
Antibacterial and antifungal activities of Myracrodruon urundeuva heartwood

Purification, Biochemical Characterization, and Cloningof Phospholipase D from Streptomyces racemochromogenesStrain 10-3
Yozo Nakazawa • Yoshimasa Sagane • Teppei Kikuchi •
Masataka Uchino • Takeshi Nagai • Hiroaki Sato •
Kazuki Toeda • Katsumi Takano
Published online: 17 November 2010
� Springer Science+Business Media, LLC 2010
Abstract We previously isolated Streptomyces racemo-
chromogenes strain 10-3, which produces a phospholipase
D (PLD) with high transphosphatidylation activity. Here,
we purified and cloned the PLD (PLD103) from the strain.
PLD103 exerted the highest hydrolytic activity at a slightly
alkaline pH, which is in contrast to the majority of known
Streptomyces PLDs that have a slightly acidic optimum pH.
PLD103 shares only 71–76% amino acid sequence identity
with other Streptomyces PLDs that have a slightly acidic
optimum pH; thus, the diversity in the primary structure
might explain the discrepancy observed in the optimum
pH. The purified PLD displayed high transphosphatidyla-
tion activity in the presence of glycerol, L-serine, and
2-aminoethanol hydrochloride with a conversion rate of
82–97% in a simple one-phase system, which was
comparable to the rate of other Streptomyces PLDs in a
complicated biphasic system.
Keywords Transphosphatidylation � Phospholipase D �Streptomyces racemochromogenes � Phospholipid
modification
Abbreviations
PLD Phospholipase D
pI Isoelectric point
PL Phospholipid
PC Phosphatidylcholine
PG Phosphatidylglycerol
PE Phosphatidylethanolamine
PA Phosphatidic acid
IEF Isoelectric focusing
MOPS 3-(N-Morpholino) propanesulfonic acid
TLC Thin-layer chromatography
2-ME 2-Mercaptoethanol
ORF Open reading frame
Rbs Ribosome binding site
1 Introduction
Phospholipids (PLs) are ubiquitous molecules that are
widely distributed among the plant and animal kingdoms.
In industrial fields, PLs can be abundantly derived from
lecithin, the mixture of PLs with other minor components
that arise as by-products from an oil-production process
from animal fats or vegetable oils. The PL content in lec-
ithin is predominantly phosphatidylcholine (PC) and small
amounts of phosphatidylserine (PS), phosphatidylglycerol
(PG), phosphatidylethanolamine (PE), and phosphatidyl-
inositol. The lecithin-derived PLs have been included in
food products, cosmetics, and pharmaceuticals because of
Electronic supplementary material The online version of thisarticle (doi:10.1007/s10930-010-9292-y) contains supplementarymaterial, which is available to authorized users.
Y. Nakazawa (&) � T. Nagai � H. Sato � K. Toeda
Department of Food and Cosmetic Science,
Faculty of Bioindustry, Tokyo University of Agriculture,
196 Yasaka, Abashiri, Hokkaido 099-2493, Japan
e-mail: [email protected]
Y. Sagane
Sars International Centre for Marine Molecular Biology,
Thormøhlensgate 55, 5008 Bergen, Norway
T. Kikuchi � M. Uchino � K. Takano
Department of Applied Biology and Chemistry, Faculty of
Applied Bioscience, Tokyo University of Agriculture, 1-1-1
Sakuragaoka, Setagaya-ku, Tokyo 156-8502, Japan
123
Protein J (2010) 29:598–608
DOI 10.1007/s10930-010-9292-y

their interfacial activity, emulsification properties, and
liposome-formation ability [7, 13, 22, 26]. In addition to
these common properties of PLs, PG can improve the
emulsification capacity of lecithin over wide ranges of pH
and salt conditions [20]. Moreover, PS, which can be
obtained from animal brain, has been proposed as a food
supplement to decrease and/or prevent senile dementia
[15, 34]. However, the yields of such PLs from lecithin and
other natural resources are limited. In particular, the use of
animal brain has been avoided because of the occurrence of
bovine spongiform encephalopathy. Even though chemi-
cally synthesized PLs are available, they are too expensive
to use in daily food products. Phospholipase D (PLD, EC
3.1.4.4) is an enzyme that catalyzes the hydrolysis of the
phosphodiester bond of PLs to generate phosphatidic acid
(PA) and its corresponding alcohol moiety. In addition to
its hydrolytic activity, PLD also catalyzes the intercon-
version of the polar head groups of PLs by a reaction called
transphosphatidylation [38]. The transphosphatidylation
reaction synthesizes various phosphatidyl derivatives of
hydroxyl compounds, such as alcohols, phenols, sugars,
nucleotides, vitamins, and others [6, 18, 19, 23, 31, 35].
Therefore, this reaction is an economical means of PL
modification. The reactions performed by PLD can occur
with different degrees of selectivity between hydrolysis
and transphosphatidylation, depending on the enzymatic
source [16, 17]. In particular, PLD from Streptomyces
predominantly displays high selectivity for the transphos-
phatidylation reaction [24, 37] and, accordingly, PLDs
from this bacterium are already used in industrial fields.
