
Rapamycin Inhibits PAI-1 Expression and Reduces Interstitial Fibrosis and Glomerulosclerosis in...
10
Rapamycin Inhibits PAI-1 Expression and Reduces Interstitial Fibrosis and Glomerulosclerosis in Chronic Allograft Nephropathy Paola Pontrelli, 1 Michele Rossini, 2 Barbara Infante, 1 Giovanni Stallone, 1 Antonio Schena, 2 Antonia Loverre, 2 Michele Ursi, 2 Raffaella Verrienti, 2 Annamaria Maiorano, 2 Gianluigi Zaza, 2 Elena Ranieri, 1 Loreto Gesualdo, 1 Pasquale Ditonno, 3 Carlo Bettocchi, 3 Francesco Paolo Schena, 2 and Giuseppe Grandaliano 2,4 Background. Chronic allograft nephropathy (CAN) is characterized by deposition of extracellular matrix (ECM) in all renal compartments. PAI-1 seems to play a pivotal role in ECM turnover in CAN. Rapamycin has been shown to improve long-term graft survival in patients with CAN. The aim of the study was to evaluate the molecular mechanisms underlying the beneficial effects of rapamycin on CAN progression at glomerular and tubulointerstitial level. Methods. After a biopsy-proven CAN diagnosis (T0), 18 patients on calcineurin inhibitors (CNI) were randomly assigned in a 2:1 ratio to continue CNI (6 patients) or to receive rapamycin (RAPA; 12 patients). After 2 years of treatment (T24), all patients underwent a second renal biopsy. Morphometric analysis was conducted at T0 and at T24. PAI-1 expression was evaluated at T0 and T24 by immunohistochemistry. We evaluated the effect of rapamycin on PAI-1 gene expression in cultured proximal tubular cells incubated with CD40L or thrombin, two potential CAN pathogenic mediators. Results.The RAPA group showed a significant regression of glomerulosclerotic lesions and only a 26% increase in interstitial fibrosis after 2 years compared to baseline, whereas the CNI group showed progression of glomerulosclerosis and 112% increase in fibrosis. Glomerular and tubulointerstitial PAI-1 expression was reduced compared to the baseline in the RAPA group, while they were unchanged in the CNI group. In vitro data showed that rapamycin significantly reduced PAI-1 gene expression induced by both CD40L and thrombin in proximal tubular epithelial cells. Conclusions. These data suggest that rapamycin may modulate ECM deposition in CAN reducing PAI-1 expression. Keywords: Rapamycin, Interstitial fibrosis, Glomerulosclerosis, Kidney allograft. (Transplantation 2008;85: 125–134) C hronic allograft nephropathy (CAN) is the main cause of renal graft failure and is histologically characterized by interstitial fibrosis, tubular atrophy, arterial intimal hyper- plasia with fibrointimal thickening, and glomerulosclerosis (1). Its pathogenic mechanisms are still largely unclear, al- though clinical and experimental observations suggest the in- volvement of several causes, including ischemia-reperfusion injury, ineffectively or untreated clinical and subclinical re- jection, and superimposed calcineurin inhibitor (CNI) neph- rotoxicity, exacerbating preexisting donor disease (2). Once established, interstitial fibrosis and glomerulosclerosis lead to a progressive graft dysfunction over the subsequent years (3). Interstitial fibrosis and glomerulosclerosis are the result of an impaired balance between extracellular matrix (ECM) depo- sition and degradation. A key role in the regulation of ECM degradation is played by the plasmin/matrix metalloprotein- ases (MMPs) system. Plasmin can degrade a broad range of ECM proteins and plays an additional role in regulating ECM deposition by activation of latent MMPs, which degrade ECM proteins in the kidney (4). Plasmin is generated by cleavage of plasminogen by either urokinase-type plasminogen activator (u-PA) or tissue-type plasminogen activator (t-PA) (5). Both u-PA and t-PA are inhibited in vivo by plasminogen activator inhibitor type 1 (PAI-1) (4). PAI-1 is produced by platelets, vascular endothelial cells, vascular smooth muscle cells, and several nonvascular cell types (6, 7). In the kidney, PAI-1 is produced by several different cells, including mesangial and tubular epithelial cells in several pathological states or under different stimuli (8). In normal kidneys, PAI-1 expression is low, but its mRNA levels increase rapidly during a variety of acute and chronic renal diseases (5). PAI-1 has been shown to play a pivotal role in ECM turnover in CAN as demonstrated by a striking increase in gene expression, with a distribution resembling the pattern of fibrin deposition (9). Rapamycin is an immunosuppressive drug that inhibits the proliferation of T-cell clones activated by alloantigens, through the interaction and subsequent inhibition of the mammalian target of rapamycin (mTOR) kinase (10). This Drs. Pontrelli and Rossini equally contributed to the study. This study was supported by the Ministero della Salute (ex art 12bis to F.P.S.), the 5th European Framework Quality of Life and Management of Living Resources (QLG1–2002-01215 to G.G.), Ministero dell’Universita ` e della Ricerca Scientifica (PRIN 2003 to L.G., PRIN 2004 to F.P.S., and PRIN 2005 to G.G.) and a grant from the Fondazione Cassa di Risparmio di Puglia (to L.G.). 1 Divisions of Nephrology and Clinical Pathology, Department of Biomedi- cal Sciences, University of Foggia, Foggia, Italy. 2 Division of Nephrology, Department of Emergency and Organ Transplan- tation, University of Bari, Bari, Italy. 3 Division of Urology, Department of Emergency and Organ Transplanta- tion, University of Bari, Bari, Italy. 4 Address correspondence to: Giuseppe Grandaliano, M.D., Division of Ne- phrology, Department of Emergency and Transplantation, University of Bari, Piazza Giulio Cesare 11, 70124 Bari, Italy. E-mail: [email protected] Received 4 June 2007. Revision requested 16 August 2007. Accepted 26 September 2007. Copyright © 2008 by Lippincott Williams & Wilkins ISSN 0041-1337/08/8501-125 DOI: 10.1097/01.tp.0000296831.91303.9a Transplantation • Volume 85, Number 1, January 15, 2008 125
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Rapamycin Inhibits PAI-1 Expression and Reduces Interstitial Fibrosis and Glomerulosclerosis in...

