
The development of behavioural sex differences in infant rhesus macaques (Macaca mulatta)
Upload
independentCategory
view
1download
0
1998, 72(7):5820. J. Virol.
Carol P. Mandell, Phillip Montbriand and Paul A. LuciwImran H. Khan, Earl T. Sawai, Erwin Antonio, Claudia J. Weber, Rhesus MacaquesNef-Associated Kinase and Simian AIDS inImmunodeficiency Virus Nef in Interaction with Role of the SH3-Ligand Domain of Simian
http://jvi.asm.org/content/72/7/5820Updated information and services can be found at:
These include:
REFERENCEShttp://jvi.asm.org/content/72/7/5820#ref-list-1This article cites 46 articles, 17 of which can be accessed free at:
CONTENT ALERTS more»cite this article),
Receive: RSS Feeds, eTOCs, free email alerts (when new articles
http://journals.asm.org/site/misc/reprints.xhtmlInformation about commercial reprint orders: http://journals.asm.org/site/subscriptions/To subscribe to to another ASM Journal go to:
on Septem
ber 28, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from
on S
eptember 28, 2013 by guest
http://jvi.asm.org/
Dow
nloaded from
on Septem
ber 28, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from
on S
eptember 28, 2013 by guest
http://jvi.asm.org/
Dow
nloaded from
on Septem
ber 28, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from
on S
eptember 28, 2013 by guest
http://jvi.asm.org/
Dow
nloaded from
on Septem
ber 28, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from
on S
eptember 28, 2013 by guest
http://jvi.asm.org/
Dow
nloaded from
on Septem
ber 28, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from
on S
eptember 28, 2013 by guest
http://jvi.asm.org/
Dow
nloaded from
on Septem
ber 28, 2013 by guesthttp://jvi.asm
.org/D
ownloaded from
on S
eptember 28, 2013 by guest
http://jvi.asm.org/
Dow
nloaded from

JOURNAL OF VIROLOGY,0022-538X/98/$04.0010
July 1998, p. 5820–5830 Vol. 72, No. 7
Copyright © 1998, American Society for Microbiology. All Rights Reserved.
Role of the SH3-Ligand Domain of Simian ImmunodeficiencyVirus Nef in Interaction with Nef-Associated Kinase
and Simian AIDS in Rhesus MacaquesIMRAN H. KHAN, EARL T. SAWAI, ERWIN ANTONIO, CLAUDIA J. WEBER,
CAROL P. MANDELL, PHILLIP MONTBRIAND, AND PAUL A. LUCIW*
Department of Medical Pathology, University of California,Davis, California 95616
Received 27 January 1998/Accepted 26 March 1998
The nef gene of the human and simian immunodeficiency viruses (HIV and SIV) is dispensable for viralreplication in T-cell lines; however, it is essential for high virus loads and progression to simian AIDS (SAIDS)in SIV-infected adult rhesus macaques. Nef proteins from HIV type 1 (HIV-1), HIV-2, and SIV contain aproline-Xaa-Xaa-proline (PxxP) motif. The region of Nef with this motif is similar to the Src homology region3 (SH3) ligand domain found in many cell signaling proteins. In virus-infected lymphoid cells, Nef interactswith a cellular serine/threonine kinase, designated Nef-associated kinase (NAK). In this study, analysis of viralclones containing point mutations in the nef gene of the pathogenic clone SIVmac239 revealed that severalstrictly conserved residues in the PxxP region were essential for Nef-NAK interaction. The results of thisanalysis of Nef mutations in in vitro kinase assays indicated that the PxxP region in SIV Nef was strikinglysimilar to the consensus sequence for SH3 ligand domains possessing the minus orientation. To test thesignificance of the PxxP motif of Nef for viral pathogenesis, each proline was mutated to an alanine to producethe viral clone SIVmac239-P104A/P107A. This clone, expressing Nef that does not associate with NAK, wasinoculated into seven juvenile rhesus macaques. In vitro kinase assays were performed on virus recovered fromeach animal; the ability of Nef to associate with NAK was restored in five of these animals as early as 8 weeksafter infection. Analysis of nef genes from these viruses revealed patterns of genotypic reversion in the mutatedPxxP motif. These revertant genotypes, which included a second-site suppressor mutation, restored the abilityof Nef to interact with NAK. Additionally, the proportion of revertant viruses increased progressively duringthe course of infection in these animals, and two of these animals developed fatal SAIDS. Taken together, theseresults demonstrated that in vivo selection for the ability of SIV Nef to associate with NAK was correlated withthe induction of SAIDS. Accordingly, these studies implicate a role for the conserved SH3 ligand domain forNef function in virally induced immunodeficiency.
The nef gene of primate lentiviruses (human immunode-ficiency virus types 1 and 2 [HIV-1 and HIV-2] and simianimmunodeficiency virus [SIV]) encodes a 27- to 35-kDa pro-tein that is myristoylated at the N terminus and localized large-ly in cell membranes (8, 41, 48). This gene is dispensable forvirus replication in vitro in cultures of CD4-positive T cells andmacrophages. Kestler et al. have shown that expression of anintact SIV nef gene was essential for the maintenance of highviral loads and progression to simian AIDS in adult rhesusmacaques (19). The importance of nef in the virus-host rela-tionship was also highlighted by the observation that somelong-term survivors (humans) of HIV-1 infection contain lowlevels of a virus with deletions in nef (9, 29). Nonetheless, inneonate macaques, the requirement of nef for pathogenesiscan be overcome by inoculation with high doses of an SIVclone with a deletion in nef (4, 51). Thus, it appears that age isone host factor that influences the role of this viral gene inimmunodeficiency disease.
Several functional properties have been ascribed to Nef ofprimate lentiviruses, including downregulation of the cellsurface receptor CD4 and major histocompatibility complex(MHC) class I molecules on T cells, enhancement of virioninfectivity, and modulation of T-cell activation (8, 41, 48). Nef
was shown to exert inhibitory effects on the induction of tran-scription factors NF-kB and AP-1, interleukin-2, and interleu-kin-2 receptor alpha chain (37). Other reports described acti-vation of T-cell proliferation by Nef, which correlated withincreased virus production (1, 32). The effect of Nef on T-cellactivation is most probably mediated through T-cell signalingpathways (1, 6, 47). An in vivo role for Nef in cell signalinghas been investigated by experiments performed with SIVvariants containing a nef allele with a signal sequence termedthe immunoreceptor tyrosine-based activation motif (ITAM)(10, 28). The presence of an ITAM in the Nef of a clone ofSIVmac239 enabled the virus to activate resting peripheralblood mononuclear cells (PBMC) and replicate at high levelsand to produce acute fatal disease in adult macaques (10).These properties of the viral clone with an ITAM, in tissueculture cells and in animals, are similar to those of SIVpbj14,which is a variant virus that also contains this ITAM in Nef(12).
A number of cell signaling proteins, including tyrosine (Lck,Hck, Src, and Lyn) and serine/threonine kinases (protein ki-nase C-theta, p21-activated kinase [PAK]), have been reportedto associate with Nef (reviewed in reference 41). However, thephysiological relevance of the interaction of Nef with thesevarious cell signaling proteins remains to be established. In ourstudies, cell extracts from HIV-1- and SIV-infected lymphoidcells were immunoprecipitated with anti-Nef antibody and theimmunoprecipitates were subsequently incubated in an in vitro
* Corresponding author. Mailing address: Department of MedicalPathology, University of California, Davis, CA 95616. Phone: (530)752-3430. Fax: (530) 752-4548. E-mail: [email protected].
5820

kinase reaction. This assay revealed two cellular proteins of 62and 72 kDa (p62 and p72, respectively) that coimmunoprecipi-tated with Nef (43, 44). The kinase in these immunoprecipi-tates is designated Nef-associated kinase (NAK). Several linesof evidence have shown that p62 belongs to the PAK family ofcellular serine kinases (27, 35, 45). However, the exact identityof p62, as well as that of p72, remains to be determined. Ad-ditional in vitro kinase assays of immunoprecipitates of in-fected cell extracts, performed with anti-PAK antibodies,demonstrated hyperphosphorylation of p72 in such immuno-precipitates; thus, Nef activates PAK (45). In vivo studies of aninfectious SIV clone with a mutation in Nef, abrogating NAKactivation, suggested that the interaction of Nef with NAK wasimportant for viral pathogenesis in juvenile rhesus macaques(45).
One of the prominent structural features within Nef, withpotential for interactions with cell signaling proteins, is a re-gion with strong homology to a binding ligand for Src homol-ogy 3 (SH3) domains found in various tyrosine kinases andsignaling adapter proteins (31, 36). The role of the SH3 liganddomain (the proline-Xaa-Xaa-proline [PxxP] motif), which ishighly conserved in Nef of primate lentiviruses, was explored invitro; this motif exhibited highly specific interactions with SH3domains of the tyrosine kinases Hck and Lyn (15, 22, 33, 42).Mutational analysis revealed that the integrity of the PxxPmotif in HIV-1 Nef was important for the enhancement ofvirion infectivity; in contrast, CD4 downregulation by Nef wasapparently unaffected by mutations in the PxxP motif (42). TheNef-mediated enhancement of HIV-1 virion infectivity hasbeen correlated with Nef-NAK interaction, which was alsoshown to depend on the integrity of the PxxP region of Nef (14,16, 50). Nevertheless, a recent study examining the role of theSH3 ligand domain of SIV Nef in rhesus macaques infectedwith a viral clone with mutations in the PxxP motif (P104
KVP107) concluded that this highly conserved motif was notimportant for progression to simian AIDS (SAIDS) (20).
We examined the SH3 ligand domain of SIV Nef (contain-ing the PxxP motif P104KVP107) in Nef-NAK interactions byanalyzing SIV clones containing point mutations in this do-main in lymphoid cell cultures. Several key amino acid residuesin this domain of Nef were shown to be necessary for Nef-NAKinteraction and PAK activation. These experiments defined aconsensus sequence, which is very similar to the sequenceshown to be important in SH3 domain/SH3 ligand interactionsin HIV-1 Nef (15, 22, 33, 42). Additionally, the in vivo roleof the SH3 ligand domain for pathogenesis was investigatedby the inoculation of seven juvenile rhesus macaques with anSIVmac239 mutant, which encodes a Nef in which both pro-lines of the SH3 ligand domain were mutated to alanines.Reversion of the Nef mutations in animals inoculated with thismutant virus revealed strong selective pressure for restorationof the SH3 ligand domain. These studies also demonstratedthat the ability of Nef to associate with NAK and activate PAKwas correlated with progression to fatal SAIDS.