The transphosphatidylation by PLD is often carried out
in a biphasic system composed of an aqueous solution of
PLD enzyme and an organic solution of the PL substrate. In
the aqueous reaction mixture, PLD is exposed not only to
substrate but also to product PLs and can competitively
react to de novo synthesized PLs as it hydrolyzes the
products. This might result in low production of the
phosphatidyl derivatives [5]. However, the biphasic sys-
tems require enormous quantities of organic solvents and
considerable energy to make an emulsion with strong
agitations. Additionally, the organic solvents in this system
often include toxic compounds, which are undesirable for
industrial food, cosmetic, and medical fields. To identify
effective reaction conditions that do not use organic sol-
vents, we recently screened six Streptomyces strains,
derived from soil samples that produce PLD with high
transphosphatidylation capacity even in the one-phase
system. In this system, the substrate PC is dispersed in
aqueous buffer with a small amount of detergent [24, 25].
The culture supernatants of the strains displayed high PG-,
PS-, and PE-producing transphosphatidylation from the
substrate PC with conversion rates ranging from 74 to 97%,
which are comparable to those of Streptomyces PLDs
assayed in biphasic systems. Furthermore, they showed low
hydrolysis activities against PG and PS. Therefore, the
PLDs produced by these strains are expected to be avail-
able for PL modification through their high transphospha-
tidylation capacity in the aqueous system. In this study, we
purified, characterized and cloned the PLD enzyme from
S. racemochromogenes strain 10-3.
2 Materials and Methods
2.1 Materials
Egg yolk lecithin (PC-98N) was kindly provided by Kewpie
Fine Chemicals (Tokyo, Japan). The PL composition of the
lecithin was the following: 98.9% PC, 0.1% lysophospha-
tidylcholine, and others, according to the manufacturer’s
specification assessment. Other PLs were obtained from
Avanti Polar Lipids (Alabaster, AL, USA).
2.2 Bacterial Strains and Cultural Conditions
Streptomyces racemochromogenes strain 10-3 was isolated
from soil samples collected in Japan as previously reported
[24, 25]. Strains were cultured in GYP medium, which was
composed of 5 g/L glucose, 5 g/L Bacto yeast extract (BD
Difco, Franklin Lakes, NJ, USA), 5 g/L Polypepton (Nihon
Pharmaceutical, Tokyo, Japan), 2 g/L K2HPO4, and 0.5 g/L
MgSO4�7H2O, with shaking at 28 �C for 2 days.
2.3 PLD Assay
Phospholipase D activity was assayed using PC as a sub-
strate and measuring the formation of choline with choline
oxidase and peroxidase [12]. A one milliliter reaction
mixture that was composed of 1 mg/mL PC-98N, 4 mM
sodium deoxycholate, 20 mM Tris–HCl buffer (pH 7.5),
10 mM CaCl2, and enzyme was incubated at 37 �C for
10 min. The reaction was immediately terminated by
adding 0.2 mL of solution (1 M Tris–HCl, pH 8.0,
100 mM EDTA) and incubating in a boiling water bath for
20 min. After chilling at room temperature, the reaction
mixture was mixed with 0.2 mL of the choline assay
solution, containing 0.1 M Tris–HCl buffer (pH 8.0), 1.25
units of choline oxidase (Sigma–Aldrich, St. Louis, MO,
USA), 1 unit of peroxidase (Wako Pure Chemicals),
1.6 lmol of phenol, and 1 lmol of 4-aminoantipyrine, and
then incubated at 37 �C for 60 min to allow for color
development. After adding 2 mL of 10 g/L Triton X-100,
the absorbance of the reaction mixture was measured at
500 nm. A calibration curve was obtained by adding a
standard solution of choline chloride to the assay mixture
instead of the enzyme solution. One unit (U) was defined as
Purification, Biochemical Characterization, and Cloning of Phospholipase D 599
123

the amount of enzyme that liberated 1 lmol of choline per
min. To determine the optimum pH, PLD activity was
assayed using reaction mixtures made with 20 mM buffer
at different pHs (pH 4, 5, and 6 = sodium acetate-acetic
acid; pH 7, 7.5, and 8 = Tris–HCl; pH 9 and 10 = boric
acid-NaOH). To determine the optimum temperature, PLD
assay experiments were carried out at several temperatures
for 10 min each.
2.4 Protein Assay
Protein concentration was determined using the Bio-Rad
(Hercules, CA, USA) Protein Assay based on the method
of Bradford [2], using bovine serum albumin as a standard.
2.5 Enzyme Purification
The cultured broth (1 L) of strain 10-3 was centrifuged,
and the supernatant was brought to 65% saturation with
(NH4)2SO4. After standing overnight at 4 �C, the sus-
pension was centrifuged at 10,000 rpm for 30 min at 4 �C.
The precipitate was dissolved and dialyzed to 20 mM
sodium acetate buffer (pH 5.5) at 4 �C. The dialyzed
solution was centrifuged to remove insoluble materials.
