
An extracellular polyhydroxybutyrate depolymerase in Thermus thermophilus HB8
Upload
independentCategory
view
0download
0
The crystal structure of ribosomal protein L22 from Thermusthermophilus: insights into the mechanism of erythromycinresistanceJ Unge1, A Åberg†, S Al-Kharadaghi1, A Nikulin2, S Nikonov2, NL Davydova1,2,N Nevskaya2, M Garber2 and A Liljas1*
Background: The ribosomal protein L22 is one of five proteins necessary for theformation of an early folding intermediate of the 23S rRNA. L22 has been foundon the cytoplasmic side of the 50S ribosomal subunit. It can also be labeled by anerythromycin derivative bound close to the peptidyl-transfer center at the interfaceside of the 50S subunit, and the amino acid sequence of an erythromycin-resistant mutant is known. Knowing the structure of the protein may resolve thisapparent conflict regarding the location of L22 on the ribosome.
Results: The structure of Thermus thermophilus L22 was solved using X-raycrystallography. L22 consists of a small α+β domain and a protruding β hairpinthat is 30 Å long. A large part of the surface area of the protein has the potentialto be involved in interactions with rRNA. A structural similarity to other RNA-binding proteins is found, possibly indicating a common evolutionary origin.
Conclusions: The extensive surface area of L22 has the characteristics of anRNA-binding protein, consistent with its role in the folding of the 23S rRNA. Theerythromycin-resistance conferring mutation is located in the protruding β hairpinthat is postulated to be important in L22–rRNA interactions. This region of theprotein might be at the erythromycin-binding site close to the peptidyl transferasecenter, whereas the opposite end may be exposed to the cytoplasm.
IntroductionTo understand protein biosynthesis, structural informationof the components involved in translation is required atthe atomic level. The structure of the ribosome and itssubunits remain at low resolution, but recent progress hasbrought us to a state where a crude model of the riboso-mal RNA (rRNA) and the associated ribosomal proteinscould be made [1]. This model is the result of a combina-tion of chemical and biophysical analysis of the ribosomewith cryo-electron microscopy of complete ribosomes[2,3], crystallography on ribosomal subunits [4] and NMRand crystallography on ribosomal components [5]. Thestructures of individual protein or RNA fragments at highresolution complement structural studies on complete ribo-somes or ribosomal subunits at lower resolution. Knowl-edge of the three dimensional structure of individual com-ponents of the translational apparatus can also shed lighton the interaction of antibiotics with their targets on theribosome. The structural studies of the ribosome and itscomponents have so far led to the determination of thethree-dimensional structure for 15 out of more than 50eubacterial ribosomal proteins: a domain homologous to theS1 RNA-binding domain, S4, S5, S6, S7, S8, S15, S17, L1,L6, L7/L12, L9, the C-terminal RNA-recognition domainof L11, L14, and L30 [5–23].
L22 is a small, 113 amino acid, core protein of the 50S ribo-somal subunit of Thermus thermophilus [24]. L22 is importantfor the folding of the 23S rRNA. The formation of an earlyintermediate in the assembly of the large ribosomal subunitin Escherichia coli depends on L22 as well as on L4, L13,L20 and L24 [25]. A mutant with a three amino acid dele-tion of E. coli L22 gives rise to resistance to erythromycin[26], an antibiotic that inhibits the elongation of the nascentpeptide after two to five residues [27]. L22 has also beenlabeled with an erythromycin derivative [28] that is thoughtto bind in the vicinity of the peptidyl-transferase center onthe interface side of the 50S subunit. Combined immuneelectron microscopy (IEM) and crosslinking data [29,30]have suggested that the protein is located on the externalsurface of the subunit. In this paper we report the refinedcrystal structure of L22 from the thermophilic bacteriumT. thermophilus at 1.9 Å resolution.
Results and discussionStructure determinationThe purification and crystallization of L22 has beendescribed earlier [24]. The structure was determined bythe multiple isomorphous replacement (MIR) methodusing data from two heavy-atom derivatives, selenomethi-onine and Na2PtCl6. All data (Table 1) were collected at
Addresses: 1Molecular Biophysics, Lund University,PO Box 124, 221 00 Lund, Sweden and2Department of Structure and Function of theRibosome, Institute of Protein Research, Pushchino,Moscow Region, Russia.
†Present address: Astra Structural ChemistryLaboratory, ASTRA Hässle AB, S-431 83 Mölndal,Sweden.
*Corresponding author.E-mail: [email protected]
Key words: erythromycin resistance, ribosomal proteinL22, ribosomes, RNA-binding, X-ray crystallography
Received: 5 August 1998Revisions requested: 10 September 1998Revisions received: 7 October 1998Accepted: 13 October 1998
Structure 15 December 1998, 6:1577–1586http://biomednet.com/elecref/0969212600601577
© Current Biology Ltd ISSN 0969-2126
Research Article 1577
the crystallographic beamline BL711 at the MAX-II syn-chrotron. Phases calculated from the two derivatives andimproved with solvent flattening gave excellent maps to2.5 Å resolution and the entire polypeptide chain, apartfrom the last three residues of the C terminus, could betraced. The model was refined at 1.9 Å resolution to Rconvof 18.9% and Rfree of 21.9%. Details are given in the Mate-rials and methods section and in Table 2.
Description of the structureThe overall structure, with the approximate dimensions of67 Å × 29 Å × 18 Å, is shown in Figure 1 and the sec-ondary structure is indicated in the sequence alignment inFigure 2. The molecule consists of a single domain withthree α helices packed against a three-stranded antiparal-lel β sheet and therefore belongs to the α+β-fold group.The connectivity of the secondary structure elements isβ1–α1–α2–α3–β2–β3. The α helices and the β sheet forma well-packed hydrophobic core without charged or polarresidues. A β hairpin formed by strands β2 and β3 pro-trudes from the core of the protein.
The globular coreThe structure of the globular core contains some featuresof interest. A β bulge is found in the β sheet at Val71, ahydrophobic residue in all sequences of L22 (Figure 2).The N termini of β1 and β3 are stabilized by a salt bridgebetween Glu2 and Lys72. The C terminus of α1 includesa distortion of residues 20–24 with a hydrogen-bondingpattern closer to a 310 helix, whereas the last residue in thehelix returns to the hydrogen-bonding pattern of the α helix.The last residue in helix α3, His62, follows instead thepattern of a π helix that widens the last turn. Whether this
is an important feature or not is not clear. Asn61, which isconserved in all eubacteria, stabilizes the sidechain of thestrictly conserved residue Asn57 via a hydrogen bond, butthis interaction can be maintained without the π turn ofhelix α3. Loop 4 has two hydrogen bonds in the form of ashort 310 helix.
The β hairpinTwo of the three strands of the β structure form theβ hairpin, which extends approximately 30 Å from the glob-ular domain. Protruding features in the form of loops or
1578 Structure 1998, Vol 6 No 12
Table 1
Data collection statistics.
