
The posterior segmental maxillary osteotomy: Recent applications
Upload
independentCategory
view
0download
0
COL4A3/COL4A4 Mutations Producing FocalSegmental Glomerulosclerosis and Renal Failure inThin Basement Membrane Nephropathy
Konstantinos Voskarides,* Loukas Damianou,† Vassos Neocleous,‡ Ioanna Zouvani,§
Stalo Christodoulidou,† Valsamakis Hadjiconstantinou,† Kyriacos Ioannou,�
Yiannis Athanasiou,� Charalampos Patsias,¶ Efstathios Alexopoulos,** Alkis Pierides,�
Kyriacos Kyriacou,‡ and Constantinos Deltas*‡
*Department of Biological Sciences, University of Cyprus, ‡Cyprus Institute of Neurology and Genetics, andDepartments of §Histopathology and �Nephrology, Nicosia General Hospital, Nicosia, Cyprus; †Department ofNephrology, Evangelismos Hospital, Athens, Greece; ¶Department of Nephrology, Larnaca General Hospital,Larnaca, Cyprus; and **Department of Nephrology, Aristotle University of Thessalloniki, Greece
ABSTRACTMutations in the COL4A3/COL4A4 genes of type IV collagen have been found in �40% of cases of thin basementmembrane nephropathy, which is characterized by microscopic hematuria and is classically thought to causeproteinuria and chronic renal failure rarely. Here we report our observations of 116 subjects from 13 Cypriotfamilies clinically affected with thin basement membrane nephropathy. These families first came to our attentionbecause they segregated microscopic hematuria, mild proteinuria, and variable degrees of renal impairment, buta dual diagnosis of focal segmental glomerulosclerosis (FSGS) and thin basement membrane nephropathy wasmade in 20 biopsied cases. Molecular studies identified founder mutations in both COL4A3 and COL4A4 genesin 10 families. None of 82 heterozygous patients had any extrarenal manifestations, supporting the diagnosis ofthin basement membrane nephropathy. During follow-up of up to three decades, 31 of these 82 patients (37.8%)developed chronic renal failure and 16 (19.5%) reached end-stage renal disease. Mutations G1334E and G871Cwere detected in seven and three families, respectively, and were probably introduced by founders. We concludethat these particular COL4A3/COL4A4 mutations either predispose some patients to FSGS and chronic renalfailure, or that thin basement membrane nephropathy sometimes coexists with another genetic modifier that isresponsible for FSGS and progressive renal failure. The findings presented here do not justify the labelling of thinbasement membrane nephropathy as a benign condition with excellent prognosis.
J Am Soc Nephrol 18: 3004–3016, 2007. doi: 10.1681/ASN.2007040444
The phenotypic heterogeneity of inherited conditionsthatarecausedbymutations ineither theCOL4A3ortheCOL4A4 gene on chromosome locus 2q36–37(COL4A3 [MIM120070] and COL4A4 [MIM120131];http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?db �OMIM) extends from familial benign isolated micro-scopic hematuria as a result of thin basement membranenephropathy (TBMN), which is largely asymptomatic,to nephrotic syndrome and end-stage renal failure withsymptoms that are consistent with autosomal recessiveAlportsyndromewithprogressivehemor-rhagicnephri-tis, chronic kidney failure, sensori-neural deafness, andocular findings. Owing to their similar although hetero-geneous genetic background, it has been suggested thatthey be referred to as COL4A3/COL4A4 nephropathies.
The two genes are located head to head and code for thelong�3 and�4 collagen chains of type IV collagen that isthe main structural component of glomerular basement
Received April 13, 2007. Accepted July 2, 2007.
Published online ahead of print. Publication date available atwww.jasn.org.
L.D.’s current affiliation is Limassol General Hospital, Limassol, Cyprus.A.P.’s current affiliation is Ippokrateion Private Hospital, Nicosia, Cyprus.
Correspondence: Prof. Constantinos Deltas, Department of Bio-logical Sciences, University of Cyprus, Kallipoleos 75, 1678 Nico-sia, Cyprus. Phone: 00-357-22-892882; Fax: 00-357-22-892881;E-mail: [email protected]
Copyright © 2007 by the American Society of Nephrology
CLINICAL RESEARCH www.jasn.org
3004 ISSN : 1046-6673/1811-3004 J Am Soc Nephrol 18: 3004–3016, 2007

membranes (GBM) (for review, see Kashtan1 and Tryggvason andPatrakka2).
The autosomal recessive form of Alport Syndrome, whichaccounts for 14% of all Alport cases, was initially linked togenes COL4A3 and COL4A4 through linkage analysis and thenby identification of causative mutations.3 The majority of Al-port cases, 85%, are accounted for by mutations in theX-linked form with mutations in COL4A5.1 A small propor-tion of patients, on the order of 1%, are also accounted for bymutations in either the COL4A3 or the COL4A4 gene, exceptthat these present in heterozygosity and are described as auto-somal dominant Alport syndrome (ADAS).4 –7 ADAS is de-scribed as a milder form of the disease with later age at onsetand with not all classical symptoms.
Lemmink et al.8 were the first to describe mutations in theCOL4A4 gene in cases of benign familial hematuria. Soon after,various other groups9 –18 presented patients with a spectrum ofsymptoms and different diagnoses that were attributed to mu-tations in either of the two genes. Of particular interest is thatseveral mutations in heterozygosity can lead to isolated micro-scopic hematuria that is histologically characterized by TBMN,whereas others can occasionally lead to more severe symptomsand ADAS. TBMN is the most common cause of persistentmicroscopic hematuria in children and adults; other commoncauses include IgA nephropathy and Alport syndrome.19 –21
TBMN has a frequency of �1% in the general population,most times described as an inherited, nonprogressive disor-der.2,22 Clinically, the critical question is how to discriminate ina young patient between true benign familial hematuria causedby nonprogressive TBMN and ADAS, which could presentwith microscopic hematuria and thin basement membranes atits early stages, only to progress later to proteinuria and severeglomerular disease that nearly always leads to ESRD.19
After the first link between TBMN with familial hematuriaand COL4A3/COL4A4 mutations was made a decade ago, sev-eral groups8 –18 in different countries investigated and pub-lished cases with disparate clinical presentation. In approxi-mately 40% of families with TBMN, the phenotype has beenshown to segregate with COL4A3/COL4A4, whereas the restcould be attributed to spontaneous new mutations, preventingclear-cut linkage analysis results, or incomplete penetrance ormutations in other, as-yet-unknown genes. A predominantfeature was the significant allelic and phenotypic heterogeneityin the presence also of incomplete penetrance.2,20,23,24 Of im-portance is also that TBMN in several reports (reference 21 andreferences therein) was shown to be associated with other glo-merulopathies, such as IgA nephropathy, FSGS, minimal-change disease, mesangioproliferative glomerulonephritis,and others.
In this study, we describe a cohort of Greek-Cypriot pa-tients with renal disease of variable severity, characterized inadolescence and early adult life by familial microscopic hema-turia and TBMN in all cases in which renal biopsies were per-formed. A striking common feature in a significant percentageof affected older members was the presence of establishedchronic renal failure (CRF) or even ESRD, which on renal bi-opsy was due to advanced segmental glomerulosclerosis. Thesepatients did not develop any visual or auditory deficits; how-ever, in subsequent years, they developed proteinuria, ne-phrotic syndrome, and progressive CRF; the histologic picture
of FSGS is an important association that has only infrequentlybeen described in the past.25–27
RESULTS
Light and Electron Microscopy Observations on RenalBiopsies Revealed FSGS and TBMNThe common histologic picture of FSGS in 20 renal biopsies of13 families that segregated microscopic hematuria and CRFwas the initial main finding that prompted us to investigatethem clinically and molecularly. In patients with establishedCRF, there were sclerosed glomeruli up to 50%, and the re-maining glomeruli showed focal segmental lesions of sclerosisthat were characterized by increased matrix, causing oblitera-tion of the capillary lumina, and by hyaline material deposi-tion. Distribution of lesions within the tuft was variable, affect-ing both peripheral and perihilar segments. Sclerosedsegments formed adhesions to Bowman’s capsule (Figure 1A).
Figure 1. (A) Light microscope showing a glomerulus with FSGS.This biopsy is from a patient from family CY5303, carrying muta-tion 3854delG in COL4A4, performed at age 40. (B) Electronmicrograph showing an increase in mesangial matrix (M), as wellas partial occlusion of capillary lumina. Thinning of GBM thatmeasure 250 nm is also seen (arrows). Note the marked fusion ofpodocytes (arrowheads). This biopsy is from a patient also offamily CY5303, performed at age 47. N, nucleus; L, lumina; RBC,red blood cell. Magnifications: �250 in A; �9000 in B.