The supernatant was applied to a CM Sepharose Fast Flow
(GE Healthcare Bio-Sciences, Little Chalfont, Bucking-
ham, England) column (2.0 mm I.D. 9 80 mm) equili-
brated with 20 mM sodium acetate buffer (pH 5.5). After
washing the column with the equilibration buffer, the
bound proteins were eluted using a stepwise gradient with
0.1 M NaCl and 0.25 M NaCl in 20 mM sodium acetate
buffer (pH 5.5). The active fractions were pooled, dia-
lyzed against distilled water at 4 �C, and lyophilized. The
lyophilized material was dissolved in 2 mL of 20 mM
MOPS-NaOH buffer (pH 7.0) and loaded onto a TOYO-
PEARL DEAE-650M (Tosoh, Tokyo, Japan) column
(1.6 mm I.D. 9 50 mm) equilibrated with 20 mM MOPS-
NaOH buffer (pH 7.0). After washing the column with the
equilibration buffer, the bound proteins were eluted with
0.5 M NaCl in 20 mM MOPS-NaOH buffer (pH 7.0). The
PLD activity fractions were pooled, desalted by dialysis in
cold water, and lyophilized.
2.6 Electrophoresis Analyses
SDS–PAGE was performed using the method of Laemmli
[21] on a 12.5% polyacrylamide gel using a discontinuous
buffer system. The molecular mass under denaturing con-
ditions was determined using an LMW-SDS Marker Kit
(GE Healthcare Bio-Science). Isoelectric focusing (IEF)
electrophoresis was carried out in a Multiphor II apparatus
(GE Healthcare Bio-sciences) at 10 �C using an Ampho-
line PAG plate (pH 3.5–9.5, GE Healthcare Bio-sciences).
The isoelectric point (pI) of purified PLD was determined
using a Broad pI Kit (GE Healthcare Bio-sciences). The
proteins were detected by Coomassie blue staining.
2.7 Determination of the pH and Thermal Stability
Profiles
To determine the pH stability of the PLD, 100 lL of PLD
solution (5 U/mL) was added to 100 lL of 0.2 M buffer at
different pHs (pH 2 and 3 = glycine–HCl; pH 4, 5, 6, 7,
7.5, 8, 9 and 10 = same buffer used in pH optimum
determination; pH 11 and 12 = Na2HPO4-NaOH). Test
tubes containing the above solutions were incubated at
25 �C for 4 h. Ten microliters of the solution were then
used to determine the residual activity (Tris–HCl, pH 7.5).
To determine the thermal stability of the PLD, 100 lL
of PLD solution (2.5 U/mL) was incubated at different
temperatures (10, 20, 30, 37, 40, 50, 60, 70, and 80 �C) for
10 min, and then cooled on ice immediately. Ten micro-
liters of the solution were then used to determine the
residual activity (Tris–HCl, pH 7.5).
2.8 Amino Acid Sequencing
The NH2-terminal amino acid sequence of the purified PLD
protein was determined by the direct protein sequencing
method described by Hirano and Watanabe [11] using a
protein sequencer (model PPSQ-23A; Shimadzu, Kyoto,
Japan) equipped with online HPLC (model LC-10A;
Shimadzu).
2.9 PLD Cloning
Chromosomal DNA was prepared from S. racemochrom-
ogenes strain 10-3 according to the method described
previously [25]. Based on the NH2-terminal amino acid
sequence (NH2-ASPTPHL) of the purified PLD103, two
forward primers (N1 and N2; Supplementary Table 1) were
designed. Furthermore, a reverse primer (S300 Rv; Sup-
plementary Table 1) designed in a previous report [25] was
also employed for PCR. PCR amplification was performed
in a 50 lL reaction mixture, containing genomic DNA
(20 ng), primers (0.2 lM each), dNTP mixture (0.2 mM
each) and 1.25 U PrimeSTAR DNA Polymerase (Takara
Bio, Ohtsu, Japan), with the following cycle parameters:
denaturing for 10 s at 98 �C, primer annealing for 5 s at
55 �C, and primer extension for 1 min 30 s at 72 �C, for 30
cycles. The products were purified and then treated with T4
polynucleotide kinase. The 50 terminally phosphorylated
DNA was subcloned into the HincII-digested site of
pUC118. Plasmid DNA was subjected to cycle sequencing,
and then the sequences were analyzed. To amplify the
upstream and downstream regions of the pld gene, inverse
600 Y. Nakazawa et al.
123

PCR was performed using the circular DNA made from the
SacI or BamHI-digested DNA (50 ng) followed by self-
ligation using a T4 DNA ligase overnight at 16 �C. The
primers for the inverse PCR and nested PCR were designed
based on the nucleotide sequences of the amplified DNA
fragments (Supplementary Table 1).
The amplified PCR products were subcloned into the
pUC118 vector and subjected to cycle sequencing using the
ABI PRISM BigDye Terminator v3.1 Cycle Sequencing Kit
(Applied Biosystems), universal vector primers, and gene-
specific primers (Supplementary Table 1). The nucleotide
sequence of pld gene reported in this article has been sub-
mitted to the DDBJ/EMBL/Genbank under accession
number AB573232.
2.10 In Silico Analysis
Potential ribosome binding site (rbs) and rho-independent
transcription terminator signals were predicted using
GENETYX software (ver. 9; Genetyx, Tokyo, Japan).