MATERIALS AND METHODS
Cells and antibodies. CEMx174 cells, of the human lymphoid T/B-cell hybridcell line, which is permissive to replication of various SIVmac strains, wereprovided by James Hoxie (University of Pennsylvania). Rabbit polyclonal anti-serum was generated against SIV Nef expressed in Escherichia coli (recombinantNef protein was provided by Casey Morrow, University of Alabama). An SIV Nefmonoclonal antibody (17.2) was a generous gift from Kai Krohn (University ofTampere, Tampere, Finland); this antibody was raised against a synthetic pep-tide between amino acids 69 and 75 of SIVmac251 Nef and detects SIVmac239Nef. Rabbit polyclonal anti-rat PAK-1 antibody was purchased from Santa CruzBiotechnologies (Santa Cruz, Calif.); this antibody was raised against the N-terminal peptide (20 amino acids) of rat PAK-1 and detects human PAK-1.
Construction of SIV nef point mutants. The SIVmac239nef1 clone was pro-duced by mutating the premature stop codon in nef of the original pathogenicSIVmac239 clone (GenBank accession no. M33262) to a codon for glutamate,which is the most common codon in revertant viruses recovered from ma-caques infected with this latter viral clone (19). The nef gene and protein inSIVmac239nef1 are designated “prototype.”
The proviral genome of SIVmac239nef1 has been cloned in two halves, di-vided at the unique SphI restriction site (in the vpr gene at position 6707) for easeof handling of the proviral DNA (5). Both halves were inserted into the pGEM7vector (Promega, Madison, Wis.); the 59 half of the viral clone was designatedpVP-1, and the 39 half was designated pVP-2nef (36a). Mutants with point mu-tations in the PxxP region of Nef were generated, either by PCR or by oligonu-cleotide mutagenesis, to change codons of various amino acids to alanine codons(Fig. 1A). For PCR mutagenesis, the primers contained two convenient restric-tion sites, a BglII site (position 9375) at the 59 end of the 59 primer, and an AflIIIsite (position 9686) at the 39 end of the 39 primer. Thus, DNA fragments (311 bp)amplified with these primers contained BglII sites at their 59 ends and AflIII sitesat their 39 ends. The 39-end primer also contained the intended point mutationsin the nef gene. After PCR amplification, the amplified DNA fragments werecloned directly into the pCRII vector as specified by the manufacturer TA clon-ing kit; Invitrogen, San Diego, Calif.). All mutations were confirmed by DNAsequencing. The mutant fragments between the BglII and AflIII sites were thenused to replace the corresponding fragment in pVP-2(nef1).
For mutagenesis by oligonucleotides, a DNA fragment containing part of thenef gene between the SacI (position 9487) and NdeI (position 10008) sites inpVP-2(nef1), was subcloned into a derivative of pUC19 that lacked the AflIIIrestriction site. A unique ClaI site was then engineered at position 9600, withoutresulting in amino acid changes in Nef; this clone was designated pIK3. The PxxPregion of Nef was encoded between the new ClaI site and the unique AflIII sitein the nef gene. Mutant oligonucleotides between ClaI and AflIII sites (86 bp)were designed to contain the intended point mutations in the region encodingPxxP. The double-stranded oligonucleotides representing the mutant DNA frag-ments were used to replace the corresponding prototype ClaI-AflIII fragment inpIK3. All the mutations were confirmed by DNA sequencing. Finally, the SacI-NdeI fragment from pIK3, containing the point mutation(s) in the region en-coding PxxP, replaced the corresponding fragment in pVP-2(nef1). Completenef mutant proviruses were obtained by joining pVP-1 and the various mutantpVP-2 plasmids at the SphI site.
Production of virus stocks from proviral clones. Plasmids containing full-length SIVmac239nef1 proviral DNA and mutant proviral DNA were transfect-ed into CEMx174 cells. Transfections were performed in duplicate by electro-poration as previously described (49). Briefly, exponentially growing cells wereresuspended in serum-free RPMI medium at a concentration of 107 cells/ml.Plasmid DNA (5 mg) was mixed with 0.4 ml of this cell suspension, and electro-poration was performed in a 0.4-cm cuvette at 960 mF capacitance and 200 V byusing a gene pulser (Bio-Rad, Richmond, Calif.). Electroporated cells were cul-tured and microscopically monitored at daily intervals for virus production by theappearance of cytopathic effects such as multinucleated syncytia and giant cells.After 5 to 8 days of culture, cell-free virus stocks were obtained by removal ofcells by centrifugation (2,500 3 g for 5 min) and removal of cell debris from thesupernatant by filtration through 0.45-mm-pore-size filters. Virus stocks werestored frozen at 270°C in 1-ml aliquots. Titers of virus stocks were determinedin microtiter plates containing CEMx174 cells; the 50% tissue culture infectivedose (TCID50) was calculated by using the end-point dilution method of Reedand Muench (38).
Chronically infected cell lines. CEMx174 cells chronically infected withSIVmac239nef1 and mutant viruses were derived as outgrowths of acute infec-tion with the viruses as previously described (43). These cell lines were storedfrozen at 5 3 106 cells/ml at 2135°C in RPMI containing 20% fetal calf serumand 10% dimethyl sulfoxide.
In vitro kinase assays and anti-Nef immunoblots. In vitro kinase assays wereperformed, as described previously (43), on immunoprecipitates obtained fromlysates of virally infected CEMx174 cells by using either rabbit anti-SIV Nefantibody or rabbit anti-rat PAK-1 antibody. For anti-Nef immunoblots, proteinswere transferred from sodium dodecyl sulfate-polyacrylamide gels to polyvinyl-idene difluoride (PVDF) membranes (Bio-Rad). After three washes with Blottobuffer (5% nonfat dry milk in phosphate-buffered saline [pH 7.4]), the PVDFmembranes were incubated with anti-SIV Nef monoclonal antibody (antibody17.2). The membranes were washed three times with wash buffer (250 mM NaCl,50 mM Tris [pH 7.4], 0.1% Tween 20) and incubated with goat anti-mouse sec-ondary antibody conjugated to alkaline phosphatase (Southern BiotechnologyAssociates, Birmingham, Ala.). After being washed three times with wash bufferand once with 100 mM Tris (pH 9.5), the blots were developed with the BCIP/NBT detection kit (Vector Laboratories, Burlingame, Calif.).
Inoculation of rhesus macaques. All the animals in the study were colony-bredjuvenile rhesus macaques (Macaca mulatta) free of simian retrovirus (SRV)type D, SIV, and simian T-lymphotropic virus; these animals were housed atthe California Regional Primate Research Center at the University of California,Davis. Seven macaques were intravenously inoculated with 1,000 TCID50 ofmutant virus carrying the proline-to-alanine mutation in the two prolines withinthe SH3 ligand domain of Nef (SIVmac239-P104A/P107A). Two additional ani-mals each received an intravenous injection of 1,000 TCID50 of the pathogenic
VOL. 72, 1998 PxxP DOMAIN OF SIV Nef AND SIMIAN AIDS 5821
clone SIVmac239nef1. At fixed intervals, blood samples were drawn from theinoculated animals for virus isolation, complete blood cell count, CD4 and CD8cell count (by flow cytometry), and determination of anti-SIV antibody levels(with the HIV-2 antibody kit [Genetic Systems, Shasta, Minn.]). Virus load
measurements were made on plasma samples obtained from EDTA-anticoagu-lated blood by a branched-chain DNA (bDNA) assay developed for quantitatingSIV RNA copies in plasma (P. Dailey and J. Booth, Chiron Diagnostics, ChironReference Testing Laboratory, Emeryville, Calif.). Complete physical examina-tions were performed on the animals at regular intervals. The animals were alsomonitored for body weight and clinical signs of disease. Opportunistic infectionswere diagnosed by standard microbiological techniques performed on clinicalsamples at the Clinical Microbiology Laboratory, California Regional PrimateResearch Center. Macaques that became seriously ill and were nonresponsive totherapeutic interventions (e.g., enhanced diets and antibiotics) were humanelyeuthanized with a barbiturate overdose. Some macaques were tested for SRV atnecropsy by sensitive PCR amplification of DNA from PBMC and lymph nodeswith SRV primers (24) and by immunoblot analysis of plasma for antibodies towhole virus (23).
PCR amplification and sequencing of nef genes from infected animals. DNAwas isolated, using the Qiagen blood kit as specified by the manufacturer (QiagenInc., Chatsworth, Calif.), from either 400 ml of whole blood or 107 PBMC ob-tained from infected animals. The DNA thus obtained was used as the templatefor amplification of nef by nested PCR. Two PCR amplifications were performedper time point. In the first round (30 cycles) of PCR, the 59 and 39 primers 59-CCAGAGGCTCTCTGCGACCCTAC and 59-AGAGGGCTTTAAGCAAGCAAGCGTG, respectively, were used for nef amplification. For the second round(30 cycles), the 59-primer remained the same while the 39- primer was 59-GCCTCTCCGCAGAGCGACTGAATAC. PCR amplifications were performed in aDNA thermal cycler (Perkin-Elmer, Foster City, Calif.) with Taq polymerase(Perkin-Elmer). The final product (992 bp), containing the full-length nef gene,was directly cloned into the pCRII vector, and the DNA was sequenced on bothstrands.
Isolation of virus from PBMC of infected macaques. Virus isolations wereperformed as previously described by coculturing PBMC from infected animalswith CEMx174 cells (30). For in vitro kinase assays, acutely infected CEMx174cells, produced by cocultivation with PBMC from macaques infected withSIVmac239-P104A/P107A, were lysed as described previously (45). The lysateswere analyzed in the in vitro kinase assays as described above to test for therestoration of the ability of Nef to associate with NAK.
Histological examination of tissues from infected rhesus macaques. To assesshistopathologic changes during the course of infection, axillary and inguinallymph nodes were obtained by percutaneous biopsy under ketamine hydrochlo-ride anesthesia. Lymph node biopsy specimens and all tissues collected at nec-ropsy were fixed in 10% buffered formalin, embedded in paraffin, sectioned, andstained with hematoxylin and eosin for microscopic examination.