CLINICAL RESEARCHwww.jasn.org
J Am Soc Nephrol 18: 3004–3016, 2007 COL4A3/COL4A4 Mutations in TBMN/FSGS 3005
These changes were much milder in patients who underwentbiopsy early, when they had normal serum creatinine and onlyshowed microscopic hematuria with minimal proteinuria. Insome glomeruli, periglomerular fibrosis was evident. Immu-nofluorescence results were essentially negative. None of ninebiopsies that also were submitted for electron microscopy(EM) showed electron-dense deposits. All EM biopsy speci-mens showed mild to moderate segmental increase in mesan-gial matrix with variable folding and collapse of GBM. Themain ultrastructural features included thinning of GBM andeffacement of the podocytes foot processes (Figure 1B). Theaverage thickness of the GBM did not exceed 250 nm, whereasthe thinning of the GBM varied from extensive to focal areas ofthinning. In these attenuated GBM regions, the GBM segmentwidths did not exceed 150 nm. None of the biopsies showedsplitting or lamellation. In one patient of family CY5303, theGBM thickness had an average measurement of 300 nm, whichis borderline normal.28
DNA Linkage Analysis around Candidate LociRevealed Strong Linkage to 2q36–37, the Locus forCOL4A3/COL4A4Starting with a possible diagnosis of familial FSGS, we initiallysuspected a primary form of autosomal dominant FSGS, althoughwe did not exclude the possibility that FSGS was of secondaryetiology. Taking into consideration also the uniform existence offamilial hematuria, we proceeded with linkage analysis usingpolymorphic markers at four relevant loci: 19q13.1 for ACTN4
gene (�-actinin 4),29,30 11q22 for TRPC6,31,32 6p12.3 (CD2AP),33
and 2q36–37 (COL4A3/COL4A4) (Table 1). Suggestive linkagewas found for two families at 19q13.1 and another family at11q22, whereas none linked to 6p12.3. For families linking to19q13.1, mutation screening was performed for ACTN4 and alsofor NEPH3 and WTIP, which are important genes expressed inpodocytes and mapped in proximity to ACTN4.34,35 No muta-tions segregating with the disease were identified. One large fam-ily, CY5308, did not show linkage to any of the tested candidateloci. It is interesting that one family, CY5306, provided a positivelogarithm of odds (LOD) score suggestive of linkage at two loci,19q13.1 and 2q36–37. Five other families provided positive link-age for locus 2q36–37, which harbors the autosomal genes fortype IV collagen of the basement membranes, COL4A3/COL4A4.The maximum total LOD score at locus 2q36–37 for these sixfamilies was 5.8 at recombination fraction � � 0.06 (markerD2S1363, penetrance 0.7). Four haplotypes were constructed withmarkers D2S1363, D2S159, D2S401, and D2S439, which co-seg-regated with the disease in all six families. Haplotype K was com-mon in four families, haplotype C was found in one family, andhaplotype Ky was found also in one family (data not shown).Subsequently, haplotype K was documented in three additionalfamilies and haplotype Ky in two additional small families.
Mutation Screening and DNA Sequencing IdentifiedThree Mutations in 10 of the 13 FamiliesGenetic screening with SURVEYOR endonuclease (Trans-genomic, Cheshire, United Kingdom) and DNA sequencing of
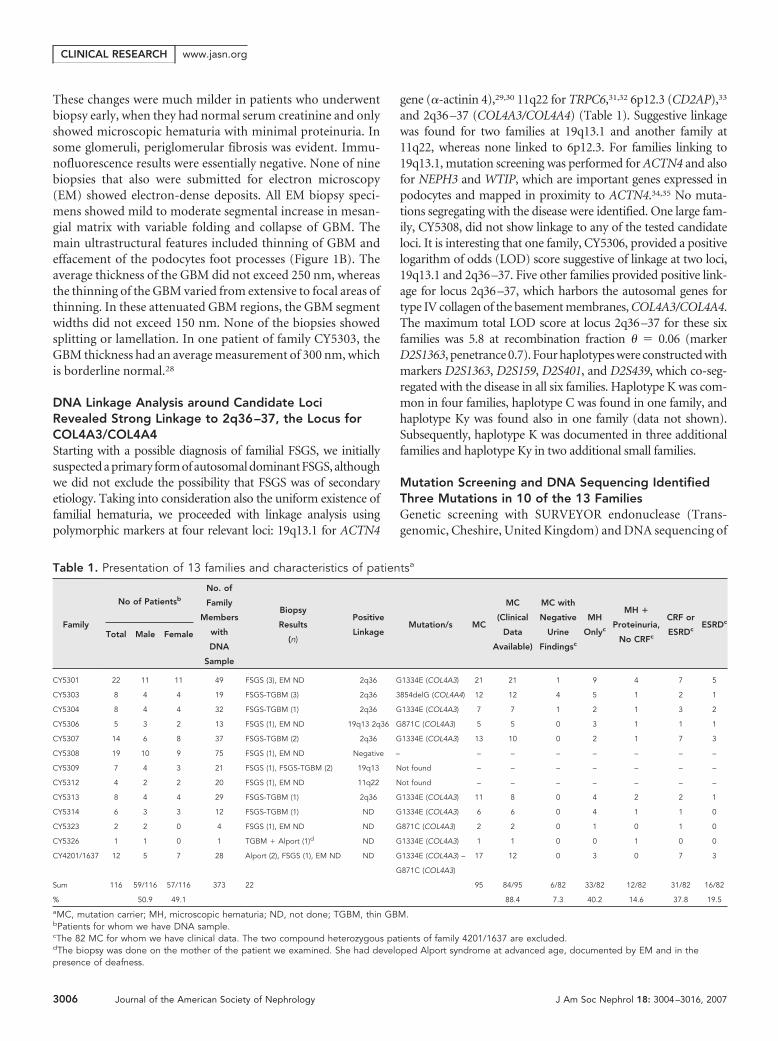
Table 1. Presentation of 13 families and characteristics of patientsa
Family
No of Patientsb
No. of
Family
Members
with
DNA
Sample
Biopsy
Results
(n)
Positive
LinkageMutation/s MC
MC
(Clinical
Data
Available)
MC with
Negative
Urine
Findingsc
MH
Onlyc
MH �
Proteinuria,
No CRFc
CRF or
ESRDcESRDc
Total Male Female
CY5301 22 11 11 49 FSGS (3), EM ND 2q36 G1334E (COL4A3) 21 21 1 9 4 7 5
CY5303 8 4 4 19 FSGS-TGBM (3) 2q36 3854delG (COL4A4) 12 12 4 5 1 2 1
CY5304 8 4 4 32 FSGS-TGBM (1) 2q36 G1334E (COL4A3) 7 7 1 2 1 3 2
CY5306 5 3 2 13 FSGS (1), EM ND 19q13 2q36 G871C (COL4A3) 5 5 0 3 1 1 1
CY5307 14 6 8 37 FSGS-TGBM (2) 2q36 G1334E (COL4A3) 13 10 0 2 1 7 3
CY5308 19 10 9 75 FSGS (1), EM ND Negative – – – – – – – –
CY5309 7 4 3 21 FSGS (1), FSGS-TGBM (2) 19q13 Not found – – – – – – –
CY5312 4 2 2 20 FSGS (1), EM ND 11q22 Not found – – – – – – –
CY5313 8 4 4 29 FSGS-TGBM (1) 2q36 G1334E (COL4A3) 11 8 0 4 2 2 1
CY5314 6 3 3 12 FSGS-TGBM (1) ND G1334E (COL4A3) 6 6 0 4 1 1 0
CY5323 2 2 0 4 FSGS (1), EM ND ND G871C (COL4A3) 2 2 0 1 0 1 0
CY5326 1 1 0 1 TGBM � Alport (1)d ND G1334E (COL4A3) 1 1 0 0 1 0 0
CY4201/1637 12 5 7 28 Alport (2), FSGS (1), EM ND ND G1334E (COL4A3) –
G871C (COL4A3)
17 12 0 3 0 7 3
Sum 116 59/116 57/116 373 22 95 84/95 6/82 33/82 12/82 31/82 16/82
% 50.9 49.1 88.4 7.3 40.2 14.6 37.8 19.5
aMC, mutation carrier; MH, microscopic hematuria; ND, not done; TGBM, thin GBM.bPatients for whom we have DNA sample.cThe 82 MC for whom we have clinical data. The two compound heterozygous patients of family 4201/1637 are excluded.dThe biopsy was done on the mother of the patient we examined. She had developed Alport syndrome at advanced age, documented by EM and in thepresence of deafness.