Rapamycin Inhibits PAI-1 Expression and ReducesInterstitial Fibrosis and Glomerulosclerosis in
Chronic Allograft Nephropathy
Paola Pontrelli,1 Michele Rossini,2 Barbara Infante,1 Giovanni Stallone,1 Antonio Schena,2
Antonia Loverre,2 Michele Ursi,2 Raffaella Verrienti,2 Annamaria Maiorano,2 Gianluigi Zaza,2
Elena Ranieri,1 Loreto Gesualdo,1 Pasquale Ditonno,3 Carlo Bettocchi,3 Francesco Paolo Schena,2
and Giuseppe Grandaliano2,4
Background. Chronic allograft nephropathy (CAN) is characterized by deposition of extracellular matrix (ECM) in allrenal compartments. PAI-1 seems to play a pivotal role in ECM turnover in CAN. Rapamycin has been shown toimprove long-term graft survival in patients with CAN. The aim of the study was to evaluate the molecular mechanismsunderlying the beneficial effects of rapamycin on CAN progression at glomerular and tubulointerstitial level.Methods. After a biopsy-proven CAN diagnosis (T0), 18 patients on calcineurin inhibitors (CNI) were randomlyassigned in a 2:1 ratio to continue CNI (6 patients) or to receive rapamycin (RAPA; 12 patients). After 2 years oftreatment (T24), all patients underwent a second renal biopsy. Morphometric analysis was conducted at T0 and at T24.PAI-1 expression was evaluated at T0 and T24 by immunohistochemistry. We evaluated the effect of rapamycin onPAI-1 gene expression in cultured proximal tubular cells incubated with CD40L or thrombin, two potential CANpathogenic mediators.Results.The RAPA group showed a significant regression of glomerulosclerotic lesions and only a 26% increase ininterstitial fibrosis after 2 years compared to baseline, whereas the CNI group showed progression of glomerulosclerosisand 112% increase in fibrosis. Glomerular and tubulointerstitial PAI-1 expression was reduced compared to thebaseline in the RAPA group, while they were unchanged in the CNI group. In vitro data showed that rapamycinsignificantly reduced PAI-1 gene expression induced by both CD40L and thrombin in proximal tubular epithelial cells.Conclusions. These data suggest that rapamycin may modulate ECM deposition in CAN reducing PAI-1 expression.
Keywords: Rapamycin, Interstitial fibrosis, Glomerulosclerosis, Kidney allograft.
(Transplantation 2008;85: 125–134)
Chronic allograft nephropathy (CAN) is the main causeof renal graft failure and is histologically characterized
by interstitial fibrosis, tubular atrophy, arterial intimal hyper-plasia with fibrointimal thickening, and glomerulosclerosis(1). Its pathogenic mechanisms are still largely unclear, al-though clinical and experimental observations suggest the in-volvement of several causes, including ischemia-reperfusioninjury, ineffectively or untreated clinical and subclinical re-jection, and superimposed calcineurin inhibitor (CNI) neph-rotoxicity, exacerbating preexisting donor disease (2). Once
established, interstitial fibrosis and glomerulosclerosis lead toa progressive graft dysfunction over the subsequent years (3).Interstitial fibrosis and glomerulosclerosis are the result of animpaired balance between extracellular matrix (ECM) depo-sition and degradation. A key role in the regulation of ECMdegradation is played by the plasmin/matrix metalloprotein-ases (MMPs) system. Plasmin can degrade a broad range ofECM proteins and plays an additional role in regulating ECMdeposition by activation of latent MMPs, which degrade ECMproteins in the kidney (4). Plasmin is generated by cleavage ofplasminogen by either urokinase-type plasminogen activator(u-PA) or tissue-type plasminogen activator (t-PA) (5). Bothu-PA and t-PA are inhibited in vivo by plasminogen activatorinhibitor type 1 (PAI-1) (4). PAI-1 is produced by platelets,vascular endothelial cells, vascular smooth muscle cells, andseveral nonvascular cell types (6, 7). In the kidney, PAI-1 isproduced by several different cells, including mesangial andtubular epithelial cells in several pathological states or underdifferent stimuli (8). In normal kidneys, PAI-1 expression islow, but its mRNA levels increase rapidly during a variety ofacute and chronic renal diseases (5). PAI-1 has been shown toplay a pivotal role in ECM turnover in CAN as demonstratedby a striking increase in gene expression, with a distributionresembling the pattern of fibrin deposition (9).
Rapamycin is an immunosuppressive drug that inhibitsthe proliferation of T-cell clones activated by alloantigens,through the interaction and subsequent inhibition of themammalian target of rapamycin (mTOR) kinase (10). This
Drs. Pontrelli and Rossini equally contributed to the study.This study was supported by the Ministero della Salute (ex art 12bis to F.P.S.),
the 5th European Framework Quality of Life and Management of LivingResources (QLG1–2002-01215 to G.G.), Ministero dell’Universita e dellaRicerca Scientifica (PRIN 2003 to L.G., PRIN 2004 to F.P.S., and PRIN2005 to G.G.) and a grant from the Fondazione Cassa di Risparmio diPuglia (to L.G.).
1 Divisions of Nephrology and Clinical Pathology, Department of Biomedi-cal Sciences, University of Foggia, Foggia, Italy.
2 Division of Nephrology, Department of Emergency and Organ Transplan-tation, University of Bari, Bari, Italy.
3 Division of Urology, Department of Emergency and Organ Transplanta-tion, University of Bari, Bari, Italy.
4 Address correspondence to: Giuseppe Grandaliano, M.D., Division of Ne-phrology, Department of Emergency and Transplantation, University ofBari, Piazza Giulio Cesare 11, 70124 Bari, Italy.
E-mail: [email protected] 4 June 2007. Revision requested 16 August 2007.Accepted 26 September 2007.Copyright © 2008 by Lippincott Williams & WilkinsISSN 0041-1337/08/8501-125DOI: 10.1097/01.tp.0000296831.91303.9a
Transplantation • Volume 85, Number 1, January 15, 2008 125

serine-threonine kinase, activated by Akt, is involved in intra-cellular signaling pathways leading to cell growth and prolif-eration (10). In addition, mTOR plays a major role in theregulation of collagen type I gene expression in dermal fibro-blasts (11). Rapamycin has been successfully used in the car-bon tetrachloride model of hepatic fibrosis (12) and in theinhibition of intimal hyperplasia, medial ECM accumulation,and vascular remodelling in rat aortic allografts (13). More-over, the administration of rapamycin has been proved toreduce the interstitial fibrosis after unilateral ureteral ob-struction in rats (14) and in a rat model of membranousnephropathy (15). Rapamycin may also affect the functional-structural glomerular remodeling in chronic kidney diseasewith renal mass reduction. It has been shown that rapamycinreduces the compensatory hypertrophy after unilateral ne-phrectomy in mice (16). We have recently demonstrated thata switch from calcineurin inhibitors (CNIs) to rapamycinmay significantly improve the clinical outcome and the tubu-lointerstitial lesions of CAN (17).
Aim of the present study was to evaluate the molecularmechanisms underlying the beneficial effects of rapamycinon CAN progression at glomerular and tubulointerstitiallevel with a particular focus on the rapamycin effects on pro-teolytic systems.
