
Structures of two natural product methyltransferases reveal the basis for substrate specificity in...
9
articles Methylation of oxygen (O-methylation), nitrogen (N-methylation) and carbon (C-methylation) is a universal process critical to all organisms. In plants, the O-methyl- ation patterns of polyhydroxylated small molecules are cru- cial to determining final product distribution via multiple branched biosynthetic pathways using the same or similar intermediates and substrates. The secondary metabolic pathway of phenylpropanoid biosynthesis uses cinnamate and acetate units to construct a diverse set of hydroxylated, polycyclic aromatic compounds, which are used for regula- tory, structural and functional purposes in plants, including protection against UV photodamage, pigmentation, fertil- ization, signaling, gene induction 1,2 , antimicrobial defense 3,4 , chemoattraction 5 and structural support 6 . Site specific methylation of flavonoid and isoflavonoid derivatives modu- lates their in vivo activity by limiting the number of reactive hydroxyl groups, altering the solubility properties of the nature structural biology • volume 8 number 3 • march 2001 271 Structures of two natural product methyltransferases reveal the basis for substrate specificity in plant O-methyltransferases Chloe Zubieta 1 , Xian-Zhi He 2 , Richard A. Dixon 2 and Joseph P. Noel 1 Chalcone O-methyltransferase (ChOMT) and isoflavone O-methyltransferase (IOMT) are S-adenosyl-L-methionine (SAM) dependent plant natural product methyltransferases involved in secondary metabolism in Medicago sativa (alfalfa). Here we report the crystal structure of ChOMT in complex with the product S-adenosyl-L-homocysteine and the substrate isoliquiritigenin (4,2′,4′-trihydroxychalcone) refined to 1.8 Å as well as the crystal structure of IOMT in complex with the products S-adenosyl-L-homocysteine and isoformononetin (4′-hydroxy-7- methoxyisoflavone) refined to 1.4 Å. These two OMTs constitute the first plant methyltransferases to be structurally characterized and reveal a novel oligomerization domain and the molecular determinants for substrate selection. As such, this work provides a structural basis for understanding the substrate specificity of the diverse family of plant OMTs and facilitates the engineering of novel activities in this extensive class of natural product biosynthetic enzymes. Fig. 1 Minimal phenylpropanoid biosynthetic pathway in M. sativa L (alfalfa) 48 . Carbon flow begins in primary metabolism with phenyl- alanine, which ultimately serves as the building block for a diverse class of plant secondary metabolites. The enzymes depicted are: PAL, phenylalanine-ammonia lyase; CA4H, cinnamic acid 4-hydroxylase; 4CL, 4-coumarate:coenzyme A ligase; CHS, chalcone synthase; CHR, chalcone reductase; ChOMT, 4,2′,4′-trihydroxychalcone 2′-hydroxyl- O-methyltransferase; CHI, chalcone isomerase; IFS, isoflavone syn- thase; IOMT, isoflavone-O-methyltransferase (isoflavanone-O- methyltransferase); IFOH, isoflavone 2′-hydroxylase; IFR, isoflavone reductase; and PTS, pterocarpan synthase. The reaction depicted in the solid black box occurs in vitro and likely represents a cryptic activity of IOMT, which normally would methylate an isoflavanone intermediate. The depicted dehydration step can spontaneously occur in solution over time or is catalyzed by a specific dehydratase enzyme. A-rings are derived from the head-to-tail condensation of malonyl-CoA derived acetyl groups and the B-rings are derived from the p-coumaryl moiety. 1 Structural Biology Laboratory, The Salk Institute for Biological Studies, 10010 North Torrey Pines Road, La Jolla, California 92037, USA. 2 Plant Biology Division, Samuel Roberts Noble Foundation, P.O. Box 2180, Ardmore, Oklahoma 73402, USA. Correspondence should be addressed to J.P.N. email: [email protected] © 2001 Nature Publishing Group http://structbio.nature.com © 2001 Nature Publishing Group http://structbio.nature.com
-
Upload
grenoble-univ -
Category
Documents
-
view
1 -
download
0
Transcript of Structures of two natural product methyltransferases reveal the basis for substrate specificity in...

articles
Methylation of oxygen (O-methylation), nitrogen (N-methylation) and carbon (C-methylation) is a universalprocess critical to all organisms. In plants, the O-methyl-ation patterns of polyhydroxylated small molecules are cru-cial to determining final product distribution via multiplebranched biosynthetic pathways using the same or similarintermediates and substrates. The secondary metabolicpathway of phenylpropanoid biosynthesis uses cinnamateand acetate units to construct a diverse set of hydroxylated,polycyclic aromatic compounds, which are used for regula-tory, structural and functional purposes in plants, includingprotection against UV photodamage, pigmentation, fertil-ization, signaling, gene induction1,2, antimicrobial defense3,4,chemoattraction5 and structural support6. Site specificmethylation of flavonoid and isoflavonoid derivatives modu-lates their in vivo activity by limiting the number of reactivehydroxyl groups, altering the solubility properties of the
nature structural biology • volume 8 number 3 • march 2001 271
Structures of two natural productmethyltransferases reveal the basis for substrate specificity in plant O-methyltransferasesChloe Zubieta1, Xian-Zhi He2, Richard A. Dixon2 and Joseph P. Noel1
Chalcone O-methyltransferase (ChOMT) and isoflavone O-methyltransferase (IOMT) are S-adenosyl-L-methionine(SAM) dependent plant natural product methyltransferases involved in secondary metabolism in Medicago sativa(alfalfa). Here we report the crystal structure of ChOMT in complex with the product S-adenosyl-L-homocysteineand the substrate isoliquiritigenin (4,2′,4′-trihydroxychalcone) refined to 1.8 Å as well as the crystal structure ofIOMT in complex with the products S-adenosyl-L-homocysteine and isoformononetin (4′-hydroxy-7-methoxyisoflavone) refined to 1.4 Å. These two OMTs constitute the first plant methyltransferases to bestructurally characterized and reveal a novel oligomerization domain and the molecular determinants forsubstrate selection. As such, this work provides a structural basis for understanding the substrate specificity ofthe diverse family of plant OMTs and facilitates the engineering of novel activities in this extensive class ofnatural product biosynthetic enzymes.
Fig. 1 Minimal phenylpropanoid biosynthetic pathway in M. sativa L(alfalfa)48. Carbon flow begins in primary metabolism with phenyl-alanine, which ultimately serves as the building block for a diverseclass of plant secondary metabolites. The enzymes depicted are: PAL,phenylalanine-ammonia lyase; CA4H, cinnamic acid 4-hydroxylase;4CL, 4-coumarate:coenzyme A ligase; CHS, chalcone synthase; CHR,chalcone reductase; ChOMT, 4,2′,4′-trihydroxychalcone 2′-hydroxyl-O-methyltransferase; CHI, chalcone isomerase; IFS, isoflavone syn-thase; IOMT, isoflavone-O-methyltransferase (isoflavanone-O-methyltransferase); IFOH, isoflavone 2′-hydroxylase; IFR, isoflavonereductase; and PTS, pterocarpan synthase. The reaction depicted inthe solid black box occurs in vitro and likely represents a crypticactivity of IOMT, which normally would methylate an isoflavanoneintermediate. The depicted dehydration step can spontaneouslyoccur in solution over time or is catalyzed by a specific dehydrataseenzyme. A-rings are derived from the head-to-tail condensation ofmalonyl-CoA derived acetyl groups and the B-rings are derived fromthe p-coumaryl moiety.
1Structural Biology Laboratory, The Salk Institute for Biological Studies, 10010 North Torrey Pines Road, La Jolla, California 92037, USA. 2Plant Biology Division, SamuelRoberts Noble Foundation, P.O. Box 2180, Ardmore, Oklahoma 73402, USA.
Correspondence should be addressed to J.P.N. email: [email protected]
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://s
tru
ctb
io.n
atu
re.c
om
© 2001 Nature Publishing Group http://structbio.nature.com

articles
resulting products and ultimately determining whether a partic-ular small molecule will interact with cellular receptors.
Plant small molecule O-methyltransferases (OMTs) use S-adenosyl-L-methionine (SAM) as a methyl source, yielding S-adenosyl-L-homocysteine (SAH) and the methyl ether deriva-tives as products. This family of enzymes must conserve SAMbinding while affording a sufficient degree of active site diversity tobind and correctly position a variety of disparate small molecules.Substrate discrimination by these plant OMTs is considerablegiven that plants synthesize several thousand phenylpropanoidcompounds often with multiple hydroxyl groups7.