RESULTS
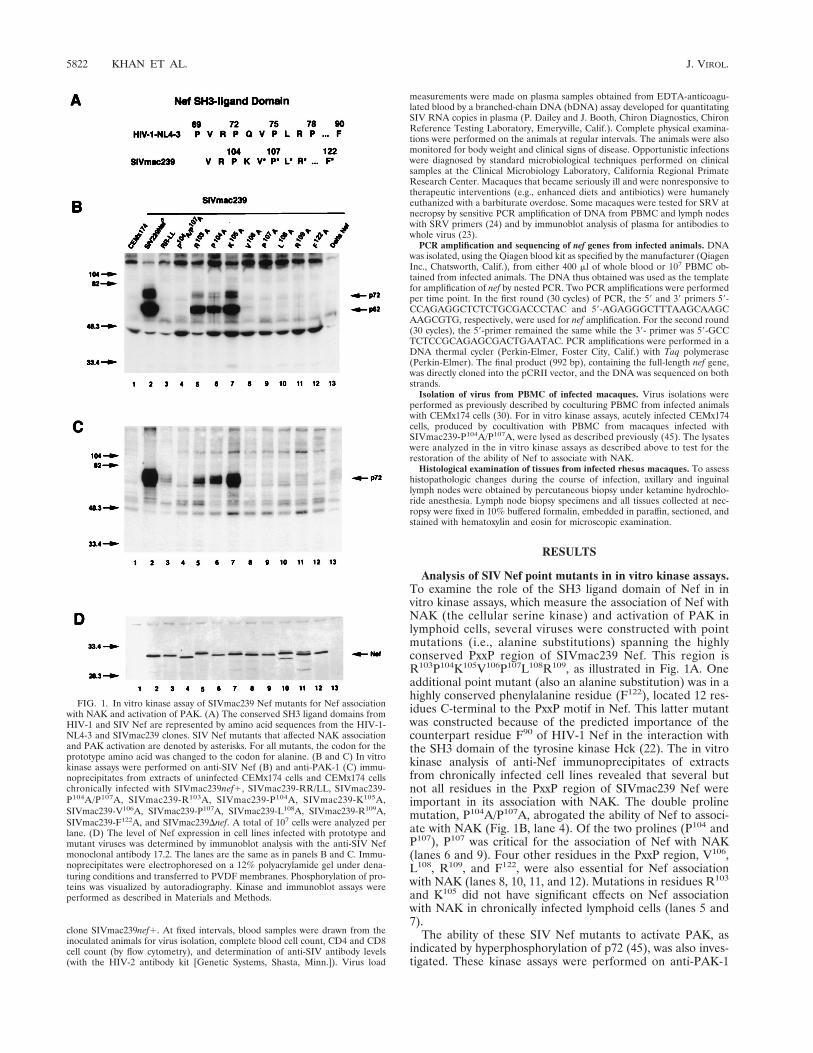
Analysis of SIV Nef point mutants in in vitro kinase assays.To examine the role of the SH3 ligand domain of Nef in invitro kinase assays, which measure the association of Nef withNAK (the cellular serine kinase) and activation of PAK inlymphoid cells, several viruses were constructed with pointmutations (i.e., alanine substitutions) spanning the highlyconserved PxxP region of SIVmac239 Nef. This region isR103P104K105V106P107L108R109, as illustrated in Fig. 1A. Oneadditional point mutant (also an alanine substitution) was in ahighly conserved phenylalanine residue (F122), located 12 res-idues C-terminal to the PxxP motif in Nef. This latter mutantwas constructed because of the predicted importance of thecounterpart residue F90 of HIV-1 Nef in the interaction withthe SH3 domain of the tyrosine kinase Hck (22). The in vitrokinase analysis of anti-Nef immunoprecipitates of extractsfrom chronically infected cell lines revealed that several butnot all residues in the PxxP region of SIVmac239 Nef wereimportant in its association with NAK. The double prolinemutation, P104A/P107A, abrogated the ability of Nef to associ-ate with NAK (Fig. 1B, lane 4). Of the two prolines (P104 andP107), P107 was critical for the association of Nef with NAK(lanes 6 and 9). Four other residues in the PxxP region, V106,L108, R109, and F122, were also essential for Nef associationwith NAK (lanes 8, 10, 11, and 12). Mutations in residues R103
and K105 did not have significant effects on Nef associationwith NAK in chronically infected lymphoid cells (lanes 5 and7).
The ability of these SIV Nef mutants to activate PAK, asindicated by hyperphosphorylation of p72 (45), was also inves-tigated. These kinase assays were performed on anti-PAK-1
FIG. 1. In vitro kinase assay of SIVmac239 Nef mutants for Nef associationwith NAK and activation of PAK. (A) The conserved SH3 ligand domains fromHIV-1 and SIV Nef are represented by amino acid sequences from the HIV-1-NL4-3 and SIVmac239 clones. SIV Nef mutants that affected NAK associationand PAK activation are denoted by asterisks. For all mutants, the codon for theprototype amino acid was changed to the codon for alanine. (B and C) In vitrokinase assays were performed on anti-SIV Nef (B) and anti-PAK-1 (C) immu-noprecipitates from extracts of uninfected CEMx174 cells and CEMx174 cellschronically infected with SIVmac239nef1, SIVmac239-RR/LL, SIVmac239-P104A/P107A, SIVmac239-R103A, SIVmac239-P104A, SIVmac239-K105A,SIVmac239-V106A, SIVmac239-P107A, SIVmac239-L108A, SIVmac239-R109A,SIVmac239-F122A, and SIVmac239Dnef. A total of 107 cells were analyzed perlane. (D) The level of Nef expression in cell lines infected with prototype andmutant viruses was determined by immunoblot analysis with the anti-SIV Nefmonoclonal antibody 17.2. The lanes are the same as in panels B and C. Immu-noprecipitates were electrophoresed on a 12% polyacrylamide gel under dena-turing conditions and transferred to PVDF membranes. Phosphorylation of pro-teins was visualized by autoradiography. Kinase and immunoblot assays wereperformed as described in Materials and Methods.
5822 KHAN ET AL. J. VIROL.
immunoprecipitates from CEMx174 cells chronically infectedwith all of the Nef mutant viruses described above. The sameNef mutants which were defective in association with NAK(P104A/P107A, V106A, P107A, L108A, R109A and F122A [Fig.1B]) were also defective in activation of PAK (Fig. 1C, lanes 4and 8 through 12). This finding was consistent with our previ-ously reported results showing that Nef mutants with muta-tions in the double-arginine motif (R137R138) were defective inboth NAK association and activation of PAK (Fig. 1B and C,lanes 3) (45). Within the SH3 ligand domain of SIV Nef, theresidue P104 could be altered to A without affecting associationor activation of PAK in the in vitro kinase assays (Fig. 1B andC, lanes 6). Because prolines in SH3 ligand domains of cellularproteins contribute to SH3 binding through hydrophobic in-teractions (25), it is possible that in the absence of P104 in Nef,some other neighboring hydrophobic residue(s) (e.g., V102)contributes to the interaction with NAK. Immunoblot analysisdemonstrated that in CEMx174 cell lines chronically infectedwith prototype SIVmac239 or various Nef mutant viruses, all ofthe mutant Nef proteins were expressed at levels similar tothose in the cells infected with prototype Nef (Fig. 1D). Takentogether, the analysis of these SIV mutants in the NAK asso-ciation assay (on immunoprecipitates prepared with anti-Nefantibody) and the PAK activation assay (on immunoprecipi-tates prepared with anti-PAK-1 antibody) demonstrate the im-portance of V106P107L108R109, and F122 in the SH3 ligand do-main of Nef.
Clinical outcome of macaques infected with SIVmac239-P104A/P107A. All the rhesus macaques for this study werejuvenile animals, ranging in age from 27 to 49 months atthe time of inoculation. Seven macaques received a cell-freepreparation of SIVmac239-P104A/P107A at 1,000 TCID50by the intravenous route. To serve as comparisons, two ma-caques were inoculated with the pathogenic molecular cloneSIVmac239nef1. All the animals were monitored for virusload, antiviral antibodies, and hematological and clinical signsof immunodeficiency.
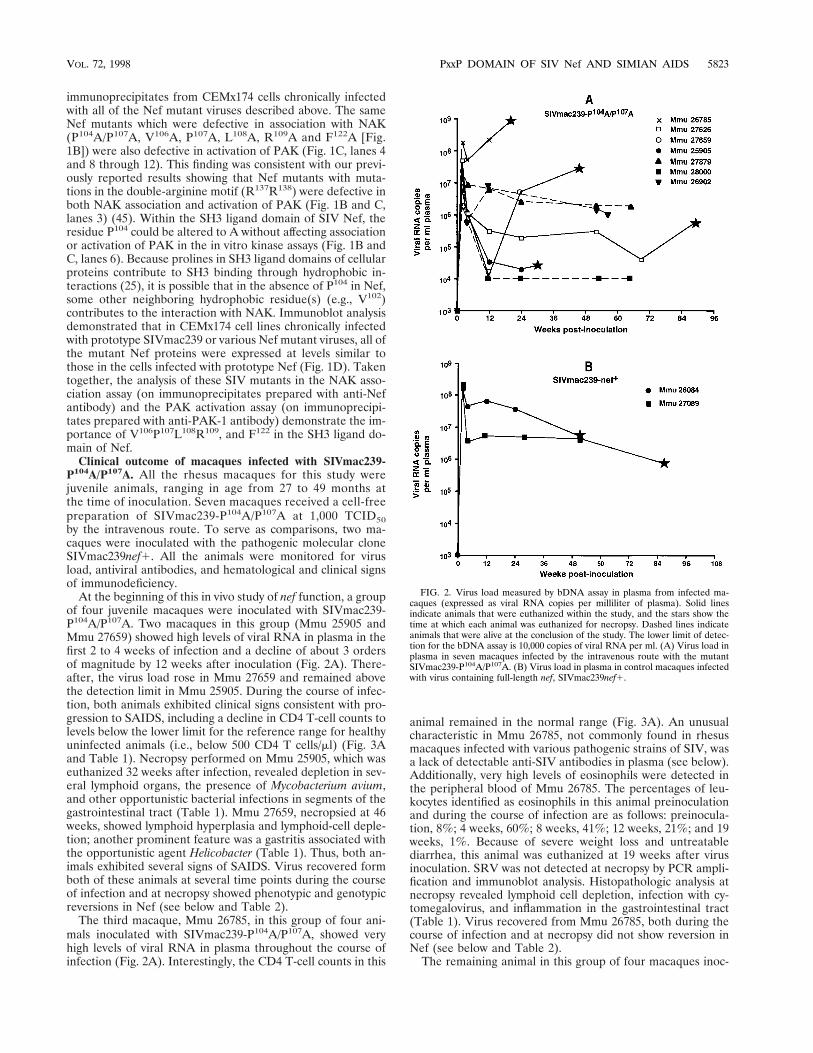
At the beginning of this in vivo study of nef function, a groupof four juvenile macaques were inoculated with SIVmac239-P104A/P107A. Two macaques in this group (Mmu 25905 andMmu 27659) showed high levels of viral RNA in plasma in thefirst 2 to 4 weeks of infection and a decline of about 3 ordersof magnitude by 12 weeks after inoculation (Fig. 2A). There-after, the virus load rose in Mmu 27659 and remained abovethe detection limit in Mmu 25905. During the course of infec-tion, both animals exhibited clinical signs consistent with pro-gression to SAIDS, including a decline in CD4 T-cell counts tolevels below the lower limit for the reference range for healthyuninfected animals (i.e., below 500 CD4 T cells/ml) (Fig. 3Aand Table 1). Necropsy performed on Mmu 25905, which waseuthanized 32 weeks after infection, revealed depletion in sev-eral lymphoid organs, the presence of Mycobacterium avium,and other opportunistic bacterial infections in segments of thegastrointestinal tract (Table 1). Mmu 27659, necropsied at 46weeks, showed lymphoid hyperplasia and lymphoid-cell deple-tion; another prominent feature was a gastritis associated withthe opportunistic agent Helicobacter (Table 1). Thus, both an-imals exhibited several signs of SAIDS. Virus recovered formboth of these animals at several time points during the courseof infection and at necropsy showed phenotypic and genotypicreversions in Nef (see below and Table 2).
