
The Stem Cell Marker CD133 (Prominin-1) Is Expressed in Various Human Glandular Epithelia
Upload
independentCategory
view
2download
0
Simultaneous Quantification of Human GlandularKallikrein 2 and Prostate-Specific Antigen mRNAs inPeripheral Blood from Prostate Cancer Patients
Alice Ylikoski,* Matti Karp,* Kim Pettersson,*Hans Lilja,† and Timo Lovgren*
From the Department of Biotechnology,* University of Turku,
Turku, Finland; and the Department of Laboratory Medicine,†
Division of Clinical Chemistry, Lund University, University
Hospital Malmo, Malmo, Sweden
We present a multiplexed and internally calibratedquantitative reverse transcription-PCR (QRT-PCR) as-say to detect human glandular kallikrein 2 (hK2) andprostate-specific antigen (PSA) transcripts in bloodsamples from healthy subjects and prostate cancer(PC) patients. The assay detected 50 copies of hK2 andPSA mRNA, and 1 PSA- and 10 hK2-expressing LNCaPcells in the presence of 2.5 � 106 PSA- and hK2-negative cells. In PC patients, 20 of 25 and 19 of 25gave detectable PSA and hK2 mRNAs, respectively.Number of hK2 mRNA copies was significantly higherthan that of PSA mRNA copies in patients with biochem-ically progressive (P � 0.02) PC, and with locally ad-vanced and metastasized (P � 0.004) PC. Patients withrapidly progressive and hormone refractory PC gavedetectable hK2 mRNA only in 2 of 8 and PSA mRNA in 3of 8 patients. Neither PSA nor hK2 mRNAs were detectedin 16 healthy subjects. PSA and hK2 discriminated PCpatients with biochemically progressive and advanceddisease from the controls and from the aggressive dis-tant metastatic disease. The assay provides a reliablequantification of the number of hK2 and PSA mRNAcopies, allows to discriminate PC cases from healthysubjects, and offers a tool for further studies on molec-ular staging of PC. (J Mol Diag 2001, 3:111–122)
Prostate cancer (PC) is one of the most common malig-nancies in men in western countries. Recently, the inci-dence of PC has increased due to enhanced diagnostictools and efficient screening programs that are usuallybased on digital rectal examination and serum prostate-specific antigen (PSA) test. The serum PSA screening iscapable of detecting organ-confined or localized can-cers that may be cured surgically.1 Detection of ad-vanced cancers is also important because metastaticmalignancies require different therapeutic approachesthan the organ-confined cancers.2 However, the numberof “micrometastatic” circulating tumor cells can be very
small and therefore the minimal spread of the cancer isdifficult to detect by routine examinations. Traditionally,metastatic cancer cells have been detected in lymphnode samples using histological and immuno-histochem-ical techniques,3 but this has led to a need for newmethods capable of detecting even smaller number ofcirculating cancer cells to identify more accurately pa-tients with extraprostatic disease.
During the last few years, new reverse transcription-polymerase chain reaction (RT-PCR) assays have beendeveloped aiming to identify extraprostatic tumor cells bydetecting mRNA markers such as PSA,4–9 human glan-dular kallikrein 2 (hK2),10–12 and prostate-specific mem-brane antigen (PSMA)13,14 to mention the most com-monly used markers. The assays have been validatedwith different biological samples, such as peripheralblood, lymph nodes, and bone marrow from PC patients.Most of the assays have been designed to amplify thereverse transcribed target with one round of PCR or withnested-PCR consisting of two rounds of PCR and severaltens of amplification cycles. Thereafter, the amplificationproducts are detected by gel electrophoresis and radio-active labeling. These assays provide qualitative results(positive or negative result in respect to the PCR product)and commonly use the expression of a housekeepinggene to evaluate whether the quality of RNA is goodenough for the RT-PCR amplification. Hence the resultsfrom different RT-PCR studies aiming to prove the clinicalusefulness of the method have been controversial.15–21
Many of the research groups have stated that the largevariations in the results may be due to the various RT-PCR assays each designed and validated differently inthe absence of the basis of international standardizationof the procedures, thus proposing the need for morestandardized and quantitative RT-PCR (QRT-PCR) as-says.22–24
The first QRT-PCR assays exploited endogenous RNAstandards expressed in the cell.25 Usually the endoge-
Supported by grants from the Biomed 2 Program, area 4.1.7. (ContractBMH4-CT96–0453); the Swedish Cancer Society (project number 3555);the Faculty of Medicine at Lund University Hospital, Malmo; the CrafoordFoundation; the Gunnar, Arvid and Elisabeth Nilsson Foundation; Funda-cion Frederico S.A; and Academy of Finland (project 45252).
Accepted for publication May 25, 2001.
Address reprint requests to Professor Timo Lovgren, Department ofBiotechnology, University of Turku, Tykistokatu 6 A 6th floor, FIN-20520Turku, Finland. E-mail: [email protected].
Journal of Molecular Diagnostics, Vol. 3, No. 3, August 2001
Copyright © American Society for Investigative Pathology
and the Association for Molecular Pathology
111

nous standard is mRNA expressed by a housekeepinggene, such as �-actin or glyceraldehyde 3-phosphatedehydrogenase. The endogenous standard can be co-amplified with the target mRNA using their own primers inthe same RT-PCR reaction, thus controlling the amplifi-cation steps. The amount of target mRNA is expressed asa ratio of target to standard signals, because the numberof endogenous mRNA standard copies are not known;therefore, this approach is often called as semiquantita-tive. In addition to the fact that the exact number of targetmRNA molecules cannot be shown, another drawback isthat it will not be reproducible and accurate to determine therare or low copy number target mRNA using the common orhigh copy number endogenous standard mRNA.
Lately, the commonly used standard has been an ex-ogenous RNA or DNA containing sequences of the targetmRNA hence allowing co-amplification of the standardand the target with the same primers and the sameamplification efficiency of the two. The first assays withthe exogenous standard used an approach in whichserial dilutions of standard with a known amount of mol-ecules were mixed with a constant amount of sam-ple.26–28 This approach requires multiple tubes for theanalysis of one sample, and the quantification of thetarget is based on the determination of an equivalencypoint between the amount of the exogenous standardand the target amplification products. At present, some ofthe developed assays exploit an external calibrationcurve and need only one tube for the analysis of onesample.29–31 In this case, a constant amount of exoge-nous standard is mixed with the samples and the calibra-tors, and the target transcripts are quantified after ampli-fication by calculating the target to standard ratio in thesample and comparing the ratio to that in a calibrationcurve. In addition, if the exogenous mRNA standard isused, it will be possible to control the variations of anassay starting from the RNA extraction step.9
We have recently presented a QRT-PCR assay for thedetection of PSA mRNA9,24 and our present aim was todevelop a QRT-PCR assay for the simultaneous detectionof hK2 and PSA mRNAs in blood samples. The QRT-PCRassay uses an external calibration curve and two highlytarget-like exogenous, internal standard (IS) mRNAs, IS-hK2 and IS-PSA for the specific quantification of hK2 andPSA mRNAs, respectively. To study the detection of hK2and PSA in biological samples the multiplexed QRT-PCRassay was applied on cultured LNCaP cell samples andblood samples from 16 healthy volunteers and 25 PCpatients.
Materials and Methods
Cell Lines
The PSA- and hK2-expressing human prostatic carci-noma cell line LNCaP and the non-PSA- and non-hK2-producing mouse myeloma cell line SP2/0 were obtainedfrom the American Type Culture Collection. The cell lineswere cultured in flasks containing Dulbecco’s modifiedEagle’s medium (Life Technologies, GIBCO BRL, Grand
Island, NY) with 100 ml/L fetal bovine serum (Hyclone,Logan, UT), and maintained in a 5% CO2 incubator at37°C. The medium for LNCaP culture was supplementedwith 1 nmol/L synthetic hormone methyltrienolone(R1881) (New England Nuclear, Boston, MA). The cellswere grown until near confluency, detached, washed withphosphate-buffered saline, and counted. Unlike theSP2/0 cells, the LNCaP cells were detached by trypsin-EDTA treatment. The SP2/0 samples of 2.5 � 106 cellsand the dilutions of LNCaP cells were stored at �70°Cuntil RNA extraction.
Blood Specimens
Blood samples (EDTA) of 5 ml were collected from 25patients with PC, 9 healthy women, and 7 healthy menvolunteers. The healthy volunteers were included as con-trols in the study. The patients were divided into threegroups based on their disease stage. Group A (n � 4)consisted of patients with biochemically progressive PCas detected by rising serum PSA. Patients in group Awere under watchful waiting and no metastases hadbeen detected. Group B (n � 13) consisted of patientswith locally advanced and metastasized (regional lymphnode and bone metastases) disease under hormonaltreatment. Group C consisted of hormonally treated pa-tients (n � 8) with rapidly progressive, hormone refrac-tory PC and multiple distant metastases. The study pro-tocol was in accordance with Helsinki Declaration of1975, as revised in 1983. Nucleated blood cells wereisolated as described earlier,9 and after the isolation thecell pellets were snap-frozen in liquid nitrogen and storedat �70°C until RNA extraction. Serum total PSA valueswere available for all prostate cancer patients except forone patient in group B and for two patients in group C.
