
Single-round infectious particles enhance immunogenicity of a DNA vaccine against West Nile virus
7
Single-round infectious particles enhance immunogenicity of a DNA vaccine against West Nile virus David C Chang 1 , Wen J Liu 1 , Itaru Anraku 2 , David C Clark 1 , Christopher C Pollitt 3 , Andreas Suhrbier 2 , Roy A Hall 1 & Alexander A Khromykh 1 DNA vaccines encoding replication-defective viruses are safer than inactivated or live attenuated viruses but may fail to stimulate an immune response sufficient for effective vaccination. We augment the protective capacity of a capsid-deleted flavivirus DNA vaccine by co-expressing the capsid protein from a separate promoter. In transfected cells, the capsid-deleted RNA transcript is replicated and translated to produce secreted virus-like particles lacking the nucleocapsid. This RNA is also packaged with the help of co-expressed capsid protein to form secreted single-round infectious particles (SRIPs) that deliver the RNA into neighboring cells. In SRIP-infected cells, the RNA is replicated again and produces additional virus-like particles, but in the absence of capsid RNA no SRIPs are formed and no further spread occurs. Compared with an otherwise identical construct that does not encode capsid, our vaccine offers better protection to mice after lethal West Nile virus infection. It also elicits virus-neutralizing antibodies in horses. This approach may enable vaccination against pathogenic flaviviruses other than West Nile virus. Flaviviruses are a large group of pathogenic, positive-strand RNA viruses that cause 450 million human infections annually. Owing primarily to efficacy and safety concerns associated with inactivated or live-attenuated vaccines 1 , there are no commercial human vaccines available for medically important flaviviruses, such as West Nile virus and dengue virus. Prototype DNA-based vaccine candidates 2 have been developed for St. Louis encephalitis virus 3 , Japanese encephalitis virus 4 , Murray Valley encephalitis virus 5 , dengue virus-2 (ref. 6), tick- borne encephalitis virus 7 and West Nile virus 8 . All use a similar strategy to express the pre-membrane (prM) and envelope (E) proteins to produce highly immunogenic secreted prM-E particles and show promise for both human and veterinary medicine. For instance, 1 mg of a DNA vaccine expressing genes encoding West Nile virus prM and E proteins generates sufficient levels of neutralizing antibody to protect horses against West Nile virus challenge by infected mosquitoes 8 . However, unlike immunization with live virus, the current DNA vaccines do not replicate and spread within the immunized host. Therefore, relatively high or multiple doses are required to induce protective immunity. Furthermore, these strategies cannot induce humoral or cell-mediated immune responses to the nonstructural viral proteins, which may be important for long-term immunity 9 . Using plasmid DNAs, encoding self-replicating RNAs allows substantial reduction of the dose of DNA vaccines 10 . Flavivirus RNAs that contain large deletions in the capsid gene are unable to produce infectious virions but retain the ability to replicate and express prM and E proteins, which are secreted as highly immuno- genic prM-E particles 11 . These can be delivered as naked RNA 11 , as plasmid DNA 12 or as pseudo-infectious virus particles 13 and were shown to be effective in mice. Here we illustrate a concept in flavivirus DNA vaccine design, which differs from other flavivirus vaccine developments and combines the advantage of replicon-based DNA technology, the ability to generate pseudo-infectious virus particles and the immunogenicity of capsid- deleted flavivirus RNAs. A naturally attenuated Australian strain of West Nile virus, Kunjin MRM61C 14 that has had an additional attenuating mutation engineered into the gene encoding NS1 ( 250 Pro to Leu), which we call WNV KUN 15 , exemplifies this vaccination strategy. We previously demonstrated that immunization with WNV KUN or with the plasmid DNA pKUN1, which encodes an infectious full-length cDNA copy of WNV KUN RNA under the control of cytomegalovirus (CMV) promoter, induced efficient immunity against the virulent New York 99 strain of West Nile virus (WNV NY99 ) 16 . As the next step in developing effective but safer West Nile virus vaccine, we generated the plasmid pKUNdC from pKUN1 DNA by deleting a large portion (codons 18–100) of the gene encoding capsid protein, while retaining the cyclization sequence. To enhance the immunogenicity of the pKUNdC vaccine, we generated a second plasmid pKUNdC/C, which is capable of simultaneously transcribing two separate RNA species from two copies of the CMV promoter configured in a back-to-back orientation (Fig. 1a). One Received 27 February; accepted 28 March; published online 20 April 2008; doi:10.1038/nbt1400 1 School of Molecular and Microbial Sciences, University of Queensland, St. Lucia, Brisbane, Queensland 4072, Australia. 2 Queensland Institute of Medical Research, PO Royal Brisbane Hospital, Brisbane, Queensland 4029, Australia. 3 School of Veterinary Sciences, University of Queensland, St. Lucia, Brisbane, Queensland 4072, Australia. Correspondence should be addressed to A.A.K. ([email protected]). NATURE BIOTECHNOLOGY VOLUME 26 NUMBER 5 MAY 2008 571 LETTERS
Transcript of Single-round infectious particles enhance immunogenicity of a DNA vaccine against West Nile virus

Single-round infectious particles enhanceimmunogenicity of a DNA vaccine againstWest Nile virusDavid C Chang1, Wen J Liu1, Itaru Anraku2, David C Clark1, Christopher C Pollitt3, Andreas Suhrbier2,Roy A Hall1 & Alexander A Khromykh1
DNA vaccines encoding replication-defective viruses are safer
than inactivated or live attenuated viruses but may fail to
stimulate an immune response sufficient for effective
vaccination. We augment the protective capacity of a
capsid-deleted flavivirus DNA vaccine by co-expressing the
capsid protein from a separate promoter. In transfected cells,
the capsid-deleted RNA transcript is replicated and translated
to produce secreted virus-like particles lacking the
nucleocapsid. This RNA is also packaged with the help of
co-expressed capsid protein to form secreted single-round
infectious particles (SRIPs) that deliver the RNA into
neighboring cells. In SRIP-infected cells, the RNA is
replicated again and produces additional virus-like particles,
but in the absence of capsid RNA no SRIPs are formed and no
further spread occurs. Compared with an otherwise identical
construct that does not encode capsid, our vaccine offers
better protection to mice after lethal West Nile virus infection.
It also elicits virus-neutralizing antibodies in horses. This
approach may enable vaccination against pathogenic
flaviviruses other than West Nile virus.
