
Revisiting protein aggregation as pathogenic in sporadic ...
10
MEDICAL HYPOTHESIS Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases Alberto J. Espay, MD, MSc, Joaquin A. Vizcarra, MD, Luca Marsili, MD, PhD, Anthony E. Lang, MD, FRCPC, David K. Simon, MD, PhD, Aristide Merola, MD, PhD, Keith A. Josephs, MD, MST, MSc, Alfonso Fasano, MD, PhD, Francesca Morgante, MD, PhD, Rodolfo Savica, MD, MSc, J. Timothy Greenamyre, MD, PhD, Franca Cambi, MD, PhD, Tritia R. Yamasaki, MD, PhD, Caroline M. Tanner, MD, PhD, Ziv Gan-Or, MD, PhD, Irene Litvan, MD, Ignacio F. Mata, PhD, Cyrus P. Zabetian, MD, MS, Patrik Brundin, MD, PhD, Hubert H. Fernandez, MD, David G. Standaert, MD, PhD, Marcelo A. Kauffman, MD, PhD, Michael A. Schwarzschild, MD, PhD, S. Pablo Sardi, PharmD, PhD, Todd Sherer, PhD, George Perry, PhD, and James B. Leverenz, MD Neurology ® 2019;92:329-337. doi:10.1212/WNL.0000000000006926 Correspondence Dr. Espay [email protected] Abstract The gold standard for a definitive diagnosis of Parkinson disease (PD) is the pathologic finding of aggregated α-synuclein into Lewy bodies and for Alzheimer disease (AD) aggregated amyloid into plaques and hyperphosphorylated tau into tangles. Implicit in this clinicopathologic-based nosology is the assumption that pathologic protein aggregation at autopsy reflects pathogenesis at disease onset. While these aggregates may in exceptional cases be on a causal pathway in humans (e.g., aggregated α-synuclein in SNCA gene multiplication or aggregated β-amyloid in APP mutations), their near universality at postmortem in sporadic PD and AD suggests they may alternatively represent common outcomes from upstream mechanisms or compensatory responses to cellular stress in order to delay cell death. These 3 conceptual frameworks of protein aggregation (pathogenic, epiphenomenon, protective) are difficult to resolve because of the inability to probe brain tissue in real time. Whereas animal models, in which neither PD nor AD occur in natural states, consistently support a pathogenic role of protein aggregation, indirect evidence from human studies does not. We hypothesize that (1) current biomarkers of protein aggregates may be relevant to common pathology but not to subgroup pathogenesis and (2) disease-modifying treatments targeting oligomers or fibrils might be futile or delete- rious because these proteins are epiphenomena or protective in the human brain under mo- lecular stress. Future precision medicine efforts for molecular targeting of neurodegenerative diseases may require analyses not anchored on current clinicopathologic criteria but instead on biological signals generated from large deeply phenotyped aging populations or from smaller but well-defined genetic–molecular cohorts. From the UC Gardner Neuroscience Institute and Gardner Family Center for Parkinson’s Disease and Movement Disorders (A.J.E., J.A.V., L.M., A.M.), Department of Neurology, University of Cincinnati, OH; Edmond J. Safra Program in Parkinson’s Disease and the Morton and Gloria Shulman Movement Disorders Clinic (A.E.L., A.F.), Toronto Western Hospital, University of Toronto; Krembil Research Institute (A.E.L., A.F.), Toronto, Canada; Parkinson’s Disease and Movement Disorders Center (D.K.S.), Department of Neurology, Beth Israel Deaconess Medical Center and Harvard Medical School, Boston, MA; College of Medicine (K.A.J.), Mayo Clinic, Rochester, MN; Institute of Molecular and Clinical Sciences (F.M.), St George’s University of London, UK; Division of Movement Disorders (R.S.), Department of Neurology and Department of Health Science Research, Mayo Clinic College of Medicine, Rochester, MN; Department of Neurology and the Pittsburgh Institute for Neurodegenerative Diseases (J.T.G., F.C.), University of Pittsburgh, PA; Department of Neurology (T.R.Y.), University of Kentucky, Lexington; Parkinson’s Disease Research, Education and Clinical Center (C.M.T.), Neurology, San Francisco Veterans Affairs Medical Center; Department of Neurology (C.M.T.), University of California–San Francisco; Department of Neurology & Neurosurgery, Montreal Neurological Institute, and Department of Human Genetics (Z.G.-O.), McGill University, Canada; Parkinson & Other Movement Disorders Center UC San Diego (I.L.), Department of Neurosciences, Altman Clinical Translational Research Institute, La Jolla, CA; VA Puget Sound Health Care System and Department of Neurology (I.F.M., CP.Z.), University of Washington, Seattle; Department of Neurology (I.F.M.), University of Washington School of Medicine, Seattle; Center for Neurodegenerative Science (P.B.), Van Andel Research Institute, Grand Rapids, MI; Center for Neurological Restoration (H.H.F.) and Lou Ruvo Center for Brain Health, Neurological Institute (J.B.L.), Cleveland Clinic, OH; Department of Neurology (D.G.S.), University of Alabama at Birmingham; Consultorio y Laboratorio de Neurogen´ etica (M.A.K.), Centro Universitario de Neurolog´ ıa “Jos´ e Mar´ ıa Ramos Mej´ ıa” y Divisi´ on Neurolog´ ıa, Hospital JM Ramos Mej´ ıa, Facultad de Medicina, UBA; Programa de Medicina de Precision y Genomica Clinica (M.A.K.), Instituto de Investigaciones en Medicina Traslacional, Facultad de Ciencias Biom´ edicas, Universidad Austral-CONICET, Buenos Aires, Argentina; Department of Neurology (M.A.S.), Massachusetts General Hospital, Boston; Division of Neuroscience (S.P.S.), Sanofi-Genzyme, Framingham, MA; Michael J. Fox Foundation for Parkinson’s Research (T.S.), New York, NY; and College of Sciences (G.P.), University of Texas at San Antonio. Go to Neurology.org/N for full disclosures. Funding information and disclosures deemed relevant by the authors, if any, are provided at the end of the article. Copyright © 2019 American Academy of Neurology 329 Copyright ª 2019 American Academy of Neurology. Unauthorized reproduction of this article is prohibited.
-
Upload
khangminh22 -
Category
Documents
-
view
1 -
download
0
Transcript of Revisiting protein aggregation as pathogenic in sporadic ...

MEDICAL HYPOTHESIS
Revisiting protein aggregation as pathogenic insporadic Parkinson and Alzheimer diseasesAlberto J Espay MD MSc Joaquin A Vizcarra MD Luca Marsili MD PhD Anthony E Lang MD FRCPC
David K SimonMD PhD AristideMerolaMD PhD Keith A JosephsMDMSTMSc Alfonso FasanoMD PhD
FrancescaMorganteMD PhD Rodolfo SavicaMDMSc J TimothyGreenamyreMD PhD Franca CambiMD
PhD Tritia R Yamasaki MD PhD Caroline M Tanner MD PhD Ziv Gan-Or MD PhD Irene Litvan MD
Ignacio F Mata PhD Cyrus P Zabetian MD MS Patrik Brundin MD PhD Hubert H Fernandez MD
David G Standaert MD PhD Marcelo A Kauffman MD PhD Michael A Schwarzschild MD PhD
S Pablo Sardi PharmD PhD Todd Sherer PhD George Perry PhD and James B Leverenz MD
Neurologyreg 201992329-337 doi101212WNL0000000000006926
Correspondence
Dr Espay
albertoespayucedu
AbstractThe gold standard for a definitive diagnosis of Parkinson disease (PD) is the pathologic findingof aggregated α-synuclein into Lewy bodies and for Alzheimer disease (AD) aggregated amyloidinto plaques and hyperphosphorylated tau into tangles Implicit in this clinicopathologic-basednosology is the assumption that pathologic protein aggregation at autopsy reflects pathogenesisat disease onset While these aggregates may in exceptional cases be on a causal pathway inhumans (eg aggregated α-synuclein in SNCA gene multiplication or aggregated β-amyloid inAPP mutations) their near universality at postmortem in sporadic PD and AD suggests theymay alternatively represent common outcomes from upstream mechanisms or compensatoryresponses to cellular stress in order to delay cell death These 3 conceptual frameworks ofprotein aggregation (pathogenic epiphenomenon protective) are difficult to resolve because ofthe inability to probe brain tissue in real time Whereas animal models in which neither PD norAD occur in natural states consistently support a pathogenic role of protein aggregationindirect evidence from human studies does not We hypothesize that (1) current biomarkers ofprotein aggregates may be relevant to common pathology but not to subgroup pathogenesisand (2) disease-modifying treatments targeting oligomers or fibrils might be futile or delete-rious because these proteins are epiphenomena or protective in the human brain under mo-lecular stress Future precision medicine efforts for molecular targeting of neurodegenerativediseases may require analyses not anchored on current clinicopathologic criteria but instead onbiological signals generated from large deeply phenotyped aging populations or from smallerbut well-defined geneticndashmolecular cohorts
From the UC Gardner Neuroscience Institute and Gardner Family Center for Parkinsonrsquos Disease and Movement Disorders (AJE JAV LM AM) Department of NeurologyUniversity of Cincinnati OH Edmond J Safra Program in Parkinsonrsquos Disease and the Morton and Gloria Shulman Movement Disorders Clinic (AEL AF) Toronto Western HospitalUniversity of Toronto Krembil Research Institute (AEL AF) Toronto Canada Parkinsonrsquos Disease and Movement Disorders Center (DKS) Department of Neurology Beth IsraelDeaconess Medical Center and Harvard Medical School Boston MA College of Medicine (KAJ) Mayo Clinic Rochester MN Institute of Molecular and Clinical Sciences (FM) StGeorgersquos University of London UK Division of Movement Disorders (RS) Department of Neurology and Department of Health Science Research Mayo Clinic College of MedicineRochester MN Department of Neurology and the Pittsburgh Institute for Neurodegenerative Diseases (JTG FC) University of Pittsburgh PA Department of Neurology (TRY)University of Kentucky Lexington Parkinsonrsquos Disease Research Education and Clinical Center (CMT) Neurology San Francisco Veterans Affairs Medical Center Department ofNeurology (CMT) University of CaliforniandashSan Francisco Department of Neurology amp Neurosurgery Montreal Neurological Institute and Department of Human Genetics (ZG-O)McGill University Canada Parkinson ampOther Movement Disorders Center UC San Diego (IL) Department of Neurosciences Altman Clinical Translational Research Institute La JollaCA VA Puget Sound Health Care System and Department of Neurology (IFM CPZ) University of Washington Seattle Department of Neurology (IFM) University of WashingtonSchool of Medicine Seattle Center for Neurodegenerative Science (PB) Van Andel Research Institute Grand Rapids MI Center for Neurological Restoration (HHF) and Lou RuvoCenter for Brain Health Neurological Institute (JBL) Cleveland Clinic OH Department of Neurology (DGS) University of Alabama at Birmingham Consultorio y Laboratorio deNeurogenetica (MAK) Centro Universitario de Neurologıa ldquoJose Marıa Ramos Mejıardquo y Division Neurologıa Hospital JM Ramos Mejıa Facultad de Medicina UBA Programa deMedicina de Precision yGenomica Clinica (MAK) Instituto de Investigaciones enMedicina Traslacional Facultad de Ciencias Biomedicas Universidad Austral-CONICET Buenos AiresArgentina Department of Neurology (MAS) Massachusetts General Hospital Boston Division of Neuroscience (SPS) Sanofi-Genzyme FraminghamMAMichael J Fox Foundationfor Parkinsonrsquos Research (TS) New York NY and College of Sciences (GP) University of Texas at San Antonio
Go to NeurologyorgN for full disclosures Funding information and disclosures deemed relevant by the authors if any are provided at the end of the article
Copyright copy 2019 American Academy of Neurology 329
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
A cognitive dissonance in research on biomarkers and disease-modifying treatments for Parkinson disease (PD) and Alz-heimer disease (AD) is the dual acceptance of 2 oppositetenets that their clinical heterogeneity reflects several diseasessubsumed within each and that we are on the verge of findingthe set of perfect biomarkers that will explain their collectiveprogression and response to therapy1 Recent review articleson biomarkers and precision medicine start with the standarddisclaimer that a major challenge is the existence of manydiseases included under PD and AD (eg ldquohelliptrying to makeone drug work for all PD patientshellip is wrong because (1) PDis not a single disease and (2) no 2 individuals have the samebiological makeuprdquo2) only to revert to traditional form byreviewing or proposing analyses of a large set of clinical andbiological data collected on cohorts of clinically diagnosedindividuals to overcome heterogeneity3 Enormous financialand logistical resources have been devoted to protein-basedbiomarkers and anti-β-amyloid (Aβ) treatments with littlereturn on investment Therefore it is imperative to review thedisease framework on which biomarker development and thedesign of disease-modifying therapies are anchored
Protein aggregation as causal ofa single disease BradfordHill assessmentMutations in and multiplications of α-synuclein- and Aβ-related genes cause certain forms of PD and AD in affectedfamilies with these genetic abnormalities45 Overexpressionof these proteins coupled to excessive aggregations has beenclearly shown to cause neuronal dysfunction and death innumerous models67
To examine the causality of α-synucleinAβtau aggregation inhuman sporadic PDAD (ie without the point mutations orgene multiplication in the families where protein aggregation isassumed to be directly causal) we utilized the Bradford Hillcriteria for causality assessment89 These are a set of 9 criteriadeveloped by Sir Austin BradfordHill to provide epidemiologicevidence of a causal relationship between an apparent causeand an observed effect We tested the current disease modelunder which α-synuclein and Aβtau aggregations are thoughtto be causal to PD and AD respectively by compiling allthe published evidence from studies on humans available andcategorizing it according to each of the criteria
Search strategy and selection criteriaWe conducted a search in MEDLINE and PubMed for articlespublished until June 6 2018 using the search terms ldquoproteinaggregationrdquo ldquoalpha synucleinrdquo ldquooligomersrdquo ldquofibrilsrdquo ldquoamyloidrdquo
ldquosenile plaquerdquo ldquophospho-taurdquo ldquoLewy bodyrdquo ldquoParkinson dis-easerdquo ldquoAlzheimer diseaserdquo ldquobiomarkerrdquo and ldquopathologyrdquoWealso searched references and ClinicalTrialsgov for relevantstudies No language restrictions were applied The final ref-erence list was generated on the basis of relevance to the topicscovered in this Hypothesis article
We conducted a systematic review in accordance with thePreferred Reporting Items for Systematic Reviews andMeta-analyses (PRISMA) guidelines10 Eligible epidemiologic mo-lecular (eg neuroinflammation mitochondrial dysfunctionoxidative stress ubiquitin-proteasome system dysfunction cal-cium signaling dysregulation autophagy dysfunction synapticdysfunction cholesterol metabolism alteration)1112 pathologicautopsy imaging and interventional studies on α-synuclein Aβand tau were included We excluded animal models and vascu-lar dementiaparkinsonism studies Electronic search of articlespublished up to January 2018was conducted using theCochraneCentral Register of Controlled Trials (CENTRAL) EMBASEPubMed and references from relevant articles Search strategyincluded free text and Medical Subject Headings (MeSH)terms (table e-1 doiorg105061dryadg1nq02r) No restric-tions were applied to sex language or sample size Titles andabstracts of all studies identified were screened for inclusionand exclusion criteria Full-text studies of eligible articleswere obtained to confirm eligibility and discard duplicates Adata collection form was used to extract variables of interestfor the selected studies Given the heterogeneity of studydesigns risk of bias of individual studies was appraised uti-lizing the National Heart Lung and Blood Institute tools13
following the Cochrane handbook recommendations14 Stud-ies with high risk of bias were excluded from further analysisRemaining studies underwent data extraction and BradfordHillcriteriandashbased classification These procedures were conductedindependently by 2 authors and disagreements were settled byconsensus among authors
Out of the 1194 records derived from the initial search strat-egy 54 studies15ndash24e1ndashe41 met full search criteria (figure 1)Three studiese42ndashe44 were classified as having high risk of biasand were excluded from further analysis Agreement was metbetween evaluators in all cases A total of 2 and 4 of the 9Bradford Hill criteria supported causality for PD and ADrespectively (tables 1 and e-2)89 Altogether human studiesof protein aggregation in AD and PD do not meet a mini-mum set of Bradford Hillrsquos criteria to support a causal roleAlthough the absence of support for causality in the litera-ture of protein aggregation in human studies does not es-tablish absence of causality25 these findings encourage theexamination of alternative pathogenic hypotheses in PDand AD
GlossaryAβ = β-amyloid AD = Alzheimer disease DLB = dementia with Lewy bodies PD = Parkinson disease
330 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
Current model of diseaseand challengesOngoing work on biomarker development and disease-modifying therapies is partly built on a model of proteinmisfolding and subsequent aggregation as an initial process ina cascade of subsequent serial events held as causal to a singledisease In PD it is proposed that α-synuclein monomersassemble into oligomers which in turn give rise to α-synucleinfibrils the earliest disease-causing abnormalities and the majorprotein contribution of Lewy bodies and Lewy neurites (figure 2upper panel)26 It is surmised that α-synuclein aggregationleads to neuronal dysfunction loss of connectivity and neu-ronal death In AD it is proposed that the amyloid precursorprotein cleavage product and hyperphosphorylated tau proteinaggregate into extracellular plaques and intracellular neurofi-brillary tangles respectively12 These plaques and tangles maythen elicit a variety of secondary metabolic and molecularchanges which in turn lead to cellular dysfunction that (1)perpetuates the formation of additional pathogenic proteinaggregates or (2) directly accelerates or magnifies the effecton cell death through these molecular changes (ldquokindlingrdquophenomenon)11 (figure 2 lower panel) Nevertheless 3 mainarguments challenge these models
First challenge Uncertain temporality andbiological gradientUnlike other fields in medicine brain tissue is not readilyaccessible for diagnostic or research purposes Instead re-search endeavors have relied on specimens obtained from
2 sources (1) extracranial sources (eg biopsies of skinsalivary gland gut samples of blood and CSF) collected duringlife and (2) the brain itself harvested postmortem many yearsafter events of interest have taken place Even pathology studiesthat include samples from patients with different disease se-verity or ldquostagesrdquo are inherently cross-sectional In an attemptto assess the temporality of pathologic events AD biomarker-to-autopsy validation studies have yielded extraordinary prog-ress with in vivo CSF sampling and PET imaging of Aβtaunow accepted as valid surrogates for protein brain deposition27
These advances have led to 2 observations in AD that Aβ andtau sequentially or concurrently may be placed at the start ofa chain of events before the onset of cognitive dysfunction atwhat we assume to be time zero (see ldquoTemporalityrdquo criteria intables 1 and e-2)15ndash17 and that the co-presence of Aβ and tauleads to greater cognitive decline than either alone (see ldquoBi-ological gradientrdquo criteria in tables 1 and e-2 [doiorg105061dryadg1nq02r])1516 There is no corresponding neuroimagingor CSF biomarker counterpart for idiopathic PD While this invivo visualization has been assumed to represent time zero it isalso plausible that disease pathogenesis begins before the ag-gregation of proteins becomes detectable rendering them asintermediate rather than initiating events in the pathway tocell death
Second challenge Incidental pathologycopathology and nonspecific pathologyWithout the ability to serially examine living brain tissue forpotentially dynamic components in cellular proteostatic mech-anisms diseases are classified as proteinopathies (eg synu-cleinopathy) based on protein aggregates as end products Forsynucleinopathies for instance this is the case in PD multiplesystem atrophy and dementia with Lewy bodies (DLB) aswell as nonparkinsonian (peripheral) synucleinopathies suchas pure autonomic failure and atypical synucleinopathies in-cluding selected neurodegenerations with brain iron accumu-lation such as mitochondrial membrane protein-associatedneurodegeneration28 the parkinsonian lysosomal disordersKufor-Rakeb syndrome Gaucher disease andChediak-Higashisyndrome and the nonparkinsonian Sanfilippo syndrome2930
The α-synuclein ubiquity permeates into AD Lewy body pa-thology accumulates in over 50 of autopsy-proven ADbrains31
and conversely 77 of autopsy-proven PD dementia or DLBcases exhibit ADpathology32 Separately the classic ADproteinsAβ and tau-positive neurofibrillary tangles have been re-spectively reported in 13 and 48 of sporadic Creutzfeldt-Jakob disease cases24 Given their widespread appearancein a variety of phenotypically diverse diseases pathologies ofα-synuclein andAβtaumay not be pathogenic (see ldquoSpecificityrdquocriteria in tables 1 and e-2 [doiorg105061dryadg1nq02r])
The term ldquoincidental pathologyrdquo has been applied to the findingof protein aggregation on postmortem studies of neurologicallynormal individuals In the framework of α-synuclein- and Aβtau-based proteins as AD and PD ldquopathologyrdquo it is often as-sumed that when found on autopsy these might representprodromal individuals on the road to clinical disease had they
Figure 1 Flow chart of study selection
CENTRAL = Central Register of Controlled Trials
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 331
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
lived long enough However supra-surviving individuals (theover-90 or oldest-old) exhibit high frequency of AD pathologywithout dementia (50)3334 and PD pathology withoutparkinsonism (25)35 Notably no cognitive tests amongthe oldest-old without dementia can predict AD neuropathol-ogy within 3 years before death36 In fact neuropathologystudies in the oldest-old have either shown no correlation be-tween premortem cognitive function and AD neuropathol-ogy37 or even a paradoxical association between normalcognition and increased AD neuropathology38 The 90+ Study(183 brain autopsies from individuals who died at or after theage of 90 years) yielded robust support for an inverse re-lationship between AD pathology and clinical dementia evenbeyond the simple observation that intermediate to high AD
pathology clusters similarly in people with (23) and withoutdementia (28)39 Further analysis of the data suggests a pro-tective effect of AD pathology Compared to non-AD pathol-ogy (microinfarcts and white matter disease dementia n = 19no dementia n = 6) AD pathology (dementia n = 23 nodementia n = 24) was associated with a significantly reducedrisk of dementia (odds ratio 03 95 confidence interval01ndash09 p = 003 analysis not in the original publication)39
Therefore it is plausible that these individuals may have livedwell beyond a normal lifespan without neurologic symptomsbecause of not despite AD pathology Under this frameworkAβ and tau aggregation may have served as a mechanism tocompensate for active disease-causing biological abnormalities(while minimizing the presumably toxic soluble fibrils required
Table 1 Bradford Hill criteria applied to human studies of α-synuclein (α-syn) and β-amyloid (Aβ)tau aggregation inParkinson disease (PD) and Alzheimer disease (AD)
Criteria definition PD AD
Strength α-SynAβtau aggregation significantlycorrelates with PDAD using appropriatemethodology and accounting for confoundingfactors
Supporting 3e1ndashe3 Supporting 10e4ndashe13
Opposing 0 Opposing 0
Conclusion Support Conclusion Support
Consistency(replication)
Multiple studies using several locationspopulations and methods show a consistentcorrelation between α-synAβtau aggregateswith PDAD
Supporting 3e1ndashe3 Supporting 10e4ndashe13
Opposing 0 Opposing 0
Conclusion Support Conclusion Support
Temporality Cause preceding the effect time intervalbetween α-synAβtau aggregation ascertainedin vivo before symptom development in PDAD
Supporting No studies Supporting 315ndash17
Opposing No studies Opposing 0
Conclusion No support Conclusion Support
Experiment Interventions targeted against α-synAβtauaggregates decrease PDAD incidence severityor rate of progression
Supporting 0 Supporting 0
Opposing 0 Opposing 7e1418ndash23
Conclusion No support Conclusion No support
Biologicalgradient
Dose-response curve between α-synAβtauaggregates concentration and PDAD incidenceseverity or rate of progression
Supporting 0 Supporting 5e15ndashe1817
Opposing 2e1e2 Opposing 0
Conclusion No support Conclusion Support
Plausibility α-SynAβtau aggregates affect molecularpathways associated with PDAD
Supporting 2e19e20 Supporting 5e22ndashe26
Opposing 1e21 Opposing 4e27ndashe30
Conclusion Uncertain Conclusion Equivocal
Analogy If α-synAβtau aggregates cause PDAD similarprotein aggregations could also cause PDAD
A pathologic definition of PD precludesthis analysis to avoid circular reasoning
A pathologic definition of AD precludesthis analysis to avoid circular reasoning
Specificity If α-synAβtau aggregates cause PDAD theseproteins should not be present in diseasesother than PDAD or healthy controls
Supporting 0 Supporting 0
Opposing 10e31e32e34ndashe41 Opposing 1024e33ndashe41
Conclusion No support Conclusion No support
Coherence Causendasheffect should be consistent across allavailable evidence without contradictions ordiscrepancies
Supporting 0 Supporting 0
Opposing 13e1e2e21e31e32e34ndashe41 Opposing 21e1418minus24e27ndashe30e33ndashe41
Conclusion No support Conclusion No support
Adapted from references 8 9 and 25 Individual characteristics of supporting and opposing studies are supplied in table e-2 (doiorg105061dryadg1nq02r)
332 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
to form them) with clinical disease appearing after thismechanism is overwhelmed (see ldquoSpecificityrdquo criteria in tables 1e-2 [doiorg105061dryadg1nq02r])
Third challenge Experimental failure in anti-amyloid therapiesAnti-amyloid therapies that target insoluble Aβ forms (egAN1792 gantenerumab bapineuzumab) soluble forms(eg solanezumab) or prevent its formation (eg sem-agacestat γ-secretase inhibitor) have failed to producecognitive benefits in AD18ndash23 Some agents have been as-sociated with adverse events (eg AN1792 aseptic me-ningoencephalitis18 gantenerumab and bapineuzumabAβ-related abnormalities1920 semagacestat skin cancer andinfections)21 possibly due to immunologic mechanisms (egAN1792 T-cell response)18 or impaired protein processing(eg semagacestat Notch protein)21 in selected cases Al-though unsuccessful or insufficient Aβ load reduction mayaccount for clinical futility an issue that may be definitivelyanswered after the ongoing phase III aducanumab trial resultsare reported (ClinicalTrialsgov Identifier NCT02477800)one can argue that protein aggregation is not the culprit for cell
