
Intestinal Antiinflammatory Effects of Thiazolidenedione Peroxisome Proliferator-Activated...
9
ORIGINAL ARTICLE Intestinal Antiinflammatory Effects of Thiazolidenedione Peroxisome Proliferator-Activated Receptor- Ligands on T Helper Type 1 Chemokine Regulation Include Nontranscriptional Control Mechanisms Katherine L. Schaefer, PhD,* Svetlana Denevich,* Chen Ma, MD,* Shane R. Cooley,* Atsushi Nakajima, MD,‡ Koichiro Wada, PhD,§ Jennifer Schlezinger, PhD,† David Sherr, PhD,† and Lawrence J. Saubermann, MD* Abstract: Crohn’s disease is associated with an excessive T helper (T H ) type 1 inflammatory immune response. Reducing the influx of disease-associated CD4 + T H 1 cells into the inflamed intestine is likely to be beneficial in preventing a disease flare-up and even possibly in reducing the effect of acute disease. Thiazolidenedione (TZD) li- gands, which activate peroxisome proliferator-activated receptor- (PPAR), have been shown to reduce T H 1 inflammation in murine models of colitis, primarily in a preventative fashion. To determine whether PPAR ligands reduce this inflammation in part by reducing T H 1 chemoattractant levels in vivo, the TZD pioglitazone was tested for its effects on a T H 1 chemokine (CXCL10) in 2 models of colitis (i.e., dextran sodium sulfate and 2,4,6-dinitrobenzene sulfonic acid- mediated colitis). In both models, CXCL10 levels were significantly reduced by pioglitazone. Because TZDs can affect gene expression either directly, by regulating the binding of PPAR to consensus pro- moter elements, or indirectly, by modulating other signaling path- ways that can affect gene transcription, the regulation of CXCL10 by TZDs was investigated in vitro in both HT-29 colon epithelial cells and THP-1 monocyte/macrophage cells. TZDs significantly reduced CXCL10 protein levels from activated HT-29 cells and THP-1- derived macrophages in a dose-dependent manner at nanomolar con- centrations. However, TZDs did not affect messenger RNA levels or nuclear factor-B activation at these concentrations in these cells. These findings imply the existence of a novel posttranscriptional regulatory antiinflammatory mechanism by TZDs that is not associ- ated with reductions in nuclear factor-B activation. Key Words: colitis, Crohn’s disease, CXCL10, peroxisome prolif- erator-activated receptor-, posttranscriptional regulation (Inflamm Bowel Dis 2005;11:244–252) I nflammatory bowel disease, including Crohn’s disease (CD) and ulcerative colitis, are characterized by relapsing-and- remitting episodes of intestinal inflammation. 1,2 This inflam- mation is thought to result from an inappropriate response to normal bacterial flora, involving both innate and adaptive im- mune systems. One of the major goals of therapy in patients with inflammatory bowel disease is to reduce the level of in- flammation and prevent the initiation of further episodes. CD is mediated by an excessive T helper (T H ) type 1 response, 3 as evidenced by increased levels of the T H 1 inflam- matory cytokines tumor necrosis factor (TNF)- and inter- feron (IFN)- at the affected mucosal site. 4,5 Macrophages iso- lated from inflamed tissue overproduce interleukin (IL)-12, a key cytokine involved in initiating and perpetuating T H 1- mediated inflammation, and T cells isolated from inflamed tis- sues exhibit a T H 1 bias, including the expression of the IL-12 receptor, Stat4 activation, and increased T-bet expression. Similar responses are seen in many mouse models of T H 1 in- flammation and colitis. 6 Interfering with the influx of T H 1 cells into the colon represents an important approach to therapy and is being in- vestigated in human clinical trials. 7 In mouse models of colitis, interference with integrin attachment to its cognate receptor can reduce leukocyte trafficking to the colon and can decrease disease severity. 8–12 This reduction in disease severity has been shown to be directly related to reduced T-cell influx in the CD45Rbhi colitis model. 8 Another approach to reducing T- cell infiltration involves reducing the levels of chemoattractant cytokines (chemokines) or interfering with the chemokine re- ceptor itself, which then prevents the chemotactic response by the leukocyte. In the IL-10 knockout T H 1-mediated model of Received for publication November 5, 2004; accepted November 12, 2004. From the *Section of Gastroenterology, Boston University Medical Center, and the †Department of Environmental Health, Boston University School of Public Health, Boston, Massachusetts; the ‡Department of Pharmacol- ogy, Osaka University, Osaka, Japan; and the §Division of Gastroenterol- ogy, Yokohama City University School of Medicine, Yokohama, Japan. Supported by the Broad Medical Research Program of The Eli and Edythe L. Broad Foundation. (LJS) and NiH grant DK066928 (KLS). Reprints: Lawrence J. Saubermann, MD, Boston Medical Center, EBRC 529, 650 Albany Street, Boston, Massachusetts 02118 (e-mail: lawrence. [email protected]) Copyright © 2005 by Lippincott Williams & Wilkins 244 Inflamm Bowel Dis • Volume 11, Number 3, March 2005
Transcript of Intestinal Antiinflammatory Effects of Thiazolidenedione Peroxisome Proliferator-Activated...

ORIGINAL ARTICLE
Intestinal Antiinflammatory Effects of ThiazolidenedionePeroxisome Proliferator-Activated Receptor-� Ligands on T
Helper Type 1 Chemokine Regulation IncludeNontranscriptional Control Mechanisms
Katherine L. Schaefer, PhD,* Svetlana Denevich,* Chen Ma, MD,* Shane R. Cooley,*Atsushi Nakajima, MD,‡ Koichiro Wada, PhD,§ Jennifer Schlezinger, PhD,† David Sherr, PhD,†
and Lawrence J. Saubermann, MD*
Abstract: Crohn’s disease is associated with an excessive T helper(TH) type 1 inflammatory immune response. Reducing the influx ofdisease-associated CD4+ TH1 cells into the inflamed intestine is likelyto be beneficial in preventing a disease flare-up and even possibly inreducing the effect of acute disease. Thiazolidenedione (TZD) li-gands, which activate peroxisome proliferator-activated receptor-�(PPAR�), have been shown to reduce TH1 inflammation in murinemodels of colitis, primarily in a preventative fashion. To determinewhether PPAR� ligands reduce this inflammation in part by reducingTH1 chemoattractant levels in vivo, the TZD pioglitazone was testedfor its effects on a TH1 chemokine (CXCL10) in 2 models of colitis(i.e., dextran sodium sulfate and 2,4,6-dinitrobenzene sulfonic acid-mediated colitis). In both models, CXCL10 levels were significantlyreduced by pioglitazone. Because TZDs can affect gene expressioneither directly, by regulating the binding of PPAR� to consensus pro-moter elements, or indirectly, by modulating other signaling path-ways that can affect gene transcription, the regulation of CXCL10 byTZDs was investigated in vitro in both HT-29 colon epithelial cellsand THP-1 monocyte/macrophage cells. TZDs significantly reducedCXCL10 protein levels from activated HT-29 cells and THP-1-derived macrophages in a dose-dependent manner at nanomolar con-centrations. However, TZDs did not affect messenger RNA levels ornuclear factor-�B activation at these concentrations in these cells.These findings imply the existence of a novel posttranscriptionalregulatory antiinflammatory mechanism by TZDs that is not associ-ated with reductions in nuclear factor-�B activation.
Key Words: colitis, Crohn’s disease, CXCL10, peroxisome prolif-erator-activated receptor-�, posttranscriptional regulation
(Inflamm Bowel Dis 2005;11:244–252)
Inflammatory bowel disease, including Crohn’s disease (CD)and ulcerative colitis, are characterized by relapsing-and-
remitting episodes of intestinal inflammation.1,2 This inflam-mation is thought to result from an inappropriate response tonormal bacterial flora, involving both innate and adaptive im-mune systems. One of the major goals of therapy in patientswith inflammatory bowel disease is to reduce the level of in-flammation and prevent the initiation of further episodes.
