated with
reduced risk of lung cancer in patients with diabetes
(Govindarajan et al. 2007), a possible association
between cancer and the use of TZD has been reported
later in type 2 diabetic patients (Ramos-Nino et al.
2007). Taken together, our results consistently support
www.endocrinology-journals.org

Endocrine-Related Cancer (2008) 15 325–335
the conclusion that IR gene may be considered a new
anticancer target for PPARg, providing further
evidence for the use of TZDs as anti-proliferative
agents in selected tumors overexpressing the IR.
Acknowledgements
The authors would like to thank K S Chang, K
Fujimori, T Funahashi, H Nishizawa and M A Lazar
for providing reagents. They would also like to thank
A Malta, G Ceravolo, T Rossano and G Grandinetti
for secretarial help. Grant support: Ministero dell’Uni-
versita e della Ricerca, Italy protocol 2004062059-002
and Telethon, Italy grant GGP04245 (A Brunetti).
The authors declare that there is no conflict of
interest that would prejudice the impartiality of this
scientific work.
References
Allred CD & Kilgore MW 2005 Selective activation of
PPARg in breast, colon, and lung cancer cell lines.
Molecular and Cellular Endocrinology 235 21–29.
Araki E, Shimada F, Uzawa H, Mori M & Ebina Y 1987
Characterization of the promoter region of the human
insulin receptor gene. Journal of Biological Chemistry
262 16186–16191.
Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P,
Chien KR, Koder A& Evans RM 1999 PPARg is required
for placental, cardiac, and adipose tissue development.
Molecular Cell 4 585–595.
Berger J & Moller DE 2002 The mechanisms of action of
PPARs. Annual Review of Medicine 53 409–435.
Brunetti A, Maddux BA, Wong KY & Goldfine ID 1989
Muscle cell differentiation is associated with increased
insulin receptor biosynthesis and messenger RNA levels.
Journal of Clinical Investigation 83 192–198.
Brunetti A, Foti D & Goldfine ID 1993 Identification of
inique nuclear regulatory proteins for the insulin receptor
gene, which appear during myocyte and adipocyte
differentiation. Journal of Clinical Investigation 92
1288–1295.
Brunetti A, Manfioletti G, Chiefari E, Goldfine ID & Foti D
2001 Transcriptional regulation of human insulin receptor
gene by the high-mobility-group protein HMGI-Y.
FASEB Journal 15 492–500.
Burgermeister E & Seger R 2007 MAPK kinases as nucleo-
cytoplasmic shuttles for PPARgamma. Cell Cycle 6
1539–1548.
Chou FS, Wng PS, Kulp S & Pinzone JJ 2007 Effects of
thiazolidinediones on differentiation, proliferation, and
apoptosis. Molecular Cancer Research 5 523–530.
Desvergne B & Wahli W 1999 Peroxisome proliferator-
activated receptors: nuclear control of metabolism.
Endocrine Reviews 20 649–688.
www.endocrinology-journals.org
Elstner E, Muller C, Koshizuka K, Williamson EA, Park D,
Asou H, Shintaku P, Said JW, Heber D & Koeffler HP
1998 Ligands for peroxisome proliferator-activated
receptor gamma and retinoic acid inhibit growth and
induce apoptosis of human breast cancer cells in vitro and
in BNX mice. PNAS 95 8806–8811.
Farid NR, Shi Y & Zou M 1994 Molecular basis of thyroid
cancer. Endocrine Reviews 15 202–232.
Foti D, Iuliano R, Chiefari E & Brunetti A 2003 A
nucleoprotein complex containing Sp1, C/EBPb, and
HMGI-Y controls human insulin receptor gene
transcription. Molecular and Cellular Biology 23
2720–2732.
Foti D, Chiefari E, Fedele M, Iuliano R, Brunetti L, Paonessa
F, Manfioletti G, Barbetti F, Brunetti A, Croce CM et al.
2005 Lack of the architectural factor HMGA1 causes
severe insulin resistance and diabetes in humans and
mice. Nature Medicine 11 765–773.
Goldfine ID 1987 The insulin receptor: molecular biology
and transmembrane signalling. Endocrine Reviews 8
235–255.
Govindarajan R, Ratnasinghe L, Simmons DL, Siegel ER,
Midathada MV, Kim L, Kim PJ, Owens RJ & Lang NP
2007 Thiazolidinediones and the risk of lung, prostate,
and colon cancer in patients with diabetes. Journal of
Clinical Oncology 25 1476–1481.
Grommes C, Landreth GE & Heneka MT 2004 Antineo-
plastic effects of peroxisome proliferator-activated
receptor agonists. Lancet Oncology 5 419–429.
