
Proteins as biomarkers of oxidative/nitrosative stress in diseases: The contribution of redox...
45
PROTEINS AS BIOMARKERS OF OXIDATIVE/NITROSATIVE STRESS IN DISEASES: THE CONTRIBUTION OF REDOX PROTEOMICS Isabella Dalle-Donne, 1 * Andrea Scaloni, 2 Daniela Giustarini, 3 Eleonora Cavarra, 4 Gianluca Tell, 5 Giuseppe Lungarella, 4 Roberto Colombo, 1 Ranieri Rossi, 3 and Aldo Milzani 1 1 Department of Biology, University of Milan, via Celoria 26, I-20133, Milan, Italy 2 Proteomics and Mass Spectrometry Laboratory, I.S.P.A.A.M., National Research Council, via Argine 1085, I-80147, Naples, Italy 3 Department of Neuroscience, University of Siena, via A. Moro 4, I-53100, Siena, Italy 4 Department of Physiopathology and Experimental Medicine, University of Siena, via A. Moro 4, I-53100, Siena, Italy 5 Department of Biomedical Sciences and Technologies, University of Udine, P.le Kolbe 4, 33100 Udine, Italy Received 24 November 2003; revised 22 December 2003; accepted 29 December 2003 Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/mas.20006 I. Introduction ...................................................................... 56 II. Reactive Oxygen and Nitrogen Species ......................... ......................... 57 III. Biological Markers of Oxidative/Nitrosative Stress ........................................... 58 IV. Oxidative/Nitrosative Stress and Protein Modifications ......................................... 60 A. Oxidative/Nitrosative Modification of Protein Thiols ....................................... 60 B. Oxidative/Nitrosative Modification of Tyrosine ........................................... 61 C. Oxidative Modification of Methionine ................................................. 62 D. Protein Carbonylation ............................................................ 63 E. Oxidative Modification of Histidine and Tryptophan ....................................... 63 V. MS Approaches for the Molecular Characterization of Oxidatively/Nitrosatively Modified Proteins ......... 63 A. Analysis of Oxidized/Nitrosated Products of Protein Thiols .................................. 64 B. Analysis of Oxidized/Nitrated Products of Tyrosine Residues ................................. 65 C. Analysis of Oxidized Products of Methionine Residues ..................................... 67 D. Analysis of Protein Carbonylation Products ............................................. 67 E. Analysis of Oxidized Products of Tryptophan Residues ..................................... 68 F. Analysis of Oxidized Products of Histidine Residues ...................................... 69 VI. Proteomic Strategies for the Identification of ROS/RNS Targets in Complex Protein Mixtures ............. 69 VII. Selected Human Diseases Associated with Oxidative/Nitrosative Stress ............................. 72 A. Acute (Adult) Respiratory Distress Syndrome ........................................... 72 B. Alzheimer’s Disease ............................................................. 73 C. Amyotrophic Lateral Sclerosis ...................................................... 74 D. Asthma ...................................................................... 75 E. Atherosclerosis ................................................................. 75 F. Chronic Obstructive Pulmonary Diseases ............................................... 75 G. Diabetes Mellitus ............................................................... 76 H. HIV Infection .................................................................. 77 Mass Spectrometry Reviews, 2005, 24, 55– 99 # 2004 by Wiley Periodicals, Inc. ———— *Correspondence to: Isabella Dalle-Donne, PhD, Department of Biology, University of Milan, via Celoria 26, I-20133 Milan, Italy. E-mail: [email protected] ———— Contract grant sponsor: FIRST 2003 (Fondo Interno Ricerca Scientifica e Tecnologica, University of Milan); Contract grant sponsor: FIRB 2001 (Fondo di Incentivazione alla Ricerca di Base; Contract grant number: RBAU01T97W_003; Contract grant sponsor: Regione Campania (LR 41/94); Contract grant number: 4847316.
Transcript of Proteins as biomarkers of oxidative/nitrosative stress in diseases: The contribution of redox...

PROTEINS AS BIOMARKERS OF OXIDATIVE/NITROSATIVESTRESS IN DISEASES: THE CONTRIBUTION OFREDOX PROTEOMICS
Isabella Dalle-Donne,1* Andrea Scaloni,2 Daniela Giustarini,3 Eleonora Cavarra,4
Gianluca Tell,5 Giuseppe Lungarella,4 Roberto Colombo,1 Ranieri Rossi,3
and Aldo Milzani11Department of Biology, University of Milan, via Celoria 26, I-20133, Milan, Italy2Proteomics and Mass Spectrometry Laboratory, I.S.P.A.A.M.,National Research Council, via Argine 1085, I-80147, Naples, Italy3Department of Neuroscience, University of Siena, via A. Moro 4, I-53100, Siena, Italy4Department of Physiopathology and Experimental Medicine, University of Siena,via A. Moro 4, I-53100, Siena, Italy5Department of Biomedical Sciences and Technologies, University of Udine,P.le Kolbe 4, 33100 Udine, Italy
Received 24 November 2003; revised 22 December 2003; accepted 29 December 2003
Published online in Wiley InterScience (www.interscience.wiley.com). DOI 10.1002/mas.20006
I. Introduction . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 56
II. Reactive Oxygen and Nitrogen Species . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 57
III. Biological Markers of Oxidative/Nitrosative Stress . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 58
IV. Oxidative/Nitrosative Stress and Protein Modifications . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60A. Oxidative/Nitrosative Modification of Protein Thiols . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 60B. Oxidative/Nitrosative Modification of Tyrosine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 61C. Oxidative Modification of Methionine . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 62D. Protein Carbonylation . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63E. Oxidative Modification of Histidine and Tryptophan . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 63
V. MS Approaches for the Molecular Characterization of Oxidatively/Nitrosatively Modified Proteins . . . . . . . . . 63A. Analysis of Oxidized/Nitrosated Products of Protein Thiols . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 64B. Analysis of Oxidized/Nitrated Products of Tyrosine Residues . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 65C. Analysis of Oxidized Products of Methionine Residues . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67D. Analysis of Protein Carbonylation Products . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 67E. Analysis of Oxidized Products of Tryptophan Residues . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 68F. Analysis of Oxidized Products of Histidine Residues . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 69
VI. Proteomic Strategies for the Identification of ROS/RNS Targets in Complex Protein Mixtures . . . . . . . . . . . . . 69
VII. Selected Human Diseases Associated with Oxidative/Nitrosative Stress . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72A. Acute (Adult) Respiratory Distress Syndrome . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 72B. Alzheimer’s Disease . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 73C. Amyotrophic Lateral Sclerosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 74D. Asthma . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75E. Atherosclerosis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75F. Chronic Obstructive Pulmonary Diseases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 75G. Diabetes Mellitus . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 76H. HIV Infection . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77
Mass Spectrometry Reviews, 2005, 24, 55– 99# 2004 by Wiley Periodicals, Inc.
————*Correspondence to: Isabella Dalle-Donne, PhD, Department of
Biology, University of Milan, via Celoria 26, I-20133 Milan, Italy.
E-mail: [email protected]
————Contract grant sponsor: FIRST 2003 (Fondo Interno Ricerca
Scientifica e Tecnologica, University of Milan); Contract grant
sponsor: FIRB 2001 (Fondo di Incentivazione alla Ricerca di Base;
Contract grant number: RBAU01T97W_003; Contract grant sponsor:
Regione Campania (LR 41/94); Contract grant number: 4847316.

I. Preeclampsia . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 77J. Rheumatoid Arthritis . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78K. Transmissible Spongiform Encephalopathies . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 78
VIII. Oxidatively Modified Proteins in Human Diseases . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 79
IX. Concluding Remarks and Future Perspectives . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 85
Acknowledgments . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86
Abbreviations . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 86
References . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . 87
Reactive oxygen species (ROS) and reactive nitrogen species(RNS) contribute to the pathogenesis and/or progression ofseveral human diseases. Proteins are important molecularsignposts of oxidative/nitrosative damage. However, it isgenerally unresolved whether the presence of oxidatively/nitrosatively modified proteins has a causal role or simplyreflects secondary epiphenomena. Only direct identification andcharacterization of the modified protein(s) in a given patho-physiological condition can decipher the potential roles playedby ROS/RNS-induced protein modifications. During the last fewyears, mass spectrometry (MS)-based technologies have con-tributed in a significant way to foster a better understanding ofdisease processes. The study of oxidative/nitrosative modifica-tions, investigated by redox proteomics, is contributing toestablish a relationship between pathological hallmarks ofdisease and protein structural and functional abnormalities.MS-based technologies promise a contribution in a new era ofmolecular medicine, especially in the discovery of diagnosticbiomarkers of oxidative/nitrosative stress, enabling early detec-tion of diseases. Indeed, identification and characterization ofoxidatively/nitrosativelymodified proteins in human diseases hasjust begun. # 2004 Wiley Periodicals, Inc., Mass Spec Rev24:55–99, 2005Keywords: biological markers; mass spectrometry; methioninesulfoxide; nitrosothiols; nitrotyrosine; protein carbonylation;protein oxidation; proteomics; reactive nitrogen species;reactive oxygen species; S-glutathionylation; S-nitrosation
I. INTRODUCTION
Reactive oxygen species (ROS) and reactive nitrogenspecies (RNS) play important physiological functions andcan also cause extensive cellular damage. The balancebetween physiological functions and damage is deter-mined by the relative rates of formation and removal ofROS/RNS. Normally, these species are removed rapidlybefore they cause cell dysfunction and eventual death.Oxidative/nitrosative stress, an imbalance between thegeneration of ROS/RNS and the antioxidant defensecapacity of the body, can affect major cellular components,
including lipids, proteins, carbohydrates, and DNA. Thisphenomenon has closely been associated with a numberof human diseases. Accumulating evidence points tomany inter-related mechanisms during pathogenesis thatincrease the production of ROS/RNS or decrease theantioxidant protection against oxidative/nitrosative insult,although the exact contribution of such mechanisms isnot entirely clear. Information regarding the nature ofROS/RNS, as well as the localization and the effects ofoxidative/nitrosative stress, may be gleaned from theanalysis of discrete biomarkers isolated from tissuesand biological fluids. Biomarkers are cellular indicatorsof the physiological state and changes during a diseaseprocess, at a specific time. However, the presence ofoxidatively/nitrosatively damagedmolecules could simplyreflect secondary epiphenomena rather than having acausal role. A clear delineation of the causal connectionscannot be given at present, but a growing body of evidenceindicates that high levels of ROS/RNS induce distinctpathological consequences that greatly amplify andpropagate injury, leading to irreversible cell and tissuedegeneration.
Here,we overview the application of redox proteomicsto basic research in human diseases associated withoxidative/nitrosative stress. A description of the reactivespecies, the modification reactions, the appropriate strate-gies and experimental techniques used for redox proteomicanalysis will be reported. In addition, a report of specifichuman diseases in which the involvement of oxidative/nitrosative insult has been inferred by detecting oxida-tively/nitrosatively modified proteins will be provided.Although the recent advent of redox proteomics translatesto only a sporadic selection of current human disorderresearch, exampleswill increase dramatically over the nextfew years. On this basis, integration of redox proteomicswith functional data from established biochemical andphysiological methods should lead, in the near future, tothe development of functional proteomics, clarifyingproteome dynamics in human diseases associated withoxidative/nitrosative stressing events.
& DALLE-DONNE ET AL.
56

II. REACTIVE OXYGEN AND NITROGEN SPECIES
Mitochondria are the major source of cellular ROS(Table 1) in non-phagocytic cells. Potentially toxic oxygenmetabolites are physiologically generated at a low levelin cells and tissues during oxidative phosphorylation(Mikkelsen & Wardman, 2003). The resulting moderatelevels of ROS play an integral role in the modulation ofseveral physiological functions of the cell, including geneexpression, signal transduction, and defense againstinvading pathogens. Under normal conditions, it isestimated that up to 1% of the mitochondrial electron flowleadsprimarily to the formationof superoxideanion (Fig. 1,reaction a). Interference with electron transport candramatically increase superoxide production. Superoxideis rapidly converted within the cell to H2O2 and O2 by thesuperoxide dismutases (SODs) (Fig. 1, reaction b). H2O2
can react with reduced transition metals, via the Fentonreaction, to produce the highly reactive hydroxyl radical(Fig. 1, reaction c), a far more damaging molecule to thecell. Alternatively, H2O2 may be converted into water bythe enzymes catalase and glutathione peroxidase (Fig. 1,reaction d). In addition, H2O2 produced by inflammatorycells oxidizes myeloperoxidase to a higher oxidation state(a ferryl-oxo complex) that oxidizes Cl� to hypochlorous
acid (Fig. 1, reaction e), which is capable of oxidizingor chlorinating cellular macromolecules (Winterbourn,Vissers, & Kettle, 2000). HOCl reacts rapidly with thiolsand methionyl residues, making them biologically impor-tant both as scavengers and as targets (Winterbourn,Vissers, & Kettle, 2000). It also reacts with amines to formchloramines or with Cl� to form Cl2 gas; the latterchlorinates DNA or proteins. A similar cascade ofreactions is triggered by eosinophil peroxidase (bromoper-oxidase) that uses H2O2 to produce hypobromous acid,which brominates proteins (Henderson et al., 2001).Myeloperoxidase is also able to form RNS with proteinnitrating activity when both nitrite and H2O2 are present(Eiserich et al., 1998).
Reactive nitrogen species (Table 1) such as nitricoxide, nitrite, and peroxynitrite are both physiologicallynecessary and potentially destructive (Mikkelsen &Ward-man, 2003). Nitric oxide has been identified as a source ofoxidative/nitrosative stress with special relevance topathological conditions. The generation of NO. from theNADPH-dependent oxidation of L-arginine is catalyzed bythree isoforms of nitric oxide synthase, neuronal NOsynthase (nNOS), endothelial NO synthase (eNOS), andinducible NO synthase (iNOS). Although originallydescribed as endothelium-derived vasorelaxing factor,NO. has been recognized to present other beneficial andmalign biochemical properties. Most of the signalingfunctions inherent to NO. are manifested through itsreaction with heme prosthetic groups or by reversibleS-nitrosation of protein thiols. In contrast, the cytopathicnature of NO. is evident under conditions where it reactswith ROS and converts to more reactive redox derivatives
FIGURE 1. Main pathways for the formation of reactive oxidants/
nitrosants from superoxide anion and nitric oxide radicals. MPO,
myeloperoxidase; SOD, superoxide dismutase; CAT, catalase; GPX,
glutathione peroxidase.
TABLE 1. Reactive Oxygen Species (ROS) and Reactive
Nitrogen Species (RNS) Generated in Cells and Tissues
Reactive oxygen species (ROS)Superoxide (O2
.�)Hydroxyl radical (HO.)Peroxyl radical (RO2
.)Alkoxyl radical (RO.)Hydroperoxyl radical (HO2
.)Hydrogen peroxide (H2O2)Hypochlorous acid (HOCl)Hypobromous acid (HOBr)Ozone (O3)Singlet oxygen (1O2)
Reactive nitrogen species (RNS)Nitric oxide (.NO)Nitrogen dioxide (.NO2)Nitrous acid (HNO2)Nitrosyl cation (NOþ)Nitrosyl anion (NO�)Dinitrogen tetroxide (N2O4)Dinitrogen trioxide (N2O3)Peroxynitrite (ONOO�)Peroxynitrous acid (ONOOH)Alkyl peroxynitrites (ROONO)Nitronium cation (NO2
þ)Nitryl chloride (NO2Cl)
REDOX PROTEOMICS AND DISEASE &
57

that can attack proteins, lipids, and DNA. In fact, whenNO. concentration increases into the range of SOD tissuelevels, NO. competes with this enzyme for the removal ofO2.� by forming ONOO� (Fig. 1, reaction f ), since O2
.�
reacts with NO. with a rate constant that is three times therate of its reaction with SOD. Peroxynitrite exists in fast,dynamic equilibrium with its conjugated acid, ONOOH(Fig. 1, reaction g). In the absence of carbon dioxide,ONOO�/ONOOH decays producing harmless nitrate(Fig. 1, reaction h) as the main product, reactive NO2
.
and .OH (Fig. 1, reaction i). Significant amounts ofNO2
. may also be formed from the H2O2-dependentmyeloperoxidase reaction in the presence of low micro-molar levels of nitrite (Eiserich et al., 1998), or from thereaction of O2 with NO. (Fig. 1, reaction j). The mostpotent effects of ONOO� and RNS appear to be thiolmodifications that either affect the function of signalingsystems or result in the production of tissue-derived donorsof NO.. Oxidized NO-derived species, including ONOO�,NO2, and N2O3, readily interact with glutathione (GSH)and other thiols, including protein thiols, to cause theiroxidation or the formation of nitrosated thiols. Peroxyni-trite-mediated reactions are enhanced by a number ofsubstances including CO2. The reaction of ONOO� withCO2 (Fig. 1, reaction l) is very rapid at physiologicalcarbonate concentration (>1 mM); thus, nitrosoperoxo-carbonate, ONO2CO2
�, may be the most physiologicallyrelevant nitrating agent. However, it is very short-lived(<3 ms) and approximately one-third decomposes to yieldNO2
. and the carbonate radical, .CO3� (Fig. 1, reaction m);
the remaining two-thirds decompose to NO3� and CO2
(Fig. 1, reaction n). Peroxynitrite and its protonated formreact with cellular nucleophiles or oxidize heme-proteinsto ferryl-oxo derivatives. Both ONOOH and ferryl-oxocomplexes are strong oxidants (Hodges & Ingold, 1999;Mikkelsen & Wardman, 2003).
Naturally occurring enzymatic and non-enzymaticsystems exist to protect cells and tissues against thecontinuous production of ROS/RNS during normalmetabolism. These include antioxidant enzymes such ascatalases, SODs, peroxiredoxins, and glutathione perox-idases. Two non-enzymatic proteins, ferritin, which bindsiron in the cytoplasm of mammalian cells, and cerulo-plasmin, which binds copper in plasma, are thought tocontribute a significant antioxidant capacity to body fluids,binding transition metals involved in both metal-catalyzed(auto)oxidation and reactions leading to .OH productionfrom O2
.�. Other important and widespread antioxidantsare low molecular weight compounds such as vitamin E(a-tocopherol), a major membrane-bound antioxidant,vitamin C (ascorbic acid), and glutathione (L-g-glutamyl-L-cysteinyl-glycine). However, when ROS/RNS levelsexceed the antioxidant capacity, a deleterious conditionknown as oxidative/nitrosative stress occurs. It describes a
status in which cellular antioxidant defenses are insuffi-cient to keep the levels of ROS/RNS below a toxicthreshold. This may be either because of excessiveproduction of ROS/RNS, loss of antioxidant defenses inthe cell, or both (Dalle-Donne et al., 2003c). Unchecked,excessive ROS/RNS generation can lead to the destructionof cellular components including proteins, and ultimatelycell death via apoptosis or necrosis (Hensley et al., 2000).Frequently, different reactive species coexist in thecellular environment making it difficult to identify un-equivocally which agent is responsible for a givenbiological effect.
III. BIOLOGICAL MARKERS OF OXIDATIVE/NITROSATIVE STRESS
The significance of oxidative/nitrosative stress has becomeincreasingly recognized to the point that it is nowconsidered to be a component of virtually every disease.However, inmost disorders, oxidative/nitrosative stress is aconsequence and not a cause of the primary disease process(Halliwell &Gutteridge, 1999; Dalle-Donne et al., 2003c).
To investigate the role of oxidative/nitrosative stressand ROS/RNS in the pathogenesis and/or progression ofdiseases, optimization of appropriate analytical proceduresis necessary. Sensitive techniques for the analysis of ROS/RNS are now available;measurements ofNO., O2
.�, H2O2,and ONOO� have recently been reviewed (Tarpey &Fridovich, 2001). However, direct determination of ROS/RNS is difficult for several reasons; ROS/RNS aregenerally too reactive and/or have a too brief half-life(seconds) to measure them directly in cells/tissues or bodyfluids. Anyway, the concomitant presence of ROS/RNSand other biomolecules always yields specific products,generating a sort of specific chemical footprint of theiroccurrence. For instance, halogenated Tyr residues inproteins are specific markers of hypohalous acids. Sincemolecular products from oxidative/nitrosative stress aregenerally more stable than oxidants and nitrosantsthemselves, i.e., oxidized lipids (such as aldehydes andketones), proteins (such as carbonyl-labeled amino acidresidues), and nucleic acids (such as 8-oxo-20-deoxy-guanosine) are more stable than the reactive species thateffected theirmodification, ROS/RNSmeasurements ofteninvolve determining levels of their oxidation targetproducts (Pryor, 2001; Griffiths et al., 2002). On this basis,a variety of biological markers are available for determina-tion of oxidative/nitrosative stress and have been discussedin a number of published papers and books (e.g., Davieset al., 1999; Halliwell & Gutteridge, 1999; Pryor, 2001;Griffiths et al., 2002; Dalle-Donne et al., 2003b; Giustariniet al., 2003a,b). Furthermore, biomarkers of ROS/RNS
& DALLE-DONNE ET AL.
58