CD is mediated by an excessive T helper (TH) type 1response,3 as evidenced by increased levels of the TH1 inflam-matory cytokines tumor necrosis factor (TNF)-� and inter-feron (IFN)-� at the affected mucosal site.4,5 Macrophages iso-lated from inflamed tissue overproduce interleukin (IL)-12, akey cytokine involved in initiating and perpetuating TH1-mediated inflammation, and T cells isolated from inflamed tis-sues exhibit a TH1 bias, including the expression of the IL-12receptor, Stat4 activation, and increased T-bet expression.Similar responses are seen in many mouse models of TH1 in-flammation and colitis.6
Interfering with the influx of TH1 cells into the colonrepresents an important approach to therapy and is being in-vestigated in human clinical trials.7 In mouse models of colitis,interference with integrin attachment to its cognate receptorcan reduce leukocyte trafficking to the colon and can decreasedisease severity.8–12 This reduction in disease severity hasbeen shown to be directly related to reduced T-cell influx in theCD45Rbhi colitis model.8 Another approach to reducing T-cell infiltration involves reducing the levels of chemoattractantcytokines (chemokines) or interfering with the chemokine re-ceptor itself, which then prevents the chemotactic response bythe leukocyte. In the IL-10 knockout TH1-mediated model of
Received for publication November 5, 2004; accepted November 12, 2004.From the *Section of Gastroenterology, Boston University Medical Center,
and the †Department of Environmental Health, Boston University Schoolof Public Health, Boston, Massachusetts; the ‡Department of Pharmacol-ogy, Osaka University, Osaka, Japan; and the §Division of Gastroenterol-ogy, Yokohama City University School of Medicine, Yokohama, Japan.
Supported by the Broad Medical Research Program of The Eli and Edythe L.Broad Foundation. (LJS) and NiH grant DK066928 (KLS).
Reprints: Lawrence J. Saubermann, MD, Boston Medical Center, EBRC 529,650 Albany Street, Boston, Massachusetts 02118 (e-mail: [email protected])
Copyright © 2005 by Lippincott Williams & Wilkins
244 Inflamm Bowel Dis • Volume 11, Number 3, March 2005

colitis, blocking the TH1 chemoattractant CXCL10 [IFN-inducible protein 10 (IP-10)] was shown to prevent colitis en-tirely13 and to reduce its severity in the dextran sodium sulfate(DSS) model.14 Because TH1 chemokines like CXCL10 areinducibly expressed in the setting of inflammation, medica-tions or compounds that are recognized to reduce the activityof inflammatory nuclear gene transcription factors wouldlikely reduce CXCL10 and TH1 inflammation.
One promising strategy for reducing T-cell infiltrationinvolves the activation of the nuclear hormone receptor per-oxisome proliferator-activated receptor-� (PPAR�). PPAR� ishighly expressed in the intestinal and colonic mucosa by bothepithelial and macrophage cells.15–17 PPAR� can be activatedby a class of compounds known as thiazolidenediones (TZDs),and these compounds have been shown to reduce inflamma-tion in a number of systems, including the intestine.18 PPAR�normally functions to transcriptionally regulate a number offatty acid and metabolism-related genes by forming a heterodi-meric complex with retinoid X receptor � and binding to aspecific response element [peroxisome proliferator responseelement (PPRE)]. However, PPAR� activation can also resultin the transrepression of a number of other genes through in-teractions with a variety of nuclear hormone receptors, such asnuclear factor (NF)-�B, and it is this transrepressive mecha-nism that is thought to be its primary antiinflammatory mecha-nism.19
TZDs have been shown to reduce in vitro production ofTH1 chemoattractants (i.e., chemokines) from a number of celltypes, including endothelial cells,20 glial cells,21 monocyte-derived dendritic cells,22 and macrophages.23 In addition, theyhave been shown to prevent colitis in the DSS model24–26 andto have both preventative and therapeutic effects in the 2,4,6-trinitrobenzene sulfonic acid mouse model of colitis.27 Al-though TZDs reduce TH1 inflammation, in general, as noted bycytokine reductions and reductions in overall colonic NF-�Bactivation levels, it has not been determined whether TZDsalso reduce TH1 chemokine production in mouse colitis mod-els and whether this is mediated by their transrepressionmechanism.
To determine whether TZD treatment reduces chemo-kine expression in the context of a TH1-mediated inflammationlike CD, the effects of the TZD pioglitazone on the productionof the TH1 chemokine CXCL10 (IP-10) were determined inboth the DSS and 2,4,6-dinitrobenzene sulfonic acid (DNBS)murine models of colitis. To further examine the mechanism ofchemokine regulation in human cells, and to determine wheth-er TZDs affect TH1 chemokine production in both macro-phages and intestinal epithelial cells, the effects of TZDs onchemokine production from these cells in response to TNF-�,IFN-�, and lipopolysaccharide (LPS) were determined.CXCL10 production in response to DSS and DNBS was re-duced by pretreatment with TZDs. In addition, TZDs preventCXCL10 protein production from both intestinal epithelial cell
and macrophage cell lines in a concentration-dependent man-ner, with inhibition of the median effective concentration oc-curring at similar nanomolar concentrations. However, a closecomparison of quantitative messenger RNA (mRNA) and pro-tein levels indicated a novel, nontranscriptional regulation ef-fect by TZDs on TH1 chemokines, which is not associated witha reduction in NF-�B activation.
MATERIALS AND METHODS
ChemicalsThe pioglitazone used in the mouse experiments was a
gift of Dr. Atsushi Nakajima (Yokohama City UniversitySchool of Medicine, Yokohama, Japan). Rosiglitazone, pio-glitazone, and 15-deoxy-�12,14-prostaglandin J2 (15-�PGJ2)for the in vitro experiments were purchased from CaymanChemicals (Ann Arbor, Mich.) and were dissolved in dimeth-ylsulfoxide (DMSO). DMSO and LPS were purchased fromSigma-Aldrich Chemicals (St. Louis, Mo.). TNF-� and IFN-�were purchased from PeproTech (Rocky Hill, NJ).
MiceFor both colitis models, C57BL/6 mice between the ages
of 8 and 12 weeks (Jackson Laboratories, Bar Harbor, Me)were used. All animals were maintained in clean room facili-ties, with access to water and standard laboratory chow ad li-bitum, with a 12-hour light-dark cycle. All mice were treatedhumanely in accordance with the National Institutes of Health,Institutional Animal Use and Care Committee, the Panel ofEuthanasia of the American Veterinary Medical Association,and institutional guidelines.
Animal Models of ColitisPioglitazone was dissolved in 2% carboxymethylcellu-
lose and was administered by gavage at 20 mg/kg/d, beginning1 day before starting the induction of DSS or DNBS, and everyother day thereafter. DSS colitis was induced with 2.5% DSSin water, given ad libitum [lot 4470c; ICN Biomedicals (nowMP Pharmaceuticals), Aurora, Ohio]. For DSS experiments,the use of 6 mice (C − Pio and C + Pio), 11 mice (DSS − Pio),and 12 mice (DSS + Pio) occurred over 3 independent experi-ments. DNBS hapten-mediated colitis was induced with 5 mgof DNBS (Fisher Scientific, Pittsburgh, Pa.) in 0.1 mL of a50% ethanol solution, which was administered intrarectally bythe insertion of a catheter 4.0 cm into the rectum. For DNBSexperiments, the use of 4 mice (C-Pio and C + Pio), 11 mice(DSS-Pio), and 9 mice (DSS + Pio) were spread over 2 inde-pendent experiments. On day 5 (DSS) or day 21 (DNBS), micewere killed, and tissue samples were snap-frozen for lysates,placed in RNAlater (Qiagen, Valencia, Calif.), or were embed-ded in TissueTek (Sakura Finetek, Torrance, Calif.) freezingmedium and snap-frozen.