Dataset* Native SeMet1 SeMet2 Na2PtCl6†
X-ray source BL711 BL711 BL711 BL711Wavelength (Å) 1.012 1.012 0.96 1.012No. of unique reflections 9261 11,664 4178 3168Overall
Resolution (Å) 2.0 1.9 2.5 2.5Multiplicity 3.3 4.2 3.8 3.4Completeness (%) 96.8 98.1 96.5 91.4I/σ(I) 6.4 5.8 9.7 6.9Rsym 0.07 0.053 0.049 0.075
Highest resolution shellResolution (Å) 2.06–2.00 1.97–1.90 2.56–2.50 2.56–2.50Multiplicity 3.1 4.6 3.9 3.4Completeness (%) 96.3 97.1 97.1 91.3I/σ(I) 3.5 2.9 6.7 3.0Rsym
‡ 0.21 0.17 0.095 0.21Riso
§ (10.0–2.5 Å) 0.083 0.093 0.272Rcullis
# 0.77 0.81 0.75Phasing power¶ 1.37 1.19 1.48
*Dataset abbreviations: SeMet is selenomethionine. †Derivative of an SeMet L22 crystal. ‡Rsym = Σhkl Σi | Ihkl,i – ⟨Ihkl⟩ /Σhkl Σi Ihkl,i.§Riso = Σhkl || FPH | – | FP || / Σhkl | FP |. #Rcullis = Σhkl || FPH ± FP | – | FH, calc || / Σhkl | FPH ± FP |. ¶Phasing power = Σhkl | FH |2 / (Σhkl ( | FPH, obs | − | FPH, calc | )2 )1/2.
Table 2
Refinement statistics.
Resolution (Å) 10.0–1.90Number of reflections 11,536Rconv* (%) 18.9Rfree
† (%) 21.9Rms deviations from ideal geometry
Bonds (Å) 0.008Angles (°) 1.38Dihedrals (°) 26.32
Ramachandran plot statisticsResidues in most favored regions (%) 95.8Residues in additional allowed regions (%) 4.21Residues in disallowed regions 0
Number of non-hydrogen atoms 883Number of water molecules 63Average B factor
Wholechain (Å2) 29.7Mainchain atoms (Å2) 23.3Sidechains and waters (Å2) 35.3
*Rconv = Σ || Fobs | – k | Fcalc | /Σ | Fobs | × 100% for reflections used inrefinement, Rfree is defined similarly on a disjunct test set. †Calculatedfor 5% of data in test set.
β hairpins have frequently been observed in nucleic-acid-binding proteins, including ribosomal proteins [31]. In somecases they are stabilized by hydrophobic interactions andhydrogen bonds [22,32], but β hairpins may also be flexiblein the absence of the corresponding nucleic acid [33].
The β hairpin has regular hydrogen bonds along its length,except for two β bulges at proline residues 80 and 87.These give the extended region of the β hairpin a uniqueconformation that is more twisted than the β sheet. This isparticularly due to residues 78–80 as can be seen fromtheir unusually high φ and ϕ angles in comparison toaverage values of –139 and +135, respectively, for anantiparallell β sheet [34,35]. Glu78 has a φ angle of –75; asmaller value would be sterically hindered as a result ofthe sidechain of Pro14 pointing towards it. The ϕ angle ofGly79 is –174; this residue could not be substituted forother residues in the present conformation. The turn ofthe β hairpin contains a strictly conserved glycine (Gly91).Glycines are commonly found in tight β hairpin turns forstereochemical reasons [36].
The question of whether the extended β hairpin has thesame conformation in the ribosome as in the crystal is dif-ficult to answer. Strand β2 of the L22 hairpin packs
against helices α3 and α2 of a symmetry-related molecule.Thus the entire stretch of the β hairpin is stabilized in thecrystal environment. The contacts are mainly via side-chains or are indirect contacts via hydrogen-bonded watermolecules. This is probably a reason why the electron-density maps are of good quality even for this region ofthe structure, with temperature factors close to thosefound for other parts of the structure. The β hairpin maybe stabilized to take on a conformation that is differentfrom the one it has in vivo.
The β hairpin in L22 may also be more flexible in solu-tion. However, a small hydrophobic cluster formed by Val85,Leu86, Ile95 and Ile96 and many bulky polar sidechainsexposed to the solvent will stabilize the structure. Judgingby the sequence conservation of the highly twisted part ofthe β structure, it is likely to be a selected feature for itsrole in the ribosome. Thus the β hairpin structure may berestrained to a conformation in solution and in the rRNAcomplex that is not very different from the one observedin the crystal structure.
Structural homologies of L22If one ignores α1 and α2, L22 superposes well onto thefirst split β-α-β motif of S6 [8]. In fact, the most prominent
Research Article Ribosomal protein L22 from Thermus thermophilus Unge et al. 1579
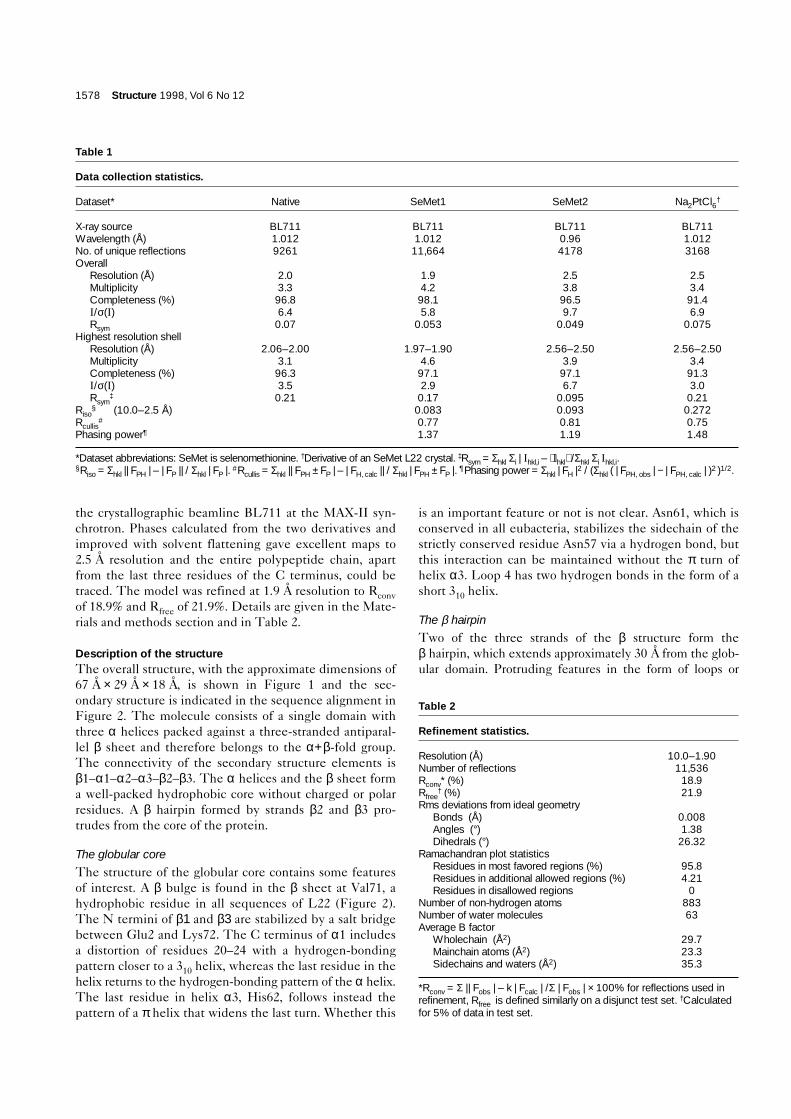
Figure 1
P80
N terminus
P80
N terminus
T100
R90
N60
T100
V10
R90
C terminus
K110
N60
V10
V50
C terminus
K110
Y70
V50
Y70
V20 V20
E30 E30
N40 N40
(a) (b)
Structure
The overall fold of L22. (a) An α-carbon trace of L22 in stereo with every tenth residue marked. The three residues marked in red (residues 82–84)correspond to the three residues deleted in the mutant protein that confers erythromycin resistance. (b) A ribbon presentation in the sameorientation as (a). Figures were produced with MOLSCRIPT [60].
loop in S6 (between β2 and β3), which in the crystal struc-ture is partly folded over the β sheet, coincides topologi-cally with the β hairpin of L22 (Figure 3). This structuralmotif is also found in the small nuclear ribonucleoparticleprotein (snRNP) complexes, as, for instance, in U1A [37].Thus the loop between strand 2 and 3 in U1A, which makes
extensive contacts with the RNA, corresponds topologi-cally to the long β hairpin of L22.