MATERIALS AND METHODS
PatientsFrom June 2002 to July 2003, a total of 18 consecutive
patients with biopsy-proven CAN, receiving cyclosporine asbase immunosuppressive therapy, were randomly assigned(T0) in a 2:1 ratio to either CNI withdrawal and rapamycin
introduction (RAPA group, n�12) or to continue with theCNI-based regimen (CNI group, n�6). Renal specimens, ob-tained by needle-core biopsies (16 gauge) that were per-formed under ultrasonographic guidance, were fixed in 4%formaldehyde, paraffin-embedded, and then processed forroutine histologic staining (hematoxylin-eosin, periodicacid-Schiff, silver methenamine, and Masson’s trichrome).Semiquantitative scoring for acute and chronic tubular, in-terstitial, vascular, and glomerular changes was performedand biopsies reports issued according to the Banff 1997 crite-ria (18). Immunohistochemistry for C4d was done in all casesretrospectively on paraffin-embedded tissue and was negativein peritubular and glomerular capillaries in all cases. No rel-evant histologic signs of chronic cellular or humoral rejection(i.e., transplant glomerulopathy, glomerulitis, inflammatorycells in peritubular capillaries, allograft vasculopathy, tubuli-tis in nonatrophic tubuli) were present. Main demographicsand clinical characteristics of the patients enrolled are reportedin Table 1. At randomization, there was no difference in the doseadministered and in the trough levels of cyclosporine betweenthe two groups. No patient was on tacrolimus. In patients ran-domized to receive rapamycin, cyclosporine was abruptly dis-continued and rapamycin introduced with a loading dose of 0.1mg/kg/day and a maintaining dose aiming at through levels of6–10 ng/ml. All patients presented with grade I CAN accordingto Banff criteria (18). Age, time from transplantation, and serumcreatinine at the time of CAN diagnosis were similar in the twogroups. After 2 years of treatment, all patients underwent a sec-ond graft biopsy (T24). The study was carried out according toDeclaration of Helsinki and approved by our institutional reviewboard.
TABLE 1. Patient characteristics
CNI group RAPA group P value
N 6 12
Donor age (years) 38.2�18.4 44.4�13.8 NS
Donor creatinine clearance (ml/min) 99.8�16.1 87.1�18.3 NS
Recipient age (years) 33.6�9.5 37.6�13.8 NS
Cold ischemia time (hours) 9.8�5.5 11.8�9.7 NS
HLA mismatches (n) 3.4�0.5 3.5�0.8 NS
Serum creatinine (mg/dl)
At T0 1.60�0.52 1.48�0.36 NS
At T24 2.33�1.88 1.56�0.55 NS
Proteinuria (g/24 h)
At T0 0.25�0.10 0.22�0.17 NS
At T24 1.07�0.51 1.25�1.49 NS
Immunosuppression
Cyclosporine dose (mg/kg/day) 3.4�1.0 2.8�1.1 NS
Cyclosporine T0 (ng/ml) 135.6�82.5 139�125.8 NS
Azathioprine/mycophenolate mofetil 3/3 3/9 NS
ACE and/or ARB (yes/no)
At T0 5/1 9/3 NS
At T24 5/1 10/2 NS
Statins (yes/no)
At T0 0/6 3/9 NS
At T24 0/6 7/5 �0.05
126 Transplantation • Volume 85, Number 1, January 15, 2008

Morphometric AnalysisMorphometric analysis was carried out to analyze glo-
merular sclerosis and interstitial fibrosis. For each biopsy allglomeruli, including sclerotic and nonsclerotic, were ana-lyzed. Extreme tangential sections, defined as glomeruli withdiscontinuous capillary loops, were excluded. An average of 13glomeruli/biopsy were present (range 10–26). Digital images ofeach glomerulus were captured with an �20 objective using aLeika (Leitz, Wetzlar, Germany) microscope fitted with a Cool-pix 990 digital camera (Nikon). Acquired images were analyzedusing the Image J software 1.33u (http://rsb.info.nih.gov/ij/).Total glomerular area and area of sclerosis were expressed aspixel2. Sclerosis index (SI) was defined as area of sclerosis/totalglomerular area. Patients with a �SI (SI T24 – SI T0) �0 weredefined as “regressors”; those with a �SI �0 were defined as“progressors.”
Extent of interstitial fibrosis was assessed on Masson’strichrome stained slides using Adobe Photoshop 8.0.1(Adobe Systems Corporation, San Jose, CA) (19). The corti-cal area of the entire biopsy was analyzed in a stepwise fashionas a series of consecutive fields, with a �20 objective, avoidingthe capsule, the subcapsular areas, and the arterial adventitia.The percentage of blue (fibrous tissue) stained area/total areawas measured. Values from all consecutive images for eachbiopsy were averaged. Percentage variations from T0 to T24were then calculated.
ImmunohistochemistryImmunohistochemical evaluation of PAI-1, TIMP-1,
matrix metalloproteinase (MMP) MMP-2, and MMP-9 pro-tein expression in CAN patients and normal controls wascarried out on paraffined renal biopsy sections. Thin (2-�m)sections of paraffin-embedded tissue were deparaffinized andhydrated through graded alcohol series. The endogenous per-oxidase activity was quenched by incubation in a solution ofH2O2 3% for 15 min. After blocking with 2% normal serum atroom temperature (RT) for 20 min, the slides were incubatedwith the following primary antibodies: rabbit polyclonalantihuman PAI-1 antibody (1:50, overnight, Santa Cruz Bio-technology, Santa Cruz, CA); mouse monoclonal anti-TIMP-1 antibody (1:20, 30 min at RT, Novus Biologicals,Littleton, CO); mouse monoclonal anti-MMP2 antibody (4�g/ml, overnight incubation at 4°C); mouse monoclonal an-ti-MMP9 antibody (6 �g/ml, overnight incubation at 4°C,Novus Biologicals).
For PAI-1, the binding of the secondary biotinylated an-tibody was detected by the Vectastain ABC system according tothe manufacturer’s instructions (Vector, Burlingame, CA). Thediaminobenzidine tetrahydrochloride solution (Dako, Carpin-tera, CA) was used as chromogen and the slides were counter-stained with hematoxylin.
For TIMP-1, MMP-2, and MMP-9, the binding of thesecondary biotinylated antibodies was detected by the EnVisionG/2 System/AP kit, according to the manufacturer’s instructions(Dako). Visualization of alkaline phosphatase was performed byincubation in Permanent Red Chromogen solution supple-mented with levamisole, giving a red precipitate. The sectionswere counterstained with Mayers hematoxylin. Negative con-trols were performed omitting primary antibody.
To quantify PAI-1 and TIMP-1 immunostaining at thetubulointerstitial level, for each biopsy the entire cortical re-
gion was analyzed, avoiding the capsule, the subcapsular ar-eas, the arterial adventitia and the glomeruli, in a stepwisefashion as a series of consecutive fields with a �20 objective.Acquired images were analyzed using Image J software. Eachacquired color image was converted into an 8-bit grayscaleimage. For each biopsy, a background image of a blank area ofthe slide was initially obtained and a background correctionwas performed in real time to adjust for irregularities in theillumination of the microscope field. A threshold was set be-low which nonstained areas were excluded and the area of theremaining positive staining was recorded. The percentage ofpositive stained area/total area was then calculated. Valuesfrom all consecutive images for each biopsy were averaged.PAI-1 glomerular expression was evaluated using a semi-quantitative score by two independent observers. Scores of 0to 4 represent negative, trace, 10%, 10 –25%, and 25% stain-ing, respectively, in each glomerulus. For MMP-2 andMMP-9, the number of positive cells/glomerulus and thenumber of positive tubular epithelial cells and positive inter-stitial cells for high power field were counted.