Inflamm Bowel Dis • Volume 11, Number 3, March 2005 Intestinal Antiinflammatory Effects of Thiazolidenedione
© 2005 Lippincott Williams & Wilkins 245

Cell Lines and Stimulation ProtocolsThe human colorectal adenocarcinoma cell line HT-29
and the human monocyte cell line THP-1 were obtained fromthe American Type Culture Collection (Manassas, Va.). Insome experiments, THP-1 cells were differentiated into mac-rophages28,29 with the addition of 200 nM phorbol myristateacetate (PMA) to the cell culture medium for 16 to 24 hours,followed by washing. For the intestinal epithelial experiments,1 × 105 HT-29 or Caco-2 cells in 24-well plates were incubatedfor 16 hours with the indicated concentration of rosiglitazone.Control wells contained an equal concentration of DMSO.Fresh medium containing the same concentration of rosi-glitazone, combined with IFN-� (100 to 300 µm/mL) orTNF-� (100 to 300 ng/mL) was then added, and supernatantswere collected 8 hours later. THP-1 experiments were per-formed in a similar fashion, except that IFN-� (100 µm/mL) orLPS (1 µg/mL) were added to 3 × 105 THP-1 cells or 1 × 105
differentiated (macrophage) THP-1 cells in 48-well plates.
Enzyme-Linked Immunosorbent Assay AnalysisLysates from murine colon samples were made by ho-
mogenizing tissue samples in RIPA buffer (Boston Bioprod-ucts, Boston, Mass.) supplemented with protease inhibitors(Complete protease inhibitor system, Roche Diagnostics, In-dianapolis, Ind.). Protein concentrations were determined us-ing the BCA assay kit (Pierce, Rockford, Ill.). MurineCXCL10 concentrations were determined using the Duo-Setenzyme-linked immunosorbent assay (ELISA) system fromR&D systems (Minneapolis, Minn.), and concentrations ob-tained from the ELISA were normalized for total protein con-centration. The concentrations of human CXCL10 in superna-tants from the in vitro stimulation experiments were deter-mined using the OptEIA murine CXCL10 ELISA kit (BDBiosciences, San Diego, Calif.).
Real-time Quantitative Polymerase ChainReaction Analysis
Total RNA was made from murine tissue samples andcultured cell lines using the Rneasy mini kit (Qiagen).Complementary DNA was made using Superscript III reversetranscriptase (Invitrogen, Carlsbad, Calif.) from 2 µg of RNA(murine experiments) or 0.6 to 1 µg of RNA (cell lines). Real-time PCR was performed using the Quantitect SYBR greenPCR kit (Qiagen) using primers specific for actin (5�CGAG-GCCCAGAGCAAGAGAG and 5�CGGTTGGCCT-TAGGGTTCAG; amplifies both murine and human actin),human CXCL10 (5� CCCATCACTTCCCTACATGG and 5�GAAGATGGGAAAGGTGAG), or murine CXCL10(5�ATCGATGGACAGCAGAGAGC and 5ACCATGGCTT-GACCATCATC). CXCL10 quantities were expressed nor-malized to actin concentration. In all cases, the primers ampli-fied a single fragment. Purified PCR fragments (MinElute;
Qiagen) made using these primers were used as concentrationcontrols.
ImmunohistochemistryFive-micrometer sections were made from frozen tissue
and were fixed in methanol at −80°C. Endogenous peroxidaseactivity was blocked with hydrogen peroxide, and nonspecificbinding was blocked with 1% immunoglobulin G-free bovineserum (Jackson Immunoresearch, West Grove, Pa.) in 0.01%Tween 20/phosphate-buffered saline solution (Fisher Scien-tific) and the streptavidin/biotin blocking kit (Vector Labora-tories, Burlingame, Calif.). Antimouse-CD4 (clone L3T4; BDBiosciences, San Jose, Calif.) at 1.5 µg/mL and peroxidase-conjugated antirat F(ab2) fragment (Jackson Immunoresearch)at 1.5 µg/mL were used as the primary and secondary antibod-ies, respectively. Staining was detected using the DAB peroxi-dase substrate kit (Vector laboratories), counterstained withGill’s hematoxylin no. 2 (Fisher Scientific).
Transient Transfections and Reporter AssaysTransient transfections of HT-29 cells were performed
with Lipofectamine 2000 (Invitrogen). After 4 hours of trans-fection, cells were incubated for 16 hours with the indicatedconcentration of TZD. Luciferase activity was measured usinga luciferase assay kit (Promega, Madison, Wis.). As all re-porter plasmids tested were themselves affected by TZD con-centrations, the results were not normalized to a reporter plas-mid. The variability of transfection using the pRL reporterplasmid was assessed in parallel and was found to be <20%(data not shown).30 The tandem 5X NF-kB luciferase constructwas from Stratagene (La Jolla, Calif.).
Electrophoretic Mobility Shift Assay AnalysisElectrophoretic mobility shift assay analysis was per-
formed in HT-29 cells as described previously, using the 32P-labeled oligonucleotides containing the NF-�B1 (5-CGCTTTGGAAAGTGAAACCTAC) and NF-�B2 (5�GAG-CAGAGGGAAATTCCGTAACTT) sites from the humanIP-10 promoter.20
Statistical AnalysisDifferences were assessed using the 1-tailed Student t
test. Differences were considered significant if P < 0.05.
RESULTS
TZD Preadministration Significantly ReducesCXCL10 Levels in Two Animal Models of Colitis
Because TZDs are known to reduce TH1 cytokines, wedetermined whether TZDs are also able to reduce TH1 chemo-attractant levels and CD4+ T cells in the colon. Two differentmodels of colitis were used. The acute-phase DSS model isassociated with severe disease, proinflammatory TH1 cytokine
Schaefer et al Inflamm Bowel Dis • Volume 11, Number 3, March 2005
246 © 2005 Lippincott Williams & Wilkins
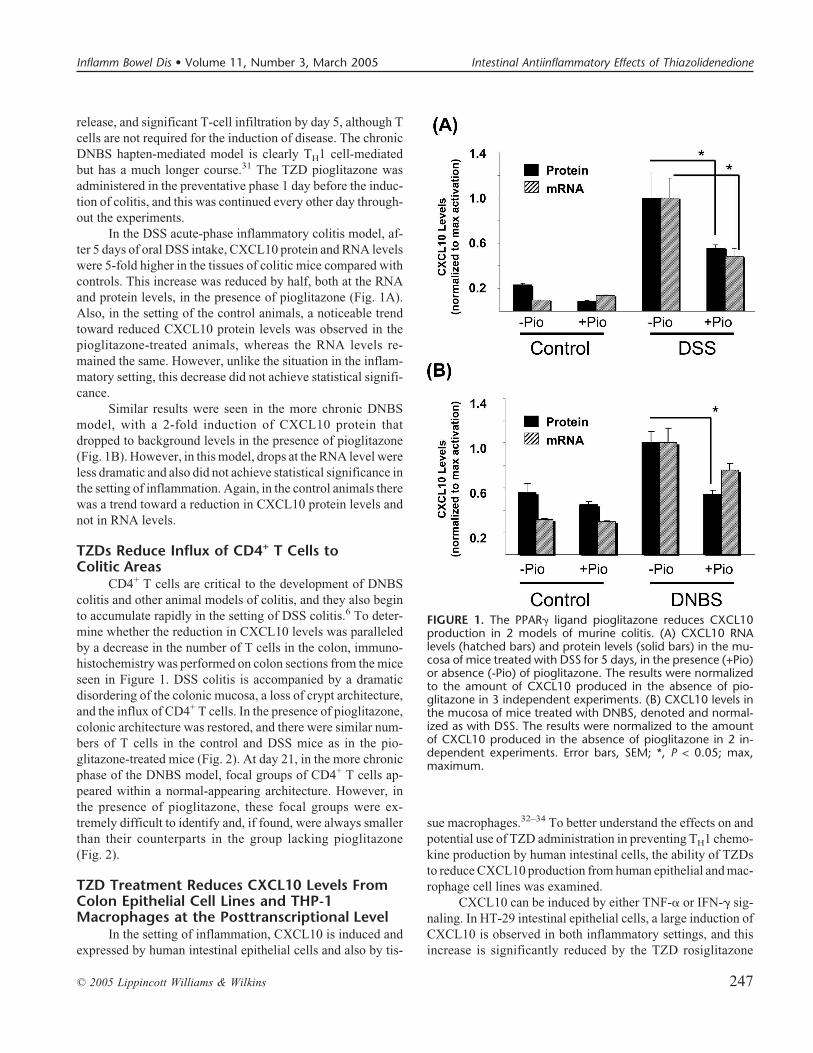
release, and significant T-cell infiltration by day 5, although Tcells are not required for the induction of disease. The chronicDNBS hapten-mediated model is clearly TH1 cell-mediatedbut has a much longer course.31 The TZD pioglitazone wasadministered in the preventative phase 1 day before the induc-tion of colitis, and this was continued every other day through-out the experiments.