Ribosomal proteins belong to a limited set of folding pat-terns. This could suggest that they have evolved from asmall set of polypeptides of a precursor to the modern
1580 Structure 1998, Vol 6 No 12
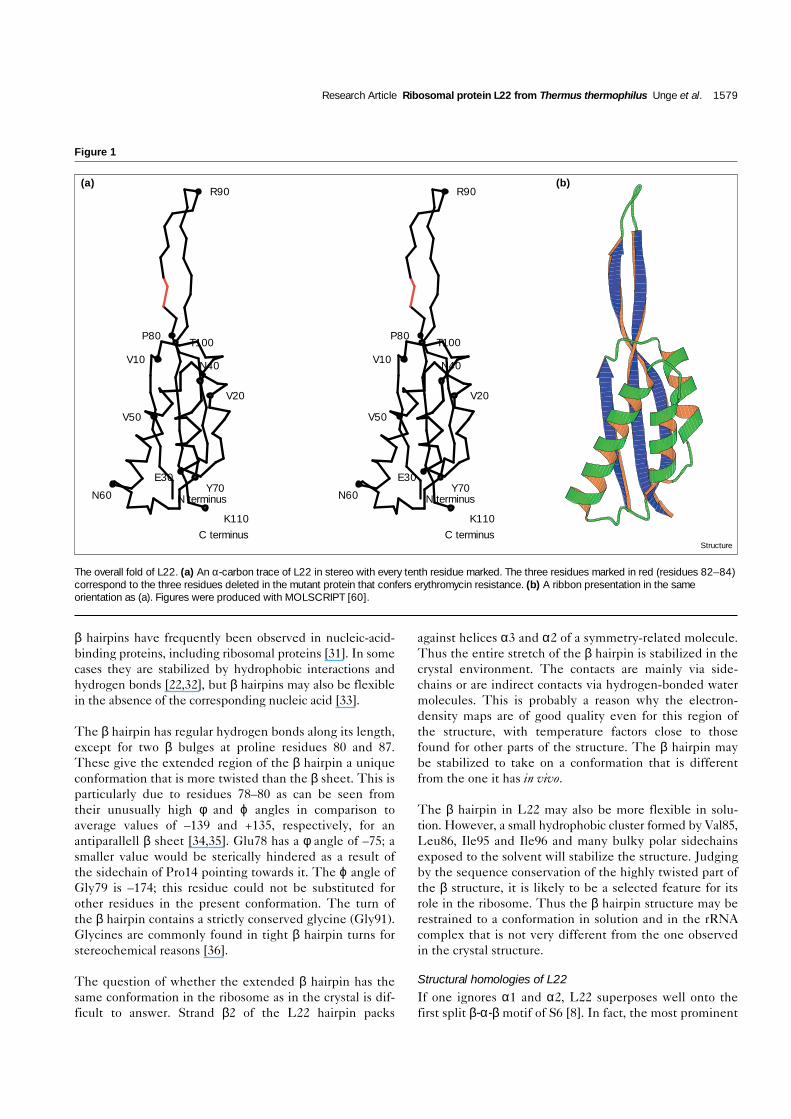
Figure 2
THETH .................MEAKAIARYVRISPRKVRLVVDLIRGKSLEEARNILRYT................................NKRGAYFVAKVL 51 HAEIN .................METIAKHRYARTSAQKARLVADLIRGKKVAQALEILTFT................................NKKAAALVKKVL 51 ECOLI .................METIAKHRHARSSAQKVRLVADLIRGKKVSQALDILTYT................................NKKAAVLVKKVL 51 BACST .................MQAKAVARTVRIAPRKARLVIDLIRGKEVGERFAILRHT................................PKAASPIIEKVL 51 MARPO ..........MQTNTSNKKIRAVAKHIHMSPHKVRRVVSQIRGRSYEQALMILEFM................................PYRACNPILQLL 58 TOBAC ...........MLKKKKTEVYALGEHISMSADKARRVINQIRGRSYEETLMILELM................................PYRACYPILKLI 57 HALHA .....GISYSVD.VDSEASAKAMLRERSISLKHSKAIAREISGETVADAKEYLQAVIDEERSVPFKQHNSGVGHRNDID..GWDAGRYPEKASKDFLKLL 92 METJA MIMMGKLKYKIQ.VNPEKTARAMGRNIPISRKHAREICKSINGMKLDEAIKFLEDVIAMRRPVLFRRHCKKVGHRKGK..LGWPAGRYPVKAAKAILKIL 97 HUMAN .....MVRYSLDPENPTKSCKSRGSNLRVHFKNTRETAQAIKGMHIRKATKYLKDVTLQKQCVPFRRYNGGVGRCAQAKQWGWTQGRWPKKSAEFLLHML 94 RAT .....MVRYSLDPENPTKSCKSRGSNLRVHFKNTRETAQAIKGMHIRKATKYLKDVTLKKQCVPFRRYNGGVGRCAQAKQWGWTQGRWPKKSAEFLLHML 94 THETH ESAAANAVNNHDMLEDRLYVKAAYVDEGPALKRVLPRARGRADIIKKRTSHITVILGEKHGK............ ......................... 113 HAEIN ESAIANAEHNDGADIDDLKVAKIFVDEGPSMKRVMPRAKGRADRILKRTSHITVVVSDR......................................... 110 ECOLI ESAIANAEHNDGADIDDLKVTKIFVDEGPSMKRIMPRAKGRADRILKRTSHITVVVSDR..... ................................... 110 BACST KSAVANAEHNYDMDVNNLVISQAYVDEGPTLKRFRPRAMGRASAINKRTSHITIVVSEKKEG........ ............................. 113 MARPO SSAAANANHNFGLSKTNLFISEIQVNKGTFFKRFQPRAQGRGYPIHKPTCHITIVLNILPK..................................... 119 TOBAC YSAAANASYNMGSSEANLVISKAEVNGGTTVKKLKPRARGRSFPIKRSTCHITIVMKDISLDDEYVEMYSLKKTRWKKKSTAMPYRDMYNSGGLWDKK 155 HALHA SNVSNNADQ.QGFDADEMVIEHVAPHKVGESQGRKPRAMGRATTWNATLCDVEIVVTETEEVTA.................................. 155 METJA QHAKANAEY.KGLNTEKLRIKHISTNKGITIKRYMPRAFGRATPKFQETVHIQVILEEYH...................................... 156 HUMAN KNAESNAEL.KGLDVDSLVIEHIQVNKAPKMRRRTYRAHGRINPYMSSPCHIEMILTEKEQIVPKPEEEVAQKKKISQKKLKKQKLMARE........ 184 RAT KNAESNAEL.KGLDVDSLVIEHIQVNKAPKMRRRTYRAHGRINPYMSSPCHIEMILTEKEQIVPKPEEEVAQKKKISQKKLKKQKLMARE........ 184
(a)
Structure
(b) (c)
Sequence conservation in L22. (a) Alignment of L22 sequencesselected from the three kingdoms: the first four are from eubacteria,two are from chloroplasts, two from archeabacteria and the final twoare from eukaryotes (cytoplasmic). The alignment was performed withthe program PILEUP from the GCG package [61] using 33 sequencesshowing homology to L22. Red boxes highlight positions of strictlyconserved amino acids in eubacterial sequences. Only three aminoacids are strictly conserved in all species and are marked in redthroughout. Yellow boxes indicate amino acids with similar propertiesin eubacteria (I/V, L/M, D/E/N/Q or R/K). The secondary structureelements are indicated above the sequences by arrows (β strands) andcylinders (α helices). Four highly conserved regions are observed: I,
helix α1 and loop2; II, C terminus of helix α3; III, parts of β strands 2and 3 in the protruding β hairpin; IV, the tip of the β hairpin.Abbreviations: THETH, Thermus thermophilus; HAEIN, Haemophilusinfluenzae; ECOLI, Escherichia coli; BACST, Bacillusstearothermophilus; MARPO, Marchantia polymorpha; TOBAC,Nicotiana tabacum; HALHA, Halobacterium halobium; METJA,Methanococcus jannaschii; HUMAN, Homo sapiens; and RAT, Rattusnorvegicus. (b) and (c) Conserved residues at the protein surfacecolor-coded with the same color scheme as in (a). The molecule isviewed from its left and right side in (b) and (c), respectively, ascompared to the orientation in Figure 1. The figures were preparedusing the program GRASP [62].