Oligonucleotides
The synthesis of PCR primers, detection probes and DNAtargets, and the biotinylation of the 3� PCR primer and theDNA targets were carried out as reported previously(Table 1).9,24 The detection probes for hybridization as-say contained additional diaminohexanedeoxycytidinesto be labeled with lanthanide chelate as described ear-lier.32–34 The detection probes for hK2 and PSA werelabeled with the Eu3� chelate, and the probes for theIS-hK2 and IS-PSA were labeled with the Tb3� chelate.
Construction of pGEM3-hK2 and pGEM3-IS-hK2 cDNA Plasmids
Full-length hK2 cDNA was amplified by PCR (primers Aand B, Table 1) from human prostate cDNA library with�gt11 clones encoding hK2.35 The 5� (A) and 3� (B)primers were designed to contain EcoRI and KpnI restric-tion sites in the 5� end of their sequence, respectively. A100-�l PCR reaction contained 20 mmol/L Tris-HCl, pH8.8; 10 mmol/L KCl; 10 mmol/L (NH4)2SO4; 2 mmol/LMgSO4; 1 ml/L Triton X-100; 1 mg/ml nuclease-free BSA;
112 Ylikoski et alJMD August 2001, Vol. 3, No. 3

0.4 �mol/L 5� primer A; 0.4 �mol/L 3� primer B; 200�mol/L each dNTP (Pharmacia Biotech, Uppsala, Swe-den); and 2.5 U of Pfu DNA polymerase (Promega Cor-poration, Madison, WI). The amplification was carried outwith Perkin-Elmer Cetus DNA Thermal Cycler using thefollowing program: initial denaturation of 5 minutes at95°C followed by 35 cycles of 95°C for 30 s, 46°C for 2minutes and 72°C for 2 minutes 30 s. A final extension of5 minutes at 72°C was carried out after the 35 amplifica-tion cycles. The amplified cDNA insert was cloned intothe EcoRI and KpnI sites of the plasmid vector, pGEM3(Promega Corporation, Madison WI), and the constructedpGEM3-hK2 plasmid was transformed into Escherichiacoli XL2 Blue cells (Stratagene, La Jolla, CA). The se-quence of hK2 cDNA was confirmed by nucleic acidsequencing of the plasmid.
For the preparation of the IS-hK2 cDNA construct, 2 bp(nucleotides 601–602 from the hK2 sequence) were de-leted from pGEM3-hK2 cDNA plasmid using PCR primersA, C, D and E. The deletion was introduced with a genesplicing by overlap extension technique36 that we usedalso previously to construct the pGEM-IS-PSA9 from thepGEM-PSA plasmid template.35 The pGEM3-IS-hK2plasmid was transformed into E. coli XL-2 Blue cells(Stratagene, La Jolla, CA, USA) and the 2-bp deletionwas confirmed by nucleic acid sequencing of the hK2sequence.
In Vitro Production and Purification of hK2, IS-hK2, PSA, and IS-PSA mRNA
In vitro production of the calibrator (hK2 and PSA) andIS (IS-hK2 and IS-PSA) mRNA was carried out withAmpliScribe T7 transcription kit (Epicentre Technologies,Madison, WI). Linearized pGEM3-hK2, pGEM3-IS-hK2,pGEM3-PSA, and pGEM3-IS-PSA plasmids served astemplates in the transcription, and the in vitro mRNAproductions were purified as described previously.9 Thepurified mRNA pellet was dissolved in diethylpyrocarbon-ate (DEPC) treated water and stored in aliquots at �70°C.The amount of the mRNA was quantified using RiboGreenRNA Quantitation Kit (Molecular Probes, Leiden, The Neth-erlands) and the quality of the mRNA was checked byagarose gel electrophoresis. The pure mRNA samples wereused to optimize the RT-PCR amplification, to control vari-ations during the sample analyses (from the beginning ofthe RNA extraction to the detection of amplification prod-ucts), and to generate calibration curves for the multiplexedPSA and hK2 RT-PCR assay.
Total RNA Extraction
Total RNA was extracted from the pelleted LNCaP, SP2/0,and blood nucleated cells using a TRIZOL reagent (Life
Table 1. Oligonucleotides Used in This Study
Oligonucleotide Name Sequence from 5� to 3� endNucleotideposition* Label
Labels/oligo-
nucleotide
Construction of pGEM3-hK25� primer A ATATGAATTCATGTGGGACCTGGT‡ 43–56 —† —3� primer B CTATGGTACCTTTTTTTTTTTTTTTTTTTTTAACCAC‡ 1497–1523 — —
Construction of IS-hK25� primer§ A ATATGAATTCATGTGGGACCTGGT3� primer C GATGGTGTCCTTGATCCACTT 793–813 — —5� mutation primer D TGTGCTAGATTACTCTGAGAAGGTGA 592–600 — —
603–6193� mutation primer E TTCTCAGAGTAATCTAGCACACATGTC 546–600 — —
603–614PCR
hK2 and IS-hK2 5� primer F GAACCAGAGGAGTTCTTGCG 523–542 — —hK2 and IS-hK2 3� primer G (modC)CCCAGAATCACCCCCACAA¶ 666–684 Biotin 1PSA and IS-PSA 5� primer H TGAACCAGAGGAGTTCTTGAC 523–543 — —PSA and IS-PSA 3� primer I (modC)CCCAGAATCACCCGAGCAG 667–685 — —PSA and IS-PSA 3� primer J (modC)CCCAGAATCACCCGAGCAG 667–685 Biotin 1
Hybridization assayshK2 probe K (modC)20GCTAGAGCTTACTCTGA 595–611 Eu3� 11IS-hK2 probe L (modC)20GCTAGATTACTCTGAG 595–600 Tb3� 11
603–612hK2 target DNA M (modC)CCTTCTCAGAGTAAGCTCTAGCACACA 590–616 Biotin 1IS-hK2 target N (modC)CCTTCTCAGAGTAATCTAGCACACATG 589–600 Biotin 1
603–616PSA probe O (modC)20GCGCAAGTTCACCCTCA 596–612 Eu3� 17IS-PSA probe P (modC)20GTGCGCAATCACCCTC 594–601 Tb3� 22
604–611
*Nucleotide positions of the oligonucleotides are based on hK2 and PSA mRNA sequences published by Riegman et al37 and Lundwall et al35,respectively.
†—, no label.‡Italic letters denote bases that are not complementary to the target. Bold letters denote bases that serve as detection and cleavage sites for EcoRI
in the primer A and KpnI in the primer B.§The 5� primer for the construction of IS-hK2 was the same primer (A) that was used for the construction of pGEM3-hK2.¶modC, diaminohexanedeoxycytidine.
Multiplexed QRT-PCR for hK2 and PSA mRNAs 113JMD August 2001, Vol. 3, No. 3

Technologies, Inc., Grand Island, NY). The RNA extrac-tion was performed as described earlier.24 A constantamount of IS-hK2 and IS-PSA mRNA (5 � 104 moleculesof each) were added into each sample after denaturationof the pelleted cells. Samples containing only 2.5 � 106
SP2/0 cells were put up to serve as negative controls inRNA extraction. Denatured LNCaP and SP2/0 cells werecombined to have a calculated amount of 1, 5, 10, 50,100, 500 and 1000 denatured LNCaP cells in 2.5 � 106
SP2/0 cells. The RNA samples were stored at �70°C untilthey were analyzed.
RT-PCR Amplification
The cDNA synthesis was carried out with First-StrandcDNA Synthesis Kit using the NotI-d(T)18 primer (Amer-sham Pharmacia Biotech AB, Uppsala, Sweden). In ad-dition to the reactions for the samples, each cDNA syn-thesis contained reactions for the calibration curve thatwas built using the in vitro produced and purified calibra-tor and IS mRNAs diluted in an inert carrier solution of 0.2g/L E. coli tRNA (Boehringer Mannheim GmbH, Mann-heim, Germany). This carrier solution was also used as asample in the negative control reaction for the cDNAsynthesis. The cDNA synthesis was performed in a finalreaction volume of 15 �l. After the synthesis a 7.5-�lcDNA sample was amplified in a 100-�l PCR reaction,which consisted of 10 mmol/L Tris-HCl, pH 8.8; 50mmol/L KCl; 1 ml/L Triton X-100; 3.5 mmol/L MgCl2; 400�mol/L each dNTP (Pharmacia Biotech, Uppsala, Swe-den); and 1 U DynaZyme II recombinant DNA polymer-ase (Finnzymes Oy, Espoo, Finland). The concentrationsof the primers were 0.15 �mol/L of primer F, and 0.15�mol/L of biotinylated primer G for the amplification ofhK2 and IS-hK2, and 0.2 �mol/L of primer H, 0.065�mol/L unlabeled primer I, and 0.035 �mol/L biotinylatedprimer J for the amplification of PSA and IS-PSA. Beforeamplification the PCR reactions were kept on ice. ThePCR amplification was performed in a PTC-200 DNAEngine (MJ Research, Inc., Watertown) using a programof 94°C for 30 s (2 minutes for the first cycle), 62°C for30 s, and 72°C for 45 s (10 minutes 45 s for the last cycle)for 30 cycles.