Chalcone O-methyltransferase (ChOMT), found in Medicagosativa L (alfalfa), methylates the 2′-hydroxyl of isoliquiritigenin(4,2′,4′-trihydroxychalcone), converting it to 4,4′-dihydroxy-2′-methoxychalcone (Fig. 1). This latter compound serves as apotent nodulation (nod) gene inducer of soil rhizobia8,9. Amongthe diverse compounds released from alfalfa roots, 4,4′-di-hydroxy-2′-methoxychalcone acts as the most efficient tran-scriptional activator of nod genes, activating nodABC throughinteraction with the transcriptional regulators nodD1 and nodD2of Rhizobium meliloti8. Additionally, ChOMT-mediated methy-lation of isoliquiritigenin prevents the chalcone isomerase (CHI)catalyzed cyclization of isoliquiritigenin to the flavanone liquirit-igenin (7,4′-dihydroxyflavanone)10. Subsequent reactions con-vert flavanones into a variety of structurally diverse naturalproducts, including anthocyanins, flavonols, pterocarpans,flavones and isoflavones.
272 nature structural biology • volume 8 number 3 • march 2001
Isoflavone O-methyltransferase (IOMT)is essential for the biosynthesis ofmedicarpin, the major phytoalexin of alfal-fa11. In vivo studies demonstrate that IOMTis necessary for the formation of for-mononetin. In vitro assays using daidzeinas substrate and in vivo studies conductedin the absence of fungal elicitation ofIOMT-overexpressing plants yield thecompound isoformononetin12. This com-pound is rarely found in plants and has noknown biological role in plant physiology.However, when elicited with CuCl2 orinfection with Phoma medicaginis, IOMT-overexpressing plants accumulate the 4′-O-methylated isoflavonoid formononetin andthe downstream phytoalexin derived fromit, medicarpin (Fig. 1).
Here we report the X-ray crystal struc-tures of ChOMT (Table 1) and IOMT
(Table 2), two SAM dependent OMTs from M. sativa L. ChOMTand IOMT are 40 kDa proteins and exist as homodimers in solu-tion. These methyltransferases possess SAM binding domains thatalign structurally with characterized viral13, bacterial14–20, archae-bacterial21 and mammalian22–24 OMTs. The fold of the catalyticSAM binding domain is conserved throughout all classes of SAM-dependent methyltransferases25–27. Unique features of plant O-methyltransferases include the presence of a second domaininvolved in dimerization and the contribution of the dimer inter-face to the substrate binding site. The structures presented here incomplex with substrates and products reveal a characteristicmechanism for methyl transfer by plant OMTs. Furthermore,these studies provide the first structural understanding of sub-strate discrimination displayed by the large family of plant OMTs.
Overall structures of ChOMT and IOMTRecombinant proteins were expressed in Escherichia coli as N-ter-minal polyhistidine-tagged proteins and purified by Ni+2 affinitychromatography and gel filtration. ChOMT and IOMT possessedspecific activities comparable to published values9,11. Both ChOMTand IOMT were crystallized from polyethylene glycol (PEG) solu-tions in the presence of a two-fold molar excess of SAM or SAH.Structures of ChOMT and IOMT were determined with seleno-methionine (SeMet) substituted proteins using multiwavelengthanomalous dispersion (MAD) phasing. Additional structures ofsubstrate and product complexes were determined using differenceFourier analysis based on the SeMet derived structures (Fig. 2a–c).
Fig. 2 Architecture of the ChOMT and IOMTmonomers. a, Cα traces of the backbones of theChOMT and IOMT monomers. Every 20th Cαatom is numbered and the N-termini and C-ter-mini are labeled. The disordered loop inChOMT between residues 160 and 173 is shownas a dashed coil. b, Stereo view of the final SIGMAA weighted 2|Fo - Fc| electron densitymap of the ChOMT active site encompassingthe bound SAH and isoliquiritigenin (two con-formations shown) molecules. Putative hydro-gen bonds are shown as dashed greencylinders. Single letter amino acid code is used.The map is contoured at 1.5 σ. c, Stereo view ofthe final SIGMAA weighted 2|Fo - Fc| electrondensity map of the IOMT active site encompass-ing the bound SAH and isoformononetin mole-cules. The map is contoured at 1.5 σ.
a
b
c
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://s
tru
ctb
io.n
atu
re.c
om
© 2001 Nature Publishing Group http://structbio.nature.com
articles
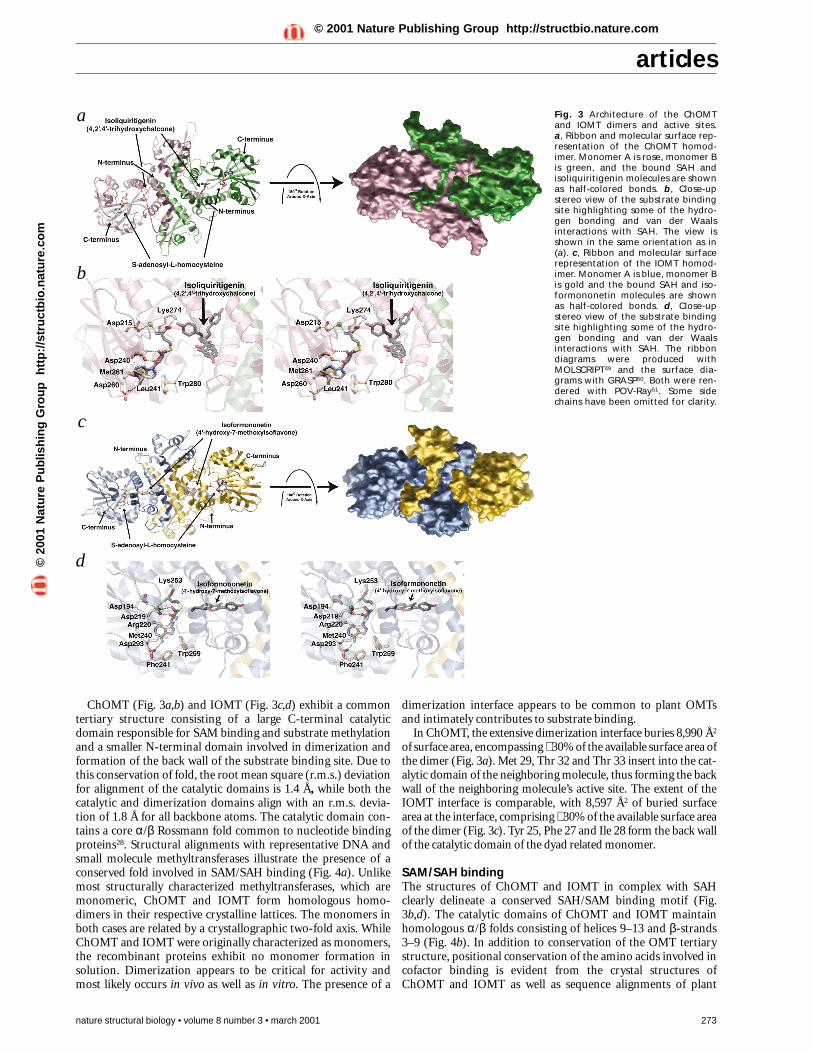
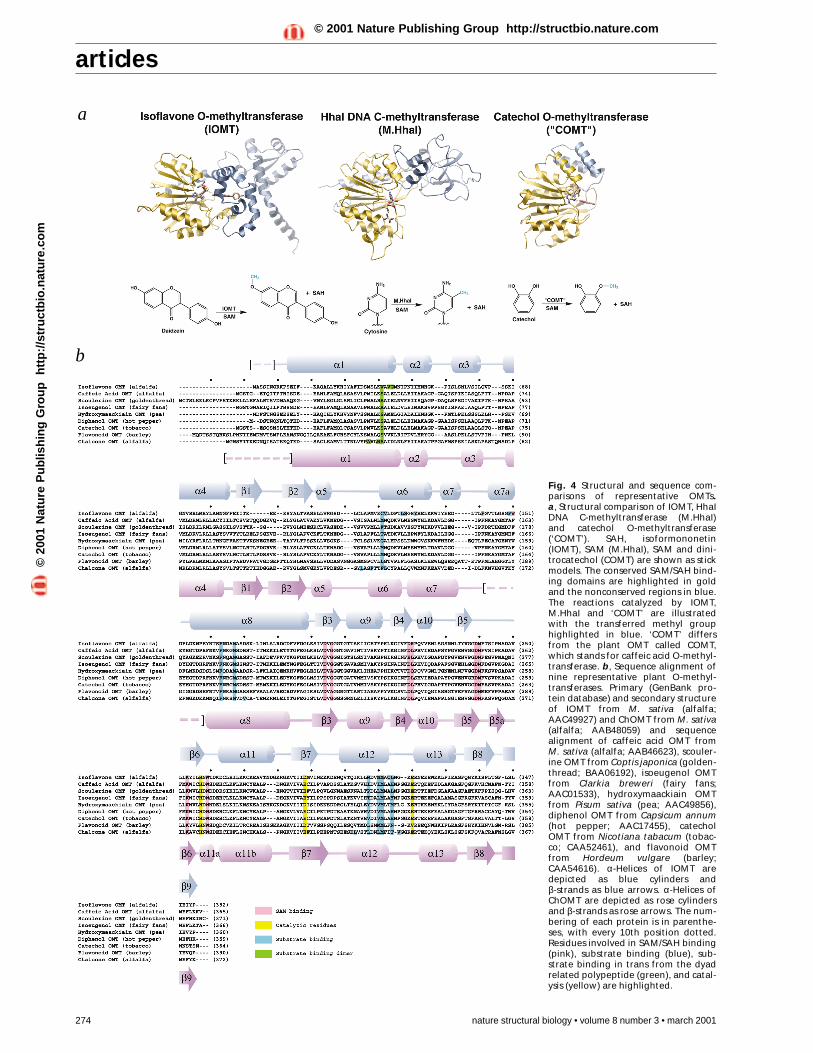
ChOMT (Fig. 3a,b) and IOMT (Fig. 3c,d) exhibit a commontertiary structure consisting of a large C-terminal catalyticdomain responsible for SAM binding and substrate methylationand a smaller N-terminal domain involved in dimerization andformation of the back wall of the substrate binding site. Due tothis conservation of fold, the root mean square (r.m.s.) deviationfor alignment of the catalytic domains is 1.4 Å, while both thecatalytic and dimerization domains align with an r.m.s. devia-tion of 1.8 Å for all backbone atoms. The catalytic domain con-tains a core α/β Rossmann fold common to nucleotide bindingproteins28. Structural alignments with representative DNA andsmall molecule methyltransferases illustrate the presence of aconserved fold involved in SAM/SAH binding (Fig. 4a). Unlikemost structurally characterized methyltransferases, which aremonomeric, ChOMT and IOMT form homologous homo-dimers in their respective crystalline lattices. The monomers inboth cases are related by a crystallographic two-fold axis. WhileChOMT and IOMT were originally characterized as monomers,the recombinant proteins exhibit no monomer formation insolution. Dimerization appears to be critical for activity andmost likely occurs in vivo as well as in vitro. The presence of a
nature structural biology • volume 8 number 3 • march 2001 273
dimerization interface appears to be common to plant OMTsand intimately contributes to substrate binding.