Han SW, Ritzenthaler JD, Rivera HN & Roman J 2005
Peroxisome proliferator-activated receptor-gamma
ligands suppress fibronectin gene expression in
human lung carcinoma cells: involvement of both
CRE and Sp1. American Journal of Physiology.
Lung Cellular and Molecular Physiology 289
L419–L428.
Hilakivi-Clarke L, Wang C, Kalil M, Riggins R &
Pestell RG 2004 Nutritional modulation of the cell
cycle and breast cancer. Endocrine-Related Cancer 11
603–622.
Hong J, Samudio I, Liu S, Abdelrahim M & Safe S 2004
Peroxisome proliferator-activated receptor g-dependent
activation of p21 in Panc-28 pancreatic cancer cells
involves Sp1 and Sp4 proteins. Endocrinology 145
5774–5785.
Keller H, Dreyer C, Medin J, Mahfoudi A, Ozato K &
Wahli W 1993a Fatty acids and retinoids control lipid
metabolism through the activation of peroxisome
proliferator-activated receptors heterodimers. PNAS 90
2160–2164.
Keller H, Mahfoudi A, Dreyer C, Hihi AK, Medin J, Ozato K
& Wahli W 1993b Peroxisome proliferator-activated
receptors and lipid metabolism. Annals of the New York
Academy of Sciences 684 157–173.
Kersten S, Desvergne B & Wahli W 2000 Roles of PPARs in
health and disease. Nature 405 421–424.
333

V Costa, D Foti et al.: Effects of PPARg and TZD-PPARg on IR expression
KliewerSA,XuHE,LambertMH&WillsonTM2001Peroxisome
proliferator-activated receptors: from genes to physiology.
Recent Progress in Hormone Research 56 239–263.
Krey G, Mahfoudi A & Wahli W 1995 Functional
interactions of peroxisome proliferator-activated receptor,
retinoid-X receptor, and Sp1 in the transcriptional
regulation of the acyl-coenzyme-A oxidase promoter.
Molecular Endocrinology 9 219–231.
Lehmann JM, Moore LB, Smith-Oliver TA, Wilkinson WO,
Willson TM & Kliewer SA 1995 An antidiabetic
thiazolidinedione is a high affinity ligand for peroxisome
proliferator-activated receptorg (PPARg). Journal of
Biological Chemistry 270 12953–12956.
Maloney CA & Rees WD 2005 Gene–nutrient interactions
during fetal development. Reproduction 130 401–410.
Mamula PW, McDonald AR, Brunetti A, Okabayashi Y,
Wong KY, Maddux BA, Logsdon C & Goldfine ID 1990
Regulating insulin-receptor-gene expression by differen-
tiation and hormones. Diabetes Care 13 288–301.
Martelli ML, Iuliano R, Le Pera I, Sama I, Monaco C,
Cammarota S, Kroll T, Chiariotti L, Santoro M& Fusco A
2002 Inhibitory effects of peroxisome proliferator-
activated receptor gamma on thyroid carcinoma
cell growth. Journal of Clinical Endocrinology and
Metabolism 87 4728–4735.
Milazzo G, Giorgino F, Damante G, Sung C, Stampfer MR,
Vigneri R, Goldfine ID & Belfiore A 1992 Insulin
receptor expression and function in human breast cancer
cell lines. Cancer Research 52 3924–3930.
Mueller E, Sarraf P, Tontonoz P, Evans RM, Martin KJ,
Zhang M, Fletcher C, Singer S & Spiegelman BM 1998
Terminal differentiation of human breast cancer through
PPARg. Molecular Cell 1 465–470.
Mulholland DJ, Dedhar S, Coetzee GA & Nelson CC 2005
Interaction of nuclear receptors with the Wnt/beta-
catenin/Tcf signaling axis: Wnt you like to know?
Endocrine Reviews 26 898–915.
Nishizawa H, Yamagata K, Shimomura I, Takahashi M,
Kuriyama H, Kishida K, Hotta K, Nagaretani H, Maeda N,
Matsuda M et al. 2002 Small heterodimer partner, an
orphan nuclear receptor, augments peroxisome prolifera-
tor-activated receptor gamma transactivation. Journal of
Biological Chemistry 277 1586–1592.
Nunez NP, Liu H & Meadows GG 2005 PPAR-gamma
ligands and amino acid deprivation promote apoptosis of
melanoma, prostate, and breast cancer cells. Cancer
Letters 236 133–141.
Osborne CK, Monaco ME, Lippman ME & Kahn CR 1978
Correlation among insulin binding, degradation, and
biological activity in human breast cancer cells in long-
term tissue culture. Cancer Research 38 94–102.