Solution Hybridization
The products of the PCR amplification were analyzed bydual-label hybridization assay based on time-resolvedfluorometry (TRF). The hybridization was performed asreported previously.24 Briefly, a 10-�l aliquot of PCRproduct and 50 �l of buffer containing 1 mol/L NaCl wereadded into each streptavidin-coated microtitration well(InnoTrac Diagnostics Oy, Turku, Finland). Each PCRproduct was added into two wells and the target (PSA orhK2) and IS (IS-PSA or IS-hK2) products were detectedfrom the same wells with the Eu3�-labeled target andTb3�-labeled IS probes, respectively. PCR amplificationand the biotinylated 3� primer (G or J) resulted in biotin-ylated PCR products, which were captured onto thestreptavidin-coated microtitration wells by incubating at
room temperature with slow shaking for 30 minutes. Afterthe capture reaction, the wells were washed three timeswith wash solution (PerkinElmer Life Sciences, Wallac Oy,Turku, Finland), and 100 �l of 50 mmol/L NaOH wasadded into each well for the denaturation of the double-stranded PCR products. Denaturation was carried out byincubating at room temperature with slow shaking for 5minutes, the denatured DNA strand was then removed bywashing three times as described above. The capturedDNA strand was detected by adding 100 �l of hybridiza-tion solution containing detection probes, nonfat milkpowder, and 1 mol/L NaCl. The hybridization solution forthe detection of hK2 and IS-hK2 contained 0.05% nonfatmilk powder, and 20 pg/�l of both the hK2 and IS-hK2detection probes (K and L), and the solution for thedetection of PSA and IS-PSA contained 0.1% nonfat milkpowder, 20 pg/�l of the PSA probe (O), and 10 pg/�l ofthe IS-PSA probe (P). After hybridization at �40°C (hK2,IS-hK2) or �50°C (PSA, IS-PSA) for 2 hours the wellswere washed six times with �40°C (hK2, IS-hK2) or�55°C (PSA, IS-PSA) wash solution. Then 200 �l of en-hancement solution for Eu3� (PerkinElmer Life Sciences)was added into each well and after shaking for 30 min-utes at room temperature the Eu3� fluorescence wasmeasured with 1420 Victor™ Multilabel Counter(PerkinElmer Life Sciences). After the measurement ofthe Eu3� fluorescence, 50 �l of enhancement solution forTb3� (PerkinElmer Life Sciences) was added into eachwell and after a 5 minute incubation the Tb3� fluores-cence was measured.
Results
pGEM3-hK2 and pGEM3-IS-hK2
The hK2 cDNA sequence in the pGEM3-hK2 plasmid wasconfirmed for the lack of secondary mutations possiblyoriginating from the PCR amplification with Pfu DNA poly-merase and for the lack of alternatively spliced hK2form.37 The hK2 cDNA was found to correspond thecorrect hK2 mRNA sequence encoding the previouslypublished 237-amino-acid protein.38 Furthermore, thepGEM3-IS-hK2 plasmid was confirmed to contain thedesired 2-bp deletion in the middle of the binding areasof the hK2 and IS-hK2 hybridization probes. The pGEM3-hK2 and pGEM3-IS-hK2 constructs were used, in com-bination with the pGEM3-PSA35 and pGEM3-IS-PSA9
constructs, as templates to produce mRNA (see Materi-als and Methods) for the calibration and validation of themultiplexed hK2 and PSA RT-PCR assay.
Assay Design
In the assay developed, the basis for the quantification ofthe target mRNAs (hK2 and PSA) in the cell pellet is theuse of an external calibration curve in RT-PCR and theaddition of the target-like IS mRNAs (IS-hK2 and IS-PSA)to the samples at the beginning of the RNA extraction.The principle of the assay is shown in Figure 1 (A and B).Compared with the wild-type hK2 and PSA, the IS-hK2
114 Ylikoski et alJMD August 2001, Vol. 3, No. 3

and IS-PSA mRNAs contain a 2-bp deletion in the middleof the binding area of the detection probes for the PCRproducts. The calibration curve covering the range of50–106 copies of hK2 and PSA mRNAs contain a con-stant number of IS-hK2 and IS-PSA mRNAs (5000 copies)mixed with the varying number of the calibrator (target)mRNAs. For the quantification of hK2 and PSA mRNAs ina sample, a constant number of IS-hK2 and IS-PSAmRNA copies (50,000 copies, i.e., 10 times more than inthe calibration curve) are added to the sample at thebeginning of the RNA extraction. The RNA pellet is dis-solved into 80 �l of sterile RNase-free water and 10% (8�l) of each sample (corresponding to 5000 copies ofIS-PSA and IS-hK2 mRNAs in theory) is analyzed with theRT-PCR assay. The target and the IS mRNAs in thesample and in the external calibration curve are co-am-plified by RT-PCR in the same amplification mixture (Fig-ure 1A). The cDNA synthesis is carried out using anuniversal oligo-d(T)18 -primer and the PCR mixture con-tains two specific primer pairs, one for the amplification ofhK2 and IS-hK2 and the other for the amplification of PSAand IS-PSA. The PCR conditions will selectively producehK2 and IS-hK2 or PSA and IS-PSA based on the pres-ence of the template. After the 30-cycle PCR, theamounts of the target and IS amplification products arequantified by dual-label solution hybridization assays andTRF using specific Eu3� and Tb3� chelate labeled de-tection probes for the target and the IS, respectively(Figure 1B). The amount of the target mRNA in the sam-ple can be then calculated by comparing the back-ground-corrected target-to-IS fluorescence ratio in thesample to the ratio in the calibration curve. In addition,the use of the IS-hK2 and IS-PSA mRNAs allows thecontrol of variations during the QRT-PCR assay from theRNA extraction to the detection of amplification productsby solution hybridization.
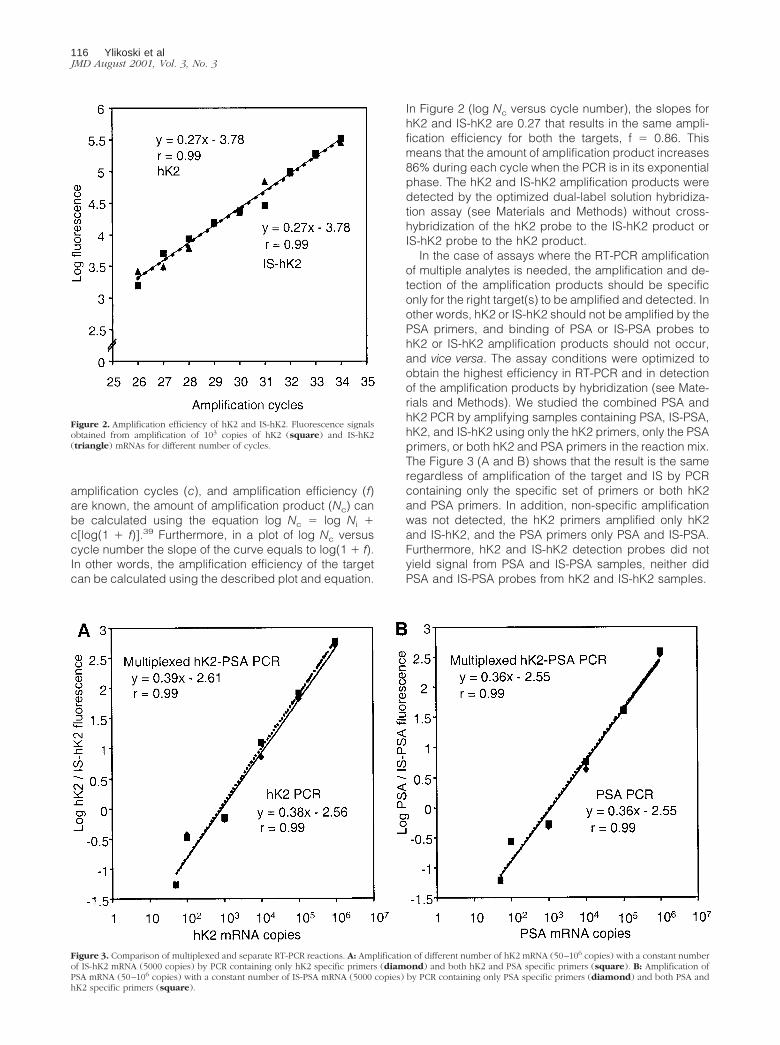
To obtain a reliable quantification of the target mRNAby QRT-PCR with exogenous IS, there are at least twomain factors to consider in the development of the assay:equal amplification efficiency of the target and the corre-sponding IS, and means of specific detection of thetarget and IS amplification products. We have previouslyshown that PSA and IS-PSA are amplified with the equalefficiency, and that the PSA and IS-PSA amplificationproducts are detected without cross-hybridization.9 Theamplification efficiencies of hK2 and IS-hK2 were studiedby amplifying 1000 mRNA copies of each for differentnumber of amplification cycles (Figure 2). It has beenshown that if the initial amount of target (Ni), number of
Figure 1. Schematic presentation of the principle of the multiplexed QRT-PCR assay for the detection of hK2 and PSA mRNAs. A: A known amount ofIS-hK2 and IS-PSA mRNAs are added to the denatured nucleated blood cellsbefore total RNA extraction. PSA and hK2 mRNAs in the sample and theIS-hK2 and IS-PSA mRNAs are co-amplified by RT-PCR. A PCR reactioncontains specific primers for the amplification of the hK2 (and IS-hK2) andPSA (and IS-PSA). Biotinylated 3� primers generate biotinylated amplificationproducts that are captured on streptavidin-coated microtitration wells. B: Theamplification products are detected by specific hybridization so that the hK2and IS-hK2 products are detected in one well and the PSA and IS-PSAproducts in the other. The target specific hybridization probes are labeledwith Eu3�-chelate and the IS probes with Tb3�-chelate. BIO, biotin; SA,streptavidin.