In ChOMT, the extensive dimerization interface buries 8,990 Å2
of surface area, encompassing ∼ 30% of the available surface area ofthe dimer (Fig. 3a). Met 29, Thr 32 and Thr 33 insert into the cat-alytic domain of the neighboring molecule, thus forming the backwall of the neighboring molecule’s active site. The extent of theIOMT interface is comparable, with 8,597 Å2 of buried surfacearea at the interface, comprising ∼ 30% of the available surface areaof the dimer (Fig. 3c). Tyr 25, Phe 27 and Ile 28 form the back wallof the catalytic domain of the dyad related monomer.
SAM/SAH bindingThe structures of ChOMT and IOMT in complex with SAHclearly delineate a conserved SAH/SAM binding motif (Fig.3b,d). The catalytic domains of ChOMT and IOMT maintainhomologous α/β folds consisting of helices 9–13 and β-strands3–9 (Fig. 4b). In addition to conservation of the OMT tertiarystructure, positional conservation of the amino acids involved incofactor binding is evident from the crystal structures ofChOMT and IOMT as well as sequence alignments of plant
Fig. 3 Architecture of the ChOMTand IOMT dimers and active sites. a, Ribbon and molecular surface rep-resentation of the ChOMT homod-imer. Monomer A is rose, monomer Bis green, and the bound SAH andisoliquiritigenin molecules are shownas half-colored bonds. b, Close-upstereo view of the substrate bindingsite highlighting some of the hydro-gen bonding and van der Waalsinteractions with SAH. The view isshown in the same orientation as in(a). c, Ribbon and molecular surfacerepresentation of the IOMT homod-imer. Monomer A is blue, monomer Bis gold and the bound SAH and iso-formononetin molecules are shownas half-colored bonds. d, Close-upstereo view of the substrate bindingsite highlighting some of the hydro-gen bonding and van der Waalsinteractions with SAH. The ribbondiagrams were produced withMOLSCRIPT49 and the surface dia-grams with GRASP50. Both were ren-dered with POV-Ray51. Some sidechains have been omitted for clarity.
a
b
c
d©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://s
tru
ctb
io.n
atu
re.c
om
© 2001 Nature Publishing Group http://structbio.nature.com
articles
Fig. 4 Structural and sequence com-parisons of representative OMTs. a, Structural comparison of IOMT, HhaIDNA C-methyltransferase (M.HhaI)and catechol O-methyltransferase(‘COMT’). SAH, isoformononetin(IOMT), SAM (M.HhaI), SAM and dini-trocatechol (COMT) are shown as stickmodels. The conserved SAM/SAH bind-ing domains are highlighted in goldand the nonconserved regions in blue.The reactions catalyzed by IOMT,M.HhaI and ‘COMT’ are illustratedwith the transferred methyl grouphighlighted in blue. ‘COMT’ differsfrom the plant OMT called COMT,which stands for caffeic acid O-methyl-transferase. b, Sequence alignment ofnine representative plant O-methyl-transferases. Primary (GenBank pro-tein database) and secondary structureof IOMT from M. sativa (alfalfa;AAC49927) and ChOMT from M. sativa(alfalfa; AAB48059) and sequencealignment of caffeic acid OMT from M. sativa (alfalfa; AAB46623), scouler-ine OMT from Coptis japonica (golden-thread; BAA06192), isoeugenol OMTfrom Clarkia breweri (fairy fans;AAC01533), hydroxymaackiain OMTfrom Pisum sativa (pea; AAC49856),diphenol OMT from Capsicum annum(hot pepper; AAC17455), catecholOMT from Nicotiana tabacum (tobac-co; CAA52461), and flavonoid OMTfrom Hordeum vulgare (barley;CAA54616). α-Helices of IOMT aredepicted as blue cylinders and β-strands as blue arrows. α-Helices ofChOMT are depicted as rose cylindersand β-strands as rose arrows. The num-bering of each protein is in parenthe-ses, with every 10th position dotted.Residues involved in SAM/SAH binding(pink), substrate binding (blue), sub-strate binding in trans from the dyadrelated polypeptide (green), and catal-ysis (yellow) are highlighted.
a
b
274 nature structural biology • volume 8 number 3 • march 2001
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://s
tru
ctb
io.n
atu
re.c
om
© 2001 Nature Publishing Group http://structbio.nature.com
articles
OMTs (Fig. 4b). SAH binding within the active site pocket ofChOMT is mediated through a network of hydrogen bonds aswell as van der Waals interactions (Fig. 3b). IOMT binds SAHthrough a similar set of interactions (Fig. 3d). The residuesinvolved in hydrogen bonding and van der Waals interactionswith SAM/SAH are spatially equivalent in both methyltrans-ferases. The two structures of ChOMT and IOMT highlight theanalogous orientation of the bound SAH as well as the commonchemical features of the SAM/SAH binding motif.