Alignments of the nucleotide sequences of the pld genes
and the amino acid sequences of PLDs were generated
using CLUSTAL X [36]. A 3D model of the PLD103 was
predicted by utilizing structure of Streptomyces sp. strain
PMF PLD (PMF-PLD; PDB ID: 1V0W) as template, using
the homology-modeling server SWISS-MODEL [9] at
http://swissmodel.expasy.org/. Template structure was
downloaded from the Protein Data Bank. The image was
generated using the UCSF Chimera version 1.4.1 (available
at http://www.cgl.ucsf.edu/chimera/) [28] to display ribbon
diagrams of 3D structure of the PLDs. The model exhibited
good geometry having 95.8% of favored regions according
to Ramachandran plots generated using MolProbity
(available at http://molprobility.biochem.duke.edu) [4].
2.11 Transphosphatidylation Analysis
Six milliliters of reaction mixture composed of 1 mg/mL
PC-98N, 4 mM sodium deoxycholate, 20 mM MOPS-
NaOH buffer (pH 7.5), 10 mM CaCl2, 0.12 U PLD, and
200 mg/mL glycerol, L-serine, or 2-aminoethanol hydro-
chloride were incubated at 37 �C. One milliliter of reaction
mixture was transferred at intervals of 30–300 min into the
test tube containing 0.2 mL of 0.1 M NaOH to terminate
the enzyme reaction. The PLs in the reaction mixture were
extracted using the method of Bligh and Dyer [1]. The
chloroform phase was separated and evaporated, and the
resultant lipid residue was dissolved with 100 lL of
chloroform. After centrifugation, 10 lL of the clear chlo-
roform phase was applied onto a silica gel 60 thin-layer
chromatography (TLC) plate (Merck, Darmstdt, Germany).
The plate was developed with chloroform/methanol/acetic
acid (40:15:6, v/v). Spots on the plate were visualized by
iodine vapor, and the intensities of the visualized spots
were measured with the Gel-Pro Analyzer software (ver-
sion 3.1; Media Cybernetics, Silver Spring, MD, USA).
The transphosphatidylation conversion rate (%) was
defined as: [PX] 9 100/[PX] ? [PC] ? [PA], and the
selectivity (%) was defined as: [PX] 9 100/[PX] ? [PA],
where the PX in the formula represents the transphospha-
tidylation product.
3 Results
3.1 Purification and Characterization of PLD Produced
by S. racemochromogenes 10-3
Phospholipase D secreted by S. racemochromogenes 10-3
(PLD103) was purified from the culture supernatant
by anion- and cation-exchange chromatographic steps
(Table 1). The purity of the enzyme was confirmed by both
of SDS–PAGE and IEF. The active fractions eluted from the
final chromatography ran as a single band at 55 kDa on
SDS–PAGE in the presence of 2-mercaptoethanol (Fig. 1a,
lane 4) and at pI 8.0 on IEF (Fig. 1b, lane 4). Starting with
1 L of the culture medium, 300 lg of the purified PLD103,
with a specific activity of 374 U/mg, was obtained. The
NH2-terminal amino acid sequence of PLD103 was deter-
mined to be NH2-ASPTPHLDSVEQTLRQVSPG. This
sequence displayed high homology with other streptomy-
cete PLD sequences (60–85% identity).
The PC-hydrolytic activity of PLD103 is stable in the
pH 4–9 range; over 70% of the original activity remained
Table 1 Summary of the results of the purification of PLD from S. racemochromogenes 10-3
Purification step Protein (mg) Activity (U) Specific activity (U/mg) Purification (-fold) Yield (%)
Culture supernatant 17.1 ± 0.7 365.9 ± 10.5 21.5 ± 0.6 1 100
(NH4)2SO4 precipitation 2.0 ± 0.1 290.5 ± 22.0 142.3 ± 3.3 6.7 ± 0.3 79.9 ± 8.5
CM Sepharose FF 0.5 ± 0.1 200.6 ± 11.1 344.7 ± 7.1 16.1 ± 0.6 55.0 ± 4.2
TOYOPEARL DEAE 650M 0.3 ± 0.02 118.3 ± 5.1 374.3 ± 12.0 17.4 ± 0.5 32.5 ± 2.3
Values are represented as mean ± SE of three independent preparations in 1,000 mL of culture
Purification, Biochemical Characterization, and Cloning of Phospholipase D 601
123

after a 4-h exposure to this pH range (Fig. 2a). The opti-
mum reaction temperature of PLD103 was 50 �C, and the
enzyme remained active with over 80% of the original
activity after 10-min exposures to a 10–60 �C temperature
range (Fig. 2b). Therefore, PLD103 exhibited similarities
in pH stability, optimum temperature, thermal stability of
the hydrolytic activity, and molecular mass to previously
reported enzymes from other Streptomyces strains
(Table 2). On the other hand, PLD103 exhibited significant
differences in optimum pH for the PC-hydrolytic activity to
those observed in other Streptomyces PLDs. While the
majority of the Streptomyces PLD enzymes show the
highest PC-hydrolysis activity at pH 5–6, our enzyme was
the most active at pH 7.5. In acidic conditions (pH 5–6),
PLD103 displayed only 10–50% of the activity compared
to its activity at the optimum pH. Therefore PLD103 dis-
played the highest activity at the slightly alkaline condi-
tion, which is in contrast to the majority of Streptomyces
PLDs that exert the highest activity at slightly acidic
conditions.