Cell Isolation and CultureHuman HK2, an immortalized proximal tubular epi-
thelial cell (PTEC) line from normal adult human kidney(20), was obtained from American Type Culture Collection(ATCC, Rockville, MD). Cells were grown to confluence inDMEM/F12 (Sigma, Milan, Italy) medium supplementedwith 10% fetal bovine serum (FBS; Sigma), 100 U/ml pen-icillin, 100 �g/ml streptomycin, and 2 mM L-glutamine(all from Life Technologies, Milan, Italy). For each pas-sage, confluent cells were washed with phosphate-bufferedsaline (PBS), removed with 0.05% trypsin/0.02% ethylene-diamine-tetra-acetic acid (EDTA; Sigma) in PBS andplated in DMEM/F12.
Western BlotHuman HK2 cells were plated in 6 well dishes and
grown to confluence in DMEM/F12 (Sigma) supplementedwith 10% FBS. The cells were incubated 40 hr in serum-freemedium and, then, exposed to sCD40L (0.1 mg/ml) andsCD40L enhancer (1 mg/ml; both from Alexis, Lausen, Swit-zerland) or thrombin (5U/ml; Sigma) for the indicated timeperiods. At the end of the treatment, the cell monolayer wasrapidly rinsed twice with ice-cold PBS and lysed in 100 �l ofradio immunoprecipitation assay (RIPA) buffer (1 mM phe-nylmethylsulphonylfluoride, 5 mM EDTA, 1 mM sodium or-thovanadate, 150 mM sodium chloride, 8 �g/ml leupeptin,1.5% Nonidet P-40, 20 mM tris-HCl, pH 7.4). The lysateswere kept on ice for 30 min and centrifuged at 10,000�g at4°C for 5 min. The supernatants were collected and stored at�80°C until used.
Aliquots containing 40 �g of proteins from each lysatewere subjected to sodium dodecyl sulfate-polyacrylamide gelelectrophoresis (SDS/PAGE) on a 10% gel under reducingconditions and then electrotransferred onto nitrocellulosemembrane (Hybond, Amersham, Buckinghamshire, UK).The filter was blocked overnight at RT with 2% bovine serumalbumin in PBS containing 0.1% tween 20 (TBS) and thenincubated with the polyclonal anti-phospho-Akt1 antibody(1:500 dilution in TBS at RT for 2 hr). The membranes werewashed twice in TBS and incubated for one hour at RT with
© 2008 Lippincott Williams & Wilkins 127Pontrelli et al.

horseradish-peroxidase-conjugated sheep anti-rabbit IgG at1:1500 dilution in TBS. The membranes were washed threetimes at RT in TBS and then once with 0.1% SDS in PBS. Thesame membranes were then stripped and immunoblottedagain with antihuman Akt1 monoclonal antibody (1:500 di-lution in TBS at RT for 2 hr). The ECL chemiluminescencesystem (Amersham) was used for detection.
RNA Isolation and Northern Blot AnalysisHK2 cells were plated in 60-mm2 Petri dishes and cul-
tured as detailed above. After reaching confluence cells wereserum-starved for 40 hr and then incubated for 6 hr withsCD40L (0.1 mg/ml) and sCD40L enhancer (1 mg/ml) orthrombin (5 U/ml). In separate sets of experiments, cells werepreincubated with rapamycin (20 ng/ml) for 2 hr before addingsCD40L or thrombin. At the end of incubation, cells were lysedwith 1 ml TRIzol Reagent (Life Technologies). PAI-1 gene ex-pression was studied by northern blotting. Electrophoresis of 20�g of total RNA from each experimental condition was carriedout in 1.1% agarose gel with 2.2 M formaldehyde. The RNA wasthen transferred overnight onto a nylon membrane (Schleicher& Schuell, Dassel, Germany). The membrane was stained withethidium bromide to evaluate the 28S and 18S ribosomal bandsand prehybridized at 42°C for 4 hr in 50% Formamide, 0.5%SDS, 5� sodium chloride/sodium citrate (SSC), and 0.1 mg/mlsalmon sperm DNA. A 1253-bp fragment of the human PAI-1cDNAs were used as probes. The DNA fragment was labeled byrandom priming using a commercially available kit (Amer-sham) and [32P]dCTP (specific activity: 3,000 Ci/mmol). The
probe (106 cpm/ml) was added to 10 ml of prehybridizationsolution, and the blots were hybridized for 16 hr at 42°C. Themembranes were then washed once in 2� SSC at room temper-ature for 5 min, once in 2� SSC 0.1% SDS at room temperaturefor 20 min, once in the same buffer at 55°C for 30 min, and inSSC 0.1% SDS at 55°C for further 30 min. After drying, mem-branes were exposed to a Kodak X-OMAT film with intensifyingscreens at �70°C.
Statistical AnalysisData were expressed as media � standard deviation
and analyzed with Student t test. Dichotomous variables wereanalyzed with the chi-square test. A P value �0.05 was con-sidered statistically significant.
RESULTS
Renal Function and HistologyAt the first biopsy (T0), all patients enrolled presented
with grade 1 CAN, no evidence of acute rejection, or no re-currence of primary renal disease. Serum creatinine and 24-hproteinuria were not different between the two groups at T0and T24 (Table 1). No episode of acute rejection occurred inboth groups. Table 2 shows patients individual data on renalfunction proteinuria and cyclosporine through levels at T0.Extent of interstitial fibrosis was not statistically different be-tween the two groups at T0, although there was a trend in lessinterstitial fibrosis in patients randomized to receive CNI
TABLE 2. Patient data at baseline and 2 years
Patient
Otherimmunosuppression
at T0C0 T0
(ng/ml)C0 T2
(ng/ml)sCr T0
(mg/dl)ProteinuriaT0 (g/24 hr) �IF (%) �SI
CNI group
1 CS, AZA 60 90 1.9 0.300 80.522 0.302
2 CS, AZA 70 92 1.1 0.310 47.500 0.000
3 CS, MMF 113 103 1.1 0.330 142.190 0.071
4 CS, MMF 209 132 2.1 0.090 156.510 0.154
5 CS, MMF 262 53 1.2 0.240 130.000 0.000
6 CS, AZA 106 95 2.2 0.250 119.706 0.446
RAPA group
7 CS, AZA 146 — 1.4 0.060 10.505 0.101
8 CS, AZA 55 — 1.8 0.220 �5.702 0.172
9 CS, MMF 55 — 1.8 0.090 8.446 0.013
10 CS, MMF 237 — 1.3 0.371 21.780 0.000
11 CS, AZA 130 — 1.6 0.010 30.790 �0.044
12 CS, MMF 203 — 1.0 0.180 26.300 �0.333
13 CS, MMF 115 — 1.3 0.210 209.232 �0.076
14 CS, MMF 278 — 1.0 0.130 �15.591 �0.200
15 CS, MMF 52 — 1.8 0.370 16.836 �0.395
16 CS, MMF 130 — 1.3 0.140 26.833 0.000
17 CS, MMF 202 — 2.2 0.600 �33.559 0.000
18 CS, MMF 68 — 1.3 0.330 19.505 0.053
C0 T0, cyclosporin through level at randomization; C0 T2, cyclosporin through level at 2 years; sCr T0, serum creatinine at randomization; proteinuria T0,24 h proteinuria at randomization; �IF, percentage variation of interstitial fibrosis from T0 to T2 �(IFT2 – IFT0)�100/IFT2; �SI, sclerosis index T2 – sclerosisindex T0; AZA, azathioprine.