Hydroxylated substrate bindingBecause of the broad structural diversity of plant phenyl-propanoid compounds, most plant OMTs possess highly selectivesubstrate and positional specificity. Efficient substrate discrimi-nation and binding is achieved in ChOMT and IOMT throughshape selectivity dictated by van der Waals interactions, including
nature structural biology • volume 8 number 3 • march 2001 275
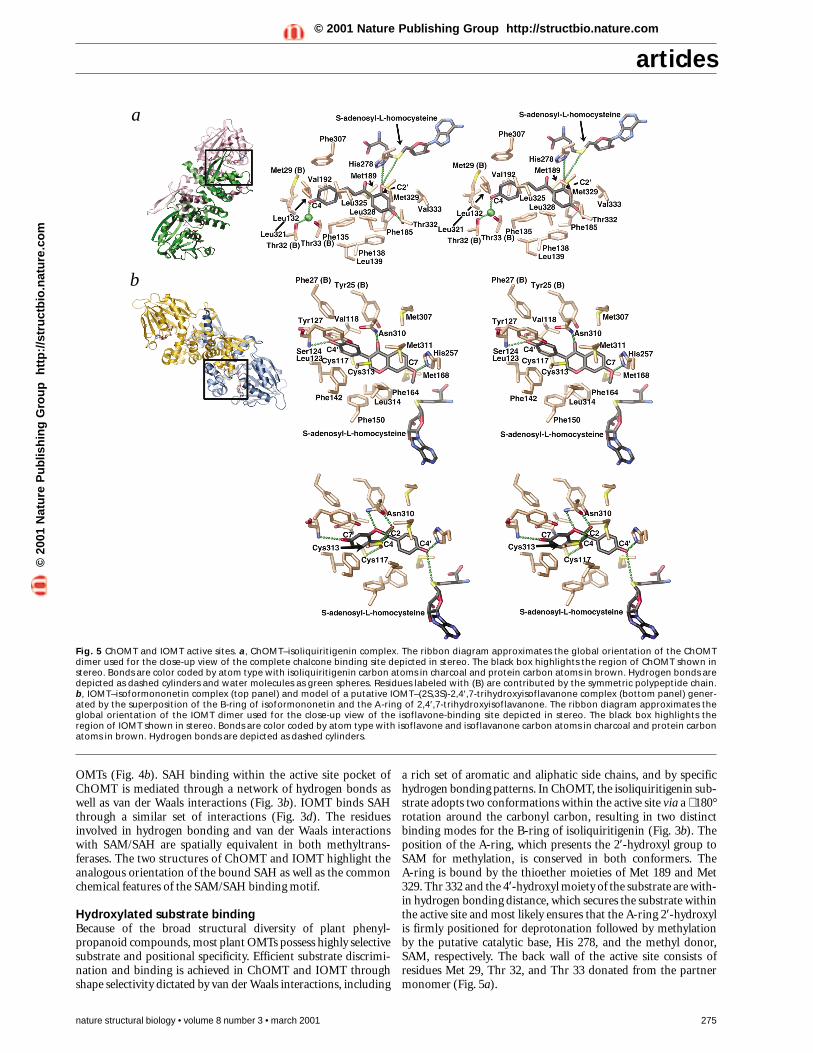
a rich set of aromatic and aliphatic side chains, and by specifichydrogen bonding patterns. In ChOMT, the isoliquiritigenin sub-strate adopts two conformations within the active site via a ∼ 180°rotation around the carbonyl carbon, resulting in two distinctbinding modes for the B-ring of isoliquiritigenin (Fig. 3b). Theposition of the A-ring, which presents the 2′-hydroxyl group toSAM for methylation, is conserved in both conformers. The A-ring is bound by the thioether moieties of Met 189 and Met329. Thr 332 and the 4′-hydroxyl moiety of the substrate are with-in hydrogen bonding distance, which secures the substrate withinthe active site and most likely ensures that the A-ring 2′-hydroxylis firmly positioned for deprotonation followed by methylationby the putative catalytic base, His 278, and the methyl donor,SAM, respectively. The back wall of the active site consists ofresidues Met 29, Thr 32, and Thr 33 donated from the partnermonomer (Fig. 5a).
a
b
Fig. 5 ChOMT and IOMT active sites. a, ChOMT–isoliquiritigenin complex. The ribbon diagram approximates the global orientation of the ChOMTdimer used for the close-up view of the complete chalcone binding site depicted in stereo. The black box highlights the region of ChOMT shown instereo. Bonds are color coded by atom type with isoliquiritigenin carbon atoms in charcoal and protein carbon atoms in brown. Hydrogen bonds aredepicted as dashed cylinders and water molecules as green spheres. Residues labeled with (B) are contributed by the symmetric polypeptide chain. b, IOMT–isoformononetin complex (top panel) and model of a putative IOMT–(2S,3S)-2,4′,7-trihydroxyisoflavanone complex (bottom panel) gener-ated by the superposition of the B-ring of isoformononetin and the A-ring of 2,4′,7-trihydroxyisoflavanone. The ribbon diagram approximates theglobal orientation of the IOMT dimer used for the close-up view of the isoflavone-binding site depicted in stereo. The black box highlights theregion of IOMT shown in stereo. Bonds are color coded by atom type with isoflavone and isoflavanone carbon atoms in charcoal and protein carbonatoms in brown. Hydrogen bonds are depicted as dashed cylinders.
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://s
tru
ctb
io.n
atu
re.c
om
© 2001 Nature Publishing Group http://structbio.nature.com

articles
The IOMT active site uses the same chemical features for sub-strate binding as ChOMT. Due to the lack of aqueous stabilityexhibited by the isoflavanone substrate 2,7,4′-trihydroxyisofla-vanone, the isoflavone daidzein was substituted in crystallizationexperiments because IOMT exhibits considerable activitytowards this compound. Cocrystallization of IOMT with SAMand daidzein resulted in the formation of a product complexconsisting of SAH and isoformononetin (Fig. 3d). Met 168 andMet 311 constrain the A-ring and help position the 7-hydroxylgroup for methylation. Given the high degree of conservation ofboth Met residues in plant OMTs, the stereochemical features ofthese interactions are most likely conserved throughout the plantOMT superfamily (Fig. 4b). Furthermore, this degree of aminoacid conservation suggests that the interaction of the Metthioether group with hydroxylated phenyl groups plays a majorenergetic role in orienting the aromatic ring so that it presents ahydroxyl group to SAM and the OMT catalytic machinery.
Tyr 25, Phe 27 and Ile 28 of the dyad related monomer ofIOMT form the back wall of the active site (Fig. 5b). Whilethese residues are contributed from the symmetrically arrangedmonomer, they do not align sequentially with the equivalentresidues in ChOMT. These contacts between the active site ofone monomer and the side chains from the symmetricallyarranged monomer have important consequences for substratespecificity. In studies of OMTs involved in berberine biosynthe-sis, for example, the high sequence conservation (93–99%identity) of four methyltransferases allowed the formation ofnon-native heterodimers. The different isoforms had differentsubstrate specificity profiles and in some cases heterodimerformation allowed these OMTs to accept new substrates29.Clearly, plant OMTs modulate the choice between chemicallysimilar substrates through variation in the dimer interface.
In the unperturbed medicarpin biosynthetic pathway, IOMTalmost certainly never encounters daidzein and thus produces noisoformononetin in vivo. While IOMT will methylate daidzein,this compound is not the in vivo substrate of IOMT. The appar-ent disparate results concerning IOMT methylation of daidzeinin vitro to form isoformononetin and the absence of this com-pound in plants strongly implicate an unstable intermediate,most likely the product of isoflavone synthase (IFS) as the truein vivo substrate of IOMT30. The putative product of thecytochrome P450 enzyme IFS31–33 and substrate for IOMT is2,7,4′-trihydroxyisoflavanone, a reactive intermediate inisoflavone biosynthesis. Because isoflavanones are unstable inaqueous solution, the full identification and characterization ofthe IFS product is incomplete34,35. However, overexpression ofIOMT in transgenic alfalfa leads to the increased production of4′-O-methylated isoflavonoids30. Alfalfa microsomes containingIOMT can convert 4′,7-dihydroxyisoflavanone to the physiolog-ical product formononetin (C. Liu & R. Dixon, unpublishedresults), thus implicating the formation of an IFS/IOMT com-plex during medicarpin biosynthesis30.
276 nature structural biology • volume 8 number 3 • march 2001
To investigate the structural basis for the apparent physiologi-cal preference of IOMT for its putative in vivo substrate, 2,4′,7-trihydroxyisoflavanone, the four possible stereoisomers of2,4′,7-trihydroxyisoflavanone were modeled in the IOMT activesite. The resulting model suggests that the optimally binding iso-mer is (2S,3S)-2,4′,7-trihydroxyisoflavanone (Fig. 5b). All fourstereoisomers were modeled by superimposing the 4′-hydroxylmoiety of the isoflavanone onto the observed location of the 7-methoxy group of isoformononetin. The resulting substratespecificity is most likely conferred by hydrogen bonding interac-tions that dictate the positioning of the physiological substrate,2,4′,7-trihydroxyisoflavanone, near the reactive methyl group ofSAM and the catalytic base, His 257. The additional hydroxylgroup located at carbon 2 and the ether oxygen at position 1 ofthe C-ring form putative hydrogen bonds with the side chaincarbonyl and side chain amide of Asn 310, respectively. In addi-tion, the 2-hydroxyl moiety of the C-ring potentially forms anadditional hydrogen bond with the side chain sulfhydryl groupof Cys 313. In a chemically similar manner, the carbonyl oxygenat carbon 4 of the C-ring forms a putative hydrogen bond withCys 117. All of these newly formed interactions are not seen inthe isoformononetin complex and likely serve to specificallysequester the isoflavanone substrate (Fig. 5b).
The accretion of hydrogen bonding interactions and the preser-vation of aromatic and hydrophobic interactions around thebound isoflavanone suggests that IOMT might display an ener-getic preference for the isoflavanone intermediate rather than thedehydrated isoflavone daidzein. Regardless of the substrate prefer-ence displayed in vitro, in vivo conditions most likely only allowfor the presence of the isoflavanone substrate. In addition, in vivoanalysis suggests that IOMT and IFS form a complex upon induc-tion of the defense response, which would provide for efficientchanneling of the isoflavanone product of IFS to IOMT30.