CLINICAL RESEARCH www.jasn.org
3006 Journal of the American Society of Nephrology J Am Soc Nephrol 18: 3004–3016, 2007

COL4A3/COL4A4 revealed the same substitution of A nucleo-tide for G at position 4001, converting glycine 1334 to gluta-mate (G1334E) in seven families, in whom the disease co-seg-regated with haplotype K (Figure 2). It is widely known that inthe collagenous domain of all collagens, the glycines are indis-pensable, because they are the smallest amino acids that canaccommodate themselves where the three chains meet to formthe triple helix. The common haplotype and same mutation inseven families raises the possibility for a founder effect.36 –38
Mutation G1334E had been previously reported in a Cypriotpatient.39
In three families who carried haplotype Ky, a substitution ofa G nucleotide at position 2611 by T resulted in conversion ofglycine 871 to cysteine (Gly871Cys; Figure 2), also strengthen-ing the hypothesis of a founder ancestor. In family CY4201/CY1637 that segregates classic autosomal Alport syndromeand microscopic hematuria, both of these mutations werefound (Figure 2C). The parents are heterozygous, whereastheir son and one daughter inherited both mutations and de-veloped classic Alport syndrome. It is interesting that themother, who is heterozygous for G1334E, also developed CRFin her late 50s after donating one of her kidneys to her son.Testing of the mutations on extended pedigrees confirmedtheir co-segregation with the disease phenotype. In addition,none of 56 or 141 unrelated healthy individuals of the generalpopulation carried mutation G871C or G1334E, respectively.Several polymorphisms were also detected (Table 2). Quickand efficient examination of samples for both mutations wasperformed by restriction enzyme digestion (Figure 2A).
Family CY5303 had haplotype C (data not shown) at locus2q36 –37, and DNA sequencing revealed a single nucleotidedeletion, 3854delG, which results in frameshift at Ser1217 andintroduces a stop codon at amino acid residue 1287. Mutation3854delG did not create or abolish a known restriction recog-nition site; therefore, a new recognition site was engineered inby designing a modified 5� primer for PCR amplification. Thismutation was previously reported in one patient.40
Molecular diagnosis of all family members revealed thatsix individuals, representing 7.3% of the 82 individuals forwhom we had detailed clinical data, had inherited the mu-tation but were healthy, with no signs of hematuria on morethan one urine examination, indicating incomplete pen-etrance of 92.7% (Table 1). In addition, three patients, twowith documented microscopic hematuria and one with doc-umented TBMN on biopsy, did not carry the relevant mu-tation, obviously representing phenocopies (Figure 3), mostprobably because their clinical condition had a differentcause.
For a comprehensive view of our patient cohort, we dividedthem into four age groups and classified them according tosymptom severity (Figure 4). Evidently, on long-term follow-up, an increasing proportion of mutation carriers progress toCRF. When we confined the observation to patients who wereolder than 51 yr, 28 (80%) of 35 had developed CRF or ESRD,
and 16 (46%) of 35 had progressed to ESRD, a finding notconforming to the term of benign familial hematuria.
DISCUSSION
We studied families and patients of the Greek-Cypriot com-munity with a population of 646,400. We presented for the firsttime detailed clinical and molecular data pertaining toCOL4A3/COL4A4 mutations, a field that in recent years hasattracted much attention, perhaps because it has been docu-mented that such mutations in homozygosity are responsiblefor the rare autosomal recessive Alport syndrome phenotype,and in heterozygosity for the more common phenotype of be-nign familial hematuria with TBMN.
Heterozygous COL4A3/COL4A4 Mutations as a Causeof TBMN Associated with FSGSIn 20 renal biopsies from patients who were aged 32 to 63 yrand belonged to 13 unrelated families with familial hematuria,there was FSGS in the presence of TBMN. In 10 (77%) of thesefamilies, mutations were found in the COL4A3/COL4A4 locus.All families had interesting clinical and genetic features with aspectrum of symptoms ranging from totally asymptomatic iso-lated microscopic hematuria to proteinuria and CRF caused byFSGS, relatively frequently leading to ESRD. There was evi-dence for phenocopies; three patients presented with hematu-ria without having inherited the respective mutation. Therewas also proof of reduced penetrance of 92.7%; six of 82 indi-viduals who carried a mutation were healthy.
All 15 patients who underwent biopsy and belonged to the10 families who segregated familial hematuria and TBMN inthe presence of COL4A3/COL4A4 mutations also had FSGS(Table 1). This association of FSGS with TBMN is a strikingfinding that allows the hypothesis that either FSGS is a second-ary development of the COL4-linked TBMN or the result of aco-inherited glomerulopathy that is actually responsible forthe progressive nature of the disease, a likelihood that also hasbeen suggested by others.27 Our data do not allow us to distin-guish with certainty between these two situations. Norby andCosio27 suggested that all patients with suspected TBMN andheavy proteinuria or renal insufficiency should undergo a renalbiopsy examination. Nieuwhof et al.25 published the results ofa prospective study with a 12-yr follow-up of 19 patients withTBMN and microscopic (18 of 19) or macroscopic (1 of 19)hematuria. They were the first to note clearly the associationbetween TBMN and late-onset renal impairment on long-termfollow-up in elderly patients. In six first-degree relatives ofthese 19 patients, ESRD was established, prompting the au-thors to conclude that TBMN predisposes to premature glo-merular obsolescence that may lead to late-onset renal insuffi-ciency. Sue et al.,41 in a retrospective review of 658 nativekidney biopsies, concluded that TBMN on its own is a benigncondition not contributing to the final renal outcome, which isdetermined only by another, coexisting glomerulopathy. To a
CLINICAL RESEARCHwww.jasn.org
J Am Soc Nephrol 18: 3004–3016, 2007 COL4A3/COL4A4 Mutations in TBMN/FSGS 3007
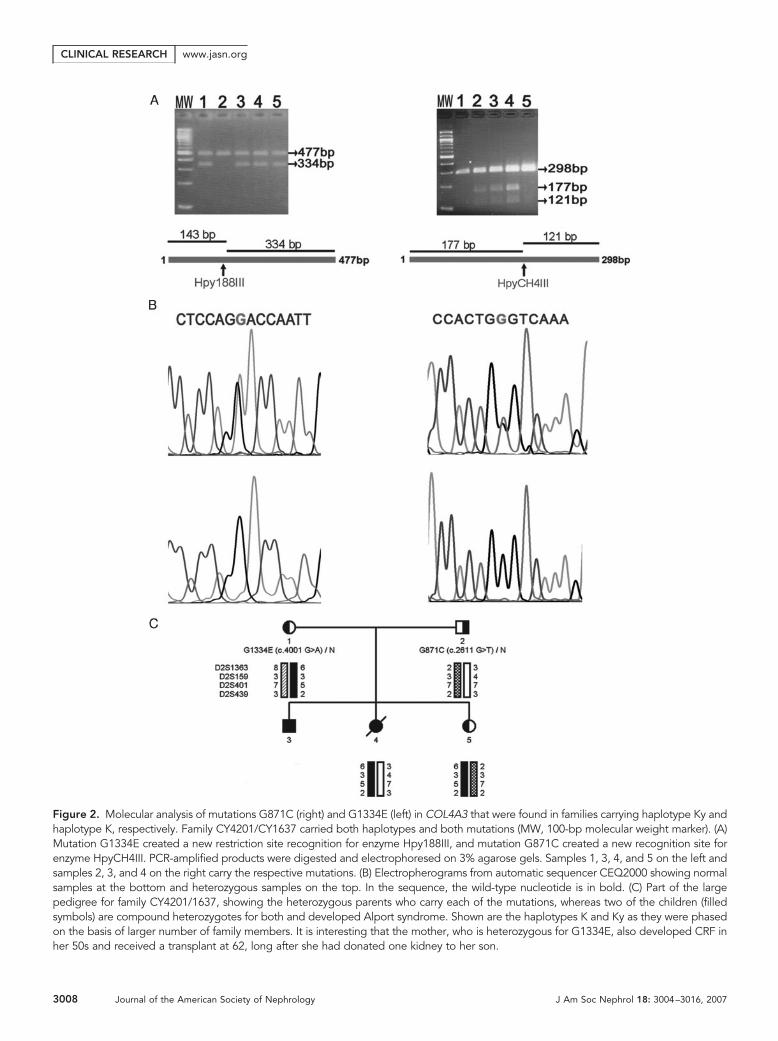
Figure 2. Molecular analysis of mutations G871C (right) and G1334E (left) in COL4A3 that were found in families carrying haplotype Ky andhaplotype K, respectively. Family CY4201/CY1637 carried both haplotypes and both mutations (MW, 100-bp molecular weight marker). (A)Mutation G1334E created a new restriction site recognition for enzyme Hpy188III, and mutation G871C created a new recognition site forenzyme HpyCH4III. PCR-amplified products were digested and electrophoresed on 3% agarose gels. Samples 1, 3, 4, and 5 on the left andsamples 2, 3, and 4 on the right carry the respective mutations. (B) Electropherograms from automatic sequencer CEQ2000 showing normalsamples at the bottom and heterozygous samples on the top. In the sequence, the wild-type nucleotide is in bold. (C) Part of the largepedigree for family CY4201/1637, showing the heterozygous parents who carry each of the mutations, whereas two of the children (filledsymbols) are compound heterozygotes for both and developed Alport syndrome. Shown are the haplotypes K and Ky as they were phasedon the basis of larger number of family members. It is interesting that the mother, who is heterozygous for G1334E, also developed CRF inher 50s and received a transplant at 62, long after she had donated one kidney to her son.