Flaviviruses are a large group of pathogenic, positive-strand RNAviruses that cause 450 million human infections annually. Owingprimarily to efficacy and safety concerns associated with inactivated orlive-attenuated vaccines1, there are no commercial human vaccinesavailable for medically important flaviviruses, such as West Nile virusand dengue virus. Prototype DNA-based vaccine candidates2 havebeen developed for St. Louis encephalitis virus3, Japanese encephalitisvirus4, Murray Valley encephalitis virus5, dengue virus-2 (ref. 6), tick-borne encephalitis virus7 and West Nile virus8. All use a similarstrategy to express the pre-membrane (prM) and envelope (E)proteins to produce highly immunogenic secreted prM-E particlesand show promise for both human and veterinary medicine. Forinstance, 1 mg of a DNA vaccine expressing genes encoding West Nilevirus prM and E proteins generates sufficient levels of neutralizingantibody to protect horses against West Nile virus challenge by
infected mosquitoes8. However, unlike immunization with live virus,the current DNA vaccines do not replicate and spread within theimmunized host. Therefore, relatively high or multiple doses arerequired to induce protective immunity. Furthermore, these strategiescannot induce humoral or cell-mediated immune responses tothe nonstructural viral proteins, which may be important forlong-term immunity9.
Using plasmid DNAs, encoding self-replicating RNAs allowssubstantial reduction of the dose of DNA vaccines10. FlavivirusRNAs that contain large deletions in the capsid gene are unable toproduce infectious virions but retain the ability to replicate andexpress prM and E proteins, which are secreted as highly immuno-genic prM-E particles11. These can be delivered as naked RNA11, asplasmid DNA12 or as pseudo-infectious virus particles13 and wereshown to be effective in mice.
Here we illustrate a concept in flavivirus DNA vaccine design, whichdiffers from other flavivirus vaccine developments and combines theadvantage of replicon-based DNA technology, the ability to generatepseudo-infectious virus particles and the immunogenicity of capsid-deleted flavivirus RNAs. A naturally attenuated Australian strain ofWest Nile virus, Kunjin MRM61C14 that has had an additionalattenuating mutation engineered into the gene encoding NS1(250Pro to Leu), which we call WNVKUN
15, exemplifies this vaccinationstrategy. We previously demonstrated that immunization withWNVKUN or with the plasmid DNA pKUN1, which encodes aninfectious full-length cDNA copy of WNVKUN RNA under the controlof cytomegalovirus (CMV) promoter, induced efficient immunityagainst the virulent New York 99 strain of West Nile virus(WNVNY99)16. As the next step in developing effective but saferWest Nile virus vaccine, we generated the plasmid pKUNdC frompKUN1 DNA by deleting a large portion (codons 18–100) of the geneencoding capsid protein, while retaining the cyclization sequence. Toenhance the immunogenicity of the pKUNdC vaccine, we generated asecond plasmid pKUNdC/C, which is capable of simultaneouslytranscribing two separate RNA species from two copies of the CMVpromoter configured in a back-to-back orientation (Fig. 1a). One
Received 27 February; accepted 28 March; published online 20 April 2008; doi:10.1038/nbt1400
1School of Molecular and Microbial Sciences, University of Queensland, St. Lucia, Brisbane, Queensland 4072, Australia. 2Queensland Institute of Medical Research,PO Royal Brisbane Hospital, Brisbane, Queensland 4029, Australia. 3School of Veterinary Sciences, University of Queensland, St. Lucia, Brisbane, Queensland 4072,Australia. Correspondence should be addressed to A.A.K. ([email protected]).
NATURE BIOTECHNOLOGY VOLUME 26 NUMBER 5 MAY 2008 57 1
L E T T E R S
©20
08 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
ureb
iote
chno
logy

CMV promoter directs transcription of KUNdC RNA, whereas theother CMV promoter directs transcription of mRNA encoding full-length mature capsid protein (codons 1–105). Together, these twoRNAs essentially express the entire viral genome, including all thenonstructural proteins required for RNA replication, and all threestructural proteins (prM, E and capsid) required for RNApackaging. The KUNdC RNA is amplified by the nonstructuralproteins, and the amplified RNA is translated to generate largeamounts of prM and E proteins, which are secreted as prM-E particles.Some of the prM-E particles, together with capsid protein, package theKUNdC RNA to generate SRIPs (Fig. 1b). Secreted SRIPs, whichcomprise prM-E, capsid and KUNdC RNA, provide a second round ofamplification by infecting new cells. In these SRIP-infected cells,KUNdC RNA is released and drives its own replication and secretionof prM-E particles (Fig. 1b). However, as the KUNdC RNA does nothave a gene encoding a functional capsid protein, which is essential forRNA packaging, no further spread of KUNdC RNA is possible and theinfectious cycle effectively terminates in SRIP-infected cells. BothpKUNdC/C DNA–transfected and SRIP-infected cells not only secreteprM-E particles that induce neutralizing antibodies, but also producethe nonstructural proteins that are often targeted by flavivirus-specificcellular immune responses17 (Fig. 1b). As a result, the DNA vaccinepresents both structural and nonstructural proteins, as well as viralRNA replication products (single-stranded and double-strandedRNA), to the immune system, thereby effectively mimicking live-virus infection without producing infectious virus.
To examine the ability of pKUNdC/C DNA to produce SRIPs andto analyze production of prM-E particles by pKUNdC/C and otherDNA vaccines, we transfected Vero cells (African green monkey kidneycell line) with the pKUNdC/C, pKUNdC (encoding capsid-deletedWNVKUN cDNA only), pcDNA-prME (encoding prM-E genes from apcDNA3.1 expression vector) and the control pKUN1 DNA (encodingthe full-length infectious cDNA of WNVKUN
14). The expression of Eprotein was then analyzed using an immunofluorescence assay andwestern blotting using anti-E-protein antibodies. The spread of SRIPswas demonstrated in cells transfected with pKUNdC/C DNA, asevidenced by foci of E-protein–positive cells (Fig. 2a, i). The infectiv-ity of SRIPs and delivery of KUNdC RNA expressing prM-E genes wasdemonstrated after transfer of the culture fluid from pKUNdC/CDNA–transfected cells to new cells, as indicated by the detection ofE-protein–expressing cells infected with SRIPs (Fig. 2a, ii). PCRanalysis of culture fluid from transfected cells to detect free DNAwas negative (not shown), ruling out potential contamination withtransfected DNA. Production of secreted prM-E particles, as shown bydetection of E protein by western blot analysis, was observed both inthe culture fluid of cells transfected with all four DNAs (althoughpKUNdC DNA–transfected cells produced less secreted E protein) aswell as in the culture fluid of cells infected with the culture mediumcollected from pKUNdC/C DNA– and pKUN1 DNA–transfected cells(Supplementary Fig. 1b,c online). Notably, reinfection of new cellswith culture fluid collected from SRIP-infected cells did not reveal thepresence of any infectious particles (Fig. 2a, iii). As expected, there
Figure 1 Structure and mode of operation of a
split genome–based DNA vaccine against a
flavivirus. (a) The pKUNdC/C DNA vaccine
contains two CMV promoters in a back-to-back
orientation. The first promoter directs the
transcription of WNVKUN replicon RNA, KUNdC,
which expresses all the nonstructural (NS) genes,
two of the structural genes, pre-membrane (prM)and envelope (E), and a truncated capsid (C)
gene (dC), which is missing codons 18 through
100. The transcription of KUNdC RNA with an
authentic 5¢ terminus is ensured by placing the
first nucleotide of WNVKUN cDNA sequence
directly after the CMV promoter. Generation of an
authentic 3¢ terminus is ensured by addition of
an antigenomic copy of hepatitis delta virus
ribozyme sequence (HDVr) followed by the SV40
poly A signal (pA) directly after the last
nucleotide of WNVKUN cDNA16. The secreted prM
and E proteins produced from pKUNdC RNA
form highly immunogenic prM-E particles.