death (see ldquoExperimentrdquo criteria in tables 1 and e-2 [doiorg105061dryadg1nq02r])
Alternative models of diseaseIf α-synuclein and Aβtau protein aggregation are not ina direct causal pathway in PD and AD they may still con-tribute to neurodegeneration or be an epiphenomenon orguard against toxic soluble forms of the proteins by seques-tering them as innocuous insoluble forms Each of these 3hypothetical models may be influenced by lifelong exposureto genetic and environmental factors that might contribute toincreased susceptibility exhaustion of adaptive responsesand ultimately cell dysfunction and death We use α-synu-clein to illustrate these alternative models (figure 3) but theyapply similarly to Aβ and tau protein aggregation
Alternative model 1 Protein aggregation asaccelerator of pathology of multiple diseasesAside from disorders where a genetic mutation causes initialaccumulation of α-synuclein (eg missense mutations andmultiplication of SNCA)4 or of Aβ (eg triplication of APP
Figure 2 Current model of protein aggregation in Parkinson disease (single disease model)
Abnormal soluble oligomers and fibrils of α-synuclein are directly pathogenic (upper panel) Alternatively or complementarily secondarymolecular changescreated after protein aggregation combine with oligomers and fibrils to hasten cell death (lower panel) T-0 = time zero α-syn = α-synuclein
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 333
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
in Down syndrome or amyloid precursor protein [APP] genemutations and duplication)5 in which these protein aggre-gates are truly pathogenic it is plausible that accumulationof α-synuclein or of Aβtau enhances other primary patho-genic molecular mechanisms of neurodegeneration in spo-radic forms of PD and AD (figure 3A) Putatively a prion-likepropagation of α-synuclein fibrils between adjacent cells29 fallsinto this category without requiring that oligomers and fibrilsact as the initial pathogenic event This model accepts het-erogeneous upstream pathogenic events but relies on solubleproteins (α-synuclein Aβ) misfolding and aggregating asa convergent driver of cellular death In this model misfoldedvariants of α-synuclein and Aβ are suitable for biomarkerdevelopment and therapeutic targeting
Alternative model 2 Protein aggregation asepiphenomena of multiple diseasesAccording to this model a range of molecular abnormalitieslead to cells being unable to adequately degrade misfoldedproteins which aggregate into α-synuclein fibrils in Lewybodies (figure 3B or from misfolded amyloid precursorprotein into misfolded oligomers then fibrils then plaques inAD) These protein aggregates become byproducts (commondenominators or epiphenomena) of several pathogenicpathways and themselves exert no pathogenic or protectiverole According to this model different underlying molecularabnormalities remain pathogenic of different molecular dis-eases their relationship to one another rests only in the typeof protein they cannot properly recycle
Alternative model 3 Protein aggregation asprotective mechanism of multiple diseasesIn this model molecular abnormalities affect the cellularmachinery that normally degrades proteins These misfoldedproteins are sequestered into Lewy bodies as a mechanism topromote maintenance of neuronal and synaptic function de-spite coexistent and actively disrupting molecular abnormal-ities (figure 3C) Each molecular abnormality is pathogenic toa specific disease which shares with others only the type ofprotein actively sequestered as a protective mechanism Suchprotective sequestration of proteins could allow neurons tofunction for decades before becoming overwhelmed delayingthe progression of neurologic disease In this model targetingsoluble oligomers or fibrils (eg using anti-aggregant thera-pies or possibly passive or active immunotherapy) mightpotentially be deleterious if the toxicity of any of the proteinspecies is outweighed by their protective role in the humanbrain under molecular stress
Implications for biomarkers andclinical trialsHypothesis 1 Biomarkers of proteinaggregates are relevant to common pathologybut not to subgroup pathogenesisBased on the challenges discussed above abnormal proteinaggregates (pathology) may represent downstream events
unhelpful to classify PD and AD with pathogenic relevanceThus far biomarker development efforts have been based ondisease models of clinico-pathologic convergence One suchconvergence biomarker is [123I]-FP-CIT SPECT imaging forstriatal dopamine deficiency It is positive in virtually everyonewith PD Because biomarker discovery has been based onclinically defined cohorts (eg tremor-dominant PD vs posturalinstability gait disorder type) significant results within andbetween cohorts have substantial overlap40
Hypothesis 2 Disease-modifying treatmentstargeting oligomers or fibrils might be futile ordeleterious because these proteins areepiphenomena or protective in the humanbrain under molecular stressTargeting these proteins across sporadic or otherwise non-genetic disease forms is unlikely to slow progression of thesedisorders nomatter how early the treatments are implemented(ie prodromal stage) unless protein aggregation acts as anaccelerator of pathology as suggested in alternative model 1The corollary is that applying molecular therapies aimed atachieving neuroprotection to clinically (pathologically) de-fined but not molecularly subtyped populations will remainunlikely to succeed The history of medicine has repeatedlyshown that single (untreatable) clinical diseases are oftenreplaced by several (treatable) molecular diseases As a conse-quence treatments designed to target specific mechanismswork best in certain disease subtypes but not in others
Relinquishing reductionism (the clinically heterogeneous butsingle-disease concepts for AD and PD) will require acceptingthat sophisticated data analyses of large cohorts will not becapable of identifying molecular disease subtypes as the anal-yses cannot overcome the fundamental flaw of existent datasetscohorts invariably assembled based on strict clinical criteria Bigdata may not easily be converted into good data (for the pur-poses of assisting precision medicine) if the data are based onbinary definitions of AD vs control and PD vs control Impor-tant insights may be derived on a smaller scale from studyingrare genetic forms of disease (eg PD-LRRK2 PD-GBA AD-PS1) for which targeted therapeutic efforts may be most likelyto succeedmdashin these subtypes On a wider scale molecularlytargetable neurodegenerative diseases may be best identifiedfrom population-based studies applying non-hypothesis-basedanalytic approaches to biospecimens from large cohorts of agingpeople carefully characterized across a spectrum of brainfunctions and deficits Thus aging cohorts ought to includeatypical clinical presentations and other ostensibly unrelatedneurologic disorders beyond the current criteria for PD andADThe analyses will need to be anchored on biological signals noton clinical phenotypes or clinical criteria and account for ge-netic and lifelong biochemical and environmental factors Mo-lecular subtypes of disease thus identified may or may not fallinto standard clinical classifications
The first proven neuroprotective therapy might only work infewer than 5 of everyone currently subsumed as ldquoADrdquo or
334 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
ldquoPDrdquo (eg GZSAR402671 in early-stage PD-GBA [Clin-icalTrialsgov Identifier NCT02906020]) or in specific at-risk populations (eg solanezumab or gantenerumab forhealthy individuals with an autosomal dominant AD-causingmutation [Dominantly Inherited Alzheimer Network TrialClinicalTrialsgov Identifier NCT01760005]) Such disease-modifying treatment in neurodegenerative disease will helpusher in the biomarker-driven disease subtyping strategy longadopted by oncology and other fields of medicine
AcknowledgmentThe authors thank the patients their parentscaregivers andhealth care professionals who participated in this studyincluding Dean Miller Ruth Wake Dionne Moat RobertMuni Lofra Teresinha Evangelista MD Nikoletta Nikolenko
MD Namita Goyal MD Sonia Nayyar MD CarolinePeyronnard MD Tim Lai MD Monica Lavian DO BrianMinton BS Patrick Tierney RPT Denise Davis RPT CathyChou RPT Nick Higgins and Ed Connor MD
Study fundingNo targeted funding reported
DisclosureThe authors report no disclosures relevant to the manuscriptGo to NeurologyorgN for full disclosures
Publication historyReceived by Neurology July 6 2018 Accepted in final form December14 2018
Figure 3 Alternative models of protein aggregation in Parkinson disease (multiple disease model)
Abnormal soluble oligomers and fibrils ofα-synuclein (α-syn) while not directly pathogenic act as accelerators of neurodegeneration (ldquofueling the firerdquo) due toearly pathogenic molecular abnormalities each representingmolecularly distinct diseases (A model 1) Alternatively abnormal soluble oligomers and fibrilsof α-synuclein aggregate into Lewy bodies as byproducts of earlier pathogenic molecular mechanisms without directly affecting the neurodegenerativeprocess brought on by eachmolecular disease (Bmodel 2) Finally α-synuclein aggregates into Lewy bodies as amechanism to protect the neuron from toxicprotein species or from the biological dysfunction that may have generated the formation of toxic species ldquocoolingrdquo progression of cell degeneration underbiological stress (C model 3) Note that each molecularly defined disease has a different time to death collectively time to neuronal death is longest indiseases corresponding to model C
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 335
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
References1 Gwinn K David KK Swanson-Fischer C et al Parkinsonrsquos disease biomarkers
perspective from the NINDS Parkinsonrsquos Disease Biomarkers Program BiomarkMed201711451ndash473
2 Mollenhauer B Caspell-Garcia CJ Coffey CS et al Longitudinal CSF biomarkers inpatients with early Parkinson disease and healthy controls Neurology 2017891959ndash1969
3 Payami H The emerging science of precision medicine and pharmacogenomics forParkinsonrsquos disease Mov Disord 2017321139ndash1146
4 Chartier-Harlin M-C Kachergus J Roumier C et al α-Synuclein locus duplication asa cause of familial Parkinsonrsquos disease Lancet 20043641167ndash1169
5 Sleegers K BrouwersNGijselinck I et al APPduplication is sufficient to cause early onsetAlzheimerrsquos dementia with cerebral amyloid angiopathy Brain 20061292977ndash2983
6 Bezard E Yue Z Kirik D Spillantini MG Animal models of Parkinsonrsquos disease limitsand relevance to neuroprotection studies Mov Disord 20132861ndash70
7 Bloom GS Amyloid-β and tau JAMA Neurol 2014715058 Bradford-Hill A The environment and disease association or causation Proc R Soc
Med 196558295ndash3009 Fedak KM Bernal A Capshaw ZA Gross S Applying the Bradford Hill criteria in the
21st century how data integration has changed causal inference in molecular epi-demiology Emerg Themes Epidemiol Biomed Cent 20151214
Appendix Authors
Name Location Role Contribution
Alberto JEspay MD MSc
University ofCincinnati OH
CorrespondingAuthor
Conception ofresearchprojectliteraturesearch writingof the firstmanuscriptdraft
Joaquin AVizcarra MD
University ofCincinnati OH
Author Literaturesearch writingof the firstmanuscriptdraft
Luca MarsiliMD PhD
University ofCincinnati
Author Literaturesearch reviewand critique ofthe manuscript
Anthony ELang MDFRCPC
University ofTorontoCanada
Author Review andcritique of themanuscript
David K SimonMD PhD
HarvardMedical SchoolBoston MA
Author Review andcritique of themanuscript
AristideMerola MDPhD
University ofCincinnati OH
Author Review andcritique of themanuscript
Keith AJosephs MDMST MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
AlfonsoFasano MDPhD
University ofTorontoCanada
Author Review andcritique of themanuscript
FrancescaMorgante MDPhD
University ofMessina Italyand St GeorgersquosUniversity ofLondon UK
Author Review andcritique of themanuscript
Rodolfo SavicaMD MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
J TimothyGreenamyreMD PhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
FrancescaCambi MDPhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
Tritia RYamasaki MDPhD
University ofKentuckyLexington
Author Review andcritique of themanuscript
Caroline MTanner MDPhD
University ofCaliforniandashSanFrancisco
Author Review andcritique of themanuscript
Ziv Gan-OrMD PhD
McGillUniversityMontrealCanada
Author Review andcritique of themanuscript
Irene LitvanMD
UC San DiegoCA
Author Review andcritique of themanuscript
Appendix (continued)
Name Location Role Contribution
Ignacio FMata PhD
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Cyrus PZabetian MDMS
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Patrik BrundinMD PhD
Van AndelResearchInstitute GrandRapids MI
Author Review andcritique of themanuscript
Hubert HFernandez MD
ClevelandClinic OH
Author Review andcritique of themanuscript
David GStandaert MDPhD
University ofAlabama atBirmingham
Author Review andcritique of themanuscript
Marcelo AKauffman MDPhD
UBA andUniversidadAustral-CONICETBuenos AiresArgentina
Author Review andcritique of themanuscript
Michael ASchwarzschildMD PhD
MassachusettsGeneralHospitalBoston
Author Review andcritique of themanuscript
S Pablo SardiPharmD PhD
Sanofi Author Review andcritique of themanuscript
Todd ShererPhD
Michael J FoxFoundation forParkinsonrsquosResearch NewYork NY
Author Review andcritique of themanuscript
George PerryPhD
University ofTexas at SanAntonio
Author Review andcritique of themanuscript
James BLeverenz MD
ClevelandClinic OH
Author Literaturesearch reviewand critique ofthe manuscript
336 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
10 Moher D Liberati A Tetzlaff J Altman DG PRISMA Group Preferred reportingitems for systematic reviews and meta-analyses the PRISMA statement Ann InternMed 2009151264ndash269 W64
11 PoeweW Seppi K Tanner CM et al Parkinson disease Nat Rev Dis Primers 2017317013
12 Querfurth HW LaFerla FM Alzheimerrsquos disease N Engl J Med 2010362329ndash34413 NHLBI Research Triangle Institute International National Heart Lung and Blood
Institute quality appraisal tools [online] Available at httpswwwnhlbinihgovhealth-topicsstudy-quality-assessment-tools Accessed December 15 2017
14 Higgins JPT Green S eds Cochrane Handbook for Systematic Reviews of Inter-ventions [online] 510 The Cochrane Collaboration 2011 Available at httpstrainingcochraneorghandbook Accessed December 16 2017
15 Vos SJ Xiong C Visser PJ et al Preclinical Alzheimerrsquos disease and its outcomea longitudinal cohort study Lancet Neurol 201312957ndash965
16 Van Harten AC Smits LL Teunissen CE et al Preclinical AD predicts decline inmemory and executive functions in subjective complaints Neurology 2013811409ndash1416
17 Burnham SC Bourgeat P Dore V et al Clinical and cognitive trajectories in cogni-tively healthy elderly individuals with suspected non-Alzheimerrsquos disease patho-physiology (SNAP) or Alzheimerrsquos disease pathology a longitudinal study LancetNeurol 2016151044ndash1053
18 Gilman S Koller M Black RS et al Clinical effects of A immunization (AN1792) inpatients with AD in an interrupted trial Neurology 2005641553ndash1562
19 Ostrowitzki S Lasser RA Dorflinger E et al A phase III randomized trial of gante-nerumab in prodromal Alzheimerrsquos disease Alzheimers Res Ther 2017995
20 Salloway S Sperling R Fox NC et al Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370322ndash333
21 Doody RS Raman R Farlow M et al A phase 3 trial of semagacestat for treatment ofAlzheimerrsquos disease N Engl J Med 2013369341ndash350
22 Doody RS Thomas RG Farlow M et al Phase 3 trials of solanezumab for mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370311ndash321
23 Honig LS Vellas B Woodward M et al Trial of solanezumab for mild dementia dueto Alzheimerrsquos disease N Engl J Med 2018378321ndash330
24 Cali I Cohen ML Haїk S et al Iatrogenic Creutzfeldt-Jakob disease with amyloid-βpathology an international study Acta Neuropathol Commun 201865
25 Howick J Glasziou P Aronson JK The evolution of evidence hierarchies what canBradford Hillrsquos ldquoguidelines for causationrdquo contribute J R SocMed 2009102186ndash194
26 Roberts RF Wade-Martins R Alegre-Abarrategui J Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinsonrsquos diseasebrain Brain 20151381642ndash1657
27 Blennow K Mattsson N Scholl M Hansson O Zetterberg H Amyloid biomarkers inAlzheimerrsquos disease Trends Pharmacol Sci 201536297ndash309
28 Hogarth P Gregory A Kruer MC et al New NBIA subtype genetic clinical path-ologic and radiographic features of MPAN Neurology 201380268ndash275
29 Olanow CW Brundin P Parkinsonrsquos disease and alpha synuclein is Parkinsonrsquosdisease a prion-like disorder Mov Disord 20132831ndash40
30 Winder-Rhodes SE Garcia-Reitbock P Ban M et al Genetic and pathological linksbetween Parkinsonrsquos disease and the lysosomal disorder Sanfilippo syndrome MovDisord 201227312ndash315
31 Brenowitz WD Keene CD Hawes SE et al Alzheimerrsquos disease neuropathologicchange Lewy body disease and vascular brain injury in clinic- and community-basedsamples Neurobiol Aging 20175383ndash92
32 Irwin DJ Grossman M Weintraub D et al Neuropathological and genetic correlatesof survival and dementia onset in synucleinopathies a retrospective analysis LancetNeurol 20171655ndash65
33 Crystal H Dickson D Fuld P et al Clinico-pathologic studies in dementia non-demented subjects with pathologically confirmed Alzheimerrsquos disease Neurology1988381682ndash1687
34 Polvikoski T Sulkava R Myllykangas L et al Prevalence of Alzheimerrsquos disease in veryelderly people a prospective neuropathological study Neurology 2001561690ndash1696
35 MarkesberyWR Jicha GA Liu H Schmitt FA Lewy body pathology in normal elderlysubjects J Neuropathol Exp Neurol 200968816ndash822
36 Balasubramanian AB Kawas CH Peltz CB Brookmeyer R Corrada MM Alzheimerdisease pathology and longitudinal cognitive performance in the oldest-old with nodementia Neurology 201279915ndash921
37 Silver MH Newell K Brady C Hedley-White ET Perls TT Distinguishing betweenneurodegenerative disease and disease-free aging correlating neuropsychologicalevaluations and neuropathological studies in centenarians Psychosom Med 200264493ndash501
38 Berlau DJ Corrada MM Head E Kawas CH APOE 2 is associated with intactcognition but increased Alzheimer pathology in the oldest old Neurology 200972829ndash834
39 Kawas CH Kim RC Sonnen JA Bullain SS Trieu T Corrada MM Multiple pa-thologies are common and related to dementia in the oldest-old Neurology 201585535ndash542
40 Espay AJ Schwarzschild MA Tanner CM et al Biomarker-driven phenotyping inParkinsonrsquos disease a translational missing link in disease-modifying clinical trialsMov Disord 201732319ndash324Data available from Dryad (Additional References References e1ndashe44)doiorg105061dryadg1nq02r
Did You Knowhellip
hellipyou can browse by subspecialty topics on Neurologyorg
Go to Neurologyorg and click on ldquoTopicsrdquo in the top navigation bar
Share Your Artistic Expressions in Neurology lsquoVisionsrsquo
AAN members are urged to submit medically or scientifically related artistic images such as photographs photomicrographsand paintings to the ldquoVisionsrdquo section of Neurologyreg These images are creative in nature rather than the medically instructiveimages published in the NeuroImages section The image or series of up to six images may be black and white or color andmust fit into one published journal page Accompanying description should be 100 words or less the title should be a maximumof 96 characters including spaces and punctuation
Please access the Author Center at NPuborgauthors for full submission information
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 337
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
DOI 101212WNL0000000000006926201992329-337 Neurology
Alberto J Espay Joaquin A Vizcarra Luca Marsili et al diseases
Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer
This information is current as of February 11 2019
ServicesUpdated Information amp
httpnneurologyorgcontent927329fullincluding high resolution figures can be found at
References httpnneurologyorgcontent927329fullref-list-1
This article cites 38 articles 10 of which you can access for free at
Citations httpnneurologyorgcontent927329fullotherarticles
This article has been cited by 3 HighWire-hosted articles
Permissions amp Licensing
httpwwwneurologyorgaboutabout_the_journalpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpnneurologyorgsubscribersadvertiseInformation about ordering reprints can be found online
rights reserved Print ISSN 0028-3878 Online ISSN 1526-632X1951 it is now a weekly with 48 issues per year Copyright copy 2019 American Academy of Neurology All
reg is the official journal of the American Academy of Neurology Published continuously sinceNeurology

A cognitive dissonance in research on biomarkers and disease-modifying treatments for Parkinson disease (PD) and Alz-heimer disease (AD) is the dual acceptance of 2 oppositetenets that their clinical heterogeneity reflects several diseasessubsumed within each and that we are on the verge of findingthe set of perfect biomarkers that will explain their collectiveprogression and response to therapy1 Recent review articleson biomarkers and precision medicine start with the standarddisclaimer that a major challenge is the existence of manydiseases included under PD and AD (eg ldquohelliptrying to makeone drug work for all PD patientshellip is wrong because (1) PDis not a single disease and (2) no 2 individuals have the samebiological makeuprdquo2) only to revert to traditional form byreviewing or proposing analyses of a large set of clinical andbiological data collected on cohorts of clinically diagnosedindividuals to overcome heterogeneity3 Enormous financialand logistical resources have been devoted to protein-basedbiomarkers and anti-β-amyloid (Aβ) treatments with littlereturn on investment Therefore it is imperative to review thedisease framework on which biomarker development and thedesign of disease-modifying therapies are anchored
Protein aggregation as causal ofa single disease BradfordHill assessmentMutations in and multiplications of α-synuclein- and Aβ-related genes cause certain forms of PD and AD in affectedfamilies with these genetic abnormalities45 Overexpressionof these proteins coupled to excessive aggregations has beenclearly shown to cause neuronal dysfunction and death innumerous models67
To examine the causality of α-synucleinAβtau aggregation inhuman sporadic PDAD (ie without the point mutations orgene multiplication in the families where protein aggregation isassumed to be directly causal) we utilized the Bradford Hillcriteria for causality assessment89 These are a set of 9 criteriadeveloped by Sir Austin BradfordHill to provide epidemiologicevidence of a causal relationship between an apparent causeand an observed effect We tested the current disease modelunder which α-synuclein and Aβtau aggregations are thoughtto be causal to PD and AD respectively by compiling allthe published evidence from studies on humans available andcategorizing it according to each of the criteria
Search strategy and selection criteriaWe conducted a search in MEDLINE and PubMed for articlespublished until June 6 2018 using the search terms ldquoproteinaggregationrdquo ldquoalpha synucleinrdquo ldquooligomersrdquo ldquofibrilsrdquo ldquoamyloidrdquo
ldquosenile plaquerdquo ldquophospho-taurdquo ldquoLewy bodyrdquo ldquoParkinson dis-easerdquo ldquoAlzheimer diseaserdquo ldquobiomarkerrdquo and ldquopathologyrdquoWealso searched references and ClinicalTrialsgov for relevantstudies No language restrictions were applied The final ref-erence list was generated on the basis of relevance to the topicscovered in this Hypothesis article
We conducted a systematic review in accordance with thePreferred Reporting Items for Systematic Reviews andMeta-analyses (PRISMA) guidelines10 Eligible epidemiologic mo-lecular (eg neuroinflammation mitochondrial dysfunctionoxidative stress ubiquitin-proteasome system dysfunction cal-cium signaling dysregulation autophagy dysfunction synapticdysfunction cholesterol metabolism alteration)1112 pathologicautopsy imaging and interventional studies on α-synuclein Aβand tau were included We excluded animal models and vascu-lar dementiaparkinsonism studies Electronic search of articlespublished up to January 2018was conducted using theCochraneCentral Register of Controlled Trials (CENTRAL) EMBASEPubMed and references from relevant articles Search strategyincluded free text and Medical Subject Headings (MeSH)terms (table e-1 doiorg105061dryadg1nq02r) No restric-tions were applied to sex language or sample size Titles andabstracts of all studies identified were screened for inclusionand exclusion criteria Full-text studies of eligible articleswere obtained to confirm eligibility and discard duplicates Adata collection form was used to extract variables of interestfor the selected studies Given the heterogeneity of studydesigns risk of bias of individual studies was appraised uti-lizing the National Heart Lung and Blood Institute tools13
following the Cochrane handbook recommendations14 Stud-ies with high risk of bias were excluded from further analysisRemaining studies underwent data extraction and BradfordHillcriteriandashbased classification These procedures were conductedindependently by 2 authors and disagreements were settled byconsensus among authors
Out of the 1194 records derived from the initial search strat-egy 54 studies15ndash24e1ndashe41 met full search criteria (figure 1)Three studiese42ndashe44 were classified as having high risk of biasand were excluded from further analysis Agreement was metbetween evaluators in all cases A total of 2 and 4 of the 9Bradford Hill criteria supported causality for PD and ADrespectively (tables 1 and e-2)89 Altogether human studiesof protein aggregation in AD and PD do not meet a mini-mum set of Bradford Hillrsquos criteria to support a causal roleAlthough the absence of support for causality in the litera-ture of protein aggregation in human studies does not es-tablish absence of causality25 these findings encourage theexamination of alternative pathogenic hypotheses in PDand AD
GlossaryAβ = β-amyloid AD = Alzheimer disease DLB = dementia with Lewy bodies PD = Parkinson disease
330 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