have the potential not only to determine the extents ofoxidative injury but also to identify the nature of theoxidant itself. For instance, .OH specifically convertsprotein phenylalanine residues to the unnatural aminoacid isomer o-tyrosine (o-Tyr). Tyrosyl radical (theoxidizing intermediate generated by peroxidases) formso,o0-dityrosine (di-Tyr) as the major product; differently,ONOO� generates 3-nitrotyrosine (NO2-Tyr). Such a bit ofinformation is important for predicting the consequencesof oxidation as well as for providing a basis for designingappropriate interventions to alleviate injury.
The usefulness of a biomarker lies in its ability toprovide early indication of disease or progression of thedisease (Fig. 2). A valid biological marker of oxidative/nitrosative stress should be:
* accessible in a target tissue or a valid surrogate oneable to quantitatively reflect the oxidative modifica-tion of the target tissue;
* specific for the reactive species involved;
* a chemically and biologically stable product, notsusceptible to artifactual induction, oxidation, or lossduring storage;
* determined by an assay that is specific, sensitive, andreproducible;
* a major product of oxidative/nitrosative modificationthat may be implicated directly in the onset and/orprogression of disease;
* representative of the balance between oxidative/nitrosative damage generation and clearance;
* free of confounding factors from dietary intake.
In addition, the target should have a high reactivityand be present at a high enough concentration for the
biomarker to be a significant product. Case–controlstudies on stored samples should be used to test theefficiency of biomarkers. Care must be taken to defineand establish references or baseline profiles from normaltissues, cells, or body fluids. The use of a panel ofbiomarkers would enhance the positive predictive valueof a test and minimize the proportion of false-positive andfalse-negative results.
There is no doubt that proteins are major targets forradicals and other oxidants when these are formed in vivoeither in intra- or extracellular environments. On the basisof published rate constants and the knowledge of therelative abundance of macromolecules within cells, it hasbeen estimated that proteins can scavenge 50–75% ofreactive radicals (Davies et al., 1999). Furthermore,proteins can retain the fingerprint of the initial oxidative/nitrosative insult that mediates damage. This contrastswith lipid peroxidation, where propagation reactionsinvolving the initial lipid oxidation products result in lossof the information about the initial oxidative insult.Amino acid oxidation products are superior to lipidoxidation products in terms of stability during samplestorage. Furthermore, assays for lipid oxidation productshave not yet been reported to routinely monitor forartifactual formation during sample storage and analysis,such as through incorporation of isotopically labeledparent lipids, as instead usually checked for amino acidoxidation products. Thus, a significant advantage ofprotein oxidation products as markers of disease risk andprogression is their possible use in banked specimens.Archival specimens from pre-existing clinical studies,which will undoubtedly play a critical role in validatingthe clinical utility of any oxidation marker, cannot beemployed for examining lipid oxidation products, unlessextensive precautionary measures were taken to prevent
FIGURE 2. Use of oxidative/nitrosative stress biomarkers in detection of disease initiation and/or
progression as well as in assessing effective therapies.
REDOX PROTEOMICS AND DISEASE &
59

artificial oxidation. Thus, stable species like 3-chloro-tyrosine (Cl-Tyr) and NO2-Tyr, whose concentration isnot affected during prolonged storage in a freezer, can beconsidered as ideal markers of oxidative/nitrosative stressin vivo. Such data, together with the knowledge that someproteins have long half-lives and, hence, are likely to ac-cumulate oxidative ‘‘hits,’’ suggest that modified residueson proteins may be considered as most sensitive markersfor oxidative damage in mammalian cells (Davies &Dean, 1997; Dean et al., 1997; Davies et al., 1999).
Besides identifying pathways of damage, proteinmarkers will be extremely useful to monitor oxidativestress in vivo if they are liberated by proteolysis andexcreted into body fluids (e.g., urine, blood). Animportant question regarding the use of oxidationproducts to monitor oxidative stress in vivo is their fatein the body. For example, di-Tyr might be re-incorporatedinto proteins or metabolized to other compounds,invalidating the relationship between its concentrationin urine and protein oxidation. Investigating this issue byinjecting purified radiolabeled di-Tyr intravenously intomice, it has been observed that di-Tyr released fromproteins is relatively resistant to metabolism and isexcreted by the kidney into urine in near-quantitativeyield (Bhattacharjee et al., 2001). Thus, to assess thepotential utility of oxidation products in humans, it willbe important to determine the fate of oxidized aminoacids and other oxidation products in vivo.
Oxidative damage to proteins is induced eitherdirectly by ROS/RNS or indirectly by reaction ofsecondary products of oxidative/nitrosative stress. It canoccur via different mechanisms, resulting in polypeptidechain cleavage, cross-linking, and modification of theside chain of virtually every amino acid (Berlett &Stadtman, 1997; Davies & Dean, 1997; Dean et al., 1997;Dalle-Donne et al., 2003c). These reactions can lead todiverse functional consequences such as inhibition ofenzymatic and binding activities, increased susceptibilityto aggregation and proteolysis, increased or decreaseduptake by cells, and altered immunogenicity. However,not all proteins are equally sensitive to oxidative damage,and oxidation susceptibility depends on the structure ofthe protein (e.g., sequence motifs, residues exposed onthe molecular surface, bound metal atoms).
A number of oxidative reactions determining thecleavage of the polypeptide backbone have beenelucidated (Berlett & Stadtman, 1997; Davies & Dean,1997; Dean et al., 1997). However, though it can beeasily detected with isolated proteins (e.g., using sodiumdodecylsulfate–polyacrylamide gel electrophoresis,SDS–PAGE, or high performance liquid chromatogra-phy, HPLC), its use as a marker of protein oxidationin vivo is very limited because of the occurrence of otherproteins in complex systems and the potential role of
proteases in polypeptide hydrolysis. Thus, backbonefragmentation is rarely used to quantify protein oxidationin complex systems. Differently, the use of stableproducts of protein side chain oxidation as potentialmarkers for assessing oxidative damage in vivo is amplydiffused and applicated in the study of human diseases(Davies & Dean, 1997; Dean et al., 1997; Davieset al., 1999; Griffiths et al., 2002; Dalle-Donne et al.,2003b,d).
IV. OXIDATIVE/NITROSATIVE STRESS ANDPROTEIN MODIFICATIONS
Oxidative/nitrosative stress may cause reversible and/orirreversible modifications on sensitive proteins (Stadtman& Berlett, 1998). ROS/RNS leading to protein oxidationinclude both radical and non-radical species (Table 1).Reversible modifications, usually at Cys residues, mayhave a dual role of protection from irreversible damageand modulation of protein function (redox regulation).Irreversible modifications induced by ROS/RNS, such di-Tyr formation, protein–protein cross-linking, Lys and Argcarbonylation, are generally associated with permanentloss of function and may lead to either the degradation ofthe damaged proteins (Berlett & Stadtman, 1997; Davies& Dean, 1997; Dean et al., 1997; Grune et al., 2003) ortheir progressive accumulation into cytoplasmic inclu-sions, as observed in age-related neurodegenerative dis-orders (Giasson et al., 2000, 2002; Butterfield & Kanski,2001).
A. Oxidative/Nitrosative Modificationof Protein Thiols
Thiol groups are easily oxidized by many ROS/RNS, theirsusceptibility being inversely influenced by their pKa
value. Protein thiol group modifications can have differentphysiological effects, depending on their reversible orirreversible nature. Mild oxidation of cysteines cangenerate sulfenic acid (P-SOH), inter- or intra-moleculardisulfides, protein mixed disulfides with low molecularweight thiols (e.g., GSH), and S-nitrosothiols. It is ageneral opinion that these reversible modifications can bepart of regulatory processes of protein functions, in whichcysteines can cycle between the oxidized and reducedstate. Sulfenic acid is extremely unstable, being frequentlyan intermediate in sulfinic and/or sulfonic acid generationor participating in redox reactions inwhich other reversibleoxidative modifications of sulfhydryl (SH) groups takeplace. On the contrary, Cys residues can be irreversiblyoxidized by strong oxidative insults to sulfinic (P-SO2H)and sulfonic acids (P-SO3H), which cannot usually bereversed by metabolic processes and can cause loss of
& DALLE-DONNE ET AL.
60

protein function. However, it has recently been demon-strated that also the sulfinic inactive form of peroxiredoxinI, produced during the exposure of cells to H2O2, is rapidlyreduced to the catalytically active thiol form through anunknown conversion process (Woo et al., 2003).
Whereas protein disulfide generation can be hamperedby steric hindrance, protein SHgroups can easily react withlow molecular weight compounds in response to anoxidative insult producing mixed disulfides. GSH is thedominant ligand in this reaction because of its highconcentration (0.5–10mM) inmammalian cells (Halliwell& Gutteridge, 1999). Different mechanisms have beenproposed for protein S-glutathionylation: by reaction ofGSH with partially oxidized reactive protein SH groups(thiyl radical or sulfenic acid intermediates), by thiol/disulfide exchange reaction, by nitrosoglutathione produc-tion and formation of glutathione S-oxide as intermediate(Cotgreave & Gerdes, 1998; Klatt & Lamas, 2000;Okamoto et al., 2001). Various enzymatic systems suchas thioredoxin, glutaredoxin, and protein disulfide iso-merase are able to reduce these disulfide bonds using GSHor NADPH as donors of reducing equivalents (Arner &Holmgren, 2000; Schwaller, Wilkinson, & Gilbert, 2003).In general, S-glutathionylation has been proposed as ameans of storing GSH during oxidative stress and, beingreversible, it has been regarded as a protective mechanismguarding against irreversible protein thiol oxidation (Klatt& Lamas, 2000; Schafer & Buettner, 2001).
As a consequence of NO. metabolism, proteincysteines can also undergo nitrosativemodifications.Nitricoxide can form adducts with SH groups producing S-nitrosothiols. These molecules are thought to be inter-mediates in the storage and delivery of NO., showing thebiological properties of NO. itself (e.g., vasodilation). Thedirect reaction between NO. and SH groups does notyield S-nitrosothiols; therefore, alternative pathways forS-nitrosothiol generation have been proposed. In particu-lar, metabolites formed during NO. autooxidation, such asN2O3 and ONOO�, are likely candidates for this reaction(Hogg, 2002). Furthermore, equilibrium reactions namedtransnitrosations, in which the NO group is exchangedfrom a low molecular weight S-nitrosothiol to a thiol, mayreversibly transfer the NOþ moiety to protein SH groups(Hogg, 2002). The reversible nature of S-nitrosation isbecause of S-nitrosothiol susceptibility to catalytic decom-position by metal-induced hemolytic cleavage or toreduction by ascorbic acid, GSH, and thioredoxin. More-over, thiols can cause the decay of S-nitrosothiols inducingprotein S-thiolation (Hogg, 2002).
Various proteins have been studied to assay thesusceptibility of their SH groups to undergo reversibleoxidative modifications. Variations in inter- or intra-protein disulfide concentration have widely been reportedas a result ofmodification. However,while it iswell-known
that these processes are involved in protein folding (Frand,Cuozzo, & Kaiser, 2000), only a few data are availableabout the relevance of thesemodifications in response to anoxidative insult (Georgiou, 2002). Although S-glutathio-nylation is usually considered a modification occurring inresponse to oxidative stress, a number of S-glutathiony-lated proteins, including enzymes, cytoskeletal proteins,and transcription factors, have been observed under basalphysiological conditions, suggesting their possible invol-vement in signaling and regulatory pathways (Cotgreave&Gerdes, 1998;Klatt et al., 1999;Klatt&Lamas, 2000; Lindet al., 2002; Eaton, Fuller, & Shattock, 2002a; Eaton et al.,2002b,c; Fratelli et al., 2002; Dalle-Donne et al., 2003a,d).However, if the Cys residue involved in mixed disulfide isfunctionally critical, S-thiolation will render the proteininactive, as in the case of glyceraldehyde 3-phosphatedehydrogenase, thus contributing to cellular dysfunctionduring oxidative stress (Klatt & Lamas, 2000; Eaton et al.,2002b). Also nitrosative modifications leading to S-nitrosothiol generation are able to modify several enzy-matic or structural functions (Dalle-Donne et al., 2000;Jaffrey et al., 2001; Stamler, Lamas, & Fang, 2001; Hogg,2002). For instance, during the analysis of proapoptoticsignaling in mammalian cells, it has recently beensuggested that S-nitrosation can mediate interactionsbetween proteins (Matsumoto et al., 2003). Likewise S-glutathionylation, the occurrence of S-nitrosation has beenshown under basal conditions too, as in the case ofconstitutively S-nitrosated caspase-3 (Mannick et al.,1999). Differently, only few evidences are available onthe physiological relevance and function of proteincysteine oxidation to sulfenic acid in cellular signaling(van Montfort et al., 2003; Wood et al., 2003).
B. Oxidative/Nitrosative Modification of Tyrosine
A powerful strategy for understanding the underlyinginvivomechanisms of tyrosine oxidative injury is to identifystable end-products of protein oxidation produced by dif-ferent reaction pathways (Heinecke, 1999a,b). In particu-lar, specific tyrosine derivatives produced by oxidationhave been characterized for: myeloperoxidase-catalyzedreaction pathways through the action of HOCl (Cl-Tyr,and 3,5-dichlorotyrosine, di-Cl-Tyr) (Domigan et al.,1995; Fu et al., 2000; Podrez, Abu-Soud, & Hazen, 2000),eosinophil peroxidase-catalyzed reaction pathwaysthrough the action of hypobromous acid (3-bromoty-rosine, Br-Tyr, and 3,5-dibromotyrosine, di-Br-Tyr) (Wuet al., 1999), free radical pathways (o-Tyr, m-Tyr, anddi-Tyr), and RNS-catalyzed pathways (NO2-Tyr, di-Tyr,3,4-dihydroxy phenylalanine, and the correspondingquinone).
Peroxynitrite is commonly implicated as the principalmediator of tyrosine nitration, and its concentrationmay be
REDOX PROTEOMICS AND DISEASE &
61

a determining factor in modification (Mallozzi, Di Stasi, &Minetti, 2001; Minetti, Mallozzi, & Di Stasi, 2002).However, other facile tyrosine nitration pathways can alsobe operative during inflammatory oxidative reactions;myeloperoxidase and other metalloproteins capable ofperoxidase activity can catalyze tyrosine nitration in thevascular compartment (Baldus et al., 2002; Gaut et al.,2002a). Because substrates supporting peroxidase-depen-dent tyrosine nitration (NO., NO2
�, and hydroperoxides)become abundantly available for myeloperoxidase-depen-dent nitration, it is likely that multiple mechanisms willaccount for tissue NO2-Tyr formation in diverse vascularinflammatory events (Eiserich et al., 1998; Van der Vlietet al., 1999).
Tyrosine nitration is selective, largely because of thelocal environment of certain Tyr residues in proteins.Modification of Tyr residues can alter protein function,because incorporation of bulky groups onto the aromaticring lowers the pKa of the phenolic group and imposes bothsteric and electronic perturbations that affect the capacityof Tyr to function in electron-transfer reactions andmaintenance of protein conformation (Eiserich et al.,1999; Zhu et al., 2000). On this basis, tyrosine nitration hasbeen reported to cause either gain or loss of function (Goleet al., 2000; Greenacre & Ischiropoulos, 2001); it can beremoved or reduced by either enzymatic or non-enzymaticmechanisms (Kamisaki et al., 1998; Balabanli et al., 1999;Davis et al., 2001). It is important to note that RNS,although yielding mainly NO2-Tyr, can also concurrentlyinduce di-Tyr formation and oxidativemodification of evenmore RNS-susceptible protein targets (e.g., Cys, FeS, orZnS complexes) that may actually be the proximal cause ofimpaired protein function (Van derVliet et al., 1999). Thus,Tyr nitration may parallel but not induce enzyme/proteindysfunction.
The existence of distinct pathways for tyrosinenitration underscores the potential significance of thisprocess in inflammation and cell signaling. Thus, this post-translational protein modification is a marker of oxidative/nitrosative injury that is frequently linked to altered proteinfunction during inflammatory conditions (MacMillan-Crow, Crow, & Thompson, 1998; Eiserich et al., 1999;Aulak et al., 2001; Aslan et al., 2003). The reversibility ofNO2-Tyr formation (Kamisaki et al., 1998; Balabanli et al.,1999;Davis et al., 2001) also implies that tyrosine nitrationmay not only represent a marker of RNS formation andlikely altered protein function, as recently demonstratedfor actin in both human sickle cell disease tissues and amurinemodel of sickle cell disease (Aslan et al., 2003), butmay also evoke protein conformational changes that,modulating protein function, mimic or affect cell signalingevents such as adenylation and tyrosine phosphorylationduring inflammation (Berlett, Levine, & Stadtman, 1998).In this regard, it is interesting to note that the yields ofNO2-
Tyr in inflammatory events are similar in magnitude to theextent of tyrosine phosphorylation during cell signaling.Protein tyrosine nitration has also been detected innumerous tissues under apparently normal physiologicalconditions (Greenacre & Ischiropoulos, 2001). In thecardiovascular system, basal protein nitration was found inall major types of cells. Basal protein nitration was alsofound in plasma proteins of healthy subjects; low levelsof NO2-Tyr were measured in albumin and low-densitylipoprotein (LDL) (Khan et al., 1998). These data areconsistent with the emerging perspective that low levelsof Tyr nitration may be a physiological regulator of asignaling pathway.
C. Oxidative Modification of Methionine
Methionine residues are highly susceptible to oxidation byalmost all ROS/RNS (Vogt, 1995). Mild oxidizingconditions determine the generation of methionine sulf-oxide (MetO), which can be further oxidized tomethioninesulfone (MetO2) under stronger oxidizing conditions(Vogt, 1995; Levine, Moskovitz, & Stadtman, 2000a).ROS/RNS lead to formation of a mixture of two diaster-eomeric forms: Met-R(O) andMet-S(O) (Moskovitz et al.,2002).
Methionine oxidation has been associated with loss ofprotein function, as in the case of a-1 protease inhibitor,a-chymotrypsin, ribonuclease, subtilisin, phosphogluco-mutase, actin, and human immunodeficiency virus (HIV)-2protease, as well as several peptide hormones (Vogt, 1995;Davis et al., 2000;Dalle-Donne et al., 2002).However,Metoxidation does not necessarily cause a loss of function in allproteins (Levine et al., 1996). This modification can bereverted by methionine sulfoxide reductases (Msrs) thatcatalyze the NAD(P)H-dependent reduction of MetO.Cells contain two different stereo-specific Msrs: onespecific for the S-isomer (MsrA) and the other specificfor the R-isomer (MsrB) (Stadtman & Berlett, 1998;Levine, Moskovitz, & Stadtman, 2000a; Moskovitz et al.,2002; Weissbach et al., 2002).
Since methionine oxidation is enzymatically re-versible, it has been proposed that Met residues may serveas an endogenous antioxidant defense, protecting thetargeted proteins from more extensive irreversible oxida-tive damage at other essential amino acids (Levine et al.,1996; Levine, Moskovitz, & Stadtman, 2000a). Thishypothesis is consistent with the observation that, unlikeother amino acid modifications, Met oxidation haslittle or no effect on the susceptibility of proteins toproteolytic degradation (Levine et al., 1996; Stadtmanet al., 2002). The role of reversible Met oxidation as adefense mechanism against oxidative/nitrosative stressis underscored by the finding that overexpression ofMsr confers resistance to oxidants both in yeast and
& DALLE-DONNE ET AL.
62

mammalian cells (Moskovitz et al., 1998). In accor-dance with this finding and with the hypothesis that Metresidues in proteins serve protective roles by preventingoxidative damage at other residues, transgenic mice with aknockout of MsrA have a reduced life span and increasedprotein carbonyl levels (Moskovitz et al., 2001). A separatestudy using a transgenic MsrA Drosophila model sup-ports these observations by demonstrating that transgenicflies overexpressing MsrA live longer, more active liveswhen compared with wild-type littermates (Ruan et al.,2002).
D. Protein Carbonylation
Protein carbonylation is an irreversible oxidative modifi-cation. Carbonylated proteins are not repaired and areeither removed by proteolytic degradation or accumulateas damaged or unfolded proteins (Stadtman & Berlett,1998). The number of carbonyl groups observed within aprotein perfectly correlates with protein damage caused byoxidative stress (Shacter et al., 1994). Thus, proteincarbonylation is an important marker of protein oxidationand its measurement is thought to be a good indicator forthe extent of oxidative damage of proteins associated withvarious human diseases (Levine et al., 2000b; Dalle-Donneet al., 2003c).
Carbonyl groups (aldehydes and ketones) may beintroduced in the protein at different sites and by differentmechanisms (Stadtman & Berlett, 1998). Carbonylmoieties are produced on protein side chains, especiallyof Pro, Arg, Lys, and Thr, when these amino acids areoxidized into ketone or aldehyde derivatives (Berlett &Stadtman, 1997). In parallel, protein carbonyl groups canalso be generated through oxidative cleavage of proteins byeither thea-amidation pathway or by oxidation of glutamylside chains, leading to formation of a peptide in which theN-terminal amino acid is blocked by an a-ketoacylderivative (Berlett & Stadtman, 1997). Protein carbonyla-tion can also occur by reaction with various reactiveproducts generated during lipid peroxidation, 4-hydrox-ynonenal (4-HNE), 2-propenal (acrolein), and malondial-dehyde (MDA). It involves Michael-addition ofthese reactive aldehydes to the nucleophilic side-chainof Cys, His, or Lys residues, determining the incorpo-ration of aldehyde/carbonyl group into the peptide chain.Finally, reactive carbonyl groups (ketoamines, keto-aldehydes, deoxyosones) can also be generated bysecondary reaction of the primary amino group of Lysresidues with reducing sugars or their oxidation pro-ducts (glycation/glycoxidation) reactions (Stadtman &Berlett, 1998). The occurrence of these carbonyl moietiesmay alter the conformation of the polypeptide chain, thusdetermining the partial or total inactivation of numerousproteins.
E. Oxidative Modification of Histidineand Tryptophan
Metal-catalyzed oxidation of proteins involves reductionof Fe(III) or Cu(II) by a suitable electron donor such asNADH, NADPH, ascorbate, or mercaptane. Fe(II) or Cu(I)ions bound to specific metal-binding sites on proteins reactwith oxidants to generate radicals (Berlett & Stadtman,1997), which immediately oxidize neighboring amino acidresidues. In general, protein metal-catalyzed oxidation is ahighly selective reaction that occurs at sites with transitionmetal-binding capability. Thus, in addition to theMet, Cys,and Tyr oxidation already described, a close proximity ofHis and Trp residues to heme or Cu(II) binding sites candetermine specific amino acid modifications. Thus, 2-oxo-histidine and 4- or 5-hydroxy-2-oxo-histidine are gener-ated from His oxidation. Similarly, hydroxytryptophan,N-formylkynurenine, kynurenine, and 3-OH-kynurenineare Trp oxidation products. Examples of protein metal-catalyzed oxidation have been reported for myoglobin(Gunther et al., 1998; Hara et al., 2001), b-amyloid peptide(Schoneich & Williams, 2002), Cu,Zn-SOD (Kurahashiet al., 2001), and recombinant prion protein (Requena et al.,2001b). Moreover, the occurrence of proteins in closeproximity to a general source ofROS, as normally respiringtissues or UV radiation, can determine His or Trp modi-fication. In fact, N-formylkynurenine generation has beenreported in normal conditions for lens proteins (Finleyet al., 1998) and for a series of cardiac mitochondrialproteins (Taylor et al., 2003). All of these reactions typi-cally result in structural alterations and loss of enzymaticactivity implicated in a variety of diseases, includingseveral neurodegenerative ailments as well as in aging.
V. MS APPROACHES FOR THE MOLECULARCHARACTERIZATION OF OXIDATIVELY/NITROSATIVELY MODIFIED PROTEINS
A series of analytical strategies based on mass spectro-metry (MS) techniques has been reported in the literaturefor the detection of oxidation products in isolated proteins.In general, all of these methodologies are based on theobservation that these reactions, causing a covalentmodification of amino acids, determine a specific mole-cular mass variation in the products, easily detectable bymass spectrometric measurements. Depending on adductnature, different MS approaches have been developed forthe detection of oxidatively/nitrosatively modified specieseither in intact proteins, in peptide mixtures generatedfollowing digestion with proteases or reagents with highspecificity, or in amino acid hydrolysates produced byextensive enzymic or chemical hydrolysis. In all cases, topreserve the stability of the modified amino acids, specificexperimental conditions have to be carefully chosen for
REDOX PROTEOMICS AND DISEASE &
63