KUNdC RNA also encodes nonstructural proteins
that have RNA replicase activity, which amplifies
KUNdC RNA. The second CMV promoter directs
transcription of full-length mRNA of the WNVKUN
capsid gene, which encodes the full-length (105
amino acids) mature C protein. (b) In combi-nation, the two RNAs produce all three WNVKUN
structural proteins (prM, E and capsid) required
to package the amplified KUNdC RNA into
secreted SRIPs. SRIPs infect more cells and
release KUNdC RNA into the cytoplasm. The
KUNdC RNA again amplifies itself, producing
more prM-E particles and nonstructural proteins.
As the KUNdC RNA does not code for the full-length C gene, no additional SRIPs are produced in SRIP-infected cells and further spread is thereby
prevented. Both pKUNdC/C DNA–transfected cells and SRIP-infected cells stimulate humoral immunity through secretion of immunogenic prM-E particles
(eliciting virus-neutralizing antibodies) as well as NS1 protein (eliciting protective cytolytic antibodies). These cells also stimulate cellular immunity
(e.g., CD8+ cytotoxic T lymphocytes, CTLs) specific for intracellular nonstructural proteins.
Structural Nonstructural (NS)pKUN1
a
b
5’U
TR
3’U
TR
prM
prM
18
prM NS1-5
NS1-5
RNAs
Proteins
CMV
CMV CMVC
C E
E
EC
pKUNdC/C
Capsid mRNA
pKUNdC/C
ProtectiveNS1 Ab
ProtectiveNS1 Ab
pKUNdC/C transfected cell Secreted SRIPs SRIP infected cellNo further
viral spreadSRIP infection
prM
prMprM
NS1-5
NS1-5NS1-5
NS1-5
EprM NS1-5E
EEprM NS1-5
NS1-5E
E
NS
CTLs
NS
NS
CTLs
NS1 NS1
Nucleus
Cytoplasm
Nucleus
DNA
C
Cytoplasm
NS
KUNdC RNArelease
KUNdC RNApackaging
NeutralizingAb
SRIPformation
prM
NS
EC
Self-replicating KUNdC RNA
dC100
1 3 5pA
pA
HDVr
HDVr
2A 2B 4A 4B
Self-replicationSelf-replication
prME
ENS1-5
Secreted prMEimmunogenic particles
prMprM
5 72 VOLUME 26 NUMBER 5 MAY 2008 NATURE BIOTECHNOLOGY
L E T T E R S
©20
08 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
ureb
iote
chno
logy
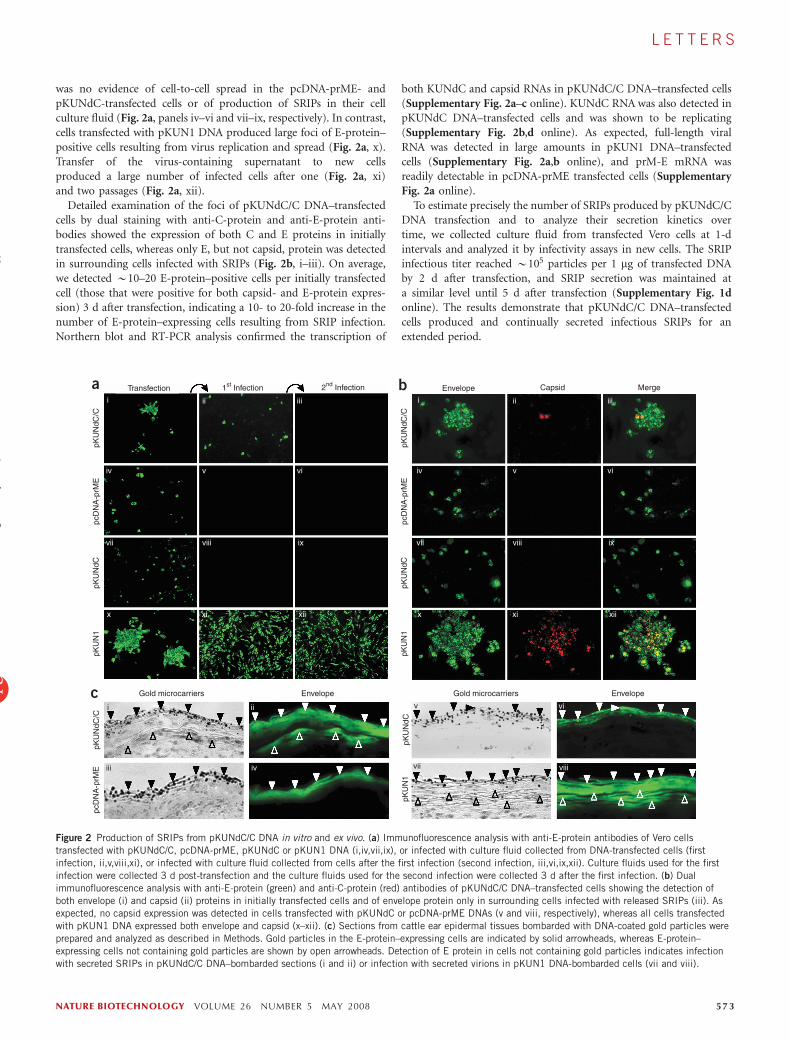
was no evidence of cell-to-cell spread in the pcDNA-prME- andpKUNdC-transfected cells or of production of SRIPs in their cellculture fluid (Fig. 2a, panels iv–vi and vii–ix, respectively). In contrast,cells transfected with pKUN1 DNA produced large foci of E-protein–positive cells resulting from virus replication and spread (Fig. 2a, x).Transfer of the virus-containing supernatant to new cellsproduced a large number of infected cells after one (Fig. 2a, xi)and two passages (Fig. 2a, xii).