CLINICAL RESEARCH www.jasn.org
3008 Journal of the American Society of Nephrology J Am Soc Nephrol 18: 3004–3016, 2007

similar conclusion had arrived Nogueira et al.,26 who had pre-sented eight patients with TBMN, five of whom had heavyproteinuria and nephrotic syndrome as a consequence of asso-ciated FSGS.
van Paassen et al.,42 in their comprehensive elegant work,recognized that in a series of 22 patients who originally hadbeen classified as having primary FSGS with microscopic he-maturia, 50% had secondary FSGS as a result of TBMN,
thereby admitting that FSGS can develop on the genetic back-ground of TBMN. On the contrary, Haas43 reported that noneof 37 biopsies of patients who had TBMN and presented withpersistent or recurrent hematuria showed FSGS, whereas inseven of 10 patients who had FSGS and were incidentally dis-covered to have TBMN, their FSGS was likely of primary na-ture.
In our cohort, in a total of 82 patients from 10 families
Table 2. Mutations and polymorphisms found in the studied genesa
Type of Variant/Gene Nucleotide Change Exon Reference/Database
MutationCOL4A3
G871C 2611 G�T 32 This studyG1334E 4001 G�A 45 39
COL4A43854delG (frameshift at Ser1217, Stop at 1287) 39 40
PolymorphismCOL4A3
IVS5 �73C/T Intron 5 Ensembl databaseP141L 422 C/T 7 12
E162G 485 A/G 9 39
IVS15 �30G/A Intron 15 Ensembl databaseIVS17 �80T/C Intron 17 This studyK834R 2501 A/G 32 15
IVS41 �110T/G Intron 41 This studyIVS42 �66C/T Intron 42 This studyQ1495R 4484 A/G 49 12
IVS50 �67delA Intron 50 Ensembl databaseCOL4A4
P482S 1444 C/T 21 11
G545A 1634 G/C 23 40
L1004P 3011 T/C 33 40
G1198G 3594 G/A 39 11
K1228K 3684 G/A 39 11
IVS40 �9G/C Intron 40 Ensembl databaseIVS41 �34T/C Intron 41 Ensembl databaseM1327V 3979 A/G 42 40
P1360P 4080 A/G 42 11
IVS43 �36G/A Intron 43 Ensembl databaseP1403S 4207 C/T 44 40
IVS44 �24C/T Intron 44 Ensembl databaseV1516V 4548 A/G 47 11
ACTN4P179P 537 G/A 5 Ensembl database
TRPC6IVS3 �100G/A Intron 3 Ensembl databaseN561N 1683 T/C 6 Ensembl databaseIVS10 �138C/T Intron 10 Ensembl databaseIVS12 �20–22delCTT Intron 12 NCBI database
NEPH3�170 508 G/A 4 Ensembl database�351� 1053 G/T 8 Ensembl databaseV353M 1057 G/A 9 Ensembl database
WTIPIVS4 �8G/C Intron 4 This studyIVS5 �25C/T Intron 5 Ensembl
aNucleotide number 1 is the A of the first ATG codon of translation. NCBI, National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/)
CLINICAL RESEARCHwww.jasn.org
J Am Soc Nephrol 18: 3004–3016, 2007 COL4A3/COL4A4 Mutations in TBMN/FSGS 3009

with detailed clinical information, 31 (38%) patients hadprogressed to CRF and 16 (19.5%) had progressed to ESRDthat required renal replacement therapy (Table 1). Confin-ing the statistics to patients who were older than 51 yr, 28
(80%) of 35 had developed CRF or ESRD and 16 (46%) of 35had progressed to ESRD (Figure 4), representing the highestpercentages of patients who had TBMN and progressed toFSGS and ESRD, to our knowledge.19 These high figures are
Figure 3. CY5301 is one of our largest pedigrees carrying mutation G1334E. Carriers of this same mutation present with a spectrumof symptoms ranging from microscopic hematuria in the presence of TBMN to ESRD in five patients (see also Table 1). Samples wereavailable from individuals in generations III and IV. It is interesting that we noticed that individual III4 represents phenocopy, becauseshe has biopsy-proven TBMN in the presence of microscopic hematuria and in the absence of the mutation. Individual IV25 carries theG1334E mutation but is asymptomatic (reduced penetrance). Monozygotic twins IV9 and IV10, who were shown to carry the mutation,exhibited identical phenotype. Some individuals of the youngest generation who seem healthy have not been examined clinically ormolecularly.
CLINICAL RESEARCH www.jasn.org
3010 Journal of the American Society of Nephrology J Am Soc Nephrol 18: 3004–3016, 2007

not the norm in existing literature, where most times TBMNis described as a benign condition with excellent prognosis.Another likely hypothesis is that the mutations presentedhere are more pathogenic and predispose a subset of pa-tients to severe phenotype. Perhaps future work could focuson detailed examination of patients carrying the same mu-tation to evaluate carefully the intrafamilial variability andassess the impact of the mutation on pathology. Anotherapproach is to create transgenic mice with this and anothermutation and assess their long-term phenotypes; however,many similar mutations substituting the essential glycineresidues have been described by others,24 without notingsuch high percentage of adverse outcome.
Kashtan, in his review,1 asked the rhetorical question ofwhether TBMN and benign familial hematuria should be con-sidered synonymous, and he provided the answer: “Probablynot.” Our work here enhances this propensity; in fact, our datasuggest that an ever-increasing percentage of COL4A3/COL4A4 mutation carriers may shift to the group of patientswith CRF by advanced age (Figure 4). It is reasonable to hy-pothesize that those who progress to CRF do so under theinfluence of yet-unknown modifier genes that on the back-ground of TBMN predispose to FSGS and CRF, although onecannot exclude that the modifier gene effect creates an age-dependent penetrance, as suggested by the results in Figure 4.Another explanation for not seeing this adverse outcome moreoften in many patients with TBMN could be the short fol-low-up of these patients, considering that in most cases thedisease course takes more than two decades to cause protein-uria and detectable renal impairment. One advantage in oursetup is our small communities, the frequent and systematicfollow-up, and the controlled delivery of services by a smallteam of specialists.
Mutation Screening, DNA Sequencing, and FounderEffectsIt is worth commenting that the strategy used for mutationscreening of these multiexon large genes, with the systematicuse of SURVEYOR enzyme accompanied by direct automatedDNA sequencing, is very reliable and effective and may be con-sidered for use in routine clinical diagnostic set-up. Themethod is suitable for larger DNA fragments (we applied it tofragments of nearly 800 bp), and it allows accurate localizationof the mismatch upon electrophoresis. The future develop-ment of an automated method for mutation screening willprove to be instrumental in cases in which a kidney biopsy isnot possible, and the noninvasive molecular diagnosis may re-place renal biopsy and EM for the diagnosis of TBMN. Theavailability of fast and reliable molecular testing will be evenmore helpful in distinguishing between IgA nephropathy andTBMN or Alport syndrome in early stages, and it will be ofparamount importance in cases of potential living-related kid-ney donors.