ribosome [5]. A recurring motif is the split β-α-β motif [38].This motif is structurally homologous to the widespreadRNA-recognition motif (RRM) [39]. The RRM in U1A alsoexists in proteins binding messenger RNA (mRNA), pre-mRNA, nuclear RNAs and small nuclear RNAs [39]. Thewidespread prevalence of the RRM in animal, plant, fungaland bacterial cells in nearly all organelles where RNA ispresent suggests that it is an ancient protein structure withimportant and early evolutionary functions. However, thetwo short consensus sequences RNP1 and RNP2 thatdefine the RRM domain have not been found in L22, norhave they been found in other ribosomal proteins [39].
Functional aspectsSequence conservationFigure 2 presents an alignment of 10 selected L22sequences of the 33 available from procarya, chloroplasts,archaea and eucarya. Invariant residues and conservedresidues with similar chemical properties in eubacteria areclustered in four regions of the protein that consist of:helix α1 and a part of loop 2 (I); the C-terminal half of
helix α3 (II); the base of the extended β hairpin where itemerges from the globular part of the structure (III); andthe tip of the β hairpin (IV). The β hairpin is of a constantlength in all species. It is therefore likely to be an essentialfeature of L22.
Only three residues are invariant in all sequences: Leu36 ispart of the hydrophobic core; Asn57 is located in region II ofthe conserved residues at the C terminus of helix α3, point-ing into the solvent and hydrogen bonded to Asp61; andGly91 is located in the β-hairpin turn (region IV). Asn57may be involved in functional interactions with ribosomalproteins or the 23S RNA. Gly91, which probably is impor-tant for the turn of the β hairpin, is preceded by the β-bulgeresidue Pro87 (conserved in 22 sequences out of 24 fromeubacteria and chloroplasts). Pro14, Gly79 and Pro80 seemto be important for the high local twist of the β hairpin andshow significant conservation as well. Gly79 is conservedin 22 eubacteria and chloroplasts but Pro80 is slightly lessconserved. Pro14 is conserved in 75% of the eubacterialsequences of L22.
Research Article Ribosomal protein L22 from Thermus thermophilus Unge et al. 1581
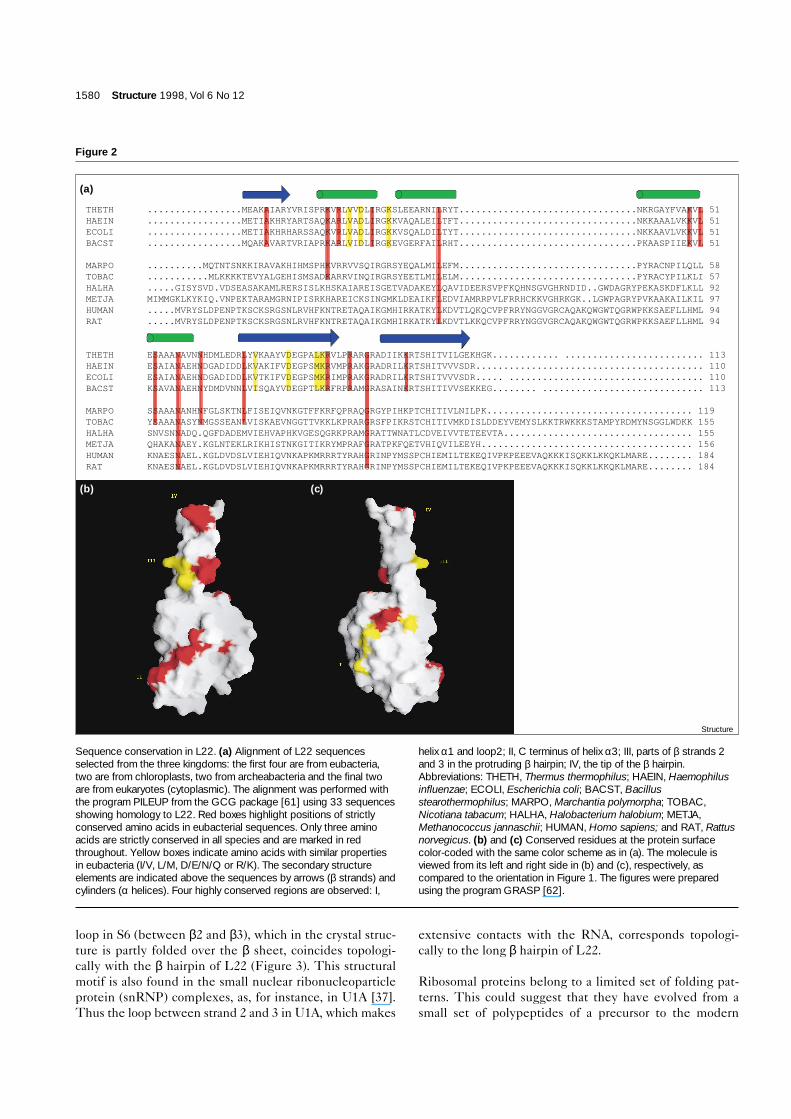
Figure 3
A ribbon diagram of the structures of (a) S6(PDB code 1ris [8]) and (b) L22. The threeβ strands of L22 and one helix thatsuperimpose well on the first split β-α-β unitof S6 are shown in red. The extendedβ hairpin of L22 and the bent loop of S6 aretopologically equivalent.