Detailed examination of the foci of pKUNdC/C DNA–transfectedcells by dual staining with anti-C-protein and anti-E-protein anti-bodies showed the expression of both C and E proteins in initiallytransfected cells, whereas only E, but not capsid, protein was detectedin surrounding cells infected with SRIPs (Fig. 2b, i–iii). On average,we detected B10–20 E-protein–positive cells per initially transfectedcell (those that were positive for both capsid- and E-protein expres-sion) 3 d after transfection, indicating a 10- to 20-fold increase in thenumber of E-protein–expressing cells resulting from SRIP infection.Northern blot and RT-PCR analysis confirmed the transcription of
both KUNdC and capsid RNAs in pKUNdC/C DNA–transfected cells(Supplementary Fig. 2a–c online). KUNdC RNA was also detected inpKUNdC DNA–transfected cells and was shown to be replicating(Supplementary Fig. 2b,d online). As expected, full-length viralRNA was detected in large amounts in pKUN1 DNA–transfectedcells (Supplementary Fig. 2a,b online), and prM-E mRNA wasreadily detectable in pcDNA-prME transfected cells (SupplementaryFig. 2a online).
To estimate precisely the number of SRIPs produced by pKUNdC/CDNA transfection and to analyze their secretion kinetics overtime, we collected culture fluid from transfected Vero cells at 1-dintervals and analyzed it by infectivity assays in new cells. The SRIPinfectious titer reached B105 particles per 1 mg of transfected DNAby 2 d after transfection, and SRIP secretion was maintained ata similar level until 5 d after transfection (Supplementary Fig. 1donline). The results demonstrate that pKUNdC/C DNA–transfectedcells produced and continually secreted infectious SRIPs for anextended period.
Transfectiona b
c Gold microcarriers Envelope Gold microcarriers Envelope
pKU
NdC
/Cpc
DN
A-p
rME
pcD
NA
-prM
EpK
UN
dCpK
UN
dC/C
pKU
N1
pKU
N1
pKU
NdC
pKU
NdC
/Cpc
DN
A-p
rME
pKU
NdC
pKU
N1
Envelope Capsid Merge1st Infection 2nd Infection
i
i ii
iii
v
viiiv
vi
viii
ii iii
iv v vi
vii viii ix
x xi xii
i ii iii
iv v vi
vii viii ix
x xi xii
Figure 2 Production of SRIPs from pKUNdC/C DNA in vitro and ex vivo. (a) Immunofluorescence analysis with anti-E-protein antibodies of Vero cells
transfected with pKUNdC/C, pcDNA-prME, pKUNdC or pKUN1 DNA (i,iv,vii,ix), or infected with culture fluid collected from DNA-transfected cells (first
infection, ii,v,viii,xi), or infected with culture fluid collected from cells after the first infection (second infection, iii,vi,ix,xii). Culture fluids used for the first
infection were collected 3 d post-transfection and the culture fluids used for the second infection were collected 3 d after the first infection. (b) Dual
immunofluorescence analysis with anti-E-protein (green) and anti-C-protein (red) antibodies of pKUNdC/C DNA–transfected cells showing the detection of
both envelope (i) and capsid (ii) proteins in initially transfected cells and of envelope protein only in surrounding cells infected with released SRIPs (iii). Asexpected, no capsid expression was detected in cells transfected with pKUNdC or pcDNA-prME DNAs (v and viii, respectively), whereas all cells transfected
with pKUN1 DNA expressed both envelope and capsid (x–xii). (c) Sections from cattle ear epidermal tissues bombarded with DNA-coated gold particles were
prepared and analyzed as described in Methods. Gold particles in the E-protein–expressing cells are indicated by solid arrowheads, whereas E-protein–
expressing cells not containing gold particles are shown by open arrowheads. Detection of E protein in cells not containing gold particles indicates infection
with secreted SRIPs in pKUNdC/C DNA–bombarded sections (i and ii) or infection with secreted virions in pKUN1 DNA-bombarded cells (vii and viii).
NATURE BIOTECHNOLOGY VOLUME 26 NUMBER 5 MAY 2008 57 3
L E T T E R S
©20
08 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
ureb
iote
chno
logy

The delivery of plasmid DNA to host cells by gene-gun bombard-ment with DNA-coated gold particles is more efficient than intra-muscular (i.m.) injection, with substantially less DNA required togenerate the same immune response18,19. We used initial ex vivotesting involving cattle ear epidermal cells to evaluate the suitabilityof gene-gun immunization for the delivery of pKUNdC/C DNA intohost cells. Light microscopy of tissue sections prepared from smallareas of excised ear epidermis bombarded with pKUNdC/C DNA–coated gold particles and cultivated for 2 d revealed gold particlesembedded in the epidermal layer (Fig. 2c, i). Immunofluorescenceassays using anti-E-protein antibodies clearly showed the expression ofE protein in gold particle–containing cells (Fig. 2c, ii), indicatingsuccessful delivery of pKUNdC/C DNA and expression of encodedproteins. In addition, some adjacent cells not containing gold particleswere positive for E-protein expression (Fig. 2c, ii), indicating infectionof these cells with secreted SRIPs. As expected, bombardment withpcDNA-prME or pKUNdC DNA–coated gold particles resulted inexpression of E protein in transfected cells containing gold particlesbut no signs of E protein in cells not containing gold particles (Fig. 2c,iii–iv and v–vi, respectively). E protein was detected in large areas,both in cells that contained and in cells that did not contain goldparticles (Fig. 2c, vii–viii), after cells were bombarded with pKUN1DNA–coated gold particles, illustrating efficient virus spread tosurrounding tissues.
The immunogenicity of gene gun–delivered pKUNdC/C DNA wasfirst evaluated in BALB/c mice after a single gene-gun immunizationwith 0.02, 0.002 and 0.0002 pmol of pKUNdC/C, pKUNdC, pKUN1or pcDNA-prME DNA. Levels of WNVKUN-specific antibodies(determined by enzyme-linked immunosorbent assay (ELISA) onfixed WNVKUN-infected cells) in mice immunized with 0.02 and0.002 pmol of pKUNdC/C and pKUN1 DNA vaccines were on averageB10- to 20-fold higher than those from pcDNA-prME–immunizedmice, whereas immunization with pKUNdC DNA resulted in very lowtiters of antibodies at all doses (Fig. 3a). Sera of mice immunized withthe two higher doses of each DNA vaccine was also analyzed for
WNVKUN-neutralizing antibodies (Table 1). The levels of neutralizingantibodies after pKUN1 and pKUNdC/C immunizations were onaverage higher than after pcDNA-prME immunizations at corre-sponding DNA doses, particularly for the 0.002 pmol dose. Neutraliz-ing antibodies were below the level of detection after pKUNdCvaccination with both doses of DNA. NS3-specific CD8+ T-cellresponses were also detected after vaccination with the two higherdoses of pKUNdC/C and pKUN1 DNA (Fig. 3b). As expected,no responses to the NS3 epitope were detected in pcDNA-prME–immunized mice (Fig. 3b). CD8+ T-cell responses after immunizationwith pKUNdC DNA were not analyzed.