In the DSS acute-phase inflammatory colitis model, af-ter 5 days of oral DSS intake, CXCL10 protein and RNA levelswere 5-fold higher in the tissues of colitic mice compared withcontrols. This increase was reduced by half, both at the RNAand protein levels, in the presence of pioglitazone (Fig. 1A).Also, in the setting of the control animals, a noticeable trendtoward reduced CXCL10 protein levels was observed in thepioglitazone-treated animals, whereas the RNA levels re-mained the same. However, unlike the situation in the inflam-matory setting, this decrease did not achieve statistical signifi-cance.
Similar results were seen in the more chronic DNBSmodel, with a 2-fold induction of CXCL10 protein thatdropped to background levels in the presence of pioglitazone(Fig. 1B). However, in this model, drops at the RNA level wereless dramatic and also did not achieve statistical significance inthe setting of inflammation. Again, in the control animals therewas a trend toward a reduction in CXCL10 protein levels andnot in RNA levels.
TZDs Reduce Influx of CD4+ T Cells toColitic Areas
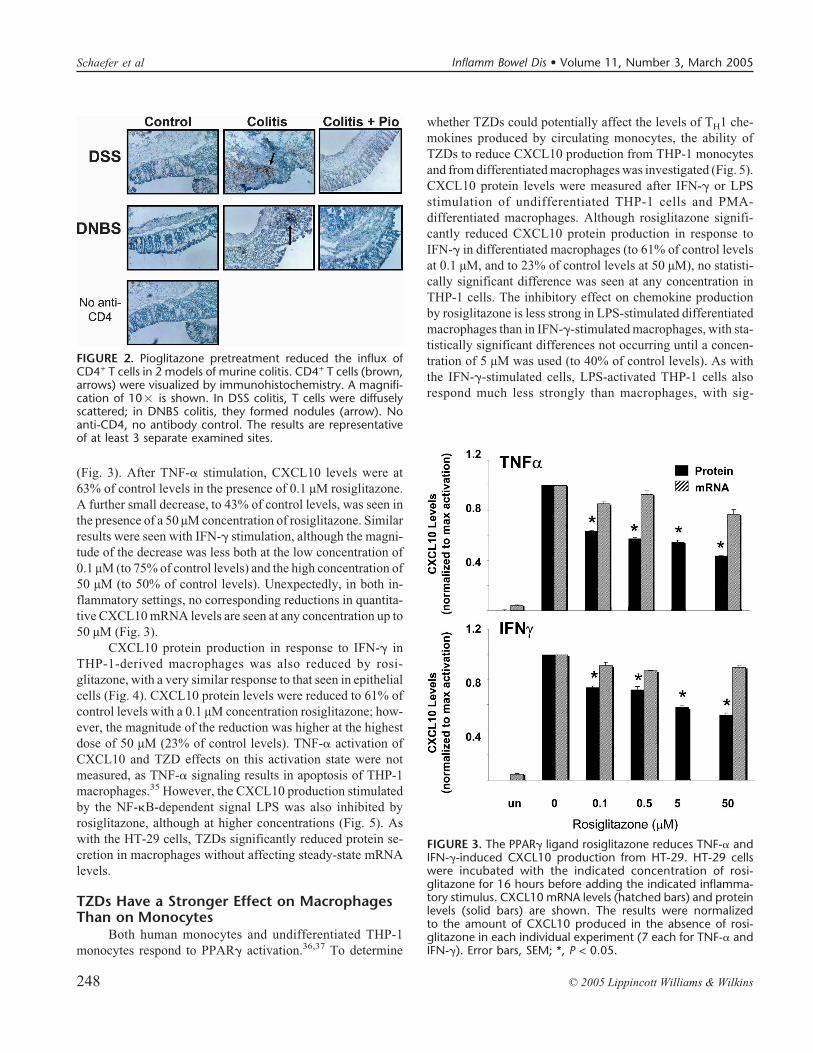
CD4+ T cells are critical to the development of DNBScolitis and other animal models of colitis, and they also beginto accumulate rapidly in the setting of DSS colitis.6 To deter-mine whether the reduction in CXCL10 levels was paralleledby a decrease in the number of T cells in the colon, immuno-histochemistry was performed on colon sections from the miceseen in Figure 1. DSS colitis is accompanied by a dramaticdisordering of the colonic mucosa, a loss of crypt architecture,and the influx of CD4+ T cells. In the presence of pioglitazone,colonic architecture was restored, and there were similar num-bers of T cells in the control and DSS mice as in the pio-glitazone-treated mice (Fig. 2). At day 21, in the more chronicphase of the DNBS model, focal groups of CD4+ T cells ap-peared within a normal-appearing architecture. However, inthe presence of pioglitazone, these focal groups were ex-tremely difficult to identify and, if found, were always smallerthan their counterparts in the group lacking pioglitazone(Fig. 2).
TZD Treatment Reduces CXCL10 Levels FromColon Epithelial Cell Lines and THP-1Macrophages at the Posttranscriptional Level
In the setting of inflammation, CXCL10 is induced andexpressed by human intestinal epithelial cells and also by tis-
sue macrophages.32–34 To better understand the effects on andpotential use of TZD administration in preventing TH1 chemo-kine production by human intestinal cells, the ability of TZDsto reduce CXCL10 production from human epithelial and mac-rophage cell lines was examined.
CXCL10 can be induced by either TNF-� or IFN-� sig-naling. In HT-29 intestinal epithelial cells, a large induction ofCXCL10 is observed in both inflammatory settings, and thisincrease is significantly reduced by the TZD rosiglitazone
FIGURE 1. The PPAR� ligand pioglitazone reduces CXCL10production in 2 models of murine colitis. (A) CXCL10 RNAlevels (hatched bars) and protein levels (solid bars) in the mu-cosa of mice treated with DSS for 5 days, in the presence (+Pio)or absence (-Pio) of pioglitazone. The results were normalizedto the amount of CXCL10 produced in the absence of pio-glitazone in 3 independent experiments. (B) CXCL10 levels inthe mucosa of mice treated with DNBS, denoted and normal-ized as with DSS. The results were normalized to the amountof CXCL10 produced in the absence of pioglitazone in 2 in-dependent experiments. Error bars, SEM; *, P < 0.05; max,maximum.