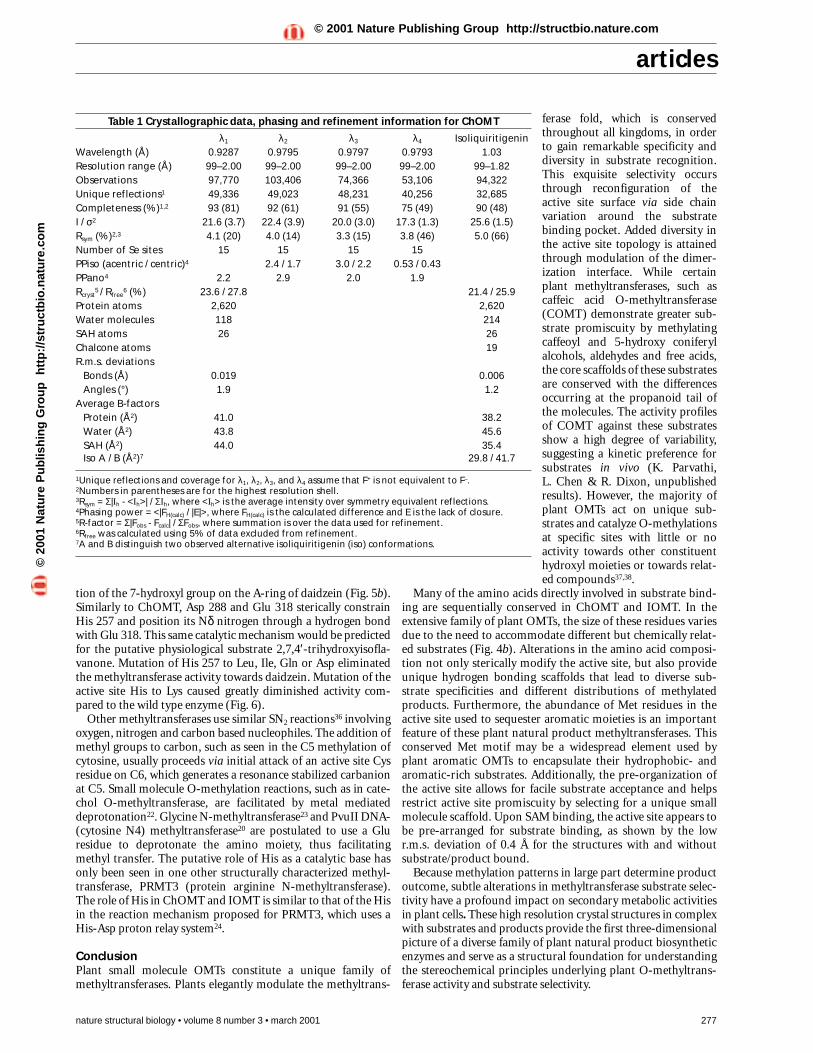
Reaction mechanismBased both upon the structures of ChOMT and IOMT andsequence alignments with the large family of plant OMTs,methylation most likely proceeds via base-assisted deprotona-tion of the hydroxyl group followed by a nucleophilic attack ofthe newly generated phenolate anion of the substrate on thereactive methyl group of SAM. In ChOMT, deprotonation of the2′-hydroxyl group of the A-ring by His 278 sets up the subse-quent attack by the hydroxyl anion on the methyl group of SAM.Because the sulfur of SAM is positively charged, the transmethyl-ation process is easily facilitated by the deprotonation step. Glu 306 and Glu 337 bracket the catalytic His residue, and theNδ nitrogen of this His makes a hydrogen bonding interaction tothe carboxylate group of Glu 337 (Fig. 2b). This interactionensures the optimal orientation of the imidazole group fordeprotonation of the 2′-hydroxyl of the isoliquiritigenin sub-strate by the Nε nitrogen of His 278 (Fig. 5a). Mutation of His278 to Leu, Ala, Gln, Lys or Asn completely eliminated methyl-transferase activity, further implicating His 278 as an importantcatalytic residue (Fig. 6).
Catalysis in IOMT proceeds through a comparable mecha-nism, with His 257 serving as the base responsible for deprotona-
Fig. 6 Thin layer chromatography assay of ChOMT and IOMT catalytic Hismutants. Lanes 1–6 of the left panel refer to wild type ChOMT, and themutants H278L, H278A, H278Q, H278K, and H278N, respectively. 14C-methylated 4,4′-dihydroxy-2′-methoxychalcone is labeled (*). Lanes 1–6of the right panel correspond to wild type IOMT, and the mutants H257L,H257I, H257Q, H257K, and H257D, respectively. 14C-methylated product 4′-hydroxy-7-methoxyisoflavone (isoformononetin) is labeled (**).
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://s
tru
ctb
io.n
atu
re.c
om
© 2001 Nature Publishing Group http://structbio.nature.com
articles
tion of the 7-hydroxyl group on the A-ring of daidzein (Fig. 5b).Similarly to ChOMT, Asp 288 and Glu 318 sterically constrainHis 257 and position its Nδ nitrogen through a hydrogen bondwith Glu 318. This same catalytic mechanism would be predictedfor the putative physiological substrate 2,7,4′-trihydroxyisofla-vanone. Mutation of His 257 to Leu, Ile, Gln or Asp eliminatedthe methyltransferase activity towards daidzein. Mutation of theactive site His to Lys caused greatly diminished activity com-pared to the wild type enzyme (Fig. 6).
Other methyltransferases use similar SN2 reactions36 involvingoxygen, nitrogen and carbon based nucleophiles. The addition ofmethyl groups to carbon, such as seen in the C5 methylation ofcytosine, usually proceeds via initial attack of an active site Cysresidue on C6, which generates a resonance stabilized carbanionat C5. Small molecule O-methylation reactions, such as in cate-chol O-methyltransferase, are facilitated by metal mediateddeprotonation22. Glycine N-methyltransferase23 and PvuII DNA-(cytosine N4) methyltransferase20 are postulated to use a Gluresidue to deprotonate the amino moiety, thus facilitatingmethyl transfer. The putative role of His as a catalytic base hasonly been seen in one other structurally characterized methyl-transferase, PRMT3 (protein arginine N-methyltransferase).The role of His in ChOMT and IOMT is similar to that of the Hisin the reaction mechanism proposed for PRMT3, which uses aHis-Asp proton relay system24.
ConclusionPlant small molecule OMTs constitute a unique family ofmethyltransferases. Plants elegantly modulate the methyltrans-
nature structural biology • volume 8 number 3 • march 2001 277
ferase fold, which is conservedthroughout all kingdoms, in orderto gain remarkable specificity anddiversity in substrate recognition.This exquisite selectivity occursthrough reconfiguration of theactive site surface via side chainvariation around the substratebinding pocket. Added diversity inthe active site topology is attainedthrough modulation of the dimer-ization interface. While certainplant methyltransferases, such ascaffeic acid O-methyltransferase(COMT) demonstrate greater sub-strate promiscuity by methylatingcaffeoyl and 5-hydroxy coniferylalcohols, aldehydes and free acids,the core scaffolds of these substratesare conserved with the differencesoccurring at the propanoid tail ofthe molecules. The activity profilesof COMT against these substratesshow a high degree of variability,suggesting a kinetic preference forsubstrates in vivo (K. Parvathi, L. Chen & R. Dixon, unpublishedresults). However, the majority ofplant OMTs act on unique sub-strates and catalyze O-methylationsat specific sites with little or noactivity towards other constituenthydroxyl moieties or towards relat-ed compounds37,38.
Many of the amino acids directly involved in substrate bind-ing are sequentially conserved in ChOMT and IOMT. In theextensive family of plant OMTs, the size of these residues variesdue to the need to accommodate different but chemically relat-ed substrates (Fig. 4b). Alterations in the amino acid composi-tion not only sterically modify the active site, but also provideunique hydrogen bonding scaffolds that lead to diverse sub-strate specificities and different distributions of methylatedproducts. Furthermore, the abundance of Met residues in theactive site used to sequester aromatic moieties is an importantfeature of these plant natural product methyltransferases. Thisconserved Met motif may be a widespread element used byplant aromatic OMTs to encapsulate their hydrophobic- andaromatic-rich substrates. Additionally, the pre-organization ofthe active site allows for facile substrate acceptance and helpsrestrict active site promiscuity by selecting for a unique smallmolecule scaffold. Upon SAM binding, the active site appears tobe pre-arranged for substrate binding, as shown by the lowr.m.s. deviation of 0.4 Å for the structures with and withoutsubstrate/product bound.
Because methylation patterns in large part determine productoutcome, subtle alterations in methyltransferase substrate selec-tivity have a profound impact on secondary metabolic activitiesin plant cells. These high resolution crystal structures in complexwith substrates and products provide the first three-dimensionalpicture of a diverse family of plant natural product biosyntheticenzymes and serve as a structural foundation for understandingthe stereochemical principles underlying plant O-methyltrans-ferase activity and substrate selectivity.