To determine the protective efficacy of the DNA vaccinations,mice were challenged intraperitoneally with WNVNY99. All controlmice (100%) showed signs of morbidity and 66% were euthanizedowing to the development of symptoms associated with centralnervous system (CNS) disease, such as a hunched posture, flaccidhind leg paralysis or moribundity. All mice vaccinated with all doses ofpKUN1 DNA and the two highest doses of pKUNdC/C or pcDNA-prME DNA did not develop CNS disease (Table 1). None of the miceimmunized with the two highest doses of pKUNdC/C DNA developedany signs of disease, whereas 16% and 25% of mice immunizedwith 0.02 pmol and 0.002 pmol of pcDNA-prME DNA, respectively,showed signs of infection. At the lowest dose of 0.0002 pmol,33% of mice immunized with pKUNdC/C DNA and 50% ofmice immunized with pcDNA-prME DNA developed CNS disease.All animals immunized with all three doses of pKUNdC DNAshowed signs of infection, with 33%, 42% and 75% progressing toCNS disease at 0.02 pmol, 0.002 pmol and 0.0002 pmol doses,respectively. Overall, the results show that the pKUNdC/C DNAvaccine provided improved protection over DNA-based vaccinesthat do not produce SRIPs, particularly when lower doses of DNAare used for immunization.
Having established the efficacy of the pKUNdC/C DNA immuniza-tion in mice, the immunogenicity of pKUNdC/C DNA vaccine wasthen evaluated in horses to assess whether the same effects are evident
Figure 3 Immunogenicity of pKUNdC/C DNA in
mice and horses. (a) Titers of WNVKUN-specific
antibodies in sera of mice immunized with
pKUN1, pKUNdC/C, pKUNdC and pcDNA-prME
DNA vaccines. Four-week-old female BALB/c
mice (n ¼ 12) were immunized once by gene-
gun bombardment of gold particles coated with
indicated doses of plasmid DNAs and analyzedfor antibody responses at 2 weeks after
immunization by ELISA on fixed WNVKUN-
infected cells. (b) CD8+ T-cell responses in mice
immunized with pKUN1, pKUNdC/C and pcDNA-
prME DNA vaccines. Splenocytes from
immunized mice (n ¼ 3) were assayed for CD8+
T cells specific for the NS3 epitope GYISTRVEL30
by ex vivo ELISPOT. (c,d) WNVKUN- and
WNVNY99-specific neutralizing antibody responses
(c and d, respectively) in serum from pKUNdC/C
DNA–immunized horses. Groups of two horses
were immunized two or three times by
intramuscular (i.m.) inoculation or gene gun–
mediated bombardment with indicated doses of
pKUNdC/C DNA and serum analyzed at 4 weeks
after each immunization for neutralizing
antibodies using a microneutralization protocol16.
Asterisks indicate horses that did not receive the
third immunization. Control sera in this assay included (i) serum from horse R injected i.m. with 50 pmol of pcDNA3, (ii) serum from horse 6, whichreceived gene gun–mediated bombardment with 0.5 pmol of pcDNA3 and (iii) serum from a horse M naturally infected with WNVKUN (KUN+).
12,000a
c d
b600
500
400
300
200
100
0
10,000Mice Mice
Rec
ipro
cal E
LIS
A e
nd-p
oint
tite
rR
ecip
roca
l neu
tral
izin
g A
b tit
er
IFN
Y s
pots
/106 s
plen
ocyt
es
8,000
6,000
4,000
2,000
640Horses HorsesWNVKUN WNVNY99
1ximm2ximm3ximmKUN+
1ximm2ximm3ximmKUN+
320
16080
40
2010
5
200 40I.M. G.G.
0.4 Controls2F* 3* J 5 9 12 S 8 R 6 M
200 40I.M. G.G.
0.4 Controls2F* 3* J 5 9 12 S 8 R 6 M
<5
Rec
ipro
cal n
eutr
aliz
ing
Ab
titer 640
320
160
8040
2010
5<5
pKUN1 pKUNdC/C pKUNdC pcDNA-prME
pKUN1 pKUNdC/C pcDNA-prME
0.02
0.02
0.00
2
0.00
2
0.02
0.00
2
0.02
0.00
2
0.00
02
0.02
0.00
20.
0002
0.02
0.00
20.
0002
0.02
0.00
20.
0002
Con
trol
0
5 74 VOLUME 26 NUMBER 5 MAY 2008 NATURE BIOTECHNOLOGY
L E T T E R S
©20
08 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
ureb
iote
chno
logy

in a large animal. As horses are readily infected with WNVNY99 andcan develop potentially lethal symptoms20, vaccinating horses againstWNVNY99 is an important veterinary consideration. Two differentinoculation routes, gene-gun bombardment and i.m. injection, andtwo different DNA doses for each immunization route, 2 pmol or0.4 pmol for gene gun and 200 pmol or 40 pmol for i.m. injection,were used to immunize groups of two horses. Five hundred p.s.i. wasselected as the optimal pressure for gene gun–mediated delivery ofgold particles to the epidermis (Supplementary Fig. 3 online). Bloodsamples were collected 4 weeks after each immunization and assayedfor neutralizing antibodies specific for WNVNY99 and WNVKUN. Allimmunized horses showed responses to both viruses and the higherdoses of DNA induced neutralizing antibody responses againstWNVKUN after only a single immunization (Fig. 3c). The titers ofneutralizing antibodies increased with each vaccination and after twoand three gene-gun inoculations neutralization titers were broadlysimilar for WNVNY99 and WNVKUN (Fig. 3c,d). After three gene gun–vaccinations, these titers were only two- to four-fold lower than theneutralizing antibody titer detected in the horse naturally infectedwith WNVKUN (Fig. 3c, horse M). Based on other West Nile virusequine vaccination studies8,21, the levels of neutralizing antibodiesachieved even after a single immunization with pKUNdC/C DNA arehighly likely to protect horses from WNVNY99 infection. Immuniza-tion with pKUNdC/C DNA also elicited high titers of antibodies toNS1 (Supplementary Fig. 4 online). Antibodies to flavivirus NS1protein have been shown to mediate protective immunity throughcomplement-dependent cytolysis of infected cells and/or complement-and Fc-g receptor–independent mechanisms22,23. In addition toprotective antibodies to NS1, pKUNdC/C DNA vaccination alsoelicited cytotoxic T-cell responses to other nonstructural proteins(e.g., NS3; Fig. 3b), thus providing additional mechanisms of protec-tion compared with conventional nonreplicating DNA and inactivatedvirus vaccines.