3.2 Nucleotide Sequence of the PLD103 Gene
To clarify the primary structure of PLD103 and compare it
to other Streptomyces PLDs, we isolated the PLD103 gene
from the genomic DNA of strain 10-3. We previously
cloned a 274-bp partial fragment of the pld103 gene [25].
In this study, to isolate the entire ORF of pld103, we
designed degenerate primers based on the NH2-terminal
amino acid sequence of the purified enzyme. PCR using
degenerate primers (Supplementary Table 1) yields a
1,390-bp product. To amplify the downstream and
upstream regions of the PCR product, inverse PCR was
performed. Finally, we determined the nucleotide sequence
of a 2,770-bp genomic DNA fragment. In the sequence,
there is a potential rbs (AAGGAA), but the general start
codon, ATG, could not be found near the binding site.
Instead, a TTG codon was located 5-bp downstream from
the rbs and could be a good candidate for the start codon.
The rho-independent transcription terminator signal [29],
consisting of a 36-bp stem-loop sequence, was also found
37-bp downstream of the pld103 gene. Therefore, the
determined sequence contained the entire putative 1,587-bp
ORF that encodes 528 amino acid residues (Fig. 3). The
NH2-terminal amino acid sequence of the purified PLD103
exactly matched with the nucleotide-deduced amino acid
sequence starting at Ala-27. The SignalP 3.0 sever program
[8] predicted that the signal cleavage site is between Ala-26
and Ala-27, which coincides with the NH2-terminal amino
acid sequence of the purified PLD103. Computer-assisted
motif analysis using the protein family database, InterPro,
predicted that the enzyme contains a set of dual PLD active
Fig. 1 Electrophoretic analysis of the purified Streptomyces racemo-chromogenes 10-3 strain phospholipase D (PLD103). a SDS–PAGE
banding profiles of the PLD103 preparation at each purification step.
The molecular mass of the purified enzyme was estimated to be
55 kDa based on the electrophoretic mobility of the purified PLD103
(lane 4) and molecular mass standard proteins (lane M). Lane 1culture supernatant, lane 2 ammonium sulfate precipitation, lane 3
elutant from CM Sepharose and lane 4 elutant from TOYOPEARL
DEAE. b Isoelectric focusing (IEF) banding profile of the purified
PLD103. The purified enzyme eluted from TOYOPEARL DEAE
corresponds to lane 4 in panel a. The isoelectric point (pI) of PLD103
was estimated to be 8.0 based on the electrophoretic mobility of the
pI standard proteins (lane M) concomitantly applied to the IEF
602 Y. Nakazawa et al.
123

site HKD motifs at the residue segments His-192–Asp-199
and His-462–Asp-469.
A comparison of the sequences of Streptomyces PLDs,
whose enzymatic properties have been published (Table 2),
using CLUSTAL X multiple-sequence alignment analysis
indicated that PLD103 shares relatively low sequence
homology (71–76% identical) with other Streptomyces
PLDs (Fig. 4a). Hence, the difference in the optimum pH
for hydrolytic activity observed among enzymes might
result from the diversity of their primary structures. To find
out the difference in 3D structures of the PLD103 and the
other Streptomyces PLDs, the computer modeled 3D
structures of the enzymes were generated (Fig. 4b). The
comparison of the models indicated that the diversity in the
primary structure of the PLD103 generates the disruption
of one of 18 a helices.
3.3 PG-, PS-, and PE-Producing
Transphosphatidylation by PLD103
The PG-, PS-, and PE-producing transphosphatidylation
activities of PLD103 were examined in an aqueous one-
phase reaction mixture in which 1 mg/mL egg yolk lecithin
(98.9% PC) was used as a substrate and was dispersed with
a small amount of detergent (4 mM deoxycholate). In the
PG-producing reaction, 97.1% of the substrate PC was
converted into PG by using glycerol as the acceptor. The
selectivity for transphosphatidylation instead of hydrolysis
was significantly high; 97.7% of the product resulted from
transphosphatidylation; only 2.3% was the hydrolytic
product PA (Fig. 5a). In the PS-producing transphosphati-
dylation reaction, 82.5% of the substrate PC was converted
into PS. Of the reaction products, 88.9% resulted from the
Fig. 2 Effect of pH (a) and temperature (b) on the activity and
stability of PLD103. The maximal enzyme activities observed were