The third macaque, Mmu 26785, in this group of four ani-mals inoculated with SIVmac239-P104A/P107A, showed veryhigh levels of viral RNA in plasma throughout the course ofinfection (Fig. 2A). Interestingly, the CD4 T-cell counts in this
animal remained in the normal range (Fig. 3A). An unusualcharacteristic in Mmu 26785, not commonly found in rhesusmacaques infected with various pathogenic strains of SIV, wasa lack of detectable anti-SIV antibodies in plasma (see below).Additionally, very high levels of eosinophils were detected inthe peripheral blood of Mmu 26785. The percentages of leu-kocytes identified as eosinophils in this animal preinoculationand during the course of infection are as follows: preinocula-tion, 8%; 4 weeks, 60%; 8 weeks, 41%; 12 weeks, 21%; and 19weeks, 1%. Because of severe weight loss and untreatablediarrhea, this animal was euthanized at 19 weeks after virusinoculation. SRV was not detected at necropsy by PCR ampli-fication and immunoblot analysis. Histopathologic analysis atnecropsy revealed lymphoid cell depletion, infection with cy-tomegalovirus, and inflammation in the gastrointestinal tract(Table 1). Virus recovered from Mmu 26785, both during thecourse of infection and at necropsy did not show reversion inNef (see below and Table 2).
The remaining animal in this group of four macaques inoc-
FIG. 2. Virus load measured by bDNA assay in plasma from infected ma-caques (expressed as viral RNA copies per milliliter of plasma). Solid linesindicate animals that were euthanized within the study, and the stars show thetime at which each animal was euthanized for necropsy. Dashed lines indicateanimals that were alive at the conclusion of the study. The lower limit of detec-tion for the bDNA assay is 10,000 copies of viral RNA per ml. (A) Virus load inplasma in seven macaques infected by the intravenous route with the mutantSIVmac239-P104A/P107A. (B) Virus load in plasma in control macaques infectedwith virus containing full-length nef, SIVmac239nef1.
VOL. 72, 1998 PxxP DOMAIN OF SIV Nef AND SIMIAN AIDS 5823
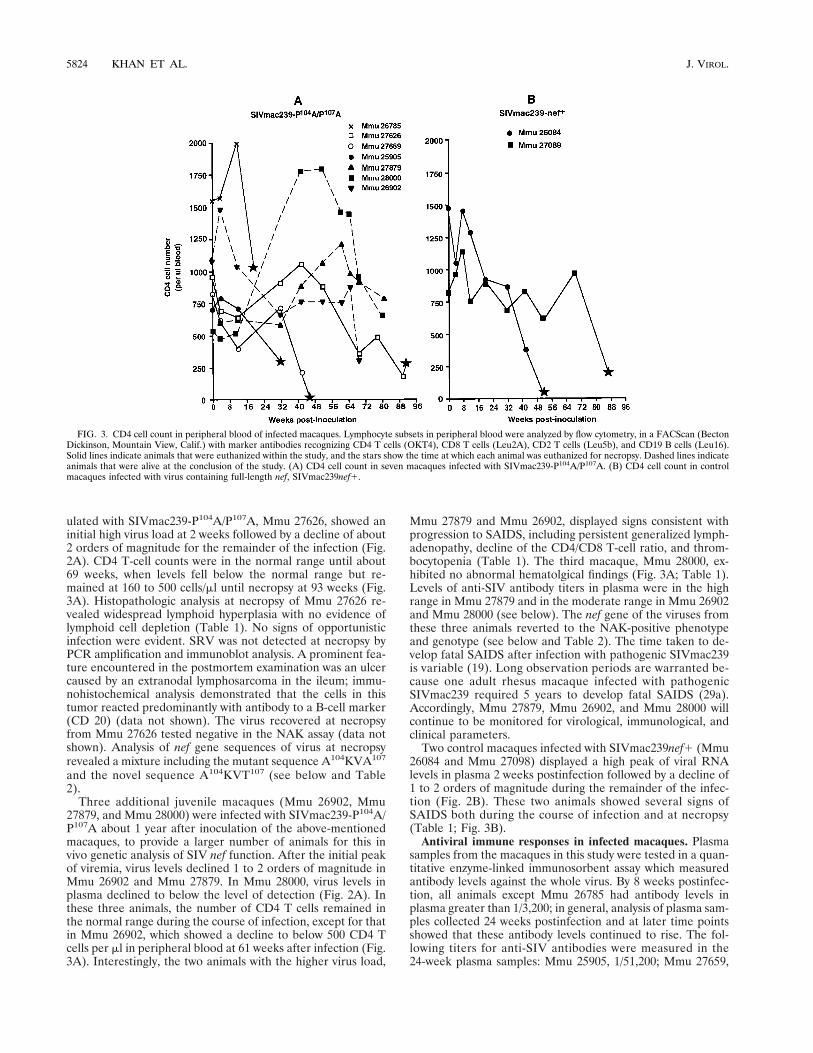
ulated with SIVmac239-P104A/P107A, Mmu 27626, showed aninitial high virus load at 2 weeks followed by a decline of about2 orders of magnitude for the remainder of the infection (Fig.2A). CD4 T-cell counts were in the normal range until about69 weeks, when levels fell below the normal range but re-mained at 160 to 500 cells/ml until necropsy at 93 weeks (Fig.3A). Histopathologic analysis at necropsy of Mmu 27626 re-vealed widespread lymphoid hyperplasia with no evidence oflymphoid cell depletion (Table 1). No signs of opportunisticinfection were evident. SRV was not detected at necropsy byPCR amplification and immunoblot analysis. A prominent fea-ture encountered in the postmortem examination was an ulcercaused by an extranodal lymphosarcoma in the ileum; immu-nohistochemical analysis demonstrated that the cells in thistumor reacted predominantly with antibody to a B-cell marker(CD 20) (data not shown). The virus recovered at necropsyfrom Mmu 27626 tested negative in the NAK assay (data notshown). Analysis of nef gene sequences of virus at necropsyrevealed a mixture including the mutant sequence A104KVA107
and the novel sequence A104KVT107 (see below and Table2).
Three additional juvenile macaques (Mmu 26902, Mmu27879, and Mmu 28000) were infected with SIVmac239-P104A/P107A about 1 year after inoculation of the above-mentionedmacaques, to provide a larger number of animals for this invivo genetic analysis of SIV nef function. After the initial peakof viremia, virus levels declined 1 to 2 orders of magnitude inMmu 26902 and Mmu 27879. In Mmu 28000, virus levels inplasma declined to below the level of detection (Fig. 2A). Inthese three animals, the number of CD4 T cells remained inthe normal range during the course of infection, except for thatin Mmu 26902, which showed a decline to below 500 CD4 Tcells per ml in peripheral blood at 61 weeks after infection (Fig.3A). Interestingly, the two animals with the higher virus load,
Mmu 27879 and Mmu 26902, displayed signs consistent withprogression to SAIDS, including persistent generalized lymph-adenopathy, decline of the CD4/CD8 T-cell ratio, and throm-bocytopenia (Table 1). The third macaque, Mmu 28000, ex-hibited no abnormal hematolgical findings (Fig. 3A; Table 1).Levels of anti-SIV antibody titers in plasma were in the highrange in Mmu 27879 and in the moderate range in Mmu 26902and Mmu 28000 (see below). The nef gene of the viruses fromthese three animals reverted to the NAK-positive phenotypeand genotype (see below and Table 2). The time taken to de-velop fatal SAIDS after infection with pathogenic SIVmac239is variable (19). Long observation periods are warranted be-cause one adult rhesus macaque infected with pathogenicSIVmac239 required 5 years to develop fatal SAIDS (29a).Accordingly, Mmu 27879, Mmu 26902, and Mmu 28000 willcontinue to be monitored for virological, immunological, andclinical parameters.
Two control macaques infected with SIVmac239nef1 (Mmu26084 and Mmu 27098) displayed a high peak of viral RNAlevels in plasma 2 weeks postinfection followed by a decline of1 to 2 orders of magnitude during the remainder of the infec-tion (Fig. 2B). These two animals showed several signs ofSAIDS both during the course of infection and at necropsy(Table 1; Fig. 3B).
Antiviral immune responses in infected macaques. Plasmasamples from the macaques in this study were tested in a quan-titative enzyme-linked immunosorbent assay which measuredantibody levels against the whole virus. By 8 weeks postinfec-tion, all animals except Mmu 26785 had antibody levels inplasma greater than 1/3,200; in general, analysis of plasma sam-ples collected 24 weeks postinfection and at later time pointsshowed that these antibody levels continued to rise. The fol-lowing titers for anti-SIV antibodies were measured in the24-week plasma samples: Mmu 25905, 1/51,200; Mmu 27659,
FIG. 3. CD4 cell count in peripheral blood of infected macaques. Lymphocyte subsets in peripheral blood were analyzed by flow cytometry, in a FACScan (BectonDickinson, Mountain View, Calif.) with marker antibodies recognizing CD4 T cells (OKT4), CD8 T cells (Leu2A), CD2 T cells (Leu5b), and CD19 B cells (Leu16).Solid lines indicate animals that were euthanized within the study, and the stars show the time at which each animal was euthanized for necropsy. Dashed lines indicateanimals that were alive at the conclusion of the study. (A) CD4 cell count in seven macaques infected with SIVmac239-P104A/P107A. (B) CD4 cell count in controlmacaques infected with virus containing full-length nef, SIVmac239nef1.
5824 KHAN ET AL. J. VIROL.
1/102,400; Mmu 27626, 1/102,400; Mmu 27879, 1/409,600;Mmu 28000, 1/51,200; and Mmu 26902, 1/204,800. In strikingcontrast, antibody titers in plasma samples collected fromMmu 26785 at 8, 12, and 19 weeks were below the detectionlimit of this assay (,1/100). Other investigators have also re-
ported undetectable antiviral antibody levels in adult rhesusmacaques exhibiting rapid disease progression after infectionwith pathogenic strains of SIV (17, 53).