protein manipulation and/or hydrolysis. Although allstrategies can provide quantitative information on themodification extent, only mass spectrometric analysis ofthe modified peptides can be uniquely used for the assign-ment of the modification to specific amino acid residues.
A. Analysis of Oxidized/Nitrosated Productsof Protein Thiols
Formation of inter-molecular disulfides following oxida-tive/nitrosative insult generates macroscopic variation ofprotein molecular mass; on this basis, it has usually beendetected by low-resolution techniques as SDS–PAGEunder not reducing conditions. Differently, the occurrenceof mixed disulfides with lowmolecular weight compoundsor intra-molecular disulfides, determining limited variationin molecular mass of intact proteins, has been revealedby conventional MS procedures. In the case of S-gluta-thionylated, S-cysteinyl-glycinylated, S-cysteinylated, andS-sulfonated proteins, the occurrence of S-conjugatedspecies has been ascertained by direct electrosprayionization (ESI)measurements of intact proteins, detectingthe corresponding adducts with amass difference ofþ305,þ176, þ119, and þ80 Da, respectively (Hanson et al.,1999; Naito & Niwa, 2000; Lim et al., 2003). As expected,the mass spectra of species containing mixed disulfideswere totally affected by reducing agent treatment. In thecase of intra-molecular disulfides, the limited variation inmolecular mass of intact proteins compared with notstressed species (Dm¼�2 Da for each S–S bond)determined a need of additional measurements (Caselliet al., 1998). For this reason, a modification of the MSstrategy conventionally used for the titration of free thiolsin proteins has been applied for the detection of oxidizedcysteines. Simply comparing the molecular mass valueof the intact protein in its native and stressed state, beforeand following extensive alkylation with iodoacetamideunder denaturing not reducing conditions, the number ofthe Cys residues involved in oxidative/nitrosative insultand the nature of the modification can be inferred (Vilardoet al., 2001; Cecconi et al., 2002). In fact, cysteinesinvolved in disulfides will not react with iodoacetamide,thus not generating the corresponding mass increase(Dm¼þ57 Da for each available SH), easily detectableby ESI measurements. Assuming a comparable ionizationtendency for all of the different species obtained followingalkylation, this procedure can be successfully applied toevaluate the quantitative extent of the oxidative insult.Recently, this approach has been used for the molecularcharacterization of the products generated from theoxidative modification of bovine lens aldose reductaseinduced by cupper ions or by intermediates of GSHturnover, as illustrated in Figure 3 (Vilardo et al., 2001;Cecconi et al., 2002).
Mixed disulfide assignment to specific Cys residuesoccurring in the polypeptide chain can be obtained bymassmapping experiments on peptide mixtures generatedfrom carboxamidomethylated species following alkyla-tion under denaturing not reducing conditions. A carefulevaluation of experimental conditions suitable to avoidscrambling phenomena during protein hydrolysis isstrongly recommended. Identification of the modifiedresidues has been obtained by liquid chromatography-electrospray ionization (LC-ESI) or matrix-assisted laserdesorption/ionization (MALDI) mapping experiments,detecting the peptides bearing a mass difference ofþ305 Da (S-glutathionylated), þ176 Da (S-cysteinyl-gly-cinylated), þ119 Da (S-cysteinylated), and þ80 Da (S-sulfonated), and eventually confirmed by collision induceddissociation (CID) measurements (Vilardo et al., 2001;Lim et al., 2003). Similarly, cysteine pairing identificationin species containing intra-molecular disulfides as a resultof oxidative/nitrosative insult are derived by using themassmapping and tandem MS approaches conventionally usedfor the assignment of disulfides in native polypeptidespecies (Tell et al., 1998; Zheng, Aslund, & Storz, 1998;Song et al., 2000;Vilardo et al., 2001; Cecconi et al., 2002).
Irreversible oxidation of cysteines to sulfinic andsulfonic acids has been determined in proteins bymeasuring the occurrence of adducts with Dm¼þ32 andþ48Da, respectively, in the ESI-MS spectrum of the intactmolecules (Yang et al., 2002; Woo et al., 2003). Thesespecies are stable and insensitive to treatment withreducing agents. The selectivity of this modificationtoward Cys and not Met residues was verified followingspecific labeling with iodoacetamide. Identification of themodified cysteines was obtained by MALDI or LC-ESImass mapping experiments on protein digests, specificallyrevealing peptides bearing these mass increases, andconfirmed by tandem MS analysis (Rabilloud et al.,2002; Yang et al., 2002).
The occurrence of protein S-nitrosation has beendetected by ESI-MS measurement, as reported in the caseof hemoglobin, caspase-3 subunits, and Ca-ATPase(Ferranti et al., 1997; Zech et al., 1999; Viner, Williams,& Schoneich, 1999). The occurrence in the spectra ofadducts presenting a Dm¼þ29 Da was indicative of NO.
addiction to intact molecular species. In general, acidconditions are strongly recommended for the purification,digestion, and analysis of S-nitrosated species, as a result oftheir well-known instability (Ferranti et al., 1997; Viner,Williams, & Schoneich, 1999). For this reason, peptidemixtures for mass mapping experiments are convention-ally generated by pepsin hydrolysis and their analysis isperformed using soft ionization techniques (LC-ESI).Confirmation of signal assignment to S-nitrosated peptidesis inferred by selective fragmentation at their S–NO bondby increasing the cone voltage. These notices allowed the
& DALLE-DONNE ET AL.
64

identification of the unique S-nitrosated cysteine inhemoglobin following treatment with NOS or nitroso-cysteine (Ferranti et al., 1997).
B. Analysis of Oxidized/Nitrated Productsof Tyrosine Residues
In vivo modification of tyrosine residues is a post-translational modification mediated by reactive oxygenand nitrogen radical species that often has been implicatedin the pathogenesis of a number of diseases. Depending ondifferent oxidative/nitrosative pathways and active radicalsinvolved in these processes, a series of stable end-products
of protein modification reactions has been identifiedthrough in vitro and in vivo studies (see above) in specifictissues or isolated proteins. Therefore, Cl-Tyr, di-Cl-Tyr,Br-Tyr, di-Br-Tyr, NO2-Tyr, di-Tyr, and the unnaturalisomers m-Tyr and o-Tyr (derived from protein–Pheresidues following reaction with hydroxyl radicals) havebeen all selected as amino acid products stable to acidhydrolysis, making them useful markers for proteinoxidation/nitration studies (Heinecke et al., 1999b,c). Onthis basis, specific procedures for the detection of thesederivatives in amino acid hydrolysates or biological fluids,following precolumn derivatization, have been optimizedby using direct HPLC quantification (Shigenaga et al.,
FIGURE 3. Electrospray mass spectrometric analysis of aldose reductase (AR) products generated from
the reaction of the enzyme with intermediates of GSH turnover or cupper ions. Native AR (Panel A), ARwith 0.4 mM cystine at 258C (Panel B), AR with 0.4 mM cystine at 378C (Panel C), AR with 0.4 mM
CysGly disulfide at 258C (Panel D), AR with 0.4 mM CysGly disulfide at 378C (Panel E), and AR with
7mMCuCl2 at 258C (Panel F). All reactionswere performed in 100mMphosphate buffer, for 5 h, at pH 6.8.
Samples were analyzed following alkylation with 1.1 M iodoacetamide in 0.25 M Tris-HCl, 1.25 mM
EDTA, 6 M guanidinium chloride, pH 7, under not reducing conditions, for 1 min, at 258C. Samples were
desalted by reversed phase HPLC. CAM, carboxamidomethyl group; CysAR, Cys-AR mixed disulfide;
CysGlyAR, CysGly-AR mixed disulfide; AR-SS, AR containing an intramolecular disulfide.
REDOX PROTEOMICS AND DISEASE &
65

1997) and gas chromatography (GC)- or liquid chromato-graphy (LC)-MS analysis (Heinecke et al., 1999b,c; Wuet al., 1999). However, when compared with the UV-or fluorescence-measurement counterpart, MS-basedap proaches provided structural information, therebyreducing the potential for confusion with extraneouscompounds coeluting with target analytes during chroma-tography, and allowing to perform selective ionmonitoring(SIM) experiments for quantification of trace quantities. Inaddition, MS-based analyses permitted the use of stable,isotopically labeled internal standards essential for correc-tion associated with analyte loss during processing andprecision of quantitative measurements (Heinecke et al.,1999b,c). On this basis, GC-MS analysis of n-propyl-heptafluorobutiryl-amino acid derivatives has successfullybeen used for the study of the oxidative pathwaysassociated with Parkinson’s disease, atherosclerosis, andthe occurrence of extracellular metal ions (Heinecke,1999b,c; Pennathur et al., 1999). Similarly, LC-MSprocedures have been used for the characterization of thehalogenated products generated by HOCl treatment oractivated eosinophils (Wu et al., 1999; Fu et al., 2000).Recently, different GC- or LC-tandem MS-basedapproaches have been proposed for the very accuratequantification of basal levels ofNO2-Tyr, di-Tyr, o-Tyr, andNO2-Tyr-containing proteins in plasma and tissues (Frost,Halliwell, & Moore, 2000; Yi et al., 2000; Gaut et al.,2002b; Marvin et al., 2003; Tsikas et al., 2003). Thesestudies highlighted the possibility of artifactual forma-tion of nitrated tyrosine during sample extraction andderivatization.
Contrary to the above-mentioned procedures directedto ascertain the occurrence of Tyr-directed oxidative/nitrosative insults by GC- or LC-MS analysis of themodified amino acids recovered in protein hydrolysates,methodologies for the detection of these modifications bydirect ESI- or MALDI-MS analysis of intact proteins ortheir peptide digests have found a positive application onlyin the case of NO2-Tyr-, di-Tyr-containing proteins, andTyr radical species (Minetti et al., 2000; Petersson et al.,2001; Sarver et al., 2001). Before the introduction ofdedicated MS procedures, proteins containing modifiedtyrosine residues have been detected by measuring thespecific absorbance at 365 nm (NO2-Tyr), the specificfluorescence at 410 nm (excitation 315 nm) (di-Tyr), andthe specific ESR absorbance in the presence of spin-trapping compounds (Tyr radicals).
In the case of NO2-Tyr-containing proteins, the occur-rence of nitration events has been ascertained by directmeasurements of intact molecules, detecting the corre-sponding adducts presenting a mass difference ofþ45 Da.However, when a comparative analysis of polypeptidescontaining NO2-Tyr was performed by MALDI- and ESI-MS techniques, it was evident that MALDI measurements
yielded unexpected significant underestimation of themodification extent, as a result of a prompt fragmentationinvolving the nitro group (Petersson et al., 2001; Sarveret al., 2001). This phenomenon has been associated witha series of photodecomposition reactions determiningthe formation of 3NO-Tyr, 3NHOH-Tyr, and 3NH2-Tyradducts, respectively. The effect of laser shots, laser power,and peptide concentration on the formation of these pho-todecomposition fragments was evaluated. These investi-gations ascertained that fragmentation of NO2-Tyr cannotbe controlled, thus highlighting the unreliability of thismethodology for the sensitive detection of nitrationproducts. On the contrary, ESI-MS measurements did notshow this phenomenon, allowing a complete evaluation ofthe protein modification extent. Moreover, site-specificidentification of NO2-Tyr in proteins has been reported byLC-ESI or MALDI mapping experiments, detectingpeptide species bearing the expected mass difference(Minetti et al., 2000). However, on the basis of theconsiderations reported above, most of the applicationsreported in the literature used ESI sources for the analysisof the nitrated peptides. Unequivocal assignment of NO2-Tyr was determined by tandemMS experiments (MacMil-lan-Crow, Crow, & Thompson, 1998; Petersson et al.,2001; Aslan et al., 2003; Murray et al., 2003). The use ofprecursor ion scanning for the specific immonium ion atm/z 181.06 combined with ESI-MS–MS measurements isthe general approach that found a broader application in theidentification of nitrated peptides by LC-MS from peptidedigests (Petersson et al., 2001).
The occurrence of intermolecular cross-linked di-Tyrresidues in proteins following oxidative/nitrosative insult,determining macroscopic variation of protein molecularmass, has been usually detected by low-resolutiontechniques such as SDS–PAGE under reducing conditions(MacMillan-Crow, Crow, & Thompson, 1998; Lardinois,Medzihradszky, & Ortiz de Montellano, 1999). In factthese species, contrary to disulfide cross-linked polypep-tides, are not sensitive to incubation with reducing agents.The occurrence of globin dimers following peroxynitritetreatment has been ascertained also by ESI measurements(Minetti et al., 2000). Cross-linking assignment to specificTyr residues has been obtained by mass mapping experi-ments on peptide digests using either MALDI- or ESI-MSprocedures (Lardinois, Medzihradszky, & Ortiz de Mon-tellano, 1999; Minetti et al., 2000) and confirmed by CIDexperiments. In some cases, a selective isolation of the di-Tyr-containing peptides by a preliminary HPLC purifica-tion step has been performed.
The generation of tyrosyl radicals following oxidativeinsult of heme-containing proteins has been detected bytrapping these species with specific spin-trapping agentsand measuring the addition of the modifying moiety tothe intact molecules by direct ESI or MALDI analysis
& DALLE-DONNE ET AL.
66

(Gunther et al., 1998; Lardinois, Medzihradszky, & Ortizde Montellano, 1999; Zhang, He, & Mauk, 2002).Depending on the nature of the compound used, a massincrease of þ344 Da (3,5-dibromo-4-nitrosobenzenesul-fonic acid), þ113 Da (5,5-dimethyl-1-pyrroline N-oxide),or þ72 Da (2-methyl-2-nitrosopropane) was observed forthe corresponding adducts. Investigation on the nature ofthe trapped derivatives obtained under different experi-mental conditions allowed researchers to elucidate themechanism of interaction with heme group for differentoxidative/nitrosative agents (Gunther et al., 1998; Pietra-forte et al., 2002). Also in this case, identification of thetyrosine residues subjected to spin-trapping agent additionwas obtained by LC-ESI–MS or MALDI mappingexperiments on protein digests and confirmed by ESI-MS–MS or post-source decay (PSD) analysis of themodified peptides (Lardinois, Medzihradszky, & Ortiz deMontellano, 1999; Zhang, He, & Mauk, 2002).
C. Analysis of Oxidized Productsof Methionine Residues
Methionine residues and their oxidized forms are becom-ing more and more important in view of their role ininfluencing the biological activity of proteins. Determina-tion of methionine oxidation products in protein hydro-lysates by conventional chromatographic procedures hasbeen discouraged as a result of the reducing conditionsused during acid hydrolysis, determining the spontane-ous conversion of the oxidized products back to Met. Aseries of methodologies has been proposed to limit theextent of these side-reactions, although a robust procedurefor the determination of methionine redox state has notbeen developed (Sochaski et al., 2001). Oxidation ofmethionine thioether group to the corresponding sulfoxideand sulfone derivatives can be easily detected in the ESI orMALDI mass spectra of intact molecular species, byrevealing the corresponding adductswith amass differenceof þ16 and þ32 Da, respectively (Hanson et al., 1999,2000). In general, a numerical evaluation of the oxygenatoms introduced into a protein can be determined bycounting the multiple addition of 16 mass units comparedwith the unmodified species. The selectivity of this modi-fication toward Met and not Cys residues can be easilyverified following mass measurement of the alkylationproducts obtained with thiol-specific reagents. Moreover,the occurrence of MetO residues in oxidized proteins hasalso been verified by assaying the limited succeptibility ofthis species to cleavage by cyanogen bromide (Milzaniet al., 2000).
Identification of modified Met residues has beenobtained in different proteins by LC-ESI or MALDI massmapping experiments, by detecting the peptides specifi-cally bearing these mass differences (Hanson et al., 1999,
2000; Taggart et al., 2000). In most cases, it has beenobserved that the oxidized peptides usually elute earlierthan the unmodified ones in an RP-chromatographyseparation.Very recently, a solid-phase isolation procedurefor the selective enrichment of protein digests in MetO-containing peptides has been proposed (Grunert et al.,2003). In general, the occurrence of oxidized componentspresenting MetO or MetO2 at specific positions is easilyverified by low-energy CID experiments, revealing thecharacteristic loss ofmethanesulfenic acid (�64or�32Dafor singly or doubly protonated ions, respectively) ormethanesulfonic acid (�80 or�40 Da for singly or doublyprotonated ions, respectively) from the side chain ofoxidized Met derivatives (Lagerwerf et al., 1996; Guan,Yates, & Bakhtiar, 2003). The correct assignment of themodification to a specific Met residue has easily beenobtained by database search routines after the necessaryadjustment in the parameter file to account for the massshift associated with the modification.
However, methionine oxidation in peptides andproteins occurs in vivo or may be an artifact resulting fromsample manipulation during analytical characterization.To solve these difficulties, a dedicated procedure based onprotein N-terminal acetylation, selective hydrolysis at Metresidues by CNBr, and specific labeling of the newlygenerated amino groups with a bromine-containingcompound has been proposed (Hollemeyer, Heinzle, &Tholey, 2002). This procedure allows the unequivocallocalization of oxidized methionines even in complexpeptide mixtures.
D. Analysis of Protein Carbonylation Products
Protein carbonylation can occur at different sites (Pro, Arg,Lys, andThr) and through a series of differentmechanisms.Depending on the nature of the generated derivatives andtheir relative stability to drastic hydrolysis conditions,carbonylated adducts have been revealed either in proteinand fluid hydrolysates by GC- and LC-ESI–MS analysis,or by direct ESI- or MALDI-MS measurements on intactproteins or their peptide digests.
The occurrence of Pro, Arg, and Lys residues in closeproximity to the highly reactive .OH can directly convertthese amino acids in carbonyl-containing derivatives(Requena et al., 2001a). The generated glutamic semi-aldehyde (Pro and Arg) and aminoadipic semialdehyde(Lys) products occurring in the polypeptide chain areusually detected by GC-MS measurement of the 5-hydroxy-2-aminovaleric acid (HAVA) and 6-hydroxy-2-caproic acid (HACA) obtained following reductivestabilization and extensive acid hydrolysis (Requenaet al., 2001a). Amino acids are usually converted to theirN,O-trifluoroacetyl methyl esters or N(O)-ethoxycarbonylethyl esters by precolumn derivatization. The general
REDOX PROTEOMICS AND DISEASE &
67