Table 1 Crystallographic data, phasing and refinement information for ChOMT
λ1 λ2 λ3 λ4 IsoliquiritigeninWavelength (Å) 0.9287 0.9795 0.9797 0.9793 1.03Resolution range (Å) 99–2.00 99–2.00 99–2.00 99–2.00 99–1.82Observations 97,770 103,406 74,366 53,106 94,322Unique reflections1 49,336 49,023 48,231 40,256 32,685Completeness (%)1,2 93 (81) 92 (61) 91 (55) 75 (49) 90 (48)I / σ2 21.6 (3.7) 22.4 (3.9) 20.0 (3.0) 17.3 (1.3) 25.6 (1.5)Rsym (%)2,3 4.1 (20) 4.0 (14) 3.3 (15) 3.8 (46) 5.0 (66)Number of Se sites 15 15 15 15PPiso (acentric / centric)4 2.4 / 1.7 3.0 / 2.2 0.53 / 0.43PPano4 2.2 2.9 2.0 1.9Rcryst
5 / Rfree6 (%) 23.6 / 27.8 21.4 / 25.9
Protein atoms 2,620 2,620Water molecules 118 214SAH atoms 26 26Chalcone atoms 19R.m.s. deviations
Bonds (Å) 0.019 0.006Angles (°) 1.9 1.2
Average B-factorsProtein (Å2) 41.0 38.2Water (Å2) 43.8 45.6SAH (Å2) 44.0 35.4Iso A / B (Å2)7 29.8 / 41.7
1Unique reflections and coverage for λ1, λ2, λ3, and λ4 assume that F+ is not equivalent to F–.2Numbers in parentheses are for the highest resolution shell.3Rsym = Σ|Ih - <Ih>| / ΣIh, where <Ih> is the average intensity over symmetry equivalent reflections.4Phasing power = <|FH(calc) / |E|>, where FH(calc) is the calculated difference and E is the lack of closure.5R-factor = Σ|Fobs - Fcalc| / ΣFobs, where summation is over the data used for refinement.6Rfree was calculated using 5% of data excluded from refinement.7A and B distinguish two observed alternative isoliquiritigenin (iso) conformations.
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://s
tru
ctb
io.n
atu
re.c
om
© 2001 Nature Publishing Group http://structbio.nature.com
articles
MethodsMaterials. The pET-15b expression vector and E. coli strainBL21(DE3) were purchased from Novagen. Ni2+-NTA resin was pur-chased from Qiagen. Benzamidine Sepharose and Superdex 200FPLC columns were obtained from Pharmacia. SeMet, thrombin, S-adenosyl-L-methionine and S-adenosyl-L-homocysteine wereobtained from Sigma. All oligonucleotides were purchased fromOperon, Inc. Adenosyl-L-methionine-S-(methyl-14C) was purchasedfrom New England Nuclear (NEN). 4,2′,4′-trihydroxychalcone and4′,7-dihydroxyisoflavone were acquired from Indofine.
Expression, purification and mutagenesis. The alfalfa ChOMTgene (GenBank accession number L10211) and IOMT gene(GenBank accession number AF000976) were inserted into the E. coli expression vector pHIS839 (ChOMT) or pET-15b (IOMT). ChOMTand IOMT constructs were transformed into E. coli BL21(DE3).Transformed E. coli were grown at 37 °C in terrific broth (TB) con-taining 50 µg ml-1 kanamycin (ChOMT) or 100 µg ml-1 ampicillin(IOMT) until A600nm = 1.0. After induction with 0.5 mM isopropyl 1-thio-β-galactopyranoside (IPTG), the cultures were grown for 6 hat 25 °C. Cells were pelleted, harvested, and resuspended in lysisbuffer (50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 20 mM imidazole (pH8.0), 20 mM β-mercaptoethanol, 10% (v/v) glycerol and 1% (v/v)Tween-20). After sonication and centrifugation, the supernatantwas passed over a Ni2+-NTA column, washed with 10 bed volumes oflysis buffer, 10 bed volumes of wash buffer (50 mM Tris-HCl (pH 8.0),500 mM NaCl, 20 mM imidazole (pH 8.0), 20 mM β-mercaptoethanoland 10% (v/v) glycerol), then the His tagged protein was eluted withelution buffer (50 mM Tris-HCl (pH 8.0), 500 mM NaCl, 250 mM imi-dazole (pH 8.0), 20 mM β-mercaptoethanol, and 10% (v/v) glycerol).Incubation with thrombin during dialysis for 24 h at 4 °C against25 mM HEPES (pH 7.5), 100 mM NaCl, 1 mM dithiothreitol (DTT)
278 nature structural biology • volume 8 number 3 • march 2001
removed the N-terminal His tag. Dialyzed proteinwas reloaded onto a Ni2+-NTA column to removecleaved His tag followed by thrombin depletionusing a benzamidine Sepharose column. Gel filtra-tion on a Superdex 200 column equilibrated with25 mM HEPES (pH 7.5), 100 mM NaCl, 1 mM DTTresulted in homogenous and active ChOMT andIOMT. Fractions containing the protein of interestwere pooled and concentrated to ∼ 25 mg ml-1 andstored at -80 °C. SeMet substituted protein wasobtained from E. coli grown in minimal media withappropriate amino acids and SeMet added40.Expression and purification steps were as above. Allmutants were generated with the QuikChange(Stratagene) protocol. Automated nucleotidesequencing confirmed the fidelity of the PCR prod-ucts (Salk Institute DNA sequencing facility). Allmutants were expressed as described above.
Enzyme activity assays. Mutant enzymes werepurified by Ni+2 affinity chromatography, dialyzedagainst 25 mM HEPES (pH 7.5), 100 mM NaCl, 2 mMDTT, and concentrated to ∼ 2 mg ml-1. Qualitativeactivity assays were performed using 20 µg of pro-tein, 500 µM substrate (2′,4,4′-trihydroxychalcone forChOMT and 4′,7-dihydroxyisoflavone for IOMT), and500 µM adenosyl-L-methionine-S-(methyl-14C), in 50 µl of 250 mM HEPES (pH 7.5), 100 mM NaCl.Reactions were allowed to proceed for 2 h at roomtemperature after which time the reaction productswere extracted into ethyl acetate and applied to aWhatman LK6D silica TLC plate. Chromatogramswere developed in ethyl actetate:hexane (50:50, v/v).The products were visualized by autoradiography.
Crystallography. Crystals of ChOMT and IOMTwere grown by vapor diffusion in hanging dropscontaining a 1:1 mixture of protein and crystalliza-tion buffer (ChOMT, 12% (w/v) PEG 8000, 0.05 MHEPES (pH 7.5), 0.3 M ammonium acetate, 2 mM
DTT at 4 °C ; IOMT, 17% (w/v) PEG 8000, 0.05 M TAPS (pH 8.25), 0.35M lithium sulfate, 2 mM DTT, 15 °C). Crystals for both proteins grewin space group C2 with one molecule per asymmetric unit. Unit celldimensions for ChOMT were a = 127.19 Å, b = 53.79 Å, c = 73.55 Å, β = 125.55°. IOMT cell dimensions were a = 145.56 Å, b = 50.54 Å, c = 63.82 Å, β = 106.69°. Diffraction data were collected from singlecrystals mounted in a cryoloop and flash frozen in a nitrogen streamat 105 K. All diffraction data were collected at the StanfordSynchrotron Radiation Facility, beamline 9-2 (IOMT data and ChOMTSeMet data) on a Quantum 4 CCD detector and beamline 7-1(ChOMT–isoliquiritigenin complex) on a 30 cm MAR imaging plate.All images were indexed and scaled using DENZO41 and the reflec-tions merged with SCALEPACK41. The ChOMT and IOMT structureswere determined using multiple wavelength anomalous dispersion(MAD) phasing on the SeMet substituted protein. Initial heavy atomsites were found with SOLVE42. SHARP43 was used to refine the initialsites and to locate additional sites. MAD phases were improved withSOLOMON44. Subsequent complexes were solved by the differenceFourier method. All refinements were carried out using CNS45.During refinements, structure factors obtained from intensity datawere used to generate SIGMAA weighted 2Fo - Fc and Fo - Fcelectron density maps with phases calculated from the structure ofthe in-progress model. Inspection of the electron density maps andmodel building was performed in O46. The quality of all models wasassessed using the program PROCHECK47. For the ChOMT–isoliquirit-igenin, complex 92.6%, 6.4%, 0.7%, and 0.3% of the residues werefound in the most favored, the allowed, the generously allowed, andthe disallowed regions of the Ramachandran plot, respectively, witha G factor of 0.39. For the IOMT–isoformononetin complex, 91%,8%, and 1% of the residues were found in the most favored, theallowed, and the generously allowed regions of the Ramachandranplot, respectively, with a G factor of 0.30.