Structure
(a) (b)

RNA-binding properties
Clusters of positively charged residues, primarily interact-ing with the negatively charged phosphate groups of theRNA, and aromatic residues, which may stack onto thebases of the RNA, contribute frequently to RNA interac-tions in the known structures of nucleic-acid–protein com-plexes. In addition, polar residues can act as specifichydrogen-bond donors and acceptors, whereas hydropho-bic patches of the protein surface may interact withhydrophobic parts of the RNA.
The electrostatic surface potential of L22 shows a posi-tively charged, concave area where the β hairpin extendsfrom the core of the protein (Figure 4). Several residuescontribute to this charged surface: Arg11, Arg15, Lys16,Arg42, Arg84 and Lys98, forming, with additional posi-tively charged residues, a collar around the β hairpin.There is also a positively charged area at the tip of theβ hairpin that is due to arginines 88, 90 and 92.
In L22 from T. thermophilus there are five tyrosines(residues 9, 38, 45, 70, 75) and one phenylalanine (Phe46)in total. These aromatic sidechains are all exposed to thesolvent at the protein surface (Figure 5), but are generallynot well conserved. Two out of the three most conservedtyrosines in eubacteria (Tyr70 and Tyr75) are part of strandβ2. The third tyrosine (Tyr38) is located at the end of α2 ina surface area where there are no obvious indications offunctional interactions. Tyr9 is only conserved in five out of
twelve eubacteria and is located at the conserved patch IIof the β sheet. Several hydrophobic residues, especiallyvalines, isoleucines and leucines, are found on the surfaceof the protein and are partly or fully exposed to solvent(Figure 5). Again, these residues are clustered at the fourconserved regions, including the exposed surface of theβ sheet. In contrast, Tyr45 and Phe46 are present in onlytwo of the twelve available eubacterial sequences. However,residue 46 is a nonpolar residue in all eubacteria. These lesswell conserved features may form additional interactionareas with rRNA or ribosomal proteins on the surface ofL22. If the variation in the types of hydrophobic residue isdue to differences in the RNA environment in the ribo-some or if it is a way of enhancing the thermostability of theribosomes in T. thermophilus is not known.
An analysis of the sequence alignment shows that all thearomatic residues in L22 of other species are found on thesurface of the protein, provided that a given amino acidposition in the T. thermophilus structure has an equivalentposition in the other structures. Seven of these residues arelocated in the β hairpin and three others are located in theβ sheet (the lower part in the figures) and thus contribute tothe surfaces already recognized as being involved in interac-tions. Gly26 is highly conserved. Its mainchain dihedralangles will not permit the accommodation of larger side-chains. Thus its conservation as a glycyl residue is primarilystructural. However, the lack of a sidechain will allow a snugfit for possible interacting protein or RNA molecules.
L22 from eukaryotes and archeabacteria contains a thirty-residue insertion at T. thermophilus residue 39. The highcontent of conserved aromatic and basic residues, mainlyarginines, in this insertion suggests an RNA-binding func-tion. This insertion is located at the end of helix α2 andwould extend outwards, close to the protruding hairpin.Nucleic-acid-binding proteins with two extending loopshave been observed before [33].
Antibiotic resistanceErythromycin inhibits peptidyl transfer once two to fiveresidues have been synthesized [27,40]. L4 and L22 bindto domain I of the 23S RNA [41]. Mutations in domains IIand V of E. coli 23S RNA lead to erythromycin resistance[42,43]. The deletion of Met82–Lys83–Arg84 in E. coliL22 also confers erythromycin resistance to the cells [26].Residues 83 and 84 are well conserved in the knownsequences and are located on one strand of the β hairpin.It should be noted that this deletion occurs in the β strandwhere there are two β bulges at proline residues 80 and 87.Even if the protein could incorporate this deletion withoutany major changes of the mainchain hydrogen bonds, thisdeletion would nevertheless change the twist and stronglydistort the structure of the hairpin. As the β hairpin is pos-tulated to be involved in RNA binding, the mutation couldlead to a local distortion or even larger rearrangement of
1582 Structure 1998, Vol 6 No 12
Figure 4
Electrostatic surface potential distribution of L22 calculated usingGRASP [62]. Positive potential is in blue and negative is in red. Thearea at the stem of the β-hairpin extension, loop 2 and the tip of theβ hairpin show pronounced positive charges. The end of the moleculeopposite the extended β hairpin is primarily negatively charged.
Structure

parts of the RNA. Ribosomes containing the mutated L22bind erythromycin without being inhibited [40]. In addi-tion, affinity labeling experiments with erythromycin deriv-atives have shown a strong labeling of L22 and a weakerlabeling of proteins L2, L4 and L15 [28].
Role of L22 in assembly of 50S subunits and subunitinteractions
As discussed above, a large part of the protein may con-tribute to interactions with the rRNA and ribosomal pro-teins. These interacting regions are helix α1 and loop1,the C terminal half of helix α3 and a large portion of theβ sheet, including the β-hairpin extension. This indicatesthat L22 may make extensive interactions with severalregions of the 23S RNA; this is in line with assemblystudies where L22 is one of the proteins that is importantfor the architecture of the 23S RNA [25]. Erythromycinnot only inhibits the elongation of short peptides, but alsointerferes with the assembly of 50S subunits. This inhibi-tion is significantly reduced in the erythromycin-resistantmutants with altered L4 or L22 proteins [44].
A mutation of the highly conserved Arg8→Cys in E. coliL22 makes the ribosomes temperature sensitive [45]. Ribo-somes with the mutant protein have several functionaldefects at elevated temperatures, loss of subunit re-associ-ation among them [46]. This can obviously be related tothe observation that L22 can be crosslinked to the 16S
RNA [47]. Ribosomes from E. coli cells expressing bothmutant and wild-type L22 contained only wild-type L22at low and high temperatures, indicating a preferentialinclusion of wild-type L22 during assembly [45]. Arg8 issituated at the C-terminal end of β1 opposite the concave,basic surface where the β hairpin emerges from the globu-lar core. It is part of the collar of basic residues that sur-rounds the root of the β hairpin. Thus the β hairpin and itsbase, which probably are intimately involved in interac-tions with 23S RNA, play, directly or indirectly, importantroles in the assembly of the 50S subunit as well as in inter-actions between the subunits.
The chloroplast protein corresponding to L22 has variableN- and C-terminal extensions. Spinach CL22 binds to 5SRNA [48]. Mutants lacking the C-terminal extension werenot affected in binding to 5S RNA. However, if the centralportion of CL22, corresponding to the complete bacterialL22, was removed, the binding capacity for 5S RNA waslost [49]. The suggestion that the structure correspondingto bacterial L22 not only binds to 23S RNA but also to 5SRNA may be premature. The central portion of CL22 mightbe needed to stabilize a binding conformation of the N-ter-minal extension with the 5S RNA. Nevertheless, it is aninteresting observation that L22 is a protein that not onlyplays an important role in the proper folding of 23S RNA,but that it is in close proximity to the 5S RNA [48,49] aswell as to the 16S RNA [47].
Research Article Ribosomal protein L22 from Thermus thermophilus Unge et al. 1583
Figure 5
Stereo diagram showing the hydrophobicresidues that are not part of the internal coreof the structure. These are likely to be part ofan RNA-binding surface. Areas with residuesidechains shown in this figure overlap withareas in Figure 4. Yellow spheres representsulphur atoms and red spheres representoxygen atoms. Stereoviews were prepared inMOLSCRIPT [60].