Inflamm Bowel Dis • Volume 11, Number 3, March 2005 Intestinal Antiinflammatory Effects of Thiazolidenedione
© 2005 Lippincott Williams & Wilkins 247
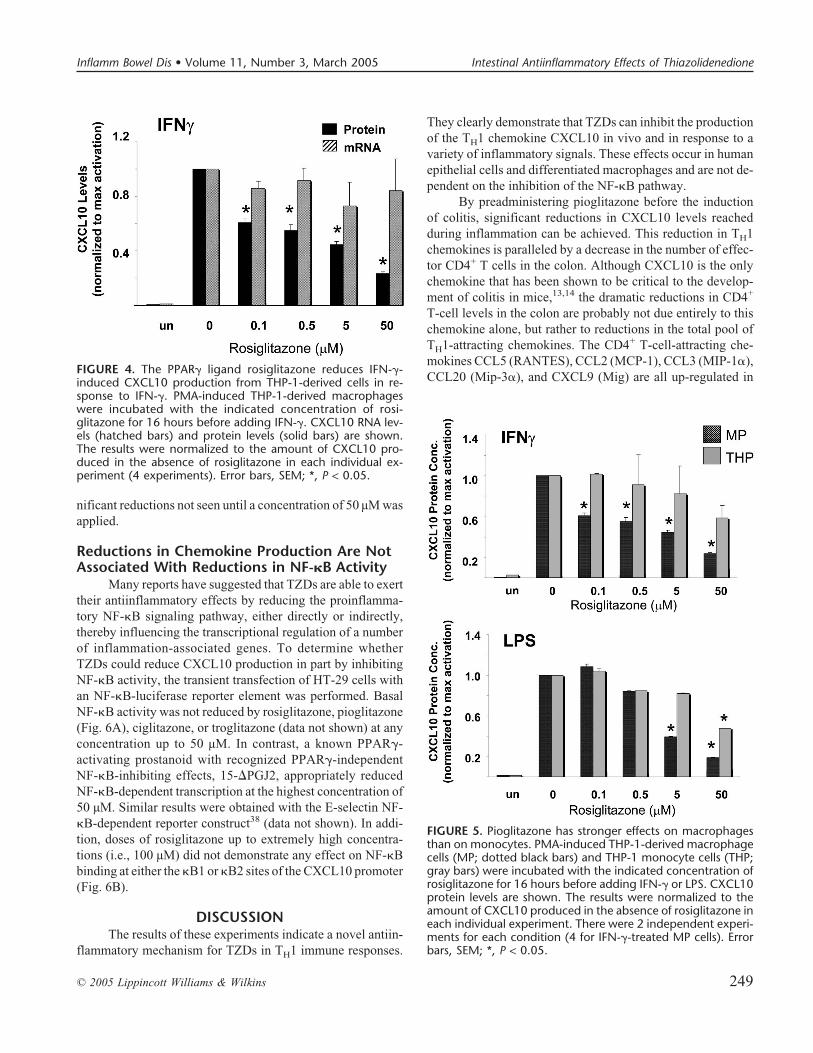
(Fig. 3). After TNF-� stimulation, CXCL10 levels were at63% of control levels in the presence of 0.1 µM rosiglitazone.A further small decrease, to 43% of control levels, was seen inthe presence of a 50 µM concentration of rosiglitazone. Similarresults were seen with IFN-� stimulation, although the magni-tude of the decrease was less both at the low concentration of0.1 µM (to 75% of control levels) and the high concentration of50 µM (to 50% of control levels). Unexpectedly, in both in-flammatory settings, no corresponding reductions in quantita-tive CXCL10 mRNA levels are seen at any concentration up to50 µM (Fig. 3).
CXCL10 protein production in response to IFN-� inTHP-1-derived macrophages was also reduced by rosi-glitazone, with a very similar response to that seen in epithelialcells (Fig. 4). CXCL10 protein levels were reduced to 61% ofcontrol levels with a 0.1 µM concentration rosiglitazone; how-ever, the magnitude of the reduction was higher at the highestdose of 50 µM (23% of control levels). TNF-� activation ofCXCL10 and TZD effects on this activation state were notmeasured, as TNF-� signaling results in apoptosis of THP-1macrophages.35 However, the CXCL10 production stimulatedby the NF-�B-dependent signal LPS was also inhibited byrosiglitazone, although at higher concentrations (Fig. 5). Aswith the HT-29 cells, TZDs significantly reduced protein se-cretion in macrophages without affecting steady-state mRNAlevels.
TZDs Have a Stronger Effect on MacrophagesThan on Monocytes
Both human monocytes and undifferentiated THP-1monocytes respond to PPAR� activation.36,37 To determine
whether TZDs could potentially affect the levels of TH1 che-mokines produced by circulating monocytes, the ability ofTZDs to reduce CXCL10 production from THP-1 monocytesand from differentiated macrophages was investigated (Fig. 5).CXCL10 protein levels were measured after IFN-� or LPSstimulation of undifferentiated THP-1 cells and PMA-differentiated macrophages. Although rosiglitazone signifi-cantly reduced CXCL10 protein production in response toIFN-� in differentiated macrophages (to 61% of control levelsat 0.1 µM, and to 23% of control levels at 50 µM), no statisti-cally significant difference was seen at any concentration inTHP-1 cells. The inhibitory effect on chemokine productionby rosiglitazone is less strong in LPS-stimulated differentiatedmacrophages than in IFN-�-stimulated macrophages, with sta-tistically significant differences not occurring until a concen-tration of 5 µM was used (to 40% of control levels). As withthe IFN-�-stimulated cells, LPS-activated THP-1 cells alsorespond much less strongly than macrophages, with sig-
FIGURE 3. The PPAR� ligand rosiglitazone reduces TNF-� andIFN-�-induced CXCL10 production from HT-29. HT-29 cellswere incubated with the indicated concentration of rosi-glitazone for 16 hours before adding the indicated inflamma-tory stimulus. CXCL10 mRNA levels (hatched bars) and proteinlevels (solid bars) are shown. The results were normalizedto the amount of CXCL10 produced in the absence of rosi-glitazone in each individual experiment (7 each for TNF-� andIFN-�). Error bars, SEM; *, P < 0.05.
FIGURE 2. Pioglitazone pretreatment reduced the influx ofCD4+ T cells in 2 models of murine colitis. CD4+ T cells (brown,arrows) were visualized by immunohistochemistry. A magnifi-cation of 10� is shown. In DSS colitis, T cells were diffuselyscattered; in DNBS colitis, they formed nodules (arrow). Noanti-CD4, no antibody control. The results are representativeof at least 3 separate examined sites.
Schaefer et al Inflamm Bowel Dis • Volume 11, Number 3, March 2005
248 © 2005 Lippincott Williams & Wilkins
nificant reductions not seen until a concentration of 50 µM wasapplied.
Reductions in Chemokine Production Are NotAssociated With Reductions in NF-�B Activity
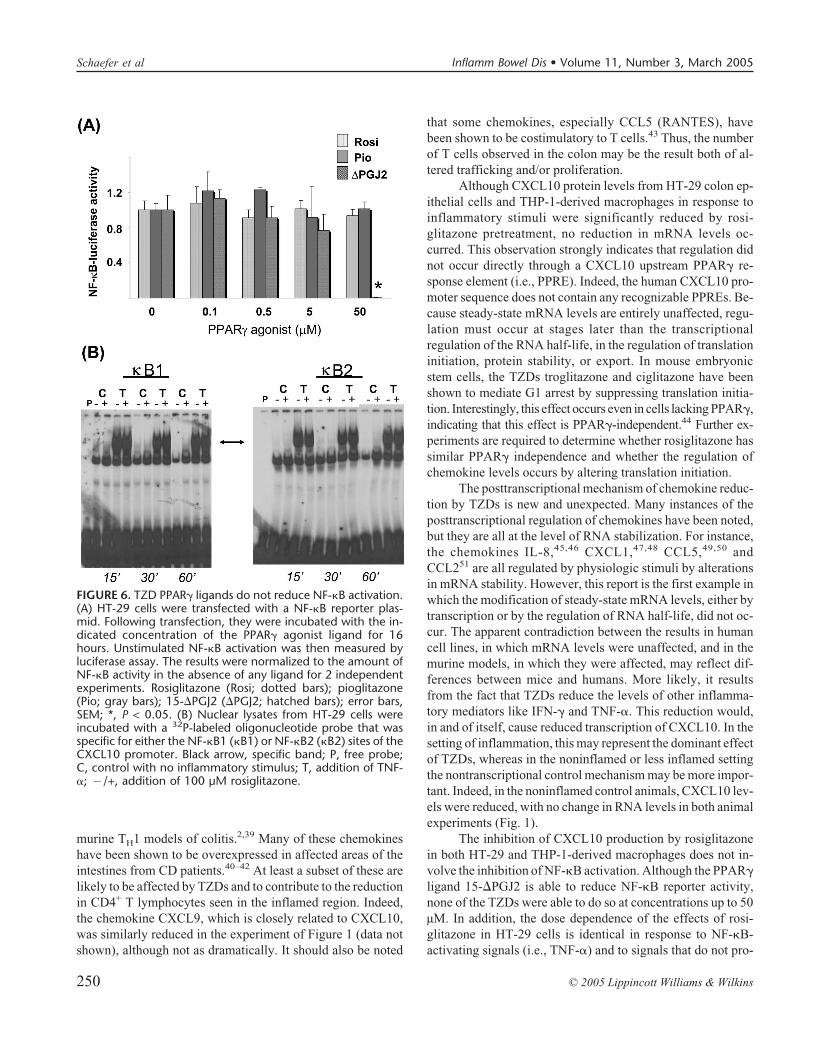
Many reports have suggested that TZDs are able to exerttheir antiinflammatory effects by reducing the proinflamma-tory NF-�B signaling pathway, either directly or indirectly,thereby influencing the transcriptional regulation of a numberof inflammation-associated genes. To determine whetherTZDs could reduce CXCL10 production in part by inhibitingNF-�B activity, the transient transfection of HT-29 cells withan NF-�B-luciferase reporter element was performed. BasalNF-�B activity was not reduced by rosiglitazone, pioglitazone(Fig. 6A), ciglitazone, or troglitazone (data not shown) at anyconcentration up to 50 µM. In contrast, a known PPAR�-activating prostanoid with recognized PPAR�-independentNF-�B-inhibiting effects, 15-�PGJ2, appropriately reducedNF-�B-dependent transcription at the highest concentration of50 µM. Similar results were obtained with the E-selectin NF-�B-dependent reporter construct38 (data not shown). In addi-tion, doses of rosiglitazone up to extremely high concentra-tions (i.e., 100 µM) did not demonstrate any effect on NF-�Bbinding at either the �B1 or �B2 sites of the CXCL10 promoter(Fig. 6B).