The strategy developed here to deliver separate complementing viralelements with a single plasmid DNA offers an attractive alternative to
conventional vaccine approaches as it closelymimics live viral infection without producingan infectious virus. Our approach exploits theconvenience of manufacturing and deliveringDNA vaccines, the ability of flavivirus repli-cons to replicate autonomously, the immuno-genicity of flavivirus prM-E particles and theability to produce single-round infectiousparticles. Production of infectious virus bythe pKUNdC/C system or similar systemsbased on other flaviviruses by recombinationbetween replicating (KUNdC) and nonrepli-cating (capsid) RNAs produced frompKUNdC/C DNA is unlikely. Despite numer-ous complementation experiments by us andothers, recombination leading to generation ofinfectious recombinant flaviviruses has neverbeen demonstrated24,25. Furthermore, there isno evidence of recombination in the naturalenvironment between different strains of WestNile virus, yellow fever virus or tick-borneencephalitis virus26, and data suggestingrecombination of other flaviviruses havebeen criticized27. With the high degree ofsimilarity in genome structure and similarRNA replication and packaging strategies
among flaviviruses28, our split-genome vaccine approach should bereadily amenable to other pathogenic flaviviruses. It may proveparticularly useful for the design of DNA vaccines against denguevirus and Japanese encephalitis virus.
METHODSAll animal experiments were approved by the University of Queensland Animal
Ethics Committee (SMMS/455/06/NIH/NHMRC and SMMS/703/07).
Plasmid DNA construction. pKUNdC/C and pKUNdC DNA were constructed
from pKUN1 plasmid16 encoding full-length cDNA of WNVKUN RNA using
standard recombinant DNA techniques. pcDNA-prME plasmid was con-
structed by cloning WNVKUN prM-E genes into pcDNA3.1 expression vector
(Invitrogen). The details of construction can be obtained from corresponding
author upon request.
DNA transfection and immunofluorescence assay. Vero cells at 80–90%
confluency were transfected with plasmid DNAs using FuGENE 6 transfection
reagent (Roche) following the protocol recommended by the manufacture.
Transfected cells were maintained in Gibco DMEM (Invitrogen) with 2%
(vol/vol) Gibco FBS (Invitrogen) for 2–5 d in a humidified 5% CO2 chamber.
Culture fluid from transfected cells were either collected and kept in –80 1C until
further use or discarded before fixing cells with acetone/methanol (50/50) at
–20 1C for 20 min. Fixed cells were stained with primary antibodies for envelope
and/or capsid protein(s), followed by goat anti-mouse Alexa Fluor 488 (Invitro-
gen) or goat anti-rabbit Texas Red (Invitrogen) secondary antibodies, respec-
tively. Fluorescence images were viewed with Carl Zeiss microscope (Carl Zeiss).
Infectivity assay of secreted particles. Infectious titers of SRIPs were deter-
mined by immunofluorescence assays using anti-E-protein antibodies of Vero
cells infected with serial tenfold dilutions of culture fluid collected from
transfected cells. Cells positive for E protein were counted at appropriate
dilution and the number of SRIPs per 1 mg of transfected pKUNdC/C
DNA were calculated based on the following formula: SRIPs/1 mg pKUNdC/C
DNA ¼ N � dilution factor � 1.77 � 6, where N is a number of E-protein–
positive cells.
Histology. To detect WNVKUN protein expression in cattle ear epidermal cells,
tissue sections were excised from the small areas of cattle ear bombarded with
Table 1 Neutralizing antibody induction and protection from challenge with WNVNY99
Vaccination groupa
% Morbidity and mortality after intraperitoneal challenge
with WNVNY99c
DNA
Dose
(pmol)
WNVKUN-neutralizing
antibody titerb% Morbidityd
(sick/challenged)
% Mortalitye
(dead/challenged)
Control 0 ND 100 (12/12) 66 (8/12)
pKUN1 0.02 160, 80, 40 0 (0/12) 0 (0/12)
0.002 40, 40, 20 0 (0/11) 0 (0/11)
0.0002 ND 0 (0/12) 0 (0/12)
pKUNdC/C 0.02 160, 80, 40 0 (0/12) 0 (0/12)
0.002 80, 80, 20 0 (0/11) 0 (0/11)
0.0002 ND 100 (12/12) 33 (4/12)
pKUNdC 0.02 o10, o10, o10 100 (12/12) 33 (4/12)
0.002 o10, o10, o10 100 (12/12) 42 (5/12)
0.0002 ND 100 (12/12) 75 (9/12)
pcDNA-prME 0.02 80, 40, 20 16 (2/12) 0 (0/12)
0.002 20, 20, 10 25 (3/12) 0 (0/12)
0.0002 ND 100 (12/12) 50 (6/12)
aMice were immunized with the indicated doses of pKUN1, pKUNdC/C, pKUNdC or pcDNA-prME DNAs at 4 weeks of age.bSerum from each group were pooled (n ¼ 3–4) into three samples. The WNVKUN-neutralizing antibody titers were measured asdescribed in Methods. The reciprocal of the neutralizing antibody titer is shown for each of the three pooled samples. ND, notdetermined. cMice were challenged intraperitoneally with 100 infectious units of NY99-4132 strain of West Nile virus 3 weeksafter single immunization. dPercentage of mice that developed disease symptoms (severely ruffled fur and lethargy). ePercentageof mice that developed CNS disease symptoms (hunched posture, flaccid hind leg paralysis or moribundity), requiring that theanimals be euthanized.
NATURE BIOTECHNOLOGY VOLUME 26 NUMBER 5 MAY 2008 57 5
L E T T E R S
©20
08 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
ureb
iote
chno
logy

pKUNdC/C DNA–coated gold particles and maintained for 2 d in DMEM
with 10% (vol/vol) FBS. The tissues were then fixed in 10% buffered formalin
overnight and water in cells was progressively replaced with 45 1C ethanol in a
series of increasing ethanol concentrations (70%, 90%, 95% and absolute).
Xylene (100%) was then used to replace the ethanol, before embedding with
paraffin wax. Sections (4–5 mm) were prepared from paraffin-embedded tissues
and mounted on electrostatic Super Frost Plus slides (Menzel-Glaser). Gold
microcarriers were observed with standard white light microscopy, whereas
expression of E protein was detected by immunofluorescence assays using anti-
E-protein antibody.
Gene-gun cartridge preparation. Cartridges with pKUNdC/C, pKUNdC,
pKUN1 or pcDNA-prME DNA–coated gold particles were prepared according
to the standard procedure recommended by the manufacturer (Bio-Rad).