Finally, mutations G1334E and G871C on common haplo-types in seven and three families, respectively, suggest foundereffects, something not uncommon among the Greek-Cypriotpopulation. There are two good examples for dominant dis-eases. One is the PKD2 mutation R742X in a family with thetype 2 variant of polycystic kidney disease, with �80 affectedindividuals in Lythrodontas village.37 The second refers tomedullary cystic kidney disease 1, for which we reported on sixlarge families in whom the disease segregates with a commonMCKD1 gene haplotype. All patients are from the area of Pafosnear the west coast, geographically clustered in a triangle ofthree neighboring villages.44 For a recessive disorder, a classicexample is the CFTR mutation F508del, with a carrier fre-quency of 1:14 in the village of Athienou, near Nicosia, the
Figure 4. Diagrammatic representation of 76 patients who are heterozygous for a COL4A3/COL4A4 mutation (the two compoundheterozygous patients of family CY4201/1637 are excluded). They are classified in four age intervals on the basis of three clinicalsymptoms, namely microscopic hematuria, proteinuria, and CRF. In age interval 11 to 30, 18 patients; 31 to 50, 23 patients; 51 to 70,28 patients; 71 to 90, seven patients. Note that 80% (28 of 35) of patients aged 51 to 90 yr have progressed to CRF or even ESRD. Thesix patients who carried a mutation but were asymptomatic were not included in this representation.
CLINICAL RESEARCHwww.jasn.org
J Am Soc Nephrol 18: 3004–3016, 2007 COL4A3/COL4A4 Mutations in TBMN/FSGS 3011

capital36 (for an extensive description of founder effects inCyprus, see Deltas38).
Many publications emphasize that TBMN co-segregatingwith heterozygous COL4A3/COL4A4 mutations is a cause ofbenign familial hematuria with excellent prognosis. Our ex-perience with 82 patients from 10 families was that 38% ofpatients of all ages developed CRF and 19.5% progressed toESRD, thereby suggesting that the term “benign” is a mis-nomer, at least in our population. That 15 biopsies withFSGS were from families with familial hematuria, TBMN,and heterozygous COL4A3/COL4A4 mutations allows thehypothesis of a causal relationship. This is also supported bysimilar findings in a COL4A3�/� knockout murine modelthat develops TBMN coinciding with focal glomeruloscle-rosis and other renal pathology.45 In addition, the presenceof phenotypic heterogeneity allows the hypothesis that ge-netic modifiers and/or a co-inherited glomerulopathy maycontribute to the disease course. One way to deal with thesignificant variable expressivity in carriers of such muta-tions might be to refer to them collectively as COL4A3/COL4A4 nephropathies, thereby treating them under a uni-fying molecular terminology.23
CONCISE METHODS
PatientsPatients and healthy individuals belonged to 13 families from Cyprus
who had no documented relationship. A distant, unknown relation-
ship to one or more common ancestors cannot be excluded; in fact, it
is highly suggested on the basis of the molecular findings. All families
were initially selected through a proband because of FSGS in their
renal biopsies performed after the development of proteinuria and/or
mild renal failure, in the presence of microscopic hematuria. Addi-
tional family members were recruited on the basis of the observation
of familial microscopic hematuria as well as other renal symptoms,
such as proteinuria and/or renal failure. Under certain circumstances
(Table 1), several patients from the same family underwent biopsy.
Patients with clinical or histologic evidence of systemic lupus ery-
thematosus, vasculitis, collagen diseases, and drug consumption were
excluded. Patients with HIV or hepatitis B or C infection were also
excluded. CRF was defined as an elevated serum creatinine �1.5 mg/
100 ml. In total, 20 biopsies were performed percutaneously at ages
that ranged from 32 to 63 yr (mean age 45.5 yr). In one family, a renal
biopsy was available on the mother of the patient (family CY5326;
Table 1). In family CY4201/1637, two additional biopsies were done
in two young siblings who proved to be compound heterozygotes with
mutations in the COL4A3. They both developed early-onset Alport
syndrome with sensorineural deafness and underwent kidney trans-
plantation. Their mother carried mutation G1334E, and after donat-
ing one of her kidneys, she also gradually developed end-stage renal
failure and received a transplant at age 60. Her husband and his sib-
lings carried mutation G871C, showed microscopic hematuria, and
developed various degrees of CRF. The symptoms of the patients in
our cohort varied significantly, ranging from asymptomatic isolated
microscopic hematuria to additional proteinuria, up to CRF and
ESRD that required hemodialysis or kidney transplantation, perhaps
justifying the diagnosis of ADAS, although with an incomplete spec-
trum of symptoms.
Immunofluorescence, Light Microscopy, and EMRenal biopsy samples were processed for light microscopy and EM
and for direct immunofluorescence. For light microscopy, paraffin
sections were stained with hematoxylin and eosin, periodic acid-
Schiff, silver methenamine, and Masson’s trichrome. For immuno-
fluorescence, cryostat sections were incubated with a panel of FITC-
linked mouse antibodies against human IgA, IgG, IgM, C1q, C3, and
fibrin. For EM, specimens were processed as described previously28
and photographed in a JEM-1010 transmission electron microscope
(JEOL 1010, Tokyo, Japan). The diagnosis of TBMN was made at the
ultrastructural level as described previously46 and considering the
thickness of the GBM to be 300 to 400 nm in normal adults.
DNA Linkage AnalysisAll individuals were informed and gave their consent for participa-
tion. DNA from peripheral blood of 373 individuals in total, 116 of
whom were classified as affected on the basis of clinical criteria, was
isolated by one of two methods: Either a salting-out procedure47 or
the Qiagen kit (Qiagen, Hilden, Germany). DNA linkage analysis at
locus 2q36 –37 that contains the COL4A3/COL4A4 genes was per-
formed with polymorphic microsatellite markers D2S1363-0.8, Mb-
D2S159-0.43, Mb-D2S401-0.92, and Mb-D2S439 (Ensemble Data-
base; http://www.ensembl.org/Homo_sapiens/index.html), and the
results were processed for two-point linkage analysis using the
MLINK program of the LINKAGE package of programs.48 Statistical
analysis and LOD score estimation were performed using disease pen-
etrance varying from 0.5 to 1.0. Linkage analysis was also performed
for loci 19q13, 11q22, and 6p12.3, which contain genes, mutations in
which are responsible for primary FSGS.29 –33
Mutation Screening and DNA SequencingAll exons of candidate genes were screened for mutations using the
SURVEYOR endonuclease, which cleaves double-stranded DNA at
positions of mismatches.49 For this, the PCR-amplified genomic se-
quences were subjected to denaturation by heating to 95°C for 5 min.
Then the DNA was allowed to renature by cooling to 65°C for 30 min
and 25°C for 30 min. The renatured PCR-amplified DNA were incu-
bated with 0.5 �l of SURVEYOR enzyme at 42°C for 20 min and then
examined by electrophoresis on high-resolution Eurobio 3:1 agarose
gels, 3%. When cleavage was evident, DNA sequencing was per-
formed using a kit for Dye Terminator Cycle DNA sequencing from
Beckman Coulter and fractionated on an automatic DNA sequencer
(CEQ2000XL; Beckman Coulter, Fullerton, CA). The oligonucleotide
primers were designed to include single or multiple exonic coding
sequences,39,40 as well as at least 60 bp of splice junctions and intronic
regions (Primer_3 software; http://www-genome.wi.mit.edu/cgi-bin/
primer/primer3_www.cgi) (Tables 3 and 4).
Mutation G871C in COL4A3 created a new recognition site for
restriction enzyme HpyCH4III, and mutation G1334E created a rec-
ognition site for enzyme Hpy188III (New England Biolabs, Boston,
CLINICAL RESEARCH www.jasn.org
3012 Journal of the American Society of Nephrology J Am Soc Nephrol 18: 3004–3016, 2007

MA). These enzymes were used for easy examination of additional
family members and for testing a number of healthy individuals in the
general population to ascertain whether they are pathogenic muta-
tions or neutral variants. Mutation 3854delG in COL4A4 was tested
by amplification and digestion of the relevant sequence in exon 39.
Amplification was with the modified forward primer 3854delG-F:
ggaatacctgggctaaaaggggagagaggagaccctcg and the reverse primer
3854delG-R: tctggaggaatgtgggaccagagagtggcagaatagaag. The forward
primer was modified by substitution of the penultimate nucleotide of
G by C (italics) so that upon amplification of the mutant sequence, a
restriction recognition site for XhoI is created.