Multiplexed QRT-PCR for hK2 and PSA mRNAs 115JMD August 2001, Vol. 3, No. 3
amplification cycles (c), and amplification efficiency (f)are known, the amount of amplification product (Nc) canbe calculated using the equation log Nc � log Ni �c[log(1 � f)].39 Furthermore, in a plot of log Nc versuscycle number the slope of the curve equals to log(1 � f).In other words, the amplification efficiency of the targetcan be calculated using the described plot and equation.
In Figure 2 (log Nc versus cycle number), the slopes forhK2 and IS-hK2 are 0.27 that results in the same ampli-fication efficiency for both the targets, f � 0.86. Thismeans that the amount of amplification product increases86% during each cycle when the PCR is in its exponentialphase. The hK2 and IS-hK2 amplification products weredetected by the optimized dual-label solution hybridiza-tion assay (see Materials and Methods) without cross-hybridization of the hK2 probe to the IS-hK2 product orIS-hK2 probe to the hK2 product.
In the case of assays where the RT-PCR amplificationof multiple analytes is needed, the amplification and de-tection of the amplification products should be specificonly for the right target(s) to be amplified and detected. Inother words, hK2 or IS-hK2 should not be amplified by thePSA primers, and binding of PSA or IS-PSA probes tohK2 or IS-hK2 amplification products should not occur,and vice versa. The assay conditions were optimized toobtain the highest efficiency in RT-PCR and in detectionof the amplification products by hybridization (see Mate-rials and Methods). We studied the combined PSA andhK2 PCR by amplifying samples containing PSA, IS-PSA,hK2, and IS-hK2 using only the hK2 primers, only the PSAprimers, or both hK2 and PSA primers in the reaction mix.The Figure 3 (A and B) shows that the result is the sameregardless of amplification of the target and IS by PCRcontaining only the specific set of primers or both hK2and PSA primers. In addition, non-specific amplificationwas not detected, the hK2 primers amplified only hK2and IS-hK2, and the PSA primers only PSA and IS-PSA.Furthermore, hK2 and IS-hK2 detection probes did notyield signal from PSA and IS-PSA samples, neither didPSA and IS-PSA probes from hK2 and IS-hK2 samples.
Figure 2. Amplification efficiency of hK2 and IS-hK2. Fluorescence signalsobtained from amplification of 103 copies of hK2 (square) and IS-hK2(triangle) mRNAs for different number of cycles.
Figure 3. Comparison of multiplexed and separate RT-PCR reactions. A: Amplification of different number of hK2 mRNA (50–106 copies) with a constant numberof IS-hK2 mRNA (5000 copies) by PCR containing only hK2 specific primers (diamond) and both hK2 and PSA specific primers (square). B: Amplification ofPSA mRNA (50–106 copies) with a constant number of IS-PSA mRNA (5000 copies) by PCR containing only PSA specific primers (diamond) and both PSA andhK2 specific primers (square).
116 Ylikoski et alJMD August 2001, Vol. 3, No. 3
Detection Limit and Reproducibility of theMultiplexed hK2 and PSA Assay
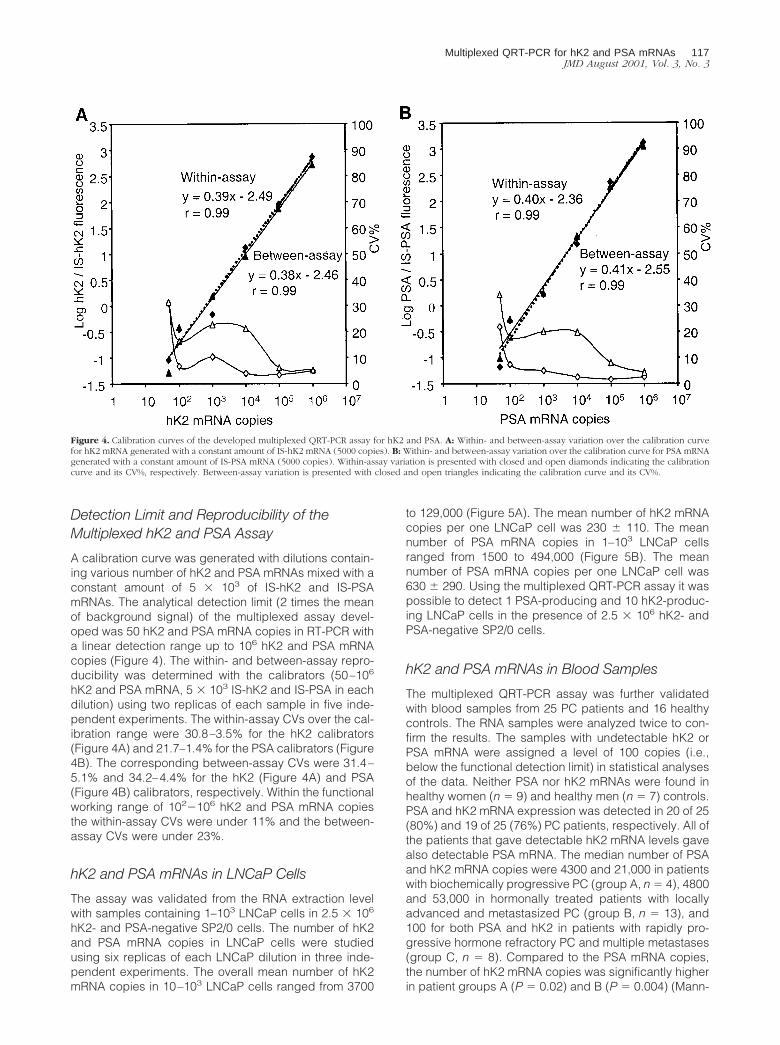
A calibration curve was generated with dilutions contain-ing various number of hK2 and PSA mRNAs mixed with aconstant amount of 5 � 103 of IS-hK2 and IS-PSAmRNAs. The analytical detection limit (2 times the meanof background signal) of the multiplexed assay devel-oped was 50 hK2 and PSA mRNA copies in RT-PCR witha linear detection range up to 106 hK2 and PSA mRNAcopies (Figure 4). The within- and between-assay repro-ducibility was determined with the calibrators (50–106
hK2 and PSA mRNA, 5 � 103 IS-hK2 and IS-PSA in eachdilution) using two replicas of each sample in five inde-pendent experiments. The within-assay CVs over the cal-ibration range were 30.8–3.5% for the hK2 calibrators(Figure 4A) and 21.7–1.4% for the PSA calibrators (Figure4B). The corresponding between-assay CVs were 31.4–5.1% and 34.2–4.4% for the hK2 (Figure 4A) and PSA(Figure 4B) calibrators, respectively. Within the functionalworking range of 102�106 hK2 and PSA mRNA copiesthe within-assay CVs were under 11% and the between-assay CVs were under 23%.
hK2 and PSA mRNAs in LNCaP Cells
The assay was validated from the RNA extraction levelwith samples containing 1–103 LNCaP cells in 2.5 � 106
hK2- and PSA-negative SP2/0 cells. The number of hK2and PSA mRNA copies in LNCaP cells were studiedusing six replicas of each LNCaP dilution in three inde-pendent experiments. The overall mean number of hK2mRNA copies in 10–103 LNCaP cells ranged from 3700
to 129,000 (Figure 5A). The mean number of hK2 mRNAcopies per one LNCaP cell was 230 � 110. The meannumber of PSA mRNA copies in 1–103 LNCaP cellsranged from 1500 to 494,000 (Figure 5B). The meannumber of PSA mRNA copies per one LNCaP cell was630 � 290. Using the multiplexed QRT-PCR assay it waspossible to detect 1 PSA-producing and 10 hK2-produc-ing LNCaP cells in the presence of 2.5 � 106 hK2- andPSA-negative SP2/0 cells.