Current model of diseaseand challengesOngoing work on biomarker development and disease-modifying therapies is partly built on a model of proteinmisfolding and subsequent aggregation as an initial process ina cascade of subsequent serial events held as causal to a singledisease In PD it is proposed that α-synuclein monomersassemble into oligomers which in turn give rise to α-synucleinfibrils the earliest disease-causing abnormalities and the majorprotein contribution of Lewy bodies and Lewy neurites (figure 2upper panel)26 It is surmised that α-synuclein aggregationleads to neuronal dysfunction loss of connectivity and neu-ronal death In AD it is proposed that the amyloid precursorprotein cleavage product and hyperphosphorylated tau proteinaggregate into extracellular plaques and intracellular neurofi-brillary tangles respectively12 These plaques and tangles maythen elicit a variety of secondary metabolic and molecularchanges which in turn lead to cellular dysfunction that (1)perpetuates the formation of additional pathogenic proteinaggregates or (2) directly accelerates or magnifies the effecton cell death through these molecular changes (ldquokindlingrdquophenomenon)11 (figure 2 lower panel) Nevertheless 3 mainarguments challenge these models
First challenge Uncertain temporality andbiological gradientUnlike other fields in medicine brain tissue is not readilyaccessible for diagnostic or research purposes Instead re-search endeavors have relied on specimens obtained from
2 sources (1) extracranial sources (eg biopsies of skinsalivary gland gut samples of blood and CSF) collected duringlife and (2) the brain itself harvested postmortem many yearsafter events of interest have taken place Even pathology studiesthat include samples from patients with different disease se-verity or ldquostagesrdquo are inherently cross-sectional In an attemptto assess the temporality of pathologic events AD biomarker-to-autopsy validation studies have yielded extraordinary prog-ress with in vivo CSF sampling and PET imaging of Aβtaunow accepted as valid surrogates for protein brain deposition27
These advances have led to 2 observations in AD that Aβ andtau sequentially or concurrently may be placed at the start ofa chain of events before the onset of cognitive dysfunction atwhat we assume to be time zero (see ldquoTemporalityrdquo criteria intables 1 and e-2)15ndash17 and that the co-presence of Aβ and tauleads to greater cognitive decline than either alone (see ldquoBi-ological gradientrdquo criteria in tables 1 and e-2 [doiorg105061dryadg1nq02r])1516 There is no corresponding neuroimagingor CSF biomarker counterpart for idiopathic PD While this invivo visualization has been assumed to represent time zero it isalso plausible that disease pathogenesis begins before the ag-gregation of proteins becomes detectable rendering them asintermediate rather than initiating events in the pathway tocell death
Second challenge Incidental pathologycopathology and nonspecific pathologyWithout the ability to serially examine living brain tissue forpotentially dynamic components in cellular proteostatic mech-anisms diseases are classified as proteinopathies (eg synu-cleinopathy) based on protein aggregates as end products Forsynucleinopathies for instance this is the case in PD multiplesystem atrophy and dementia with Lewy bodies (DLB) aswell as nonparkinsonian (peripheral) synucleinopathies suchas pure autonomic failure and atypical synucleinopathies in-cluding selected neurodegenerations with brain iron accumu-lation such as mitochondrial membrane protein-associatedneurodegeneration28 the parkinsonian lysosomal disordersKufor-Rakeb syndrome Gaucher disease andChediak-Higashisyndrome and the nonparkinsonian Sanfilippo syndrome2930
The α-synuclein ubiquity permeates into AD Lewy body pa-thology accumulates in over 50 of autopsy-proven ADbrains31
and conversely 77 of autopsy-proven PD dementia or DLBcases exhibit ADpathology32 Separately the classic ADproteinsAβ and tau-positive neurofibrillary tangles have been re-spectively reported in 13 and 48 of sporadic Creutzfeldt-Jakob disease cases24 Given their widespread appearancein a variety of phenotypically diverse diseases pathologies ofα-synuclein andAβtaumay not be pathogenic (see ldquoSpecificityrdquocriteria in tables 1 and e-2 [doiorg105061dryadg1nq02r])
The term ldquoincidental pathologyrdquo has been applied to the findingof protein aggregation on postmortem studies of neurologicallynormal individuals In the framework of α-synuclein- and Aβtau-based proteins as AD and PD ldquopathologyrdquo it is often as-sumed that when found on autopsy these might representprodromal individuals on the road to clinical disease had they
Figure 1 Flow chart of study selection
CENTRAL = Central Register of Controlled Trials
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 331
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
lived long enough However supra-surviving individuals (theover-90 or oldest-old) exhibit high frequency of AD pathologywithout dementia (50)3334 and PD pathology withoutparkinsonism (25)35 Notably no cognitive tests amongthe oldest-old without dementia can predict AD neuropathol-ogy within 3 years before death36 In fact neuropathologystudies in the oldest-old have either shown no correlation be-tween premortem cognitive function and AD neuropathol-ogy37 or even a paradoxical association between normalcognition and increased AD neuropathology38 The 90+ Study(183 brain autopsies from individuals who died at or after theage of 90 years) yielded robust support for an inverse re-lationship between AD pathology and clinical dementia evenbeyond the simple observation that intermediate to high AD
pathology clusters similarly in people with (23) and withoutdementia (28)39 Further analysis of the data suggests a pro-tective effect of AD pathology Compared to non-AD pathol-ogy (microinfarcts and white matter disease dementia n = 19no dementia n = 6) AD pathology (dementia n = 23 nodementia n = 24) was associated with a significantly reducedrisk of dementia (odds ratio 03 95 confidence interval01ndash09 p = 003 analysis not in the original publication)39
Therefore it is plausible that these individuals may have livedwell beyond a normal lifespan without neurologic symptomsbecause of not despite AD pathology Under this frameworkAβ and tau aggregation may have served as a mechanism tocompensate for active disease-causing biological abnormalities(while minimizing the presumably toxic soluble fibrils required
Table 1 Bradford Hill criteria applied to human studies of α-synuclein (α-syn) and β-amyloid (Aβ)tau aggregation inParkinson disease (PD) and Alzheimer disease (AD)
Criteria definition PD AD
Strength α-SynAβtau aggregation significantlycorrelates with PDAD using appropriatemethodology and accounting for confoundingfactors
Supporting 3e1ndashe3 Supporting 10e4ndashe13
Opposing 0 Opposing 0
Conclusion Support Conclusion Support
Consistency(replication)
Multiple studies using several locationspopulations and methods show a consistentcorrelation between α-synAβtau aggregateswith PDAD
Supporting 3e1ndashe3 Supporting 10e4ndashe13
Opposing 0 Opposing 0
Conclusion Support Conclusion Support
Temporality Cause preceding the effect time intervalbetween α-synAβtau aggregation ascertainedin vivo before symptom development in PDAD
Supporting No studies Supporting 315ndash17
Opposing No studies Opposing 0
Conclusion No support Conclusion Support
Experiment Interventions targeted against α-synAβtauaggregates decrease PDAD incidence severityor rate of progression
Supporting 0 Supporting 0
Opposing 0 Opposing 7e1418ndash23
Conclusion No support Conclusion No support
Biologicalgradient
Dose-response curve between α-synAβtauaggregates concentration and PDAD incidenceseverity or rate of progression
Supporting 0 Supporting 5e15ndashe1817
Opposing 2e1e2 Opposing 0
Conclusion No support Conclusion Support
Plausibility α-SynAβtau aggregates affect molecularpathways associated with PDAD
Supporting 2e19e20 Supporting 5e22ndashe26
Opposing 1e21 Opposing 4e27ndashe30
Conclusion Uncertain Conclusion Equivocal
Analogy If α-synAβtau aggregates cause PDAD similarprotein aggregations could also cause PDAD
A pathologic definition of PD precludesthis analysis to avoid circular reasoning
A pathologic definition of AD precludesthis analysis to avoid circular reasoning
Specificity If α-synAβtau aggregates cause PDAD theseproteins should not be present in diseasesother than PDAD or healthy controls
Supporting 0 Supporting 0
Opposing 10e31e32e34ndashe41 Opposing 1024e33ndashe41
Conclusion No support Conclusion No support
Coherence Causendasheffect should be consistent across allavailable evidence without contradictions ordiscrepancies
Supporting 0 Supporting 0
Opposing 13e1e2e21e31e32e34ndashe41 Opposing 21e1418minus24e27ndashe30e33ndashe41
Conclusion No support Conclusion No support
Adapted from references 8 9 and 25 Individual characteristics of supporting and opposing studies are supplied in table e-2 (doiorg105061dryadg1nq02r)
332 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
to form them) with clinical disease appearing after thismechanism is overwhelmed (see ldquoSpecificityrdquo criteria in tables 1e-2 [doiorg105061dryadg1nq02r])
Third challenge Experimental failure in anti-amyloid therapiesAnti-amyloid therapies that target insoluble Aβ forms (egAN1792 gantenerumab bapineuzumab) soluble forms(eg solanezumab) or prevent its formation (eg sem-agacestat γ-secretase inhibitor) have failed to producecognitive benefits in AD18ndash23 Some agents have been as-sociated with adverse events (eg AN1792 aseptic me-ningoencephalitis18 gantenerumab and bapineuzumabAβ-related abnormalities1920 semagacestat skin cancer andinfections)21 possibly due to immunologic mechanisms (egAN1792 T-cell response)18 or impaired protein processing(eg semagacestat Notch protein)21 in selected cases Al-though unsuccessful or insufficient Aβ load reduction mayaccount for clinical futility an issue that may be definitivelyanswered after the ongoing phase III aducanumab trial resultsare reported (ClinicalTrialsgov Identifier NCT02477800)one can argue that protein aggregation is not the culprit for cell
death (see ldquoExperimentrdquo criteria in tables 1 and e-2 [doiorg105061dryadg1nq02r])
Alternative models of diseaseIf α-synuclein and Aβtau protein aggregation are not ina direct causal pathway in PD and AD they may still con-tribute to neurodegeneration or be an epiphenomenon orguard against toxic soluble forms of the proteins by seques-tering them as innocuous insoluble forms Each of these 3hypothetical models may be influenced by lifelong exposureto genetic and environmental factors that might contribute toincreased susceptibility exhaustion of adaptive responsesand ultimately cell dysfunction and death We use α-synu-clein to illustrate these alternative models (figure 3) but theyapply similarly to Aβ and tau protein aggregation
Alternative model 1 Protein aggregation asaccelerator of pathology of multiple diseasesAside from disorders where a genetic mutation causes initialaccumulation of α-synuclein (eg missense mutations andmultiplication of SNCA)4 or of Aβ (eg triplication of APP
Figure 2 Current model of protein aggregation in Parkinson disease (single disease model)
Abnormal soluble oligomers and fibrils of α-synuclein are directly pathogenic (upper panel) Alternatively or complementarily secondarymolecular changescreated after protein aggregation combine with oligomers and fibrils to hasten cell death (lower panel) T-0 = time zero α-syn = α-synuclein
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 333
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
in Down syndrome or amyloid precursor protein [APP] genemutations and duplication)5 in which these protein aggre-gates are truly pathogenic it is plausible that accumulationof α-synuclein or of Aβtau enhances other primary patho-genic molecular mechanisms of neurodegeneration in spo-radic forms of PD and AD (figure 3A) Putatively a prion-likepropagation of α-synuclein fibrils between adjacent cells29 fallsinto this category without requiring that oligomers and fibrilsact as the initial pathogenic event This model accepts het-erogeneous upstream pathogenic events but relies on solubleproteins (α-synuclein Aβ) misfolding and aggregating asa convergent driver of cellular death In this model misfoldedvariants of α-synuclein and Aβ are suitable for biomarkerdevelopment and therapeutic targeting
Alternative model 2 Protein aggregation asepiphenomena of multiple diseasesAccording to this model a range of molecular abnormalitieslead to cells being unable to adequately degrade misfoldedproteins which aggregate into α-synuclein fibrils in Lewybodies (figure 3B or from misfolded amyloid precursorprotein into misfolded oligomers then fibrils then plaques inAD) These protein aggregates become byproducts (commondenominators or epiphenomena) of several pathogenicpathways and themselves exert no pathogenic or protectiverole According to this model different underlying molecularabnormalities remain pathogenic of different molecular dis-eases their relationship to one another rests only in the typeof protein they cannot properly recycle
Alternative model 3 Protein aggregation asprotective mechanism of multiple diseasesIn this model molecular abnormalities affect the cellularmachinery that normally degrades proteins These misfoldedproteins are sequestered into Lewy bodies as a mechanism topromote maintenance of neuronal and synaptic function de-spite coexistent and actively disrupting molecular abnormal-ities (figure 3C) Each molecular abnormality is pathogenic toa specific disease which shares with others only the type ofprotein actively sequestered as a protective mechanism Suchprotective sequestration of proteins could allow neurons tofunction for decades before becoming overwhelmed delayingthe progression of neurologic disease In this model targetingsoluble oligomers or fibrils (eg using anti-aggregant thera-pies or possibly passive or active immunotherapy) mightpotentially be deleterious if the toxicity of any of the proteinspecies is outweighed by their protective role in the humanbrain under molecular stress
Implications for biomarkers andclinical trialsHypothesis 1 Biomarkers of proteinaggregates are relevant to common pathologybut not to subgroup pathogenesisBased on the challenges discussed above abnormal proteinaggregates (pathology) may represent downstream events
unhelpful to classify PD and AD with pathogenic relevanceThus far biomarker development efforts have been based ondisease models of clinico-pathologic convergence One suchconvergence biomarker is [123I]-FP-CIT SPECT imaging forstriatal dopamine deficiency It is positive in virtually everyonewith PD Because biomarker discovery has been based onclinically defined cohorts (eg tremor-dominant PD vs posturalinstability gait disorder type) significant results within andbetween cohorts have substantial overlap40
Hypothesis 2 Disease-modifying treatmentstargeting oligomers or fibrils might be futile ordeleterious because these proteins areepiphenomena or protective in the humanbrain under molecular stressTargeting these proteins across sporadic or otherwise non-genetic disease forms is unlikely to slow progression of thesedisorders nomatter how early the treatments are implemented(ie prodromal stage) unless protein aggregation acts as anaccelerator of pathology as suggested in alternative model 1The corollary is that applying molecular therapies aimed atachieving neuroprotection to clinically (pathologically) de-fined but not molecularly subtyped populations will remainunlikely to succeed The history of medicine has repeatedlyshown that single (untreatable) clinical diseases are oftenreplaced by several (treatable) molecular diseases As a conse-quence treatments designed to target specific mechanismswork best in certain disease subtypes but not in others
Relinquishing reductionism (the clinically heterogeneous butsingle-disease concepts for AD and PD) will require acceptingthat sophisticated data analyses of large cohorts will not becapable of identifying molecular disease subtypes as the anal-yses cannot overcome the fundamental flaw of existent datasetscohorts invariably assembled based on strict clinical criteria Bigdata may not easily be converted into good data (for the pur-poses of assisting precision medicine) if the data are based onbinary definitions of AD vs control and PD vs control Impor-tant insights may be derived on a smaller scale from studyingrare genetic forms of disease (eg PD-LRRK2 PD-GBA AD-PS1) for which targeted therapeutic efforts may be most likelyto succeedmdashin these subtypes On a wider scale molecularlytargetable neurodegenerative diseases may be best identifiedfrom population-based studies applying non-hypothesis-basedanalytic approaches to biospecimens from large cohorts of agingpeople carefully characterized across a spectrum of brainfunctions and deficits Thus aging cohorts ought to includeatypical clinical presentations and other ostensibly unrelatedneurologic disorders beyond the current criteria for PD andADThe analyses will need to be anchored on biological signals noton clinical phenotypes or clinical criteria and account for ge-netic and lifelong biochemical and environmental factors Mo-lecular subtypes of disease thus identified may or may not fallinto standard clinical classifications
The first proven neuroprotective therapy might only work infewer than 5 of everyone currently subsumed as ldquoADrdquo or
334 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
ldquoPDrdquo (eg GZSAR402671 in early-stage PD-GBA [Clin-icalTrialsgov Identifier NCT02906020]) or in specific at-risk populations (eg solanezumab or gantenerumab forhealthy individuals with an autosomal dominant AD-causingmutation [Dominantly Inherited Alzheimer Network TrialClinicalTrialsgov Identifier NCT01760005]) Such disease-modifying treatment in neurodegenerative disease will helpusher in the biomarker-driven disease subtyping strategy longadopted by oncology and other fields of medicine
AcknowledgmentThe authors thank the patients their parentscaregivers andhealth care professionals who participated in this studyincluding Dean Miller Ruth Wake Dionne Moat RobertMuni Lofra Teresinha Evangelista MD Nikoletta Nikolenko
MD Namita Goyal MD Sonia Nayyar MD CarolinePeyronnard MD Tim Lai MD Monica Lavian DO BrianMinton BS Patrick Tierney RPT Denise Davis RPT CathyChou RPT Nick Higgins and Ed Connor MD
Study fundingNo targeted funding reported
DisclosureThe authors report no disclosures relevant to the manuscriptGo to NeurologyorgN for full disclosures
Publication historyReceived by Neurology July 6 2018 Accepted in final form December14 2018
Figure 3 Alternative models of protein aggregation in Parkinson disease (multiple disease model)
Abnormal soluble oligomers and fibrils ofα-synuclein (α-syn) while not directly pathogenic act as accelerators of neurodegeneration (ldquofueling the firerdquo) due toearly pathogenic molecular abnormalities each representingmolecularly distinct diseases (A model 1) Alternatively abnormal soluble oligomers and fibrilsof α-synuclein aggregate into Lewy bodies as byproducts of earlier pathogenic molecular mechanisms without directly affecting the neurodegenerativeprocess brought on by eachmolecular disease (Bmodel 2) Finally α-synuclein aggregates into Lewy bodies as amechanism to protect the neuron from toxicprotein species or from the biological dysfunction that may have generated the formation of toxic species ldquocoolingrdquo progression of cell degeneration underbiological stress (C model 3) Note that each molecularly defined disease has a different time to death collectively time to neuronal death is longest indiseases corresponding to model C
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 335
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
References1 Gwinn K David KK Swanson-Fischer C et al Parkinsonrsquos disease biomarkers
perspective from the NINDS Parkinsonrsquos Disease Biomarkers Program BiomarkMed201711451ndash473
2 Mollenhauer B Caspell-Garcia CJ Coffey CS et al Longitudinal CSF biomarkers inpatients with early Parkinson disease and healthy controls Neurology 2017891959ndash1969
3 Payami H The emerging science of precision medicine and pharmacogenomics forParkinsonrsquos disease Mov Disord 2017321139ndash1146
4 Chartier-Harlin M-C Kachergus J Roumier C et al α-Synuclein locus duplication asa cause of familial Parkinsonrsquos disease Lancet 20043641167ndash1169
5 Sleegers K BrouwersNGijselinck I et al APPduplication is sufficient to cause early onsetAlzheimerrsquos dementia with cerebral amyloid angiopathy Brain 20061292977ndash2983
6 Bezard E Yue Z Kirik D Spillantini MG Animal models of Parkinsonrsquos disease limitsand relevance to neuroprotection studies Mov Disord 20132861ndash70
7 Bloom GS Amyloid-β and tau JAMA Neurol 2014715058 Bradford-Hill A The environment and disease association or causation Proc R Soc
Med 196558295ndash3009 Fedak KM Bernal A Capshaw ZA Gross S Applying the Bradford Hill criteria in the
21st century how data integration has changed causal inference in molecular epi-demiology Emerg Themes Epidemiol Biomed Cent 20151214
Appendix Authors
Name Location Role Contribution
Alberto JEspay MD MSc
University ofCincinnati OH
CorrespondingAuthor
Conception ofresearchprojectliteraturesearch writingof the firstmanuscriptdraft
Joaquin AVizcarra MD
University ofCincinnati OH
Author Literaturesearch writingof the firstmanuscriptdraft
Luca MarsiliMD PhD
University ofCincinnati
Author Literaturesearch reviewand critique ofthe manuscript
Anthony ELang MDFRCPC
University ofTorontoCanada
Author Review andcritique of themanuscript
David K SimonMD PhD
HarvardMedical SchoolBoston MA
Author Review andcritique of themanuscript
AristideMerola MDPhD
University ofCincinnati OH
Author Review andcritique of themanuscript
Keith AJosephs MDMST MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
AlfonsoFasano MDPhD
University ofTorontoCanada
Author Review andcritique of themanuscript
FrancescaMorgante MDPhD
University ofMessina Italyand St GeorgersquosUniversity ofLondon UK
Author Review andcritique of themanuscript
Rodolfo SavicaMD MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
J TimothyGreenamyreMD PhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
FrancescaCambi MDPhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
Tritia RYamasaki MDPhD
University ofKentuckyLexington
Author Review andcritique of themanuscript
Caroline MTanner MDPhD
University ofCaliforniandashSanFrancisco
Author Review andcritique of themanuscript
Ziv Gan-OrMD PhD
McGillUniversityMontrealCanada
Author Review andcritique of themanuscript
Irene LitvanMD
UC San DiegoCA
Author Review andcritique of themanuscript
Appendix (continued)
Name Location Role Contribution
Ignacio FMata PhD
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Cyrus PZabetian MDMS
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Patrik BrundinMD PhD
Van AndelResearchInstitute GrandRapids MI
Author Review andcritique of themanuscript
Hubert HFernandez MD
ClevelandClinic OH
Author Review andcritique of themanuscript
David GStandaert MDPhD
University ofAlabama atBirmingham
Author Review andcritique of themanuscript
Marcelo AKauffman MDPhD
UBA andUniversidadAustral-CONICETBuenos AiresArgentina
Author Review andcritique of themanuscript
Michael ASchwarzschildMD PhD
MassachusettsGeneralHospitalBoston
Author Review andcritique of themanuscript
S Pablo SardiPharmD PhD
Sanofi Author Review andcritique of themanuscript
Todd ShererPhD
Michael J FoxFoundation forParkinsonrsquosResearch NewYork NY
Author Review andcritique of themanuscript
George PerryPhD
University ofTexas at SanAntonio
Author Review andcritique of themanuscript
James BLeverenz MD
ClevelandClinic OH
Author Literaturesearch reviewand critique ofthe manuscript
336 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
10 Moher D Liberati A Tetzlaff J Altman DG PRISMA Group Preferred reportingitems for systematic reviews and meta-analyses the PRISMA statement Ann InternMed 2009151264ndash269 W64
11 PoeweW Seppi K Tanner CM et al Parkinson disease Nat Rev Dis Primers 2017317013
12 Querfurth HW LaFerla FM Alzheimerrsquos disease N Engl J Med 2010362329ndash34413 NHLBI Research Triangle Institute International National Heart Lung and Blood
Institute quality appraisal tools [online] Available at httpswwwnhlbinihgovhealth-topicsstudy-quality-assessment-tools Accessed December 15 2017
14 Higgins JPT Green S eds Cochrane Handbook for Systematic Reviews of Inter-ventions [online] 510 The Cochrane Collaboration 2011 Available at httpstrainingcochraneorghandbook Accessed December 16 2017
15 Vos SJ Xiong C Visser PJ et al Preclinical Alzheimerrsquos disease and its outcomea longitudinal cohort study Lancet Neurol 201312957ndash965
16 Van Harten AC Smits LL Teunissen CE et al Preclinical AD predicts decline inmemory and executive functions in subjective complaints Neurology 2013811409ndash1416
17 Burnham SC Bourgeat P Dore V et al Clinical and cognitive trajectories in cogni-tively healthy elderly individuals with suspected non-Alzheimerrsquos disease patho-physiology (SNAP) or Alzheimerrsquos disease pathology a longitudinal study LancetNeurol 2016151044ndash1053
18 Gilman S Koller M Black RS et al Clinical effects of A immunization (AN1792) inpatients with AD in an interrupted trial Neurology 2005641553ndash1562
19 Ostrowitzki S Lasser RA Dorflinger E et al A phase III randomized trial of gante-nerumab in prodromal Alzheimerrsquos disease Alzheimers Res Ther 2017995
20 Salloway S Sperling R Fox NC et al Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370322ndash333
21 Doody RS Raman R Farlow M et al A phase 3 trial of semagacestat for treatment ofAlzheimerrsquos disease N Engl J Med 2013369341ndash350
22 Doody RS Thomas RG Farlow M et al Phase 3 trials of solanezumab for mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370311ndash321
23 Honig LS Vellas B Woodward M et al Trial of solanezumab for mild dementia dueto Alzheimerrsquos disease N Engl J Med 2018378321ndash330
24 Cali I Cohen ML Haїk S et al Iatrogenic Creutzfeldt-Jakob disease with amyloid-βpathology an international study Acta Neuropathol Commun 201865
25 Howick J Glasziou P Aronson JK The evolution of evidence hierarchies what canBradford Hillrsquos ldquoguidelines for causationrdquo contribute J R SocMed 2009102186ndash194
26 Roberts RF Wade-Martins R Alegre-Abarrategui J Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinsonrsquos diseasebrain Brain 20151381642ndash1657
27 Blennow K Mattsson N Scholl M Hansson O Zetterberg H Amyloid biomarkers inAlzheimerrsquos disease Trends Pharmacol Sci 201536297ndash309
28 Hogarth P Gregory A Kruer MC et al New NBIA subtype genetic clinical path-ologic and radiographic features of MPAN Neurology 201380268ndash275
29 Olanow CW Brundin P Parkinsonrsquos disease and alpha synuclein is Parkinsonrsquosdisease a prion-like disorder Mov Disord 20132831ndash40
30 Winder-Rhodes SE Garcia-Reitbock P Ban M et al Genetic and pathological linksbetween Parkinsonrsquos disease and the lysosomal disorder Sanfilippo syndrome MovDisord 201227312ndash315
31 Brenowitz WD Keene CD Hawes SE et al Alzheimerrsquos disease neuropathologicchange Lewy body disease and vascular brain injury in clinic- and community-basedsamples Neurobiol Aging 20175383ndash92
32 Irwin DJ Grossman M Weintraub D et al Neuropathological and genetic correlatesof survival and dementia onset in synucleinopathies a retrospective analysis LancetNeurol 20171655ndash65
33 Crystal H Dickson D Fuld P et al Clinico-pathologic studies in dementia non-demented subjects with pathologically confirmed Alzheimerrsquos disease Neurology1988381682ndash1687