set as 100% relative activity. a To determine the optimum pH
(indicated with closed circles), enzyme activity was measured under
the standard assay conditions with various pH values. To determine
the pH stability of PLD103 (indicated with open circles), the enzyme
was incubated in buffers with various pHs at 25 �C for 4 h prior to the
standard assay. b To determine the optimum temperature (indicated
with closed circles), enzyme activity was measured under standard
assay conditions at varying temperatures. To determine the heat
stability of PLD103 (indicated with open circles), the enzyme was
incubated at different temperatures for 10 min prior to the standard
assay. Both experiments were repeated in triplicate, and the errorbars represent the standard error of the means
Table 2 Enzymatic properties of streptomycete PLD enzymes
Specific activity
(U/mg)
Molecular
mass (kDa)
pI Optimum pH Optimum
temperature (�C)
pH stability Thermal
stability (�C)
References
PLD103 392 55 8.0 7.5 50 4.0–9.0 \60 This work
PMF-PLD 42 54 9.1 5.0 60 4.0–9.0 ND [3]
TH2-PLD 46 55 6.5 5.0 55 4.0–9.0 \60 [10]
Sci PLD 468 54 ND 6.0 50 ND ND [27]
San PLD 1,437 64 6.5 5.5 60 4.0–8.0 \50 [30]
PLD103, Streptomyces racemochromogenes strain 10-3 PLD; PMF-PLD, Streptomyces sp. strain PMF PLD; TH2-PLD, Streptomyces septatusstrain TH-2 PLD; Sci PLD, Streptovelticillium cinnamoneum PLD; San PLD, Streptomyces antibioticus strain S-170
Purification, Biochemical Characterization, and Cloning of Phospholipase D 603
123

transphosphatidylation reaction, and 10.3% resulted from
the hydrolytic reaction. Thus, the apparent transphospha-
tidylation efficiency for PS production was inferior to that
for PG production. Moreover, PS production reached a
plateau after 120 min, indicating that increased production
of PS could not be expected even with the additional
reaction time (Fig. 5b). Recently, Iwasaki et al. [14]
attempted efficient PS production in various aqueous
Fig. 3 Genomic nucleotide sequence of the S. racemochromogenes10-3 pld gene open reading frame (ORF) and the deduced amino acid
sequence of PLD103. The potential ribosome binding site (rbs) is
underlined upstream of the ORF. A putative initiation TTG codon is
boxed. Arrows below the nucleotide sequence show the region of
dyad symmetry for the rho-independent transcription terminator
signal, which consists of a stem-loop sequence. A double underlinebelow the deduced amino acid sequence indicates the determined
NH2-terminal amino acid sequence based on the direct sequencing of
purified PLD103. The dotted lines below the deduced amino acid
sequence indicate the HKD motifs in the PLD enzyme. The putative
signal cleavage site predicted is indicated with an arrowhead
604 Y. Nakazawa et al.
123

a
b
Purification, Biochemical Characterization, and Cloning of Phospholipase D 605
123
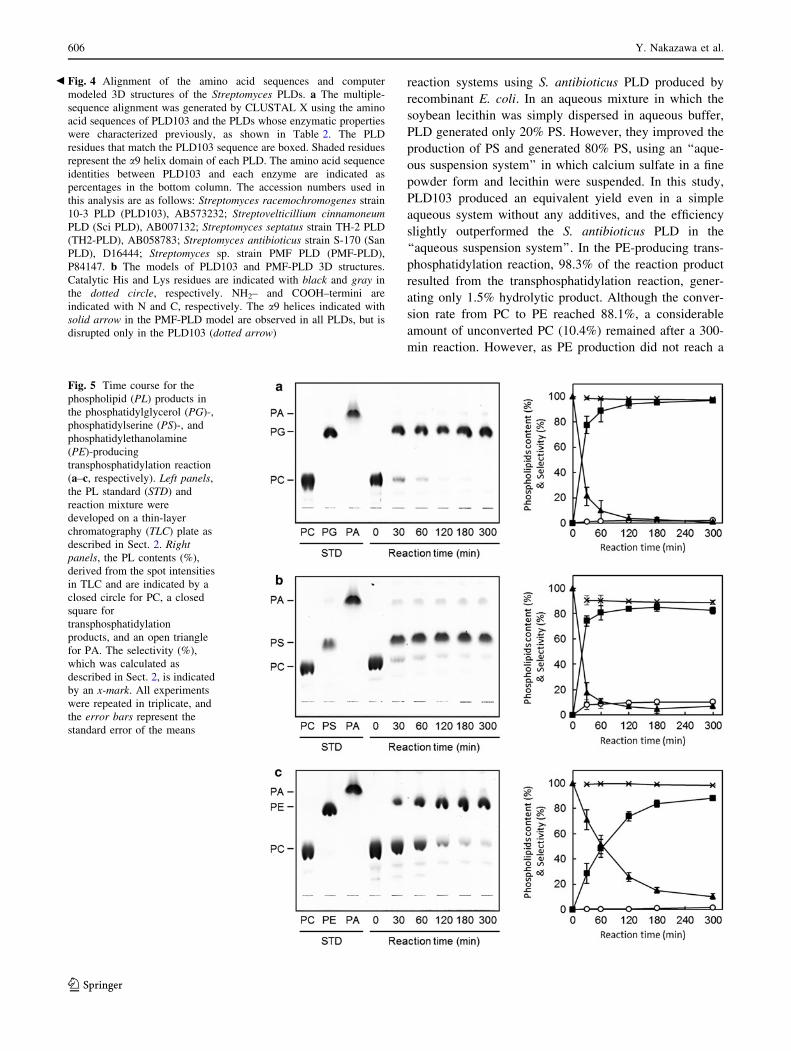
reaction systems using S. antibioticus PLD produced by
recombinant E. coli. In an aqueous mixture in which the
soybean lecithin was simply dispersed in aqueous buffer,
PLD generated only 20% PS. However, they improved the
production of PS and generated 80% PS, using an ‘‘aque-
ous suspension system’’ in which calcium sulfate in a fine
powder form and lecithin were suspended. In this study,
PLD103 produced an equivalent yield even in a simple
aqueous system without any additives, and the efficiency
slightly outperformed the S. antibioticus PLD in the
‘‘aqueous suspension system’’. In the PE-producing trans-
phosphatidylation reaction, 98.3% of the reaction product
resulted from the transphosphatidylation reaction, gener-
ating only 1.5% hydrolytic product. Although the conver-
sion rate from PC to PE reached 88.1%, a considerable
amount of unconverted PC (10.4%) remained after a 300-
min reaction. However, as PE production did not reach a
Fig. 5 Time course for the
phospholipid (PL) products in
the phosphatidylglycerol (PG)-,
phosphatidylserine (PS)-, and
phosphatidylethanolamine
(PE)-producing
transphosphatidylation reaction
(a–c, respectively). Left panels,
the PL standard (STD) and
reaction mixture were
developed on a thin-layer
chromatography (TLC) plate as
described in Sect. 2. Rightpanels, the PL contents (%),
derived from the spot intensities