approach described by Stadtman and coworkers allowed toperform SIM experiments for the quantification of tracequantities in biological samples as well as the use ofdeuterated internal standards for analyte loss correctionand precise quantitative measurements. On this basis, thequantification of HAVA and HACA was obtained for aseries of model proteins and mammalian tissues undernormal and stressing conditions.
Non-enzymatic glycation of proteins, also designatedasMaillard reaction, is initiated by the reaction of reducingcarbohydrates with Lys or N-terminal residues, yieldingAmadori compounds (aminoketoses) as primary products.These products have been detected by ESI or MALDImeasurements of intact proteins, revealing the correspond-ing adducts presenting a mass difference of þ162 Da(glucose and fructose) (Peterson et al., 1998; Saraswathi,Nakanishi, & Shimizu, 1999; Lapolla, Fedele, & Traldi,2000). The occurrence of glycooxidized products was alsodetected (Lapolla, Fedele, & Traldi, 2000). Glycationassignment to specific Lys residues has been obtained bymass mapping experiments on peptide digests by eitherMALDI- or ESI-MS experiments (Miyata et al., 1994;Takahashi et al., 1995;Marotta et al., 2003;McKillop et al.,2003), using a strategy similar to that used for theassignment of lactosylation sites in milk proteins (Scaloniet al., 2002). The Amadori compounds are slowlyoxidatively degradated, in complex reaction pathways viadicarbonyl intermediates (3-deoxyglucosone, dideoxy-sones, methylglyoxal, and glyoxal), to a plethora ofcross-linked derivatives as crosslines, N-carboxymethyl-lysine (CML), 6-[1-(5-ammonio-6-oxido-6-oxohexyl)imi-dazolium-3-yl]-L-norleucinate (GOLD), 6-[1-(5-ammonio-6-oxido-6-oxohexyl)-4-methylimidazolium-3-yl]-L-norle-ucinate (MOLD),N6-[2-(4-ammonio-5-oxido-5-oxopentyl)amino]-5-(2,3,4-trihydroxybutyl)-3,5-dihydro-4H-imida-zol-4-ylidene-L-lysinate (DOGDIC), N6-[2-(4-ammonio-5-oxido-5-oxopentyl)amino]-5-(2,3,4-dihydroxypropyl)-3,5-dihydro-4H-imidazol-4-ylidene-L-lysinate (DOPDIC),N6-glycoloyl-lisine (GALA), N6-[2-(5-ammino-5-carbox-ypentyl)amino]-2-oxoethyl-lysine (GOLA), and others.Contrary to Amadori compounds, these derivatives havebeen detected in vitro or in vivo following extensiveprotein/tissue acid or enzymic hydrolysis by dedicatedGC-MS or LC-ESI-MS analytical procedures (Biemelet al., 2001; Glomb & Pfahler, 2001; Biemel, Friedl, &Lederer, 2002). Also in this case, the synthesis of 13C-containing internal standards and the possibility to per-form SIM experiments allowed an accurate evaluationof trace quantities in human serum albumin and lensproteins.
Proteinmodification by 4-HNEproceeds primarily viaa Michael addition to Lys, His, and Cys residues. Thisreaction can be monitored for intact proteins by ESI- orMALDI-MS analysis, detecting the corresponding adducts
with a mass increase of þ156 Da (Bennaars-Eiden et al.,2002; Crabb et al., 2002; Alderton et al., 2003). Theidentification of modified residues has been obtained indifferent proteins by LC-ESI or MALDI-mass mappingexperiments, by detecting the peptides specifically bearingthis mass difference (Bennaars-Eiden et al., 2002; Crabbet al., 2002; Alderton et al., 2003). The nature of themodified amino acid was definitively ascertained by MS-MS-based investigations.Alternatively, the extent of lysinemodification has been evaluated by GC-MS analysis,reavealing the 3-(Ne-lysino)-4-hydroxynonal-1-ol gener-ated in protein exaustive hydrolysates following reductionwith NaBH4 (Requena et al., 1997). The use of deuteratedinternal standards allowed an accurate measurement. Thisapproach was also applied to the quantification of themodified lysines in proteins following MDA treatment.This reaction proceeds via a Schiff-base adduct formation.In this case, the quantification of the 3-(Ne-lysino)propan-1-ol and 1,3-di(Ne-lysino)propane led to an evaluation ofthe cross-linked adducts (Requena et al., 1997).
E. Analysis of Oxidized Productsof Tryptophan Residues
The oxidation of tryptophan has been known for decades,since the inactivation of lysozyme by oxidants modifying acritical Trp residue was reported (Previero, Coletti-Previero, & Jolles, 1967). These early reports relied onidentification of tryptophan oxidation products only bycharacteristic electronic absorbance spectra. Few yearsago, the first complete MS characterization of a proteinfrom bovine lens, a-crystallin, presenting oxidized Trpresidues as a result of exposition to oxidative Fenton insulthas been described. The occurrence of hydroxytryptophan,N-formylkynurenine, kynurenine, and 3OH-kynurenine inreaction products was ascertained by direct ESI measure-ments, detecting the corresponding adducts with a massincrease of þ16, þ32, þ4, and þ20 Da, respectively(Finley et al., 1998). The identification of the modifiedresidues was obtained by MALDI mass mapping experi-ments combined with ESI-MS–MS analysis of theoxidized peptides. Although it could not be demonstratedunequivocally whether oxidation occurred during samplehandling or in vivo, the same Trp residues were foundoxidized during the 2D-LC-MS–MS analysis of humancataract lens digests (MacCoss et al., 2002). Later on, it hasbeen reported the oxidation of a critical Trp residue in thechloroplast photosystem II protein CP43, providing thefirst example of this selective modification in vivo(Anderson et al., 2002). Very recently, a massive analysisof tryptophan oxidation in cardiac mitochondrial proteinshas been reported. The ESI-MS–MS detection of N-formylkynerenine in a series of target proteins suggested
& DALLE-DONNE ET AL.
68

that Trp modification is obtained in vivo as a result of theclose proximity of these polypeptides to a source of ROS(Taylor et al., 2003).
Recently, the heme-assisted oxidation of tryptophanin myoglobin mutants to 2,6-dihydro-2,6-dioxoindole and2,6-dihydro-2-imino-6-oxoindole derivatives has beenreported by a combined MALDI-mass mapping, PSDanalysis, and 1H/13C nuclear magnetic resonance (NMR)spectroscopy approach. The oxidized peptides occurring inthe digest presented a selective mass difference ofþ30 Dacompared with the unmodified residue (Hara et al., 2001).
F. Analysis of Oxidized Products of Histidine Residues
Similarly to other amino acid residues, the occurrence ofhistidines at sites with transition metal-binding capacitycan determine His modification as a result of metal-catalyzed protein oxidation. 2-Oxo-His and 4- or 5-hydroxy-2-oxo-His have been proposed as main reactionproducts (Schey & Finley, 2000; Requena et al., 2001b).Although the occurrence of 2-oxo-histidine in proteinhydrolysates has been detected by conventional amino acidanalysis procedures (Requena et al., 2001b), the presenceof oxidized His derivatives can also be ascertained bydirect ESI or MALDI analysis of intact molecules,detecting the corresponding adducts with a mass increaseof þ16 and þ32 Da, respectively (Kurahashi et al., 2001;Schoneich & Williams, 2002). The assignment of themodified residues in the polypeptide sequence can beobtained by MALDI or ESI mass mapping experiments,revealing the peptides specifically bearing this massdifference or ESI-MS–MS precursor ion scanning for thespecific immonium ion atm/z 126 or 142, respectively. Thenature of the modified amino acid can be definitivelyascertained by ESI tandem MS analysis. These experi-ments led to the identification of 2-oxo-histidine and 4- or5-hydroxy-2-oxo-histidine in the oxidation products of b-amyloid peptide, Cu,Zn-SOD, and recombinant prionprotein, allowing to draw a general scheme of reaction(Kurahashi et al., 2001; Requena et al., 2001b; Schoneich& Williams, 2002). In these works, the detection of theoxidation products for other amino acids in close proximityto His in sites with transition metal-binding capability wasalso reported.
VI. PROTEOMIC STRATEGIES FOR THEIDENTIFICATION OF ROS/RNS TARGETSIN COMPLEX PROTEIN MIXTURES
Oxidative/nitrosative damage by reactive radical speciesappears central to the pathogenesis of many disorders.Specific modified proteins are generated following stres-sing insults and accumulate in different degenerated tissuesand fluids, determining in some cases altered organ
functionalities. Proteome analysis, providing researcherswith a general approach to describe all protein componentsat specific cellular moments, is an ideal choice forrevealing all the polypeptide modifications because of aparticular stressing condition or disease. Therefore, toobtain a comprehensive decription of oxidative insults,different proteomic approaches have been developed andused for the detection and identification of ROS/RNSprotein targets among all the species present in a biologicalsample (Ghezzi & Bonetto, 2003). These investigationshave recently been ascribed to the general term of redoxproteomics.
Whether the goal is the identification of differentlymodified or expressed proteins in oxidatively stressed ordisease samples, accurate resolution of thousands ofproteins is an absolute requirement. For this reason, two-dimensional (2D) gel electrophoresis approaches havewidely been used to separate proteins according to their pIand mass value. Detection of differently modified/expressed proteins is achieved by comparing 2D electro-phoretic maps of samples subjected to oxidative/nitro-sative insult with the corresponding controls (differentialproteomics). As an example, Figure 4 reports the case ofhuman epithelial cells following treatment with H2O2
(Paron et al., 2003). Adequate image analysis has beenessential to properly map modified protein spots. Specia-lized image analysis softwares are now available to dealwith the complexity of protein spot patterns. Suchsoftwares fulfill a number of requirements, includingprotein spot detection and quantification, and the abilitiesto perform both multiple image alignments and imagecomparisons (Panek & Vohradsky, 1999). Furthermore,massive screening and selective detection of oxidatively/nitrosatively modified proteins separated by 2D electro-phoresis have been facilitated by the availability of specificstaining compounds or commercial antibodies allowingtheir immunodetection (Fig. 5). Samples are separated ondifferent 2Dgels that are stained in parallelwith derivativesspecific for ROS/RNS-modified species and with aspecificstaining chemicals. Again, adequate image analysis isessential to properly detect modified protein spots andassociate them with identified protein species. Similarly,the incorporation of radioactivity by using specific radio-active precursors of modifying groups has been used todetect ROS/RNS-modified proteins. Nowadays, this gen-eral methodology has been widely applicated in thedescription of ROS/RNS-related stressing events ordiseases, demonstrating that, in a specific tissue or fluid,selected proteins are modified to a much greater extentthan others. Alternatively, approaches based on specificimmuno-affinity enrichment or purification before electro-phoretic separation have been used to selectively bindpossible ROS/RNS targets to derivatized beads (Lind et al.,2002; Eaton et al., 2003). Also in this case, the detection of
REDOX PROTEOMICS AND DISEASE &
69

the oxidatively modified proteins has been obtained byspecific staining or Western blotting procedures.
On this basis, protein carbonyls have been detectedand quantified on 2D gels, measuring the incorporation oftritium following reduction with [3H]-borohydride (Yan &Sohal, 1998), or following derivatization with fluoresceinhydrazide (Ahn, Rhee, & Stadtman, 1987) or 2,4-dinitrophenylhydrazine (DNPH) (Levine et al., 1990).The latter procedure, based on the pioneering analysis ofstable 2,4-dinitrophenyl (DNP) hydrazone products byLevine, Stadtman, and co-workers, has become the mostwidely utilized methodology for the measurement ofprotein oxidation in several human diseases (Dalle-Donneet al., 2003b,c), experimental models (Powell, Gurzenda,& Wahezi, 2001), and purified proteins (Milzani et al.,2000; Dalle-Donne et al., 2001). The detection ofcarbonylated proteins has also been facilitated by therecent introduction of anti-DNP antibodies allowingcarbonyl detection by Western blotting analysis (oxyblot
analysis; e.g., Castegna et al., 2002a,b). Similarly, anantibody raised against glycated RNAse has been used forimmuno-detection of advanced glycation end-products(Poggioli, Bakala, & Friguet, 2002).
Analogous methodologies have been developed fordetectingNO2-Tyr in complex proteinmixtures as amarkerof nitrosative insult and irreversible protein damage. All ofthese methodologies are based on the relative stability ofthis modification. A first approach uses immunochemicaltechniques for the specific detection of NO2-Tyr-contain-ing proteins following 2D-gel electrophoresis by anti-NO2-Tyr antibodies (Ye et al., 1996).A secondprocedure isbased on the release of the modified Tyr in proteinhydrolysates and its detection by LC-UV, LC-ECD, GC,and GC-MS techniques (Herce-Pagliai, Kotecha, &Shuker, 1998).
Similarly, proteomics methods for the detection of S-glutathionylated proteins have been devised either using[35S]-labeled GSH for the specific detection of [35S]
FIGURE 4. Proteomic analysis of human epithelial lens cells in response to H2O2. CD5A cells were
incubated with and without 500 mMH2O2 for 30 min, at 378C. Cells were harvested, lysed and intracellularproteins were analyzed by 2D-electrophoresis with subsequent silver staining (Panel A). Regions
comprising statistical significative differences were cropped (Panel B). Quantitative analysis of reduced(right) and oxidized (left) peroxyredoxin I species detected following exposition to indicated doses ofH2O2
is reported (Panel C).
& DALLE-DONNE ET AL.
70

incorporation into proteins resolved on 2D gels (Fratelliet al., 2002) or biotinylated GSH ester for the selectiveimmuno-affinity purification of S-glutathionylated pro-teins (Sullivan et al., 2000; Eaton, Fuller, & Shattock,2002a; Eaton et al., 2002b, 2003). In the first case, S-glutathionylated proteins are identified by incubation ofcells with radiolabeled Cys in the absence of proteinsynthesis, exposure to oxidants, isolation, and detection ofradiolabeled (i.e., glutathionylated) proteins (Fratelli et al.,2002). This methodology applied to the study of stressed Tlymphocytes has revealed that numerous proteins aretargets for redox-dependent modification. Several of theenzymes identified were inactivated during in vitro assays,implying that S-glutathionylation of proteins upon altera-tion of the cellular redox status is a potentialmechanism forregulating the activity of many targets. In the second
approach, the amino terminus of cysteine is tagged withbiotin and loaded into cells or tissues. When oxidizingchanges occur, formation of a disulfide bond betweenredox-sensitive protein cysteines and biotin-cysteine isinduced. These oxidized proteins carrying a biotin tag aredetected using non-reducing Western blotting and strepta-vidin-HRP or purified by streptavidin-agarose affinitychromatography. The efficiency of purification can beimproved with the use of gel-filtration chromatography forseparating proteins from free biotin-cysteine in homo-genates, which otherwise competes with S-thiolatedproteins for column binding. These proteins are thenseparated by SDS–PAGE and stained by aspecificcompounds.
An elegant approachwas reported for the isolation ofS-nitrosated proteins (Jaffrey et al., 2001). Using this method,
FIGURE 5. A general strategy for the proteomic analysis of oxidatively/nitrosatively stressed tissues/body
fluids by 2D-electrophoresis and specific immunodetection. Samples are separated on different 2D gels that
are stained in parallel with derivatives or commercial antibodies specific for ROS/RNS-modified proteins
andwith aspecific staining chemicals. Spots fromgels subjected to aspecific staining are analyzed by peptide
mass fingerprint or tandem mass spectrometry analysis. Images are compared by dedicated analysis
softwares.
REDOX PROTEOMICS AND DISEASE &
71

free SH groups were blocked with methyl methanethio-sulfonate. Protein-S-nitrosocysteine groups were succes-sively reduced to free thiols by treatment with ascorbate.The newly released protein thiols were derivatized with acleavable biotin-containing reagent, affinity purified usingstreptavidin agarose beads and released for SDS–PAGEseparation. This methodology was validated by confirma-tion that many targets of exogenous nitrosating agents arein vivo targets, as revealed by the lack of labeling in brainlysates from mice deficient in nNOS (Jaffrey et al., 2001).
In all the cases reported above, protein spots or bandspresenting increased radioactivity or positive staining byspecific chemicals or antibodies have to be furtheranalyzed to identify their nature. Accordingly, proteinspecies are excised, in-gel digested, and their peptidedigests analyzed by MALDI-MS or LC-ESI-MS proce-dures. In fact, as a result of the dramatic technologicaldevelopments affecting sensitivity, resolution, and accu-racy properties,MShas increasingly become themethod ofchoice for the analysis of separated protein components.Peptide-mass fingerprint and tandem mass spectrometry-based peptide sequencing approaches have been usedextensively for the identification of the oxidatively/nitrosatively modified proteins as well as for assignmentof themodification to specific amino acid residues (Pandey& Mann, 2000). The achievement of this second issue isstrictly related to the possibility to obtain a detailedstructural characterization of the entire protein primarystructure, including modifications, even using the pooramounts recovered from gels or beads. In this sense, alltechnological improvements for sample manipulation andminiaturization of the chromatographic devices coupled tomass spectrometers have been essential for an extensiveprotein characterization. Examination of the measuredmass and fragmentation spectra obtained for a singleprotein via manual or computer-assisted interpretation ledto the assignment of oxidized/nitrosated/nitrated aminoacids using the criteria described in the previous section.
On this basis, high-throughput methodologies haverecently been applied to the examination of global proteinoxidation in oxidatively stressed human epithelial cells(Paron et al., 2003), S-glutathionylation in oxidativelystressed human T lymphocytes (Fratelli et al., 2002) and inheart or kidney following ischemia and reperfusion (Eatonet al., 2003), protein carbonylation in Alzheimer’s disease(Castegna et al., 2002a,b; Choi et al., 2002, 2003;Korolainen et al., 2002) and in aged liver mitochondria(Rabek, Boylston, & Papaconstantinou, 2003), S-nitrosa-tion in brain lysates (Jaffrey et al., 2001), and Tyr nitrationin Alzheimer’s and sickle cell diseases (Aslan et al., 2003;Castegna et al., 2003) or during inflammation (Aulaket al., 2001). Various protein targets of ROS/RNS insulthave been identified, although only in very few casesthe assignment of the modified amino acids has been
achieved (Aslan et al., 2003; Paron et al., 2003). As anexample, Figure 6 reports the ascertained modification ofperoxiredoxin I Cys52 to cysteic acid in oxidativelystressed human epithelial cells (Paron et al., 2003).
Recently, attempts have been made to define proteinmodifications on a proteome-wide scale without the use of2D gel electrophoresis. Given the difficulties of identifyingall modifications even in a single protein, it is clear that, atpresent, scanning for proteome-wide modifications is notcomprehensive. Nevertheless, a large amount of biologi-cally useful information can, in principle, be generated bythis approach. One of the strategies used is essentially anextension of the approach used to analyze protein mixturesby 2D LC-MS–MS procedures (MacCoss et al., 2002).Instead of searching the database only for non-modifiedpeptides, the database search algoritm is instructed to alsomatch potentially modified peptides. To avoid a ‘‘combi-natorial explosion’’ resulting from the need to consider allthe possible modifications for all the peptides in thedatabase, the experiment is usually divided into identifica-tion of a set of proteins on the basis of non-modifiedpeptides, followed by searching only those proteins formodified peptides. An a priori oriented strategy focuses onthe search for one selected type of modification on all theproteins present in a sample. This approach has success-fully been used for the examination of the occurrence ofN-formylkynurenine, a product of Trp oxidation, through themitochondrial proteome of normal human heart tissues(Taylor et al., 2003). Thirty-seven different proteins wereidentified as ‘‘hot spots’’ for oxidation in close proximity toa source of ROS.
VII. SELECTED HUMAN DISEASES ASSOCIATEDWITH OXIDATIVE/NITROSATIVE STRESS
We shall now examine briefly some selected diseasesstrictly related to oxidative/nitrosative stress, in which theextensive detection and characterization of differentoxidatively/nitrosatively modified proteins will contributeto understand the dysfunction mechanisms.
A. Acute (Adult) Respiratory Distress Syndrome
Acute (adult) respiratory distress syndrome (ARDS) is asevere hypoxia caused by pulmonary edema, characterizedby diffuse inflammation in the lung parenchyma. Increasedproduction of ROS/RNS combined with decreased anti-oxidant activity in the lung contributes to this pathology(Lamb et al., 1999; Lang et al., 2002).
In ARDS patients and patients at-risk for ARDS, theepithelial lining fluid recovered as bronchoalveolar lavage(BAL) fluid is characterized by increased neutrophils,which generate O2
.� and release inflammation mediators.Furthermore, in response to various inflammatory stimuli,
& DALLE-DONNE ET AL.
72

lung endothelial cells, alveolar cells, and airway epithelialcells, as well as activated alveolar macrophages, produceperoxynitrite, which can nitrate and oxidize various lungproteins, such as surfactant protein A (Gole et al., 2000),inhibiting their functions. The results from various studies(reviewed in Lang et al., 2002) demonstrate increasedlevels of both ROS/RNS and reactive intermediates in boththe BAL fluid and edema fluid of patients who are at riskof developing ARDS or who have established ARDS.Levels of reactive species correlate both with the outcomeof the disease and the severity of the injury to the alveolarepithelium.
B. Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common senileneurodegenerative disorder estimated to affect approxi-mately 15 million people worldwide. As more people liveto old age, AD is becoming a greater medical and social
problem. Clinically, AD is characterized by progressiveloss of memory, language, and other cognitive functionsaccompanied by concomitant behavioral, emotional, andsocial deterioration, all leading to dementia. The neuro-pathological hallmarks of the brain of AD patients arenumerous extracellular senile plaques containing amyloid-b and neurofibrillary tangles (NFTs), which occur in pyra-midal neurons of the cerebral cortex and hippocampus.
There is abundant evidence proving that oxidative/nitrosative stress has a major role in the pathogenesis ofAD, manifested by protein oxidation and lipid peroxida-tion, ROS formation, among other markers of oxidativedamage. Amyloid-b, the main constituent of senileplaques, induces or exacerbates oxidative stress, producingH2O2 and other ROS, which cause peroxidation of cellmembranes and ultimately lead to cell death (Butterfieldet al., 2001, 2002). Furthermore, administration of selegi-line (a monoamino-oxidase inhibitor) and vitamin E leadsto a delay in disease progression (Sano et al., 1997).
FIGURE 6. MALDI-TOFmass spectrometry analysis of the reduced and oxidized forms of PrxI reported in
Figure 5, following carboxamidomethylation and trypsin or endoprotease AspN hydrolysis. The figure
reports the spectrum of the tryptic digest fraction eluted from mZipTip devices with 40% acetonitrile for
reduced (Panel A) and oxidized PrxI (Panel B) as well as the endoprotease AspN digest fraction elutedwith
50% acetonitrile for reduced (Panel C) and oxidized PrxI (Panel D). CAM, carboxamidomethyl.
REDOX PROTEOMICS AND DISEASE &
73