34 Polvikoski T Sulkava R Myllykangas L et al Prevalence of Alzheimerrsquos disease in veryelderly people a prospective neuropathological study Neurology 2001561690ndash1696
35 MarkesberyWR Jicha GA Liu H Schmitt FA Lewy body pathology in normal elderlysubjects J Neuropathol Exp Neurol 200968816ndash822
36 Balasubramanian AB Kawas CH Peltz CB Brookmeyer R Corrada MM Alzheimerdisease pathology and longitudinal cognitive performance in the oldest-old with nodementia Neurology 201279915ndash921
37 Silver MH Newell K Brady C Hedley-White ET Perls TT Distinguishing betweenneurodegenerative disease and disease-free aging correlating neuropsychologicalevaluations and neuropathological studies in centenarians Psychosom Med 200264493ndash501
38 Berlau DJ Corrada MM Head E Kawas CH APOE 2 is associated with intactcognition but increased Alzheimer pathology in the oldest old Neurology 200972829ndash834
39 Kawas CH Kim RC Sonnen JA Bullain SS Trieu T Corrada MM Multiple pa-thologies are common and related to dementia in the oldest-old Neurology 201585535ndash542
40 Espay AJ Schwarzschild MA Tanner CM et al Biomarker-driven phenotyping inParkinsonrsquos disease a translational missing link in disease-modifying clinical trialsMov Disord 201732319ndash324Data available from Dryad (Additional References References e1ndashe44)doiorg105061dryadg1nq02r
Did You Knowhellip
hellipyou can browse by subspecialty topics on Neurologyorg
Go to Neurologyorg and click on ldquoTopicsrdquo in the top navigation bar
Share Your Artistic Expressions in Neurology lsquoVisionsrsquo
AAN members are urged to submit medically or scientifically related artistic images such as photographs photomicrographsand paintings to the ldquoVisionsrdquo section of Neurologyreg These images are creative in nature rather than the medically instructiveimages published in the NeuroImages section The image or series of up to six images may be black and white or color andmust fit into one published journal page Accompanying description should be 100 words or less the title should be a maximumof 96 characters including spaces and punctuation
Please access the Author Center at NPuborgauthors for full submission information
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 337
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
DOI 101212WNL0000000000006926201992329-337 Neurology
Alberto J Espay Joaquin A Vizcarra Luca Marsili et al diseases
Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer
This information is current as of February 11 2019
ServicesUpdated Information amp
httpnneurologyorgcontent927329fullincluding high resolution figures can be found at
References httpnneurologyorgcontent927329fullref-list-1
This article cites 38 articles 10 of which you can access for free at
Citations httpnneurologyorgcontent927329fullotherarticles
This article has been cited by 3 HighWire-hosted articles
Permissions amp Licensing
httpwwwneurologyorgaboutabout_the_journalpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpnneurologyorgsubscribersadvertiseInformation about ordering reprints can be found online
rights reserved Print ISSN 0028-3878 Online ISSN 1526-632X1951 it is now a weekly with 48 issues per year Copyright copy 2019 American Academy of Neurology All
reg is the official journal of the American Academy of Neurology Published continuously sinceNeurology

Current model of diseaseand challengesOngoing work on biomarker development and disease-modifying therapies is partly built on a model of proteinmisfolding and subsequent aggregation as an initial process ina cascade of subsequent serial events held as causal to a singledisease In PD it is proposed that α-synuclein monomersassemble into oligomers which in turn give rise to α-synucleinfibrils the earliest disease-causing abnormalities and the majorprotein contribution of Lewy bodies and Lewy neurites (figure 2upper panel)26 It is surmised that α-synuclein aggregationleads to neuronal dysfunction loss of connectivity and neu-ronal death In AD it is proposed that the amyloid precursorprotein cleavage product and hyperphosphorylated tau proteinaggregate into extracellular plaques and intracellular neurofi-brillary tangles respectively12 These plaques and tangles maythen elicit a variety of secondary metabolic and molecularchanges which in turn lead to cellular dysfunction that (1)perpetuates the formation of additional pathogenic proteinaggregates or (2) directly accelerates or magnifies the effecton cell death through these molecular changes (ldquokindlingrdquophenomenon)11 (figure 2 lower panel) Nevertheless 3 mainarguments challenge these models
First challenge Uncertain temporality andbiological gradientUnlike other fields in medicine brain tissue is not readilyaccessible for diagnostic or research purposes Instead re-search endeavors have relied on specimens obtained from
2 sources (1) extracranial sources (eg biopsies of skinsalivary gland gut samples of blood and CSF) collected duringlife and (2) the brain itself harvested postmortem many yearsafter events of interest have taken place Even pathology studiesthat include samples from patients with different disease se-verity or ldquostagesrdquo are inherently cross-sectional In an attemptto assess the temporality of pathologic events AD biomarker-to-autopsy validation studies have yielded extraordinary prog-ress with in vivo CSF sampling and PET imaging of Aβtaunow accepted as valid surrogates for protein brain deposition27
These advances have led to 2 observations in AD that Aβ andtau sequentially or concurrently may be placed at the start ofa chain of events before the onset of cognitive dysfunction atwhat we assume to be time zero (see ldquoTemporalityrdquo criteria intables 1 and e-2)15ndash17 and that the co-presence of Aβ and tauleads to greater cognitive decline than either alone (see ldquoBi-ological gradientrdquo criteria in tables 1 and e-2 [doiorg105061dryadg1nq02r])1516 There is no corresponding neuroimagingor CSF biomarker counterpart for idiopathic PD While this invivo visualization has been assumed to represent time zero it isalso plausible that disease pathogenesis begins before the ag-gregation of proteins becomes detectable rendering them asintermediate rather than initiating events in the pathway tocell death
Second challenge Incidental pathologycopathology and nonspecific pathologyWithout the ability to serially examine living brain tissue forpotentially dynamic components in cellular proteostatic mech-anisms diseases are classified as proteinopathies (eg synu-cleinopathy) based on protein aggregates as end products Forsynucleinopathies for instance this is the case in PD multiplesystem atrophy and dementia with Lewy bodies (DLB) aswell as nonparkinsonian (peripheral) synucleinopathies suchas pure autonomic failure and atypical synucleinopathies in-cluding selected neurodegenerations with brain iron accumu-lation such as mitochondrial membrane protein-associatedneurodegeneration28 the parkinsonian lysosomal disordersKufor-Rakeb syndrome Gaucher disease andChediak-Higashisyndrome and the nonparkinsonian Sanfilippo syndrome2930
The α-synuclein ubiquity permeates into AD Lewy body pa-thology accumulates in over 50 of autopsy-proven ADbrains31
and conversely 77 of autopsy-proven PD dementia or DLBcases exhibit ADpathology32 Separately the classic ADproteinsAβ and tau-positive neurofibrillary tangles have been re-spectively reported in 13 and 48 of sporadic Creutzfeldt-Jakob disease cases24 Given their widespread appearancein a variety of phenotypically diverse diseases pathologies ofα-synuclein andAβtaumay not be pathogenic (see ldquoSpecificityrdquocriteria in tables 1 and e-2 [doiorg105061dryadg1nq02r])
The term ldquoincidental pathologyrdquo has been applied to the findingof protein aggregation on postmortem studies of neurologicallynormal individuals In the framework of α-synuclein- and Aβtau-based proteins as AD and PD ldquopathologyrdquo it is often as-sumed that when found on autopsy these might representprodromal individuals on the road to clinical disease had they
Figure 1 Flow chart of study selection
CENTRAL = Central Register of Controlled Trials
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 331
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
lived long enough However supra-surviving individuals (theover-90 or oldest-old) exhibit high frequency of AD pathologywithout dementia (50)3334 and PD pathology withoutparkinsonism (25)35 Notably no cognitive tests amongthe oldest-old without dementia can predict AD neuropathol-ogy within 3 years before death36 In fact neuropathologystudies in the oldest-old have either shown no correlation be-tween premortem cognitive function and AD neuropathol-ogy37 or even a paradoxical association between normalcognition and increased AD neuropathology38 The 90+ Study(183 brain autopsies from individuals who died at or after theage of 90 years) yielded robust support for an inverse re-lationship between AD pathology and clinical dementia evenbeyond the simple observation that intermediate to high AD
pathology clusters similarly in people with (23) and withoutdementia (28)39 Further analysis of the data suggests a pro-tective effect of AD pathology Compared to non-AD pathol-ogy (microinfarcts and white matter disease dementia n = 19no dementia n = 6) AD pathology (dementia n = 23 nodementia n = 24) was associated with a significantly reducedrisk of dementia (odds ratio 03 95 confidence interval01ndash09 p = 003 analysis not in the original publication)39
Therefore it is plausible that these individuals may have livedwell beyond a normal lifespan without neurologic symptomsbecause of not despite AD pathology Under this frameworkAβ and tau aggregation may have served as a mechanism tocompensate for active disease-causing biological abnormalities(while minimizing the presumably toxic soluble fibrils required
Table 1 Bradford Hill criteria applied to human studies of α-synuclein (α-syn) and β-amyloid (Aβ)tau aggregation inParkinson disease (PD) and Alzheimer disease (AD)
Criteria definition PD AD
Strength α-SynAβtau aggregation significantlycorrelates with PDAD using appropriatemethodology and accounting for confoundingfactors
Supporting 3e1ndashe3 Supporting 10e4ndashe13
Opposing 0 Opposing 0
Conclusion Support Conclusion Support
Consistency(replication)
Multiple studies using several locationspopulations and methods show a consistentcorrelation between α-synAβtau aggregateswith PDAD
Supporting 3e1ndashe3 Supporting 10e4ndashe13
Opposing 0 Opposing 0
Conclusion Support Conclusion Support
Temporality Cause preceding the effect time intervalbetween α-synAβtau aggregation ascertainedin vivo before symptom development in PDAD
Supporting No studies Supporting 315ndash17
Opposing No studies Opposing 0
Conclusion No support Conclusion Support
Experiment Interventions targeted against α-synAβtauaggregates decrease PDAD incidence severityor rate of progression
Supporting 0 Supporting 0
Opposing 0 Opposing 7e1418ndash23
Conclusion No support Conclusion No support
Biologicalgradient
Dose-response curve between α-synAβtauaggregates concentration and PDAD incidenceseverity or rate of progression
Supporting 0 Supporting 5e15ndashe1817
Opposing 2e1e2 Opposing 0
Conclusion No support Conclusion Support
Plausibility α-SynAβtau aggregates affect molecularpathways associated with PDAD
Supporting 2e19e20 Supporting 5e22ndashe26
Opposing 1e21 Opposing 4e27ndashe30
Conclusion Uncertain Conclusion Equivocal
Analogy If α-synAβtau aggregates cause PDAD similarprotein aggregations could also cause PDAD
A pathologic definition of PD precludesthis analysis to avoid circular reasoning
A pathologic definition of AD precludesthis analysis to avoid circular reasoning
Specificity If α-synAβtau aggregates cause PDAD theseproteins should not be present in diseasesother than PDAD or healthy controls
Supporting 0 Supporting 0
Opposing 10e31e32e34ndashe41 Opposing 1024e33ndashe41
Conclusion No support Conclusion No support
Coherence Causendasheffect should be consistent across allavailable evidence without contradictions ordiscrepancies
Supporting 0 Supporting 0
Opposing 13e1e2e21e31e32e34ndashe41 Opposing 21e1418minus24e27ndashe30e33ndashe41
Conclusion No support Conclusion No support
Adapted from references 8 9 and 25 Individual characteristics of supporting and opposing studies are supplied in table e-2 (doiorg105061dryadg1nq02r)
332 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
to form them) with clinical disease appearing after thismechanism is overwhelmed (see ldquoSpecificityrdquo criteria in tables 1e-2 [doiorg105061dryadg1nq02r])
Third challenge Experimental failure in anti-amyloid therapiesAnti-amyloid therapies that target insoluble Aβ forms (egAN1792 gantenerumab bapineuzumab) soluble forms(eg solanezumab) or prevent its formation (eg sem-agacestat γ-secretase inhibitor) have failed to producecognitive benefits in AD18ndash23 Some agents have been as-sociated with adverse events (eg AN1792 aseptic me-ningoencephalitis18 gantenerumab and bapineuzumabAβ-related abnormalities1920 semagacestat skin cancer andinfections)21 possibly due to immunologic mechanisms (egAN1792 T-cell response)18 or impaired protein processing(eg semagacestat Notch protein)21 in selected cases Al-though unsuccessful or insufficient Aβ load reduction mayaccount for clinical futility an issue that may be definitivelyanswered after the ongoing phase III aducanumab trial resultsare reported (ClinicalTrialsgov Identifier NCT02477800)one can argue that protein aggregation is not the culprit for cell
death (see ldquoExperimentrdquo criteria in tables 1 and e-2 [doiorg105061dryadg1nq02r])
Alternative models of diseaseIf α-synuclein and Aβtau protein aggregation are not ina direct causal pathway in PD and AD they may still con-tribute to neurodegeneration or be an epiphenomenon orguard against toxic soluble forms of the proteins by seques-tering them as innocuous insoluble forms Each of these 3hypothetical models may be influenced by lifelong exposureto genetic and environmental factors that might contribute toincreased susceptibility exhaustion of adaptive responsesand ultimately cell dysfunction and death We use α-synu-clein to illustrate these alternative models (figure 3) but theyapply similarly to Aβ and tau protein aggregation
Alternative model 1 Protein aggregation asaccelerator of pathology of multiple diseasesAside from disorders where a genetic mutation causes initialaccumulation of α-synuclein (eg missense mutations andmultiplication of SNCA)4 or of Aβ (eg triplication of APP
Figure 2 Current model of protein aggregation in Parkinson disease (single disease model)
Abnormal soluble oligomers and fibrils of α-synuclein are directly pathogenic (upper panel) Alternatively or complementarily secondarymolecular changescreated after protein aggregation combine with oligomers and fibrils to hasten cell death (lower panel) T-0 = time zero α-syn = α-synuclein
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 333
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
in Down syndrome or amyloid precursor protein [APP] genemutations and duplication)5 in which these protein aggre-gates are truly pathogenic it is plausible that accumulationof α-synuclein or of Aβtau enhances other primary patho-genic molecular mechanisms of neurodegeneration in spo-radic forms of PD and AD (figure 3A) Putatively a prion-likepropagation of α-synuclein fibrils between adjacent cells29 fallsinto this category without requiring that oligomers and fibrilsact as the initial pathogenic event This model accepts het-erogeneous upstream pathogenic events but relies on solubleproteins (α-synuclein Aβ) misfolding and aggregating asa convergent driver of cellular death In this model misfoldedvariants of α-synuclein and Aβ are suitable for biomarkerdevelopment and therapeutic targeting
Alternative model 2 Protein aggregation asepiphenomena of multiple diseasesAccording to this model a range of molecular abnormalitieslead to cells being unable to adequately degrade misfoldedproteins which aggregate into α-synuclein fibrils in Lewybodies (figure 3B or from misfolded amyloid precursorprotein into misfolded oligomers then fibrils then plaques inAD) These protein aggregates become byproducts (commondenominators or epiphenomena) of several pathogenicpathways and themselves exert no pathogenic or protectiverole According to this model different underlying molecularabnormalities remain pathogenic of different molecular dis-eases their relationship to one another rests only in the typeof protein they cannot properly recycle
Alternative model 3 Protein aggregation asprotective mechanism of multiple diseasesIn this model molecular abnormalities affect the cellularmachinery that normally degrades proteins These misfoldedproteins are sequestered into Lewy bodies as a mechanism topromote maintenance of neuronal and synaptic function de-spite coexistent and actively disrupting molecular abnormal-ities (figure 3C) Each molecular abnormality is pathogenic toa specific disease which shares with others only the type ofprotein actively sequestered as a protective mechanism Suchprotective sequestration of proteins could allow neurons tofunction for decades before becoming overwhelmed delayingthe progression of neurologic disease In this model targetingsoluble oligomers or fibrils (eg using anti-aggregant thera-pies or possibly passive or active immunotherapy) mightpotentially be deleterious if the toxicity of any of the proteinspecies is outweighed by their protective role in the humanbrain under molecular stress
Implications for biomarkers andclinical trialsHypothesis 1 Biomarkers of proteinaggregates are relevant to common pathologybut not to subgroup pathogenesisBased on the challenges discussed above abnormal proteinaggregates (pathology) may represent downstream events
unhelpful to classify PD and AD with pathogenic relevanceThus far biomarker development efforts have been based ondisease models of clinico-pathologic convergence One suchconvergence biomarker is [123I]-FP-CIT SPECT imaging forstriatal dopamine deficiency It is positive in virtually everyonewith PD Because biomarker discovery has been based onclinically defined cohorts (eg tremor-dominant PD vs posturalinstability gait disorder type) significant results within andbetween cohorts have substantial overlap40
Hypothesis 2 Disease-modifying treatmentstargeting oligomers or fibrils might be futile ordeleterious because these proteins areepiphenomena or protective in the humanbrain under molecular stressTargeting these proteins across sporadic or otherwise non-genetic disease forms is unlikely to slow progression of thesedisorders nomatter how early the treatments are implemented(ie prodromal stage) unless protein aggregation acts as anaccelerator of pathology as suggested in alternative model 1The corollary is that applying molecular therapies aimed atachieving neuroprotection to clinically (pathologically) de-fined but not molecularly subtyped populations will remainunlikely to succeed The history of medicine has repeatedlyshown that single (untreatable) clinical diseases are oftenreplaced by several (treatable) molecular diseases As a conse-quence treatments designed to target specific mechanismswork best in certain disease subtypes but not in others
Relinquishing reductionism (the clinically heterogeneous butsingle-disease concepts for AD and PD) will require acceptingthat sophisticated data analyses of large cohorts will not becapable of identifying molecular disease subtypes as the anal-yses cannot overcome the fundamental flaw of existent datasetscohorts invariably assembled based on strict clinical criteria Bigdata may not easily be converted into good data (for the pur-poses of assisting precision medicine) if the data are based onbinary definitions of AD vs control and PD vs control Impor-tant insights may be derived on a smaller scale from studyingrare genetic forms of disease (eg PD-LRRK2 PD-GBA AD-PS1) for which targeted therapeutic efforts may be most likelyto succeedmdashin these subtypes On a wider scale molecularlytargetable neurodegenerative diseases may be best identifiedfrom population-based studies applying non-hypothesis-basedanalytic approaches to biospecimens from large cohorts of agingpeople carefully characterized across a spectrum of brainfunctions and deficits Thus aging cohorts ought to includeatypical clinical presentations and other ostensibly unrelatedneurologic disorders beyond the current criteria for PD andADThe analyses will need to be anchored on biological signals noton clinical phenotypes or clinical criteria and account for ge-netic and lifelong biochemical and environmental factors Mo-lecular subtypes of disease thus identified may or may not fallinto standard clinical classifications
The first proven neuroprotective therapy might only work infewer than 5 of everyone currently subsumed as ldquoADrdquo or
334 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
ldquoPDrdquo (eg GZSAR402671 in early-stage PD-GBA [Clin-icalTrialsgov Identifier NCT02906020]) or in specific at-risk populations (eg solanezumab or gantenerumab forhealthy individuals with an autosomal dominant AD-causingmutation [Dominantly Inherited Alzheimer Network TrialClinicalTrialsgov Identifier NCT01760005]) Such disease-modifying treatment in neurodegenerative disease will helpusher in the biomarker-driven disease subtyping strategy longadopted by oncology and other fields of medicine
AcknowledgmentThe authors thank the patients their parentscaregivers andhealth care professionals who participated in this studyincluding Dean Miller Ruth Wake Dionne Moat RobertMuni Lofra Teresinha Evangelista MD Nikoletta Nikolenko
MD Namita Goyal MD Sonia Nayyar MD CarolinePeyronnard MD Tim Lai MD Monica Lavian DO BrianMinton BS Patrick Tierney RPT Denise Davis RPT CathyChou RPT Nick Higgins and Ed Connor MD
Study fundingNo targeted funding reported
DisclosureThe authors report no disclosures relevant to the manuscriptGo to NeurologyorgN for full disclosures
Publication historyReceived by Neurology July 6 2018 Accepted in final form December14 2018
Figure 3 Alternative models of protein aggregation in Parkinson disease (multiple disease model)
Abnormal soluble oligomers and fibrils ofα-synuclein (α-syn) while not directly pathogenic act as accelerators of neurodegeneration (ldquofueling the firerdquo) due toearly pathogenic molecular abnormalities each representingmolecularly distinct diseases (A model 1) Alternatively abnormal soluble oligomers and fibrilsof α-synuclein aggregate into Lewy bodies as byproducts of earlier pathogenic molecular mechanisms without directly affecting the neurodegenerativeprocess brought on by eachmolecular disease (Bmodel 2) Finally α-synuclein aggregates into Lewy bodies as amechanism to protect the neuron from toxicprotein species or from the biological dysfunction that may have generated the formation of toxic species ldquocoolingrdquo progression of cell degeneration underbiological stress (C model 3) Note that each molecularly defined disease has a different time to death collectively time to neuronal death is longest indiseases corresponding to model C
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 335
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
References1 Gwinn K David KK Swanson-Fischer C et al Parkinsonrsquos disease biomarkers
perspective from the NINDS Parkinsonrsquos Disease Biomarkers Program BiomarkMed201711451ndash473
2 Mollenhauer B Caspell-Garcia CJ Coffey CS et al Longitudinal CSF biomarkers inpatients with early Parkinson disease and healthy controls Neurology 2017891959ndash1969
3 Payami H The emerging science of precision medicine and pharmacogenomics forParkinsonrsquos disease Mov Disord 2017321139ndash1146
4 Chartier-Harlin M-C Kachergus J Roumier C et al α-Synuclein locus duplication asa cause of familial Parkinsonrsquos disease Lancet 20043641167ndash1169
5 Sleegers K BrouwersNGijselinck I et al APPduplication is sufficient to cause early onsetAlzheimerrsquos dementia with cerebral amyloid angiopathy Brain 20061292977ndash2983
6 Bezard E Yue Z Kirik D Spillantini MG Animal models of Parkinsonrsquos disease limitsand relevance to neuroprotection studies Mov Disord 20132861ndash70
7 Bloom GS Amyloid-β and tau JAMA Neurol 2014715058 Bradford-Hill A The environment and disease association or causation Proc R Soc
Med 196558295ndash3009 Fedak KM Bernal A Capshaw ZA Gross S Applying the Bradford Hill criteria in the
21st century how data integration has changed causal inference in molecular epi-demiology Emerg Themes Epidemiol Biomed Cent 20151214
Appendix Authors
Name Location Role Contribution
Alberto JEspay MD MSc
University ofCincinnati OH
CorrespondingAuthor
Conception ofresearchprojectliteraturesearch writingof the firstmanuscriptdraft
Joaquin AVizcarra MD
University ofCincinnati OH
Author Literaturesearch writingof the firstmanuscriptdraft
Luca MarsiliMD PhD
University ofCincinnati
Author Literaturesearch reviewand critique ofthe manuscript
Anthony ELang MDFRCPC
University ofTorontoCanada
Author Review andcritique of themanuscript
David K SimonMD PhD
HarvardMedical SchoolBoston MA
Author Review andcritique of themanuscript
AristideMerola MDPhD
University ofCincinnati OH
Author Review andcritique of themanuscript
Keith AJosephs MDMST MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
AlfonsoFasano MDPhD
University ofTorontoCanada
Author Review andcritique of themanuscript
FrancescaMorgante MDPhD
University ofMessina Italyand St GeorgersquosUniversity ofLondon UK
Author Review andcritique of themanuscript
Rodolfo SavicaMD MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
J TimothyGreenamyreMD PhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
FrancescaCambi MDPhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
Tritia RYamasaki MDPhD
University ofKentuckyLexington
Author Review andcritique of themanuscript
Caroline MTanner MDPhD
University ofCaliforniandashSanFrancisco
Author Review andcritique of themanuscript
Ziv Gan-OrMD PhD
McGillUniversityMontrealCanada
Author Review andcritique of themanuscript
Irene LitvanMD
UC San DiegoCA
Author Review andcritique of themanuscript
Appendix (continued)
Name Location Role Contribution
Ignacio FMata PhD
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Cyrus PZabetian MDMS
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Patrik BrundinMD PhD
Van AndelResearchInstitute GrandRapids MI
Author Review andcritique of themanuscript
Hubert HFernandez MD
ClevelandClinic OH
Author Review andcritique of themanuscript
David GStandaert MDPhD
University ofAlabama atBirmingham
Author Review andcritique of themanuscript
Marcelo AKauffman MDPhD
UBA andUniversidadAustral-CONICETBuenos AiresArgentina
Author Review andcritique of themanuscript
Michael ASchwarzschildMD PhD
MassachusettsGeneralHospitalBoston
Author Review andcritique of themanuscript
S Pablo SardiPharmD PhD
Sanofi Author Review andcritique of themanuscript
Todd ShererPhD
Michael J FoxFoundation forParkinsonrsquosResearch NewYork NY
Author Review andcritique of themanuscript
George PerryPhD