hK2 and PSA mRNAs in Blood Samples
The multiplexed QRT-PCR assay was further validatedwith blood samples from 25 PC patients and 16 healthycontrols. The RNA samples were analyzed twice to con-firm the results. The samples with undetectable hK2 orPSA mRNA were assigned a level of 100 copies (i.e.,below the functional detection limit) in statistical analysesof the data. Neither PSA nor hK2 mRNAs were found inhealthy women (n � 9) and healthy men (n � 7) controls.PSA and hK2 mRNA expression was detected in 20 of 25(80%) and 19 of 25 (76%) PC patients, respectively. All ofthe patients that gave detectable hK2 mRNA levels gavealso detectable PSA mRNA. The median number of PSAand hK2 mRNA copies were 4300 and 21,000 in patientswith biochemically progressive PC (group A, n � 4), 4800and 53,000 in hormonally treated patients with locallyadvanced and metastasized PC (group B, n � 13), and100 for both PSA and hK2 in patients with rapidly pro-gressive hormone refractory PC and multiple metastases(group C, n � 8). Compared to the PSA mRNA copies,the number of hK2 mRNA copies was significantly higherin patient groups A (P � 0.02) and B (P � 0.004) (Mann-
Figure 4. Calibration curves of the developed multiplexed QRT-PCR assay for hK2 and PSA. A: Within- and between-assay variation over the calibration curvefor hK2 mRNA generated with a constant amount of IS-hK2 mRNA (5000 copies). B: Within- and between-assay variation over the calibration curve for PSA mRNAgenerated with a constant amount of IS-PSA mRNA (5000 copies). Within-assay variation is presented with closed and open diamonds indicating the calibrationcurve and its CV%, respectively. Between-assay variation is presented with closed and open triangles indicating the calibration curve and its CV%.
Multiplexed QRT-PCR for hK2 and PSA mRNAs 117JMD August 2001, Vol. 3, No. 3
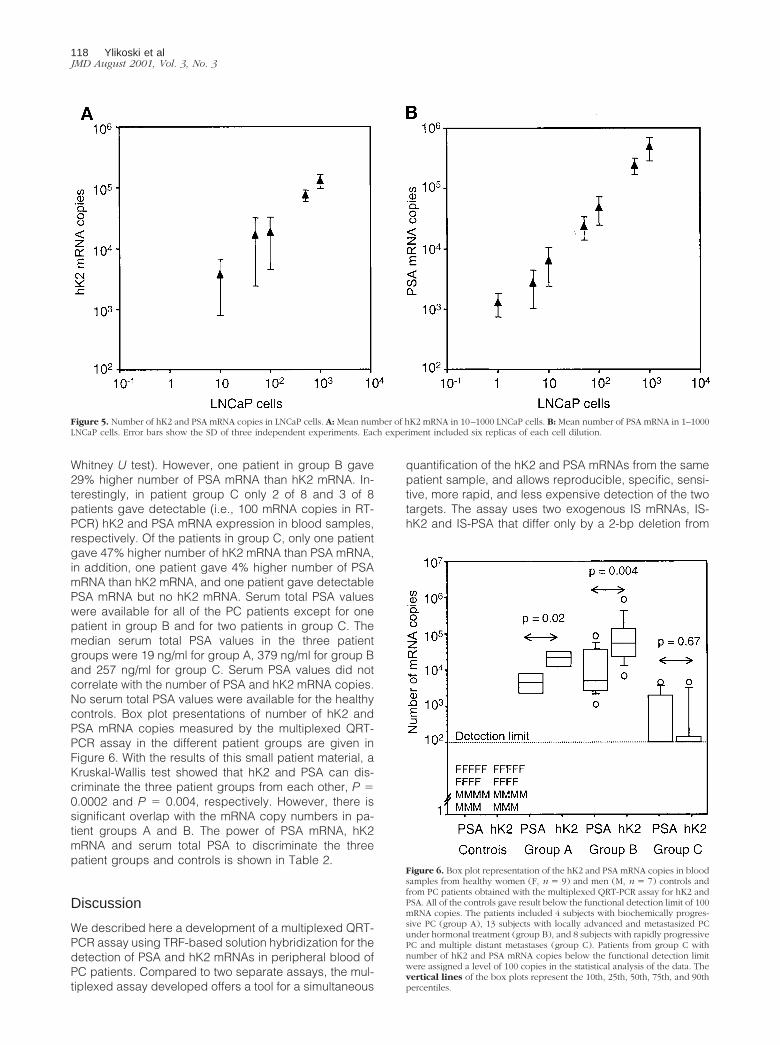
Whitney U test). However, one patient in group B gave29% higher number of PSA mRNA than hK2 mRNA. In-terestingly, in patient group C only 2 of 8 and 3 of 8patients gave detectable (i.e., 100 mRNA copies in RT-PCR) hK2 and PSA mRNA expression in blood samples,respectively. Of the patients in group C, only one patientgave 47% higher number of hK2 mRNA than PSA mRNA,in addition, one patient gave 4% higher number of PSAmRNA than hK2 mRNA, and one patient gave detectablePSA mRNA but no hK2 mRNA. Serum total PSA valueswere available for all of the PC patients except for onepatient in group B and for two patients in group C. Themedian serum total PSA values in the three patientgroups were 19 ng/ml for group A, 379 ng/ml for group Band 257 ng/ml for group C. Serum PSA values did notcorrelate with the number of PSA and hK2 mRNA copies.No serum total PSA values were available for the healthycontrols. Box plot presentations of number of hK2 andPSA mRNA copies measured by the multiplexed QRT-PCR assay in the different patient groups are given inFigure 6. With the results of this small patient material, aKruskal-Wallis test showed that hK2 and PSA can dis-criminate the three patient groups from each other, P �0.0002 and P � 0.004, respectively. However, there issignificant overlap with the mRNA copy numbers in pa-tient groups A and B. The power of PSA mRNA, hK2mRNA and serum total PSA to discriminate the threepatient groups and controls is shown in Table 2.
Discussion
We described here a development of a multiplexed QRT-PCR assay using TRF-based solution hybridization for thedetection of PSA and hK2 mRNAs in peripheral blood ofPC patients. Compared to two separate assays, the mul-tiplexed assay developed offers a tool for a simultaneous
quantification of the hK2 and PSA mRNAs from the samepatient sample, and allows reproducible, specific, sensi-tive, more rapid, and less expensive detection of the twotargets. The assay uses two exogenous IS mRNAs, IS-hK2 and IS-PSA that differ only by a 2-bp deletion from
Figure 5. Number of hK2 and PSA mRNA copies in LNCaP cells. A: Mean number of hK2 mRNA in 10–1000 LNCaP cells. B: Mean number of PSA mRNA in 1–1000LNCaP cells. Error bars show the SD of three independent experiments. Each experiment included six replicas of each cell dilution.
Figure 6. Box plot representation of the hK2 and PSA mRNA copies in bloodsamples from healthy women (F, n � 9) and men (M, n � 7) controls andfrom PC patients obtained with the multiplexed QRT-PCR assay for hK2 andPSA. All of the controls gave result below the functional detection limit of 100mRNA copies. The patients included 4 subjects with biochemically progres-sive PC (group A), 13 subjects with locally advanced and metastasized PCunder hormonal treatment (group B), and 8 subjects with rapidly progressivePC and multiple distant metastases (group C). Patients from group C withnumber of hK2 and PSA mRNA copies below the functional detection limitwere assigned a level of 100 copies in the statistical analysis of the data. Thevertical lines of the box plots represent the 10th, 25th, 50th, 75th, and 90thpercentiles.
118 Ylikoski et alJMD August 2001, Vol. 3, No. 3

their target mRNAs (hK2 and PSA). A known amount ofthe synthetic IS-hK2 and IS-PSA mRNAs are added intoeach collected and denatured cell pellet at the beginningof the RNA extraction to enable a standardization correct-ing for any variations starting from the beginning of theRNA extraction to the final detection of the amplificationproducts by solution hybridization. In addition, the use ofan external calibration curve, consisting of the in vitroproduced and purified hK2, PSA, IS-hK2, and IS-PSAmRNAs, allows the quantification of hK2 and PSA mRNAcopies in the sample by comparing the target-to-IS fluo-rescence ratio of the sample to the ratio in the calibrationcurve.
After the RNA extraction, the target and the IS mRNAsin the samples and calibrator reactions were co-amplifiedby RT-PCR. Instead of using a large number of amplifi-cation cycles and nested-PCR approach, we used anoptimized 30-cycle PCR and two pairs of primers to spe-cifically amplify hK2 and IS-hK2 cDNAs with one pair andPSA and IS-PSA cDNAs with the other pair in the sameamplification reaction. Thereafter, the amplification prod-ucts were detected by dual-label solution hybridizationbased on TRF technology in two separate microtitrationwells. The TRF offers a sensitive, nonradioactive, andrapid method for the quantification of the amplificationproducts.40 The QRT-PCR assay developed gave an an-alytical detection limit of 50 (i.e., 2 times the mean ofbackground signal) and a functional detection limit of 102
(i.e., between-assay CVs under 23%) target mRNA cop-ies with a linear range up to 106 mRNA copies.