ACKNOWLEDGMENTS
This work was funded mainly through a grant by the Cyprus Research
Promotion Foundation (�� X/0505/02) and partly by the Cyprus
Ministry of Health, The Cyprus Kidney Association (scholarship to
Table 3. Oligonucleotide primers and conditions for PCR amplification and DNA sequencing of COL4A3 exons
Exon Sense (5�33�) Antisense (5�33�)PCR AnnealingTemperature
(oC)
PCR ProductSize (bp)
1 GACCGAGCCCTACAAAACC CTCAGCCTCGCCACTAGC 68.5 3582 GTCACCTGGCTCCTAACAGATAGT GGGAAATGGAACAACATACTGC 62 2973 TGCAAAGAGTCACCATGAAGTC GATGTGGAATTCTCTCCAGGTT 66 2964 AGCTTGGCATGTGACTGAGACT ACCACTCCTTTGACATCTACCA 66 3005 GCTATTCCCAGTTATAGTGGAGGA ATCCTGTCCTGCTTGTATTTCAGG 65 3746 GATAAGTCTTCATATCAGTCAGCCTGT CCTAAAGGAATGTTTTCAAGGGAAG 62 3727 CATGACCCAGTAGCCATAAGGAAT CTACCCCAGACTTCCCTAAAGTCAT 65 3078 GAGAGGATACACAATAGCAGAGAGG CTGAATGATTAAGGTCTGGATTATG 58 2489 CTCTGAGTACATAACTTGAAAAGCA GAAAAGAGAGACGAAGTAAAGAGAGG 58 28710 to 11 GAGGTGTTATGCTCTTCTGATTTTA CTAACAGTGACCCACATGAAGAAG 58 43912 to 13 ATTGGAACTTTGATGGGTTTAG GTAAAAGAGGTGCAGGGTCACA 60 49914 GTTAACACGAGGCACATTCATAGTT ATAGCTGCTCTGCCTGTGGAGTA 58 33815 TTCTCTCTTAGATGGCTCAGCACT GAGCTAGTTTGAGTTGGGCATCTT 65 29016 to 17 CCTGAGGTCAAGAGGAGATGAT CCTTGTACTTAGGTCCAGGGTTG 65 56018 GTAGATGGGATTCCTTACCCAGATAC GTCAAAGGGACACATAGTGACCAG 65 40319 GGGCCAAAATGAAGAAACTG CTTTCCTCACCCATGTCAATTC 55 27920 TCTGATATTTGTCTAGGTGAGAGTAGG AGCCTGGAAACACTTCTAGATCA 65 31121 GTGTTATGTACCTCTCCATTGTGC AAACCACACAGACTGATGGCTAAG 60 39822 ACCATGCTTTCTCAGTTGCAG CATCACTGACTGGTAGCTATTTGG 63 35723 GTTCTTTCTGAGGACTCAATGTAGCTT CTTCCAGTGTATTGACCCTTTTGT 60 21324 CTGTCTACTTAGAGTTGGCGTTCA AAGGCACCAAACTGGTAAGTGT 63 22225 TGAAAAGTTCTTGTCCACACTG GTTTCATTCTGCAGTTAGAGTGATG 63 39826 GATCTCAATGACAGCCTAACTGGT GAGAAGCTTCGTTAGCTCTAGAAACC 63 34827 TCGTATTTTCCGCTATCGTC AAGAGAGTCAGGGTGTTGTGTGT 63 22728 ATGAAGGAAAGTTGCTGATGTG GAACATCTTCTAAATATCCACAACAAA 63 21129 GCATCTCTAGCTGGTTGAGAGATAAG ATCTAGAGTGCTCTGCCAGATAAGTG 63 28330 to 31 CTCCATAACAGAGAGTCAATATCAGTG AGTCTCATGTCTCCTGCCCTTCT 63 69332 GTTAGTAGGGGAAAGCATTTGTGG CTATGTACAGTTGACAGAGCCACCT 64 29833 to 34 CGACCCATCTCCTAGACTAATACAGT CCTCACTGTCCTTATTTGTCAACC 64 84135 TCCCACGTAGCTGGGATTAC CTGCTGGAACACTATCAGAACAGA 64 30036 CATGCATGCAAGCAACAAGT ACACAGAAAATCTGGAGTCCTCAC 63 24137 AGCCATTCTTGGGAGAAAGC ACACCGAATCTGGGTACTTGTT 58 32138 GTGCTGGCAGATAGCAGATACTAA GATTTCAGGAGGGCTATACTCTGA 63 32339 CAGACCGTTTCAGTCACTGTTGTA CAGAAATCCAAACCTCAGGAGA 63 23040 to 41 CACTGGCAAGAAATAAATCCTCTTT ATATTCAACTGCTTTCTCTCCTTGA 63 48842 GAGTCTCAACTCAGTTTTTGTAGCTCT GGAAAGAAGAAGCAACAAGGTAACA 65.5 47743 CTCCCTGGCTGGCAATACT AACATCATGAGAATGGACTAATACAG 62 29344 GTTCTAAGTGGCCAGAGGAGTTATAG GTACTAACCCCAGCTGTCTGTACTTC 65 35445 to 46 GCACACTTCTAGTATTTGTCCTTAGAGTC GAAGTTGTATCAGCTGTTTCCAAAG 65 47747 AGGTGACTAATCATCCTAGCTTGC CATTTGATCCTTGGCCTTTG 55 39948 CCACTCTTCTCTAGGATTGCTTTC GGCCAGTGTTTTATTTGCTCTC 65 48749 to 50 GTTGTCTTTGTCCAGCTTTTGC GACCCTGGGAATCACATTTTAC 63 62151 CTCCCCTTTCTTTACTCACAGTTG AAATCATGGCTACTCGTTGACC 65 37752 ACGCCCGACCCTGTAATAAAT CAGATTAGAGACCCAGATCACAGAAC 65 397
CLINICAL RESEARCHwww.jasn.org
J Am Soc Nephrol 18: 3004–3016, 2007 COL4A3/COL4A4 Mutations in TBMN/FSGS 3013

K.V.), the University of Cyprus Research Activities 3/312, and Efsta-
thios Constantinides.
Parts of this article were presented in abstract form at the 13th
PanHellenic Conference of Nephrology; Rhodos, Greece; June 16 –19,
2004; the 38th European Human Genetics Conference; Amsterdam,
Netherlands; May 6 –9, 2006; and the 39th European Human Genetics
Conference; Nice, France; June 16 –19, 2007.
We thank the patients and their families for participating in this
study. We also thank Michalis Markou (PhD Cand, University of
Cyprus) for the installation and use of linkage analysis software and
Gregory Papagregoriou (PhD Cand, University of Cyprus) for assis-
tance with figure preparation.
DISCLOSURESNone.