128 Transplantation • Volume 85, Number 1, January 15, 2008
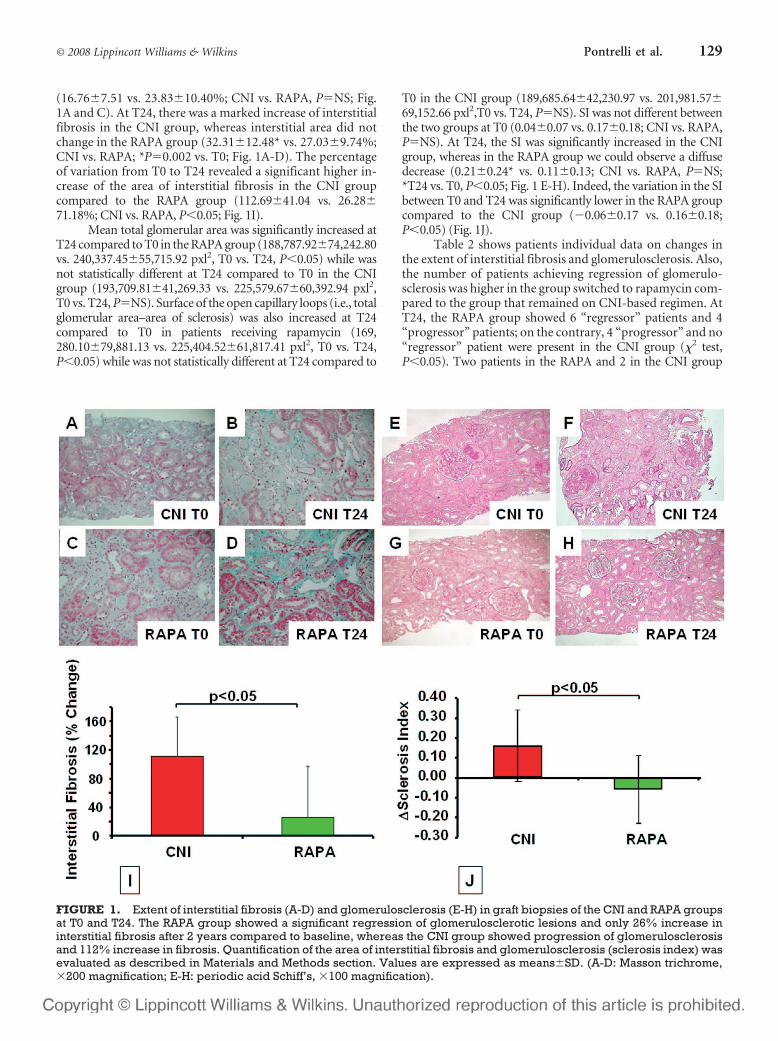
(16.76�7.51 vs. 23.83�10.40%; CNI vs. RAPA, P�NS; Fig.1A and C). At T24, there was a marked increase of interstitialfibrosis in the CNI group, whereas interstitial area did notchange in the RAPA group (32.31�12.48* vs. 27.03�9.74%;CNI vs. RAPA; *P�0.002 vs. T0; Fig. 1A-D). The percentageof variation from T0 to T24 revealed a significant higher in-crease of the area of interstitial fibrosis in the CNI groupcompared to the RAPA group (112.69�41.04 vs. 26.28�71.18%; CNI vs. RAPA, P�0.05; Fig. 1I).
Mean total glomerular area was significantly increased atT24 compared to T0 in the RAPA group (188,787.92�74,242.80vs. 240,337.45�55,715.92 pxl2, T0 vs. T24, P�0.05) while wasnot statistically different at T24 compared to T0 in the CNIgroup (193,709.81�41,269.33 vs. 225,579.67�60,392.94 pxl2,T0 vs. T24, P�NS). Surface of the open capillary loops (i.e., totalglomerular area–area of sclerosis) was also increased at T24compared to T0 in patients receiving rapamycin (169,280.10�79,881.13 vs. 225,404.52�61,817.41 pxl2, T0 vs. T24,P�0.05) while was not statistically different at T24 compared to
T0 in the CNI group (189,685.64�42,230.97 vs. 201,981.57�69,152.66 pxl2,T0 vs. T24, P�NS). SI was not different betweenthe two groups at T0 (0.04�0.07 vs. 0.17�0.18; CNI vs. RAPA,P�NS). At T24, the SI was significantly increased in the CNIgroup, whereas in the RAPA group we could observe a diffusedecrease (0.21�0.24* vs. 0.11�0.13; CNI vs. RAPA, P�NS;*T24 vs. T0, P�0.05; Fig. 1 E-H). Indeed, the variation in the SIbetween T0 and T24 was significantly lower in the RAPA groupcompared to the CNI group (�0.06�0.17 vs. 0.16�0.18;P�0.05) (Fig. 1J).
Table 2 shows patients individual data on changes inthe extent of interstitial fibrosis and glomerulosclerosis. Also,the number of patients achieving regression of glomerulo-sclerosis was higher in the group switched to rapamycin com-pared to the group that remained on CNI-based regimen. AtT24, the RAPA group showed 6 “regressor” patients and 4“progressor” patients; on the contrary, 4 “progressor” and no“regressor” patient were present in the CNI group (�2 test,P�0.05). Two patients in the RAPA and 2 in the CNI group
FIGURE 1. Extent of interstitial fibrosis (A-D) and glomerulosclerosis (E-H) in graft biopsies of the CNI and RAPA groupsat T0 and T24. The RAPA group showed a significant regression of glomerulosclerotic lesions and only 26% increase ininterstitial fibrosis after 2 years compared to baseline, whereas the CNI group showed progression of glomerulosclerosisand 112% increase in fibrosis. Quantification of the area of interstitial fibrosis and glomerulosclerosis (sclerosis index) wasevaluated as described in Materials and Methods section. Values are expressed as means�SD. (A-D: Masson trichrome,�200 magnification; E-H: periodic acid Schiff’s, �100 magnification).
© 2008 Lippincott Williams & Wilkins 129Pontrelli et al.

showed a �SI�0. There were no significant differences betweenprogressors and regressors for mycophenolate mofetil (MMF;P�0.198), angiotensin-converting enzyme (ACE) inhibitorsand/or angiotensin receptor blockers (ARBs; P�0.186), and sta-tin (P�0.533) use during the 2-year follow-up period.