We evaluated the multiplexed QRT-PCR assay on cul-tured LNCaP cells and samples from PC patients (n � 25)and healthy volunteers (n � 16). Based on the Northernblot analyses of PSA and hK2 mRNAs using non-cancer-ous prostatic tissue samples, it has been reported thatthe level of hK2 mRNA expression relative to that of PSAmRNA is approximately 10 to 20%.41 In the analysis ofLNCaP cells, we showed that the mean hK2 mRNA ex-pression relative to the mean PSA mRNA expression was
28.8 to 36.5%. In our previous reports we have found thatthe mean number of PSA mRNA per one LNCaP cellranged from 500 to 2100 copies9 and from 670 to 1100copies.24 In this paper we found the mean number of PSAmRNA per one LNCaP cell was from 340 to 920 copies,which is slightly smaller than the results obtained previ-ously. Despite the identical culture conditions, this varia-tion in the number of PSA mRNA copies is most probablydue to normal batch variation in LNCaP cell cultures.Furthermore, some of the variations may be due to diffi-culties in diluting very small amounts of LNCaP cells(1–10 cells), which affects the final calculated number oftarget copies in the sample. These issues in turn demon-strate how important it is to develop RT-PCR assays thatdo not use LNCaP cells as a calibrator material. Con-versely, properly calibrated QRT-PCR assays should beused as tools to study the different levels of target mRNAexpression in the cultured cells.
There are four previously published reports on quali-tative hK2 RT-PCR assays for the detection of PC cells inpatient samples. Young et al 12 were the first to report thedetection of hK2 mRNA together with that of PSA mRNAby two qualitative RT-PCR assays.7,12 They detected hK2mRNA in 1 of 1 PC patient with prostate-confined dis-ease, 2 of 3 patients with locally advanced disease and 2of 2 patients with distant metastatic disease. PSA mRNAwas detected only in 1 of 2 patients with distant meta-static disease. However, they did not describe the exper-imental details of their hK2 assay. It remained unclear, forexample, what were the sequences of the PCR primersand whether the hK2 assay detected only hK2 and notPSA mRNA. Neither PSA nor hK2 were detected in thecontrols. Corey et al 8,10 used two separate RT-PCRassays to detect hK2 and PSA from both the peripheralblood and bone marrow samples from patients with ad-vanced PC (n � 13) and patients with clinically localizeddisease (n � 63). All blood and bone marrow samplesfrom controls were found to be negative for hK2 and PSA.In blood samples from patients with clinically localizedPC, detectable PSA and hK2 mRNAs were found in 12 of63 and 8 of 63, respectively. In blood samples frompatients with advanced PC, 6 of 13 were positive for PSAand 4 of 13 were positive for hK2. These results showedthat the patient samples were more often positive for PSAmRNA than for hK2 mRNA. Corey et al 8,10 detected PSAand hK2 mRNAs also in bone marrow samples from thesame patients. They found that the number of patientsthat gave positive result for PSA and hK2 mRNAs washigher in the bone marrow samples than in the bloodsamples. Kawakami et al 11 studied expression of hK2and PSA mRNA in blood samples from PC patients (n �41) and controls. Neither PSA nor hK2 amplification prod-ucts were found in the control samples from 20 healthyvolunteers and 7 patients with benign prostatic hyperpla-sia. They detected PSA mRNA in 6 of 7 patients withnonpalpable cancer, 2 of 5 patients with prostate-con-fined disease, 4 of 8 patients with locally advanced dis-ease, 3 of 4 patients with pelvic lymph node involvement,and 14 of 17 patients with distant metastatic disease. Ofall of the PC patients, hK2 mRNA was found only in 7 of17 patients with distant metastatic disease. Based on
Table 2. The p Values Obtained by Mann-Whitney U Testafter Comparison of PSA mRNA, hK2 mRNA andSerum Total PSA Values of Various Patient Groupsand Healthy Controls
AnalytePatientgroups A B C
PSA mRNA AB NS*C 0.0415† 0.0014
Controls 0.0025 �0.0001 NShK2 mRNA A
B NSC 0.0066 0.0002
Controls 0.0025 �0.0001 NSSerum total PSA A
B 0.029C 0.05 NS
Controls —‡ — —
*NS, not significant.†A p value equal to 0.05 or less was considered significant.‡—, serum total PSA values were not available for the controls.Group A, biochemical progression; group B, metastatic; group C,
metastatic, hormone refractory.
Multiplexed QRT-PCR for hK2 and PSA mRNAs 119JMD August 2001, Vol. 3, No. 3

their finding that hK2 mRNA was only found in patientswith distant metastatic PC, Kawakami et al 11 concludedthat hK2 mRNA is associated with the metastatic progres-sion of PC and is an indicator of a poor prognosis. Slawinet al 42 developed a splice variant-specific RT-PCR assayfor hK2 mRNA and evaluated the assay on blood sam-ples from healthy men (n � 14), patients with metastaticPC (n � 7) and patients undergoing radical prostatec-tomy for clinically localized PC (n � 228). The assaydetected two forms of hK2 mRNA: the native transcript,which encodes for the full-length hK2 protein, and analternatively spliced transcript, which contains 37 addi-tional nucleotides downstream from the native splice do-nor site in intron IV.37 The native hK2 mRNA and thealternatively spliced hK2 mRNA were detected in 57 of228 and 58 of 228 patients undergoing radical prostatec-tomy, respectively. The results showed that the preoper-ative expression of the native hK2 but not the alternativelyspliced hK2 mRNA added prognostic information to theprediction of lymph node-positive disease in patientsundergoing radical prostatectomy. Native and alterna-tively spliced hK2 mRNAs were detected in 5 of 7 and 1of 7 patients with metastatic PC, respectively. However,native and alternatively spliced hK2 mRNAs were alsodetected in 2 of 14 and 5 of 14 healthy controls, respec-tively.
Interestingly, we found that the number of hK2 mRNAcopies was significantly higher than the number of PSAmRNA copies in PC patients with biochemically progres-sive disease (group A, n � 4, P � 0.02) and with locallyadvanced and metastasized disease (group B, n � 13,P � 0.004). It was also found that hK2 mRNA expressionwas lower than PSA mRNA expression only in two pa-tients that gave detectable result. One patient with locallyadvanced and metastasized PC had 29% hK2 mRNAexpression relative to PSA mRNA expression, and theother patient with hormone refractory and distantly met-astatic PC had 4% hK2 mRNA expression relative to PSAmRNA expression. Furthermore, the preliminary resultsshowed that the number of hK2 and PSA mRNA copiesdifferentiated the patients with biochemically progressivedisease and with advanced disease from the patientswith fast progressive hormone refractory disease andfrom the controls.
Our results showed that the overall number of patientsthat gave positive result for PSA mRNA (80%) was higherthan that for hK2 mRNA (76%). This result is in accor-dance with the results obtained by Corey et al 10 andKawakami et al 11 but not with the results of Young et al.12
The patients that gave positive result for hK2 mRNA werealso positive for PSA mRNA. Only in patients with hor-mone refractory PC, we found one patient who expressedPSA mRNA in blood while no hK2 mRNA was detected.However, contrary to the previous reports,10–12 we didnot find patient samples that would have given detect-able hK2 mRNA and no PSA mRNA. This may be due tothe small number of patient samples in the preliminaryevaluation of the assay.
The results of Young et al 12 showed hK2 and PSAmRNA expression in 83% and 17% of all of the patientsstudied, respectively. It was surprising that PSA mRNA
was not detectable in the blood samples from the pa-tients with organ-confined and with locally advanced PC.Our assay detected PSA and hK2 mRNA copies in 100%of the blood samples from the patients with the biochem-ically progressive PC (no metastases detected) and withthe advanced PC. However, only 38% and 25% of thepatients with hormone refractory PC were positive for PSAand hK2 mRNA, respectively. Corey et al 10 detectedPSA and hK2 in 19% and 13% of the patients with clini-cally localized PC, and in 46% and 31% of the patientswith metastatic PC, respectively. Furthermore, Slawin etal 42 detected the expression of the native hK2 mRNA in23% of the patients with organ-confined PC, 30% of thepatients with locally advanced PC, 71% of the patientswith distant metastatic PC, and 14% of the healthy mencontrols. Kawakami et al 11 found PSA mRNA in 67% ofthe patients with organ-confined PC, 58% of the patientswith advanced PC, and 82% of the patients with distantmetastatic PC, whereas hK2 mRNA was found only in41% of the patients with distant metastatic PC. From thepatients with distant metastatic PC, 4 were refractory tothe hormonal therapy. PSA and hK2 mRNAs were foundin 3 of 4 (75%) and 4 of 4 (100%) of these patients,respectively. Contrary to the results of Kawakami et al, wefound decreased expression of PSA and hK2 mRNA inthe patients with hormone refractory PC. The reason forthe high number of hK2 and PSA negative results mayowe to the late stage of the disease of these patients. Itmust be noted, however, that these results derive from asmall number of patients. Further studies with a largernumber of samples and long-term follow-up will be re-quired to confirm this phenomenon. However, the differ-ences in the results may also be due to the various assayconcepts and PCR primers, which result in amplificationproducts from different forms of PSA and hK2 mRNAs.