REFERENCES
1. Kashtan CE: Alport syndrome and thin basement membrane disease.Curr Diagn Pathol 8: 349–360, 2002
Table 4. Oligonucleotide primers and conditions for PCR amplification and DNA sequencing of COL4A4 exons
Exon Sense (5�33�) Antisense (5�33�)PCR Annealing
Temperature (oC)PCR Product
Size (bp)
2 GGAAAGATTGTTGAAGTTACCCTTT TTCAGACGGTTTACATCCATAGAAC 60 3713 GCTAGTAGGAGCAGCCTCATAGAAAT GGTAAGAAAATTGAGTCCCAGAGG 58 2774 TCTTCTCATGGAGTCAAAGGTAAAC GTGAAAAACTTCGGCTGTGAAATAC 58 3275 AGTCTTCTGTAGGCCTGAATGAGT GTGCAGTAGTGCTGGTGAGTCTT 60 2816 AAAAATAGGTGACAATGACTGCTC TAAGGATTTTGATGAGTACTTCTGC 58 2337 CTACGTAGCCTTTTGGGGTAAAG CCAGGCACACTTGTATTAACTCTG 58 3758 GGAAGAGGATACAGAAATGTCTTCTTG ATCCTGTCTGAGTATTTCCTGCAT 64 2929 ACTGCAGTTAGATGAGTCCAGATAA CTGGGATTAAAACGTGGATCATAG 58 21610 to 12 CACACGCAACTCTTTCTGTGA ACACACGTCACCATCTGCTC 62 82213 CCATGACACATAGGATTGGAAGC CTAGTGCAGTGATGCATAGAAAGACC 64 29314 to 15 CTGAAGGAGATGGAATTCAGTATGT GTGGGACTACTGACCTGGTTTTAT 56 56116 to 17 AAATGATGCACTGAGCTGGTT CTTGAATGATTCCTGGCAATAC 59 57318 TAATACTGGTTTAATGCCAGGCAAC CTAGTGTGCAAGCTATGGAAATACTG 64 24719 GTCTGAGGAAGAGGCACAGC TCAGCTACAGTTGAAAGCTACTGG 64 37120 CGATAACCTAAGCAAGTGTGTACC GGATGTGAAAGTCCAACTTCAG 65 42821 GAGGATGTGAGTGCAAAAGTCAC GTGGGTGACTAAGGTGAGTAACAA 65 30022 GTGCTCTCAATTGTCTGCAAGAAG CCCATACAGAACTGCGAGTCAAT 65 43923 GAGATTTAGGAGGGAAGAAGAGAGAG ACAGGGGAAATTACTGCAAGAG 63 25024a ACTTTACCCTCTGCTGATAA GGGAAATAGTTGTTTGTATG 50 22325a GACATTCAGTGGTTGGTAAT TAAACACTTGTACCCCAAAG 57 28026 TCTTGAGGAAGTTGTCATTGAG TAGTCTGAGGATTCACTCTTGGTTT 63 24827 GGAATAGGCAACATCCTTACTTC CCTGGACCATTTCCTCAATG 63 35528 CTGCTCTTAATTTTAGTAAGTACCCCTGTG AATCTAAACGTTCTTCAGTGACCTG 65 42329a TGGGCCATCTGTATAGTTTT TAATAGTAAGTAGGGTAAGC 57 26930 TTTCCTGCTGTGTGTGAAGC GAAGGACAAAGCCAGACTAACAGT 63 35531a TCCTAAAACTTTATGCTCTC TCAAATACCAGAAACAAATG 57 22132 GAGCTTCGTAGGTTAAGTGGGAAT GGGGAAAGTTGCCTAACAAATG 63 29733a TTTCAGCAGAGACCTGTAAC AAGAACAGAAAGGTTTTATT 52 27134 GTTACCACTGCAGATTACAAAAACC GGTGTTGCAGTTTCTTTGATACAC 63 30035a TGAGACCAAATTAAATTGTC TCATTGCCAGCTAGAAGTAA 52 21036 GAAAAGAATAGGGTGGGCTGAACT ACTAGCTTATAGGTGAGGCATAGGTG 65 36537 ATTAGACAGTGTTCTCCGTTACCC GGCTAAGGAAGAGTAGGAAGATGCTA 65 37038 GTTTGTGGCTAGAGTGAGTACCG GGTACCACTTTCTCTCATGAACCA 65 20539 to 40 GGTTCCTTTTCAGGCACTATAACAG CTTCTATTCTGCCACTCTCTGGTC 65 50041 GTCAAGCAGCCAGTATACACATTC ACAGTACCCCAGACCCTCAAG 65 29542 AGGCTAATCTTGACTCTCAGGCTA GTGCGGCCTGAAAGAAATATACTC 64 39543 TTCTGCTTGTCATTGATGCTG CAAAGTTTAGCTCACACATACAGAG 64 25044 TAACCAGAGACTGTAGAAGGAGCTTG TGCTTTCTCCTCAGTAGTTTAGGC 64 31345 GGCATACGGTATAAGCACGGTAA GAAAGCCACTTGAGAGATCAGAG 64 27946 GCCAGAACAGAGGTGCTTAACATA AGGTTAGTGATCATTTAAGGTGCTG 61 36247 CCTCCAGATTTCAGAACACGTATC GTGCTCAGAATTACTGTCCAATCC 64 49248 ATGTTAAGATCAGACCCAAACCTG ACCATGACATCTCTTAGCACAGTCT 67 445aPrimers according to Boye et al.40
CLINICAL RESEARCH www.jasn.org
3014 Journal of the American Society of Nephrology J Am Soc Nephrol 18: 3004–3016, 2007

2. Tryggvason K, Patrakka J: Thin basement membrane nephropathy.J Am Soc Nephrol 17: 813–822, 2006
3. Mochizuki T, Lemmink HH, Mariyama M, Antignac C, Gubler MC,Pirson Y, Verellen-Dumoulin C, Chan B, Schroder CH, Smeets HJ, etal.: Identification of mutations in the alpha 3(IV) and alpha 4(IV) colla-gen genes in autosomal recessive Alport syndrome. Nat Genet 8:77–81, 1994
4. Jefferson JA, Lemmink HH, Hughes AE, Hill CM, Smeets HJ, DohertyCC, Maxwell AP: Autosomal dominant Alport syndrome linked to thetype IV collage alpha 3 and alpha 4 genes (COL4A3 and COL4A4).Nephrol Dial Transplant 12: 1595–1599, 1997
5. van der Loop FT, Heidet L, Timmer ED, van den Bosch BJ, LeinonenA, Antignac C, Jefferson JA, Maxwell AP, Monnens LA, Schroder CH,Smeets HJ: Autosomal dominant Alport syndrome caused by aCOL4A3 splice site mutation. Kidney Int 58: 1870–1875, 2000
6. Ciccarese M, Casu D, Ki Wong F, Faedda R, Arvidsson S, Tonolo G,Luthman H, Satta A: Identification of a new mutation in the alpha4(IV)collagen gene in a family with autosomal dominant Alport syndromeand hypercholesterolaemia. Nephrol Dial Transplant 16: 2008–2012,2001
7. Pescucci C, Mari F, Longo I, Vogiatzi P, Caselli R, Scala E, AbaterussoC, Gusmano R, Seri M, Miglietti N, Bresin E, Renieri A: Autosomal-dominant Alport syndrome: Natural history of a disease due toCOL4A3 or COL4A4 gene. Kidney Int 65: 1598–1603, 2004
8. Lemmink HH, Nillesen WN, Mochizuki T, Schroder CH, Brunner HG,van Oost BA, Monnens LA, Smeets HJ: Benign familial hematuria dueto mutation of the type IV collagen �4 gene. J Clin Invest 98: 1114–1118, 1996
9. Buzza M, Wang YY, Dagher H, Babon JJ, Cotton RG, Powell H,Dowling J, Savige J: COL4A4 mutation in thin basement membranedisease previously described in Alport syndrome. Kidney Int 60: 480–483, 2001
10. Ozen S, Ertoy D, Heidet L, Cohen-Solal L, Ozen H, Besbas N, Bakkao-glu A, Antignac C: Benign familial hematuria associated with a novelCOL4A4 mutation. Pediatr Nephrol 16: 874–877, 2001
11. Badenas C, Praga M, Tazon B, Heidet L, Arrondel C, Armengol A,Andres A, Morales E, Camacho JA, Lens X, Davila S, Mila M, AntignacC, Darnell A, Torra R: Mutations in the COL4A4 and COL4A3 genescause familial benign hematuria. J Am Soc Nephrol 13: 1248–1254,2002
12. Longo I, Porcedda P, Mari F, Giachino D, Meloni I, Deplano C, BruscoA, Bosio M, Massella L, Lavoratti G, Roccatello D, Frasca G, MazzuccoG, Muda AO, Conti M, Fasciolo F, Arrondel C, Heidet L, Renieri A, DeMarchi M: COL4A3/COL4A4 mutations: From benign familial hema-turia to autosomal dominant or recessive Alport syndrome. Kidney Int61: 1947–1956, 2002
13. Buzza M, Dagher H, Wang YY, Wilson D, Babon JJ, Cotton RG, SavigeJ: Mutations in the COL4A4 gene in thin basement membrane dis-ease. Kidney Int 63: 447–453, 2003
14. Gross O, Netzer KO, Lambrecht R, Seibold S, Weber M: NovelCOL4A4 splice defect and in-frame deletion in a large consanguinefamily as a genetic link between benign familial haematuria and au-tosomal Alport syndrome. Nephrol Dial Transplant 18: 1122–1127,2003
15. Tazon Vega B, Badenas C, Ars E, Lens X, Mila M, Darnell A, Torra R:Autosomal recessive Alport’s syndrome and benign familial hematuriaare collagen type IV diseases. Am J Kidney Dis 42: 952–959, 2003
16. Wang YY, Rana K, Tonna S, Llin T, Sin L, Savige J: COL4A3 mutationsand their clinical consequences in thin basement membrane nephrop-athy (TBMN). Kidney Int 65: 786–790, 2004
17. Rana K, Tonna S, Wang YY, Sin L, Lin T, Shaw E, Mookerjee I, SavigeJ: Nine novel COL4A3 and COL4A4 mutations and polymorphismsidentified in inherited membrane diseases. Pediatr Nephrol 22: 652–657, 2007
18. Slajpah M, Gorinsek B, Berginc G, Vizjak A, Ferluga D, Hvala A, MeglicA, Jaksa I, Furlan P, Gregoric A, Kaplan-Pavlovcic S, Ravnik-Glavac M,