University ofTexas at SanAntonio
Author Review andcritique of themanuscript
James BLeverenz MD
ClevelandClinic OH
Author Literaturesearch reviewand critique ofthe manuscript
336 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
10 Moher D Liberati A Tetzlaff J Altman DG PRISMA Group Preferred reportingitems for systematic reviews and meta-analyses the PRISMA statement Ann InternMed 2009151264ndash269 W64
11 PoeweW Seppi K Tanner CM et al Parkinson disease Nat Rev Dis Primers 2017317013
12 Querfurth HW LaFerla FM Alzheimerrsquos disease N Engl J Med 2010362329ndash34413 NHLBI Research Triangle Institute International National Heart Lung and Blood
Institute quality appraisal tools [online] Available at httpswwwnhlbinihgovhealth-topicsstudy-quality-assessment-tools Accessed December 15 2017
14 Higgins JPT Green S eds Cochrane Handbook for Systematic Reviews of Inter-ventions [online] 510 The Cochrane Collaboration 2011 Available at httpstrainingcochraneorghandbook Accessed December 16 2017
15 Vos SJ Xiong C Visser PJ et al Preclinical Alzheimerrsquos disease and its outcomea longitudinal cohort study Lancet Neurol 201312957ndash965
16 Van Harten AC Smits LL Teunissen CE et al Preclinical AD predicts decline inmemory and executive functions in subjective complaints Neurology 2013811409ndash1416
17 Burnham SC Bourgeat P Dore V et al Clinical and cognitive trajectories in cogni-tively healthy elderly individuals with suspected non-Alzheimerrsquos disease patho-physiology (SNAP) or Alzheimerrsquos disease pathology a longitudinal study LancetNeurol 2016151044ndash1053
18 Gilman S Koller M Black RS et al Clinical effects of A immunization (AN1792) inpatients with AD in an interrupted trial Neurology 2005641553ndash1562
19 Ostrowitzki S Lasser RA Dorflinger E et al A phase III randomized trial of gante-nerumab in prodromal Alzheimerrsquos disease Alzheimers Res Ther 2017995
20 Salloway S Sperling R Fox NC et al Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370322ndash333
21 Doody RS Raman R Farlow M et al A phase 3 trial of semagacestat for treatment ofAlzheimerrsquos disease N Engl J Med 2013369341ndash350
22 Doody RS Thomas RG Farlow M et al Phase 3 trials of solanezumab for mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370311ndash321
23 Honig LS Vellas B Woodward M et al Trial of solanezumab for mild dementia dueto Alzheimerrsquos disease N Engl J Med 2018378321ndash330
24 Cali I Cohen ML Haїk S et al Iatrogenic Creutzfeldt-Jakob disease with amyloid-βpathology an international study Acta Neuropathol Commun 201865
25 Howick J Glasziou P Aronson JK The evolution of evidence hierarchies what canBradford Hillrsquos ldquoguidelines for causationrdquo contribute J R SocMed 2009102186ndash194
26 Roberts RF Wade-Martins R Alegre-Abarrategui J Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinsonrsquos diseasebrain Brain 20151381642ndash1657
27 Blennow K Mattsson N Scholl M Hansson O Zetterberg H Amyloid biomarkers inAlzheimerrsquos disease Trends Pharmacol Sci 201536297ndash309
28 Hogarth P Gregory A Kruer MC et al New NBIA subtype genetic clinical path-ologic and radiographic features of MPAN Neurology 201380268ndash275
29 Olanow CW Brundin P Parkinsonrsquos disease and alpha synuclein is Parkinsonrsquosdisease a prion-like disorder Mov Disord 20132831ndash40
30 Winder-Rhodes SE Garcia-Reitbock P Ban M et al Genetic and pathological linksbetween Parkinsonrsquos disease and the lysosomal disorder Sanfilippo syndrome MovDisord 201227312ndash315
31 Brenowitz WD Keene CD Hawes SE et al Alzheimerrsquos disease neuropathologicchange Lewy body disease and vascular brain injury in clinic- and community-basedsamples Neurobiol Aging 20175383ndash92
32 Irwin DJ Grossman M Weintraub D et al Neuropathological and genetic correlatesof survival and dementia onset in synucleinopathies a retrospective analysis LancetNeurol 20171655ndash65
33 Crystal H Dickson D Fuld P et al Clinico-pathologic studies in dementia non-demented subjects with pathologically confirmed Alzheimerrsquos disease Neurology1988381682ndash1687
34 Polvikoski T Sulkava R Myllykangas L et al Prevalence of Alzheimerrsquos disease in veryelderly people a prospective neuropathological study Neurology 2001561690ndash1696
35 MarkesberyWR Jicha GA Liu H Schmitt FA Lewy body pathology in normal elderlysubjects J Neuropathol Exp Neurol 200968816ndash822
36 Balasubramanian AB Kawas CH Peltz CB Brookmeyer R Corrada MM Alzheimerdisease pathology and longitudinal cognitive performance in the oldest-old with nodementia Neurology 201279915ndash921
37 Silver MH Newell K Brady C Hedley-White ET Perls TT Distinguishing betweenneurodegenerative disease and disease-free aging correlating neuropsychologicalevaluations and neuropathological studies in centenarians Psychosom Med 200264493ndash501
38 Berlau DJ Corrada MM Head E Kawas CH APOE 2 is associated with intactcognition but increased Alzheimer pathology in the oldest old Neurology 200972829ndash834
39 Kawas CH Kim RC Sonnen JA Bullain SS Trieu T Corrada MM Multiple pa-thologies are common and related to dementia in the oldest-old Neurology 201585535ndash542
40 Espay AJ Schwarzschild MA Tanner CM et al Biomarker-driven phenotyping inParkinsonrsquos disease a translational missing link in disease-modifying clinical trialsMov Disord 201732319ndash324Data available from Dryad (Additional References References e1ndashe44)doiorg105061dryadg1nq02r
Did You Knowhellip
hellipyou can browse by subspecialty topics on Neurologyorg
Go to Neurologyorg and click on ldquoTopicsrdquo in the top navigation bar
Share Your Artistic Expressions in Neurology lsquoVisionsrsquo
AAN members are urged to submit medically or scientifically related artistic images such as photographs photomicrographsand paintings to the ldquoVisionsrdquo section of Neurologyreg These images are creative in nature rather than the medically instructiveimages published in the NeuroImages section The image or series of up to six images may be black and white or color andmust fit into one published journal page Accompanying description should be 100 words or less the title should be a maximumof 96 characters including spaces and punctuation
Please access the Author Center at NPuborgauthors for full submission information
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 337
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
DOI 101212WNL0000000000006926201992329-337 Neurology
Alberto J Espay Joaquin A Vizcarra Luca Marsili et al diseases
Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer
This information is current as of February 11 2019
ServicesUpdated Information amp
httpnneurologyorgcontent927329fullincluding high resolution figures can be found at
References httpnneurologyorgcontent927329fullref-list-1
This article cites 38 articles 10 of which you can access for free at
Citations httpnneurologyorgcontent927329fullotherarticles
This article has been cited by 3 HighWire-hosted articles
Permissions amp Licensing
httpwwwneurologyorgaboutabout_the_journalpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpnneurologyorgsubscribersadvertiseInformation about ordering reprints can be found online
rights reserved Print ISSN 0028-3878 Online ISSN 1526-632X1951 it is now a weekly with 48 issues per year Copyright copy 2019 American Academy of Neurology All
reg is the official journal of the American Academy of Neurology Published continuously sinceNeurology
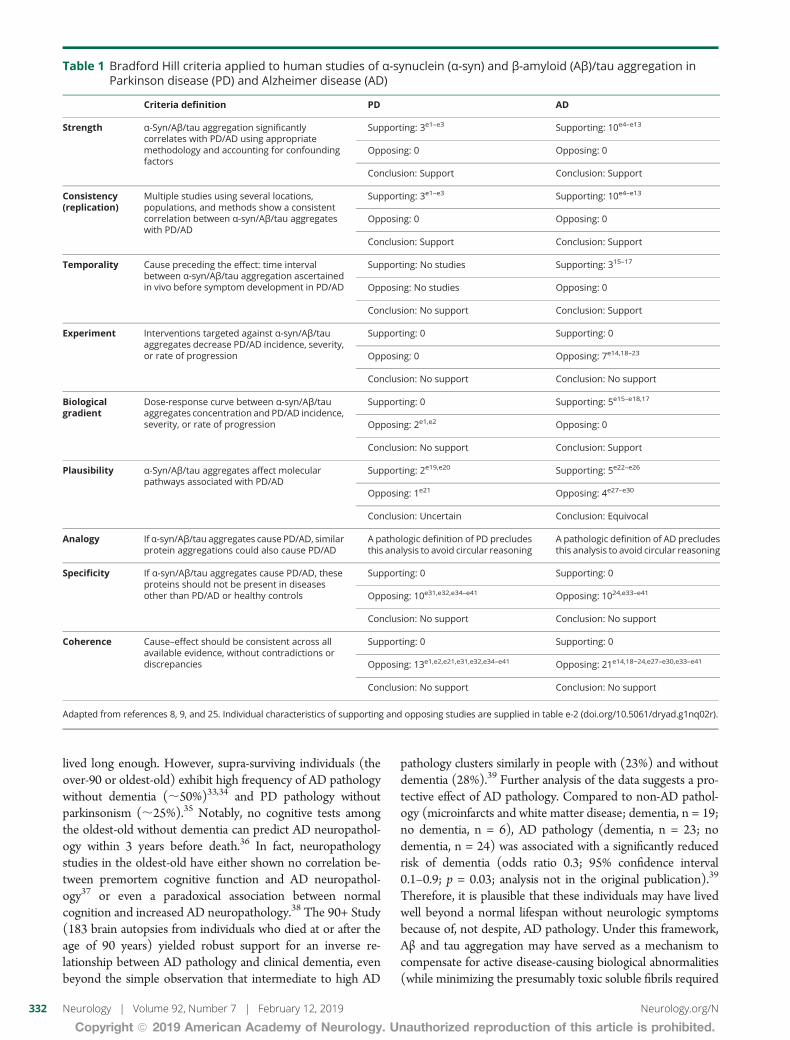
lived long enough However supra-surviving individuals (theover-90 or oldest-old) exhibit high frequency of AD pathologywithout dementia (50)3334 and PD pathology withoutparkinsonism (25)35 Notably no cognitive tests amongthe oldest-old without dementia can predict AD neuropathol-ogy within 3 years before death36 In fact neuropathologystudies in the oldest-old have either shown no correlation be-tween premortem cognitive function and AD neuropathol-ogy37 or even a paradoxical association between normalcognition and increased AD neuropathology38 The 90+ Study(183 brain autopsies from individuals who died at or after theage of 90 years) yielded robust support for an inverse re-lationship between AD pathology and clinical dementia evenbeyond the simple observation that intermediate to high AD
pathology clusters similarly in people with (23) and withoutdementia (28)39 Further analysis of the data suggests a pro-tective effect of AD pathology Compared to non-AD pathol-ogy (microinfarcts and white matter disease dementia n = 19no dementia n = 6) AD pathology (dementia n = 23 nodementia n = 24) was associated with a significantly reducedrisk of dementia (odds ratio 03 95 confidence interval01ndash09 p = 003 analysis not in the original publication)39
Therefore it is plausible that these individuals may have livedwell beyond a normal lifespan without neurologic symptomsbecause of not despite AD pathology Under this frameworkAβ and tau aggregation may have served as a mechanism tocompensate for active disease-causing biological abnormalities(while minimizing the presumably toxic soluble fibrils required
Table 1 Bradford Hill criteria applied to human studies of α-synuclein (α-syn) and β-amyloid (Aβ)tau aggregation inParkinson disease (PD) and Alzheimer disease (AD)
Criteria definition PD AD
Strength α-SynAβtau aggregation significantlycorrelates with PDAD using appropriatemethodology and accounting for confoundingfactors
Supporting 3e1ndashe3 Supporting 10e4ndashe13
Opposing 0 Opposing 0
Conclusion Support Conclusion Support
Consistency(replication)
Multiple studies using several locationspopulations and methods show a consistentcorrelation between α-synAβtau aggregateswith PDAD
Supporting 3e1ndashe3 Supporting 10e4ndashe13
Opposing 0 Opposing 0
Conclusion Support Conclusion Support
Temporality Cause preceding the effect time intervalbetween α-synAβtau aggregation ascertainedin vivo before symptom development in PDAD
Supporting No studies Supporting 315ndash17
Opposing No studies Opposing 0
Conclusion No support Conclusion Support
Experiment Interventions targeted against α-synAβtauaggregates decrease PDAD incidence severityor rate of progression
Supporting 0 Supporting 0
Opposing 0 Opposing 7e1418ndash23
Conclusion No support Conclusion No support
Biologicalgradient
Dose-response curve between α-synAβtauaggregates concentration and PDAD incidenceseverity or rate of progression
Supporting 0 Supporting 5e15ndashe1817
Opposing 2e1e2 Opposing 0
Conclusion No support Conclusion Support
Plausibility α-SynAβtau aggregates affect molecularpathways associated with PDAD
Supporting 2e19e20 Supporting 5e22ndashe26
Opposing 1e21 Opposing 4e27ndashe30
Conclusion Uncertain Conclusion Equivocal
Analogy If α-synAβtau aggregates cause PDAD similarprotein aggregations could also cause PDAD
A pathologic definition of PD precludesthis analysis to avoid circular reasoning
A pathologic definition of AD precludesthis analysis to avoid circular reasoning
Specificity If α-synAβtau aggregates cause PDAD theseproteins should not be present in diseasesother than PDAD or healthy controls
Supporting 0 Supporting 0
Opposing 10e31e32e34ndashe41 Opposing 1024e33ndashe41
Conclusion No support Conclusion No support
Coherence Causendasheffect should be consistent across allavailable evidence without contradictions ordiscrepancies
Supporting 0 Supporting 0
Opposing 13e1e2e21e31e32e34ndashe41 Opposing 21e1418minus24e27ndashe30e33ndashe41
Conclusion No support Conclusion No support
Adapted from references 8 9 and 25 Individual characteristics of supporting and opposing studies are supplied in table e-2 (doiorg105061dryadg1nq02r)
332 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
to form them) with clinical disease appearing after thismechanism is overwhelmed (see ldquoSpecificityrdquo criteria in tables 1e-2 [doiorg105061dryadg1nq02r])
Third challenge Experimental failure in anti-amyloid therapiesAnti-amyloid therapies that target insoluble Aβ forms (egAN1792 gantenerumab bapineuzumab) soluble forms(eg solanezumab) or prevent its formation (eg sem-agacestat γ-secretase inhibitor) have failed to producecognitive benefits in AD18ndash23 Some agents have been as-sociated with adverse events (eg AN1792 aseptic me-ningoencephalitis18 gantenerumab and bapineuzumabAβ-related abnormalities1920 semagacestat skin cancer andinfections)21 possibly due to immunologic mechanisms (egAN1792 T-cell response)18 or impaired protein processing(eg semagacestat Notch protein)21 in selected cases Al-though unsuccessful or insufficient Aβ load reduction mayaccount for clinical futility an issue that may be definitivelyanswered after the ongoing phase III aducanumab trial resultsare reported (ClinicalTrialsgov Identifier NCT02477800)one can argue that protein aggregation is not the culprit for cell
death (see ldquoExperimentrdquo criteria in tables 1 and e-2 [doiorg105061dryadg1nq02r])
Alternative models of diseaseIf α-synuclein and Aβtau protein aggregation are not ina direct causal pathway in PD and AD they may still con-tribute to neurodegeneration or be an epiphenomenon orguard against toxic soluble forms of the proteins by seques-tering them as innocuous insoluble forms Each of these 3hypothetical models may be influenced by lifelong exposureto genetic and environmental factors that might contribute toincreased susceptibility exhaustion of adaptive responsesand ultimately cell dysfunction and death We use α-synu-clein to illustrate these alternative models (figure 3) but theyapply similarly to Aβ and tau protein aggregation
Alternative model 1 Protein aggregation asaccelerator of pathology of multiple diseasesAside from disorders where a genetic mutation causes initialaccumulation of α-synuclein (eg missense mutations andmultiplication of SNCA)4 or of Aβ (eg triplication of APP
Figure 2 Current model of protein aggregation in Parkinson disease (single disease model)
Abnormal soluble oligomers and fibrils of α-synuclein are directly pathogenic (upper panel) Alternatively or complementarily secondarymolecular changescreated after protein aggregation combine with oligomers and fibrils to hasten cell death (lower panel) T-0 = time zero α-syn = α-synuclein
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 333
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
in Down syndrome or amyloid precursor protein [APP] genemutations and duplication)5 in which these protein aggre-gates are truly pathogenic it is plausible that accumulationof α-synuclein or of Aβtau enhances other primary patho-genic molecular mechanisms of neurodegeneration in spo-radic forms of PD and AD (figure 3A) Putatively a prion-likepropagation of α-synuclein fibrils between adjacent cells29 fallsinto this category without requiring that oligomers and fibrilsact as the initial pathogenic event This model accepts het-erogeneous upstream pathogenic events but relies on solubleproteins (α-synuclein Aβ) misfolding and aggregating asa convergent driver of cellular death In this model misfoldedvariants of α-synuclein and Aβ are suitable for biomarkerdevelopment and therapeutic targeting
Alternative model 2 Protein aggregation asepiphenomena of multiple diseasesAccording to this model a range of molecular abnormalitieslead to cells being unable to adequately degrade misfoldedproteins which aggregate into α-synuclein fibrils in Lewybodies (figure 3B or from misfolded amyloid precursorprotein into misfolded oligomers then fibrils then plaques inAD) These protein aggregates become byproducts (commondenominators or epiphenomena) of several pathogenicpathways and themselves exert no pathogenic or protectiverole According to this model different underlying molecularabnormalities remain pathogenic of different molecular dis-eases their relationship to one another rests only in the typeof protein they cannot properly recycle
Alternative model 3 Protein aggregation asprotective mechanism of multiple diseasesIn this model molecular abnormalities affect the cellularmachinery that normally degrades proteins These misfoldedproteins are sequestered into Lewy bodies as a mechanism topromote maintenance of neuronal and synaptic function de-spite coexistent and actively disrupting molecular abnormal-ities (figure 3C) Each molecular abnormality is pathogenic toa specific disease which shares with others only the type ofprotein actively sequestered as a protective mechanism Suchprotective sequestration of proteins could allow neurons tofunction for decades before becoming overwhelmed delayingthe progression of neurologic disease In this model targetingsoluble oligomers or fibrils (eg using anti-aggregant thera-pies or possibly passive or active immunotherapy) mightpotentially be deleterious if the toxicity of any of the proteinspecies is outweighed by their protective role in the humanbrain under molecular stress
Implications for biomarkers andclinical trialsHypothesis 1 Biomarkers of proteinaggregates are relevant to common pathologybut not to subgroup pathogenesisBased on the challenges discussed above abnormal proteinaggregates (pathology) may represent downstream events
unhelpful to classify PD and AD with pathogenic relevanceThus far biomarker development efforts have been based ondisease models of clinico-pathologic convergence One suchconvergence biomarker is [123I]-FP-CIT SPECT imaging forstriatal dopamine deficiency It is positive in virtually everyonewith PD Because biomarker discovery has been based onclinically defined cohorts (eg tremor-dominant PD vs posturalinstability gait disorder type) significant results within andbetween cohorts have substantial overlap40
Hypothesis 2 Disease-modifying treatmentstargeting oligomers or fibrils might be futile ordeleterious because these proteins areepiphenomena or protective in the humanbrain under molecular stressTargeting these proteins across sporadic or otherwise non-genetic disease forms is unlikely to slow progression of thesedisorders nomatter how early the treatments are implemented(ie prodromal stage) unless protein aggregation acts as anaccelerator of pathology as suggested in alternative model 1The corollary is that applying molecular therapies aimed atachieving neuroprotection to clinically (pathologically) de-fined but not molecularly subtyped populations will remainunlikely to succeed The history of medicine has repeatedlyshown that single (untreatable) clinical diseases are oftenreplaced by several (treatable) molecular diseases As a conse-quence treatments designed to target specific mechanismswork best in certain disease subtypes but not in others
Relinquishing reductionism (the clinically heterogeneous butsingle-disease concepts for AD and PD) will require acceptingthat sophisticated data analyses of large cohorts will not becapable of identifying molecular disease subtypes as the anal-yses cannot overcome the fundamental flaw of existent datasetscohorts invariably assembled based on strict clinical criteria Bigdata may not easily be converted into good data (for the pur-poses of assisting precision medicine) if the data are based onbinary definitions of AD vs control and PD vs control Impor-tant insights may be derived on a smaller scale from studyingrare genetic forms of disease (eg PD-LRRK2 PD-GBA AD-PS1) for which targeted therapeutic efforts may be most likelyto succeedmdashin these subtypes On a wider scale molecularlytargetable neurodegenerative diseases may be best identifiedfrom population-based studies applying non-hypothesis-basedanalytic approaches to biospecimens from large cohorts of agingpeople carefully characterized across a spectrum of brainfunctions and deficits Thus aging cohorts ought to includeatypical clinical presentations and other ostensibly unrelatedneurologic disorders beyond the current criteria for PD andADThe analyses will need to be anchored on biological signals noton clinical phenotypes or clinical criteria and account for ge-netic and lifelong biochemical and environmental factors Mo-lecular subtypes of disease thus identified may or may not fallinto standard clinical classifications
The first proven neuroprotective therapy might only work infewer than 5 of everyone currently subsumed as ldquoADrdquo or
334 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
ldquoPDrdquo (eg GZSAR402671 in early-stage PD-GBA [Clin-icalTrialsgov Identifier NCT02906020]) or in specific at-risk populations (eg solanezumab or gantenerumab forhealthy individuals with an autosomal dominant AD-causingmutation [Dominantly Inherited Alzheimer Network TrialClinicalTrialsgov Identifier NCT01760005]) Such disease-modifying treatment in neurodegenerative disease will helpusher in the biomarker-driven disease subtyping strategy longadopted by oncology and other fields of medicine
AcknowledgmentThe authors thank the patients their parentscaregivers andhealth care professionals who participated in this studyincluding Dean Miller Ruth Wake Dionne Moat RobertMuni Lofra Teresinha Evangelista MD Nikoletta Nikolenko
MD Namita Goyal MD Sonia Nayyar MD CarolinePeyronnard MD Tim Lai MD Monica Lavian DO BrianMinton BS Patrick Tierney RPT Denise Davis RPT CathyChou RPT Nick Higgins and Ed Connor MD
Study fundingNo targeted funding reported
DisclosureThe authors report no disclosures relevant to the manuscriptGo to NeurologyorgN for full disclosures
Publication historyReceived by Neurology July 6 2018 Accepted in final form December14 2018
Figure 3 Alternative models of protein aggregation in Parkinson disease (multiple disease model)
Abnormal soluble oligomers and fibrils ofα-synuclein (α-syn) while not directly pathogenic act as accelerators of neurodegeneration (ldquofueling the firerdquo) due toearly pathogenic molecular abnormalities each representingmolecularly distinct diseases (A model 1) Alternatively abnormal soluble oligomers and fibrilsof α-synuclein aggregate into Lewy bodies as byproducts of earlier pathogenic molecular mechanisms without directly affecting the neurodegenerativeprocess brought on by eachmolecular disease (Bmodel 2) Finally α-synuclein aggregates into Lewy bodies as amechanism to protect the neuron from toxicprotein species or from the biological dysfunction that may have generated the formation of toxic species ldquocoolingrdquo progression of cell degeneration underbiological stress (C model 3) Note that each molecularly defined disease has a different time to death collectively time to neuronal death is longest indiseases corresponding to model C
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 335
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
References1 Gwinn K David KK Swanson-Fischer C et al Parkinsonrsquos disease biomarkers
perspective from the NINDS Parkinsonrsquos Disease Biomarkers Program BiomarkMed201711451ndash473
2 Mollenhauer B Caspell-Garcia CJ Coffey CS et al Longitudinal CSF biomarkers inpatients with early Parkinson disease and healthy controls Neurology 2017891959ndash1969
3 Payami H The emerging science of precision medicine and pharmacogenomics forParkinsonrsquos disease Mov Disord 2017321139ndash1146
4 Chartier-Harlin M-C Kachergus J Roumier C et al α-Synuclein locus duplication asa cause of familial Parkinsonrsquos disease Lancet 20043641167ndash1169
5 Sleegers K BrouwersNGijselinck I et al APPduplication is sufficient to cause early onsetAlzheimerrsquos dementia with cerebral amyloid angiopathy Brain 20061292977ndash2983
6 Bezard E Yue Z Kirik D Spillantini MG Animal models of Parkinsonrsquos disease limitsand relevance to neuroprotection studies Mov Disord 20132861ndash70
7 Bloom GS Amyloid-β and tau JAMA Neurol 2014715058 Bradford-Hill A The environment and disease association or causation Proc R Soc
Med 196558295ndash3009 Fedak KM Bernal A Capshaw ZA Gross S Applying the Bradford Hill criteria in the
21st century how data integration has changed causal inference in molecular epi-demiology Emerg Themes Epidemiol Biomed Cent 20151214
Appendix Authors
Name Location Role Contribution
Alberto JEspay MD MSc
University ofCincinnati OH
CorrespondingAuthor
Conception ofresearchprojectliteraturesearch writingof the firstmanuscriptdraft
Joaquin AVizcarra MD
University ofCincinnati OH
Author Literaturesearch writingof the firstmanuscriptdraft
Luca MarsiliMD PhD
University ofCincinnati
Author Literaturesearch reviewand critique ofthe manuscript
Anthony ELang MDFRCPC
University ofTorontoCanada
Author Review andcritique of themanuscript
David K SimonMD PhD
HarvardMedical SchoolBoston MA
Author Review andcritique of themanuscript
AristideMerola MDPhD
University ofCincinnati OH
Author Review andcritique of themanuscript
Keith AJosephs MDMST MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
AlfonsoFasano MDPhD
University ofTorontoCanada
Author Review andcritique of themanuscript
FrancescaMorgante MDPhD
University ofMessina Italyand St GeorgersquosUniversity ofLondon UK
Author Review andcritique of themanuscript
Rodolfo SavicaMD MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
J TimothyGreenamyreMD PhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
FrancescaCambi MDPhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
Tritia RYamasaki MDPhD
University ofKentuckyLexington
Author Review andcritique of themanuscript
Caroline MTanner MDPhD
University ofCaliforniandashSanFrancisco
Author Review andcritique of themanuscript
Ziv Gan-OrMD PhD
McGillUniversityMontrealCanada
Author Review andcritique of themanuscript
Irene LitvanMD
UC San DiegoCA
Author Review andcritique of themanuscript
Appendix (continued)
Name Location Role Contribution
Ignacio FMata PhD
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Cyrus PZabetian MDMS
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Patrik BrundinMD PhD
Van AndelResearchInstitute GrandRapids MI
Author Review andcritique of themanuscript
Hubert HFernandez MD
ClevelandClinic OH
Author Review andcritique of themanuscript
David GStandaert MDPhD
University ofAlabama atBirmingham
Author Review andcritique of themanuscript
Marcelo AKauffman MDPhD
UBA andUniversidadAustral-CONICETBuenos AiresArgentina
Author Review andcritique of themanuscript
Michael ASchwarzschildMD PhD
MassachusettsGeneralHospitalBoston
Author Review andcritique of themanuscript
S Pablo SardiPharmD PhD
Sanofi Author Review andcritique of themanuscript
Todd ShererPhD
Michael J FoxFoundation forParkinsonrsquosResearch NewYork NY
Author Review andcritique of themanuscript
George PerryPhD
University ofTexas at SanAntonio
Author Review andcritique of themanuscript
James BLeverenz MD
ClevelandClinic OH
Author Literaturesearch reviewand critique ofthe manuscript
336 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
10 Moher D Liberati A Tetzlaff J Altman DG PRISMA Group Preferred reportingitems for systematic reviews and meta-analyses the PRISMA statement Ann InternMed 2009151264ndash269 W64
11 PoeweW Seppi K Tanner CM et al Parkinson disease Nat Rev Dis Primers 2017317013
12 Querfurth HW LaFerla FM Alzheimerrsquos disease N Engl J Med 2010362329ndash34413 NHLBI Research Triangle Institute International National Heart Lung and Blood
Institute quality appraisal tools [online] Available at httpswwwnhlbinihgovhealth-topicsstudy-quality-assessment-tools Accessed December 15 2017