Regardless of whether oxidative stress is a primary orsecondary event, it is an important neurodegenerativeelement, which may contribute to neuronal loss inducingapoptosis or necrosis (Smith et al., 2000; Butterfield et al.,2001, 2002; Butterfield & Lauderback, 2002; Beal, 2002).Increasing experimental evidence supports the hypothesisthat neuronal death may occur primarily by apoptoticmechanisms in AD, likewise in Parkinson’s disease andamyotrophic lateral sclerosis; in accordance with thishypothesis, clinical evidence shows signs of apoptosis inthese patients (Esposito et al., 2002).
A number of contributory sources are thought to playan important role in free radical production in AD(reviewed in Smith et al., 2000). (1) Redox-active iron isincreased in AD brains, both in NFTs and in amyloid-bdeposits; it catalyzes hydroxyl radical formation fromH2O2. (2) Activated microglia surrounding senile plaquesare a source of peroxynitrite, leaving NO2-Tyr as anidentifiable marker, and respond with increased radicalproduction after an oxidative challenge. (3) Amyloid-b isalso directly implicated in oxidative stress through freeradical formation, by means of mechanisms involving itsMet35 (Varadarajan et al., 2000; Butterfield et al., 2001;Butterfield&Lauderback, 2002). Amyloid-b can fragmentand generate free radical peptides, resulting in proteinoxidation and lipid peroxidation in neurons or synaptoso-mal membranes of the neocortex, ROS formation, cellulardysfunctions, and subsequent neuronal death (Butterfield &Lauderback, 2002). However, a recent alternative hy-pothesis views amyloid-b as a protective response elementto oxidative stress, which functions as a primary line ofantioxidant defense (Smith et al., 2002). (4) Advancedglycation end-products in the presence of transition metalscan undergo redox cycling with consequent production ofreactive oxygen. (5) Abnormalities in the mitochondrialgenome or deficiencies in key metabolic enzymes suggestthat metabolic abnormalities affecting mitochondria maybe the major, and possibly initiating, source of reactiveoxygen in AD. In particular, the decreased expressionof complex I (NADH/ubiquinone oxidoreductase) mayresult in generation of ROS, through the departure ofelectrons from their carrier molecules (Butterfield &Lauderback, 2002), suggesting an alternative rationaliza-tion for the well-documented existence of oxidative stressin AD.
Evidence supporting the notion of ROS/RNS produc-tion in the brain of AD patients and their implication in theAD pathogenesis (Markesbery, 1997) include: increasedprotein carbonyls in both hippocampus and the inferiorparietal lobule, but not in the cerebellum, consistent withthe regional pattern of histopathology in AD (reviewed inDalle-Donne et al., 2003c); increased NO2-Tyr and di-Tyrin AD post-mortem brain tissue and ventricular cere-brospinal fluid; decreased activity of oxidation-prone
enzymes such as glutamine synthetase and creatine kinaseand decreased energy metabolism (Beal, 2002); increasedlipid peroxidation detected in the brain by decreased levelsof polyunsaturated fatty acids and increased levels of thelipid peroxidation products, 4-HNE, acrolein, thiobarbi-turic acid reactive substances (TBARS), iso- and neuro-prostanes (Butterfield &Kanski, 2001); increased levels of4-HNE found in post-mortem cerebrospinal fluid of ADpatients (Pratico et al., 1998b). Interestingly, decreasedrepair activity of Msr has been demonstrated in AD brains(Gabbita et al., 1999).
C. Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS), also known as LouGehring’s disease, is a fatal neurodegenerative disease thataffects primarily motor neurons in the cerebral cortex,spinal cord, and brain stem, and that invariably leads todeath within 3–5 years from the onset of symptoms.Approximately 10% of the cases are inherited in anautosomal dominant manner. Approximately 15–20% ofpatients with familial ALS, clinically indistinguishablefrom the more common sporadic ALS, carry mutations inthe Cu/Zn-SOD (SOD-1) gene (more than 90 differentmutations reported), associated with a decreased enzymicactivity (20–50%). This suggested that the disease wasbecause of ROS-induced damage associated with themutated gene, leading to accumulation of toxic O2
.�.However, no deletions of SOD-1 gene have been found infamilial ALS families, which implies that expression of themutated protein is required for pathogenesis. Studies intransgenicmice suggested that, rather than causing a loss offunction, the mutations of SOD-1 in familial ALS patientscause a gain of function that results in neuronal degenera-tion (reviewed in Valentine, 2002). This hypothesis issustained by the notion thatmutations change the affinity ofthe enzyme to substrates, impair its ability to bind zinc orincrease its aggregation in neurons (Zelko, Mariani, &Folz, 2002). Either way, mutation in the Cu/Zn-SOD genecauses neuronal death by apoptosis through the sequentialactivation of caspase-1 and caspase-3 (Pasinelli et al.,2000). Notably, it has recently been presented the first caseof a novel SOD-1 mutation in a patient with geneticallyproven sporadic ALS (Alexander et al., 2002).
Anyway, there is a large amount of clinical studiesimplicating oxidative damage to proteins in ALS patho-genesis. Increased protein carbonyls and protein nitrationhave been found in brain tissues of both sporadic andfamilial ALS patients (Beal, 2002). Furthermore, markedincreases in both free NO2-Tyr and nitrated Mn-SOD havebeen shown in the cerebrospinal fluid of sporadic ALSpatients (Tohgi et al., 1999; Aoyama et al., 2000). Inaddition, fibroblasts from ALS patients were found to bemore sensitive to oxidative stress (Esposito et al., 2002). A
& DALLE-DONNE ET AL.
74

potential source of superoxide anion in ALS is the pro-inflammatory enzyme cyclooxygenase-2, whose activity isdramatically increased in spinal cord tissue of sporadicALS patients (Almer et al., 2001).
D. Asthma
Asthma is a chronic airway inflammation characterized byvariable and reversible airflow obstruction and bronchialhyper-responsiveness, because of an excessive airwaynarrowing in response to a variety of apparently unrelatedstimuli. Abnormal histopathological lesions includingedema, epithelial cell desquamation, and infiltration ofinflammatory cells (eosinophils, CD4þ T-helper 2 lym-phocytes, mast cells, neutrophils, and macrophages) arefound in all asthmatic airways. In addition, subepithelialfibrosis, increased smooth muscle mass, hypertrophy ofmucous glands, and goblet cell hyperplasia result in airwayremodeling (Andreadis et al., 2003).
A genetic basis for airway hyper-responsiveness hasbeen hypothesized suggesting that asthma is a complexmultigenic disease with a strong environmental contribu-tion (Holloway et al., 2003). Many studies have empha-sized the multifactorial nature of asthma with interactionsamong neural mechanisms, inflammatory cells, intrinsicabnormalities of the arachidonic acid pathways, and smoothmuscle (Synek et al., 1996; Bandeira-Melo, Bozza, &Weller, 2002). The inflammatory cells infiltrating the airwaysrelease a variety of mediators, including ROS/RNS, whichcontribute to the pathophysiological features of asthma.
Evidence of increased oxidative stress in asthma isshown by high levels of ROS/RNS (Rahman et al., 1996;Dworski, 2000; Bowler & Crapo, 2002); increasedproduction of lipid peroxidation products, protein carbo-nyls, total nitrites and nitrates in plasma (Rahman et al.,1996; Nadeem et al., 2003) as well as Br-Tyr and Cl-Tyr inBAL proteins (Wu et al., 2000); higher levels of NO. aswell as 10-fold increases in NO2-Tyr content in proteinsrecovered from airways (Dweik et al., 2001; MacPhersonet al., 2001); increased oxidized glutathione in BAL fluid(Kelly et al., 1999); increased production of NO. inexhaled air (Kharitonov et al., 1994). In addition, changesin antioxidant defenses have been reported (Rahman et al.,1996; Kelly et al., 1999; Bowler & Crapo, 2002; Nadeemet al., 2003).
E. Atherosclerosis
Atherosclerosis is amultifactorial disease characterized byhardening and thickening of the arterial wall. The vascularareas affected by this disease contain mononuclear cells,proliferating smooth muscle cells, and extracellular matrixcomponents. Lipid-laden foam cells derived from macro-phages are the cellular hallmark of the early atheroscleroticlesion.
Atherosclerosis is commonly viewed as a chronicinflammatory disease and is associated with certain riskfactors such as hypercholesterolemia, smoking, aging,diabetes, and hypertension that increase production ofROS/RNS by endothelial, vascular smooth muscle, andadventitial cells (Kunsch & Medford, 1999; Cai &Harrison, 2000; Harrison et al., 2003). ROS/RNS initiateseveral processes involved in atherogenesis, includingexpression of intercellular adhesion molecules and proteinkinases, stimulation of vascular smooth muscle prolifera-tion andmigration, apoptosis in the endothelium, oxidationof lipids and proteins resulting in reactive products (Fuet al., 1998; Baynes & Thorpe, 2000), activation of matrixmetalloproteinases, and altered vasomotor activity (Harri-son et al., 2003). Several enzyme systems contribute toexcess production of ROS/RNS in vascular tissues (Chen,Thomas, & Keaney, 2003; Harrison et al., 2003). Super-oxide production increases as a function of clinical riskfactors for atherosclerosis, suggesting that NADPHoxidase plays a causal role in the disease. Furthermore, arecent study has linked mitochondrial oxidant productionto the early atherosclerotic lesion development (Chen,Thomas, & Keaney, 2003). Physical forces also regulatevascular production of ROS. Oscillatory shear, which ispresent, vice laminar shear, at sites where atherosclerosisdevelops, seems a particularly potent stimulus of O2
.� andH2O2 production by endothelial cells. Furthermore,oscillatory shear leads to a decrease in endothelial celllevels of total GSH (Harrison et al., 2003).
The invasion of the artery wall by monocytes and Tlymphocytes is one of the earliest events in the developmentof atherosclerotic lesions. Monocytes, macrophages, andsmooth muscle cells possess the so-called scavengerreceptor for oxidized LDL. Binding of oxidized LDL leadsto the activation of monocytes and macrophages andstimulates the expression of Mn-SOD which, in turn,increases the concentration of H2O2 by perturbing thesteady-state levels of ROS (Kinscherf et al., 1997, 1998).This process is associated with massive macrophageapoptosis and, thereby, contributes to the formation ofatherosclerotic lesions (Kinscherf et al., 1999). The processmaybe further enhanced by cytokines and other factors suchas tumor necrosis factor-a (TNF-a), interleukin-1b, angio-tensin II, and interferon-g, which induce superoxideproduction by the membrane-bound NADPH oxidase inendothelial cells (Chen, Thomas, & Keaney, 2003).
F. Chronic Obstructive Pulmonary Diseases
Chronic obstructive pulmonary disease (COPD) is definedas a disease state characterized by the presence of airflowobstruction because of chronic bronchitis or emphysema(ATS Committee, 1995). Both conditions contribute to avariable degree to COPD in individual patients. The
REDOX PROTEOMICS AND DISEASE &
75

primary cause ofCOPD is exposure to tobacco smoke,withatmospheric pollution as an additional contributory factor.Although>90% of patients with COPD are smokers, only15–20% of smokers develop the disease. In a cigarettesmoke-induced emphysema model, it has been demon-strated that individual factors such as low serum anti-protease levels and/or an inherited sensitivity to oxidantspredisposemouse strains to smoking-induced lung damage(Cavarra et al., 2001). Another important risk factor ishomozygous a1-antitrypsin deficiency, which accounts forless than 1% of COPD cases (Rijcken & Britton, 1998).The injury to extracellular matrix in pulmonary emphy-sema has been ascribed to an imbalance of protease/antiprotease activities. Evidence from studies on humansand experimental animal models support the elastasetheory of human emphysema (Lungarella, Cavarra, &Martorana, 1999).
Increasing evidences suggest that an oxidant/antiox-idant imbalance occurs in smokers and patientswithCOPD(Comhair & Erzurum, 2002; Langen, Korn, & Wouters,2003). Cigarette smoke is themajor source of oxidants, andthe airways of COPD patients contain increased numbersof neutrophils able to produce free radicals (Saetta et al.,2001). Thus, the oxidative burden produced by cigarettesmoke can be enhanced by ROS released by neutrophilsand macrophages recruited in the lung (Morrison et al.,1999). In endothelial and epithelial cells, oxidative burdenmay also impair macromolecular barrier function andincrease leukocyte adhesion to endothelium via activationof NF-kB-mediated transcription of integrin genes (Rah-man & MacNee, 1996). The up-regulation of redox-sensitive transcription factors such as NF-kB causesincreased expression of genes for many inflammatorymediators, such asNO., interleukin-8, andTNF-a,markersof inflammation that have been shown to be elevated in thesputum of COPD patients (Rahman & MacNee, 1998).Moreover, oxidants cause inactivation of a1-antitrypsin byoxidation of theMet residue at its active site, a critical eventfor the protease/antiprotease imbalance.
Biomarkers of oxidative stress have been demon-strated in the epithelial lining fluid, in the breath, and in theurine of cigarette smokers and COPD patients. Inparticular, COPD patients show: higher levels of H2O2
and NO. in the breath (Dekhuijzen et al., 1996; Maziaket al., 1998); higher levels of xanthine/xanthine oxidase inBAL fluid (Pinamonti et al., 1996); an increased F2-isoprostane content in the urine (Pratico et al., 1998a);higher levels of 4-HNE-modified proteins in airwayepithelial cells and endothelial cells (Rahman et al.,2002); increased levels ofNO2-Tyr in airway inflammatorycells (Ichinose et al., 2000).Moreover, patientswith COPDshow evidence of systemic oxidative stress: increasedproduction of O2
.� by peripheral blood neutrophils duringacute exacerbation (Rahman, Skwarska, & MacNee,
1997); higher plasma levels of lipid peroxidation products(Rahman et al., 1996; Morrison et al., 1999); nitratedfibrinogen, transferrin, plasminogen, and ceruloplasmin(Pignatelli et al., 2001).
G. Diabetes Mellitus
Diabetesmellitus is a very common chronic disease causedby the impaired production of insulin by pancreatic isletb-cells and/or by diminished tissue responses to insulin(insulin resistance). Diabetes is a major worldwide healthproblem predisposing to markedly increased cardio-vascular mortality and serious morbidity and mortalityrelated to development of nephropathy, neuropathy, andretinopathy. Hyperglycemia (elevated blood glucose) is acommon hallmark of both insulin-dependent (type 1,juvenile onset) and non-insulin-dependent (type 2) dia-betes mellitus, together with increased urinary glucoseexcretion as well as derangement in carbohydrate and lipidmetabolism.
There is emerging, substantial evidence that ROS/RNS make a significant contribution to the progression ofdiabetes and its complications (Rosen et al., 2001; Yorek,2003). Elevated glucose levels are associated withincreased production of ROS by several different mechan-isms (Baynes, 1991; Nishikawa et al., 2000; Yorek, 2003),including superoxide generation by the process of glucoseauto-oxidation that is associated with the formation ofglycated proteins in the plasma of diabetic patients(Baynes, 1991).
The increase in glycoxidation and lipoxidationproducts in plasma and tissue proteins suggests thatoxidative stress is increased in diabetes. However, someof these products are formed independent on oxidationchemistry and may also result from elevated levels ofoxidizable substrates.Moreover, there is also an increase inproducts of reaction of proteins with dicarbonyl com-pounds formed by nonoxidative mechanisms (Baynes &Thorpe, 1999). The increased chemical modification ofproteins by carbohydrates and lipids in diabetes and otherdiseases such as uremia may therefore be viewed as theresult of increased carbonyl stress (carbonyl overload),which is caused by a generalized increase in the con-centration of reactive carbonyl precursors of advancedglycation/lipoxidation end-products (AGEs/ALEs), gly-coxidation and lipoxidation products. Carbonyl stress mayresult from an increase in substrate stress and/or a decreasein the efficiency of detoxification of reactive carbonylcompounds, i.e., an imbalance between the rates of pro-duction and detoxification of reactive carbonyls. Com-pared with oxidative stress (a condition in which carbonylsare derived exclusively from oxidative reactions), carbonylstress is a more comprehensive term, since it includesincreases in carbonyls derived from both oxidative and
& DALLE-DONNE ET AL.
76

nonoxidative reactions (Baynes & Thorpe, 1999; Metzet al., 2003).
The increased oxidative/carbonyl stress as well as theaccumulation of AGEs/ALEs in tissue proteins are thoughtto contribute to the development of diabetic complicationssuch as atherosclerosis, vascular and neural dysfunction,and ophthalmic complications (Baynes, 1991; Altomareet al., 1997; Grattagliano et al., 1998; Baynes & Thorpe,1999; Thornalley, 2002; Metz et al., 2003; Yorek, 2003).Moreover, the interaction of AGEswith corresponding cellsurface receptors stimulatesROSproduction and decreasesintracellular GSH levels (Yan et al., 1994). GSH metabo-lism is also altered in both type 1 and type 2 diabeteserythrocytes, leading to GSH depletion in the clinicalcourse of the disease (Dominguez et al., 1998).
H. HIV Infection
Human immunodeficiency virus (HIV) infection is asso-ciated with progressive deterioration of the immunesystem, leading eventually to lethal opportunistic infec-tions. Monocytes and macrophages are susceptible targetsfor HIV-1 infection and have a pivotal role in latency anddiffusion of the infection. Relatively early in the course ofHIV infection, a decrease in various functional activities oflymphoid cells is observed, followed by a strong decreasein CD4þ T lymphocyte number. In the late stages of thedisease, patients often suffer frommassive skeletal musclewasting.
HIV-infected individuals suffer from systemic chronicoxidative stress (Nakamura, Masutani, & Yodoi, 2002),which has been implicated in the pathophysiology of HIVinfection. This effect is subsequent to depletion ofendogenous antioxidant molecules and to increasedproduction of ROS. In addition, this response is enhancedby the chronic inflammation that is associated withactivation of lymphocytes and phagocytes, and is accom-panied by direct or indirect effects of several opportunisticpathogens (Mollace et al., 2001). Neutrophils and mono-cytes from HIV-infected patients spontaneously produceincreased basal amounts of H2O2 (Elbim et al., 2001). Thisincreased H2O2 production is associated with changes inthe expression of antiapoptopic/antioxidant compounds(Bcl-2 and thioredoxin) along the course of the disease aswell as alterations of adhesion molecule expression at thecell surface, which also reflects basal activation ofphagocytes. In monocytes, basal H2O2 production directlycorrelates with plasma viral load.
Total and reduced glutathione levels decrease inplasma and BAL fluid in HIV-infected individuals.Intracellular GSH levels in T cells decrease during theprogression of acquired immunodeficiency syndrome(AIDS). Inflammatory cyokines such as TNF-a playimportant roles in AIDS progression, stimulating replica-
tion of HIV and activation of NF-kB, which controls thetranscription of genes for HIV replication. ROS serve asintracellular second messengers for the activation of NF-kB, which augments the replication of HIV. These findingssuggested that oxidative stress caused by elevatedinflammatory cytokines and decreased GSH-dependentantioxidant functions promotes NF-kB activation and HIVreplication, resulting in the disease progression associatedwith the CD4þ T-cell loss, immunodeficiency, andopportunistic infections. Another redox-regulating mole-cule, thioredoxin, which in the reduced state catalyzes thereduction of protein disulfides, is also transiently down-regulated inmonocytes by acute HIVinfection. In contrast,plasma levels of thioredoxin are elevated in the late stage ofHIV infection. Then, intracellular GSH and plasmathioredoxin, most of which is shown to be in the oxidizedform (Nakamura,Masutani,&Yodoi, 2002), can be used asbiomarkers to predict the prognosis of the disease.
Evidence for oxidative stress was present both in brain(lipid peroxidation) and in cerebrospinal fluid (increasedprotein carbonylation) of HIV-infected patients withdementia when compared with HIV-infected patientswithout dementia and normal controls (Turchan et al.,2003). Thus, oxidative stress seems to play a role also in thepathogenesis of HIV dementia, as in other neurodegenera-tive diseases (Butterfield & Kanski, 2001; Dalle-Donneet al., 2003c; and this review). Nitrite production fromblood mononuclear cells and polymorphonuclear leuko-cytes is raised in AIDS patients with opportunisticinfections; similarly, nitrite and nitrate concentrations areincreased in the serum and cerebrospinal fluid of AIDSpatients with complications in the central nervous system(Torre, Pugliese, & Speranza, 2002). Increased productionofNO.has been confirmed in the sera of childrenwithHIV-1 infection, which also show increased levels of circulatingcytokines, such as interleukin-1b, TNF-a, and interferon g(Torre, Pugliese, & Speranza, 2002).
I. Preeclampsia
Preeclampsia is a pregnancy specific syndrome whichoccurs in 5–7% of all deliveries and is an important causeofmaternalmorbidity andmortality. It is usually diagnosedin the presence of hypertension, normally occurring afterthe 20thweek of gestation, and proteinuria. Ischemia of thefeto-placental unit triggers signals to increase its ownblood supply, which paradoxically causes vasoconstrictionin vital organs of the mother (Page et al., 2000).
The etiology of preeclampsia is unknown. Thetoxemia theory, which proposes that the compromisedplacenta produces substances leading to the maternalsyndrome, remains the favored hypothesis. The pathophy-siology seems linked to a decrease in perfusion to virtuallyall organs (Roberts&Cooper, 2001), which is secondary to
REDOX PROTEOMICS AND DISEASE &
77