in TLC and are indicated by a
closed circle for PC, a closed
square for
transphosphatidylation
products, and an open triangle
for PA. The selectivity (%),
which was calculated as
described in Sect. 2, is indicated
by an x-mark. All experiments
were repeated in triplicate, and
the error bars represent the
standard error of the means
Fig. 4 Alignment of the amino acid sequences and computer
modeled 3D structures of the Streptomyces PLDs. a The multiple-
sequence alignment was generated by CLUSTAL X using the amino
acid sequences of PLD103 and the PLDs whose enzymatic properties
were characterized previously, as shown in Table 2. The PLD
residues that match the PLD103 sequence are boxed. Shaded residues
represent the a9 helix domain of each PLD. The amino acid sequence
identities between PLD103 and each enzyme are indicated as
percentages in the bottom column. The accession numbers used in
this analysis are as follows: Streptomyces racemochromogenes strain
10-3 PLD (PLD103), AB573232; Streptovelticillium cinnamoneumPLD (Sci PLD), AB007132; Streptomyces septatus strain TH-2 PLD
(TH2-PLD), AB058783; Streptomyces antibioticus strain S-170 (San
PLD), D16444; Streptomyces sp. strain PMF PLD (PMF-PLD),
P84147. b The models of PLD103 and PMF-PLD 3D structures.
Catalytic His and Lys residues are indicated with black and gray in
the dotted circle, respectively. NH2– and COOH–termini are
indicated with N and C, respectively. The a9 helices indicated with
solid arrow in the PMF-PLD model are observed in all PLDs, but is
disrupted only in the PLD103 (dotted arrow)
b
606 Y. Nakazawa et al.
123

plateau and the hydrolytic product was scarcely observed
after 300 min, further PE production would be achieved
after additional reaction time (Fig. 5c).
The transphosphatidylation conversion rates of the com-
mercially available Streptomyces sp. PLD-P (Asahi-Kasei,
Tokyo, Japan) and S. chromofuscus PLD (Asahi-Kasei) in
the same aqueous one-phase system were 54.1 and 10.7% for
PG-, 80.4 and 0.6% for PS-, and 75.4 and 4.5% for the PE-
producing reactions after a 300 min incubation, respectively
(data not shown). Therefore, the transphosphatidylation
conversion rates of PLD103 were much higher than already
known Streptomyces PLDs in the aqueous one-phase system.
4 Discussion
In this study, we identified diversity among the amino acid
sequences of PLD103 and other Streptomyces PLDs, which
might affect on the optimum pH of the enzyme. Notably,
the diversity found in the a9 helix domain generates the
significant alternation of the secondary structure in the
computer modeled 3D structure. The a9 helix, which is
disrupted in the PLD103 model, is located on opposite side
of the active pocket. Therefore, this domain would not
directly contact to the substrate molecule, but may be
responsible for the accessibility to the substrate due to
structural change of the enzyme in a pH dependent manner.
Very recently, Simkhada et al. [32, 33] isolated alkaline
PLDs, exerting highest activity at pH 8, from Streptomyces
species. Although the primary structures of these PLDs are
not available at present, comparison of the amino acid
sequences of our PLD103 and these enzymes would be
worthwhile to clarify the relation of the primary structure
and pH optimum of the enzyme.
PLD103 transfers lecithin-derived PC into PG, PS, and
PE with high efficiency and selectivity even in the one-
phase aqueous system at a rate that is comparable to the
efficiency of other Streptomyces PLDs, which were
assayed in biphasic systems. Because of its ubiquitous
distribution, the in vivo functions of plant and animal PLD
have been well investigated. Furthermore, the dual reac-
tions performed by PLD have been a longstanding and
well-known property of the enzyme, regardless of the
source. Despite this, elucidating the detailed reaction
mechanism of transphosphatidylation and hydrolysis by
PLD had been hampered partially due to low productivity
of the enzyme. Further investigation of PLD103, including
overexpression in proper bacterium, may not only con-
tribute to its use in industrial fields because of its unique
property of transphosphatidylation activity in an aqueous
system, but may also reveal the transphosphatidylation
mechanism of the PLD enzyme.