In vivo genotypic and phenotypic reversions in nef. To in-vestigate potential reversions in the nef sequence (genotypic
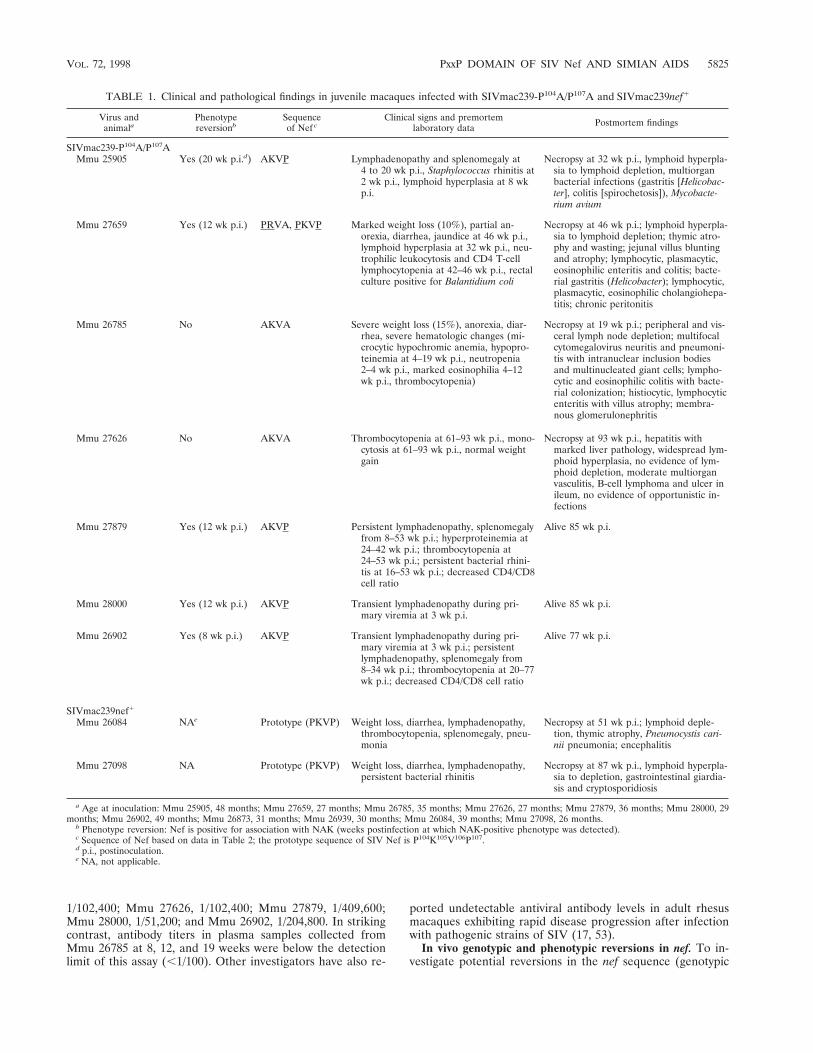
TABLE 1. Clinical and pathological findings in juvenile macaques infected with SIVmac239-P104A/P107A and SIVmac239nef1
Virus andanimala
Phenotypereversionb
Sequenceof Nef c
Clinical signs and premortemlaboratory data Postmortem findings
SIVmac239-P104A/P107AMmu 25905 Yes (20 wk p.i.d) AKVP Lymphadenopathy and splenomegaly at
4 to 20 wk p.i., Staphylococcus rhinitis at2 wk p.i., lymphoid hyperplasia at 8 wkp.i.
Necropsy at 32 wk p.i., lymphoid hyperpla-sia to lymphoid depletion, multiorganbacterial infections (gastritis [Helicobac-ter], colitis [spirochetosis]), Mycobacte-rium avium
Mmu 27659 Yes (12 wk p.i.) PRVA, PKVP Marked weight loss (10%), partial an-orexia, diarrhea, jaundice at 46 wk p.i.,lymphoid hyperplasia at 32 wk p.i., neu-trophilic leukocytosis and CD4 T-celllymphocytopenia at 42–46 wk p.i., rectalculture positive for Balantidium coli
Necropsy at 46 wk p.i.; lymphoid hyperpla-sia to lymphoid depletion; thymic atro-phy and wasting; jejunal villus bluntingand atrophy; lymphocytic, plasmacytic,eosinophilic enteritis and colitis; bacte-rial gastritis (Helicobacter); lymphocytic,plasmacytic, eosinophilic cholangiohepa-titis; chronic peritonitis
Mmu 26785 No AKVA Severe weight loss (15%), anorexia, diar-rhea, severe hematologic changes (mi-crocytic hypochromic anemia, hypopro-teinemia at 4–19 wk p.i., neutropenia2–4 wk p.i., marked eosinophilia 4–12wk p.i., thrombocytopenia)
Necropsy at 19 wk p.i.; peripheral and vis-ceral lymph node depletion; multifocalcytomegalovirus neuritis and pneumoni-tis with intranuclear inclusion bodiesand multinucleated giant cells; lympho-cytic and eosinophilic colitis with bacte-rial colonization; histiocytic, lymphocyticenteritis with villus atrophy; membra-nous glomerulonephritis
Mmu 27626 No AKVA Thrombocytopenia at 61–93 wk p.i., mono-cytosis at 61–93 wk p.i., normal weightgain
Necropsy at 93 wk p.i., hepatitis withmarked liver pathology, widespread lym-phoid hyperplasia, no evidence of lym-phoid depletion, moderate multiorganvasculitis, B-cell lymphoma and ulcer inileum, no evidence of opportunistic in-fections
Mmu 27879 Yes (12 wk p.i.) AKVP Persistent lymphadenopathy, splenomegalyfrom 8–53 wk p.i.; hyperproteinemia at24–42 wk p.i.; thrombocytopenia at24–53 wk p.i.; persistent bacterial rhini-tis at 16–53 wk p.i.; decreased CD4/CD8cell ratio
Alive 85 wk p.i.
Mmu 28000 Yes (12 wk p.i.) AKVP Transient lymphadenopathy during pri-mary viremia at 3 wk p.i.
Alive 85 wk p.i.
Mmu 26902 Yes (8 wk p.i.) AKVP Transient lymphadenopathy during pri-mary viremia at 3 wk p.i.; persistentlymphadenopathy, splenomegaly from8–34 wk p.i.; thrombocytopenia at 20–77wk p.i.; decreased CD4/CD8 cell ratio
Alive 77 wk p.i.
SIVmac239nef1
Mmu 26084 NAe Prototype (PKVP) Weight loss, diarrhea, lymphadenopathy,thrombocytopenia, splenomegaly, pneu-monia
Necropsy at 51 wk p.i.; lymphoid deple-tion, thymic atrophy, Pneumocystis cari-nii pneumonia; encephalitis
Mmu 27098 NA Prototype (PKVP) Weight loss, diarrhea, lymphadenopathy,persistent bacterial rhinitis
Necropsy at 87 wk p.i., lymphoid hyperpla-sia to depletion, gastrointestinal giardia-sis and cryptosporidiosis
a Age at inoculation: Mmu 25905, 48 months; Mmu 27659, 27 months; Mmu 26785, 35 months; Mmu 27626, 27 months; Mmu 27879, 36 months; Mmu 28000, 29months; Mmu 26902, 49 months; Mmu 26873, 31 months; Mmu 26939, 30 months; Mmu 26084, 39 months; Mmu 27098, 26 months.
b Phenotype reversion: Nef is positive for association with NAK (weeks postinfection at which NAK-positive phenotype was detected).c Sequence of Nef based on data in Table 2; the prototype sequence of SIV Nef is P104K105V106P107.d p.i., postinoculation.e NA, not applicable.
VOL. 72, 1998 PxxP DOMAIN OF SIV Nef AND SIMIAN AIDS 5825

reversion) and Nef function (phenotypic reversion), virus wasisolated at various time points from PBMC obtained frommacaques infected with the mutant clone SIVmac239-P104A/P107A. In these recovered viruses, DNA from the infected cellswas used for PCR amplification to determine the sequence ofnef genes and Nef function was analyzed in in vitro kinaseassays.
Reversions in the nef sequence were first detected at thefollowing time points: Mmu 26902 at 8 weeks postinoculation;Mmu 27659, Mmu 27879, and Mmu 28000 at 12 weeks; andMmu 25905 at 20 weeks (Table 2). The virus isolated from thisgroup of macaques, except Mmu 27659, displayed a changeof A107 to P. This pattern of change, which produced the se-quence A104KVP107, was a revertant phenotype, as demon-strated in the previous analysis of mutants with point muta-tions in the PxxP region in the in vitro kinase assay (Fig. 1B andC); this assay revealed that the genotype A104KVP107 waspositive for NAK association and PAK activation. In most ofthese animals, the proportion of nef clones with the sequencepositive for Nef-NAK interaction increased at subsequent timepoints (Table 2). As an example of phenotypic reversion forthe A104KVP107 genotype, the results of an in vitro kinase
assay are shown for virus isolated from Mmu 25905 at 20 and30 weeks postinoculation; strong phosphorylation of both p62and p72 was observed (Fig. 4A, lanes 4 and 5). Taken together,these findings demonstrated significant selection pressure invivo for the ability of Nef to associate with NAK.
The pattern of genotypic reversions observed in the virusisolated from Mmu 27659 was more complex than the patternin the viruses from the other animals. At 12 weeks after inoc-ulation of Mmu 27659, 3 of 10 nef clones displayed reversion atposition 104 (changing A104 to P) whereas alanine at position107 (A107) remained unchanged (Table 2). Based on the anal-ysis of point mutants in the in vitro kinase assay, this change ofA104 to P alone would not result in association of Nef withNAK (Fig. 1B and C). However, all three of the Nef clonescontaining the A104 to P reversion also contained a second-sitemutation at position 105, changing K105 to R (Table 2). To de-termine whether Nef with this revertant sequence was ca-pable of association with NAK, this sequence, P104R105VA107,was introduced into the SIVmac239nef1 clone to produce themutant clone designated SIVmac239-K105R/P107A. This mu-tant virus revealed a low but positive level of Nef associationwith NAK (Fig. 4C, lane 3). The level of Nef-NAK interac-tion for this mutant Nef was similar to the level detected invirus isolated from Mmu 27659 at 24 weeks postinoculation(Fig. 4A, lane 7), at which time point the mutant sequenceP104RVA107 predominated (Table 2). Additionally, the nefclones in Mmu 27659 exhibited further evolution during thecourse of infection. Importantly, at 42 weeks, 6 of 12 nef clonesdisplayed reversion to the prototype sequence (P104KVP107)whereas the other 6 nef clones still contained the P104R105VA107
sequence (Table 2); both of these genotypes are positive forthe NAK association phenotype. This increase in the propor-tion of virus with the P104KVP107 genotype was reflected in thestriking increase in the phosphorylation of p62 and p72 in thein vitro kinase assay performed on virus isolated at the sametime point (Fig. 4B, lane 4). The proportion of nef clones con-taining the prototype sequence (P104KVP107) increased from50% at 42 weeks to 70% at 46 weeks (Table 2). Taken to-gether, the pattern of reversions in Mmu 27659 not only re-inforced the significance of in vivo selection of Nef with theability to associate with NAK but also highlighted the impor-tance of Nef residue P104 in the SH3 ligand domain.
Analysis of virus recovered from the animal displaying veryrapid disease progression, Mmu 26785, showed that the mutantnef gene did not revert from the mutant sequence either duringthe course of infection or at necropsy at 19 weeks postinocu-lation (Table 2). Thus, the lack of genotypic revertants wasconsistent with the NAK-negative phenotype of virus recov-ered from this animal at necropsy (data not shown).