an intense vasospasmbecause of an increased sensitivity ofthe vasculature to any pressor agent. This phenomenon isfurther emphasized by activation of the coagulationcascade, particularly of platelets, with consequent micro-thrombi formation. A defect in placental trophoblastinvasion into maternal spiral arteries leads to a persistentplacental underperfusion, which in turn provokes therelease of some factors able to gain access to maternalcirculation with the effect of vascular dysfunction (Zhou,Damsky, & Fisher, 1997). Several studies have shown thatthe maternal vascular endothelium is the ultimate target(Roberts&Redman, 1993). Oxidative stressmay provide aplausible explanation for the endothelial dysfunction andsubsequent pathophysiological changes during preeclamp-sia. It has been hypothesized that the reduced placentalperfusion results in the generation of ROS and RNS, whichcan enter the systemic circulation and damage theendothelium. Neutrophils and monocytes, which couldbe activated during their passage through the placentalcirculation by local inflammatory agents, hypoxic condi-tions or oxidative stress, can also generate ROS on contactwith the endothelium (Roberts & Hubel, 1999). Evidencesfor the presence of oxidative stress in preeclampsia aregiven by increased levels of S-nitrosothiols, NO2-Tyr, andprotein carbonyls in blood, plasma, and placenta ofpregnant preeclamptic women (Zusterzeel et al., 2000,2001; Barden et al., 2001; Tyurin et al., 2001). Clinicaltrials testing antioxidant administration to women in earlypregnancy have shown decreased oxidative stress, endo-thelial activation, and lower prevalence of preeclampsia(Roberts & Cooper, 2001).
J. Rheumatoid Arthritis
Whereas the enhancement of immune reactivity by pro-oxidative conditions may be critically important for theimmune system to control and defeat rapidly multiplyingpathogens, such enhancement also bears the risk ofinducing autoimmune processes. Rheumatoid arthritis(RA) is a systemic autoimmune disease characterized bychronic joint inflammation, especially in hands and legs,which eventually leads to cartilage destruction andsubsequent bone erosion. The proliferated synovial liningof the inflamed joints is invaded by T and B lymphocytes,neutrophils, monocytes/macrophages, and dendritic cells.
Several lines of evidence suggest that production ofROS/RNS at the site of inflammation may contributedecisively to the pathogenesis of RA (Davies, Blake, &Winyard, 2001). In the inflamed rheumatoid joint, O2
.�
is generated from oxygen in reaction catalyzed by theNADPH oxidase within the plasma membrane of activatedpolymorphonuclear leukocytes sequestered within theinflamed joint space, and by the synovial membraneendothelial cells (Davies, Blake, & Winyard, 2001).
Furthermore, the synovial endothelial cells and osteoblastsexpress eNOS, whilst iNOS is present within the macro-phage-like cells of the chronically inflamed synovium.Although the formation of O2
.� and NO. has clearbeneficial effects (e.g., antimicrobial), it is thought thatoverproduction of highly reactive oxidants, such asperoxynitrite, are responsible for cellular and extracellularmatrix destruction within the joint (MacPherson &Ralston, 2000). The migration of lymphocytes andmonocytes into the RA synovium is mediated by theabnormal expression of several adhesion moleculesincluding ELAM-1, VCAM-1, ICAM-1, and ICAM-2(Veale &Maple, 1996), an effect that may be explained bythe abnormal induction of redox-sensitive signaling path-ways.
Thioredoxin, a cellular catalyst induced by oxidativestress, is over-expressed in synovial cells and tissues ofpatients with RA (Maurice et al., 1999), and co-stimulatesthe TNF-a-induced synthesis of IL-6 and IL-8 by synovialfibroblast-like cells; it also activates the NF-kB pathway(Schett et al., 2001). Although several antioxidativefunctions are present in the synovial membrane of RA(i.e., metallothioneines, thioredoxin reductase, and glu-tathione reductase), these activities are thought not to fullycounterbalance local oxidative stress, not preventing theexposure of synovial cells to ROS (Schett et al., 2001).
K. Transmissible Spongiform Encephalopathies
Transmissible spongiform encephalopathies (TSEs), alsoknown as prion disorders, are a group of rare fatalneurodegenerative diseases affecting humans (e.g., Creutz-feldt-Jakob disease, fatal familial insomnia) and otheranimals (e.g., bovine spongiform encephalopathy or madcow disease in cattle, scrapie in sheep and goats, and felinespongiformencephalopathy in cat). TSEs are characterizedby the conversion of the prion protein (PrPC), a protease-sensitive, primarily a-helical species found predominantlyin the central nervous system, into the scrapie isoform ofthe protein (PrPSc), conformationally modified, b-sheet-rich, and protease-resistant (Prusiner et al., 1998). As aresult of its secondary structure, PrPSc tends to aggregateforming insoluble amyloid deposits in the brain, some-times generating amyloid plaques. As described for AD,the presence of protein aggregates is associated withneuronal cell death by apoptosis. The accumulation ofPrPSc in the central nervous system is thought to beresponsible for neuronal loss and/or astrocytosis (Collinge,2001).
The function of PrPC and its role in neurodegenerativeprocesses remain enigmatic. Even though there is not yet adefinitive demonstration that PrPSc constitutes, by itself,the agent responsible for transmission of the disease, thereis no doubt that transition from PrPC to PrPSc is a crucial
& DALLE-DONNE ET AL.
78

pathogenic event, as clearly exemplified by the fact thatPrP-null mice are resistant to the disease. Recent studieshave provided evidence that normal prion protein mighthave a physiological function in neuroprotective signaling,suggesting that loss of prion protein activity might contri-bute to the pathogenesis of prion disease. However,studies using knockout animals do not support the loss-of-function hypothesis and argue that prion neurodegen-eration might be associated with a gain of neurotoxicactivity by themisfolded prion protein (Hetz,Maundrell,&Soto, 2003).
Various factors such as oxidative stress, mitochondrialdysfunction, and disruption of calcium homeostasis (thelatter usually related to oxidative stress, apoptosis, andmitochondrial damage) are associated with the pathogen-esis of prion diseases (Milhavet & Lehmann, 2002). It hasbeen proposed that PrPC may play a role in the control ofthe cellular oxidative state through a regulation of thecopper transport/metabolism and/or modification of Cu/Zn-SOD activity (Milhavet & Lehmann, 2002). It has beendemonstrated that prion-infected neuronal cells displayeda higher sensitivity to oxidative stress, increased lipidperoxidation, and a dramatic decrease in the activities ofglutathione peroxidase, glutathione reductase, and Mn-SOD as well as in SOD protein levels (Milhavet et al.,2000). Increased levels of heme oxygenase-1 (a markerof oxidative stress), NO2-Tyr, and MDA have beenshown in the brain of infected mice (Milhavet & Lehmann,2002). In addition, a recent study in knockout mice hasshown the presence of several markers of oxidative stressand the animals were more sensitive to neurologicaldamage. No tissue degeneration was observed, but cellsderived from these animals were hypersensitive tooxidative stress (Brown, Nicholas, & Canevari, 2002).Collectively, these findings suggest that prion infectioncompromises the cellular resistance to ROS. It hasbeen hypothesized that PrPC could act as a stress sensorinvolved in the regulation of the cellular response tooxidative stress through signal transduction (Milhavet &Lehmann, 2002).
VIII. OXIDATIVELY MODIFIED PROTEINS INHUMAN DISEASES
Substantial evidence suggesting that oxidative/nitrosativeevents contribute to the pathogenesis and/or progression ofmost human diseases comes from studies reportingelevated levels of protein oxidation markers in tissues/fluids from affected individuals (Table 2). More impor-tantly, much of the progress in defining the molecularmechanisms accounting for protein oxidation arises frommarkers, whose structure conveys information aboutoxidation pathways leading to their formation, as pre-
viously reported in the case of NO2-Tyr, Cl-Tyr, Br-Tyr, di-Tyr, o-Tyr, and m-Tyr. These compounds are informativein determining oxidation mechanisms occurring in vivo.However, their detection does not clarify how oxidizedproteins might affect cellular metabolism and, ultimately,lead to cell death. Identification of proteins specificallymodified by oxidative/nitrosative injury in diseased tissueswill allow researchers to determine proteins more prone toinactivation byROS/RNS, thus representing a fundamentalstep in linking well-established pathological hallmarks tooxidative/nitrosative events.
Invitro studies indicate that Cl-Tyr is a highly sensitiveand specific marker for protein oxidation by HOCl (Hazen& Heinecke, 1997). Moreover, chlorinate tyrosines areideal biomarkers for HOCl since they are stable underthe acid conditions required to hydrolyse proteins(Winterbourn & Kettle, 2000). Confirmation of thesuitability of Cl-Tyr as a biomarker of oxidative injuryhas come from studies in which isotope dilution GC/MSwas utilized for examining oxidized LDL from thearterial wall of human atherosclerotic aortic lesionscompared with LDL from periferal blood (Hazen &Heinecke, 1997; Heinecke, 1999a) as well as plasmaproteins of patients on chronic hemodialysis therapy(Himmelfarb et al., 2001).
Using isotope dilution GC/MS, it has been quantifiedthat LDL isolated from atherosclerotic lesions containedan 80-fold increase in NO2-Tyr amount compared withcirculating LDL; similarly, the concentration of di-Tyr inlesion LDL was 100-fold higher than in circulating LDL(Leeuwenburgh et al., 1997a,b). Notably, di-Tyr increasedsignificantly with disease (Upston et al., 2002). Biologicalnitration of protein tyrosine is associated with humandiseases including lung infection, central nervous systeminflammation, shock, cancer, neurological disorders (e.g.,ALS, AD, Parkinson’s disease, and stroke), and asthma(Aulak et al., 2001; Esposito et al., 2002; Andreadis et al.,2003). Interestingly, Tyr nitration is one of the earliestmarkers found in AD brains, in the brain plaques generatedby multiple sclerosis, and in degenerating upper and lowermotor neurons in ALS patients. Its formation might reallyrepresent a useful clinical parameter of the occurrence ofoxidative/nitrosative stress in neurodegenerative diseases.In general, protein nitration is typically indicated indiseases presenting an inflammatory component, possiblycaused by up-regulation of iNOS. It has been detected inmultiple organ systems and cell types during both acuteand chronic inflammation (Greenacre & Ischiropoulos,2001). Although NO2-Tyr is formed in diverse inflamma-tory diseases, only a few specific proteins have beenidentified as in vivo targets of NO-dependent modification(MacMillan-Crow et al., 1996; Gole et al., 2000; Zhu et al.,2001; Aslan et al., 2003). Moreover, a recent clinical studyhas demonstrated clinical utility in monitoring systemic
REDOX PROTEOMICS AND DISEASE &
79

TABLE
2.Summarized
DataonOxidative/NitrosativeProtein
Modificationsin
Human
Diseases
& DALLE-DONNE ET AL.
80

(Continued
)
REDOX PROTEOMICS AND DISEASE &
81

TABLE
2.(Continued
)
& DALLE-DONNE ET AL.
82
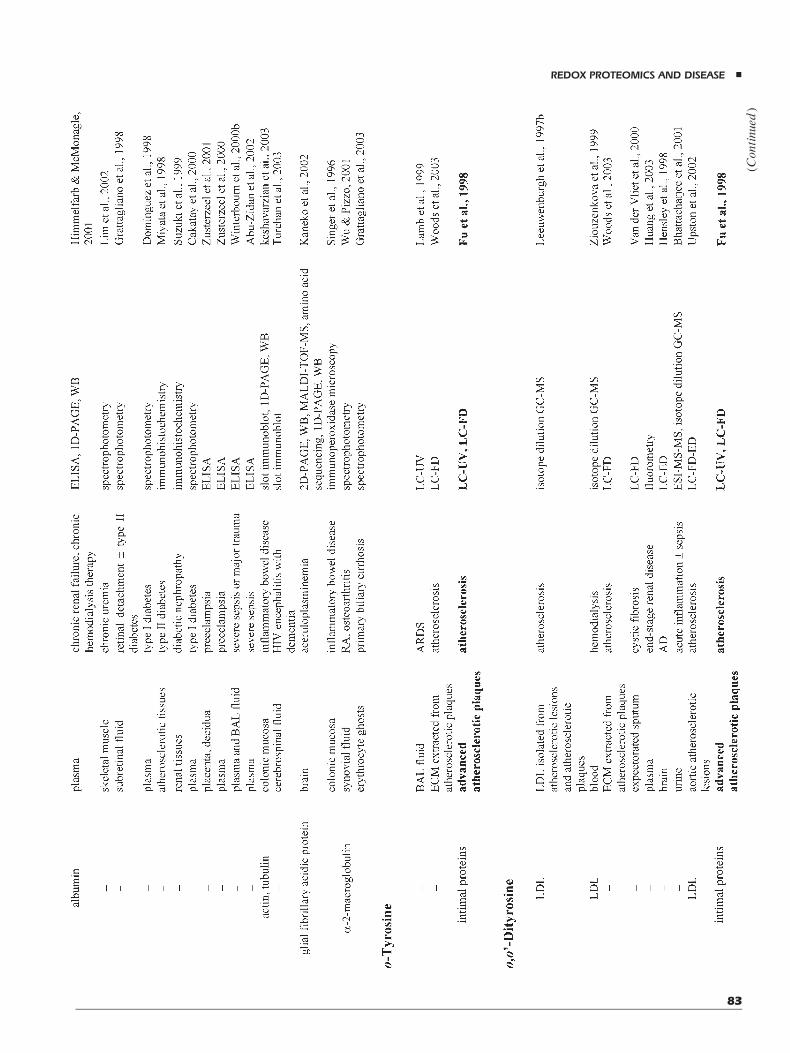
(Continued
)
REDOX PROTEOMICS AND DISEASE &
83

TABLE
2.(C
ontinued
)
HIV,human
immunodeficiency
virus;AD,A
lzheimer’sdisease;BAL,brochoalveolarlavage;ALI,acutelunginjury;ARDS,acute(adult)respiratory
distresssyndrome;ALS,amyotrophic
lateralsclerosis;LDL,low-density-lipoprotein;RA,rheumatoid
arthritis;ECM,extracellularmatrix;FD,fluorometricdetector;ED,electrochem
icaldetector.
& DALLE-DONNE ET AL.
84

levels of protein-bound NO2-Tyr as a predictor of risk forcoronary artery disease, atherosclerotic burden, andresponse to statin therapy (Shishehbor et al., 2003). Stableisotope dilution ESI-MS-based analyses in a case controlstudy with over 200 patients revealed protein NO2-Tyr as arobust predictor of cardiovascular risk, independent ofestablished risk factors and other inflammatory markerslike C-reactive protein. In addition, the strength of theassociation between NO2-Tyr levels and atherosclerosisincreased with increasing clinical evidence of athero-sclerotic burden. The same report revealed that statintherapy, known to reduce cardiovascular risk, resulted in amarked reduction of NO2-Tyr levels in plasma proteins.This effect was comparable in magnitude to reductions intotal cholesterol and LDL particle number (Shishehboret al., 2003).
Both S-nitrosated and S-glutathionylated proteins havebeen investigated as possible markers of oxidative/nitrosative stress in correlation with patho-physiologicalconditions. In particular, since alterations in some bloodparameters can reflect alterations even in hardly achievablecompartments of the body, hemoglobin, and albumin havebeen analyzed in some pathologies (Table 2).
Protein carbonyl content is actually taken as the mostgeneral and widely used biomarker of severe oxidativeprotein damage. In the case of AD, it is particularlyinteresting that specific carbonylated proteins have beenobserved in blood plasma of patients (Conrad et al., 2000).In a subsequent proteomic study, 2D-PAGE analysiscoupled to Western blotting with anti-DNP antibody andmass fingerprint experiments identified isoforms offibrinogen g-chain precursor protein and of a1-anti-trypsinas specific targets of modification. Their oxidation levelswere 2–6-fold higher in plasma from AD patients whencompared with controls (Choi et al., 2002). The observa-tion of these carbonylated proteins in plasma—a body fluideasily obtainable without invasive procedures and, moreinteresting and unlike brain samples, before the death ofthe subject—suggests that these oxidized species may beuseful as pre-symptomatic diagnostic biomarkers for AD.In a very recent study, Winterbourn and co-workers havedetermined that patients with acute pancreatitis had signi-ficantly increased concentration of protein carbonyls inplasma, persisting over 5 days on admission to hospital,and that protein carbonyls were related to disease severity,thus confirming that this protein modification could be auseful plasma marker of oxidative injury (Winterbournet al., 2003).
IX. CONCLUDING REMARKS ANDFUTURE PERSPECTIVES
Redox proteomics is currently a nascent but rapidlygrowing field of research. The opportunities for identifica-
tion of proteins involved in diseases strictly linked tooxidative/nitrosative stress are clear and compelling. Sinceproteins are involved in virtually every cellular function,proteome dictates functional phenotype in each tissue ororgan. This phenotype varies continuously under normalconditions (depending on differentiation, age, and cellcycle stage) and can change as a result of oxidative/nitrosative stress, contributing to initiation and/or progres-sion of acute or chronic diseases. Usually, acute insults leadto rapid post-translational modifications of proteins,whereas in chronic diseases co-translational (isoformswitching) and post-translational protein modificationsoccur in concert with altered gene expression, leading tovaried protein levels. For specific proteins, disease-inducedmodifications will substantially affect function, which inturn has the potential to affect other proteins. The result is adynamic, ongoing process of protein expression andmodification.
Identification of oxidatively/nitrosatively modifiedproteins in a few human diseases has just begun (Table 2).New proteomic tools, in the near future, will facilitate theidentification of protein markers still unknown, allowingtheir complete characterization in a given pathophysio-logical condition. These findings will contribute toestablish relationships between pathological hallmarksof the disease and protein functional and/or structuralalterations. Furthermore, comparison of the molecularfingerprints obtained in cells or body fluids with thoseproduced by various in vitro oxidation/nitrosation/nitra-tion systems will indicate biochemical pathways creatingdamage in vivo. All these data will decipher the potentialroles played by ROS/RNS-induced modifications inhuman disorders. In this sense, redox proteomics isopening new scenarios to gain insight into molecularmechanisms involved in human pathologies. This infor-mation will establish new hypotheses for the mechanismof disease-induced cell and tissue alterations. However,hierarchies of altered proteins (i.e., which proteinalteration is the primary event and which are secondaryphenomena) have to be established. Consequently, otherapproaches (e.g., animal models with over-expressed,knocked-in, or knocked-out proteins identified as poten-tially important in a certain disease) need to be selectedto complement MS-based redox proteomic investiga-tions. As for any laboratory-based assay that is used inepidemiological research, methodological issues suchas reproducibility, inter- versus intra-person variability,sensitivity, and specificity, have to be considered. A keyissue concerns the potential for artifacts in estimationof modified protein baseline levels, associated withuncorrect sample handling, processing, and analysis.This issue has plagued investigation on many differentbiomarkers of protein oxidation. Quantitative proteomicswill have a leading role in the solution of this specific
REDOX PROTEOMICS AND DISEASE &
85

problem as well as in the evaluation of all remainingissues.
The finding of ROS/RNS-induced damage in humandiseases suggests the possible use of antioxidants inslowing down disease progression. Development ofprocedures to ameliorate excess ROS/RNS productionand control protein oxidation damage is fundamental and,in the near future, may be one of the central issues inresearch on oxidative/nitrosative stress-related diseases.Biomarkers of oxidative/nitrosative stress (Fig. 2) will bepossibly used to evaluate the efficacy of antioxidanttherapy. These markers may yield information on threeprogressive levels of disease outcome: (i) as measurableend-points of damage to biomolecules such as proteins/amino acids, DNA bases, lipids; (ii) as functional markers,for example, of blood flow, platelet aggregation, orcognitive function; and (iii) as end-points related tospecific diseases. Whereas the clinical symptoms of adisease are end-points in themselves, they are not suitable,in many cases, for early detection and, therefore, preven-tion of diseases associatedwith oxidative/nitrosative stress.A series of biomarkers of oxidative/nitrosative stress wouldbe preferred (Halliwell & Gutteridge, 1999; Dalle-Donneet al., 2003c), each validated in sequence. To this end, theassociation between a biomarker and a disease should bedefined.
As described in this review, a battery of markers ofprotein oxidation/nitrosation/nitration is available or underdevelopment. The challenge now facing this research fieldis sorting out which markers or combinations of markersare predictive of human diseases associatedwith oxidative/nitrosative stress. This issue will require an answer to acentral question still unresolved: does the formation ofoxidized proteins has a significant, direct physiological orpathological impact, or is it a secondary phenomenon?Many evidences indicate that ROS/RNS-induced proteinmodification may be of physiological significance, andthat in some pathologies it is not solely a secondaryconsequence. In this sense, we do believe that redoxproteomics will have a central role in the definition ofredox molecular mechanisms associated with humanpathologies.
ACKNOWLEDGMENTS
Contract grant sponsors: FIRST 2003 (Fondo InternoRicerca Scientifica e Tecnologica, University of Milan);FIRB 2001 (Fondo di Incentivazione alla Ricerca di Base;Contract grant number: RBAU01T97W_003); andRegione Campania (LR 41/94, n. 4847316). Owing tospace limitations, we could not cite all the articles relevantto the presented topic. We sincerely apologize to thoseauthors whose work we could not include.
ABBREVIATIONS
ROS reactive oxygen speciesRNS reactive nitrogen speciesMS mass spectrometrySOD superoxide dismutaseDNA deoxyribonucleic acidNADPH reduced nicotinamide adenine dinucleotide
phosphatenNOS neuronal NO synthaseeNOS endothelial NO synthaseiNOS inducible NO synthaseGSH glutathioneo-Tyr o-tyrosinedi-Tyr o,o0-dityrosineNO2-Tyr 3-nitrotyrosineCl-Tyr 3-chlorotyrosineSDS–PAGE sodium dodecylsulfate–polyacrylamide
gel electrophoresisHPLC high performance liquid chromatographySH sulfhydryldi-Cl-Tyr 3,5-dichlorotyrosineBr-Tyr 3-bromotyrosinedi-Br-Tyr 3,5-dibromotyrosineLDL low-density lipoproteinMetO methionine sulfoxideMetO2 methionine sulfoneHIV human immunodeficiency virusMsr methionine sulfoxide reductase4-HNE 4-hydroxynonenalacrolein 2-propenalMDA malondialdehydeESI electrospray ionizationLC-ESI liquid chromatography-electrospray
ionizationMALDI matrix-assisted laser
desorption/ionizationCID collision induced dissociationGC gas chromatographyLC liquid chromatographySIM selective ion monitoringPSD post-source decayHAVA 5-hydroxy-2-aminovaleric acidHACA 6-hydroxy-2-caproic acidNMR nuclear magnetic resonance2D two-dimensionalDNPH 2,4-dinitrophenylhydrazineDNP 2,4-dinitrophenylHRP horseradish peroxidaseARDS acute (adult) respiratory distress syndromeBAL bronchoalveolar lavageAD Alzheimer’s diseaseNFTs neurofibrillary tanglesTBARS thiobarbituric acid reactive substances
& DALLE-DONNE ET AL.
86