Acknowledgments This work was partially supported by Grant-in-
aid for Scientific Research from the Kieikai Foundation (Tokyo,
Japan), which is greatly appreciated. We thank Mr. K. Hirabayashi
and Mr. S. Sakurai for their technical assistances. We also thank
Professor T. Nagashima (Tokyo University of Agriculture, Japan) for
his excellent contributions to this work.
References
1. Bligh EG, Dyer WJ (1959) Can J Biochem Physiol 37:911–917
2. Bradford MM (1977) Anal Biochem 72:248–254
3. Carrea G, D’Arrigo P, Piergianni V, Roncaglio S, Secundo F,
Servi S (1995) Biochim Biophys Acta 1255:273–279
4. Chen VB, Arendall WB III, Headd JJ, Keedy DA, Immormino
RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC
(2010) Acta Crystallogr D66:12–21
5. Comfurius P, Bevers EM, Zwaal RFA (1990) J Lipid Res
31:1719–1721
6. D’Arrigo P, de Ferra L, Piergianni V, Ricci A, Scarcelli D, Servi
S (1994) J Chem Soc Chem Commun 14:1709–1710
7. Doig SD, Diks RMM (2003) Eur J Lipid Sci Technol 105:
368–376
8. Emanuelsson O, Brunak S, von Heijne G, Nielsen H (2007) Nat
Protoc 2:953–971
9. Guex N, Peitsch MC (1997) Electrophoresis 18:2714–2723
10. Hatanaka T, Negishi T, Kubota-Akizawa M, Hagishita T (2002)
Enzyme Microb Technol 31:233–241
11. Hirano H, Watanabe T (1990) Electrophoresis 11:573–580
12. Imamura S, Horiuti Y (1979) J Biochem 83:677–680
13. Ishii F, Nii T (2005) Colloids Surf B Biointerfaces 41:257–262
14. Iwasaki Y, Mizumoto Y, Okada T, Yamamoto T, Tsutsumi K,
Yamamoto T (2003) J Am Oil Chem Soc 80:653–657
15. Jorissen BL, Brouns F, Van Boxtel MPJ, Riedel WJ (2002) Nutr
Neurosci 5:337–343
16. Juneja LR, Kazuoka T, Goto N, Yamane T, Shimizu S (1989)
Biochim Biophys Acta 1003:277–283
17. Juneja LR, Kazuoka T, Yamane T, Shimizu S (1988) Biochim
Biophys Acta 960:334–341
18. Koga T, Nagao A, Terao J, Sawada K, Mukai K (1994) Lipids
29:83–89
19. Kokusho Y, Tsunoda A, Kato S, Machida H, Iwasaki S (1993)
Biosci Biotechnol Biochem 57:1302–1305
20. Kudo S, Kuroda A (1990) Bio Ind 7:494–500 (in Japanese)
21. Laemmli UK (1970) Nature 227:680–685
22. Miura S, Tanaka M, Suzuki A, Sato K (2004) J Am Oil Chem Soc
81:97–100
23. Nagao A, Ishida N, Terao J (1991) Lipids 26:390–394
24. Nakazawa Y, Uchino M, Sagane Y, Sato H, Takano K (2009)
Microbiol Res 164:43–48
25. Nakazawa Y, Suzuki R, Uchino M, Sagane Y, Kudo T, Nagai T,
Sato H, Takano K (2010) Curr Microbiol 60:365–372
26. Nii T, Ishii F (2004) Colloids Surf B Biointerfaces 39:57–63
27. Ogino C, Negi Y, Matsumiya T, Nakaoka K, Kondo A, Kuroda S,
Tokuyama S, Kikkawa U, Yamane T, Fukuda H (1999) J Bio-
chem 125:263–269
28. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt
DM, Meng EC, Ferrin TE (2004) J Comput Chem 25:1605–1612
29. Rosenberg M, Court D (1979) Annu Rev Genet 13:319–353
30. Shimbo K, Iwasaki Y, Yamane T, Ina K (1993) Biosci Biotechnol
Biochem 57:1946–1948
31. Shuto S, Ueda S, Imamura S, Fukukawa K, Matsuda A, Ueda T
(1987) Tetrahedron Lett 28:199–202
Purification, Biochemical Characterization, and Cloning of Phospholipase D 607
123

32. Simkhada JR, Lee HJ, Jang SY, Cho SS, Park EJ, Sohng JK, Yoo
JC (2009) Biotechnol Lett 31:429–435
33. Simkhada JR, Cho SS, Choi HS, Kim SW, Lee HC, Sohng JK,
Yoo JC (2010) Biotechnol Bioprocess Eng 15:595–602
34. Suzuki S, Yamatoya H, Sakai M, Kataoka A, Furushiro M, Kudo
S (2001) J Nutr 131:2951–2956
35. Takami M, Hidaka N, Suzuki Y (1994) Biosci Biotechnol Bio-
chem 58:2140–2144
36. Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins
DG (1997) Nucleic Acids Res 25:4876–4882
37. Ulbrich-Hofmann R (2003) Eur J Lipid Sci Technol 105:305–308
38. Yang SF, Freer S, Benson AA (1966) J Biol Chem 242:477–484
608 Y. Nakazawa et al.
123
Copyright © 2022 FDOKUMEN




