14 Higgins JPT Green S eds Cochrane Handbook for Systematic Reviews of Inter-ventions [online] 510 The Cochrane Collaboration 2011 Available at httpstrainingcochraneorghandbook Accessed December 16 2017
15 Vos SJ Xiong C Visser PJ et al Preclinical Alzheimerrsquos disease and its outcomea longitudinal cohort study Lancet Neurol 201312957ndash965
16 Van Harten AC Smits LL Teunissen CE et al Preclinical AD predicts decline inmemory and executive functions in subjective complaints Neurology 2013811409ndash1416
17 Burnham SC Bourgeat P Dore V et al Clinical and cognitive trajectories in cogni-tively healthy elderly individuals with suspected non-Alzheimerrsquos disease patho-physiology (SNAP) or Alzheimerrsquos disease pathology a longitudinal study LancetNeurol 2016151044ndash1053
18 Gilman S Koller M Black RS et al Clinical effects of A immunization (AN1792) inpatients with AD in an interrupted trial Neurology 2005641553ndash1562
19 Ostrowitzki S Lasser RA Dorflinger E et al A phase III randomized trial of gante-nerumab in prodromal Alzheimerrsquos disease Alzheimers Res Ther 2017995
20 Salloway S Sperling R Fox NC et al Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370322ndash333
21 Doody RS Raman R Farlow M et al A phase 3 trial of semagacestat for treatment ofAlzheimerrsquos disease N Engl J Med 2013369341ndash350
22 Doody RS Thomas RG Farlow M et al Phase 3 trials of solanezumab for mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370311ndash321
23 Honig LS Vellas B Woodward M et al Trial of solanezumab for mild dementia dueto Alzheimerrsquos disease N Engl J Med 2018378321ndash330
24 Cali I Cohen ML Haїk S et al Iatrogenic Creutzfeldt-Jakob disease with amyloid-βpathology an international study Acta Neuropathol Commun 201865
25 Howick J Glasziou P Aronson JK The evolution of evidence hierarchies what canBradford Hillrsquos ldquoguidelines for causationrdquo contribute J R SocMed 2009102186ndash194
26 Roberts RF Wade-Martins R Alegre-Abarrategui J Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinsonrsquos diseasebrain Brain 20151381642ndash1657
27 Blennow K Mattsson N Scholl M Hansson O Zetterberg H Amyloid biomarkers inAlzheimerrsquos disease Trends Pharmacol Sci 201536297ndash309
28 Hogarth P Gregory A Kruer MC et al New NBIA subtype genetic clinical path-ologic and radiographic features of MPAN Neurology 201380268ndash275
29 Olanow CW Brundin P Parkinsonrsquos disease and alpha synuclein is Parkinsonrsquosdisease a prion-like disorder Mov Disord 20132831ndash40
30 Winder-Rhodes SE Garcia-Reitbock P Ban M et al Genetic and pathological linksbetween Parkinsonrsquos disease and the lysosomal disorder Sanfilippo syndrome MovDisord 201227312ndash315
31 Brenowitz WD Keene CD Hawes SE et al Alzheimerrsquos disease neuropathologicchange Lewy body disease and vascular brain injury in clinic- and community-basedsamples Neurobiol Aging 20175383ndash92
32 Irwin DJ Grossman M Weintraub D et al Neuropathological and genetic correlatesof survival and dementia onset in synucleinopathies a retrospective analysis LancetNeurol 20171655ndash65
33 Crystal H Dickson D Fuld P et al Clinico-pathologic studies in dementia non-demented subjects with pathologically confirmed Alzheimerrsquos disease Neurology1988381682ndash1687
34 Polvikoski T Sulkava R Myllykangas L et al Prevalence of Alzheimerrsquos disease in veryelderly people a prospective neuropathological study Neurology 2001561690ndash1696
35 MarkesberyWR Jicha GA Liu H Schmitt FA Lewy body pathology in normal elderlysubjects J Neuropathol Exp Neurol 200968816ndash822
36 Balasubramanian AB Kawas CH Peltz CB Brookmeyer R Corrada MM Alzheimerdisease pathology and longitudinal cognitive performance in the oldest-old with nodementia Neurology 201279915ndash921
37 Silver MH Newell K Brady C Hedley-White ET Perls TT Distinguishing betweenneurodegenerative disease and disease-free aging correlating neuropsychologicalevaluations and neuropathological studies in centenarians Psychosom Med 200264493ndash501
38 Berlau DJ Corrada MM Head E Kawas CH APOE 2 is associated with intactcognition but increased Alzheimer pathology in the oldest old Neurology 200972829ndash834
39 Kawas CH Kim RC Sonnen JA Bullain SS Trieu T Corrada MM Multiple pa-thologies are common and related to dementia in the oldest-old Neurology 201585535ndash542
40 Espay AJ Schwarzschild MA Tanner CM et al Biomarker-driven phenotyping inParkinsonrsquos disease a translational missing link in disease-modifying clinical trialsMov Disord 201732319ndash324Data available from Dryad (Additional References References e1ndashe44)doiorg105061dryadg1nq02r
Did You Knowhellip
hellipyou can browse by subspecialty topics on Neurologyorg
Go to Neurologyorg and click on ldquoTopicsrdquo in the top navigation bar
Share Your Artistic Expressions in Neurology lsquoVisionsrsquo
AAN members are urged to submit medically or scientifically related artistic images such as photographs photomicrographsand paintings to the ldquoVisionsrdquo section of Neurologyreg These images are creative in nature rather than the medically instructiveimages published in the NeuroImages section The image or series of up to six images may be black and white or color andmust fit into one published journal page Accompanying description should be 100 words or less the title should be a maximumof 96 characters including spaces and punctuation
Please access the Author Center at NPuborgauthors for full submission information
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 337
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
DOI 101212WNL0000000000006926201992329-337 Neurology
Alberto J Espay Joaquin A Vizcarra Luca Marsili et al diseases
Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer
This information is current as of February 11 2019
ServicesUpdated Information amp
httpnneurologyorgcontent927329fullincluding high resolution figures can be found at
References httpnneurologyorgcontent927329fullref-list-1
This article cites 38 articles 10 of which you can access for free at
Citations httpnneurologyorgcontent927329fullotherarticles
This article has been cited by 3 HighWire-hosted articles
Permissions amp Licensing
httpwwwneurologyorgaboutabout_the_journalpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpnneurologyorgsubscribersadvertiseInformation about ordering reprints can be found online
rights reserved Print ISSN 0028-3878 Online ISSN 1526-632X1951 it is now a weekly with 48 issues per year Copyright copy 2019 American Academy of Neurology All
reg is the official journal of the American Academy of Neurology Published continuously sinceNeurology

to form them) with clinical disease appearing after thismechanism is overwhelmed (see ldquoSpecificityrdquo criteria in tables 1e-2 [doiorg105061dryadg1nq02r])
Third challenge Experimental failure in anti-amyloid therapiesAnti-amyloid therapies that target insoluble Aβ forms (egAN1792 gantenerumab bapineuzumab) soluble forms(eg solanezumab) or prevent its formation (eg sem-agacestat γ-secretase inhibitor) have failed to producecognitive benefits in AD18ndash23 Some agents have been as-sociated with adverse events (eg AN1792 aseptic me-ningoencephalitis18 gantenerumab and bapineuzumabAβ-related abnormalities1920 semagacestat skin cancer andinfections)21 possibly due to immunologic mechanisms (egAN1792 T-cell response)18 or impaired protein processing(eg semagacestat Notch protein)21 in selected cases Al-though unsuccessful or insufficient Aβ load reduction mayaccount for clinical futility an issue that may be definitivelyanswered after the ongoing phase III aducanumab trial resultsare reported (ClinicalTrialsgov Identifier NCT02477800)one can argue that protein aggregation is not the culprit for cell
death (see ldquoExperimentrdquo criteria in tables 1 and e-2 [doiorg105061dryadg1nq02r])
Alternative models of diseaseIf α-synuclein and Aβtau protein aggregation are not ina direct causal pathway in PD and AD they may still con-tribute to neurodegeneration or be an epiphenomenon orguard against toxic soluble forms of the proteins by seques-tering them as innocuous insoluble forms Each of these 3hypothetical models may be influenced by lifelong exposureto genetic and environmental factors that might contribute toincreased susceptibility exhaustion of adaptive responsesand ultimately cell dysfunction and death We use α-synu-clein to illustrate these alternative models (figure 3) but theyapply similarly to Aβ and tau protein aggregation
Alternative model 1 Protein aggregation asaccelerator of pathology of multiple diseasesAside from disorders where a genetic mutation causes initialaccumulation of α-synuclein (eg missense mutations andmultiplication of SNCA)4 or of Aβ (eg triplication of APP
Figure 2 Current model of protein aggregation in Parkinson disease (single disease model)
Abnormal soluble oligomers and fibrils of α-synuclein are directly pathogenic (upper panel) Alternatively or complementarily secondarymolecular changescreated after protein aggregation combine with oligomers and fibrils to hasten cell death (lower panel) T-0 = time zero α-syn = α-synuclein
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 333
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
in Down syndrome or amyloid precursor protein [APP] genemutations and duplication)5 in which these protein aggre-gates are truly pathogenic it is plausible that accumulationof α-synuclein or of Aβtau enhances other primary patho-genic molecular mechanisms of neurodegeneration in spo-radic forms of PD and AD (figure 3A) Putatively a prion-likepropagation of α-synuclein fibrils between adjacent cells29 fallsinto this category without requiring that oligomers and fibrilsact as the initial pathogenic event This model accepts het-erogeneous upstream pathogenic events but relies on solubleproteins (α-synuclein Aβ) misfolding and aggregating asa convergent driver of cellular death In this model misfoldedvariants of α-synuclein and Aβ are suitable for biomarkerdevelopment and therapeutic targeting
Alternative model 2 Protein aggregation asepiphenomena of multiple diseasesAccording to this model a range of molecular abnormalitieslead to cells being unable to adequately degrade misfoldedproteins which aggregate into α-synuclein fibrils in Lewybodies (figure 3B or from misfolded amyloid precursorprotein into misfolded oligomers then fibrils then plaques inAD) These protein aggregates become byproducts (commondenominators or epiphenomena) of several pathogenicpathways and themselves exert no pathogenic or protectiverole According to this model different underlying molecularabnormalities remain pathogenic of different molecular dis-eases their relationship to one another rests only in the typeof protein they cannot properly recycle
Alternative model 3 Protein aggregation asprotective mechanism of multiple diseasesIn this model molecular abnormalities affect the cellularmachinery that normally degrades proteins These misfoldedproteins are sequestered into Lewy bodies as a mechanism topromote maintenance of neuronal and synaptic function de-spite coexistent and actively disrupting molecular abnormal-ities (figure 3C) Each molecular abnormality is pathogenic toa specific disease which shares with others only the type ofprotein actively sequestered as a protective mechanism Suchprotective sequestration of proteins could allow neurons tofunction for decades before becoming overwhelmed delayingthe progression of neurologic disease In this model targetingsoluble oligomers or fibrils (eg using anti-aggregant thera-pies or possibly passive or active immunotherapy) mightpotentially be deleterious if the toxicity of any of the proteinspecies is outweighed by their protective role in the humanbrain under molecular stress
Implications for biomarkers andclinical trialsHypothesis 1 Biomarkers of proteinaggregates are relevant to common pathologybut not to subgroup pathogenesisBased on the challenges discussed above abnormal proteinaggregates (pathology) may represent downstream events
unhelpful to classify PD and AD with pathogenic relevanceThus far biomarker development efforts have been based ondisease models of clinico-pathologic convergence One suchconvergence biomarker is [123I]-FP-CIT SPECT imaging forstriatal dopamine deficiency It is positive in virtually everyonewith PD Because biomarker discovery has been based onclinically defined cohorts (eg tremor-dominant PD vs posturalinstability gait disorder type) significant results within andbetween cohorts have substantial overlap40
Hypothesis 2 Disease-modifying treatmentstargeting oligomers or fibrils might be futile ordeleterious because these proteins areepiphenomena or protective in the humanbrain under molecular stressTargeting these proteins across sporadic or otherwise non-genetic disease forms is unlikely to slow progression of thesedisorders nomatter how early the treatments are implemented(ie prodromal stage) unless protein aggregation acts as anaccelerator of pathology as suggested in alternative model 1The corollary is that applying molecular therapies aimed atachieving neuroprotection to clinically (pathologically) de-fined but not molecularly subtyped populations will remainunlikely to succeed The history of medicine has repeatedlyshown that single (untreatable) clinical diseases are oftenreplaced by several (treatable) molecular diseases As a conse-quence treatments designed to target specific mechanismswork best in certain disease subtypes but not in others
Relinquishing reductionism (the clinically heterogeneous butsingle-disease concepts for AD and PD) will require acceptingthat sophisticated data analyses of large cohorts will not becapable of identifying molecular disease subtypes as the anal-yses cannot overcome the fundamental flaw of existent datasetscohorts invariably assembled based on strict clinical criteria Bigdata may not easily be converted into good data (for the pur-poses of assisting precision medicine) if the data are based onbinary definitions of AD vs control and PD vs control Impor-tant insights may be derived on a smaller scale from studyingrare genetic forms of disease (eg PD-LRRK2 PD-GBA AD-PS1) for which targeted therapeutic efforts may be most likelyto succeedmdashin these subtypes On a wider scale molecularlytargetable neurodegenerative diseases may be best identifiedfrom population-based studies applying non-hypothesis-basedanalytic approaches to biospecimens from large cohorts of agingpeople carefully characterized across a spectrum of brainfunctions and deficits Thus aging cohorts ought to includeatypical clinical presentations and other ostensibly unrelatedneurologic disorders beyond the current criteria for PD andADThe analyses will need to be anchored on biological signals noton clinical phenotypes or clinical criteria and account for ge-netic and lifelong biochemical and environmental factors Mo-lecular subtypes of disease thus identified may or may not fallinto standard clinical classifications
The first proven neuroprotective therapy might only work infewer than 5 of everyone currently subsumed as ldquoADrdquo or
334 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
ldquoPDrdquo (eg GZSAR402671 in early-stage PD-GBA [Clin-icalTrialsgov Identifier NCT02906020]) or in specific at-risk populations (eg solanezumab or gantenerumab forhealthy individuals with an autosomal dominant AD-causingmutation [Dominantly Inherited Alzheimer Network TrialClinicalTrialsgov Identifier NCT01760005]) Such disease-modifying treatment in neurodegenerative disease will helpusher in the biomarker-driven disease subtyping strategy longadopted by oncology and other fields of medicine
AcknowledgmentThe authors thank the patients their parentscaregivers andhealth care professionals who participated in this studyincluding Dean Miller Ruth Wake Dionne Moat RobertMuni Lofra Teresinha Evangelista MD Nikoletta Nikolenko
MD Namita Goyal MD Sonia Nayyar MD CarolinePeyronnard MD Tim Lai MD Monica Lavian DO BrianMinton BS Patrick Tierney RPT Denise Davis RPT CathyChou RPT Nick Higgins and Ed Connor MD
Study fundingNo targeted funding reported
DisclosureThe authors report no disclosures relevant to the manuscriptGo to NeurologyorgN for full disclosures
Publication historyReceived by Neurology July 6 2018 Accepted in final form December14 2018
Figure 3 Alternative models of protein aggregation in Parkinson disease (multiple disease model)
Abnormal soluble oligomers and fibrils ofα-synuclein (α-syn) while not directly pathogenic act as accelerators of neurodegeneration (ldquofueling the firerdquo) due toearly pathogenic molecular abnormalities each representingmolecularly distinct diseases (A model 1) Alternatively abnormal soluble oligomers and fibrilsof α-synuclein aggregate into Lewy bodies as byproducts of earlier pathogenic molecular mechanisms without directly affecting the neurodegenerativeprocess brought on by eachmolecular disease (Bmodel 2) Finally α-synuclein aggregates into Lewy bodies as amechanism to protect the neuron from toxicprotein species or from the biological dysfunction that may have generated the formation of toxic species ldquocoolingrdquo progression of cell degeneration underbiological stress (C model 3) Note that each molecularly defined disease has a different time to death collectively time to neuronal death is longest indiseases corresponding to model C
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 335
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
References1 Gwinn K David KK Swanson-Fischer C et al Parkinsonrsquos disease biomarkers
perspective from the NINDS Parkinsonrsquos Disease Biomarkers Program BiomarkMed201711451ndash473
2 Mollenhauer B Caspell-Garcia CJ Coffey CS et al Longitudinal CSF biomarkers inpatients with early Parkinson disease and healthy controls Neurology 2017891959ndash1969
3 Payami H The emerging science of precision medicine and pharmacogenomics forParkinsonrsquos disease Mov Disord 2017321139ndash1146
4 Chartier-Harlin M-C Kachergus J Roumier C et al α-Synuclein locus duplication asa cause of familial Parkinsonrsquos disease Lancet 20043641167ndash1169
5 Sleegers K BrouwersNGijselinck I et al APPduplication is sufficient to cause early onsetAlzheimerrsquos dementia with cerebral amyloid angiopathy Brain 20061292977ndash2983
6 Bezard E Yue Z Kirik D Spillantini MG Animal models of Parkinsonrsquos disease limitsand relevance to neuroprotection studies Mov Disord 20132861ndash70
7 Bloom GS Amyloid-β and tau JAMA Neurol 2014715058 Bradford-Hill A The environment and disease association or causation Proc R Soc
Med 196558295ndash3009 Fedak KM Bernal A Capshaw ZA Gross S Applying the Bradford Hill criteria in the
21st century how data integration has changed causal inference in molecular epi-demiology Emerg Themes Epidemiol Biomed Cent 20151214
Appendix Authors
Name Location Role Contribution
Alberto JEspay MD MSc
University ofCincinnati OH
CorrespondingAuthor
Conception ofresearchprojectliteraturesearch writingof the firstmanuscriptdraft
Joaquin AVizcarra MD
University ofCincinnati OH
Author Literaturesearch writingof the firstmanuscriptdraft
Luca MarsiliMD PhD
University ofCincinnati
Author Literaturesearch reviewand critique ofthe manuscript
Anthony ELang MDFRCPC
University ofTorontoCanada
Author Review andcritique of themanuscript
David K SimonMD PhD
HarvardMedical SchoolBoston MA
Author Review andcritique of themanuscript
AristideMerola MDPhD
University ofCincinnati OH
Author Review andcritique of themanuscript
Keith AJosephs MDMST MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
AlfonsoFasano MDPhD
University ofTorontoCanada
Author Review andcritique of themanuscript
FrancescaMorgante MDPhD
University ofMessina Italyand St GeorgersquosUniversity ofLondon UK
Author Review andcritique of themanuscript
Rodolfo SavicaMD MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
J TimothyGreenamyreMD PhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
FrancescaCambi MDPhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
Tritia RYamasaki MDPhD
University ofKentuckyLexington
Author Review andcritique of themanuscript
Caroline MTanner MDPhD
University ofCaliforniandashSanFrancisco
Author Review andcritique of themanuscript
Ziv Gan-OrMD PhD
McGillUniversityMontrealCanada
Author Review andcritique of themanuscript
Irene LitvanMD
UC San DiegoCA
Author Review andcritique of themanuscript
Appendix (continued)
Name Location Role Contribution
Ignacio FMata PhD
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Cyrus PZabetian MDMS
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Patrik BrundinMD PhD
Van AndelResearchInstitute GrandRapids MI
Author Review andcritique of themanuscript
Hubert HFernandez MD
ClevelandClinic OH
Author Review andcritique of themanuscript
David GStandaert MDPhD
University ofAlabama atBirmingham
Author Review andcritique of themanuscript
Marcelo AKauffman MDPhD
UBA andUniversidadAustral-CONICETBuenos AiresArgentina
Author Review andcritique of themanuscript
Michael ASchwarzschildMD PhD
MassachusettsGeneralHospitalBoston
Author Review andcritique of themanuscript
S Pablo SardiPharmD PhD
Sanofi Author Review andcritique of themanuscript
Todd ShererPhD
Michael J FoxFoundation forParkinsonrsquosResearch NewYork NY
Author Review andcritique of themanuscript
George PerryPhD
University ofTexas at SanAntonio
Author Review andcritique of themanuscript
James BLeverenz MD
ClevelandClinic OH
Author Literaturesearch reviewand critique ofthe manuscript
336 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
10 Moher D Liberati A Tetzlaff J Altman DG PRISMA Group Preferred reportingitems for systematic reviews and meta-analyses the PRISMA statement Ann InternMed 2009151264ndash269 W64
11 PoeweW Seppi K Tanner CM et al Parkinson disease Nat Rev Dis Primers 2017317013
12 Querfurth HW LaFerla FM Alzheimerrsquos disease N Engl J Med 2010362329ndash34413 NHLBI Research Triangle Institute International National Heart Lung and Blood
Institute quality appraisal tools [online] Available at httpswwwnhlbinihgovhealth-topicsstudy-quality-assessment-tools Accessed December 15 2017
14 Higgins JPT Green S eds Cochrane Handbook for Systematic Reviews of Inter-ventions [online] 510 The Cochrane Collaboration 2011 Available at httpstrainingcochraneorghandbook Accessed December 16 2017
15 Vos SJ Xiong C Visser PJ et al Preclinical Alzheimerrsquos disease and its outcomea longitudinal cohort study Lancet Neurol 201312957ndash965
16 Van Harten AC Smits LL Teunissen CE et al Preclinical AD predicts decline inmemory and executive functions in subjective complaints Neurology 2013811409ndash1416
17 Burnham SC Bourgeat P Dore V et al Clinical and cognitive trajectories in cogni-tively healthy elderly individuals with suspected non-Alzheimerrsquos disease patho-physiology (SNAP) or Alzheimerrsquos disease pathology a longitudinal study LancetNeurol 2016151044ndash1053
18 Gilman S Koller M Black RS et al Clinical effects of A immunization (AN1792) inpatients with AD in an interrupted trial Neurology 2005641553ndash1562
19 Ostrowitzki S Lasser RA Dorflinger E et al A phase III randomized trial of gante-nerumab in prodromal Alzheimerrsquos disease Alzheimers Res Ther 2017995
20 Salloway S Sperling R Fox NC et al Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370322ndash333
21 Doody RS Raman R Farlow M et al A phase 3 trial of semagacestat for treatment ofAlzheimerrsquos disease N Engl J Med 2013369341ndash350
22 Doody RS Thomas RG Farlow M et al Phase 3 trials of solanezumab for mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370311ndash321
23 Honig LS Vellas B Woodward M et al Trial of solanezumab for mild dementia dueto Alzheimerrsquos disease N Engl J Med 2018378321ndash330
24 Cali I Cohen ML Haїk S et al Iatrogenic Creutzfeldt-Jakob disease with amyloid-βpathology an international study Acta Neuropathol Commun 201865
25 Howick J Glasziou P Aronson JK The evolution of evidence hierarchies what canBradford Hillrsquos ldquoguidelines for causationrdquo contribute J R SocMed 2009102186ndash194
26 Roberts RF Wade-Martins R Alegre-Abarrategui J Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinsonrsquos diseasebrain Brain 20151381642ndash1657
27 Blennow K Mattsson N Scholl M Hansson O Zetterberg H Amyloid biomarkers inAlzheimerrsquos disease Trends Pharmacol Sci 201536297ndash309
28 Hogarth P Gregory A Kruer MC et al New NBIA subtype genetic clinical path-ologic and radiographic features of MPAN Neurology 201380268ndash275
29 Olanow CW Brundin P Parkinsonrsquos disease and alpha synuclein is Parkinsonrsquosdisease a prion-like disorder Mov Disord 20132831ndash40
30 Winder-Rhodes SE Garcia-Reitbock P Ban M et al Genetic and pathological linksbetween Parkinsonrsquos disease and the lysosomal disorder Sanfilippo syndrome MovDisord 201227312ndash315
31 Brenowitz WD Keene CD Hawes SE et al Alzheimerrsquos disease neuropathologicchange Lewy body disease and vascular brain injury in clinic- and community-basedsamples Neurobiol Aging 20175383ndash92
32 Irwin DJ Grossman M Weintraub D et al Neuropathological and genetic correlatesof survival and dementia onset in synucleinopathies a retrospective analysis LancetNeurol 20171655ndash65
33 Crystal H Dickson D Fuld P et al Clinico-pathologic studies in dementia non-demented subjects with pathologically confirmed Alzheimerrsquos disease Neurology1988381682ndash1687
34 Polvikoski T Sulkava R Myllykangas L et al Prevalence of Alzheimerrsquos disease in veryelderly people a prospective neuropathological study Neurology 2001561690ndash1696
35 MarkesberyWR Jicha GA Liu H Schmitt FA Lewy body pathology in normal elderlysubjects J Neuropathol Exp Neurol 200968816ndash822
36 Balasubramanian AB Kawas CH Peltz CB Brookmeyer R Corrada MM Alzheimerdisease pathology and longitudinal cognitive performance in the oldest-old with nodementia Neurology 201279915ndash921
37 Silver MH Newell K Brady C Hedley-White ET Perls TT Distinguishing betweenneurodegenerative disease and disease-free aging correlating neuropsychologicalevaluations and neuropathological studies in centenarians Psychosom Med 200264493ndash501
38 Berlau DJ Corrada MM Head E Kawas CH APOE 2 is associated with intactcognition but increased Alzheimer pathology in the oldest old Neurology 200972829ndash834
39 Kawas CH Kim RC Sonnen JA Bullain SS Trieu T Corrada MM Multiple pa-thologies are common and related to dementia in the oldest-old Neurology 201585535ndash542
40 Espay AJ Schwarzschild MA Tanner CM et al Biomarker-driven phenotyping inParkinsonrsquos disease a translational missing link in disease-modifying clinical trialsMov Disord 201732319ndash324Data available from Dryad (Additional References References e1ndashe44)doiorg105061dryadg1nq02r
Did You Knowhellip
hellipyou can browse by subspecialty topics on Neurologyorg
Go to Neurologyorg and click on ldquoTopicsrdquo in the top navigation bar
Share Your Artistic Expressions in Neurology lsquoVisionsrsquo
AAN members are urged to submit medically or scientifically related artistic images such as photographs photomicrographsand paintings to the ldquoVisionsrdquo section of Neurologyreg These images are creative in nature rather than the medically instructiveimages published in the NeuroImages section The image or series of up to six images may be black and white or color andmust fit into one published journal page Accompanying description should be 100 words or less the title should be a maximumof 96 characters including spaces and punctuation