ImmunohistochemistryPAI-1 protein expression, rarely detectable in control
tissue (data not shown), was significantly increased at T0 inboth groups at tubulointerstitial (Fig. 2A and C) and glomer-ular level (Fig. 2E and G). Interestingly, at T24 patientsswitched to rapamycin presented a significant reduction inthe PAI-1 protein expression at tubulointerstitial level (T016.97�6.93 vs. T24 4.36�2.01%; P�0.05) compared to pa-tients randomized to continue CNI, in which the intensityand the pattern of distribution of the PAI-1 staining was un-changed compared to the baseline (T0 23.31�11.63 vs. T2422.06�10.46%, P�NS) (Fig. 2B, D, and I). The same statis-tically significant difference between the two study groups atT24 was also observed at the glomerular level (RAPA: T0
1.53�0.13 vs. T24 0.77�0.11; P�0.05; CNI: T0 1.67�0.29 vs.T24 1.47�0.28, P�NS; Fig. 2F, H, and J).
At T0, in both groups the protein expression of MMP-2and MMP-9 was confined in few glomerular and tubularepithelial cells, and in scattered interstitial cells, mainly asso-ciated with fibrotic areas. The number of MMPs positive glo-merular cells was increased at T24 in the RAPA group com-pared to the CNI group, although the difference did not reachstatistical significance (MMP-2 T0: 0.86�0.28 vs. 0.56�0.39;MMP-2 T24: 0.61�0.23 vs. 1.27�0.71; MMP-9 T0:0.52�0.46 vs. 0.23�0.32; MMP-9 T24: 0.52�0.44 vs.0.94�0.92; CNI vs. RAPA, P�NS; Fig. 3). No differences inthe MMPs tubulointerstitial expression were observed at T24between the 2 groups, although both groups presented anincrease in the number of positive MMP-2 cells in the inter-stitium, mainly associated to areas of fibrosis (data not shown).Immunohistochemistry for TIMP-1 showed a weak tubular posi-tivity at T0 in all CAN biopsies. At T24, a slight, although not signif-icant, reduction of TIMP-1 expression was observed in those kid-neys randomized to receive rapamycin compared to T0, while
FIGURE 2. PAI-1 protein expression at the tubulointerstitial (A-D) and glomerular (E-H) level in graft biopsies of the CNIand RAPA groups at T0 and T24. Glomerular and tubulointerstitial PAI-1 expression was reduced compared to the baselinein the RAPA group while was unchanged in the CNI group. For each treatment group, all the images are from a single patientand are representative of the whole group of patients. Quantification of PAI-1 specific staining was performed as describedin Materials and Methods. Values are expressed as means � SD. (Anti-PAI-1, �400 magnification).
130 Transplantation • Volume 85, Number 1, January 15, 2008

expression was unchanged, compared to the baseline, in the CNIgroup (data not shown).
Cell CultureTo confirm that PAI-1 downregulation in CAN biop-
sies was indeed due to a direct effect of rapamycin, we evalu-ated the ability of the drug to modulate PAI-1 gene expressionin cultured PTEC. To this purpose, tubular cells were stimu-lated with soluble CD40L and thrombin, two potential CANpathogenic mediators that have been previously shown toinduce PAI-1 gene and protein expression. Both thrombinand soluble CD40L induced a significant and time-dependentincrease in Akt phosphorylation, as demonstrated by westernblotting analysis (Fig. 4A-D). Interestingly, both sCD40L-and thrombin-induced PAI-1 gene expression in culturedproximal tubular cells was significantly reduced by preincu-bation with rapamycin (Fig. 4E and F).
DISCUSSIONIn this study, we demonstrated for the first time that
rapamycin can reduce in vivo, in patients with biopsy provenCAN, and in vitro, in activated PTEC, the expression ofPAI-1, a key mediator in the development and progression ofinterstitial fibrosis and glomerulosclerosis. The role of PAI-1
in the development of organ fibrosis was demonstrated inPAI-1 null mice. In this setting, Hattori et al. showed thatPAI-1 deficiency reduced lung fibrosis induced by bleomycinby enhancing plasmin generation, and the treatment withtranexamic acid, an inhibitor of plasmin formation, reversedthe protective effect of PAI-1 absence (21). Oda et al. subse-quently demonstrated a significant reduction in renal inter-stitial fibrosis after either unilateral ureteral obstruction orprotein overload in PAI-1 null mice compared with wild-typeanimals (22). Ma et al. elegantly showed that PAI-1 was ableto induce interstitial fibrosis even in the absence of trans-forming growth factor-beta (TGF-beta) activation in beta-6null mice (23).
Progressive interstitial fibrosis is one of the main his-tological features of CAN (1, 2). The possibility that PAI-1may play a pathogenic role in the development and pro-gression of interstitial fibrosis in CAN is suggested by sev-eral observations of an increased PAI-1 expression inexperimental and human chronic allograft dysfunction (9,24 –26). We have previously reported a striking increase inPAI-1 gene expression at the tubulointerstitial, glomeru-lar, and vascular level in CAN with a pattern resemblingthe one observed in the present set of graft biopsies (9).The gene expression level of this antifibrinolytic mediator
FIGURE 3. MMP-2 (A-D) and MMP-9 (E-H) protein expression at the glomerular level in graft biopsies of CNI and RAPAgroup at T0 and T24. The number of MMPs positive glomerular cells was increased at T24 in the RAPA group compared tothe CNI group, although the difference did not reach statistical significance. Values are expressed as means�SD. (Anti-MMP-2, �400 magnification; anti-MMP-9, �400 magnification).
© 2008 Lippincott Williams & Wilkins 131Pontrelli et al.

FIGURE 4. Thrombin- (A and B) and CD40L- (C and D) induced Akt activation in proximal tubular cells. Effect of rapa-mycin on thrombin- and sCD40L-induced PAI-1 gene expression (E and F). (A and C) Confluent, quiescent HK2 cells werestimulated either with thrombin (5 U/ml) for 5, 15, 30, and 60 min or with sCD40L (0.1 mg/ml) and then lysed in RIPA buffer.Equal amounts of protein from each cell lysate (40 �g) were separated by SDS-PAGE, transferred onto nitrocellulose filter,and than blotted with rabbit polyclonal anti-phospho-Akt antibody as described in Methods (A, top panel). The samemembrane was than stripped and immunoblotted again with mouse monoclonal anti-Akt antibody (A, bottom panel). Thefigure is representative of three experiments. (B and D) Quantification of Akt phosphorylation in HK2 cells was performedas described in Materials and Methods. Results are expressed as mean�SD of pAkt/Akt from three different experiments.*P�0.05 vs. basal condition. (E) Confluent HK2 cells were incubated for 6 hr with sCD40L (0.1 mg/ml) and sCD40L enhancer(1 mg/ml) or thrombin (5U/ml) in the presence or in the absence of rapamycin (20 ng/ml). PAI-1 gene expression wasevaluated by Northern blotting (upper panel), as described in Materials and Methods. Then 28S and 18S ribosomal RNAbands on ethidium bromide-stained gel were used to control the RNA loading (lower panel). (F) Quantification of PAI-1 geneexpression was performed as described in Materials and Methods, normalized to the intensity of 28S band and expressedas PAI-1/28S. The figure is representative of three experiments. Values are expressed as means�SD. *P�0.001 vs. basal;°P�0.001 vs. Thrombin; §P�0.0001 vs. CD40L.