Both PSA and hK2 mRNA transcripts are present inseveral alternatively spliced forms.37,43–45 However, verylittle is known about the presence or function of the prod-ucts encoded by these alternative mRNAs. At least fourPSA and five hK2 mRNA forms have been characterized.Riegman et al 45 characterized three of the PSA tran-scripts. The major 1.5 kb PSA mRNA encodes for thenative PSA protein. Additional forms of PSA transcriptsincluded a truncated 0.9 kb mRNA with 145 additionalnucleotides from the intron III between the exons 2 and 3,and a 1.9 kb mRNA with 442 additional nucleotides fromthe intron IV between the exons 4 and 5. Recently, Heuzeet al 43 described a 2.1 kb alternative PSA mRNA with 644additional nucleotides from the intron IV between theexons 4 and 5. Riegman et al37 reported that in additionto the native 1.5 kb hK2 mRNA, there is an alternativelyspliced variant, which contains 37 additional nucleotidesfrom the intron IV between the exons 4 and 5. Morerecently, Liu et al 44 reported three additional species ofhK2 transcripts, including two 3.0 kb transcripts corre-sponding to the two above mentioned 1.5 kb hK2 iso-forms, but each with an additional 1.5 kb of untranslatedregion attributable to transcription through the first poly-adenylation signal to a second signal located down-stream. The third 1.5 kb transcript contains a deletion of13 nucleotides between exons 3 and 4 of the hK2 mRNA.
120 Ylikoski et alJMD August 2001, Vol. 3, No. 3

The different assays seem to detect different forms ofPSA and hK2 transcripts. The PSA and hK2 primers of ourassay were designed so that the 5� primers spanned thejunction between the exons 3 and 4, and the 3� primersspanned the junction between the exons 4 and 5. ThePSA and hK2 PCR primers amplified a 163-bp fragmentspanning the end of exon 3, exon 4, and the beginning ofthe exon 5. Therefore, the PSA primers produced theamplification product from the native 1.5 kb mRNA. ThehK2 primers amplified the 163-bp fragment from the na-tive 1.5 kb mRNA and its related 3.0 kb mRNA with the1.5 kb 3� untranslated region. Neither the PSA nor thehK2 primers did amplify the other alternatively splicedforms of the transcripts. Young et al 12 used in their studyPSA primers that were previously developed by Katz etal.7 In addition to the native PSA mRNA, these primersamplified a fragment spanning exons 3 and 4 from thethree above-mentioned alternative PSA mRNA forms. Thesequences of the hK2 primers were not reported. Coreyet al 10 used hK2 primers that amplified a fragment span-ning exons 2 and 4, and PSA primers that produced afragment spanning exons 2 and 5. Both the PSA and thehK2 primers allowed the amplification of all of the above-mentioned forms of PSA and hK2 transcripts. Kawakamiet al 11 used hK2 primers that amplified a fragment span-ning exons 2 and 3. The primers for the PSA assayshared the same sequences as reported previously byothers.13 The PSA primers amplified a fragment spanningexons 3 and 5. The PSA and hK2 primers used byKawakami et al 11 allowed also the amplification of all ofthe above-mentioned alternative PSA and hK2 tran-scripts. Slawin et al 42 studied the expression of thenative hK2 mRNA and the alternatively spliced hK2mRNA with the additional 37 nucleotides between theexons 4 and 5. The hK2 primers amplified a fragmentspanning exons 4 and 5 so that they amplified both thenative and the alternative transcripts and their related 3.0kb mRNAs with the 1.5 kb 3� untranslated region.
Lately, the immunofluorometric studies on hK2 andPSA protein expression in serum of prostate cancer pa-tients have shown that measurements of hK2 in additionto PSA have significantly enhanced the discrimination ofPC patients from patients with benign prostate disease,46
improved the identification of poorly differentiated pros-tate tumors,47 and allowed more accurate prediction oforgan-confined from nonorgan-confined disease.47,48
Furthermore, the studies on hK2 and PSA protein expres-sion on tissue level have revealed that both hK2 and PSAare down-regulated in malignant tissue compared withnormal prostate tissue.49 The ultimate additional benefitof quantitative measurements of hK2 and PSA mRNAexpression aiming to improve PC detection and molecu-lar staging awaits continued investigations. A drawbackof the RT-PCR assays is that they cannot distinguishwhether the number of target mRNA copies indicates afew cells that contain many target mRNA molecules ormany cells that contain a few target mRNA molecules.However, quantitative assays capable of detecting differ-ent levels of target mRNAs may prove to be useful indetermining the clinically significant numbers of the tar-get copies. If used in combination with cell sorting meth-
ods, quantitative RT-PCR assays may be valuable tools insolving the problem with the number of target mRNAs percancer cell.
In conclusion, the multiplexed QRT-PCR assay devel-oped provides sensitive and specific detection of thenumber of hK2 and PSA mRNA copies in blood samples.The quantification of PSA and hK2 mRNAs is based onthe use of an external calibration curve consisting oftarget mRNAs and two target-like IS mRNAs. The ISmRNAs are also used to control the variations from thebeginning of the RNA extraction to the detection of am-plification products to allow reproducible quantification ofthe targets. To our knowledge, we are the first to report amultiplexed QRT-PCR assay for the simultaneous detec-tion of hK2 and PSA mRNAs and for the quantification ofan exact number of hK2 mRNA copies in LNCaP cellsand in blood samples from PC patients. Further studiesare needed to determine the impact of PSA and hK2QRT-PCR in the molecular staging of PC.
Acknowledgments
We thank Dong Liu for the construction of pGEM3-hK2plasmid, Johanna Kolehmainen and Louise Gladdis fortheir technical assistance in the development of the hK2assay, Minna Kujanpaa for the synthesis, labeling, andpurification of oligonucleotides, Sari Lindgren for supply-ing the SP2/0 cells, and Pauliina Nurmikko for supplyingthe LNCaP cells.
References
1. Bjork T, Piironen T, Pettersson K, Lovgren T, Stenman UH, OesterlingJE, Abrahamsson PA, Lilja H: Comparison of analysis of the differentprostate-specific antigen forms in serum for detection of clinicallylocalized prostate cancer. Urology 1996, 48:882–888
2. Gomella LG, Raj GV, Moreno JG: Reverse transcriptase polymerasechain reaction for prostate specific antigen in the management ofprostate cancer. J Urol 1997, 158:326–337
3. Pelkey TJ, Frierson HFJ, Bruns DE: Molecular and immunologicaldetection of circulating tumor cells and micrometastases from solidtumors. Clin Chem 1996, 42:1369–1381
4. Moreno JG, Croce CM, Fischer R, Monne M, Vihko P, Mulholland SG,Gomella LG: Detection of hematogenous micrometastasis in patientswith prostate cancer. Cancer Res 1992, 52:6110–6112
5. Vessella RL, Riley DE, Blouke KA, Arfman EW, Lange PH: A sensitivemethod for a detection of a prostate tumor cell marker using thepolymerase chain reaction. J Urol 1992, 147:441A
6. Galvan B, Christopoulos TK, Diamandis EP: Detection of prostate-specific antigen mRNA by reverse transcription polymerase chainreaction and time-resolved fluorometry. Clin Chem 1995, 41:1705–1709
7. Katz AE, Olsson CA, Raffo AJ, Cama C, Perlman H, Seaman E,O’Toole KM, McMahon D, Benson MC, Buttyan R: Molecular stagingof prostate cancer with the use of an enhanced reverse transcriptase-PCR assay. Urology 1994, 43:765–775
8. Corey E, Arfman EW, Liu AY, Vessella RL: Improved reverse tran-scriptase polymerase chain reaction protocol with exogenous internalcompetitive control for prostate-specific antigen mRNA in blood andbone marrow. Clin Chem 1997, 43:443–452
9. Ylikoski A, Sjoroos M, Lundwall A, Karp M, Lovgren T, Lilja H, Iitia A:Quantitative reverse transcription-PCR assay with an internal stan-dard for the detection of prostate-specific antigen mRNA. Clin Chem1999, 45:1397–1407
10. Corey E, Arfman EW, Oswin MM, Melchior SW, Tindall DJ, Young CY,
Multiplexed QRT-PCR for hK2 and PSA mRNAs 121JMD August 2001, Vol. 3, No. 3