Please access the Author Center at NPuborgauthors for full submission information
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 337
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
DOI 101212WNL0000000000006926201992329-337 Neurology
Alberto J Espay Joaquin A Vizcarra Luca Marsili et al diseases
Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer
This information is current as of February 11 2019
ServicesUpdated Information amp
httpnneurologyorgcontent927329fullincluding high resolution figures can be found at
References httpnneurologyorgcontent927329fullref-list-1
This article cites 38 articles 10 of which you can access for free at
Citations httpnneurologyorgcontent927329fullotherarticles
This article has been cited by 3 HighWire-hosted articles
Permissions amp Licensing
httpwwwneurologyorgaboutabout_the_journalpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpnneurologyorgsubscribersadvertiseInformation about ordering reprints can be found online
rights reserved Print ISSN 0028-3878 Online ISSN 1526-632X1951 it is now a weekly with 48 issues per year Copyright copy 2019 American Academy of Neurology All
reg is the official journal of the American Academy of Neurology Published continuously sinceNeurology

in Down syndrome or amyloid precursor protein [APP] genemutations and duplication)5 in which these protein aggre-gates are truly pathogenic it is plausible that accumulationof α-synuclein or of Aβtau enhances other primary patho-genic molecular mechanisms of neurodegeneration in spo-radic forms of PD and AD (figure 3A) Putatively a prion-likepropagation of α-synuclein fibrils between adjacent cells29 fallsinto this category without requiring that oligomers and fibrilsact as the initial pathogenic event This model accepts het-erogeneous upstream pathogenic events but relies on solubleproteins (α-synuclein Aβ) misfolding and aggregating asa convergent driver of cellular death In this model misfoldedvariants of α-synuclein and Aβ are suitable for biomarkerdevelopment and therapeutic targeting
Alternative model 2 Protein aggregation asepiphenomena of multiple diseasesAccording to this model a range of molecular abnormalitieslead to cells being unable to adequately degrade misfoldedproteins which aggregate into α-synuclein fibrils in Lewybodies (figure 3B or from misfolded amyloid precursorprotein into misfolded oligomers then fibrils then plaques inAD) These protein aggregates become byproducts (commondenominators or epiphenomena) of several pathogenicpathways and themselves exert no pathogenic or protectiverole According to this model different underlying molecularabnormalities remain pathogenic of different molecular dis-eases their relationship to one another rests only in the typeof protein they cannot properly recycle
Alternative model 3 Protein aggregation asprotective mechanism of multiple diseasesIn this model molecular abnormalities affect the cellularmachinery that normally degrades proteins These misfoldedproteins are sequestered into Lewy bodies as a mechanism topromote maintenance of neuronal and synaptic function de-spite coexistent and actively disrupting molecular abnormal-ities (figure 3C) Each molecular abnormality is pathogenic toa specific disease which shares with others only the type ofprotein actively sequestered as a protective mechanism Suchprotective sequestration of proteins could allow neurons tofunction for decades before becoming overwhelmed delayingthe progression of neurologic disease In this model targetingsoluble oligomers or fibrils (eg using anti-aggregant thera-pies or possibly passive or active immunotherapy) mightpotentially be deleterious if the toxicity of any of the proteinspecies is outweighed by their protective role in the humanbrain under molecular stress
Implications for biomarkers andclinical trialsHypothesis 1 Biomarkers of proteinaggregates are relevant to common pathologybut not to subgroup pathogenesisBased on the challenges discussed above abnormal proteinaggregates (pathology) may represent downstream events
unhelpful to classify PD and AD with pathogenic relevanceThus far biomarker development efforts have been based ondisease models of clinico-pathologic convergence One suchconvergence biomarker is [123I]-FP-CIT SPECT imaging forstriatal dopamine deficiency It is positive in virtually everyonewith PD Because biomarker discovery has been based onclinically defined cohorts (eg tremor-dominant PD vs posturalinstability gait disorder type) significant results within andbetween cohorts have substantial overlap40
Hypothesis 2 Disease-modifying treatmentstargeting oligomers or fibrils might be futile ordeleterious because these proteins areepiphenomena or protective in the humanbrain under molecular stressTargeting these proteins across sporadic or otherwise non-genetic disease forms is unlikely to slow progression of thesedisorders nomatter how early the treatments are implemented(ie prodromal stage) unless protein aggregation acts as anaccelerator of pathology as suggested in alternative model 1The corollary is that applying molecular therapies aimed atachieving neuroprotection to clinically (pathologically) de-fined but not molecularly subtyped populations will remainunlikely to succeed The history of medicine has repeatedlyshown that single (untreatable) clinical diseases are oftenreplaced by several (treatable) molecular diseases As a conse-quence treatments designed to target specific mechanismswork best in certain disease subtypes but not in others
Relinquishing reductionism (the clinically heterogeneous butsingle-disease concepts for AD and PD) will require acceptingthat sophisticated data analyses of large cohorts will not becapable of identifying molecular disease subtypes as the anal-yses cannot overcome the fundamental flaw of existent datasetscohorts invariably assembled based on strict clinical criteria Bigdata may not easily be converted into good data (for the pur-poses of assisting precision medicine) if the data are based onbinary definitions of AD vs control and PD vs control Impor-tant insights may be derived on a smaller scale from studyingrare genetic forms of disease (eg PD-LRRK2 PD-GBA AD-PS1) for which targeted therapeutic efforts may be most likelyto succeedmdashin these subtypes On a wider scale molecularlytargetable neurodegenerative diseases may be best identifiedfrom population-based studies applying non-hypothesis-basedanalytic approaches to biospecimens from large cohorts of agingpeople carefully characterized across a spectrum of brainfunctions and deficits Thus aging cohorts ought to includeatypical clinical presentations and other ostensibly unrelatedneurologic disorders beyond the current criteria for PD andADThe analyses will need to be anchored on biological signals noton clinical phenotypes or clinical criteria and account for ge-netic and lifelong biochemical and environmental factors Mo-lecular subtypes of disease thus identified may or may not fallinto standard clinical classifications
The first proven neuroprotective therapy might only work infewer than 5 of everyone currently subsumed as ldquoADrdquo or
334 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
ldquoPDrdquo (eg GZSAR402671 in early-stage PD-GBA [Clin-icalTrialsgov Identifier NCT02906020]) or in specific at-risk populations (eg solanezumab or gantenerumab forhealthy individuals with an autosomal dominant AD-causingmutation [Dominantly Inherited Alzheimer Network TrialClinicalTrialsgov Identifier NCT01760005]) Such disease-modifying treatment in neurodegenerative disease will helpusher in the biomarker-driven disease subtyping strategy longadopted by oncology and other fields of medicine
AcknowledgmentThe authors thank the patients their parentscaregivers andhealth care professionals who participated in this studyincluding Dean Miller Ruth Wake Dionne Moat RobertMuni Lofra Teresinha Evangelista MD Nikoletta Nikolenko
MD Namita Goyal MD Sonia Nayyar MD CarolinePeyronnard MD Tim Lai MD Monica Lavian DO BrianMinton BS Patrick Tierney RPT Denise Davis RPT CathyChou RPT Nick Higgins and Ed Connor MD
Study fundingNo targeted funding reported
DisclosureThe authors report no disclosures relevant to the manuscriptGo to NeurologyorgN for full disclosures
Publication historyReceived by Neurology July 6 2018 Accepted in final form December14 2018
Figure 3 Alternative models of protein aggregation in Parkinson disease (multiple disease model)
Abnormal soluble oligomers and fibrils ofα-synuclein (α-syn) while not directly pathogenic act as accelerators of neurodegeneration (ldquofueling the firerdquo) due toearly pathogenic molecular abnormalities each representingmolecularly distinct diseases (A model 1) Alternatively abnormal soluble oligomers and fibrilsof α-synuclein aggregate into Lewy bodies as byproducts of earlier pathogenic molecular mechanisms without directly affecting the neurodegenerativeprocess brought on by eachmolecular disease (Bmodel 2) Finally α-synuclein aggregates into Lewy bodies as amechanism to protect the neuron from toxicprotein species or from the biological dysfunction that may have generated the formation of toxic species ldquocoolingrdquo progression of cell degeneration underbiological stress (C model 3) Note that each molecularly defined disease has a different time to death collectively time to neuronal death is longest indiseases corresponding to model C
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 335
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
References1 Gwinn K David KK Swanson-Fischer C et al Parkinsonrsquos disease biomarkers
perspective from the NINDS Parkinsonrsquos Disease Biomarkers Program BiomarkMed201711451ndash473
2 Mollenhauer B Caspell-Garcia CJ Coffey CS et al Longitudinal CSF biomarkers inpatients with early Parkinson disease and healthy controls Neurology 2017891959ndash1969
3 Payami H The emerging science of precision medicine and pharmacogenomics forParkinsonrsquos disease Mov Disord 2017321139ndash1146
4 Chartier-Harlin M-C Kachergus J Roumier C et al α-Synuclein locus duplication asa cause of familial Parkinsonrsquos disease Lancet 20043641167ndash1169
5 Sleegers K BrouwersNGijselinck I et al APPduplication is sufficient to cause early onsetAlzheimerrsquos dementia with cerebral amyloid angiopathy Brain 20061292977ndash2983
6 Bezard E Yue Z Kirik D Spillantini MG Animal models of Parkinsonrsquos disease limitsand relevance to neuroprotection studies Mov Disord 20132861ndash70
7 Bloom GS Amyloid-β and tau JAMA Neurol 2014715058 Bradford-Hill A The environment and disease association or causation Proc R Soc
Med 196558295ndash3009 Fedak KM Bernal A Capshaw ZA Gross S Applying the Bradford Hill criteria in the
21st century how data integration has changed causal inference in molecular epi-demiology Emerg Themes Epidemiol Biomed Cent 20151214
Appendix Authors
Name Location Role Contribution
Alberto JEspay MD MSc
University ofCincinnati OH
CorrespondingAuthor
Conception ofresearchprojectliteraturesearch writingof the firstmanuscriptdraft
Joaquin AVizcarra MD
University ofCincinnati OH
Author Literaturesearch writingof the firstmanuscriptdraft
Luca MarsiliMD PhD
University ofCincinnati
Author Literaturesearch reviewand critique ofthe manuscript
Anthony ELang MDFRCPC
University ofTorontoCanada
Author Review andcritique of themanuscript
David K SimonMD PhD
HarvardMedical SchoolBoston MA
Author Review andcritique of themanuscript
AristideMerola MDPhD
University ofCincinnati OH
Author Review andcritique of themanuscript
Keith AJosephs MDMST MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
AlfonsoFasano MDPhD
University ofTorontoCanada
Author Review andcritique of themanuscript
FrancescaMorgante MDPhD
University ofMessina Italyand St GeorgersquosUniversity ofLondon UK
Author Review andcritique of themanuscript
Rodolfo SavicaMD MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
J TimothyGreenamyreMD PhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
FrancescaCambi MDPhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
Tritia RYamasaki MDPhD
University ofKentuckyLexington
Author Review andcritique of themanuscript
Caroline MTanner MDPhD
University ofCaliforniandashSanFrancisco
Author Review andcritique of themanuscript
Ziv Gan-OrMD PhD
McGillUniversityMontrealCanada
Author Review andcritique of themanuscript
Irene LitvanMD
UC San DiegoCA
Author Review andcritique of themanuscript
Appendix (continued)
Name Location Role Contribution
Ignacio FMata PhD
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Cyrus PZabetian MDMS
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Patrik BrundinMD PhD
Van AndelResearchInstitute GrandRapids MI
Author Review andcritique of themanuscript
Hubert HFernandez MD
ClevelandClinic OH
Author Review andcritique of themanuscript
David GStandaert MDPhD
University ofAlabama atBirmingham
Author Review andcritique of themanuscript
Marcelo AKauffman MDPhD
UBA andUniversidadAustral-CONICETBuenos AiresArgentina
Author Review andcritique of themanuscript
Michael ASchwarzschildMD PhD
MassachusettsGeneralHospitalBoston
Author Review andcritique of themanuscript
S Pablo SardiPharmD PhD
Sanofi Author Review andcritique of themanuscript
Todd ShererPhD
Michael J FoxFoundation forParkinsonrsquosResearch NewYork NY
Author Review andcritique of themanuscript
George PerryPhD
University ofTexas at SanAntonio
Author Review andcritique of themanuscript
James BLeverenz MD
ClevelandClinic OH
Author Literaturesearch reviewand critique ofthe manuscript
336 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
10 Moher D Liberati A Tetzlaff J Altman DG PRISMA Group Preferred reportingitems for systematic reviews and meta-analyses the PRISMA statement Ann InternMed 2009151264ndash269 W64
11 PoeweW Seppi K Tanner CM et al Parkinson disease Nat Rev Dis Primers 2017317013
12 Querfurth HW LaFerla FM Alzheimerrsquos disease N Engl J Med 2010362329ndash34413 NHLBI Research Triangle Institute International National Heart Lung and Blood
Institute quality appraisal tools [online] Available at httpswwwnhlbinihgovhealth-topicsstudy-quality-assessment-tools Accessed December 15 2017
14 Higgins JPT Green S eds Cochrane Handbook for Systematic Reviews of Inter-ventions [online] 510 The Cochrane Collaboration 2011 Available at httpstrainingcochraneorghandbook Accessed December 16 2017
15 Vos SJ Xiong C Visser PJ et al Preclinical Alzheimerrsquos disease and its outcomea longitudinal cohort study Lancet Neurol 201312957ndash965
16 Van Harten AC Smits LL Teunissen CE et al Preclinical AD predicts decline inmemory and executive functions in subjective complaints Neurology 2013811409ndash1416
17 Burnham SC Bourgeat P Dore V et al Clinical and cognitive trajectories in cogni-tively healthy elderly individuals with suspected non-Alzheimerrsquos disease patho-physiology (SNAP) or Alzheimerrsquos disease pathology a longitudinal study LancetNeurol 2016151044ndash1053
18 Gilman S Koller M Black RS et al Clinical effects of A immunization (AN1792) inpatients with AD in an interrupted trial Neurology 2005641553ndash1562
19 Ostrowitzki S Lasser RA Dorflinger E et al A phase III randomized trial of gante-nerumab in prodromal Alzheimerrsquos disease Alzheimers Res Ther 2017995
20 Salloway S Sperling R Fox NC et al Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370322ndash333
21 Doody RS Raman R Farlow M et al A phase 3 trial of semagacestat for treatment ofAlzheimerrsquos disease N Engl J Med 2013369341ndash350
22 Doody RS Thomas RG Farlow M et al Phase 3 trials of solanezumab for mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370311ndash321
23 Honig LS Vellas B Woodward M et al Trial of solanezumab for mild dementia dueto Alzheimerrsquos disease N Engl J Med 2018378321ndash330
24 Cali I Cohen ML Haїk S et al Iatrogenic Creutzfeldt-Jakob disease with amyloid-βpathology an international study Acta Neuropathol Commun 201865
25 Howick J Glasziou P Aronson JK The evolution of evidence hierarchies what canBradford Hillrsquos ldquoguidelines for causationrdquo contribute J R SocMed 2009102186ndash194
26 Roberts RF Wade-Martins R Alegre-Abarrategui J Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinsonrsquos diseasebrain Brain 20151381642ndash1657
27 Blennow K Mattsson N Scholl M Hansson O Zetterberg H Amyloid biomarkers inAlzheimerrsquos disease Trends Pharmacol Sci 201536297ndash309
28 Hogarth P Gregory A Kruer MC et al New NBIA subtype genetic clinical path-ologic and radiographic features of MPAN Neurology 201380268ndash275
29 Olanow CW Brundin P Parkinsonrsquos disease and alpha synuclein is Parkinsonrsquosdisease a prion-like disorder Mov Disord 20132831ndash40
30 Winder-Rhodes SE Garcia-Reitbock P Ban M et al Genetic and pathological linksbetween Parkinsonrsquos disease and the lysosomal disorder Sanfilippo syndrome MovDisord 201227312ndash315
31 Brenowitz WD Keene CD Hawes SE et al Alzheimerrsquos disease neuropathologicchange Lewy body disease and vascular brain injury in clinic- and community-basedsamples Neurobiol Aging 20175383ndash92
32 Irwin DJ Grossman M Weintraub D et al Neuropathological and genetic correlatesof survival and dementia onset in synucleinopathies a retrospective analysis LancetNeurol 20171655ndash65
33 Crystal H Dickson D Fuld P et al Clinico-pathologic studies in dementia non-demented subjects with pathologically confirmed Alzheimerrsquos disease Neurology1988381682ndash1687
34 Polvikoski T Sulkava R Myllykangas L et al Prevalence of Alzheimerrsquos disease in veryelderly people a prospective neuropathological study Neurology 2001561690ndash1696
35 MarkesberyWR Jicha GA Liu H Schmitt FA Lewy body pathology in normal elderlysubjects J Neuropathol Exp Neurol 200968816ndash822
36 Balasubramanian AB Kawas CH Peltz CB Brookmeyer R Corrada MM Alzheimerdisease pathology and longitudinal cognitive performance in the oldest-old with nodementia Neurology 201279915ndash921
37 Silver MH Newell K Brady C Hedley-White ET Perls TT Distinguishing betweenneurodegenerative disease and disease-free aging correlating neuropsychologicalevaluations and neuropathological studies in centenarians Psychosom Med 200264493ndash501
38 Berlau DJ Corrada MM Head E Kawas CH APOE 2 is associated with intactcognition but increased Alzheimer pathology in the oldest old Neurology 200972829ndash834
39 Kawas CH Kim RC Sonnen JA Bullain SS Trieu T Corrada MM Multiple pa-thologies are common and related to dementia in the oldest-old Neurology 201585535ndash542
40 Espay AJ Schwarzschild MA Tanner CM et al Biomarker-driven phenotyping inParkinsonrsquos disease a translational missing link in disease-modifying clinical trialsMov Disord 201732319ndash324Data available from Dryad (Additional References References e1ndashe44)doiorg105061dryadg1nq02r
Did You Knowhellip
hellipyou can browse by subspecialty topics on Neurologyorg
Go to Neurologyorg and click on ldquoTopicsrdquo in the top navigation bar
Share Your Artistic Expressions in Neurology lsquoVisionsrsquo
AAN members are urged to submit medically or scientifically related artistic images such as photographs photomicrographsand paintings to the ldquoVisionsrdquo section of Neurologyreg These images are creative in nature rather than the medically instructiveimages published in the NeuroImages section The image or series of up to six images may be black and white or color andmust fit into one published journal page Accompanying description should be 100 words or less the title should be a maximumof 96 characters including spaces and punctuation
Please access the Author Center at NPuborgauthors for full submission information
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 337
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
DOI 101212WNL0000000000006926201992329-337 Neurology
Alberto J Espay Joaquin A Vizcarra Luca Marsili et al diseases
Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer
This information is current as of February 11 2019
ServicesUpdated Information amp
httpnneurologyorgcontent927329fullincluding high resolution figures can be found at
References httpnneurologyorgcontent927329fullref-list-1
This article cites 38 articles 10 of which you can access for free at
Citations httpnneurologyorgcontent927329fullotherarticles
This article has been cited by 3 HighWire-hosted articles
Permissions amp Licensing
httpwwwneurologyorgaboutabout_the_journalpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpnneurologyorgsubscribersadvertiseInformation about ordering reprints can be found online
rights reserved Print ISSN 0028-3878 Online ISSN 1526-632X1951 it is now a weekly with 48 issues per year Copyright copy 2019 American Academy of Neurology All
reg is the official journal of the American Academy of Neurology Published continuously sinceNeurology
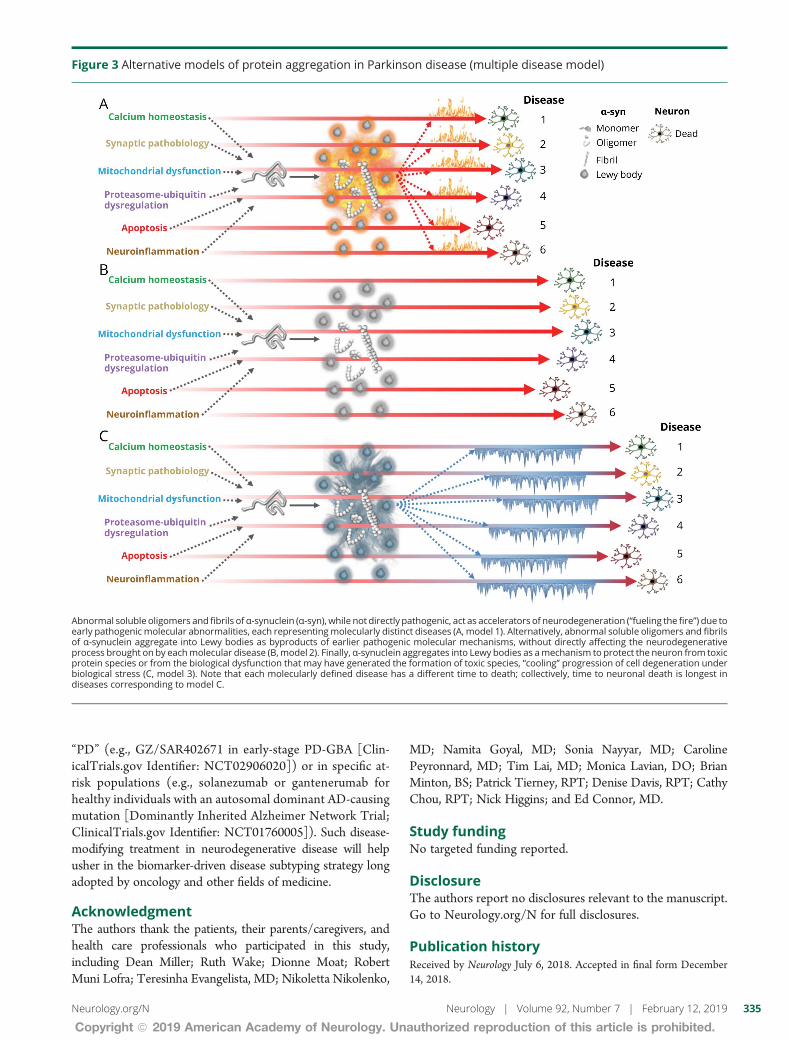
ldquoPDrdquo (eg GZSAR402671 in early-stage PD-GBA [Clin-icalTrialsgov Identifier NCT02906020]) or in specific at-risk populations (eg solanezumab or gantenerumab forhealthy individuals with an autosomal dominant AD-causingmutation [Dominantly Inherited Alzheimer Network TrialClinicalTrialsgov Identifier NCT01760005]) Such disease-modifying treatment in neurodegenerative disease will helpusher in the biomarker-driven disease subtyping strategy longadopted by oncology and other fields of medicine
AcknowledgmentThe authors thank the patients their parentscaregivers andhealth care professionals who participated in this studyincluding Dean Miller Ruth Wake Dionne Moat RobertMuni Lofra Teresinha Evangelista MD Nikoletta Nikolenko
MD Namita Goyal MD Sonia Nayyar MD CarolinePeyronnard MD Tim Lai MD Monica Lavian DO BrianMinton BS Patrick Tierney RPT Denise Davis RPT CathyChou RPT Nick Higgins and Ed Connor MD
Study fundingNo targeted funding reported
DisclosureThe authors report no disclosures relevant to the manuscriptGo to NeurologyorgN for full disclosures
Publication historyReceived by Neurology July 6 2018 Accepted in final form December14 2018
Figure 3 Alternative models of protein aggregation in Parkinson disease (multiple disease model)
Abnormal soluble oligomers and fibrils ofα-synuclein (α-syn) while not directly pathogenic act as accelerators of neurodegeneration (ldquofueling the firerdquo) due toearly pathogenic molecular abnormalities each representingmolecularly distinct diseases (A model 1) Alternatively abnormal soluble oligomers and fibrilsof α-synuclein aggregate into Lewy bodies as byproducts of earlier pathogenic molecular mechanisms without directly affecting the neurodegenerativeprocess brought on by eachmolecular disease (Bmodel 2) Finally α-synuclein aggregates into Lewy bodies as amechanism to protect the neuron from toxicprotein species or from the biological dysfunction that may have generated the formation of toxic species ldquocoolingrdquo progression of cell degeneration underbiological stress (C model 3) Note that each molecularly defined disease has a different time to death collectively time to neuronal death is longest indiseases corresponding to model C
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 335
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
References1 Gwinn K David KK Swanson-Fischer C et al Parkinsonrsquos disease biomarkers
perspective from the NINDS Parkinsonrsquos Disease Biomarkers Program BiomarkMed201711451ndash473
2 Mollenhauer B Caspell-Garcia CJ Coffey CS et al Longitudinal CSF biomarkers inpatients with early Parkinson disease and healthy controls Neurology 2017891959ndash1969
3 Payami H The emerging science of precision medicine and pharmacogenomics forParkinsonrsquos disease Mov Disord 2017321139ndash1146
4 Chartier-Harlin M-C Kachergus J Roumier C et al α-Synuclein locus duplication asa cause of familial Parkinsonrsquos disease Lancet 20043641167ndash1169
5 Sleegers K BrouwersNGijselinck I et al APPduplication is sufficient to cause early onsetAlzheimerrsquos dementia with cerebral amyloid angiopathy Brain 20061292977ndash2983
6 Bezard E Yue Z Kirik D Spillantini MG Animal models of Parkinsonrsquos disease limitsand relevance to neuroprotection studies Mov Disord 20132861ndash70
7 Bloom GS Amyloid-β and tau JAMA Neurol 2014715058 Bradford-Hill A The environment and disease association or causation Proc R Soc
Med 196558295ndash3009 Fedak KM Bernal A Capshaw ZA Gross S Applying the Bradford Hill criteria in the
21st century how data integration has changed causal inference in molecular epi-demiology Emerg Themes Epidemiol Biomed Cent 20151214
Appendix Authors
Name Location Role Contribution
Alberto JEspay MD MSc
University ofCincinnati OH
CorrespondingAuthor
Conception ofresearchprojectliteraturesearch writingof the firstmanuscriptdraft
Joaquin AVizcarra MD
University ofCincinnati OH
Author Literaturesearch writingof the firstmanuscriptdraft
Luca MarsiliMD PhD
University ofCincinnati
Author Literaturesearch reviewand critique ofthe manuscript
Anthony ELang MDFRCPC
University ofTorontoCanada
Author Review andcritique of themanuscript
David K SimonMD PhD
HarvardMedical SchoolBoston MA
Author Review andcritique of themanuscript
AristideMerola MDPhD
University ofCincinnati OH
Author Review andcritique of themanuscript
Keith AJosephs MDMST MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
AlfonsoFasano MDPhD
University ofTorontoCanada
Author Review andcritique of themanuscript
FrancescaMorgante MDPhD
University ofMessina Italyand St GeorgersquosUniversity ofLondon UK
Author Review andcritique of themanuscript
Rodolfo SavicaMD MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
J TimothyGreenamyreMD PhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
FrancescaCambi MDPhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
Tritia RYamasaki MDPhD
University ofKentuckyLexington
Author Review andcritique of themanuscript
Caroline MTanner MDPhD
University ofCaliforniandashSanFrancisco
Author Review andcritique of themanuscript
Ziv Gan-OrMD PhD
McGillUniversityMontrealCanada
Author Review andcritique of themanuscript
Irene LitvanMD
UC San DiegoCA
Author Review andcritique of themanuscript
Appendix (continued)
Name Location Role Contribution
Ignacio FMata PhD
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Cyrus PZabetian MDMS
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Patrik BrundinMD PhD
Van AndelResearchInstitute GrandRapids MI
Author Review andcritique of themanuscript
Hubert HFernandez MD
ClevelandClinic OH
Author Review andcritique of themanuscript
David GStandaert MDPhD
University ofAlabama atBirmingham
Author Review andcritique of themanuscript
Marcelo AKauffman MDPhD
UBA andUniversidadAustral-CONICETBuenos AiresArgentina
Author Review andcritique of themanuscript
Michael ASchwarzschildMD PhD
MassachusettsGeneralHospitalBoston
Author Review andcritique of themanuscript
S Pablo SardiPharmD PhD
Sanofi Author Review andcritique of themanuscript
Todd ShererPhD
Michael J FoxFoundation forParkinsonrsquosResearch NewYork NY
Author Review andcritique of themanuscript
George PerryPhD
University ofTexas at SanAntonio