L65 L65 M1
I6
Y45 F46
M1
Y9
I106
I6
Y70
Y38
Y45 F46
Y9
I35 I106
Y70
L82
Y38
V85
Y75
I35
L82
L23
L86 V85
Y75
L19
I95 I96
L86
L23
I95 I96
L19
Structure

The location and orientation of L22The data identifying the location and orientation of L22 inthe 50S subunit is too limited for definite conclusions to bemade. However, the binding site in the ribosome for ery-thromycin partly overlaps with the binding site for chloram-phenicol, which is an inhibitor of peptidyl transfer [50].Erythromycin resistance can be obtained by modificationsof the peptidyl-transfer loop of domain V of the 23S RNA[42]. Despite this close spatial relationship of erythromycinwith the peptidyl-transfer center, erythromycin does notinhibit peptidyl transfer but inhibits protein synthesis onlywhen a certain number of amino acids have been incorpo-rated into the nascent chain [27,40]. A simple interpretationis that erythromycin blocks the exit route for the nascentpeptide [27,28]. Despite the size of erythromycin and itsreactive derivative, the affinity labeling of L22 by the ery-thromycin derivative indicates that part of the protein is inthe vicinity of the peptidyl-transferase site at the subunit-interface side of the 50S particle [28], possibly close to theexit route for the nascent peptide. The most likely arrange-ment is that the β hairpin is in the vicinity of the ery-thromycin-binding site, as this is where the erythromycin-resistance conferring mutation occurs. On the other hand,combined observations from IEM and protein–proteincrosslinking studies indicate that L22 is centrally locatedclose to the cytoplasmic surface of the 50S particle beneaththe central protuberance — opposite the peptidyl-transfersite at the subunit interface [29,30,51,52]. The negativeelectrostatic potential at the end of the molecule oppositethe β hairpin may indicate that this surface of the protein isnot involved in RNA contacts. Thus the elongated shape ofL22 is compatible with a proximity to the binding site forerythromycin, as well as to the outer or cytoplasmic side ofthe 50S subunit.
Biological implicationsThe synthesis of new proteins in a cell occurs on theribosome, a large ribonucleoprotein complex. The func-tions of many ribosomal proteins are primarily architec-tural, directing the proper folding of ribosomal RNAsand maintaining their functional three-dimensionalstructures. To understand the mechanism underlyingthis complex task, detailed structural information at theatomic level is required. In E. coli, L22 is one of the fiveproteins that bind to the 23S RNA to form the firstintermediate in the assembly of the large subunit. It islocated near the peptidyl-transferase center at the inter-face side of the large subunit, but at the same time it canbe observed to be on the external side of the ribosome.
Here, we present the crystal structure of ribosomalprotein L22 from T. thermophilus at 1.9 Å resolution. Itconsists of a small domain with a long protrudingβ hairpin. Structural analysis of conserved amino acidsand patterns of amino acids on the surface typical ofRNA-interacting proteins give indications of several
rRNA-binding areas. A concave surface where theβ hairpin protrudes from the domain has a particularlyhigh density of basic residues. In addition, highly con-served and exposed hydrophobic residues are clusteredin several regions, which could be of importance forrRNA binding. Consistent with biochemical reconsti-tution studies, this gives the impression of a proteindeeply involved in the binding and folding of the localrRNA structure.
Derivatives of the antibiotic erythromycin can label L22.Furthermore, a deletion of three amino acid residues inE. coli L22 confers resistance to erythromycin. Theeffect of the mutation is that erythromycin can still bindto the ribosomes but not inhibit translation. The muta-tion is located on one strand of the protruding β hairpinstructure. This should affect the hairpin structure pre-sumed to be involved in the rRNA interaction consider-ably. The mutation would indicate that the β hairpin isclosest to the erythromycin-binding site near the pep-tidyl-transfer center. The protein is extended enough toreach the cytoplasmic surface of the large subunit withits globular part.
Materials and methodsCloning, expression and protein purificationThe protein L22 was overexpressed and purified as described else-where [24]. Incorporation of selenomethionione was performedaccording to Van Duyne et al. [53]. The incorporation of selenomethio-nine was tested in a matrix-assisted laser-desorption/ionization (MALDI)mass-spectrometry experiment on a Finnigan LASERMAT instrument.The spectra suggested an almost complete substitution of twomethionines for selenomethionines.
CrystallizationFor the crystallization, 2.0 M ammonium hydrogen phosphate was usedas precipitant at pH 4.5 and approximately 15 mg/ml L22 [24]. Thecrystals display a relatively high sensitivity to changes in pH (crystalscould only be grown within a range of 1 pH unit) and the addition of100 mM MgCl2 facilitated crystal growth. Rod-like crystals grew to amaximum size of 0.8 × 0.2 × 0.2 mm. The space group is P212121 withcell dimensions a = 32.6 Å, b = 66.0 Å, c = 67.7 Å.
Data collectionAll data were collected at beamline BL711 at the MAX-II synchrotron,Lund, Sweden. Data from native L22, a selenomethionine-substitutedL22 and a Na2PtCl6 derivative were collected at a wavelength of1.013 Å and another data set for the selenomethionine-substituted L22was collected at 0.96 Å using a MAR imaging plate. The temperatureat the crystal was approximately 0°. A total rotation of 90° was used foreach data set in 1°-rotation batches. Integration of the data was per-formed with the programs DENZO and SCALEPACK [54].
Phasing and refinementThe selenomethionine and platinum derivatives were used to obtain thephase angles for the native data set using both isomorphous andanomalous differences. The two selenomethionine data sets at differentwavelengths were used as two independent derivatives. Phase-angledetermination, refinement and map calculations were performed usingthe CCP4 [55] suite of programs. The highest peak in the platinum dif-ference Patterson map was solved by manual inspection as well as byusing automatic procedures with the program RSPS [55]. The phaseangles obtained were used in calculation of cross-difference Fourier
1584 Structure 1998, Vol 6 No 12

maps from which one selenium site could be located. MIR phases fromthe two derivatives were improved by solvent flattening. Electron-density maps, at 2.5 Å resolution after this stage, were of excellentquality and permitted chain tracing without ambiguities. The N-terminalselenomethionine could not be found from Patterson or difference-Fourier techniques. However, its position was revealed at the stage ofmodel building from a small peak in difference-Fourier maps. Thesolvent-flattened maps and skeletons produced from it, together withthe sequence marker provided by the major selenium site, made modelbuilding easy and the entire molecule, apart from the last three C-termi-nal residues, could be built into the maps.
Phase extension and refinement to a resolution of 2.0 Å was performedwith the program X-PLOR [56–59], initially with restrained MIR phases.Forty solvent molecules where added into a Fo–Fc map contoured at3σ. The final refinement was carried out using the selenomethioninedata set to 1.9 Å resolution. Bulk solvent correction as well as overallanisotropic B-factor scaling were used with a resulting decrease inRfree. For several amino acid residues the maps clearly showed two dif-ferent sidechain conformations. Residues Ser53 and Arg99 were builtwith two alternative conformations. The final model contains 110 aminoacid residues and 63 water molecules. The present Rfree is 21.9% andthe corresponding Rconv. is 18.9% for data in the resolution range from10 Å to 1.9 Å. The model has excellent stereochemistry, and there areno residues in the disallowed regions of the Ramachandran plot. Thequality of the final electron-density map is demonstrated in Figure 6.