Virus in Mmu 27626 did not exhibit reversion in the nefsequence (Table 2) or in the ability of Nef to associate withNAK as analyzed at 24 weeks (Fig. 4A, lane 8) and 42 weeks(Fig. 4B, lane 3) postinfection. At necropsy at 93 weeks, nefclones from Mmu 27626 exhibited the mutant sequenceA104KVA107 as well as a variant sequence, A104KVT107 (Ta-ble 2). Nef produced by virus recovered from this animal atnecropsy did not associate with NAK in the in vitro kinaseassay (data not shown).
DISCUSSION
Structural features of the SH3 ligand domain in Nef. Anunderstanding of the SH3 ligand domain of Nef is built on ourknowledge of the molecular basis of SH3-domain/SH3-ligandinteractions (reviewed in reference 36) as well as of structuralmodels of HIV-1 Nef (15, 22). From the analysis of cellular
TABLE 2. Genotypic reversions in Nef of viruses recoveredfrom juvenile rhesus macaques infected
with SIVmac239-P104A/P107A
Animal Time (wk)postinfectiona
No. ofclones
Amino acid sequencefrom 104 to 107b
Mmu 25905 20 16 AKVP
30 15 AKVP
Mmu 27659 12 3 PRVA11 AKVA
24 1 PKVP9 PRVA
42 6 PKVP6 PRVA
46 7 PKVP3 PRVA
Mmu 26785 19 10 AKVA
Mmu 27626 93 4 AKVA5 AKVT
Mmu 27879 12 16 AKVP4 AKVA
24 19 AKVP
Mmu 28000 12 15 AKVP9 AKVA
17 8 AKVP
Mmu 26902 8 17 AKVP
12 26 AKVP
a Time of virus recovery.b DNA sequence analysis was conducted on PCR amplified nef clones from
viruses recovered from macaques infected with the mutant virus. The genotypicreversions are presented as the deduced amino acid sequence. The prototype Nefsequence is P104K105V106P107.
5826 KHAN ET AL. J. VIROL.

proteins that bind SH3 domains, the consensus sequence Pxx-PLR was identified. The prolines in this motif make directcontacts with aromatic residues, and the arginine makes ionicor salt bridges with acidic residues within SH3 domains (2).The salient features of the SH3 ligand motif found in Nef areas follows: (i) this motif is present in all isolates of HIV-1,HIV-2, and SIV; (ii) the PxVPLR sequence is conserved inHIV-1, HIV-2, and SIV (34, 46); and (iii) this motif exists inthe “minus” orientation as determined by the location of thecritical arginine residue on the C-terminal side of the prolines(25). The in vitro kinase analysis, presented in Fig. 1B and C,clearly demonstrated that the strictly conserved amino acidresidues V106, P107, L108, and R109 of SIVmac239 Nef werecritical for both association of Nef with NAK and Nef-medi-ated PAK activation. X-ray crystallographic studies of HIV-1Nef complexed with the mutant SH3 domain of Fyn revealedthat three amino acid residues in HIV-1 Nef, V74, P75, and R77
(which correspond to V106, P107, and R109, respectively, of SIVNef), make key contacts with conserved residues within theSH3 domain (15, 22).
The specificity and affinity of binding of the SH3 domain ofHck to HIV-1 Nef depend on a tertiary interaction between astructure in Hck known as the RT loop, and a hydrophobicregion in Nef that is relatively distant from the PxxP motif,involving the F90 residue of HIV-1 Nef (21, 22). The RT loopis poorly conserved among different SH3 domains and there-fore is likely to be involved in imparting specificity in otherSH3-domain/SH3-ligand interactions as well. F90 is strictly
conserved in Nef of HIV-1, HIV-2, and SIV (34, 46). Accord-ingly, we constructed and tested a viral clone with a mutationin the corresponding residue, F122, in SIV Nef. This mutantwas defective both in association of Nef with NAK (Fig. 1B)and in activation of PAK by Nef (Fig. 1C). Thus, the require-ment for F122 for these in vitro SIV Nef functions supports arole for this amino acid residue in binding specificity as well.
Taken together, our findings, which demonstrate the impor-tance of the residues V106, P107, L108, R109, and F122 of SIVNef in the in vitro kinase assays, indicated that the SH3 liganddomain of Nef was similar to the SH3 ligand domains in cel-lular proteins involved in cell activation. Both prolines in theSIV Nef PxxP motif were not necessary, since the Nef proteinswith the sequences A104KVP107 and P104RVA107 interact withNAK and activate PAK (Fig. 1B and 4C). Interestingly, arecent structural study of Src showed that an internal SH3ligand domain in this cellular tyrosine kinase contains a singleproline (52). A speculation is that the SH3 ligand domain ofNef may mediate the interaction with a unique member of thePAK family that contains an SH3 domain or, alternatively, itcould function through an adapter molecule possessing an SH3domain. Interestingly, the adapter molecule Nck, which con-tains three SH3 domains and one SH2 domain, has been shownto associate with PAK (13, 26). Further investigations are re-quired to determine whether Nef interacts with NAK via Nckor via some other adapter molecule containing an SH3 do-main. Additionally, a combination of structural and functionalstudies are needed to determine whether the NAK-negative
FIG. 4. Phenotypic reversions of the SIVmac239-P104A/P107A mutant in macaques. In vitro kinase assays were performed on anti-Nef immunoprecipitates fromuninfected CEMx174 cells and from CEMx174 cells infected with prototype virus and viruses recovered from several macaques infected with the mutant virus.(A) Control kinase assays were done on anti-SIV Nef immunoprecipitates from extracts of uninfected CEMx174 cells and on extracts of CEMx174 cells chronicallyinfected with SIVmac239nef1 or SIVmac239-P104A/P107A. In vitro kinase assay results are shown for anti-Nef immunoprecipitates from extracts of CEMx174 cellsacutely infected with virus isolated from Mmu 25905 at weeks 20 (20wk) and 30, with virus from Mmu 27659 at weeks 8 and 24, and with virus from Mmu 27626 atweek 24 postinoculation. (B) In vitro kinase assay performed on virus isolated from Mmu 27626 and Mmu 27659 at 42 weeks (42wk) postinoculation, analyzed in thesame manner as in panel A with controls: in vitro kinase analysis of anti-SIV Nef immunoprecipitates from extracts of uninfected CEMx174 cells and CEMx174 cellschronically infected with SIVmac239nef1. (C) Association of Nef with NAK in the mutant virus, SIVmac239-K105R/P107A; the mutation in nef in this virus wasconstructed on the basis of the second-site revertant sequence in virus isolated from Mmu 27659 (Table 2). Results of in vitro kinase analysis of anti-SIV Nefimmunoprecipitates from uninfected CEMx174 cells and from CEMx174 cells chronically infected with SIVmac239nef1 and SIVmac239-K105R/P107A are shown. Atotal of 107 cells were analyzed per lane. The kinase assays were performed as described in Materials and Methods. Immunoprecipitates were electrophoresed on a12% polyacrylamide gel under denaturing conditions, and phosphorylation of proteins was visualized by autoradiography.
VOL. 72, 1998 PxxP DOMAIN OF SIV Nef AND SIMIAN AIDS 5827

phenotype of Nef mutants is due to the inability of mutantprotein (i) to bind NAK or (ii) to bind but not activate NAK.
Nef reversion and disease with the SIVmac239-P104A/P107Amutant. To test the importance of the SH3 ligand domain ofNef for the virus-host relationship, seven juvenile rhesus ma-caques were inoculated with the nef mutant virus SIVmac239-P104A/P107A and tested for virological and clinical parametersas well as for changes (i.e., reversions) in the nef mutations.Five of seven macaques showed reversion in the PxxP motifand restoration of the NAK-positive phenotype (summarizedin Table 2); this pattern of reversion supports the importanceof the SH3 ligand domain in Nef for SIV infection in vivo. Twomacaques, Mmu 25905 and Mmu 27659, developed hemato-logical abnormalities during the course of infection and pro-gressed to fatal SAIDS (Table 1). All nef clones from Mmu25905 contained A104KVP107 (Table 2), which is encoded by agenotype that is positive in the NAK assay (Fig. 4A). In Mmu27659, the pattern of reversion demonstrated greater complex-ity during the course of infection. At 12 weeks, Nef from thisanimal contained the sequence P104R105V106A107 (Table 2);the second-site mutation, converting K105 to R105 and accom-panied by reversion of A104 to P104, partially restored the Nef-NAK interaction (Fig. 4A). Interestingly, the viral load in Mmu27659, which had shown a decrease after the acute phase ofinfection, began to increase at 12 weeks postinoculation (Fig.2A). At subsequent time points, nef clones from this animalexhibited the prototype pattern P104KVP107 (Table 2), whichfully restored Nef-NAK interaction (Fig. 4C). Importantly, theproportion of clones with the prototype Nef sequence progres-sively increased from 10% at 20 weeks to 70% at 46 weeks.This genetic evidence indicates that not only was there a se-lection for Nef with full ability to interact with NAK but alsoboth prolines were important for disease progression in thisanimal.
The outcome of infection of Mmu 26785 with the SIVmac239-P104A/P107A mutant was strikingly different from that of thefive animals showing reversions in nef. Mmu 26785 displayedvery high virus loads, no detectable anti-SIV antibodies, nodetectable reversions in Nef, and a rapid disease course (Table1). Other investigators have also described a pattern of rapiddisease progression with no detectable seroconversion in adultrhesus macaques infected with pathogenic strains of SIV (17,53). It is possible that either a host genetic factor (e.g., MHC-1genotype) or a cofactor (e.g., an unidentified infectious agent)produced a situation in Mmu 26785 in which the host immuneresponse to SIV was compromised and a high virus load andrapid disease progression ensued. Unlike all the other ma-caques in our study, eosinophil levels in this animal were ele-vated at the time of virus inoculation and became very highduring the course of infection (see Results); eosinophilia hasbeen associated with parasite infections, allergy, and increasedmorbidity in HIV-1-infected individuals (7). Thus, it is possiblethat Mmu 26785 harbored an undetected parasite cofactorwhich could elicit disease by a virus with a mutation in nef. In-terestingly, in another study, two of two adult macaques infect-ed with the SIVmac239-P104A/P107A mutant clone also exhib-ited a pattern of rapid disease progression; these animals died9 and 18 weeks after infection and showed weak antibody re-sponses (20). It is difficult to draw reliable conclusions aboutthe in vivo importance of viral gene functions based on studieswith very few animals (i.e., only Mmu 26785 in our study andonly two animals in the previous study [20]). Additionally,rapid disease progression, without seroconversion and withouta chronic phase, in infected macaques is not a model for AIDScaused by HIV infection in humans (3, 40).
Another outcome of infection with the SIVmac239-P104A/P107A mutant was exemplified by Mmu 27626. This animal wassacrificed at 93 weeks because of severe hepatitis. Necropsyalso revealed evidence of lymphoid hyperplasia; however, nolymphoid-cell depletion was noted in peripheral, inguinal, andmesenteric lymph nodes or in splenic or gut-associated lym-phoid tissue (Table 1). Additionally, this animal did not showsevere depletion of CD4 T cells in peripheral blood (Fig. 3A),did not exhibit weight loss, and did not present evidence of anopportunistic infection. The virus load in Mmu 27626 wasrelatively low throughout the course of infection. The virusrecovered at necropsy was negative in the NAK assay (data notshown); the biological significance of the A104KVT107 pattern,in several nef clones obtained at necropsy, remains to be de-termined. Taken together, clinical and virological findings inMmu 27626 do not support a diagnosis of typical SAIDS.