DISCUSSIONThe results of these experiments indicate a novel antiin-
flammatory mechanism for TZDs in TH1 immune responses.
They clearly demonstrate that TZDs can inhibit the productionof the TH1 chemokine CXCL10 in vivo and in response to avariety of inflammatory signals. These effects occur in humanepithelial cells and differentiated macrophages and are not de-pendent on the inhibition of the NF-�B pathway.
By preadministering pioglitazone before the inductionof colitis, significant reductions in CXCL10 levels reachedduring inflammation can be achieved. This reduction in TH1chemokines is paralleled by a decrease in the number of effec-tor CD4+ T cells in the colon. Although CXCL10 is the onlychemokine that has been shown to be critical to the develop-ment of colitis in mice,13,14 the dramatic reductions in CD4+
T-cell levels in the colon are probably not due entirely to thischemokine alone, but rather to reductions in the total pool ofTH1-attracting chemokines. The CD4+ T-cell-attracting che-mokines CCL5 (RANTES), CCL2 (MCP-1), CCL3 (MIP-1�),CCL20 (Mip-3�), and CXCL9 (Mig) are all up-regulated in
FIGURE 5. Pioglitazone has stronger effects on macrophagesthan on monocytes. PMA-induced THP-1-derived macrophagecells (MP; dotted black bars) and THP-1 monocyte cells (THP;gray bars) were incubated with the indicated concentration ofrosiglitazone for 16 hours before adding IFN-� or LPS. CXCL10protein levels are shown. The results were normalized to theamount of CXCL10 produced in the absence of rosiglitazone ineach individual experiment. There were 2 independent experi-ments for each condition (4 for IFN-�-treated MP cells). Errorbars, SEM; *, P < 0.05.
FIGURE 4. The PPAR� ligand rosiglitazone reduces IFN-�-induced CXCL10 production from THP-1-derived cells in re-sponse to IFN-�. PMA-induced THP-1-derived macrophageswere incubated with the indicated concentration of rosi-glitazone for 16 hours before adding IFN-�. CXCL10 RNA lev-els (hatched bars) and protein levels (solid bars) are shown.The results were normalized to the amount of CXCL10 pro-duced in the absence of rosiglitazone in each individual ex-periment (4 experiments). Error bars, SEM; *, P < 0.05.
Inflamm Bowel Dis • Volume 11, Number 3, March 2005 Intestinal Antiinflammatory Effects of Thiazolidenedione
© 2005 Lippincott Williams & Wilkins 249
murine TH1 models of colitis.2,39 Many of these chemokineshave been shown to be overexpressed in affected areas of theintestines from CD patients.40–42 At least a subset of these arelikely to be affected by TZDs and to contribute to the reductionin CD4+ T lymphocytes seen in the inflamed region. Indeed,the chemokine CXCL9, which is closely related to CXCL10,was similarly reduced in the experiment of Figure 1 (data notshown), although not as dramatically. It should also be noted
that some chemokines, especially CCL5 (RANTES), havebeen shown to be costimulatory to T cells.43 Thus, the numberof T cells observed in the colon may be the result both of al-tered trafficking and/or proliferation.
Although CXCL10 protein levels from HT-29 colon ep-ithelial cells and THP-1-derived macrophages in response toinflammatory stimuli were significantly reduced by rosi-glitazone pretreatment, no reduction in mRNA levels oc-curred. This observation strongly indicates that regulation didnot occur directly through a CXCL10 upstream PPAR� re-sponse element (i.e., PPRE). Indeed, the human CXCL10 pro-moter sequence does not contain any recognizable PPREs. Be-cause steady-state mRNA levels are entirely unaffected, regu-lation must occur at stages later than the transcriptionalregulation of the RNA half-life, in the regulation of translationinitiation, protein stability, or export. In mouse embryonicstem cells, the TZDs troglitazone and ciglitazone have beenshown to mediate G1 arrest by suppressing translation initia-tion. Interestingly, this effect occurs even in cells lacking PPAR�,indicating that this effect is PPAR�-independent.44 Further ex-periments are required to determine whether rosiglitazone hassimilar PPAR� independence and whether the regulation ofchemokine levels occurs by altering translation initiation.
The posttranscriptional mechanism of chemokine reduc-tion by TZDs is new and unexpected. Many instances of theposttranscriptional regulation of chemokines have been noted,but they are all at the level of RNA stabilization. For instance,the chemokines IL-8,45,46 CXCL1,47,48 CCL5,49,50 andCCL251 are all regulated by physiologic stimuli by alterationsin mRNA stability. However, this report is the first example inwhich the modification of steady-state mRNA levels, either bytranscription or by the regulation of RNA half-life, did not oc-cur. The apparent contradiction between the results in humancell lines, in which mRNA levels were unaffected, and in themurine models, in which they were affected, may reflect dif-ferences between mice and humans. More likely, it resultsfrom the fact that TZDs reduce the levels of other inflamma-tory mediators like IFN-� and TNF-�. This reduction would,in and of itself, cause reduced transcription of CXCL10. In thesetting of inflammation, this may represent the dominant effectof TZDs, whereas in the noninflamed or less inflamed settingthe nontranscriptional control mechanism may be more impor-tant. Indeed, in the noninflamed control animals, CXCL10 lev-els were reduced, with no change in RNA levels in both animalexperiments (Fig. 1).
The inhibition of CXCL10 production by rosiglitazonein both HT-29 and THP-1-derived macrophages does not in-volve the inhibition of NF-�B activation. Although the PPAR�ligand 15-�PGJ2 is able to reduce NF-�B reporter activity,none of the TZDs were able to do so at concentrations up to 50µM. In addition, the dose dependence of the effects of rosi-glitazone in HT-29 cells is identical in response to NF-�B-activating signals (i.e., TNF-�) and to signals that do not pro-
FIGURE 6. TZD PPAR� ligands do not reduce NF-�B activation.(A) HT-29 cells were transfected with a NF-�B reporter plas-mid. Following transfection, they were incubated with the in-dicated concentration of the PPAR� agonist ligand for 16hours. Unstimulated NF-�B activation was then measured byluciferase assay. The results were normalized to the amount ofNF-�B activity in the absence of any ligand for 2 independentexperiments. Rosiglitazone (Rosi; dotted bars); pioglitazone(Pio; gray bars); 15-�PGJ2 (�PGJ2; hatched bars); error bars,SEM; *, P < 0.05. (B) Nuclear lysates from HT-29 cells wereincubated with a 32P-labeled oligonucleotide probe that wasspecific for either the NF-�B1 (�B1) or NF-�B2 (�B2) sites of theCXCL10 promoter. Black arrow, specific band; P, free probe;C, control with no inflammatory stimulus; T, addition of TNF-�; �/+, addition of 100 µM rosiglitazone.