ALS amyotrophic lateral sclerosisTNF-a tumor necrosis factor-aCOPD chronic obstructive pulmonary diseaseNF-kB nuclear factor-kBAGEs advanced glycation end-productsALEs advanced lipoxidation end-productsAIDS acquired immunodeficiency syndromeRA rheumatoid arthritisTSEs transmissible spongiform
encephalopathiesPrPC prion proteinPrPSc scrapie isoform of the prion protein
REFERENCES
Abu-Zidan FM, Plank LD, Windsor JA. 2002. Proteolysis insevere sepsis is related to oxidation of plasma protein. Eur JSurg 168:119–123.
Ahn B, Rhee SG, Stadtman ER. 1987. Use of fluoresceinhydrazide and fluorescein thiosemicarbazide reagents forthe fluorometric determination of protein carbonyl groupsand for the detection of oxidized protein on polyacrylamidegels. Anal Biochem 161:245–257.
Aksenov MY, Aksenova MV, Butterfield DA, Geddes JW,Markesbery WR. 2001. Protein oxidation in the brain inAlzheimer’s disease. Neuroscience 103:373–383.
Al-Abed Y, VanPatten S, Li H, Lawson JA, Fitzgerald GA,Manogue KR, Bucala R. 2001. Characterization of a novelhemoglobin-glutathione adduct that is elevated in diabeticpatients. Mol Med 7:619–623.
Alderton AL, Faustman C, Liebler DC, Hill DW. 2003. Inductionof redox instability of bovine myoglobin by adduction with4-hydroxy-2-nonenal. Biochemistry 42:4398–4405.
Aldridge RE, Chan T, Van Dalen CJ, Senthilmohan R, Winn M,Venge P, Town GI, Kettle AJ. 2002. Eosinophil peroxidaseproduces hypobromous acid in the airways of stableasthmatics. Free Radic Biol Med 33:847–856.
Alexander MD, Traynor BJ, Miller N, Corr B, Frost E, McQuaidS, Brett FM, Green A, Hardiman O. 2002. ‘‘True’’ sporadicALS associated with a novel SOD-1 mutation. Ann Neurol52:680–683.
AlmerG, Guegan C, Teismann P, Naini A, Rosoklija G, Hays AP,Chen C, Przedborski S. 2001. Increased expression of thepro-inflammatory enzyme cyclooxygenase-2 in amyo-trophic lateral sclerosis. Ann Neurol 49:176–185.
Altomare E, Grattagliano I, Vendemaile G, Micelli-Ferrari T,Signorile A, Cardia L. 1997. Oxidative protein damage inhuman diabetic eye: Evidence of a retinal participation. EurJ Clin Invest 27:141–147.
Anderson LB, Maderia M, Ouellette AJ, Putnam-Evans C,Higgins L, Krick T, MacCoss MJ, Lim H, Yates JR III,Barry BA. 2002. Posttranslationalmodifications in the CP43subunit of photosystem II. Proc Natl Acad Sci USA 99:14676–14681.
Andreadis AA, Hazen SL, Comhair SAA, Erzurum SC. 2003.Oxidative and nitrosative events in asthma. Free Radic BiolMed 35:213–225.
Aoyama K, Matsubara K, Fujikawa Y, Nagahiro Y, Shimizu K,Umegae N, Hayase N, Shiono H, Kobayashi S. 2000. Nitra-tion of manganese superoxide dismutase in cerebrospinalfluids is amarker for peroxynitrite-mediated oxidative stressin neurodegenerative diseases. Ann Neurol 47:524–527.
Arner ES, Holmgren A. 2000. Physiological functions of thiore-doxin and thioredoxin reductase. Eur J Biochem 267:6102–6109.
AslanM, Ryan TM, Townes TM, Coward L, KirkMC, Barnes S,Alexander CB, Rosenfeld SS, Freeman BA. 2003. Nitricoxide-dependent generation of reactive species in sickle celldisease. Actin tyrosine nitration induces defective cytoske-letal polymerization. J Biol Chem 278:4194–4204.
ATS Committee. 1995. Standards for the diagnosis and care ofpatients with chronic obstructive pulmonary disease. Am JRespir Crit Care Med 152:S77–S120.
Aulak KS, Miyagi M, Yan L, West KA, Massillon D, Crabb JW,Stuehr DJ. 2001. Proteomic method identifies proteinsnitrated in vivo during inflammatory challenge. Proc NatlAcad Sci USA 98:12056–12061.
Bagasra O, Michaels FH, Zheng YM, Bobroski LE, Spitsin SV,Fu ZF, Tawadros R, Koprowski H. 1995. Activation of theinducible form of nitric oxide synthase in the brains ofpatients with multiple sclerosis. Proc Natl Acad Sci USA92:12041–12045.
Balabanli B, Kamisaki Y, Martin E, Murad F. 1999. Require-ments for heme and thiols for the nonenzymatic modificationof nitrotyrosine. Proc Natl Acad Sci USA 96:13136–13141.
Baldus S, Eiserich JP,BrennanM-L, JacksonRM,AlexanderCB,Freeman BA. 2002. Spatial mapping of pulmonary andvascular nitrotyrosine reveals the pivotal role of myeloper-oxidase as a catalyst for tyrosine nitration in inflammatorydiseases. Free Radic Biol Med 33:1010–1019.
Bandeira-Melo C, Bozza PT, Weller PF. 2002. The cellularbiology of eosinophil eicosanoid formation and function.J Allergy Clin Immunol 109:393–400.
Barden A, Ritchie J, Walters B, Michael C, Rivera J, Mori T,Croft K, Beilin L. 2001. Study of plasma factors associatedwith neutrophil activation and lipid peroxidation inpreeclampsia. Hypertension 38:803–808.
Baynes JW. 1991. Role of oxidative stress in development ofcomplications in diabetes. Diabetes 40:405–412.
Baynes JW, Thorpe SR. 1999. Role of oxidative stress in diabeticcomplications: A new perspective on an old paradigm.Diabetes 48:1–9.
Baynes JW, Thorpe SR. 2000. Glycoxidation and lipoxidation inatherogenesis. Free Radic Biol Med 28:1708–1716.
Beal MF. 2002. Oxidatively modified proteins in aging anddisease. Free Radic Biol Med 32:797–803.
Beal MF, Ferrante RJ, Browne SE, Matthews RT, Kowall NW,Brown RH, Jr. 1997. Increased 3-nitrotyrosine in bothsporadic and familial amyotrophic lateral sclerosis. AnnNeurol 42:646–654.
REDOX PROTEOMICS AND DISEASE &
87

Behr J,Maier K, BraunB, SchwaiblmairM, Vogelmeier C. 2000.Evidence for oxidative stress in bronchiolitis obliteranssyndrome after lung and heart-lung transplantation. TheMunich Lung Transplant Group. Transplantation 69:1856–1860.
Bennaars-Eiden A, Higgins L, Hertzel AV, Kapphahn RJ,Ferrington DA, Bernlohr DA. 2002. Covalent modificationof epithelial fatty acid-binding protein by 4-hydroxynonenalin vitro and in vivo. Evidence for a role in antioxidantbiology. J Biol Chem 277:50693–50702.
Berlett BS, Levine RL, Stadtman ER. 1998. Carbon dioxidestimulates peroxynitrite-mediated nitration of tyrosineresidues and inhibits oxidation of methionine residues ofglutamine synthetase: Both modifications mimic effects ofadenylylation. Proc Natl Acad Sci USA 95:2784–2789.
Berlett BS, Stadtman ER. 1997. Protein oxidation in aging, dis-ease, and oxidative stress. J Biol Chem 272:20313–20316.
Bhattacharjee S, Pennathur S, Byun J, Crowley J, Mueller D,Gischler J, Hotchkiss RS, Heinecke JW. 2001. NADPHoxidase of neutrophils elevates o,o0-dityrosine cross-links inproteins and urine during inflammation. Arch BiochemBiophys 395:69–77.
Biemel KM, Friedl DA, Lederer MO. 2002. Identification andquantification of major maillard cross-links in human serumalbumin and lens protein. Evidence for glucosepane as thedominant compound. J Biol Chem 277:24907–24915.
Biemel KM, Reihl O, Conrad J, Lederer MO. 2001. Formationpathways for lysine-arginine cross-links derived fromhexoses and pentoses by Maillard processes: Unravelingthe structure of a pentosidine precursor. J Biol Chem 276:23405–23412.
BowlerRP,Crapo JD. 2002.Oxidative stress in airways: Is there arole for extracellular superoxide dismutase? Am J RespirCrit Care Med 166:S38–43.
BrownDR,Nicholas RS, Canevari L. 2002. Lack of prion proteinexpression results in a neuronal phenotype sensitive tostress. J Neurosci Res 67:211–224.
Buss H, Darlow BA, Winterbourn CC. 2000. Elevated proteincarbonyls and lipid peroxidation products correlating with-myeloperoxidase in tracheal aspirates from prematureinfants. Pediatr Res 47:640–645.
Buss IH, Senthilmohan R, Darlow BA, Mogridge N, Kettle AJ,Winterbourn CC. 2003. 3-Chlorotyrosine as a marker ofprotein damage by myeloperoxidase in tracheal aspiratesfrom preterm infants: Association with adverse respiratoryoutcome. Pediatr Res 53:455–462.
Butterfield DA, Kanski J. 2001. Brain protein oxidation in age-related neurodegenerative disorders that are associated withaggregated proteins. Mech Ageing Devel 122:945–962.
Butterfield DA, Lauderback CM. 2002. Lipid peroxidation andprotein oxidation in Alzheimer’s disease brain: Potentialcauses and consequences involving amyloid beta-peptide-associated free radical oxidative stress. Free Radic BiolMed32:1050–1060.
Butterfield DA, Drake J, Pocernich C, Castegna A. 2001.Evidence of oxidative damage in Alzheimer’s disease brain:
Central role for amyloid beta-peptide. Trends Mol Med7:548–554.
Butterfield DA, Castegna A, Lauderback CM, Drake J. 2002.Evidence that amyloid beta-peptide-induced lipid peroxida-tion and its sequelae in Alzheimer’s disease brain contributeto neuronal death. Neurobiol Aging 23:655–664.
Buttery LD, Springall DR, Chester AH, Evans TJ, Standfield EN,Parums DV, Yacoub MH, Polak JM. 1996. Inducible nitricoxide synthase is present within human atheroscleroticlesions and promotes the formation and activity of peroxy-nitrite. Lab Investig 75:77–85.
Cai H, Harrison DG. 2000. Endothelial dysfunction in cardio-vascular diseases: The role of oxidant stress. Circ Res 87:840–844.
Cakatay U, Telci A, Salman S, Satman L, Sivas A. 2000.Oxidative protein damage in type I diabetic patientswith andwithout complications. Endocr Res 26:365–379.
Calabrese V, Scapagnini G, Ravagna A, Bella R, Foresti R, BatesTE, Giuffrida Stella AM, Pennisi G. 2002. Nitric oxidesynthase is present in the cerebrospinal fluid of patients withactive multiple sclerosis and is associated with increases incerebrospinal fluid protein nitrotyrosine and S-nitrosothiolsand with changes in glutathione levels. J Neurosci Res 70:580–587.
Calabrese V, Scapagnini G, Ravagna A, Bella R, Butterfield DA,Calvani M, Pennisi G, Giuffrida Stella AM. 2003. Disrup-tion of thiol homeostasis and nitrosative stress in thecerebrospinal fluid of patients with activemultiple sclerosis:Evidence for a protective role of acetylcarnitine.NeurochemRes 28:1321–1328.
Caselli A, Marzocchini R, Camici G, Manao G, Moneti G,Pieraccini G, Ramponi G. 1998. The inactivationmechanism of low molecular weight phosphotyrosine-protein phosphatase by H2O2. J Biol Chem 273:32554–32560.
Castegna A, AksenovM, Aksenova M, Thongboonkerd V, KleinJ, Pierce W, Booze R, Markesbery W, Butterfield D. 2002a.Proteomic identification of oxidatively modified proteins inAlzheimer’s disease brain. Part I: Creatine kinase BB,glutamine synthase, and ubiquitin carboxy-terminalhydrolase L-1. Free Radic Biol Med 33:562–571.
Castegna A, Aksenov M, Thongboonkerd V, Klein JB, PierceWM, Booze R, Markesbery WR, Butterfield DA. 2002b.Proteomic identification of oxidatively modified proteins inAlzheimer’s disease brain. Part II: Dihydropyrimidinase-related protein 2, a-enolase and heat shock cognate 71.J Neurochem 82:1524–1532.
Castegna A, Thongboonkerd V, Klein JB, Lynn B, MarkesberyWR, Butterfield DA. 2003. Proteomic identification ofnitrated proteins in Alzheimer’s disease brain. J Neurochem85:1394–1401.
Cavarra E, Bartalesi B, Lucattelli M, Fineschi S, Lunghi B,Gambelli F, Ortiz LA, Martorana PA, Lungarella G.2001. Effects of cigarette smoke in mice with differentlevels of a1-proteinase inhibitor and sensitivity to oxidants.Am J Respir Crit Care Med 164:886–890.
& DALLE-DONNE ET AL.
88

Cecconi I, Scaloni A, Rastelli G, Moroni M, Vilardo PG,Costantino L, Cappiello M, Garland D, Carper D, PetrashJM, Del Corso A, Mura U. 2002. Oxidative modification ofaldose reductase induced by copper ion. Definition of themetal–protein interaction mechanism. J Biol Chem 277:42017–42027.
Ceriello A, Mercuri F, Quagliaro L, Assaloni R, Motz E,Tonutti L, Taboga C. 2001. Detection of nitrotyrosine in thediabetic plasma: Evidence of oxidative stress. Diabetologia44:834–838.
CerielloA,QuagliaroL, CatoneB, PasconR, PiazzolaM,BaisB,Marra G, Tonutti L, Taboga C, Motz E. 2002. Roleof hyperglycemia in nitrotyrosine postprandial generation.Diabetes Care 25:1439–1443.
Chen K, Thomas SR, Keaney JF, Jr. 2003. Beyond LDLoxidation: ROS in vascular signal transduction. Free RadicBiol Med 35:117–132.
Choi J, Malakowsky CA, Talent JM, Conrad CC, Gracy RW.2002. Identification of oxidized plasma proteins in Alzhei-mer’s disease. Biochem Biophys Res Commun 293:1566–1570.
Choi J, Malakowsky CA, Talent JM, Conrad CC, Carroll CA,Weintraub ST, Gracy RW. 2003. Anti-apoptotic proteins areoxidized by Ab25–35 in Alzheimer’s fibroblasts. BiochimBiophys Acta 1637:135–141.
Ciriolo MR, Paci M, Sette M, DeMartino A, Bozzi A, Rotilio G.1993. Transduction of reducing power across the plasmamembrane by reduced glutathione. A 1H-NMR spin-echostudy of intact human erythrocytes. Eur J Biochem 215:711–718.
Collinge J. 2001. Prion diseases of humans and animals: Theircauses and molecular basis. Annu Rev Neurosci 24:519–550.
Comhair SA, Erzurum SC. 2002. Antioxidant responses tooxidant-mediated lung diseases. Am J Physiol Lung CellMol Physiol 283:L246–L255.
Conrad CC, Marshall PL, Talent JM, Malakowsky CA, Choi J,Gracy RW. 2000. Oxidized proteins in Alzheimer’s plasma.Biochem Biophys Res Commun 275:678–681.
Cotgreave IA, Gerdes RG. 1998. Recent trends in glutathionebiochemistry-glutathione–protein interactions: A molecu-lar link between oxidative stress and cell proliferation?Biochem Biophys Res Commun 242:1–9.
Crabb JW, O’Neil J, Miyagi M, West K, Hoff HF. 2002.Hydroxynonenal inactivates cathepsin B by formingMichael adducts with active site residues. Protein Sci 11:831–840.
Dalle-Donne I,MilzaniA,GiustariniD,Di Simplicio P, ColomboR, Rossi R. 2000. S-NO-actin: S-nitrosylation kinetics andthe effect on isolated vascular smooth muscle. J Muscle ResCell Motil 21:171–181.
Dalle-Donne I, Rossi R, Giustarini D, Gagliano N, Lusini L,Milzani A, Di Simplicio P, Colombo R. 2001. Actincarbonylation: From a simple marker of protein oxidationto relevant signs of severe functional impairment. FreeRadic Biol Med 31:1075–1083.
Dalle-Donne I, Rossi R, Giustarini D, Gagliano N, Di SimplicioP, Colombo R, Milzani A. 2002. Methionine oxidation as amajor cause of the functional impairment of oxidized actin.Free Radic Biol Med 32:927–937.
Dalle-Donne I, Giustarini D, Rossi R, Colombo R, Milzani A.2003a. Reversible S-glutathionylation of Cys(374) regulatesactin filament formation by inducing structural changes inthe actin molecule. Free Radic Biol Med 34:23–32.
Dalle-Donne I, Rossi R, Giustarini D, Milzani A, Colombo R.2003b. Protein carbonyl groups as biomarkers of oxidativestress. Clin Chim Acta 329:23–38.
Dalle-Donne I, Giustarini D, Colombo R, Rossi R, Milzani A.2003c. Protein carbonylation in human diseases. TrendsMolMed 9:169–176.
Dalle-Donne I, Rossi R, Giustarini D, Colombo R, Milzani A.2003d. Actin S-glutathionylation: Evidence against a rolefor glutathione disulfide. Free Radic Biol Med 35:1185–1193.
Davies C, Blake D, Winyard P. 2001. Radicals in inflammation:Mediators and modulators. In: Fuchs J, Packer L, editors.Environmental stressors: Effects on lung, skin, eye,and immune system function. New York: Marcel Dekker.pp 17–52.
Davies MJ, Dean RT. 1997. Radical-mediated protein oxidation.From chemistry to medicine. The pathology of proteinoxidation. New York: Oxford University Press, Inc.
Davies MJ, Fu S, Wang H, Dean RT. 1999. Stable markers ofoxidant damage to proteins and their application in study ofhuman disease. Free Radic Biol Med 27:1151–1161.
Davis DA, Newcomb FM, Moskovitz J, Wingfield PT, Stahl SJ,Kaufman J, Fales HM, Levine RL, Yarchoan R. 2000. HIV-2protease is inactivated after oxidation at the dimer interfaceand activity can be partly restoredwithmethionine solfoxidereductase. Biochem J 346:305–311.
Davis KL, Martin E, Turko IV, Murad F. 2001. Novel effects ofnitric oxide. Annu Rev Pharmacol Toxicol 41:203–236.
De Andrade JA, Crow JP, Viera L, Bruce Alexander C,Randall Young K, McGiffin DC, Zorn GL, Zhu S,Matalon S, JacksonRM. 2000. Protein nitration,metabolitesof reactive nitrogen species, and inflammation inlung allografts. Am J Respir Crit Care Med 161:2035–2042.
Dean RT, Fu S, Stocker R, Davies MJ. 1997. Biochemistry andpathology of radical-mediated protein oxidation. Biochem J324:1–18.
Dekhuijzen PN, Aben KK, Dekker I, Aarts LP, Wielders PL, vanHerwaarden CL, Bast A. 1996. Increased exhalation ofhydrogen peroxide in patients with stable and unstablechronic obstructive pulmonary disease. Am J Respir CritCare Med 154:813–816.
Depre C, Havaux X, Renkin J, Vanoverschelde JL, Wijns W.1999. Expression of inducible nitric oxide synthase inhuman coronary atherosclerotic plague. Cardiovasc Res 41:465–472.
Domigan NM, Charlton TS, Duncan MW, Winterbourn CC,Kettle AJ. 1995. Chlorination of tyrosyl residues in peptides
REDOX PROTEOMICS AND DISEASE &
89