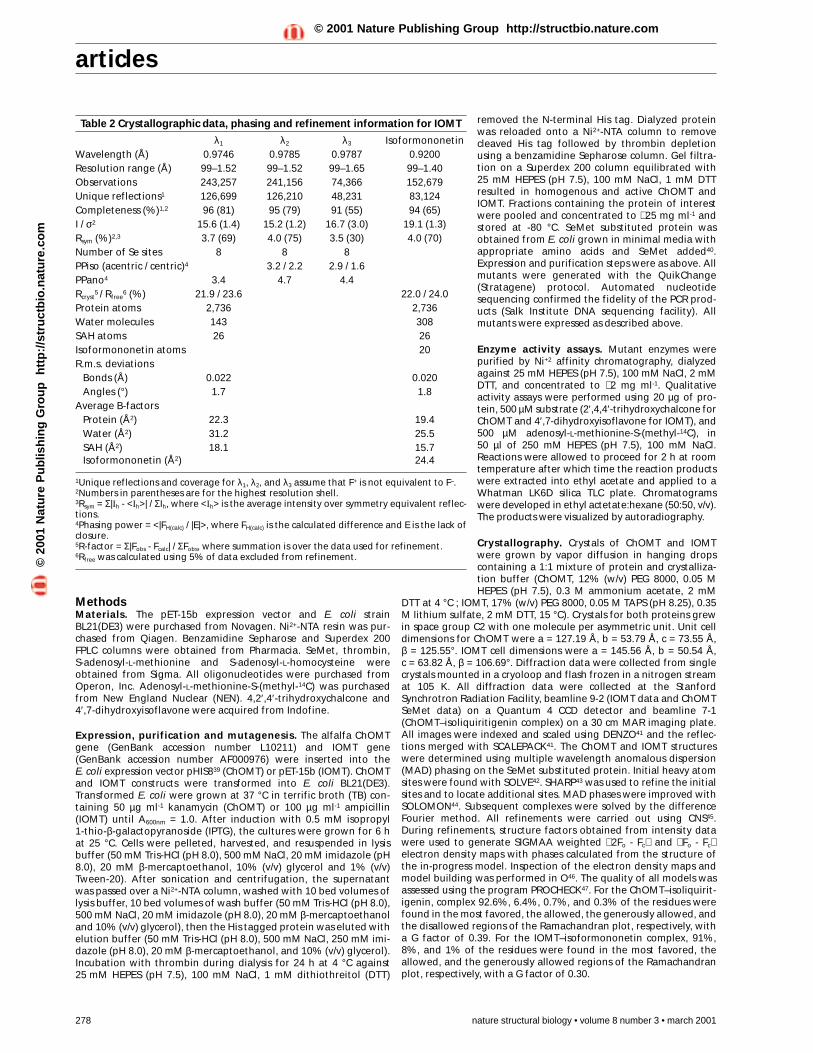
Table 2 Crystallographic data, phasing and refinement information for IOMT
λ1 λ2 λ3 IsoformononetinWavelength (Å) 0.9746 0.9785 0.9787 0.9200Resolution range (Å) 99–1.52 99–1.52 99–1.65 99–1.40Observations 243,257 241,156 74,366 152,679Unique reflections1 126,699 126,210 48,231 83,124Completeness (%)1,2 96 (81) 95 (79) 91 (55) 94 (65)I / σ2 15.6 (1.4) 15.2 (1.2) 16.7 (3.0) 19.1 (1.3)Rsym (%)2,3 3.7 (69) 4.0 (75) 3.5 (30) 4.0 (70)Number of Se sites 8 8 8PPiso (acentric / centric)4 3.2 / 2.2 2.9 / 1.6PPano4 3.4 4.7 4.4Rcryst
5 / Rfree6 (%) 21.9 / 23.6 22.0 / 24.0
Protein atoms 2,736 2,736Water molecules 143 308SAH atoms 26 26Isoformononetin atoms 20R.m.s. deviations
Bonds (Å) 0.022 0.020Angles (°) 1.7 1.8
Average B-factorsProtein (Å2) 22.3 19.4Water (Å2) 31.2 25.5SAH (Å2) 18.1 15.7Isoformononetin (Å2) 24.4
1Unique reflections and coverage for λ1, λ2, and λ3 assume that F+ is not equivalent to F–.2Numbers in parentheses are for the highest resolution shell.3Rsym = Σ|Ih - <Ih>| / ΣIh, where <Ih> is the average intensity over symmetry equivalent reflec-tions.4Phasing power = <|FH(calc) / |E|>, where FH(calc) is the calculated difference and E is the lack ofclosure.5R-factor = Σ|Fobs - Fcalc| / ΣFobs, where summation is over the data used for refinement.6Rfree was calculated using 5% of data excluded from refinement.©
2001
Nat
ure
Pu
blis
hin
g G
rou
p
htt
p:/
/str
uct
bio
.nat
ure
.co
m© 2001 Nature Publishing Group http://structbio.nature.com

articles
1. Hartwig, U.A., Maxwell, C.A., Joseph, C.M. & Phillips, D.A. Effects of alfalfa nodgene-inducing flavonoids on nodABC transcription in Rhizobium meliloti strainscontaining different nodD genes. J. Bacteriol. 172, 2769–2773 (1990).
2. Peters, N.K., Frost, J.W. & Long, S.R. A plant flavone, luteolin, induces expressionof Rhizobium meliloti genes. Science 233, 977–980 (1986).
3. Smith, D.A. & Banks, S.W. Formation and biological properties of isoflavonoidphytoalexins. Prog. Clin. Biol. Res. 213, 113–124 (1986).
4. Ingham, J.L. & Millar, R.L. Sativin: an induced isoflavan from the leaves ofMedicago sativa L. Nature 242, 125–126 (1973).
5. Dobson, H.E.M. Floral volatiles in insect biology. In Insect-plant interactions. (ed.,Bernays, E.) 47–81 (CRC Press, Boca Raton, Florida; 1993).
6. Lewis, N.G., Davin, L.B. & Sarkanen, S. The nature and function of lignins. InComprehensive natural products chemistry 3 (eds Barton, D. & Nakanishi, K.)(Elsevier Science Ltd., Amsterdam; 1999).
7. Barton, D. & Nakanishi, K. Comprehensive Natural Products Chemistry 1 (ElsevierScience Ltd., Amsterdam; 1999).
8. Maxwell, C.A., Harrison, M.J. & Dixon, R.A. Molecular characterization andexpression of alfalfa isoliquiritigenin 2’-O-methyltransferase, an enzymespecifically involved in the biosynthesis of an inducer of Rhizobium melilotinodulation genes. Plant J. 4, 971–981 (1993).
9. Maxwell, C.A., Edwards, R. & Dixon, R.A. Identification, purification, andcharacterization of S-adenosyl-L-methionine: isoliquiritigenin 2’-O-methyltransferase from alfalfa (Medicago sativa L.). Arch. Biochem. Biophys.293, 158–166 (1992).
10. Jez, J.M., Bowman, M.E., Dixon, R.A. & Noel, J.P. Structure and mechanism of theevolutionarily unique plant enzyme chalcone isomerase. Nature Struct. Biol. 7,786–791 (2000).
11. He, X-Z. & Dixon, R.A. Affinity chromatography, substrate/product specificity, andamino acid sequence analysis of an isoflavone O-methyltransferase from alfalfa(Medicago sativa L.). Arch. Biochem. Biophys. 336, 121–129 (1996).
12. He, X-Z., Reddy, J.T. & Dixon, R.A. Stress response in alfalfa (Medicago sativa L).XXII. cDNA cloning and characterization of an elicitor-inducible isoflavone 7-O-methyltranferase. Plant Mol. Biol. 36, 43–54 (1998).
13. Hodel, A.E., Gershon, P.D., Shi, X. & Quiocho, F.A. The 1.85 Å structure of vacciniaprotein VP39: a bifunctional enzyme that participates in the modification ofboth mRNA ends. Cell 85, 247–256 (1996).
14. Cheng, X., Kumar, S., Posfai, J., Pflugrath, J.W. & Roberts, R.J. Crystal structure ofthe HhaI DNA methyltranferase complexed with S-adenosyl-L-methionine. Cell74, 299–307 (1993).
15. Schroeder, S.G. & Samudzi, C.T. Structural studies of EcoRII methylase: exploringsimilarities among methylases. Protein Eng. 10, 1385–1393 (1997).
16. Djordjevic, S. & Stock, A.M. Crystal structure of the chemotaxis receptormethyltransferase CheR suggests a conserved structural motif for binding S-adenosylmethionine. Structure 5, 545–558 (1997).