Accession numbersThe coordinates and structure factors for L22 have been depositedwith the Brookhaven Protein Data Bank with accession codes 1bxeand r1bxesf, respectively.
AcknowledgementsWe are grateful to Anders Svensson and Yngve Cerenius for assistance atbeamline BL711 of the MAX-II synchrotron during data collection, and toMats Åkesson and Per Björk at Pharmacia UpJohn, Lund for the mass-spec-trometry measurements. The kind assistance of Pharmacia Upjohn with theFinnigan LASERMAT instrument is very much appreciated. This work wassupported by grants from Swedish Natural Science Research Council andEC Biotechnology (BIO4-CT97-2188), and from Russian Academy of Sci-ences and Russian Foundation for Basic Research. In addition thesestudies were supported in part by the International Research Scholar’saward of the Howard Hughes Medical Institute to MG and AL.
References1. Mueller, F. & Brimacombe, R. (1997). A new model for the three-
dimensional folding of Escherichia coli 16S ribosomal RNA. I. Fittingthe RNA to a 3D electron microscopic map at 20 Å. J. Mol. Biol. 271,524-544.
2. Frank, J., et al., & Agrawal, R.K. (1995). A model of protein synthesisbased on cryo-electron microscopy of the E. coli ribosome. Nature376, 441-444.
3. Stark, H., et al., & van Heel, M. (1995). The 70S Escherichia coliribosome at 23 Å resolution: fitting the ribosomal RNA. Structure 3,815-821.
4. Ban, N., et al., & Steitz, T. (1998). A 9 Å resolution X-ray crystallographicmap of the large ribosomal subunit. Cell 93, 1105-1115.
5. Ramakrishnan, V. & White, S.W. (1998) Ribosomal protein structures:insights into the architecture, machinery and evolution of theribosome. Trends Biochem. Sci. 23, 208-212.
6. Bycroft, M., Hubbard, T.J., Proctor, M., Freund, S.M. & Murzin, A.G.(1997). The solution structure of the S1 RNA binding domain: amember of an ancient nucleic acid-binding fold. Cell 88, 235-242.
7. Ramakrishnan, V. & White, S.W. (1992). The structure of ribosomalprotein S5 reveals sites of interaction with 16S rRNA. Nature 358,768-771.
8. Lindahl, M., et al., & Amons, R. (1994). Crystal structure of the ribosomalprotein S6 from Thermus thermophilus. EMBO J. 13, 1249-1254.
9. Wimberly, B., White, S.W. & Ramakrishnan, V. (1997). The structureof ribosomal protein S7 at 1.9 Å resolution reveals a β-hairpin motifthat binds double-stranded nucleic acids. Structure 5, 1187-1198.
10. Hosaka, H., et al., & Wakatsuki, S. (1997). Ribosomal protein S7: anew RNA-binding motif with structural similarities to a DNAarchitectural factor. Structure 5, 1199-1208.
11. Davies, C., Ramakrishnan, V. & White, S.W. (1996). Structuralevidence for specific S8–RNA and S8–protein interactions within the30S ribosomal subunit: ribosomal protein S8 from Bacillusstearothermophilus at 1.9 Å resolution. Structure 4, 1093-1104.
12. Nevskaya, N., et al., & Nikonov, S. (1998). Crystal structure ofribosomal protein S8 from Thermus thermophilus reveals a highdegree of structural conservation of a specific RNA binding site. J. Mol. Biol. 279, 233-244.
13. Berglund, H., Rak, A., Serganov, M., Garber, M. & Härd, T. (1997).Solution structure of the ribosomal RNA binding protein S15 fromThermus thermophilus. Nat. Struct. Biol. 4, 20-23.
14. Clemons, W.M., Davies, C., White, S.W. & Ramakrishnan, V. (1998)Conformational variability of the N-terminal helix in the structure ofribosomal protein S15. Structure 6, 429-438.
15. Jaishree, T.N., Ramakrishnan, V. & White, S.W. (1996). Solutionstructure of prokaryotic ribosomal protein S17 by high-resolution NMRspectroscopy. Biochemistry 35, 2845-2853.
Research Article Ribosomal protein L22 from Thermus thermophilus Unge et al. 1585
Figure 6
A stereoview of part of the electron-densitymap calculated from the final model phases.
Structure

16. Nikonov, S., et al., & Liljas, A. (1996). Crystal structure of the RNAbinding ribosomal protein L1 from Thermus thermophilus. EMBO J.15, 1350-1359.
17. Golden, B.L., Ramakrishnan, V. & White, S.W. (1993). Ribosomalprotein L6: structural evidence of gene duplication from a primitiveRNA binding protein. EMBO J. 12, 4901-4908.
18. Leijonmarck, M. & Liljas, A. (1987). Structure of the C-terminal domainof the ribosomal protein L7/L12 from Escherichia coli at 1.7 Å. J. Mol.Biol. 195, 555-579.
19. Bocharov, E.W., Gudkov, A.T. & Arseniev, A.S. (1996). Topology ofthe secondary structure of ribosomal protein L7/L12 from E. coli insolution. FEBS Lett. 379, 291-294.
20. Hoffman, D.W., Cameron, C.S., Davies, C., White, S.W. &Ramakrishnan, V. (1996). Ribosomal protein L9: a structuredetermination by the combined use of X-ray crystallography and NMRspectroscopy. J. Mol. Biol. 264, 1058-1071.
21. Hinck, A.P., et al., & Torchia, D.A. (1997). The RNA binding domain ofribosomal protein L11: Three-dimensional structure of the RNA boundform of the protein and its interaction with 23S RNA. J. Mol. Biol. 274,101-113.
22. Davies, C., White, S.W. & Ramakrishnan, V. (1996). The crystalstructure of ribosomal protein L14 reveals an important organizationalcomponent of the translational apparatus. Structure 4, 55-66.
23. Wilson, K.S., Appelt, K., Badger, J., Tanaka, I. & White, S.W. (1986).Crystal structure of a prokaryotic ribosomal protein. Proc. Natl Acad.Sci. USA 83, 7251-7255.
24. Davydova, N.L., et al., & Garber, M.B. (1995). Ribosomal protein L22from Thermus thermophilus: sequencing, overexpression andcrystallisation. FEBS Lett. 369, 229-232.
25. Nierhaus, K. (1990). Reconstitution of ribosomes. In Ribosomes andProtein Synthesis. (Spedding, G., ed.), pp. 161-189, IRL PRESS,Oxford/New York/Tokyo.
26. Chittum, H.S. & Champney, W.S. (1994). Ribosomal protein genesequence changes in erythromycin-resistant mutants of Escherichiacoli. J. Bacteriol. 176, 6192-6198.
27. Contreras, A. & Vazquez, D. (1977). Cooperative and antagonisticinteractions of peptidyl-tRNA and antibiotics with bacterial ribosomes.Eur. J. Biochem. 74, 539-547.
28. Arevalo, M.A., Tejedor, F., Polo, F. & Ballesta, J.P.G. (1988). Proteincomponents of the erythromycin binding site in bacterial ribosomes. J. Biol. Chem. 263, 58-63.