Ellis WJ, Vessella RL: Detection of circulating prostate cells by re-verse transcriptase-polymerase chain reaction of human glandularkallikrein (hK2) and prostate-specific antigen (PSA) messages. Urol-ogy 1997, 50:184–188
11. Kawakami M, Okaneya T, Furihata K, Nishizawa O, Katsuyama T:Detection of prostate cancer cells circulating in peripheral blood byreverse transcription-PCR for hKLK2. Cancer Res 1997, 57:4167–4170
12. Young CY, Seay T, Hogen K, Charlesworth MC, Roche PC, Klee GG,Tindall DJ: Prostate-specific human kallikrein (hK2) as a novel markerfor prostate cancer. Prostate Suppl 1996, 7:17–24
13. Israeli RS, Miller WHJ, Su SL, Powell CT, Fair WR, Samadi DS, HurykRF, DeBlasio A, Edwards ET, Wise GJ: Sensitive nested reversetranscription polymerase chain reaction detection of circulating pros-tatic tumor cells: comparison of prostate-specific membrane antigenand prostate-specific antigen-based assays. Cancer Res 1994, 54:6306–6310
14. Cama C, Olsson CA, Raffo AJ, Perlman H, Buttyan R, O’Toole K,McMahon D, Benson MC, Katz AE: Molecular staging of prostatecancer. II. A comparison of the application of an enhanced reversetranscriptase polymerase chain reaction assay for prostate specificantigen versus prostate specific membrane antigen. J Urol 1995,153:1373–1378
15. Diamandis EP: Prostate specific antigen: its usefulness in clinicalmedicine. Trends Endocrinol Metab 1998, 9:310–316
16. Monne M, Croce CM, Yu H, Diamandis EP: Molecular characteriza-tion of prostate-specific antigen messenger RNA expressed in breasttumors. Cancer Res 1994, 54:6344–6347
17. Olsson CA, de Vries GM, Raffo AJ, Benson MC, O’Toole K, Cao Y,Buttyan RE, Katz AE: Preoperative reverse transcriptase polymerasechain reaction for prostate specific antigen predicts treatment failurefollowing radical prostatectomy. J Urol 1996, 155:1557–1562
18. Smith MR, Biggar S, Hussain M: Prostate-specific antigen messengerRNA is expressed in non-prostate cells: implications for detection ofmicrometastases. Cancer Res 1995, 55:2640–2644
19. Sokoloff MH, Tso CL, Kaboo R, Nelson S, Ko J, Dorey F, Figlin RA,Pang S, Dekernion J, Belldegrun A: Quantitative polymerase chainreaction does not improve preoperative prostate cancer staging: aclinicopathological molecular analysis of 121 patients. J Urol 1996,156:1560–1566
20. Yu H, Diamandis EP, Levesque M, Giai M, Roagna R, Ponzone R,Sismondi P, Monne M, Croce C: Prostate specific antigen in breastcancer, benign breast disease and normal breast tissue. BreastCancer Res Treat 1996, 40:171–178
21. Zarghami N, Diamandis EP: Detection of prostate-specific antigenmRNA and protein in breast tumors. Clin Chem 1996, 42:361–366
22. Gala JL, Heusterspreute M, Loric S, Hanon F, Tombal B, Van CanghP, De Nayer P, Philippe M: Expression of prostate-specific antigenand prostate-specific membrane antigen transcripts in blood cells:implications for the detection of hematogenous prostate cells andstandardization. Clin Chem 1998, 44:472–481
23. O’Hara SM, Veltri SM, Skirpstunas P: Basal PSA mRNA levels de-tected by quantitative reverse transcriptase polymerase chain reac-tion (Q-RT-PCR-PSA) in blood from subjects without prostate cancer.J Urol 1996, 155:418A
24. Ylikoski A, Karp M, Lilja H, Lovgren T: Dual-label detection of ampli-fied products in quantitative RT-PCR assay using lanthanide-labeledprobes. Biotechniques 2001, 30:832–836,838,840–843
25. Chelly J, Kaplan JC, Maire P, Gautron S, Kahn A: Transcription of thedystrophin gene in human muscle and non-muscle tissue. Nature1988, 333:858–860
26. Becker-Andre M, Hahlbrock K: Absolute mRNA quantification usingthe polymerase chain reaction (PCR): a novel approach by a PCRaided transcript titration assay (PATTY). Nucleic Acids Res 1989,17:9437–9446
27. Gilliland G, Perrin S, Blanchard K, Bunn HF: Analysis of cytokinemRNA and DNA: detection and quantitation by competitive polymer-ase chain reaction. Proc Natl Acad Sci USA 1990, 87:2725–2729
28. Wang AM, Doyle MV, Mark DF: Quantitation of mRNA by the poly-merase chain reaction. Proc Natl Acad Sci USA 1989, 86:9717–9721
29. Bortolin S, Christopoulos TK: Quantitative RT-PCR combined with
time-resolved fluorometry for determination of BCR-ABL mRNA. ClinChem 1996, 42:1924–1929
30. Bortolin S, Christopoulos TK, Verhaegen M: Quantitative polymerasechain reaction using a recombinant DNA internal standard and time-resolved fluorometry. Anal Chem 1996, 68:834–840
31. Tsuruta H, Matsui S, Oka K, Namba T, Shinngu M, Nakamura M:Quantitation of IL-1 beta mRNA by a combined method of RT-PCRand an ELISA based on ion-sensitive field effect transistor. J ImmunolMethods 1995, 180:259–264
32. Sund C, Ylikoski J, Hurskainen P, Kwiatkoeski M: Construction ofeuropium (Eu3�)-labelled oligo DNA hybridization probes. NucleosNucleot 1988, 7:655–659
33. Hurskainen P, Dahlen P, Ylikoski J, Kwiatkowski M, Siitari H, LovgrenT: Preparation of europium-labelled DNA probes and their properties.Nucleic Acids Res 1991, 19:1057–1061
34. Dahlen PO, Iitia AJ, Skagius G, Frostell A, Nunn MF, Kwiatkowski M:Detection of human immunodeficiency virus type 1 by using thepolymerase chain reaction and a time-resolved fluorescence-basedhybridization assay. J Clin Microbiol 1991, 29:798–804
35. Lundwall A, Lilja H: Molecular cloning of human prostate specificantigen cDNA. FEBS Lett 1987, 214:317–322
36. Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR: Site-directedmutagenesis by overlap extension using the polymerase chain reac-tion. Gene 1989, 77:51–59
37. Riegman PH, Vlietstra RJ, van der Korput HA, Romijn JC, Trapman J:Identification and androgen-regulated expression of two major hu-man glandular kallikrein-1 (hGK-1) mRNA species. Mol Cell Endocri-nol 1991, 76:181–190
38. Schedlich LJ, Bennetts BH, Morris BJ: Primary structure of a humanglandular kallikrein gene. DNA 1987, 6:429–437
39. Raeymaekers L: Quantitative PCR: theoretical considerations withpractical implications. Anal Biochem 1993, 214:582–585
40. Soini E, Lovgren T: Time-resolved fluorescence of lanthanide probesand applications in biotechnology. CRC Crit Rev Anal Chem 1987,18:105–154
41. Chapdelaine P, Paradis G, Tremblay RR, Dube JY: High level ofexpression in the prostate of a human glandular kallikrein mRNArelated to prostate-specific antigen. FEBS Lett 1988, 236:205–208
42. Slawin KM, Shariat SF, Nguyen C, Leventis AK, Song W, Kattan MW,Young CYF, Tindall DJ, Wheeler TM: Detection of metastatic prostatecancer using a splice variant-specific reverse transcriptase-poly-merase chain reaction assay for human glandular kallikrein. CancerRes 2000, 60:7142–7148
43. Heuze N, Olayat S, Gutman N, Zani M-L, Courty Y: Molecular cloningof an alternative hKLK3 transcript coding for a variant protein ofprostate-specific antigen. Cancer Res 1999, 59:2820–2824
44. Liu XF, Essand M, Vasmatzis G, Lee B, Pastan I: Identification of threenew alternate human kallikrein 2 transcripts: evidence of long tran-script and alternative splicing. Biochem Biophys Res Commun 1999,264:833–839
45. Riegman PH, Klaassen P, van der Korput JA, Romijn JC, Trapman J:Molecular cloning and characterization of novel prostate antigencDNA’s. Biochem Biophys Res Commun 1988, 155:181–188
46. Becker C, Piironen T, Pettersson K, Bjork T, Wojno K, Oesterling JE,Lilja H: Discrimination of men with prostate cancer from those withbenign disease by measurements of human glandular kallikrein 2(HK2) in serum. J Urol 2000, 163:311–316
47. Recker F, Kwiatkowski MK, Piironen T, Pettersson K, Huber A, Lum-men G, Tscholl R: Human glandular kallikrein as a tool to improvediscrimination of poorly differentiated and non-organ-confined pros-tate cancer compared with prostate-specific antigen. Urology 2000,55:481–485
48. Haese A, Becker C, Noldus J, Graefen M, Huland E, Huland H, LiljaH: Human glandular kallikrein 2: a potential serum marker for predict-ing the organ confined versus non-organ confined growth of prostatecancer. J Urol 2000, 163:1491–1497
49. Magklara A, Scorilas A, Stephan C, Kristiansen GO, Hauptmann S,Jung K, Diamandis EP: Decreased concentrations of prostate-spe-cific antigen and human glandular kallikrein 2 in malignant versusnonmalignant prostatic tissue. Urology 2000, 56:527–532
122 Ylikoski et alJMD August 2001, Vol. 3, No. 3
Copyright © 2022 FDOKUMEN




