17. Schluckebier, G., Zhong, P., Stewart, K.D., Kavanaugh, T.J. & Abad-Zapatero, C.The 2.2 Å structure of the rRNA methyltransferase ErmC’ and its complexes withcofactor and cofactor analogs: implications for the reaction mechanism. J. Mol.Biol. 289, 277–291 (1999).
18. Tran, P.H., Korszun, Z.R., Cerritelli, S., Springhorn, S.S. & Lacks, S.A. Crystalstructure of the DpnM DNA adenine methyltransferase from the DpnII restrictionsystem of Streptococcus pneumoniae bound to S-adenosylmethionine. Structure6, 1563–1575 (1998).
19. Schluckebier, G., Kozak, M., Bleimling, N., Weinhold, E. & Saenger, W.Differential binding of S-adenosylmethionine S-adenosylhomocysteine andsinefungin to the adenine-specific DNA methyltransferase M.TaqI. J. Mol. Biol.265, 56–67 (1997).
20. Gong, W., O’Gara, M., Blumenthal, R.M. & Cheng, X. Structure of PvuII DNA-(cytosine N4) methyltransferase, an example of domain permutation and proteinfold assignment. Nucleic Acids Res. 25, 2702–2715 (1997).
21. Hashimoto, H., Inoue, T., Nishioka, M., Fujiwara, S., Takagi, M., Imanaka, T. & Kai,Y. Hyperthermostable protein structure maintained by intra and inter-helix ion-pairs in archaeal O6-methylguanine-DNA methyltransferase. J. Mol. Biol. 292,707–716 (1999).
22. Vidgren, J., Svensson, L. & Liljas, A. Crystal structure of catechol O-methyltransferase. Nature 368, 354–358 (1994).
23. Pattanayek, R., Newcomer, M.E. & Wagner, C. Crystal structure of apo-glycine N-methyltransferase (GNMT). Protein Sci. 7, 1326–1331 (1998).
24. Zhang, X., Zhou, L. & Cheng, X. Crystal structure of the conserved core of proteinarginine methyltransferase PRMT3. EMBO J. 19, 3509–3519 (2000).
Coordinates. Coordinates have been deposited in the Protein DataBank (accession codes 1FPQ, 1FP1, 1FPX, 1FP2 for the ChOMT–SAH,ChOMT–SAH–isoliquiritigenin, IOMT–SAH, and IOMT–SAH–isofor-mononetin complexes, respectively).
AcknowledgmentsWe acknowledge the assistance provided by members of the Noel group and thestaff of beamlines 7-1 and 9-2 at the Stanford Synchrotron Radiation Facility. The
SSRL Biotechnology Program is supported by the NIH, National Center forResearch Resources, Biomedical Technology Program, and the DOE, Office ofBiological and Environmental Research. This work was supported by funds fromthe Salk Institute and the National Science Foundation awarded to J.P.N. C.Z. wassupported by funds from the NIH Molecular Biophysics Training Grantadministered by the University of California, San Diego and funds from theSamuel Roberts Noble Foundation.
Received 21 August, 2000; accepted 22 January, 2001.
25. Kagan, R.M. & Clarke, S. Widespread occurrence of three sequence motifs indiverse S-adenosylmethionine-dependent methyltransferases suggests acommon structure for these enzymes. Arch. Biochem. Biophys. 310, 417–427(1994).
26. O’Gara, M., McCloy, K., Malone, T. & Cheng, X. Structure-based alignment ofthree Ado-Met-dependent DNA methyltransferases. Gene 157, 135–138 (1995).
27. Schluckebier, G., O’Gara, M., Saenger, W. & Cheng, X. Universal catalytic domainstructure of AdoMet-dependent methyltransferases. J. Mol. Biol. 247, 16–20(1995).
28. Rossmann, M.G., Moras, D. & Olsen, K.W. Chemical and biological evolution of anucleotide-binding protein. Nature 250, 194–199 (1974).
29. Frick, S. & Kutchan, T.M. Molecular cloning and functional expression of O-methyltransferases common to isoquinoline alkaloid and phenylpropanoidbiosynthesis. Plant J. 17, 329–339 (1999).
30. He, X-Z. & Dixon, R.A. Genetic manipulation of isoflavone 7-O-methyltransferaseenhances the biosynthesis of 4’-O-methylated isoflavonoid phytoalexins anddisease resistance in alfalfa. Plant Cell 12, 1689–1702 (2000).
31. Steele, C.L., Gijzen, M., Qutob, D. & Dixon, R.A. Molecular characterization of theenzyme catalyzing the aryl migration reaction of isoflavonoid biosynthesis. Arch.Biochem. Biophys. 367, 146–150 (1999).
32. Akashi, T., Aoki, T. & Ayabe, S. Cloning and functional expression of a cytochromeP450 cDNA encoding 2-hydroxyisoflavanone synthase involved in biosynthesis ofthe isoflavonoid skeleton in licorice. Plant Physiol. 121, 821–828 (1999).
33. Jung, W., Yu, O., Lau, S.M.C., O’Keefe, D.P., Odell, J., Fader, G. & McGonigle, B.Identification and expression of isoflavone synthase, the key enzyme forbiosynthesis of isoflavones in legumes. Nature Biotechnol. 18, 208–212 (2000).
34. Hashim, M. F., Hakamatsuka, T., Ebizuka, Y. & Sankawa U. Reaction mechanism ofoxidative rearrangement of flavanone in isoflavone biosynthesis. FEBS Lett. 271,219–222 (1990).
35. Kochs, G. & Grisebach, H. Enzymatic synthesis of isoflavones. Eur. J. Biochem. 155,311–318 (1986).
36. Walsh, C. Enzymatic reaction mechanisms. (W.H. Freeman and Company, SanFrancisco; 1979).
37. Ibrahim, R.K., Bruneau, A. & Bantignies, B. Plant O-methyltransferases: molecularanalysis, common signature and classification. Plant Mol. Biol. 36, 1–10 (1998).
38. Ibrahim, R.K. Plant O-methyltransferase signatures. Trends Plant Sci. 2, 249–250(1997).
39. Jez, J.M., Ferrer, J.L., Bowman, M.E., Dixon, R.A. & Noel, J.P. Dissection ofmalonyl-coenzyme A decarboxylation from polyketide formation in the reactionmechanism of a plant polyketide synthase. Biochemistry 39, 890–902 (2000).
40. Doublie, S. Preparation of selenomethionyl proteins for phase determination.Methods Enzymol. 276, 523–530 (1997).
41. Otwinowski, Z. & Minor, W. Processing of X-ray diffraction data collected inoscillation mode. Methods Enzymol. 276, 307–326 (1997).
42. Terwilliger, T.C. & Berendzen, J. Automated MAD and MIR structure solution.Acta Crystallogr. D 55, 849–861 (1999).
43. de la Fourtelle, E. & Bricogne, G. Maximum likelihood heavy-atom parameterrefinement for multiple isomorphous replacement and multiwavelengthanomalous diffraction methods. Methods Enzymol. 276, 472–494 (1997).
44. Abrahams, J.P. & Leslie, A.G.W. Methods used in the structure determination ofbovine mitochondrial F1 ATPase. Acta Crystallogr. D 49, 148–157 (1996).
45. Brunger, A.T. et al. Crystallography and NMR system: a new software suite formacromolecular structure dertermination. Acta Crystallogr. D 54, 905–921(1998).
46. Jones, T.A., Zou, J.Y., Cowan, S.W. & Kjeldgaard, M. Improved methods forbuilding protein models in electron density maps and the location of errors inthese models. Acta Crystallogr. D 49, 148–157 (1993).
47. Laskowski, R.A., MacArthur, M.W., Moss, D.S. & Thrornton, J.M. PROCHECK: aprogram to check the stereochemical quality of protein structures. J. Appl.Crystallogr. 26, 283–291 (1993).
48. Dixon, R.A. & Paiva, N.L. Stress-induced phenylpropanoid metabolism. Plant Cell7, 1085–1097 (1995).
49. Kraulis, P.J. MOLSCRIPT: a program to produce both detailed and schematic plotsof protein structures. J. Appl. Crystallogr. 24, 946–950 (1991).
50. Nicholls, A., Charp, K. & Honig, B. Protein folding and association: insights fromthe interfacial and thermodynamic properties of hydrocarbons. Proteins 11,281–296 (1991).
51. Amundsen, S. et al. X-POV-Team POV-Ray: persistence of vision ray-tracer.http://www.povray.org (1997).
nature structural biology • volume 8 number 3 • march 2001 279
©20
01 N
atu
re P
ub
lish
ing
Gro
up
h
ttp
://s
tru
ctb
io.n
atu
re.c
om
© 2001 Nature Publishing Group http://structbio.nature.com




