Briefly, DNA purified with Qiagen endotoxin-free mega plasmid DNA
purification kit (Qiagen) was mixed with 1 mm–diameter gold particles at
mg DNA/mg gold particles ratios of 0.01, 0.1 and 1 (depending on the DNA
dose required for a specific experiment) in sterile water containing 0.05 M
spermidine. An equal volume of 1 M CaCl2 was then added to precipitate
plasmid DNA to the gold particle surface at 22 1C for 10 min followed by three
washes with 100% ethanol to remove unbound DNA. DNA–gold particle pellet
was then resuspended in desired volume of 0.05 mg/ml of polyvinylpyrrolidone
(PVP) in 100% ethanol, injected into nitrogen pre-dried Gold-Coate tubing
(Bio-Rad) installed in the tubing preparation station, and was let to settle to the
tube surface for 3 min. Ethanol was then removed; Gold Coat tubing was
rotated for 30 s to allow DNA-gold particles to distribute evenly on the inside
surface of the tubing and was then dried with nitrogen gas for 5 min in a
continuous rotation. The DNA-gold particle–coated tube was then cut to
prepare individual gene-gun cartridges and stored at 4 1C. Each cartridge
contained predetermined amount of DNA-coated particles.
Mouse immunization and West Nile virus challenge. Groups of 11-12 BALB/c
mice 3–4 weeks of age were immunized with 0.02, 0.002 and 0.0002 pmol of
pKUNdC/C, pKUNdC, pKUN1 or pcDNA-prME DNA–coated gold particles
using Helios Gene-Gun (Bio-Rad) bombardment of shaved abdomen skin.
DNA/gold microcarrier cartridges were prepared as described above. All mice
were bled and analyzed for the presence of WNVKUN-specific antibodies by
ELISA on fixed WNVKUN-infected cells as described previously16. The same
mice were then challenged intraperitoneally with 100 infectious units of NY99-
4132 strain of West Nile virus. Challenged mice were closely monitored during
the following 14 d to score disease symptoms. Mice that showed symptoms of
CNS disease (flaccid hind leg paralysis, hunched posture and moribundity)
were euthanized. In a parallel immunization, groups of three BALB/c mice were
immunized with 0.02 and 0.002 pmol of pKUNdC/C, pKUN1, or pcDNA-
prME DNA. At 21 d after immunization, all mice were killed and their spleens
processed for analysis of CD8+ T-cell responses to WNVKUN NS3 epitope by ex
vivo enzyme-linked immunosorbent spot (ELISPOT).
Horse immunizations. Groups of two, female, standard bred horses, 4–8 years
of age, sero-negative to WNVNY99/WNVKUN, were immunized with
pKUNdC/C by gene-gun or intramuscular (i.m.) injections. Horses receiving
pKUNdC/C immunization by gene-gun had a small area on the neck trimmed
with electric clippers, followed by shaving with a razor to remove all hair which
may interfere with the gene-gun delivery efficiency. The shaved site was cleaned
with 70% ethanol and dried before gene-gun bombardment. The helium
pressure setting was set at 500 p.s.i., and DNA was delivered at 0.1 pmol per
shot. Each horse received either 4 shots (0.4-pmol dose) or 20 shots (2-pmol
dose). DNA for i.m. immunizations was prepared in 10 ml sterile PBS at
appropriate concentrations (20 pmol/ml for 200-pmol dose and 4 pmol/ml for
40-pmol dose), and each horse was injected five times with 2 ml in each injection
of PBS-DNA solution into the neck muscle. Sera was collected 4 weeks after each
immunization. The 2nd and 3rd immunizations were given at 4- and 7-week
intervals after the first immunization, respectively. The horses immunized with
the high dose of DNA (200 pmol) by i.m. route had only two immunizations.
Virus neutralization assay. Serum samples from immunized mice and horses
were tested for neutralization of WNVKUN (mice and horses) and WNVNY99
(horses) viruses using a microneutralization assay as described previously16.
Briefly, sera was heat-inactivated at 56 1C and serially diluted by twofold in cell
growth medium. We then added 50 ml of each dilution in quadruplicate to the
wells of a 96-well culture plate. An equal volume of growth medium containing
100 infectious units of virus was then added to each well, and plates were
allowed to incubate at 37 1C with occasional gentle agitation. We added 50 ml of
growth medium containing 104 Vero cells to each well, and plates were
incubated at 37 1C for 5 d. The reciprocal of the serum dilution that completely
(100%) inhibited the formation of viral cytopathic effects was deemed as the
end-point neutralization titer.
Detection of mouse CD8+ T-cell responses by ELISPOT assay. Epitope-
specific gamma interferon (IFN-g)-secreting cells were enumerated by an
enzyme-linked immunospot (ELISPOT) assay as described previously29 except
using MultiScreen-IP hydrophobic PVDF membrane microtiter plates (Milli-
pore) and recombinant human IL-2 (kindly provided by Cetus Corp.) (25 IU/
ml). The CD8+ T-cell peptide epitope, GYISTRVEL30 (AusPep Ltd.), was used,
which we have confirmed represents a CD8 T-cell epitope for WNVKUN.
Note: Supplementary information is available on the Nature Biotechnology website.
ACKNOWLEDGMENTSWe thank Jane Pollitt, Joanne Meers and Paul Mills (all from School ofVeterinary Sciences, University of Queensland) for valuable assistance indesigning and performing horse vaccination experiments; Wai Yuen Cheah,Kim Pham and Natalie Prow (University of Queensland) for assistance withmouse experiments and serological analysis of mouse and horse sera; GregSmith, Alyssa Pyke and Bruce Harrower (all from Queensland Heath Forensicand Scientific Services) for generously providing PC3 animal facilities andlogistical assistance for mouse challenge studies; and James Doecke (QIMR) forassistance with statistics.
AUTHOR CONTRIBUTIONSD.C. Chang designed experiments, performed the majority of the experiments,analyzed data and wrote the manuscript; W.J.L. designed experiments,immunized mice and analyzed data; I.A. designed, performed and analyzedELISPOT assays; D.C. Clark assisted with mice experiments; C.C.P. providedveterinary expertise and supervised horse experiments; A.S. designed theELISPOT assays and wrote the manuscript; R.A.H. designed animal experiments,analyzed data and wrote the manuscript; and A.A.K. generated the idea,designed and coordinated the research, analyzed data and wrote the manuscript.
Published online at http://www.nature.com/naturebiotechnology/
Reprints and permissions information is available online at http://npg.nature.com/
reprintsandpermissions
1. Pugachev, K.V., Guirakhoo, F., Trent, D.W. & Monath, T.P. Traditional and novelapproaches to flavivirus vaccines. Int. J. Parasitol. 33, 567–582 (2003).