Glavac D: Sixteen novel mutations identified in COL4A3, COL4A4,and COL4A5 genes in Slovenian families with Alport syndrome andbenign familial hematuria. Kidney Int 71: 1287–1295, 2007
19. Savige J, Rana K, Tonna S, Buzza M, Dagher H, Wang YY: Thinbasement membrane nephropathy. Kidney Int 64: 1169–1178, 2003
20. Kashtan CE: Familial hematurias: What we know and what we don’t.Pediatr Nephrol 20: 1027–1035, 2005
21. Gregory MC: The clinical features of thin basement membrane ne-phropathy. Semin Nephrol 25: 140–145, 2005
22. Wang YY, Savige J: The epidemiology of thin basement membranenephropathy. Semin Nephrol 25: 136–139, 2005
23. Torra R, Tazon-Vega B, Ars E, Ballarin J: Collagen type IV (alpha3-alpha4) nephropathy: From isolated haematuria to renal failure. Neph-rol Dial Transplant 19: 2429–2432, 2004
24. Rana K, Wang YY, Buzza M, Tonna S, Zhang KW, Lin T, Sin L, PadavaratS, Savige J: The genetics of thin basement membrane nephropathy.Semin Nephrol 25: 163–170, 2005
25. Nieuwhof CMG, de Heer F, de Leeuw P, van Breda Vriesman PJC: ThinGBM nephropathy: Premature glomerular obsolescence is associatedwith hypertension and late onset renal failure. Kidney Int 51: 1596–1601, 1997
26. Nogueira M, Cartwright J Jr, Horn K, Doe N, Shappell S, Barrios R,Coroneos E, Truong LD: Thin basement membrane disease with heavyproteinuria or nephrotic syndrome at presentation. Am J Kidney Dis35: E15, 2000
27. Norby SM, Cosio FG: Thin basement membrane nephropathy associ-ated with other glomerular diseases. Semin Nephrol 25: 176–179,2005
28. Marquez B, Stavrou F, Zouvani I, Anastasiades E, Patsias C, Pierides A,Kyriacou K: Thin glomerular basement membranes in patients withhematuria and minimal change disease. Ultrastruct Pathol 23: 149–156, 1999
29. Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ,Mathis BJ, Rodriguez-Perez JC, Allen PG, Beggs AH, Pollak MR:Mutations in ACTN4, encoding alpha-actinin-4, cause familial focalsegmental glomerulosclerosis. Nat Genet 24: 251–256, 2000
30. Weins A, Kenlan P, Herbert S, Le TC, Villegas I, Kaplan BS, Appel GB,Pollak MR: Mutational and biological analysis of alpha-actinin-4 infocal segmental glomerulosclerosis. J Am Soc Nephrol 16: 3694–3701, 2005
31. Winn MP, Conlon PJ, Lynn KL, Farrington MK, Creazzo T, Hawkins AF,Daskalakis N, Kwan SY, Ebersviller S, Burchette JL, Pericak-Vance MA,Howell DN, Vance JM, Rosenberg PB: A mutation in the TRPC6 cationchannel causes familial focal segmental glomerulosclerosis. Science308: 1801–1804, 2005
32. Reiser J, Polu KR, Moller CC, Kenlan P, Altintas MM, Wei C, Faul C,Herbert S, Villegas I, Avila-Casado C, McGee M, Sugimoto H, BrownD, Kalluri R, Mundel P, Smith PL, Clapham DE, Pollak MR: TRPC6 is aglomerular slit diaphragm-associated channel required for normal re-nal function. Nat Genet 37: 739–744, 2005
33. Kim JM, Wu H, Green G, Winkler CA, Kopp JB, Miner JH, Unanue ER,Shaw AS: CD2-associated protein haploinsufficiency is linked to glo-merular disease susceptibility. Science 300: 1298–1300, 2003
34. Ihalmo P, Palmen T, Ahola H, Valtonen E, Holthofer H: Filtrin is a novelmember of nephrin-like proteins. Biochem Biophys Res Commun 300:364–370, 2003
35. Srichai MB, Konieczkowski M, Padiyar A, Konieczkowski DJ, Mukher-jee A, Hayden PS, Kamat S, El-Meanawy MA, Khan S, Mundel P, LeeSB, Bruggeman LA, Schelling JR, Sedor JR: A WT1 co-regulator con-trols podocyte phenotype by shuttling between adhesion structuresand nucleus. J Biol Chem 279: 14398–14408, 2004
36. Yiallouros P, Neocleous V, Zeniou M, Adamidou T, Costi C, Christophi C,Tzetis M, Kanavakis E, Deltas C: Cystic fibrosis mutational spectrum andgenotypic/phenotypic features in Greek-Cypriots, with emphasis on de-hydration as presenting symptom. Clin Genet 71: 290–292, 2007
37. Demetriou K, Tziakouri C, Anninou K, Eleftheriou A, Koptides M,
CLINICAL RESEARCHwww.jasn.org
J Am Soc Nephrol 18: 3004–3016, 2007 COL4A3/COL4A4 Mutations in TBMN/FSGS 3015

Nicolaou A, Deltas CC, Pierides A: Autosomal dominant polycystickidney disease-type 2: Ultrasound, genetic and clinical correlations.Nephrol Dial Transplant 15: 205–211, 2000
38. Deltas C: Inherited diseases and Cyprus reality: A historico-geneticapproach. In: 30th Annals of the Cyprus Research Center (Epetirida),edited by the Ministry of Education and Culture, Nicosia, Cyprus,Ministry of Education and Culture, 2004:457–489.
39. Heidet L, Arrondel C, Forestier L, Cohen-Solal L, Mollet G, GutierrezB, Stavrou C, Gubler MC, Antignac C: Structure of the human type IVcollagen gene COL4A3 and mutations in autosomal Alport syndrome.J Am Soc Nephrol 12: 97–106, 2001
40. Boye E, Mollet G, Forestier L, Cohen-Solal L, Heidet L, Cochat P,Grunfeld JP, Palcoux JB, Gubler MC, Antignac C: Determination of thegenomic structure of the COL4A4 gene and of novel mutations caus-ing autosomal recessive Alport syndrome. Am J Hum Genet 63:1329–1340, 1998
41. Sue Y-M, Huang J-J, Hsieh R-Y, Chen F-F: Clinical features of thinbasement membrane disease and associated glomerulopathies. Ne-phrology 9: 14–18, 2004
42. van Paasen P, van Breda Vriesman PJ, van Rie H, Tervaert JW: Signsand symptoms of thin basement membrane nephropathy: A prospec-tive regional study on primary glomerular disease—The Limburg Re-nal Registry. Kidney Int 66: 909–913, 2004
43. Haas M: Thin glomerular basement membrane nephropathy: Inci-
dence in 3471 consecutive renal biopsies examined by electron mi-croscopy. Arch Pathol Lab Med 130: 699–706, 2006
44. Stavrou C, Koptides M, Tombazos C, Psara E, Patsias C, Zouvani I,Kyriacou K, Hildebrandt F, Christofides T, Pierides A, Deltas CC:Autosomal-dominant medullary cystic kidney disease type 1: Clinicaland molecular findings in six large Cypriot families. Kidney Int 62:1385–1394, 2002
45. Beirowski B, Weber M, Gross O: Chronic renal failure and shortenedlifespan in Col4a3�/� mice: An animal model for thin basementmembrane nephropathy. J Am Soc Nephrol 17: 1986–1994, 2006
46. Marquez B, Zouvani I, Karagrigoriou A, Anastasiades E, Pierides A,Kyriacou K: A simplified method for measuring the thickness of glo-merular basement membranes. Ultrastruct Pathol 27: 409–416, 2003
47. Miller SA, Dykes DD, Polesky HF: A simple salting out procedure forextracting DNA from human nucleated cells. Nucleic Acids Res 16:1215, 1988
48. Lathrop GP, Lalouel JM, Julier C, Ott J: Multilocus linkage analysis inhumans: Detection of linkage and estimation of recombination. Am JHum Genet 37: 482–498, 1985
49. Oleykowski CA, Mullins CRB, Godwin AK, Yeung AT: Mutation detec-tion using a novel plant endonuclease. Nucleic Acids Res 26: 4597–4602, 1998
See related editorial, “The Wages of Thin,” on pages 2800–2802.
CLINICAL RESEARCH www.jasn.org
3016 Journal of the American Society of Nephrology J Am Soc Nephrol 18: 3004–3016, 2007
Copyright © 2022 FDOKUMEN




