Author Review andcritique of themanuscript
James BLeverenz MD
ClevelandClinic OH
Author Literaturesearch reviewand critique ofthe manuscript
336 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
10 Moher D Liberati A Tetzlaff J Altman DG PRISMA Group Preferred reportingitems for systematic reviews and meta-analyses the PRISMA statement Ann InternMed 2009151264ndash269 W64
11 PoeweW Seppi K Tanner CM et al Parkinson disease Nat Rev Dis Primers 2017317013
12 Querfurth HW LaFerla FM Alzheimerrsquos disease N Engl J Med 2010362329ndash34413 NHLBI Research Triangle Institute International National Heart Lung and Blood
Institute quality appraisal tools [online] Available at httpswwwnhlbinihgovhealth-topicsstudy-quality-assessment-tools Accessed December 15 2017
14 Higgins JPT Green S eds Cochrane Handbook for Systematic Reviews of Inter-ventions [online] 510 The Cochrane Collaboration 2011 Available at httpstrainingcochraneorghandbook Accessed December 16 2017
15 Vos SJ Xiong C Visser PJ et al Preclinical Alzheimerrsquos disease and its outcomea longitudinal cohort study Lancet Neurol 201312957ndash965
16 Van Harten AC Smits LL Teunissen CE et al Preclinical AD predicts decline inmemory and executive functions in subjective complaints Neurology 2013811409ndash1416
17 Burnham SC Bourgeat P Dore V et al Clinical and cognitive trajectories in cogni-tively healthy elderly individuals with suspected non-Alzheimerrsquos disease patho-physiology (SNAP) or Alzheimerrsquos disease pathology a longitudinal study LancetNeurol 2016151044ndash1053
18 Gilman S Koller M Black RS et al Clinical effects of A immunization (AN1792) inpatients with AD in an interrupted trial Neurology 2005641553ndash1562
19 Ostrowitzki S Lasser RA Dorflinger E et al A phase III randomized trial of gante-nerumab in prodromal Alzheimerrsquos disease Alzheimers Res Ther 2017995
20 Salloway S Sperling R Fox NC et al Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370322ndash333
21 Doody RS Raman R Farlow M et al A phase 3 trial of semagacestat for treatment ofAlzheimerrsquos disease N Engl J Med 2013369341ndash350
22 Doody RS Thomas RG Farlow M et al Phase 3 trials of solanezumab for mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370311ndash321
23 Honig LS Vellas B Woodward M et al Trial of solanezumab for mild dementia dueto Alzheimerrsquos disease N Engl J Med 2018378321ndash330
24 Cali I Cohen ML Haїk S et al Iatrogenic Creutzfeldt-Jakob disease with amyloid-βpathology an international study Acta Neuropathol Commun 201865
25 Howick J Glasziou P Aronson JK The evolution of evidence hierarchies what canBradford Hillrsquos ldquoguidelines for causationrdquo contribute J R SocMed 2009102186ndash194
26 Roberts RF Wade-Martins R Alegre-Abarrategui J Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinsonrsquos diseasebrain Brain 20151381642ndash1657
27 Blennow K Mattsson N Scholl M Hansson O Zetterberg H Amyloid biomarkers inAlzheimerrsquos disease Trends Pharmacol Sci 201536297ndash309
28 Hogarth P Gregory A Kruer MC et al New NBIA subtype genetic clinical path-ologic and radiographic features of MPAN Neurology 201380268ndash275
29 Olanow CW Brundin P Parkinsonrsquos disease and alpha synuclein is Parkinsonrsquosdisease a prion-like disorder Mov Disord 20132831ndash40
30 Winder-Rhodes SE Garcia-Reitbock P Ban M et al Genetic and pathological linksbetween Parkinsonrsquos disease and the lysosomal disorder Sanfilippo syndrome MovDisord 201227312ndash315
31 Brenowitz WD Keene CD Hawes SE et al Alzheimerrsquos disease neuropathologicchange Lewy body disease and vascular brain injury in clinic- and community-basedsamples Neurobiol Aging 20175383ndash92
32 Irwin DJ Grossman M Weintraub D et al Neuropathological and genetic correlatesof survival and dementia onset in synucleinopathies a retrospective analysis LancetNeurol 20171655ndash65
33 Crystal H Dickson D Fuld P et al Clinico-pathologic studies in dementia non-demented subjects with pathologically confirmed Alzheimerrsquos disease Neurology1988381682ndash1687
34 Polvikoski T Sulkava R Myllykangas L et al Prevalence of Alzheimerrsquos disease in veryelderly people a prospective neuropathological study Neurology 2001561690ndash1696
35 MarkesberyWR Jicha GA Liu H Schmitt FA Lewy body pathology in normal elderlysubjects J Neuropathol Exp Neurol 200968816ndash822
36 Balasubramanian AB Kawas CH Peltz CB Brookmeyer R Corrada MM Alzheimerdisease pathology and longitudinal cognitive performance in the oldest-old with nodementia Neurology 201279915ndash921
37 Silver MH Newell K Brady C Hedley-White ET Perls TT Distinguishing betweenneurodegenerative disease and disease-free aging correlating neuropsychologicalevaluations and neuropathological studies in centenarians Psychosom Med 200264493ndash501
38 Berlau DJ Corrada MM Head E Kawas CH APOE 2 is associated with intactcognition but increased Alzheimer pathology in the oldest old Neurology 200972829ndash834
39 Kawas CH Kim RC Sonnen JA Bullain SS Trieu T Corrada MM Multiple pa-thologies are common and related to dementia in the oldest-old Neurology 201585535ndash542
40 Espay AJ Schwarzschild MA Tanner CM et al Biomarker-driven phenotyping inParkinsonrsquos disease a translational missing link in disease-modifying clinical trialsMov Disord 201732319ndash324Data available from Dryad (Additional References References e1ndashe44)doiorg105061dryadg1nq02r
Did You Knowhellip
hellipyou can browse by subspecialty topics on Neurologyorg
Go to Neurologyorg and click on ldquoTopicsrdquo in the top navigation bar
Share Your Artistic Expressions in Neurology lsquoVisionsrsquo
AAN members are urged to submit medically or scientifically related artistic images such as photographs photomicrographsand paintings to the ldquoVisionsrdquo section of Neurologyreg These images are creative in nature rather than the medically instructiveimages published in the NeuroImages section The image or series of up to six images may be black and white or color andmust fit into one published journal page Accompanying description should be 100 words or less the title should be a maximumof 96 characters including spaces and punctuation
Please access the Author Center at NPuborgauthors for full submission information
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 337
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
DOI 101212WNL0000000000006926201992329-337 Neurology
Alberto J Espay Joaquin A Vizcarra Luca Marsili et al diseases
Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer
This information is current as of February 11 2019
ServicesUpdated Information amp
httpnneurologyorgcontent927329fullincluding high resolution figures can be found at
References httpnneurologyorgcontent927329fullref-list-1
This article cites 38 articles 10 of which you can access for free at
Citations httpnneurologyorgcontent927329fullotherarticles
This article has been cited by 3 HighWire-hosted articles
Permissions amp Licensing
httpwwwneurologyorgaboutabout_the_journalpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpnneurologyorgsubscribersadvertiseInformation about ordering reprints can be found online
rights reserved Print ISSN 0028-3878 Online ISSN 1526-632X1951 it is now a weekly with 48 issues per year Copyright copy 2019 American Academy of Neurology All
reg is the official journal of the American Academy of Neurology Published continuously sinceNeurology
References1 Gwinn K David KK Swanson-Fischer C et al Parkinsonrsquos disease biomarkers
perspective from the NINDS Parkinsonrsquos Disease Biomarkers Program BiomarkMed201711451ndash473
2 Mollenhauer B Caspell-Garcia CJ Coffey CS et al Longitudinal CSF biomarkers inpatients with early Parkinson disease and healthy controls Neurology 2017891959ndash1969
3 Payami H The emerging science of precision medicine and pharmacogenomics forParkinsonrsquos disease Mov Disord 2017321139ndash1146
4 Chartier-Harlin M-C Kachergus J Roumier C et al α-Synuclein locus duplication asa cause of familial Parkinsonrsquos disease Lancet 20043641167ndash1169
5 Sleegers K BrouwersNGijselinck I et al APPduplication is sufficient to cause early onsetAlzheimerrsquos dementia with cerebral amyloid angiopathy Brain 20061292977ndash2983
6 Bezard E Yue Z Kirik D Spillantini MG Animal models of Parkinsonrsquos disease limitsand relevance to neuroprotection studies Mov Disord 20132861ndash70
7 Bloom GS Amyloid-β and tau JAMA Neurol 2014715058 Bradford-Hill A The environment and disease association or causation Proc R Soc
Med 196558295ndash3009 Fedak KM Bernal A Capshaw ZA Gross S Applying the Bradford Hill criteria in the
21st century how data integration has changed causal inference in molecular epi-demiology Emerg Themes Epidemiol Biomed Cent 20151214
Appendix Authors
Name Location Role Contribution
Alberto JEspay MD MSc
University ofCincinnati OH
CorrespondingAuthor
Conception ofresearchprojectliteraturesearch writingof the firstmanuscriptdraft
Joaquin AVizcarra MD
University ofCincinnati OH
Author Literaturesearch writingof the firstmanuscriptdraft
Luca MarsiliMD PhD
University ofCincinnati
Author Literaturesearch reviewand critique ofthe manuscript
Anthony ELang MDFRCPC
University ofTorontoCanada
Author Review andcritique of themanuscript
David K SimonMD PhD
HarvardMedical SchoolBoston MA
Author Review andcritique of themanuscript
AristideMerola MDPhD
University ofCincinnati OH
Author Review andcritique of themanuscript
Keith AJosephs MDMST MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
AlfonsoFasano MDPhD
University ofTorontoCanada
Author Review andcritique of themanuscript
FrancescaMorgante MDPhD
University ofMessina Italyand St GeorgersquosUniversity ofLondon UK
Author Review andcritique of themanuscript
Rodolfo SavicaMD MSc
Mayo ClinicMinneapolisMN
Author Review andcritique of themanuscript
J TimothyGreenamyreMD PhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
FrancescaCambi MDPhD
University ofPittsburgh PA
Author Review andcritique of themanuscript
Tritia RYamasaki MDPhD
University ofKentuckyLexington
Author Review andcritique of themanuscript
Caroline MTanner MDPhD
University ofCaliforniandashSanFrancisco
Author Review andcritique of themanuscript
Ziv Gan-OrMD PhD
McGillUniversityMontrealCanada
Author Review andcritique of themanuscript
Irene LitvanMD
UC San DiegoCA
Author Review andcritique of themanuscript
Appendix (continued)
Name Location Role Contribution
Ignacio FMata PhD
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Cyrus PZabetian MDMS
University ofWashingtonSeattle
Author Review andcritique of themanuscript
Patrik BrundinMD PhD
Van AndelResearchInstitute GrandRapids MI
Author Review andcritique of themanuscript
Hubert HFernandez MD
ClevelandClinic OH
Author Review andcritique of themanuscript
David GStandaert MDPhD
University ofAlabama atBirmingham
Author Review andcritique of themanuscript
Marcelo AKauffman MDPhD
UBA andUniversidadAustral-CONICETBuenos AiresArgentina
Author Review andcritique of themanuscript
Michael ASchwarzschildMD PhD
MassachusettsGeneralHospitalBoston
Author Review andcritique of themanuscript
S Pablo SardiPharmD PhD
Sanofi Author Review andcritique of themanuscript
Todd ShererPhD
Michael J FoxFoundation forParkinsonrsquosResearch NewYork NY
Author Review andcritique of themanuscript
George PerryPhD
University ofTexas at SanAntonio
Author Review andcritique of themanuscript
James BLeverenz MD
ClevelandClinic OH
Author Literaturesearch reviewand critique ofthe manuscript
336 Neurology | Volume 92 Number 7 | February 12 2019 NeurologyorgN
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
10 Moher D Liberati A Tetzlaff J Altman DG PRISMA Group Preferred reportingitems for systematic reviews and meta-analyses the PRISMA statement Ann InternMed 2009151264ndash269 W64
11 PoeweW Seppi K Tanner CM et al Parkinson disease Nat Rev Dis Primers 2017317013
12 Querfurth HW LaFerla FM Alzheimerrsquos disease N Engl J Med 2010362329ndash34413 NHLBI Research Triangle Institute International National Heart Lung and Blood
Institute quality appraisal tools [online] Available at httpswwwnhlbinihgovhealth-topicsstudy-quality-assessment-tools Accessed December 15 2017
14 Higgins JPT Green S eds Cochrane Handbook for Systematic Reviews of Inter-ventions [online] 510 The Cochrane Collaboration 2011 Available at httpstrainingcochraneorghandbook Accessed December 16 2017
15 Vos SJ Xiong C Visser PJ et al Preclinical Alzheimerrsquos disease and its outcomea longitudinal cohort study Lancet Neurol 201312957ndash965
16 Van Harten AC Smits LL Teunissen CE et al Preclinical AD predicts decline inmemory and executive functions in subjective complaints Neurology 2013811409ndash1416
17 Burnham SC Bourgeat P Dore V et al Clinical and cognitive trajectories in cogni-tively healthy elderly individuals with suspected non-Alzheimerrsquos disease patho-physiology (SNAP) or Alzheimerrsquos disease pathology a longitudinal study LancetNeurol 2016151044ndash1053
18 Gilman S Koller M Black RS et al Clinical effects of A immunization (AN1792) inpatients with AD in an interrupted trial Neurology 2005641553ndash1562
19 Ostrowitzki S Lasser RA Dorflinger E et al A phase III randomized trial of gante-nerumab in prodromal Alzheimerrsquos disease Alzheimers Res Ther 2017995
20 Salloway S Sperling R Fox NC et al Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370322ndash333
21 Doody RS Raman R Farlow M et al A phase 3 trial of semagacestat for treatment ofAlzheimerrsquos disease N Engl J Med 2013369341ndash350
22 Doody RS Thomas RG Farlow M et al Phase 3 trials of solanezumab for mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370311ndash321
23 Honig LS Vellas B Woodward M et al Trial of solanezumab for mild dementia dueto Alzheimerrsquos disease N Engl J Med 2018378321ndash330
24 Cali I Cohen ML Haїk S et al Iatrogenic Creutzfeldt-Jakob disease with amyloid-βpathology an international study Acta Neuropathol Commun 201865
25 Howick J Glasziou P Aronson JK The evolution of evidence hierarchies what canBradford Hillrsquos ldquoguidelines for causationrdquo contribute J R SocMed 2009102186ndash194
26 Roberts RF Wade-Martins R Alegre-Abarrategui J Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinsonrsquos diseasebrain Brain 20151381642ndash1657
27 Blennow K Mattsson N Scholl M Hansson O Zetterberg H Amyloid biomarkers inAlzheimerrsquos disease Trends Pharmacol Sci 201536297ndash309
28 Hogarth P Gregory A Kruer MC et al New NBIA subtype genetic clinical path-ologic and radiographic features of MPAN Neurology 201380268ndash275
29 Olanow CW Brundin P Parkinsonrsquos disease and alpha synuclein is Parkinsonrsquosdisease a prion-like disorder Mov Disord 20132831ndash40
30 Winder-Rhodes SE Garcia-Reitbock P Ban M et al Genetic and pathological linksbetween Parkinsonrsquos disease and the lysosomal disorder Sanfilippo syndrome MovDisord 201227312ndash315
31 Brenowitz WD Keene CD Hawes SE et al Alzheimerrsquos disease neuropathologicchange Lewy body disease and vascular brain injury in clinic- and community-basedsamples Neurobiol Aging 20175383ndash92
32 Irwin DJ Grossman M Weintraub D et al Neuropathological and genetic correlatesof survival and dementia onset in synucleinopathies a retrospective analysis LancetNeurol 20171655ndash65
33 Crystal H Dickson D Fuld P et al Clinico-pathologic studies in dementia non-demented subjects with pathologically confirmed Alzheimerrsquos disease Neurology1988381682ndash1687
34 Polvikoski T Sulkava R Myllykangas L et al Prevalence of Alzheimerrsquos disease in veryelderly people a prospective neuropathological study Neurology 2001561690ndash1696
35 MarkesberyWR Jicha GA Liu H Schmitt FA Lewy body pathology in normal elderlysubjects J Neuropathol Exp Neurol 200968816ndash822
36 Balasubramanian AB Kawas CH Peltz CB Brookmeyer R Corrada MM Alzheimerdisease pathology and longitudinal cognitive performance in the oldest-old with nodementia Neurology 201279915ndash921
37 Silver MH Newell K Brady C Hedley-White ET Perls TT Distinguishing betweenneurodegenerative disease and disease-free aging correlating neuropsychologicalevaluations and neuropathological studies in centenarians Psychosom Med 200264493ndash501
38 Berlau DJ Corrada MM Head E Kawas CH APOE 2 is associated with intactcognition but increased Alzheimer pathology in the oldest old Neurology 200972829ndash834
39 Kawas CH Kim RC Sonnen JA Bullain SS Trieu T Corrada MM Multiple pa-thologies are common and related to dementia in the oldest-old Neurology 201585535ndash542
40 Espay AJ Schwarzschild MA Tanner CM et al Biomarker-driven phenotyping inParkinsonrsquos disease a translational missing link in disease-modifying clinical trialsMov Disord 201732319ndash324Data available from Dryad (Additional References References e1ndashe44)doiorg105061dryadg1nq02r
Did You Knowhellip
hellipyou can browse by subspecialty topics on Neurologyorg
Go to Neurologyorg and click on ldquoTopicsrdquo in the top navigation bar
Share Your Artistic Expressions in Neurology lsquoVisionsrsquo
AAN members are urged to submit medically or scientifically related artistic images such as photographs photomicrographsand paintings to the ldquoVisionsrdquo section of Neurologyreg These images are creative in nature rather than the medically instructiveimages published in the NeuroImages section The image or series of up to six images may be black and white or color andmust fit into one published journal page Accompanying description should be 100 words or less the title should be a maximumof 96 characters including spaces and punctuation
Please access the Author Center at NPuborgauthors for full submission information
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 337
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
DOI 101212WNL0000000000006926201992329-337 Neurology
Alberto J Espay Joaquin A Vizcarra Luca Marsili et al diseases
Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer
This information is current as of February 11 2019
ServicesUpdated Information amp
httpnneurologyorgcontent927329fullincluding high resolution figures can be found at
References httpnneurologyorgcontent927329fullref-list-1
This article cites 38 articles 10 of which you can access for free at
Citations httpnneurologyorgcontent927329fullotherarticles
This article has been cited by 3 HighWire-hosted articles
Permissions amp Licensing
httpwwwneurologyorgaboutabout_the_journalpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpnneurologyorgsubscribersadvertiseInformation about ordering reprints can be found online
rights reserved Print ISSN 0028-3878 Online ISSN 1526-632X1951 it is now a weekly with 48 issues per year Copyright copy 2019 American Academy of Neurology All
reg is the official journal of the American Academy of Neurology Published continuously sinceNeurology

10 Moher D Liberati A Tetzlaff J Altman DG PRISMA Group Preferred reportingitems for systematic reviews and meta-analyses the PRISMA statement Ann InternMed 2009151264ndash269 W64
11 PoeweW Seppi K Tanner CM et al Parkinson disease Nat Rev Dis Primers 2017317013
12 Querfurth HW LaFerla FM Alzheimerrsquos disease N Engl J Med 2010362329ndash34413 NHLBI Research Triangle Institute International National Heart Lung and Blood
Institute quality appraisal tools [online] Available at httpswwwnhlbinihgovhealth-topicsstudy-quality-assessment-tools Accessed December 15 2017
14 Higgins JPT Green S eds Cochrane Handbook for Systematic Reviews of Inter-ventions [online] 510 The Cochrane Collaboration 2011 Available at httpstrainingcochraneorghandbook Accessed December 16 2017
15 Vos SJ Xiong C Visser PJ et al Preclinical Alzheimerrsquos disease and its outcomea longitudinal cohort study Lancet Neurol 201312957ndash965
16 Van Harten AC Smits LL Teunissen CE et al Preclinical AD predicts decline inmemory and executive functions in subjective complaints Neurology 2013811409ndash1416
17 Burnham SC Bourgeat P Dore V et al Clinical and cognitive trajectories in cogni-tively healthy elderly individuals with suspected non-Alzheimerrsquos disease patho-physiology (SNAP) or Alzheimerrsquos disease pathology a longitudinal study LancetNeurol 2016151044ndash1053
18 Gilman S Koller M Black RS et al Clinical effects of A immunization (AN1792) inpatients with AD in an interrupted trial Neurology 2005641553ndash1562
19 Ostrowitzki S Lasser RA Dorflinger E et al A phase III randomized trial of gante-nerumab in prodromal Alzheimerrsquos disease Alzheimers Res Ther 2017995
20 Salloway S Sperling R Fox NC et al Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370322ndash333
21 Doody RS Raman R Farlow M et al A phase 3 trial of semagacestat for treatment ofAlzheimerrsquos disease N Engl J Med 2013369341ndash350
22 Doody RS Thomas RG Farlow M et al Phase 3 trials of solanezumab for mild-to-moderate Alzheimerrsquos disease N Engl J Med 2014370311ndash321
23 Honig LS Vellas B Woodward M et al Trial of solanezumab for mild dementia dueto Alzheimerrsquos disease N Engl J Med 2018378321ndash330
24 Cali I Cohen ML Haїk S et al Iatrogenic Creutzfeldt-Jakob disease with amyloid-βpathology an international study Acta Neuropathol Commun 201865
25 Howick J Glasziou P Aronson JK The evolution of evidence hierarchies what canBradford Hillrsquos ldquoguidelines for causationrdquo contribute J R SocMed 2009102186ndash194
26 Roberts RF Wade-Martins R Alegre-Abarrategui J Direct visualization of alpha-synuclein oligomers reveals previously undetected pathology in Parkinsonrsquos diseasebrain Brain 20151381642ndash1657
27 Blennow K Mattsson N Scholl M Hansson O Zetterberg H Amyloid biomarkers inAlzheimerrsquos disease Trends Pharmacol Sci 201536297ndash309
28 Hogarth P Gregory A Kruer MC et al New NBIA subtype genetic clinical path-ologic and radiographic features of MPAN Neurology 201380268ndash275
29 Olanow CW Brundin P Parkinsonrsquos disease and alpha synuclein is Parkinsonrsquosdisease a prion-like disorder Mov Disord 20132831ndash40
30 Winder-Rhodes SE Garcia-Reitbock P Ban M et al Genetic and pathological linksbetween Parkinsonrsquos disease and the lysosomal disorder Sanfilippo syndrome MovDisord 201227312ndash315
31 Brenowitz WD Keene CD Hawes SE et al Alzheimerrsquos disease neuropathologicchange Lewy body disease and vascular brain injury in clinic- and community-basedsamples Neurobiol Aging 20175383ndash92
32 Irwin DJ Grossman M Weintraub D et al Neuropathological and genetic correlatesof survival and dementia onset in synucleinopathies a retrospective analysis LancetNeurol 20171655ndash65
33 Crystal H Dickson D Fuld P et al Clinico-pathologic studies in dementia non-demented subjects with pathologically confirmed Alzheimerrsquos disease Neurology1988381682ndash1687
34 Polvikoski T Sulkava R Myllykangas L et al Prevalence of Alzheimerrsquos disease in veryelderly people a prospective neuropathological study Neurology 2001561690ndash1696
35 MarkesberyWR Jicha GA Liu H Schmitt FA Lewy body pathology in normal elderlysubjects J Neuropathol Exp Neurol 200968816ndash822
36 Balasubramanian AB Kawas CH Peltz CB Brookmeyer R Corrada MM Alzheimerdisease pathology and longitudinal cognitive performance in the oldest-old with nodementia Neurology 201279915ndash921
37 Silver MH Newell K Brady C Hedley-White ET Perls TT Distinguishing betweenneurodegenerative disease and disease-free aging correlating neuropsychologicalevaluations and neuropathological studies in centenarians Psychosom Med 200264493ndash501
38 Berlau DJ Corrada MM Head E Kawas CH APOE 2 is associated with intactcognition but increased Alzheimer pathology in the oldest old Neurology 200972829ndash834
39 Kawas CH Kim RC Sonnen JA Bullain SS Trieu T Corrada MM Multiple pa-thologies are common and related to dementia in the oldest-old Neurology 201585535ndash542
40 Espay AJ Schwarzschild MA Tanner CM et al Biomarker-driven phenotyping inParkinsonrsquos disease a translational missing link in disease-modifying clinical trialsMov Disord 201732319ndash324Data available from Dryad (Additional References References e1ndashe44)doiorg105061dryadg1nq02r
Did You Knowhellip
hellipyou can browse by subspecialty topics on Neurologyorg
Go to Neurologyorg and click on ldquoTopicsrdquo in the top navigation bar
Share Your Artistic Expressions in Neurology lsquoVisionsrsquo
AAN members are urged to submit medically or scientifically related artistic images such as photographs photomicrographsand paintings to the ldquoVisionsrdquo section of Neurologyreg These images are creative in nature rather than the medically instructiveimages published in the NeuroImages section The image or series of up to six images may be black and white or color andmust fit into one published journal page Accompanying description should be 100 words or less the title should be a maximumof 96 characters including spaces and punctuation
Please access the Author Center at NPuborgauthors for full submission information
NeurologyorgN Neurology | Volume 92 Number 7 | February 12 2019 337
Copyright ordf 2019 American Academy of Neurology Unauthorized reproduction of this article is prohibited
DOI 101212WNL0000000000006926201992329-337 Neurology
Alberto J Espay Joaquin A Vizcarra Luca Marsili et al diseases
Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer
This information is current as of February 11 2019
ServicesUpdated Information amp
httpnneurologyorgcontent927329fullincluding high resolution figures can be found at
References httpnneurologyorgcontent927329fullref-list-1
This article cites 38 articles 10 of which you can access for free at
Citations httpnneurologyorgcontent927329fullotherarticles
This article has been cited by 3 HighWire-hosted articles
Permissions amp Licensing
httpwwwneurologyorgaboutabout_the_journalpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpnneurologyorgsubscribersadvertiseInformation about ordering reprints can be found online
rights reserved Print ISSN 0028-3878 Online ISSN 1526-632X1951 it is now a weekly with 48 issues per year Copyright copy 2019 American Academy of Neurology All
reg is the official journal of the American Academy of Neurology Published continuously sinceNeurology

DOI 101212WNL0000000000006926201992329-337 Neurology
Alberto J Espay Joaquin A Vizcarra Luca Marsili et al diseases
Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer
This information is current as of February 11 2019
ServicesUpdated Information amp
httpnneurologyorgcontent927329fullincluding high resolution figures can be found at
References httpnneurologyorgcontent927329fullref-list-1
This article cites 38 articles 10 of which you can access for free at
Citations httpnneurologyorgcontent927329fullotherarticles
This article has been cited by 3 HighWire-hosted articles
Permissions amp Licensing
httpwwwneurologyorgaboutabout_the_journalpermissionsits entirety can be found online atInformation about reproducing this article in parts (figurestables) or in
Reprints
httpnneurologyorgsubscribersadvertiseInformation about ordering reprints can be found online
rights reserved Print ISSN 0028-3878 Online ISSN 1526-632X1951 it is now a weekly with 48 issues per year Copyright copy 2019 American Academy of Neurology All
reg is the official journal of the American Academy of Neurology Published continuously sinceNeurology




