2. Chang, G.J., Davis, B.S., Hunt, A.R., Holmes, D.A. & Kuno, G. Flavivirus DNA vaccines:current status and potential. Ann. NY Acad. Sci. 951, 272–285 (2001).
3. Phillpotts, R.J., Venugopal, K. & Brooks, T. Immunisation with DNA polynucleotidesprotects mice against lethal challenge with St. Louis encephalitis virus. Arch. Virol.141, 743–749 (1996).
4. Konishi, E., Yamaoka, M., Khin Sane, W., Kurane, I. & Mason, P.W. Induction ofprotective immunity against Japanese encephalitis in mice by immunization with aplasmid encoding Japanese encephalitis virus premembrane and envelope genes.J. Virol. 72, 4925–4930 (1998).
5. Colombage, G., Hall, R., Pavy, M. & Lobigs, M. DNA-based and alphavirus-vectoredimmunisation with prM and E proteins elicits long-lived and protective immunityagainst the flavivirus, Murray Valley encephalitis virus. Virology 250, 151–163 (1998).
6. Kochel, T. et al. Inoculation of plasmids expressing the dengue-2 envelope gene elicitneutralizing antibodies in mice. Vaccine 15, 547–552 (1997).
7. Aberle, J.H. et al. A DNA immunization model study with constructs expressing thetick-borne encephalitis virus envelope protein E in different physical forms. J. Immunol.163, 6756–6761 (1999).
8. Davis, B.S. et al. West Nile virus recombinant DNA vaccine protects mouse and horsefrom virus challenge and expresses in vitro a noninfectious recombinant antigen thatcan be used in enzyme-linked immunosorbent assays. J. Virol. 75, 4040–4047 (2001).
9. Hall, R.A. & Khromykh, A.A. West Nile virus vaccines. Expert Opin. Biol. Ther. 4,1295–1305 (2004).
10. Anraku, I. et al. Kunjin virus replicon vaccine vectors induce protective CD8+ T-cellimmunity. J. Virol. 76, 3791–3799 (2002).
11. Kofler, R.M. et al. Mimicking live flavivirus immunization with a noninfectious RNAvaccine. Proc. Natl. Acad. Sci. USA 101, 1951–1956 (2004).
12. Seregin, A. et al. Immunogenicity of West Nile virus infectious DNA and its non-infectious derivatives. Virology 356, 115–125 (2006).
5 76 VOLUME 26 NUMBER 5 MAY 2008 NATURE BIOTECHNOLOGY
L E T T E R S
©20
08 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
ureb
iote
chno
logy

13. Mason, P.W., Shustov, A.V. & Frolov, I. Production and characterization of vaccinesbased on flaviviruses defective in replication. Virology 351, 432–443 (2006).
14. Hall, R.A., Broom, A.K., Smith, D.W. & Mackenzie, J.S. The ecology and epidemiologyof Kunjin virus. Curr. Top. Microbiol. Immunol. 267, 253–269 (2002).
15. Hall, R.A. et al. Loss of dimerisation of the nonstructural protein NS1 of Kunjin virusdelays viral replication and reduces virulence in mice, but still allows secretion of NS1.Virology 264, 66–75 (1999).
16. Hall, R.A. et al. DNA vaccine coding for the full-length infectious Kunjin virus RNAprotects mice against the New York strain of West Nile virus. Proc. Natl. Acad. Sci. USA100, 10460–10464 (2003).
17. Co, M.D., Terajima, M., Cruz, J., Ennis, F.A. & Rothman, A.L. Human cytotoxicT lymphocyte responses to live attenuated 17D yellow fever vaccine: identification ofHLA-B35-restricted CTL epitopes on nonstructural proteins NS1, NS2b, NS3, and thestructural protein E. Virology 293, 151–163 (2002).
18. Raz, E. et al. Intradermal gene immunization: the possible role of DNA uptake in theinduction of cellular immunity to viruses. Proc. Natl. Acad. Sci. USA 91, 9519–9523(1994).
19. Pertmer, T.M. et al. Gene gun-based nucleic acid immunization: elicitation of humoraland cytotoxic T lymphocyte responses following epidermal delivery of nanogramquantities of DNA. Vaccine 13, 1427–1430 (1995).
20. Castillo-Olivares, J. & Wood, J. West Nile virus infection of horses. Vet. Res. 35,467–483 (2004).
21. Ng, T. et al. Equine vaccine for West Nile virus. Dev. Biol. (Basel) 114, 221–227 (2003).
22. Schlesinger, J.J., Brandriss, M.W., Putnak, J.R. & Walsh, E.E. Cell surface expressionof yellow fever virus non-structural glycoprotein NS1: consequences of interaction withantibody. J. Gen. Virol. 71, 593–599 (1990).
23. Chung, K.M. et al. Antibodies against West Nile Virus nonstructural protein NS1prevent lethal infection through Fc gamma receptor-dependent and -independentmechanisms. J. Virol. 80, 1340–1351 (2006).
24. Khromykh, A.A., Sedlak, P.L. & Westaway, E.G. cis- and trans-acting elements inflavivirus RNA replication. J. Virol. 74, 3253–3263 (2000).
25. Lindenbach, B.D. & Rice, C.M. trans-Complementation of yellow fevervirus NS1 reveals a role in early RNA replication. J. Virol. 71, 9608–9617(1997).
26. Twiddy, S.S. & Holmes, E.C. The extent of homologous recombination in members ofthe genus Flavivirus. J. Gen. Virol. 84, 429–440 (2003).
27. Monath, T.P. et al. Recombination and flavivirus vaccines: a commentary. Vaccine 23,2956–2958 (2005).
28. Lindenbach, B.D. & Rice, C. Flaviviridae: the viruses and their replication. in FieldsVirology. 4th edn. (eds. Knipe, D.M. & Howley, P.M.) 991–1041 (Lippincott Williams &Wilkins, Philadelphia, 2001).
29. Le, T.T. et al. Cytotoxic T cell polyepitope vaccines delivered by ISCOMs. Vaccine 19,4669–4675 (2001).
30. Spaulding, A.C., Kurane, I., Ennis, F.A. & Rothman, A.L. Analysis of murine CD8(+) T-cell clones specific for the Dengue virus NS3 protein: flavivirus cross-reactivity andinfluence of infecting serotype. J. Virol. 73, 398–403 (1999).
NATURE BIOTECHNOLOGY VOLUME 26 NUMBER 5 MAY 2008 57 7
L E T T E R S
©20
08 N
atur
e P
ublis
hing
Gro
up
http
://w
ww
.nat
ure.
com
/nat
ureb
iote
chno
logy




