Padilla J, Kaur K, Harris SG & Phipps RP 2000 PPAR-
gamma-mediated regulation of normal and malignant B
lineage cells. Annals of the New York Academy of
Sciences 905 97–109.
Paonessa F, Foti D, Costa V, Chiefari E, Brunetti G, Leone F,
Luciano F, Wu F, Lee AS, Gulletta E et al. 2006 Activator
334
protein-2 overexpression accounts for increased insulin
receptor expression in human breast cancer. Cancer
Research 66 5085–5093.
Papageorgiou E, Pitulis N, Msaouel P, Lembessis P &
Koutsilieris H 2007 The non-genomic crosstalk
between PPARg ligands and ERK 1/2 in cancer cell
lines. Expert Opinion on Therapeutic Targets 11
1071–1085.
Patel L, Pass I, Coxon P, Downes CP, Smith SA & Macphee
CH 2001 Tumor suppressor and anti-inflammatory
actions of PPARgamma agonista are mediated via
upregulation of PTEN. Current Biology 11 764–768.
Ramos-Nino ME, MacLean CD & Littenberg B 2007
Association between cancer prevalence and use of
thiazolidinediones: results from the Vermont Diabetes
Information System. BMC Medicine 5 17–24.
Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K,
Milstone DS, Spiegelman BM & Mortensen RM 1999
PPARg is required for the differentiation of adipose tissue
in vivo and in vitro. Molecular Cell 4 611–617.
Saltiel AR & Olefsky JM 1996 Thiazolidinediones in the
treatment of insulin resistance and tipe II diabetes.
Diabetes 45 1661–1669.
Savkur RS & Miller AR 2006 Investigational PPAR-g
agonists for the treatment of Type 2 diabetes. Expert
Opinion on Investigational Drugs 15 763–778.
Seino S, Seino M, Nishi S & Bell GI 1989 Structure of the
human insulin receptor gene and characterization of its
promoter. PNAS 86 114–118.
Sugawara A, Uruno A, Kudo M, Ikeda Y, Sato K, Taniyama
Y, Ito S & Takeuchi K 2002 Transcription suppression of
thromboxane receptor gene by peroxisome proliferator-
activated receptor,-g via an interaction with Sp1 in
vascular smooth muscle cells. Journal of Biological
Chemistry 277 9676–9683.
Takata Y, Kitami Y, Yang Z-H, Nakamura M, Okura T &
Hiwada H 2002 Vascular inflammation is negatively
autoregulated by interaction between CCAAT/enhancer-
binding protein d and peroxisome proliferator-activated
receptor. Circulation Research 91 427–433.
Tan NS, Michalik L, Desvergne B & Wahli W 2005
Multiple expression control mechanisms of peroxisome
proliferator-activated receptors and their target genes.
Journal of Steroid Biochemistry and Molecular Biology
93 99–105.
Tontonoz PM, Hu E & Spiegelman BN 1994 Stimulation of
adipogenesis in fibroblasts by PPAR gamma 2, a lipid-
activated transcription factor. Cell 79 1147–1156.
Ullrich A, Bell JR, Chen EY, Herrera R, Petruzzelli LM, Dull
TJ, Gray A, Coussens L, Liao YC, Tsubokawa M et al.
1985 Human insulin receptor and its relationship to the
tyrosine kinase family of oncogenes. Nature 313 756–761.
Vidal-Puig AJ, Considine RV, Jimenezlinan M, Werman A,
Pories WJ, Caro JF & Flier JS 1997 Peroxisome
proliferator-activated receptor gene expression in human
tissues. Journal of Clinical Investigation 99 2416–2422.
www.endocrinology-journals.org

Endocrine-Related Cancer (2008) 15 325–335
Wang C, Fu M, D’Amico M, Albanese C, Zhou JN,
Brownlee M, Lisanti MP, Chatterjee VK, Lazar MA &
Pestell RG 2001 Inhibition of cellular proliferation
through IkB kinase-indipendent and peroxisome prolifera-
tor-activated receptor g-dependent repression of cyclin D1.Molecular and Cellular Biology 21 3057–3070.
www.endocrinology-journals.org
Wang LH, Yang XY, Zhang X & Farrar WL 2007 Inhibition
of adhesive interaction between multiple myeloma and
bone marrow stromal cells by PPARg crostalk with
NF-kB and C/EBPb. Blood 110 4373–4384.
White MF & Kahn CR 1994 The insulin signaling system.
Journal of Biological Chemistry 269 1–4.
335




