Level of revertant nef1 virus required to cause disease.Lang et al. showed nef gene revertants in two adult rhesusmacaques infected with the SIVmac239-P104A/P107A mutant(20). The total virus load in both animals was very high, andthe level of genotypic revertants, restoring P104 and P107, was5 to 10% of the level of total virus in each animal. Further-more, this previous study measured reversion only at P104 andP107. Our study demonstrated that phenotypic reversion canalso occur when a second-site suppressor mutation (under-lined) is generated, i.e., P104R105VA107 (Fig. 1B and 4C). Im-portantly, the ability of Nef to interact with NAK was notanalyzed in virus recovered from the mutant-infected animalsin the study by Lang et al. (20).
To test the ability of the NAK assay to detect a small pro-portion of NAK-positive virus in a mixture containing mutant(i.e., NAK-negative) virus, we performed a reconstructionexperiment. Kinase assays were performed on extracts frommixtures of cells containing increasing proportions of theprototype SIVmac239nef1 cell line relative to the mutantSIVmac239-P104A/P107A cell line. In this experiment, Nef-NAK association was detected down to the 2% level of proto-type to mutant virus-infected cell mixture (data not shown).Because the virus loads in the study by Lang et al. were veryhigh, it is possible that the small proportion of revertants intheir animals was sufficient to cause disease (20). Thus, a re-interpretation of the data in the previous paper is that the PxxPmotif in Nef is important for SIV pathogenesis.
The dose of the virus used to establish infection can beanother important variable for understanding the virus-hostrelationship and disease progression (39, 51). Our study used aninoculum of 1,000 TCID50 of SIVmac239-P104A/P107A, where-as the study by Lang et al. used 10,000 TCID50 of this mutantvirus (39, 51). It is possible that the rapid disease course wasinfluenced by the infecting dose of mutant virus as well as byhost factors. Although the dose range required for transmis-sion under natural conditions is not known for primate lenti-viruses, the lower virus inoculum in the macaque model in ourstudy is probably more physiologically relevant for elucidatingthe importance of viral gene functions in vivo. Nonetheless, therelationship of viral dose to outcome of infection (i.e., progres-sion to SAIDS) remains to be determined for juvenile andadult rhesus macaques infected with either prototype or mu-tant SIV clones.
Reversion frequency and implications for Nef structure andfunction. The time course of the appearance of viruses withreversions in the nef gene appears to be different in this studyof the SIVmac239-P104A/P107A mutant and our previous study,which analyzed a viral clone (SIVmac239-RR/LL) with pointmutations in two arginine residues (R137R138) in the highly
5828 KHAN ET AL. J. VIROL.

conserved central domain of Nef (45). In two animals infectedwith SIVmac239RR/LL, the majority of nef clones had re-verted to produce a functional Nef at 4 weeks after infection.In the present study, we detected revertant nef clones at 8weeks in one animal (Mmu 26902) and at 12 weeks in threeothers (Mmu 27659, Mmu 27879, and Mmu 26902). It is pos-sible that the nef mutations in the diarginine motif R137R138
cause a more pronounced effect on Nef structure and func-tion than do the mutations in the PxxP motif; thus, selectivepressures for restoring Nef function might be greater forSIVmac239-RR/LL. A clearer understanding of the roles ofthe domains of Nef in various functions ascribed to this viralprotein (i.e., downregulation of CD4 and MHC class I anti-gens, augmentation of virion infectivity and viral replication,association with cellular kinases) is required to determinewhether there is a hierarchy of Nef functions and whether eachfunction is subject to different selection pressures.
Conclusions. The analysis of SIV Nef mutants in the in vitrokinase assays, measuring Nef-NAK association and PAK acti-vation, demonstrated the importance of V106P107L108R109 andF122 in the PxxP region of Nef; this sequence conforms to theconsensus sequence requirements of SH3 ligand domains with“minus” orientation (2, 11). Additionally, our in vivo studyhighlighted the importance of the SH3 ligand domain in Nef bydemonstrating reversion of the PxxP Nef mutant virus (i.e.,SIVmac239-P104A/P107A) and disease progression in juvenilerhesus macaques. The animals in our study exhibited diverseoutcomes with respect to pathogenesis and reversion of themutations in the Nef SH3 ligand domain. Such different pat-terns in the virus-host relationship could occur because rhesusmacaques are outbred animals and therefore differ in genescontrolling antiviral immune responses (e.g., MHC class Igenes) (18, 54). It is also possible that some captive macaquesin different primate facilities contain an unidentified infectiousagent which serves as a cofactor for pathogenesis in animalsinfected with either prototype SIV or certain SIV mutants.Accordingly, for pathogenesis studies to examine viral genefunctions in outbred animals, it is essential to use a largenumber of animals to obtain an accurate assessment of viralgene function in vivo.
Sequence homology information also strongly supports theidea that the SH3 ligand domain of Nef plays a significant rolein the virus-host relationship. HIV-1, HIV-2, and SIV Nefcontain the SH3 ligand domain with several strictly conservedresidues in addition to the two prolines; these residues areP104xV106P107L108R109 and F122 in SIV Nef (34, 46). Such ahigh level of conservation clearly indicates an important func-tional role in vivo for this highly conserved feature of Nefproteins. Also, the structural and biochemical studies, whichdemonstrated the interaction of the SH3 ligand domain of Nefwith the SH3 domain of certain tyrosine kinases, strongly sup-port a role for the PxxP motif in Nef for cell activation (15, 22,33, 42); several lines of evidence show that Nef influences oneor more components of cell signaling (reviewed in reference41). In vitro studies support a model for Nef in which this viralprotein is multifunctional; Nef modulates cell activation, en-hances virion infectivity and viral replication, and downregu-lates CD4 cell surface antigen and MHC class I antigens (8, 41,48). An effect of Nef on a cell signaling molecule(s), via aninteraction through the SH3 ligand domain, could influenceone or more of these functions attributed to Nef. Although ourdata show a linkage between Nef-NAK interaction and SAIDS,these findings do not exclude the possibility that Nef interac-tion with other cellular proteins also contributes to diseaseprogression. Further analyses are required to fully define cel-lular components interacting with Nef and to assess the rela-
tive importance of each of these functions for viral pathogen-esis.
ACKNOWLEDGMENTS
We are grateful for the expert technical assistance of Karen Shawand Kim Schmidt. Murray Gardner, Ross Tarara, Chris Miller, andDon Canfield provided expertise in performing necropsies and his-topathologic analysis. The following individuals are acknowledged forhelpful discussions and critical comments on the manuscript: CeciliaCheng-Mayer (Aaron Diamond AIDS Research Center, New York,N.Y.) and K. Saksela (University of Tampere).
The research reported in this paper was supported by NIH grants(R01-AI38532 to P.A.L., R29-AI38718 to E.T.S., and RR00169 basegrant to the California Regional Primate Research Center) and apostdoctoral fellowship grant from the California UniversitywideAIDS Research Program (to I.H.K.).
ADDENDUM IN PROOF
Two juvenile rhesus macaques (Mmu 28870 and Mmu28790) were inoculated intravenously with 1,000 TCID50 ofvirus recovered from Mmu 27626 at necropsy at 93 weekspostinoculation. After 8 weeks of infeciton, Mmu 28870 exhib-ited a high virus load that was comparable to that of prototypevirus SIVmac239nef1. Analysis for NAK revealed that Nefreverted to a kinase-positive phenotype in the virus from Mmu28870. In the other macaque infected with necropsy virus fromMmu 27626, virus load has remained low and virus from thisanimal has not reverted to a kinase-positive phenotype. Bothanimals are being monitored for clinical signs.
REFERENCES
1. Alexander, L., Z. Du, M. Rosenzweig, J. U. Jung, and R. C. Desrosiers. 1997.A role for natural simian immunodeficiency virus and human immunodefi-ciency virus type 1 Nef alleles in lymphocyte activation. J. Virol. 71:6094–6099.
2. Alexandropoulos, K., G. Cheng, and D. Baltimore. 1995. Proline-rich se-quences that bind to Src homology 3 domains with individual specificities.Proc. Natl. Acad. Sci. USA 92:3110–3114.
3. Allan, J. S. 1997. Human immunodeficiency virus-related infections in ani-mal models, p. 15–27. In V. T. Devita, S. Hellman, and S. A. Rosenberg(ed.), AIDS—etiology, diagnosis, treatment and prevention. Lippincott-Raven, Philadelphia, Pa.
4. Baba, T. W., Y. S. Jeong, D. Pennick, R. Bronson, M. F. Greene, and R. M.Ruprecht. 1995. Pathogenicity of live, attenuated SIV after mucosal infectionof neonatal macaques. Science 267:1820–1825.
5. Banapour, B., M. L. Marthas, R. J. Munn, and P. A. Luciw. 1991. In vitromacrophage tropism of pathogenic and nonpathogenic molecular clones ofsimian immunodeficiency virus (SIVmac). Virology 183:12–19.
6. Baur, A. S., E. T. Sawai, P. Dazin, W. J. Fantl, C. Cheng-Mayer, and B. M.Peterlin. 1994. HIV-1 Nef leads to inhibition or activation of T cells depend-ing on its intracellular localization. Immunity 1:373–384.
7. Bentwich, Z., Z. Weisman, C. Moroz, S. Bar-Yehuda, and A. Kalinkovich.1996. Immune dysregulation in Ethiopian immigrants in Israel: relevance tohelminth infections? Clin. Exp. Immunol. 103:239–243.
8. Cullen, B. R. 1994. The role of Nef in the replication cycle of the human andsimian immunodeficiency viruses. Virology 205:1–6.
9. Deacon, N. J., A. Tsykin, A. Solomon, K. Smith, M. Ludford-Menting, D. J.Hooker, D. A. McPhee, A. L. Greenway, A. Ellett, C. Chatfield, et al. 1995.Genomic structure of an attenuated quasi species of HIV-1 from a bloodtransfusion donor and recipients. Science 270:988–991.
10. Du, Z., S. M. Lang, V. G. Sasseville, A. A. Lackner, P. O. Ilynskii, M. D.Daniel, J. U. Jung, and R. C. Desrosiers. 1995. Identification of a nef allelethat causes lymphocyte activation and acute disease in macaque monkeys.Cell 82:665–674.
11. Feng, S., J. K. Chen, H. Yu, J. A. Simon, and S. L. Schreiber. 1994. Twobinding orientations for peptides to the Src SH3 domain: development of ageneral model for SH3-ligand interactions. Science 266:1241–1247.