Schaefer et al Inflamm Bowel Dis • Volume 11, Number 3, March 2005
250 © 2005 Lippincott Williams & Wilkins

ceed via NF-�B (i.e., IFN-�). In THP-1-derived macrophages,the effects are actually greater in the context of IFN-� than inresponse to LPS, which induces NF-�B activity. Previous re-ports on the effects of PPAR� ligands on NF-�B activationhave been conflicting. Both troglitazone and 15-�PGJ2 havebeen shown to reduce NF-�B activation in rheumatoid syno-vial fibroblasts and THP-1 cells52,53; in microglia, troglitazoneand ciglitazone had no effect, whereas 15-�PGJ2 reduced NF-�B-binding activity.54 As with the microglia, in a mousemodel of experimental glomerulonephritis, troglitazone andciglitazone did not affect NF-�B activation in diseased tissue,whereas 15-�PGJ2 did.55 Finally, in 2 mouse models of colitis,pretreatment with rosiglitazone and pioglitazone was associ-ated with reductions in NF-�B-binding activity.24,27 Our re-sults in HT-29 cells suggest that rosiglitazone and pio-glitazone, unlike 15-�PGJ2, do not reduce NF-�B activity.The apparent paradox between our results and those in themouse models of colitis probably stems from the fact that over-all inflammation is reduced by TZDs, causing, among othereffects, reduced TNF-� and IFN-�26 production. This effectwould lead to a secondary reduction in the overall levels ofNF-�B activation, as was seen in the mouse models, withoutnecessarily requiring that NF-�B activation be directly re-duced by TZDs.
The dose dependence of the reduction in CXCL10 inHT-29-derived and THP-1-derived macrophages was verysimilar. An initial roughly 2-fold drop occurred with a rosi-glitazone concentration of 0.1 µM, and concentrations up to 5µM yielded a very small additional drop. An additional drop inprotein concentrations occurred at 50 µM. This biphasic re-sponse suggested that 2 separate regulatory events may be oc-curring; 1 with “low-dose” TZDs, and the other with “high-dose” TZDs. In PPAR�−/− macrophages, it has been shownthat high-dose (i.e., 50 µM) rosiglitazone stimulates PPAR� inaddition to PPAR�, and that this stimulation, not that ofPPAR�, is responsible for the reductions in rosiglitazone ofinducible nitric oxide synthase and IL-12 p40.23 Thus, PPAR�stimulation may also be involved in the regulation of CXCL10.
Rosiglitazone had a much stronger effect on THP-1-derived macrophages than THP-1 monocytes. In response toIFN-�, rosiglitazone did not affect CXCL10 production at anyof the concentrations tested; LPS-mediated CXCL10 produc-tion was inhibited only at 50 µM. LPS is known to reduce theexpression of PPAR� in human blood-derived monocytes,56
possibly accounting for the lack of responsiveness to rosi-glitazone in the context of LPS. However, the PPAR� ligand15-�PGJ2 is able to reduce CXCL10 production from LPS-stimulated THP-1 cells, most likely by its direct NF-�B inter-action, which suggests that PPAR� reduction may not be theexplanation.54 To our knowledge, no published data exist onthe effects on IFN-� on PPAR� expression in THP-1 cells.However, real-time quantitative PCR analysis of PPAR� lev-els in THP-1 cells showed no reduction in PPAR� levels in
response to rosiglitazone (data not shown). A close compari-son between differentiated and undifferentiated THP-1 cellswill be useful to better delineate the exact mechanism of thisnontranscriptional chemokine regulation pathway induced byTZDs. This mechanism may involve the regulation of othersecondary, yet important, gene products or the effects on en-zymatic functions that are involved in protein synthesis.
In summary, these experimental results indicate a novelnontranscriptional antiinflammatory role for TZDs that re-duces the TH1 immune responses and is not NF-�B-dependent.This antiinflammatory role has important consequences inTH1-mediated diseases like CD, and further investigation willbe required to elucidate the exact mechanism.
ACKNOWLEDGMENTSThe authors thank Caroline Savage and Luana Atherly
for their critical review of the manuscript.
REFERENCES1. Podolsky DK. Inflammatory bowel disease. N Engl J Med. 2002;347:
417–429.2. Ajuebor MN, Swain MG. Role of chemokines and chemokine receptors in
the gastrointestinal tract. Immunology. 2002;105:137–143.3. Bouma G, Strober W. The immunological and genetic basis of inflamma-
tory bowel disease. Nat Rev Immunol. 2003;3:521–533.4. Papadakis KA, Targan SR. Role of cytokines in the pathogenesis of in-
flammatory bowel disease. Annu Rev Med. 2000;51:289–298.5. Stallmach A, Giese T, Schmidt C, et al. Cytokine/chemokine transcript
profiles reflect mucosal inflammation in Crohn’s disease. Int J ColorectalDis. 2004;19:308–315.
6. Blumberg RS, Saubermann LJ, Strober W. Animal models of mucosalinflammation and their relation to human inflammatory bowel disease.Curr Opin Immunol. 1999;11:648–656.
7. Sandborn WJ, Yednock TA. Novel approaches to treating inflammatorybowel disease: targeting alpha-4 integrin. Am J Gastroenterol. 2003;98:2372–2382.
8. Picarella D, Hurlbut P, Rottman J, et al. Monoclonal antibodies specificfor � 7 integrin and mucosal addressin cell adhesion molecule-1(MAdCAM-1) reduce inflammation in the colon of scid mice reconsti-tuted with CD45RBhigh CD4+ T cells. J Immunol. 1997;158:2099–2106.
9. Taniguchi T, Tsukada H, Nakamura H, et al. Effects of the anti-ICAM-1monoclonal antibody on dextran sodium sulphate-induced colitis in rats.J Gastroenterol Hepatol. 1998;13:945–949.
10. Soriano A, Salas A, Sans M, et al. VCAM-1, but not ICAM-1 orMAdCAM-1, immunoblockade ameliorates DSS-induced colitis in mice.Lab Invest. 2000;80:1541–1551.
11. Kato S, Hokari R, Matsuzaki K, et al. Amelioration of murine experimen-tal colitis by inhibition of mucosal addressin cell adhesion molecule-1.J Pharmacol Exp Ther. 2000;295:183–189.
12. Bennett CF, Kornbrust D, Henry S, et al. An ICAM-1 antisense oligo-nucleotide prevents and reverses dextran sulfate sodium-induced colitis inmice. J Pharmacol Exp Ther. 1997;280:988–1000.
13. Singh UP, Singh S, Taub DD, et al. Inhibition of IFN-�-inducible protein-10 abrogates colitis in IL-10−/− mice. J Immunol. 2003;171:1401–1406.
14. Sasaki S, Yoneyama H, Suzuki K, et al. Blockade of CXCL10 protectsmice from acute colitis and enhances crypt cell survival. Eur J Immunol.2002;32:3197–3205.
15. Lefebvre M, Paulweber B, Fajas L, et al. Peroxisome proliferator-activated receptor � is induced during differentiation of colon epitheliumcells. J Endocrinol. 1999;162:331–340.
16. Mansen A, Guardiola-Diaz H, Rafter J, et al. Expression of the peroxi-some proliferator-activated receptor (PPAR) in the mouse colonic mu-cosa. Biochem Biophys Res Commun. 1996;222:844–851.
Inflamm Bowel Dis • Volume 11, Number 3, March 2005 Intestinal Antiinflammatory Effects of Thiazolidenedione
© 2005 Lippincott Williams & Wilkins 251

17. Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated re-ceptors (PPARs): nuclear receptors with functions in the vascular wall.Z Kardiol. 2001;90:125–132.
18. Chinetti G, Fruchart JC, Staels B. Peroxisome proliferator-activated re-ceptors and inflammation: from basic science to clinical applications. IntJ Obes Relat Metab Disord. 2003;27(suppl 3):S41–S45.
19. Delerive P, Fruchart JC, Staels B. Peroxisome proliferator-activated re-ceptors in inflammation control. J Endocrinol. 2001;169:453–459.
20. Marx N, Mach F, Sauty A, et al. Peroxisome proliferator-activated recep-tor-gamma activators inhibit IFN-�-induced expression of the T cell-active CXC chemokines IP-10, Mig, and I-TAC in human endothelialcells. J Immunol. 2000;164:6503–6508.
21. Kitamura Y, Taniguchi T, Kimura H, et al. Interleukin-4-inhibited mRNAexpression in mixed rat glial and in isolated microglial cultures. J Neuro-immunol. 2000;106:95–104.
22. Gosset P, Charbonnier AS, Delerive P, et al. Peroxisome proliferator-activated receptor gamma activators affect the maturation of humanmonocyte-derived dendritic cells. Eur J Immunol. 2001;31:2857–2865.