132 Transplantation • Volume 85, Number 1, January 15, 2008

was directly and significantly correlated with the extent ofinterstitial fibrosis (9). In the present study, we confirmthe existence of a direct correlation between PAI-1 expres-sion and interstitial fibrosis. Indeed, in the patientsswitched to rapamycin, the reduction in PAI-1 protein ex-pression was associated with a reduced progression of in-terstitial fibrosis, whereas in the patients continuing on theCNI-based regimen, we observed a significant increaseover time of interstitial fibrosis.
In the present study, we report for the first time thatconversion from CNI to rapamycin may cause regression ofglomerulosclerosis in renal allograft recipients. Regression ofglomerulosclerosis has been extensively documented in sev-eral animal models of renal disease (27) and has been re-ported also in humans. Fioretto et al. showed regression ofglomerulosclerosis in patients with diabetic nephropathy un-dergoing pancreas transplantation (28). Our data seem tosuggest that, at least in this setting, an increase in the ECMdegradation through a decreased glomerular PAI-1 proteinexpression may be responsible for the regression. We andothers have extensively demonstrated that several soluble me-diators potentially involved in the immune-mediated as wellas in the nonimmune-mediated pathogenic mechanisms ofCAN, including TGF-�, thrombin, CD40L, can induce invitro a striking upregulation of PAI-1 gene expression in cul-tured resident renal cells (8). Thus, the ability of rapamycin tomodulate this common molecular pathway to fibrosis mayrepresent a powerful therapeutic tool in this scenario. In ad-dition, we demonstrated that rapamycin could also induceMMPs expression at the glomerular level. Although the dif-ference did not reach statistical significance, these data fur-ther support the idea that rapamycin may move the balancebetween ECM synthesis and degradation towards the latter.
Chronic CNI nephrotoxicity contribute to a large ex-tent to the development and progression of CAN (1–3). Sev-eral authors have shown in experimental models of chronicCNI nephrotoxicity an increased expression of PAI-1 (29,30). In addition, there are also in vitro evidences that CNImay induce PAI-1 gene and protein expression in culturedrenal resident cells (31). On the basis of these observations, itis conceivable that the decreased PAI-1 protein expression,which we observed in the patients switched from a CNI to arapamycin-based immunosuppressive protocol, might bedue to the withdrawal of the CNI. However, the in vitro abil-ity of rapamycin to inhibit PAI-1 gene expression seems tosuggest a direct role for mTOR inhibition in the reduction ofPAI-1 protein levels at T24. In vitro data suggested a key rolefor mTOR in the production of ECM components in differentcell types (11, 32). Inhibitions by rapamycin of ECM accumu-lation and subsequent fibrosis have been reported in severalanimal models of progressive organ fibrosis. The first reportof this antifibrotic effect was in carbon tetrachloride model ofhepatic fibrosis in rats (12). Zhu et al. demonstrated in thismodel a significant reduction by rapamycin of the extent ofhepatic fibrosis along with inhibition of collagen and TGF-beta gene and protein expression (12). A rapamycin analoguewas also effective in preventing lung fibrosis in the bleomycinmodel, where PAI-1 pathogenic role in ECM accumulationwas originally described (33). Coming to renal interstitial fi-brosis, Jain et al. were the first to report the ability of mTORinhibition by rapamycin to reduce the decline in renal func-
tion, the increase in proteinuria and the progressive intersti-tial fibrosis after ischemia-reperfusion injury (34). Recently,rapamycin was shown to reduce the extent of interstitialfibrosis and the expression of profibrotic genes in an experi-mental model of progressive renal damage induced by pro-teinuria and reduced renal mass (15). On the other hand,Shihab et al. (35) investigated both TGF-beta and PAI-1 geneexpression in rats receiving either rapamycin alone or in com-bination with cyclosporine and, surprisingly, they reportedan increased expression for both profibrotic mediators. Thesame authors demonstrated that mycophenolate may bluntthese profibrotic effects of mTOR inhibition (36). However,it should be considered that the induction of TGF-beta at thetubular level in these studies was demonstrated in rats ex-posed for a relatively short time to rapamycin, whereas in ourstudy patients received the mTOR inhibitor for 2 years.
Our experience does not allow us to exclude an addi-tional beneficial effect of mycophenolate in preventing inter-stitial fibrosis and PAI-1 protein expression, although our invitro data suggest rapamycin as the main drug responsible forthese effects. Although we observed a clear beneficial effect attissue level of rapamycin introduction in patients with biop-sy-proven CAN, we did not find any significant change ingraft function at the same time point (2 years). There could beseveral reasons for this controversial result: 1) the limitednumber of patients included in the study; 2) the low grade ofCAN at the first biopsy; 3) the relatively short time betweenthe first and the second biopsy; and 4) the highest sensitivityof the histological approach to detect chronic graft damage.Our study was not designed to prove any clinical advantage ofthe switch to rapamycin, but to investigate the molecularmechanisms of such beneficial effects. Indeed, our group andother investigators already demonstrated that CNI with-drawal and rapamycin introduction significantly improvesnot only graft function, but also graft survival, at least in theshort term (17, 37, 38).
We recognize that the number of patients enrolled inthis study is too small to draw any definitive conclusion andthat more data are necessary to further analyze this topic.
In conclusion, the present study support the hypothesisthat the long-term beneficial effect of CNI withdrawal andrapamycin introduction on kidney graft—previously dem-onstrated in several clinical studies—may be due, at least inpart, to the ability of rapamycin to inhibit at the renal level theexpression of PAI-1, a key pathogenic mediator of interstitialfibrosis and glomerulosclerosis.
REFERENCES1. Chapman JR, O’Connell PJ, Nankivell BJ. Chronic renal allograft dys-
function. J Am Soc Nephrol 2005; 16: 3015.2. Pascual M, Theruvath T, Kawai T, et al. Strategies to improve long-
term outcomes after renal transplantation. N Engl J Med 2002; 346: 580.3. Nankivell BJ, Borrows RJ, Fung CL, et al. The natural history of chronic
allograft nephropathy. N Engl J Med 2003; 349: 2326.4. Loskutoff DJ, Quigley JP. PAI-1, fibrosis, and the elusive provisional
fibrin matrix. J Clin Invest 2000; 106: 1441.5. Eddy AA. Plasminogen activator-inhibitor-1 and the kidney. Am J
Physiol Renal Physiol 2002; 283: F209.6. Erickson LA, Hekman CM, Loskutof DJ. The primary plasminogen
activator-inhibitors in endothelial cells, platelets, serum, and plasmaare immunologically related. Procl Nat Acad Sci USA 1985; 82: 8710.