29. Walleczek, J., Schuler, D., Stöffler-Meilicke, M., Brimacombe, R. &Stöffler, G. (1988). A model for the spatial arrangement of theproteins in the large subunit of the Escherichia coli ribosome. EMBOJ. 7, 3571-3576.
30. Tischendorf, G.W., Zeichhardt, H. & Stöffler, G. (1974). Determinationof the location of proteins L14, L17, L19, L22, L23 on the surface ofthe 50S ribosomal subunit of Escherichia coli by immune electronmicroscopy. Mol. Gen. Genet. 134, 187-208.
31. Liljas, A. & Al-Karadaghi, S. (1997). Structural aspects of proteinsynthesis. Nat. Struct. Biol. 4, 767-771.
32. Muñoz, V., Thompson, P.A., Hofrichter, J. & Eaton, W.A. (1997). Foldingdynamics and mechanism of β-hairpin formation. Nature 390, 196-199.
33. Boelens, R., Vis, H., Vorgias, C.E., Wilson, K.S., & Kaptein, R. (1996).Structure and dynamics of the DNA binding protein HU from Bacillusstearothermophilus by NMR spectroscopy. Biopolymers 40, 553-559.
34. Arnott, S., Dover, S.D. & Elliott, A. (1967). Structure of beta-poly-L-alanine: refined atomic co-ordinates for an anti-parallel beta-pleatedsheet. J. Mol. Biol. 30, 201-208.
35. Ramachandran, G.N. & Sasisekharan, V. (1968). Conformations ofpolypeptides and proteins. Adv. Protein Chem. 23, 283-437.
36. Crawford, J.L., Lipscomb, W.N. & Schellman, C.G. (1973). Thereverse turn as a polypeptide conformation in globular proteins. Proc.Natl Acad. Sci. USA 70, 538-542.
37. Oubridge, C., Ito, N., Evans, P.R., Teo, C.-H. & Nagai, K. (1994).Crystal structure at 1.92Å resolution of the RNA-binding domain ofthe U1A spliceosomal protein complexed with an RNA hairpin. Nature372, 432-438.
38. Orengo, C.A. & Thornton, J.M. (1993). Alpha plus beta folds revisited:some favoured motifs. Structure 1, 105-120.
39. Burd, C.G. & Dreyfuss, G. (1994). Conserved structures and diversityof functions of RNA-binding proteins. Science 265, 615-621.
40. Cooperman, B.S., Weitzmann, C.J. & Fernández, C.L. (1990).Antibiotic probes of Escherichia coli ribosomal peptidyltransferase. InThe Ribosome, Structure Function and Evolution. (Hill, W., Dahlberg,A., Garrett, R.A., Moore, P.B., Schlessinger, D. & Warner, J.R., eds.),pp. 491-501, American Society for Microbiology, Washington D.C.
41. Liiv, A., Tenson, T. & Remme, J. (1996). Analysis of the ribosome largesubunit assembly and 23 S rRNA stability in vivo. J. Mol. Biol. 263,396-410.
42. Sigmund, C.D. & Morgan, E.A. (1982). Erythromycin resistance due toa mutation in a ribosomal RNA operon of Escherichia coli. Proc. NatlAcad. Sci. USA 79, 5602-5606.
43. Dam, M., Douthwaite, S., Tenson, T. & Mankin, A.S. (1996). Mutationsin domain II of 23 S rRNA facilitate translation of a 23S rRNA-encoded pentapeptide conferring erythromycin resistance. J. Mol.Biol. 259, 1-6.
44. Chittum, H.S. & Champney, W.S. (1995). Erythromycin inhibits theassembly of the large ribosomal subunit in growing Escherichia colicells. Curr. Microbiol. 30, 273-279.
45. Burnette-Vick, B., Champney, W.S. & Musich, P.R. (1994). Atemperature sensitive mutant of Escherichia coli with an alteration inribosomal protein L22. Genetica 94, 17-25.
46. Champney, W.S. (1989). Protein synthesis defects in temperature-sensitive mutants of Escherichia coli with altered ribosomal proteins.Biochim. Biophys. Acta 609, 464-474.
47. Chiam, C.L. & Wagner, R. (1983). Composition of the Escherichia coliribosomal interface: a cross-linking study. Biochemistry 22, 1193-1200.
48. Toukifimpa, R., Romby, P., Rozier, C., Ehresmann, C., Ehresmann, B. &Mache, R. (1989). Characterization and footprint analysis of two 5SrRNA binding proteins from spinach chloroplast ribosomes.Biochemistry 28, 5840-5846.
49. Carol, P., Rozier, C., Lazaro, E., Ballesta, J.P.G. & Mache, R. (1993).Erythromycin and 5S rRNA binding properties of the spinachchloroplast ribosomal protein CL22. Nucleic Acids Res. 21, 635-639.
50. Cundliffe, E. (1985) Involvement of specific portions of ribosomal RNAin defined ribosomal functions: a study utilizing antibiotics. InStructure, Function and Genetics of Ribosomes (Hardesty, B. &Kramer, G. eds), pp. 586-604, Springer-Verlag, New York/Berlin.
51. Stöffler-Meilicke, M. & Stöffler, G. (1990). Topography of theribosomal proteins from Escherichia coli within the intact subunits asdetermined by immunelectron microscopy and protein-proteincrosslinking. In The Ribosome, Structure Function and Evolution. (Hill,W., Dahlberg, A., Garrett, R.A., Moore, P.B., Schlessinger, D. &Warner, J.R., eds.), pp. 123-133, American Society for Microbiology,Washington D.C.
52. Morrison, C.A., Tischendorf, G., Stoffler, G. & Garrett, R.A. (1977).Accessibility of proteins in 50S ribosomal subunits of Escherichia colito antibodies: an ultracentrifugation study. Mol. Gen. Genet. 151,245-252.
53. Van Duyne, G.D., Standaert, R.F., Karplus, P.A., Schreiber, S.L. &Clardy, J. (1993). Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229, 105-124.
54. Otwinowski, Z. & Minor, W. (1997). Processing of X-ray diffractiondata collected in oscillation mode. Methods Enzymol. 276, 307-326.
55. Collaborative Computational Project (1994). The CCP4 suite:programs for protein crystallography. Acta Cryst. D 50, 760-763.
56. Brünger, A.T., Kuriyan, J. & Karplus, M. (1987). Crystallographic Rfactor refinement by molecular dynamics. Science 235, 458-460.
57. Brünger, A.T., Krukowski, A. & Erickson, J.W. (1990). Slow-coolingprotocols for crystallographic refinement by simulated annealing. ActaCryst. A 46, 585-593.
58. Brünger, A.T. (1992). The free R value: a novel statistical quantity forassessing the accuracy of crystal structures. Nature 355, 472-474.
59. Brünger, A.T. (1992). X-PLOR v 3.1 manual. Yale University, NewHaven, USA.
60. Kraulis, P. (1991). MOLSCRIPT: a program to produce both detailedand schematic plots of protein structures. J. Appl. Cryst. 24, 946-950.
61. (GCG) Wisconsin Package Version 9.0. Madison. USA.62. Nicholls, A., Sharp, K. & Honig, B. (1991). Protein folding and
association: insights from the interfacial and thermodynamicproperties of hydrocarbons. Proteins 11, 281-296.
1586 Structure 1998, Vol 6 No 12
Copyright © 2022 FDOKUMEN




