12. Fultz, P. N., H. M. McClure, D. C. Anderson, and W. M. Switzer. 1989.Identification and biologic characterization of an acutely lethal variant ofsimian immunodeficiency virus from sooty mangabeys (SIV/SMM). AIDSRes. Hum. Retroviruses 5:397–409.
13. Galisteo, M. L., J. Chernoff, Y.-C. Su, E. Y. Skolnick, and J. Schlessinger.1996. The acaptor protein Nck links receptor tyrosine kinases with theserine-threonine kinase Pak1. J. Biol. Chem. 271:20997–21000.
VOL. 72, 1998 PxxP DOMAIN OF SIV Nef AND SIMIAN AIDS 5829

14. Goldsmith, M. A., M. T. Warmerdam, R. E. Archison, M. D. Miller, andW. C. Greene. 1995. Dissociation of the CD4 downregulation and viralinfectivity enhancement functions of human immunodeficiency virus type 1Nef. J. Virol. 69:4112–4121.
15. Grzesiek, S., A. Bax, G. M. Clore, A. M. Gronenborn, S. J. Hu, J. Kaufman,I. Palmer, S. J. Stahl, and P. T. Wingfield. 1996. The solution structure ofHIV-1 Nef reveals an unexpected fold and permits delineation of the bindingsurface for the SH3 domain of Hck tyrosine protein kinase. Nat. Struct. Biol.3:340–345.
16. Iafrate, J. A., S. Bronson, and J. Skowronski. 1997. Separable functions ofNef disrupt two aspects of T cell receptor machinery: CD4 expression andCD3 signaling. EMBO J. 16:673–684.
17. Kannagi, M., M. Kiyotaki, R. C. Desrosiers, K. A. Reimann, N. W. King,L. M. Waldron, and N. L. Letvin. 1986. Humoral immune responses to T celltropic retrovirus simian T lymphotropic virus type II in monkeys with exper-imentally induced acquired immune deficiency-like syndrome. J. Clin. Invest.78:1229–1236.
18. Keet, I. P. M., M. R. Klein, J. J. Just, and R. A. Kaslow. 1996. The role ofhost genetics in the natural history of HIV-1 infection: the needles in thehaystack. AIDS 10:S59–S67.
19. Kestler, H. W., D. J. Ringler, K. Mori, D. L. Panicali, P. K. Sehgal, M. D.Daniel, and R. C. Desrosiers. 1991. Importance of the nef gene for mainte-nance of high virus loads and for the development of AIDS. Cell 65:651–662.
20. Lang, S. M., A. J. Iafrate, C. Stahl-Hennig, E. M. Kuhn, T. Nisslein, F.-J.Kaup, M. Maupt, G. Hunsmann, J. Skowronski, and F. Kirchoff. 1997.Association of simian immunodeficiency virus Nef with cellular serine/thre-onine kinases is dispensable for the development of AIDS in rhesus ma-caques. Nat. Med. 3:860–865.
21. Lee, C. H., B. Leung, M. A. Lemmon, J. Zheng, D. Cowburn, J. Kuriyan, andK. Saksela. 1995. A single amino acid in the SH3 domain of Hck determinesits high affinity and specificity in binding to HIV-1 Nef protein. EMBO J. 14:5006–5015.
22. Lee, C. H., K. Saksela, U. A. Mirza, B. T. Chait, and J. Kuriyan. 1996. Crystalstructure of the conserved core of HIV-1 Nef complexed with a src family ofSH3 domain. Cell 85:931–942.
23. Lerche, N., J. L. Yee, and M. B. Jennings. 1994. Establishing specific retro-virus-free breeding colonies of macaques: an approach to primary screeningand surveillance. Lab. Anim. Sci. 44:217–221.
24. Lerche, N. W., R. F. Cotterman, M. D. Dobson, J. L. Yee, A. N. Rosenthal,and W. M. Heneine. 1997. Screening for simian type-D retrovirus infection inmacaques, using nested polymerase chain reaction. Lab. Anim. Sci. 47:263–268.
25. Lim, W. A., F. M. Richards, and R. O. Fox. 1994. Structural determinants ofpeptide-binding orientation and of sequence specificity in SH3 domains.Nature 372:375–379.
26. Lu, W., S. Katz, R. Gupta, and B. J. Mayer. 1997. Activation of Pak bymembrane localization mediated by an SH3 domain from the adaptor pro-tein Nck. Curr. Biol. 7:85–94.
27. Lu, X., X. Wu, A. Plemenitas, H. Yu, E. T. Sawai, A. Abo, and B. M. Peterlin.1996. CDC42 and Rac1 are implicated in the activation of the Nef-associatedkinase and replication of HIV-1. Curr. Biol. 6:1677–1684.
28. Luo, W., and B. M. Peterlin. 1997. Activation of the T-cell receptor signalingpathway by Nef from an aggressive strain of simian immunodeficiency virus.J. Virol. 71:9531–9537.
29. Mariani, R., F. Kirchhoff, T. C. Greenough, J. L. Sullivan, R. C. Desrosiers,and J. Skowronski. 1996. High frequency of defective nef alleles in a long-term survivor with nonprogressive human immunodeficiency virus type 1infection. J. Virol. 70:7752–7764.
29a.Marthas, M. Personal communication.30. Marthas, M. L., R. A. Ramos, B. L. Lohman, K. Van Rompay, R. E. Unger,
C. J. Miller, B. Banapour, N. C. Pedersen, and P. A. Luciw. 1993. Viraldeterminants of simian immunodeficiency virus (SIV) virulence in rhesusmacaques assessed by using attenuated and pathogenic molecular clones ofSIVmac. J. Virol. 67:6047–6055.
31. Mayer, B. J., and R. Gupta. 1998. Functions of SH2 and SH3 domains. Curr.Top. Microbiol. Immunol. 228:1–22.
32. Miller, M. D., M. T. Warmerdam, I. Gaston, W. C. Greene, and M. B.Feinberg. 1994. The human immunodeficiency virus-1 nef gene product: a
positive factor for viral infection and replication in primary lymphocytes andmacrophages. J. Exp. Med. 179:101–113.
33. Moarefi, I., B. M. LaFevre, F. Sicheri, M. Huse, C. H. Lee, J. Kuriyan, andW. T. Miller. 1997. Activation of the Src-family tyrosine kinase Hck by SH3domain displacement. Nature 385:650–653.
34. Myers, G., B. Foley, J. W. Mellors, B. Korber, K. T. Jeang, and S. Wain-Hobson. 1996. Human retroviruses and AIDS 1996. Los Alamos NationalLaboratory, Los Alamos, N.M.
35. Nunn, M. F., and J. W. Marsh. 1996. Human immunodeficiency virus type 1Nef associates with a member of the p21-activated kinase family. J. Virol. 70:6157–6161.
36. Pawson, T., and J. Schlessinger. 1993. SH2 and SH3 domains. Curr. Biol. 3:434–442.
36a.Planelles, V., and P. A. Luciw. Unpublished data.37. Ratner, L., and T. M. J. Niederman. 1995. Nef. Curr. Top. Microbiol.
Immunol. 193:169–208.38. Reed, L. J., and H. Muench. 1938. A simple method of estimating fifty
percent endpoints. Am. J. Hyg. 27:493–497.39. Ruprecht, R. M., T. W. Baba, R. Rasmussen, Y. Hu, and P. L. Sharma. 1996.
Murine and simian retrovirus models: the threshold hypothesis. AIDS 10:S33–S40.
40. Saag, M. S. 1997. Clinical spectrum of human immunodeficiency virus dis-eases, p. 203–213. In V. T. Devita, S. Hellman, and S. A. Rosenberg (ed.),AIDS—etiology, diagnosis, treatment and prevention. Lippincott-Raven,Philadelphia, Pa.
41. Saksela, K. 1997. HIV-1 Nef and host cell protein kinases. Front. Biol. 2:606–618.
42. Saksela, K., G. Cheng, and D. Baltimore. 1995. Proline-rich (PxxP) motifs inHIV-1 Nef bind to SH3 domains of a subset of Src kinases and are requiredfor the enhanced growth of Nef1 viruses but not for down-regulation ofCD4. EMBO J. 14:484–491.
43. Sawai, E. T., A. Baur, H. Struble, B. M. Peterlin, J. A. Levy, and C. Cheng-Mayer. 1994. Human immunodeficiency virus type 1 Nef associates with acellular serine kinase in T lymphocytes. Proc. Natl. Acad. Sci. USA 91:1539–1543.
44. Sawai, E. T., A. S. Baur, B. M. Peterlin, J. A. Levy, and C. Cheng-Mayer.1995. A conserved domain and membrane targeting of Nef from HIV andSIV are required for association with a cellular serine kinase activity. J. Biol.Chem. 270:15307–15314.
45. Sawai, E. T., I. H. Khan, P. M. Montbriand, B. M. Peterlin, C. Cheng-Mayer,and P. A. Luciw. 1996. Activation of PAK by HIV and SIV Nef: importancefor AIDS in rhesus macaques. Curr. Biol. 6:1519–1527.
46. Shugars, D. C., M. S. Smith, D. G. Glueck, P. V. Nantermet, F. Seillier-Moiseiwitsch, and R. Swanstrom. 1993. Analysis of human immunodefi-ciency virus type 1 nef gene sequences present in vivo. J. Virol. 67:4639–4650.
47. Skowronski, J., D. Parks, and R. Mariani. 1993. Altered T cell activation anddevelopment in transgenic mice expressing the HIV-1 nef gene. EMBO J. 12:703–713.
48. Trono, D. 1995. HIV accessory proteins: leading roles for the supportingcast. Cell 82:189–192.
49. Unger, R. E., M. L. Marthas, E. Pratt-Lowe, P. A. Padrid, and P. A. Luciw.1992. The nef gene of simian immunodeficiency virus SIVmac1A11. J. Virol.66:5432–5442.
50. Wiskerchen, M., and C. Cheng-Mayer. 1997. HIV-1 Nef association withcellular serine kinase correlates with enhanced virion infectivity and effi-ciency of proviral DNA synthesis. Virology 224:292–301.
51. Wyand, M. S., K. H. Manson, A. A. Lackner, and R. C. Desrosiers. 1997.Resistance of neonatal monkeys to live attenuated vaccine strains of simianimmunodeficiency virus. Nat. Med. 3:32–36.
52. Xu, W., S. C. Harrison, and M. J. Eck. 1997. Three-dimensional structure ofthe tyrosine kinase c-Src. Nature 385:595–602.
53. Zhang, J., L. N. Martin, E. A. Watson, R. Montelaro, M. West, L. Epstein,and M. Murphey-Corb. 1988. Relationship of antibody responses and viralantigenemia to SIV/Delta-induce immunodeficiency disease in rhesus ma-caques. J. Infect. Dis. 158:1277–1286.
54. Zinkernagel, R. M. 1993. Immunity to viruses, p. 1211–1250. In W. E. Paul(ed.), Fundamental immunology. Raven Press, New York, N.Y.
5830 KHAN ET AL. J. VIROL.
Copyright © 2022 FDOKUMEN




