7. Reilly CF, McFall RC. Platelet-derived growth factor and transforminggrowth factor-� regulate plasminogen activator inhibitor-1 synthesisin vascular smooth muscle cells. J Biol Chem 1991; 266: 9419.
© 2008 Lippincott Williams & Wilkins 133Pontrelli et al.

8. Rerolle JP, Hertig A, Nguyen G, et al. Plasminogen activator inhibitortype 1 is a potential target in renal fibrogenesis. Kidney Int 2000; 58:1841.
9. Grandaliano G, Di Paolo S, Monno R, et al. Protease-activated receptor1 and plasminogen activator inhibitor 1 expression in chronic allograftnephropathy. Transplantation 2001; 72: 1437.
10. Saunders RN, Metcalfe MS, Nicholson ML. Rapamycin in transplanta-tion: A review of the evidence. Kidney Int 2001; 59: 3.
11. Shegogue D, Trojanowska M. Mammalian target of rapamycin posi-tively regulates collagen type I production via a phosphatidylinositol3-kinase-independent pathway. J Biol Chem 2004; 279: 23166.
12. Zhu J, Wu J, Frizell E, et al. Rapamycin inhibits hepatic stellate cellproliferation in vitro and limits fibrogenesis in an in vivo model of liverfibrosis. Gastroenterology 1999; 177: 1198.
13. Murphy GJ, Bicknell GR, Nicholson ML. Rapamycin inhibits vascularremodeling in an experimental model of allograft vasculopathy andattenuates associated changes in fibrosis-associated gene expression.J Heart Lung Transplant 2003; 22: 533.
14. Wu M-J, Wen M-C, Chiu Y-T, et al. Rapamycin attenuates unilateralureteral obstruction-induced renal fibrosis. Kidney Int 69:2029 –2036,2006.
15. Bonegio RG, Fuhro R, Wang Z, et al. Rapamycin ameliorates proteinuria-associated tubulointerstitial inflammation and fibrosis in experimental mem-branous nephropathy. J Am Soc Nephrol 2005; 16: 2063.
16. Chen JK, Chen J, Neilson EG, Harris RC. Role of mammalian target ofrapamycin signaling in compensatory renal hypertrophy. J Am SocNephrol 2005; 16: 1384.
17. Stallone G, Infante B, Schena A, et al. Rapamycin for treatment ofchronic allograft nephropathy in renal transplant patients. J Am SocNephrol 2005; 16: 3755.
18. Racusen LC, Solez K, Colvin RB, et al. The Banff 97 working classifica-tion of renal allograft pathology. Kidney Int 1999; 55: 713.
19. Dahab GM, Kheriza MM, El-Beltagi HM, et al. Digital quantification offibrosis in liver biopsy sections: Description of a new method by Pho-toshop software. J Gastroenterol Hepatol 2004; 19: 78.
20. Ryan MJ, Johnson G, Kirk J, et al. HK-2: An immortalized proximaltubule epithelial cell line from normal human kidney. Kidney Int 1994;45: 48.
21. Hattori N, Degen JL, Sisson TH, et al. Bleomycin-induced pulmonaryfibrosis in fibrinogen-null mice. J Clin Invest 2000; 106: 1341.
22. Oda T, Jung YO, Kim HS, et al. PAI-1 deficiency attenuates the fibro-genic response to ureteral obstruction. Kidney Int 2001; 60: 587.
23. Ma LJ, Yang H, Gaspert A, et al. Transforming growth factor-beta-dependentand-independentpathwaysof inductionoftubulointerstitial fibrosis in�6–/–mice. Am J Pathol 2003; 163: 1261.
24. Revelo MP, Federspiel C, Helderman H, Fogo AB. Chronic allograft
nephropathy: Expression and localization of PAI-1 and PPAR-gamma.Nephrol Dial Transplant 2005; 20: 2812.
25. Lahlou A, Peraldi MN, Thervet E, et al. Chronic graft dysfunction inrenal transplant patients: Potential role of plasminogen activator inhib-itor type 1. Transplantation 2002; 73: 1290.
26. Wang Y, Pratt JR, Hartley B, et al. Expression of tissue type plasmino-gen activator and type 1 plasminogen activator inhibitor, and persistentfibrin deposition in chronic renal allograft failure. Kidney Int 1997; 52:371.
27. Fogo A. Regression versus progression of chronic kidney disease. Neph-rol Dial Transplant 2006; 21: 281.
28. Fioretto P, Steffes MW, Sutherland DE, et al. Reversal of lesions ofdiabetic nephropathy after pancreas transplantation. N Engl J Med1998; 339: 69.
29. Duymelinck C, Deng JT, Dauwe SE, et al. Inhibition of the matrixmetalloproteinase system in a rat model of chronic cyclosporine ne-phropathy. Kidney Int 1998; 54: 804.
30. Shihab FS, Andoh TF, Tanner AM, et al. Role of transforming growthfactor-beta 1 in experimental chronic cyclosporine nephropathy. Kid-ney Int 1996; 49: 1141.
31. Waiser J, Dell K, Bohler T, et al. Cyclosporine A up-regulates the ex-pression of TGFbeta1.and its receptors type I and type II in rat mesan-gial cells. Nephrol Dial Transplant 2002; 17: 1568.
32. Ivarsson M, McWhirter A, Borg TK, Rubin K. Type I collagen synthesisin cultured human fibroblasts: regulation by cell spreading, platelet-derived growth factor and nteractions with collagen fibers. Matrix Biol1998; 16: 409.
33. Simler NR, Howell DC, Marshall RP, et al. The rapamycin analogueSDZ RAD attenuates bleomycin-induced pulmonary fibrosis in rats.Eur Respir J 2002; 19: 1124.
34. Jain S, Bicknell GR, Whiting PH, Nicholson ML. Rapamycin reducesexpression of fibrosis-associated genes in an experimental model ofrenal ischemia reperfusion injury. Transplant Proc 2001; 33: 556.
35. Shihab FS, Bennett WM, Yi H, et al. Sirolimus increases transforminggrowth factor beta1 expression and potentiates chronic cyclosporinenephrotoxicity. Kidney Int 2004; 65: 1262.
36. Shihab FS, Bennett WM, Yi H, et al. Combination therapy with siroli-mus and mycophenolate mofetil: Effects on the kidney and on trans-forming growth factor-beta1. Transplantation 2004; 77: 683.
37. Bumbea V, Kamar N, Ribes D, et al. Long-term results in renal trans-plant patients with allograft dysfunction after switching from cal-cineurin inhibitors to sirolimus. Nephrol Dial Transplant 2005; 20:2517.
38. Mulay AV, Cockfield S, Stryker R, et al. Conversion from calcineurininhibitors to sirolimus for chronic renal allograft dysfunction: A sys-tematic review of the evidence. Transplantation 2006; 82: 1153.
134 Transplantation • Volume 85, Number 1, January 15, 2008




