23. Welch JS, Ricote M, Akiyama TE, et al. PPARgamma and PPARdeltanegatively regulate specific subsets of lipopolysaccharide and IFN-� tar-get genes in macrophages. Proc Natl Acad Sci USA. 2003;100:6712–6717.
24. Takagi T, Naito Y, Tomatsuri N, et al. Pioglitazone, a PPAR-gamma li-gand, provides protection from dextran sulfate sodium-induced colitis inmice in association with inhibition of the NF-�B-cytokine cascade. RedoxRep. 2002;7:283–289.
25. Su CG, Wen X, Bailey ST, et al. A novel therapy for colitis utilizingPPAR-gamma ligands to inhibit the epithelial inflammatory response.J Clin Invest. 1999;104:383–389.
26. Saubermann LJ, Nakajima A, Wada K, et al. Peroxisome proliferator-activated receptor gamma agonist ligands stimulate a Th2 cytokine re-sponse and prevent acute colitis. Inflamm Bowel Dis. 2002;8:330–339.
27. Desreumaux P, Dubuquoy L, Nutten S, et al. Attenuation of colon inflam-mation through activators of the retinoid X receptor (RXR)/peroxisomeproliferator-activated receptor � (PPAR�) heterodimer: a basis for newtherapeutic strategies. J Exp Med. 2001;193:827–838.
28. Iwashima Y, Eto M, Hata A, et al. Advanced glycation end products-induced gene expression of scavenger receptors in cultured human mono-cyte-derived macrophages. Biochem Biophys Res Commun. 2000;277:368–380.
29. Podrez EA, Hoppe G, O’Neil J, et al. Macrophage receptors responsiblefor distinct recognition of low density lipoprotein containing pyrrole orpyridinium adducts: models of oxidized low density lipoprotein. J LipidRes. 2000;41:1455–1463.
30. Zhang B, Marcus SL, Sajjadi FG, et al. Identification of a peroxisomeproliferator-responsive element upstream of the gene encoding rat peroxi-somal enoyl-CoA hydratase/3-hydroxyacyl-CoA dehydrogenase. ProcNatl Acad Sci USA. 1992;89:7541–7545.
31. Mizoguchi A, Mizoguchi E, Bhan AK. Immune networks in animal mod-els of inflammatory bowel disease. Inflamm Bowel Dis. 2003;9:246–259.
32. Dwinell MB, Lugering N, Eckmann L, et al. Regulated production of in-terferon-inducible T-cell chemoattractants by human intestinal epithelialcells. Gastroenterology. 2001;120:49–59.
33. Shibahara T, Wilcox JN, Couse T, et al. Characterization of epithelialchemoattractants for human intestinal intraepithelial lymphocytes. Gas-troenterology. 2001;120:60–70.
34. Ferrero E, Biswas P, Vettoretto K, et al. Macrophages exposed to Myco-bacterium tuberculosis release chemokines able to recruit selected leuco-cyte subpopulations: focus on �� cells. Immunology. 2003;108:365–374.
35. Riendeau CJ, Kornfeld H. THP-1 cell apoptosis in response to mycobac-terial infection. Infect Immun. 2003;71:254–259.
36. Chinetti G, Zawadski C, Fruchart JC, et al. Expression of adiponectinreceptors in human macrophages and regulation by agonists of the nuclearreceptors PPAR�, PPAR�, and LXR. Biochem Biophys Res Commun.2004;314:151–158.
37. Shu H, Wong B, Zhou G, et al. Activation of PPAR� or � reduces secre-
tion of matrix metalloproteinase 9 but not interleukin 8 from humanmonocytic THP-1 cells. Biochem Biophys Res Commun. 2000;267:345–349.
38. Delude RL, Yoshimura A, Ingalls RR, et al. Construction of a lipopoly-saccharide reporter cell line and its use in identifying mutants defective inendotoxin, but not TNF-�, signal transduction. J Immunol. 1998;161:3001–3009.
39. Scheerens H, Hessel E, de Waal-Malefyt R, et al. Characterization of che-mokines and chemokine receptors in two murine models of inflammatorybowel disease: IL-10−/− mice and Rag-2−/− mice reconstituted withCD4+CD45RBhigh T cells. Eur J Immunol. 2001;31:1465–1474.
40. Kaser A, Ludwiczek O, Holzmann S, et al. Increased expression ofCCL20 in human inflammatory bowel disease. J Clin Immunol. 2004;24:74–85.
41. McCormack G, Moriarty D, O’Donoghue DP, et al. Tissue cytokine andchemokine expression in inflammatory bowel disease. Inflamm Res.2001;50:491–495.
42. Banks C, Bateman A, Payne R, et al. Chemokine expression in IBD. Mu-cosal chemokine expression is unselectively increased in both ulcerativecolitis and Crohn’s disease. J Pathol. 2003;199:28–35.
43. Wong MM, Fish EN. Chemokines: attractive mediators of the immuneresponse. Semin Immunol. 2003;15:5–14.
44. Palakurthi SS, Aktas H, Grubissich LM, et al. Anticancer effects of thia-zolidinediones are independent of peroxisome proliferator-activated re-ceptor gamma and mediated by inhibition of translation initiation. CancerRes. 2001;61:6213–6218.
45. Gerber A, Heimburg A, Reisenauer A, et al. Proteasome inhibitors modu-late chemokine production in lung epithelial and monocytic cells. EurRespir J. 2004;24:40–48.
46. Villarete LH, Remick DG. Transcriptional and post-transcriptional regu-lation of interleukin-8. Am J Pathol. 1996;149:1685–1693.
47. Dai Y, Datta S, Novotny M, et al. TGF� inhibits LPS-induced chemokinemRNA stabilization. Blood. 2003;102:1178–1185.
48. Tebo JM, Datta S, Kishore R, et al. Interleukin-1-mediated stabilization ofmouse KC mRNA depends on sequences in both 5�- and 3�-untranslatedregions. J Biol Chem. 2000;275:12987–12993.
49. Li QQ, Bever CT. Mechanisms underlying the synergistic effect of Th1cytokines on RANTES chemokine production by human glial cells. Int JMol Med. 2001;7:187–195.
50. Akoum A, Lemay A, Maheux R. Estradiol and interleukin-1� exert a syn-ergistic stimulatory effect on the expression of the chemokine regulatedupon activation, normal T cell expressed, and secreted in endometrioticcells. J Clin Endocrinol Metab. 2002;87:5785–5792.
51. Goppelt-Struebe M, Stroebel M. Synergistic induction of monocyte che-moattractant protein-1 (MCP-1) by platelet-derived growth factor and in-terleukin-1. FEBS Lett. 1995;374:375–378.
52. Yamasaki S, Nakashima T, Kawakami A, et al. Functional changes inrheumatoid fibroblast-like synovial cells through activation of peroxi-some proliferator-activated receptor �-mediated signalling pathway. ClinExp Immunol. 2002;129:379–384.
53. Ji JD, Cheon H, Jun JB, et al. Effects of peroxisome proliferator-activatedreceptor-gamma (PPAR-�) on the expression of inflammatory cytokinesand apoptosis induction in rheumatoid synovial fibroblasts and mono-cytes. J Autoimmun. 2001;17:215–221.
54. Si Q, Zhao ML, Morgan AC, et al. 15-deoxy-Delta12,14-prostaglandin J2inhibits IFN-inducible protein 10/CXC chemokine ligand 10 expressionin human microglia: mechanisms and implications. J Immunol. 2004;173:3504–3513.
55. Panzer U, Schneider A, Guan Y, et al. Effects of different PPAR�-agonists on MCP-1 expression and monocyte recruitment in experimentalglomerulonephritis. Kidney Int. 2002;62:455–464.
56. Hinz B, Brune K, Pahl A. 15-Deoxy-Delta(12,14)-prostaglandin J2 inhib-its the expression of proinflammatory genes in human blood monocytesvia a PPAR-�-independent mechanism. Biochem Biophys Res Commun.2003;302:415–420.
Schaefer et al Inflamm Bowel Dis • Volume 11, Number 3, March 2005
252 © 2005 Lippincott Williams & Wilkins




