by myeloperoxidase and human neutrophils. J Biol Chem270:16542–16548.
Dominguez C, Ruiz E, Gussinye M, Carrascosa A. 1998.Oxidative stress at onset and in early stages of type 1diabetes in children and adolescents. Diabetes Care 21:1736–1742.
Dong J, Atwood CS, Anderson VE, Siedlak SL, Smith MA,Perry G, Carey PR. 2003. Metal binding and oxidationof amyloid-b, within isolated senile plaque cores:Raman microscopic evidence. Biochemistry 42:2768–2773.
Duda JE, Giasson BI, Chen Q, Gur TL, Hurtig HI, Stern MB,Gollomp SM, Ischiropoulos H, Lee VMY, Trojanowski JQ.2000. Widespread nitration of pathological inclusions inneurodegenerative synucleinopathies. Am J Pathol 157:1439–1445.
Dweik RA, Comhair SA, Gaston B, Thunnissen FB, Farver C,Thomassen MJ, Kavuru M, Hammel J, Abu-Soud HM,Erzurum SC. 2001. NO chemical events in the humanairway during the immediate and late antigen-inducedasthmatic response. Proc Natl Acad Sci USA 98:2622–2627.
Dworski R. 2000. Oxidant stress in asthma. Thorax 55:S51–S53.
Eaton P, Fuller W, Shattock MJ. 2002a. S-Thiolation ofHSP27 regulates its multimeric aggregate size inde-pendently of phosphorylation. J Biol Chem 277:21189–21196.
Eaton P, Wright N, Hearse DJ, Shattock MJ. 2002b. Glycer-aldehyde phosphate dehydrogenase oxidation during car-diac ischemia and reperfusion. JMol Cell Cardiol 34:1549–1560.
Eaton P, Byers HL, Leeds N, Ward MA, Shattock MJ. 2002c.Detection, quantitation, purification, and identification ofcardiac proteins S-thiolated during ischemia and reperfu-sion. J Biol Chem 277:9806–9811.
Eaton P, Jones ME, McGregor E, Dunn MJ, Leeds N,Byers HL, Leung K-I, Ward MA, Pratt JR, Shattock MJ.2003. Reversible cysteine-targeted oxidation of proteinsduring renal oxidative stress. J Am Soc Nephrol 14:S290–S296.
Eiserich JP, Hristova M, Cross CE, Jones AD, Freeman BA,Halliwell B, van der Vliet A. 1998. Formation of nitricoxide-derived inflammatory oxidants by myeloperoxidasein neutrophils. Nature 391:393–397.
Eiserich JP, Estevez AG, Bamberg TV, Ye YZ, Chumley PH,Beckman JS, Freeman BA. 1999. Microtubule dysfunctionby posttranslational nitrotyrosination of alpha-tubulin: Anitric oxide-dependent mechanism of cellular injury. ProcNatl Acad Sci USA 96:6365–6370.
Elbim C, Pillet S, Prevost MH, Preira A, Girard PM, Rogine N,Hakim J, IsraelN,Gougerot-PocidaloMA. 2001. The role ofphagocytes in HIV-related oxidative stress. J Clin Virol 20:99–109.
Esposito E, Rotilio D, Di Matteo V, Di Giulio C, Cacchio M,Algeri S. 2002. A review of specific dietary antioxidantsand the effects on biochemical mechanisms related to
neurodegenerative processes. Neurobiol Aging 23:719–735.
Ferranti P, Malorni A, Mamone G, Sannolo N, Marino G. 1997.Characterisation of S-nitrosohaemoglobin by mass spectro-metry. FEBS Lett 400:19–24.
Finley EL, Dillon J, Crouch RK, Schey KL. 1998. Identificationof tryptophan oxidation products in bovine alpha-crystallin.Protein Sci 7:2391–2397.
Frand AR, Cuozzo JW, Kaiser CA. 2000. Pathways forprotein disulphide bond formation. Trends Cell Biol 10:203–210.
FratelliM,DemolH, PuypeM,Casagrande S, Eberini I, SalmonaM, Bonetto V, Mengozzi M, Duffieux F, Miclet E, Bachi A,Vandekerckhove J, Gianazza E, Ghezzi P. 2002. Identifica-tion by redox proteomics of glutathionylated proteins inoxidatively stressed human T lymphocytes. Proc Natl AcadSci USA 99:3505–3510.
Frost MT, Halliwell B, Moore KP. 2000. Analysis of free andprotein-bound nitrotyrosine in human plasma by a gaschromatography/ mass spectrometry method that avoidsnitration artifacts. Biochem J 345:453–458.
Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A,Maseri A,Nadal-Ginard B, Anversa P. 2000. Myocardial cell death inhuman diabetes. Circ Res 87:1123–1132.
Fu S, DaviesMJ, Stocker R, DeanRT. 1998. Evidence for roles ofradicals in protein oxidation in advanced human athero-sclerotic plaque. Biochem J 333:519–525.
Fu S, Wang H, Davies M, Dean R. 2000. Reactions ofhypochlorous acid with tyrosine and peptidyl-tyrosylresidues give dichlorinated and aldehydic products inaddition to 3-chlorotyrosine. J Biol Chem 275:10851–10858.
Fukuyama N, Takebayashi Y, Hida M, Ishida H, Ichimori K,Nakazawa H. 1997. Clinical evidence of peroxynitriteformation in chronic renal failure patients with septic shock.Free Radic Biol Med 22:771–774.
Gabbita SP, Askenov MY, Lovell MA, Markesbery WR. 1999.Decrease in peptide methionine sulfoxide reductase inAlzheimer’s disease brain. J Neurochem 73:1660–1666.
Gaut JP, Byun J, Tran HD, Lauber WM, Carroll JA, HotchkissRS, Belaaouaj A, Heinecke JW. 2002a. Myeloperoxidaseproduces nitrating oxidants in vivo. J Clin Invest 109:1311–1319.
Gaut JP, Byun J, Tran HD, Heinecke JW. 2002b. Artifact-freequantification of free 3-chlorotyrosine, 3-bromotyrosine,and 3-nitrotyrosine in human plasma by electron capture-negative chemical ionisation gas chromatography massspectrometry and liquid chromatography-electrosprayionization tandem mass spectrometry. Anal Biochem 300:252–259.
Georgiou G. 2002. How to flip the (redox) switch. Cell 111:607–610.
Ghezzi P, Bonetto V. 2003. Redox proteomics: Identification ofoxidatively modified proteins. Proteomics 3:1145–1153.
Ghezzi P, Romines B, Fratelli M, Eberini I, Gianazza E,Casagrande S, Laragione T, Mengozzi M, Herzenberg LA,
& DALLE-DONNE ET AL.
90

Herzenberg LA. 2002. Protein glutathionylation: Couplingand uncoupling of glutathione to protein thiol groups inlymphocytes under oxidative stress and HIV infection. MolImmunol 38:773–780.
Giasson BI, Duda JE, Murray IV, Chen Q, Souza JM, Hurtig HI,Ischiropoulos H, Trojanowski JQ, Lee VM. 2000. Oxidativedamage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science290:985–989.
Giasson BI, Ischiropoulos H, Lee VM, Trojanowski JQ. 2002.The relationship between oxidative/nitrative stress andpathological inclusions in Alzheimer’s and Parkinson’sdiseases. Free Radic Biol Med 32:1264–1275.
Giustarini D, Milzani A, Colombo R, Dalle-Donne I, Rossi R.2003a. Nitric oxide and S-nitrosothiols in human blood. ClinChim Acta 330:85–98.
Giustarini D, Dalle-Donne I, Colombo R, Milzani A, Rossi R.2003b.An improvedHPLCmesurement forGSHandGSSGin human blood. Free Radic Biol Med 35:1365–1372.
Gladstone IM, Jr., Levine RL. 1994. Oxidation of proteins inneonatal lungs. Pediatrics 93:764–768.
Glomb MA, Pfahler C. 2001. Amides are novel proteinmodifications formed by physiological sugars. J Biol Chem276:41638–41647.
Gole MD, Souza JM, Choi I, Hertkorn C, Malcolm S, Foust RF,Finkel B, Lanken PN, Ischiropoulos H. 2000. Plasmaproteins modified by tyrosine nitration in acute respiratorydistress syndrome. Am J Physiol Lung Cell Mol Physiol278:L961–967.
Good PF,Werner P, HsuA,OlanowCW, Perl DP. 1996. Evidencefor neuronal oxidative damage in Alzheimer’s disease. Am JPathol 149:21–28.
Good PF, Hsu A, Werner P, Perl DP, Olanow CW. 1998. Proteinnitration in Parkinson’s disease. J Neuropathol Exp Neurol57:338–342.
GowAJ, Chen Q, Hess DT, Day BJ, Ischiropoulos H, Stamler JS.2002. Basal and stimulated protein S-nitrosylation inmultiple cell types and tissues. J Biol Chem 277:9637–9640.
Grattagliano I, VendemialeG, Boscia F,Micelli-Ferrari T, CardiaL, Altomare E. 1998. Oxidative retinal products and oculardamages in diabetic patients. Free Radic Biol Med 25:369–372.
Grattagliano I, Giudetti AM,GrattaglianoV, Palmieri VO, GnoniGV, Lapadula G, Palasciano G, Vendemiale G. 2003.Structural and oxidative modifications of erythrocyte ghostsin patients with primary biliary cirrhosis: Relation with thedisease stage and effect of bile acid treatment. Eur J ClinInvest 33:868–874.
Greenacre SA, Ischiropoulos H. 2001. Tyrosine nitration:Localisation, quantification, consequences for proteinfunction and signal transduction. Free Radic Res 34:541–581.
Griffiths HR, Moller L, Bartosz G, Bast A, Bertoni-Freddari C,Collins A, CookeM, Coolen S, HaenenG,HobergAM, LoftS, Lunec J, Olinski R, Parry J, Pompella A, Poulsen H,
Verhagen H, Astley SB. 2002. Biomarkers. Mol AspectsMed 23:101–208.
Grune T, Merker K, Sandig G, Davies KJ. 2003. Selectivedegradation of oxidatively modified protein substrates bythe proteasome. Biochem Biophys Res Commun 305:709–718.
Grunert T, Pock K, Buchacher A, Allmaier G. 2003. Selectivesolid-phase isolation of methionine-containing peptides andsubsequent matrix-assisted laser desorption/ionisationmass spectrometric detection of methionine- and ofmethionine-sulfoxide-containing peptides. Rapid CommunMass Spectrom 17:1815–1824.
Guan Z, Yates NA, Bakhtiar R. 2003. Detection and character-ization of methionine oxidation in peptides by collision-induced dissociation and electron capture dissociation. JAmSoc Mass Spectrom 14:605–613.
GuntherMR, Tschirret-Guth RA,WitkowskaHE, FannYC, BarrDP, Ortiz DeMontellano PR, Mason RP. 1998. Site-specificspin trapping of tyrosine radicals in the oxidation ofmetmyoglobin by hydrogen peroxide. Biochem J 330:1293–1299.
Halliwell B, Gutteridge JMC. 1999. Free radicals inbiology and medicine. New York: Oxford University Press,Inc.
Hanson SR,ChenAA, Smith JB, LouMF. 1999. Thiolation of thegammaB-crystallins in intact bovine lens exposed tohydrogen peroxide. J Biol Chem 274:4735–4742.
Hanson SR, Hasan A, Smith DL, Smith JB. 2000. The major invivo modifications of the human water-insoluble lenscrystallins are disulfide bonds, deamidation, methionineoxidation and backbone cleavage. Exp Eye Res 71:195–207.
Hara I, Ueno T, Ozaki Si, Itoh S, Lee K, Ueyama N,Watanabe Y.2001. Oxidative modification of tryptophan 43 in the hemevicinity of the F43W/H64Lmyoglobin mutant. J Biol Chem276:36067–36070.
HarrisonD,GriendlingKK, LandmesserU,HornigB, Drexler H.2003. Role of oxidative stress in atherosclerosis. Am JCardiol 91:7A–11A.
Hazen SL, Heinecke JW. 1997. 3-Chlorotyrosine, a specificmarker of myeloperoxidase-catalyzed oxidation, is mark-edly elevated in lowdensity lipoprotein isolated fromhumanatherosclerotic intima. J Clin Invest 99:2075–2081.
Heinecke JW. 1999a. Mechanisms of oxidative damage bymyeloperoxidase in atherosclerosis and other inflammatorydisorders. J Lab Clin Med 133:321–325.
Heinecke JW. 1999b.Mass spectrometric quantification of aminoacid oxidation products in proteins: Insights into pathwaysthat promote LDL oxidation in the human artery wall.FASEB J 13:1113–1120.
Heinecke JW, Hsu FF, Crowley JR, Hazen SL, Leeuwenburgh C,Mueller DM, Rasmussen JE, Turk J. 1999c. Detectingoxidative modification of biomolecules with isotope dilu-tion mass spectrometry: Sensitive and quantitative assaysfor oxidized amino acids in proteins and tissues. MethodsEnzymol 300:124–144.
REDOX PROTEOMICS AND DISEASE &
91

Henderson JP, Byun J, Williams MV, McCormick ML, ParksWC, Ridnour LA, Heinecke JW. 2001. Bromination ofdeoxycytidine by eosinophil peroxidase: A mechanism formutagenesis by oxidative damage of nucleotide precursors.Proc Natl Acad Sci USA 98:1631–1636.
Hensley K, Hall N, Subramaniam R, Cole P, Harris M, AksenovM, Aksenova M, Gabbita SP, Wu JF, Carney JM, Lovell M,Markesbery WR, Butterfield DA. 1995. Brain regionalcorrespondence between Alzheimer’s disease histopathol-ogy and biomarkers of protein oxidation. J Neurochem 165:2146–2156.
Hensley K, Maidt ML, Yu Z, Sang H, Markesbery WR, FloydRA. 1998. Electrochemical analysis of protein nitrotyrosineand dityrosine in the Alzheimer brain indicates region-specific accumulation. J Neurosci 18:8126–8132.
Hensley K, Robinson KA, Gabbita SP, Salsman S, Floyd RA.2000. Reactive oxygen species, cell signaling, and cellinjury. Free Radic Biol Med 28:1456–1462.
Herce-Pagliai C, Kotecha S, Shuker DE. 1998. Analyticalmethods for 3-nitrotyrosine as a marker of exposure toreactive nitrogen species: A review. Nitric Oxide 2:324–336.
HetzC,Maundrell K, SotoC. 2003. Is loss of function of the prionprotein the cause of prion disorders? Trends Mol Med 9:237–243.
Hilliquin P, Borderie D, Hernvann A, Menkes CJ, Ekindjian OG.1997. Nitric oxide as S-nitrosoproteins in rheumatoidarthritis. Arthritis Rheum 40:1512–1517.
Himmelfarb J, McMonagle E, McMenamin E. 2000. Plasmaprotein thiol oxidation and carbonyl formation in chronicrenal failure. Kidney Int 58:2571–2578.
Himmelfarb J,McMonagleE. 2001.Albumin is themajor plasmaprotein target of oxidant stress in uremia. Kidney Int 60:358–363.
Himmelfarb J, McMenamin ME, Loseto G, Heinecke JW. 2001.Myeloperoxidase-catalyzed 3-chlorotyrosine formation indialysis patients. Free Radic Biol Med 31:1163–1169.
Hirsch J, Elssner A, Mazur G, Maier KL, Bittmann I, Behr J,Schwaiblmair M, Reichenspurner H, Furst H, Briegel J,Vogelmeier C. 1999. Bronchiolitis obliterans syndromeafter (heart-)lung transplantation. Impaired antiproteasedefense and increased oxidant activity. Am J Respir CritCare Med 160:1640–1646.
Hodges GR, Ingold KU. 1999. Cage escape of geminate radicalpairs can produce peroxynitrate from peroxynitrite under awide variety of experimental conditions. J Am Chem Soc121:10695–10701.
Hoeldtke RD, Bryner KD, McNeill DR, Hobbs GR, Riggs JE,Warehime SS, Christie I, Ganser G, Van Dyke K. 2002.Nitrosative stress, uric acid, and peripheral nerve function inearly type 1 diabetes. Diabetes 51:2817–2825.
Hogg N. 2002. The biochemistry and physiology of S-nitrosothiols. Annu Rev Pharmacol Toxicol 42:585–600.
Hollemeyer K, Heinzle E, Tholey A. 2002. Identification ofoxidized methionine residues in peptides containing twomethionine residues by derivatization and matrix-assisted
laser desorption/ionization mass spectrometry. Proteomics2:1524–1531.
Holloway JW, Jongepier H, Beghe B, Koppelman GH, HolgateST, Postma DS. 2003. The genetics of asthma. In: Chung F,Fabbri LM, editors. Asthma. Sheffield, UK: ERS JournalsLtd. pp 26–56.
Horiguchi T, Uryu K, Giasson BI, Ischiropoulos H, LightFoot R,Bellmann C, Richter-Landsberg C, Lee VM-Y, TrojanowskiJQ. 2003. Nitration of tau protein is linked to neurodegen-eration in tauopathies. Am J Pathol 163:1021–1031.
Huang KC, Yang CC, Lee KT, Chien CT. 2003. Reducedhemodialysis-induced oxidative stress in end-stage renaldisease patients by electrolyzed reduced water. Kidney Int64:704–714.
HunterGC,HendersonAM,WesterbandA,KobayashiH, SuzukiF, Yan ZQ, Sirsjo A, Putnam CW, Hansson GK. 1999. Thecontribution of inducible nitric oxide and cytomegalovirusto the stability of complex carotid plague. J Vasc Surg30:36–49.
Ichinose M, Sugiura H, Yamagata S, Koarai A, Shirato K. 2000.Increase in reactive nitrogen species production in chronicobstructive pulmonary disease airways. Am J Respir CritCare Med 162:701–706.
Inder T, Mocatta T, Darlow B, Spencer C, Volpe JJ, WinterbournCC. 2002. Elevated free radical products in the cerebrosp-inal fluid ofVLBWinfantswith cerebral whitematter injury.Pediatr Res 52:213–218.
Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, SnyderSH. 2001. Protein S-nitrosylation:A physiological signal forneuronal nitric oxide. Nat Cell Biol 3:193–197.
Kamisaki Y, Wada K, Bian K, Balabanli B, Davis K, Martin E,Behbod F, Lee YC, Murad F. 1998. An activity in rat tissuesthat modifies nitrotyrosine-containing proteins. Proc NatlAcad Sci USA 95:11584–11589.
Kaneko K, Nakamura A, Yoshida K, Kametani F, Higuchi K,Ikeda S-I. 2002. Glial fibrillary acidic protein is greatlymodified by oxidative stress in aceruloplasminemia brain.Free Radic Res 36:303–306.
Kelly FJ, Mudway I, Blomberg A, Frew A, Sandstrom T. 1999.Altered lung antioxidant status in patients withmild asthma.Lancet 354:482–483.
Keshavarzian A, Banan A, Farhadi A, Komanduri S, Mutlu E,Zhang Y, Fields JZ. 2003. Increases in free radicals andcytoskeletal protein oxidation and nitration in the colon ofpatients with inflammatory bowel disease. Gut 52:720–728.
Khan J, BrennandDM, BradleyN, Gao B, Bruckdorfer R, JacobsM, Brennan DM. 1998. 3-Nitrotyrosine in the proteins ofhuman plasma determined by an ELISAmethod. Biochem J330:795–801.
Kharitonov SA, Yates D, Robbins RA, Logan-Sinclair R,Shinebourne EA, Barnes PJ. 1994. Increased nitric oxidein exhaled air of asthmatic patients. Lancet 343:133–135.
Kinscherf R,DeignerHP,Usinger C, Pill J,WagnerM,KamencicH, Hou D, Schen M, Schmiedt W, Schrader M, Kovacs G,Kato K, Metz J. 1997. Induction of mitochondrialmanganese superoxide dismutase in macrophages by
& DALLE-DONNE ET AL.
92

oxidized LDL: Its relevance in atherosclerosis of humansand heritable hyperlipidemic rabbits. FASEB J 11:1317–1328.
Kinscherf R, Claus R, Wagner M, Gehrke C, Kamencic H, HouD, Nauen O, Schmiedt W, Kovacs G, Pill J, Metz J, DeignerHP. 1998. Apoptosis caused by oxidized LDL is manganesesuperoxide dismutase and p53dependent. FASEBJ 12:461–467.
Kinscherf R, Wagner M, Kamencic H, Bonaterra GA, Hou D,Schiele RA, Deigner HP, Metz J. 1999. Characterization ofapoptotic macrophages in atheromatous tissue of humansand heritable hyperlipidemic rabbits. Atherosclerosis144:33–39.
Klatt P, Lamas S. 2000. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosativestress. Eur J Biochem 267:4928–4944.
Klatt P, Molina ES, Lacoba MC, Padilla CA, Martinez-GlaisteoE, Barcena JA, Lamas S. 1999. Redox regulation of c-JunDNA binding by reversible S-glutathiolation. FASEB J13:1481–1490.
Kooy NW, Lewis SJ, Royall JA, Ye YZ, Kelly DR, Beckman JS.1997. Extensive tyrosine nitration in human myocardialinflammation: Evidence for the presence of peroxynitrite.Crit Care Med 25:812–819.
Korolainen MA, Goldsteins G, Alafuzoff I, Koistinaho J,Pirttila T. 2002. Proteomic analysis of protein oxidationin Alzheimer’s disease brain. Electrophoresis 23:3428–3433.
Kunsch C, Medford RM. 1999. Oxidative stress as a regulator ofgene expression in the vasculature. Circ Res 85:753–766.
Kurahashi T, Miyazaki A, Suwan S, Isobe M. 2001.Extensive investigations on oxidized amino acid residuesin H2O2-treated Cu,Zn-SOD protein with LC-ESI-Q-TOF-MS, MS/MS for the determination of the copper-bindingsite. J Am Chem Soc 123:9268–9278.
Lagerwerf FM, van deWeert M, HeermaW, Haverkamp J. 1996.Identification of oxidized methionine in peptides. RapidCommun Mass Spectrom 10:1905–1910.
LambNJ, Gutteridge JM, Baker C, Evans TW,QuinlanGJ. 1999.Oxidative damage to proteins of bronchoalveolar lavagefluid in patients with acute respiratory distress syndrome:Evidence for neutrophil-mediated hydroxylation, nitration,and chlorination. Crit Care Med 27:1738–1744.
Lang JD, McArdle PJ, O’Reilly PJ, Matalon S. 2002. Oxidant-antioxidant balance in acute lung injury. Chest 122:314S–320S.
Langen RC, Korn SH, Wouters EF. 2003. ROS in the local andsystemic pathogenesis of COPD. Free Radic Biol Med 35:226–235.
Lapolla A, Fedele D, Traldi P. 2000. The role of massspectrometry in the study of non-enzymatic protein glyca-tion in diabetes. Mass Spectrom Rev 19:279–304.
Lardinois OM, Medzihradszky KF, Ortiz de Montellano PR.1999. Spin trapping and protein cross-linking of thelactoperoxidase protein radical. J Biol Chem 274:35441–35448.
Leeuwenburgh C, Hardy MM, Hazen SL, Wagner P, Ohishi S,Steinbrecher UP, Heinecke JW. 1997a. Reactive nitrogenintermediates promote low density lipoprotein oxidation inhuman atherosclerosis. J Biol Chem 272:1433–1436.
Leeuwenburgh C, Rasmussen JE, Hsu FF, Mueller DM,Pennathur S, Heinecke JW. 1997b. Mass spectrometricquantification of markers for protein oxidation by tyrosylradical, copper, and hydroxyl radical in low densitylipoprotein isolated from human atherosclerotic plaques.J Biol Chem 272:3520–3526.
Levine RL, Moskovitz J, Stadtman ER. 2000a. Oxidation ofmethionine in proteins: Roles in antioxidant defense andcellular regulation. IUBMB Life 50:301–307.
Levine RL, Garland D, Oliver CN, Amici A, Climent I, Lenz A,Ahn B, Shaltiel S, Stadtman ER. 1990. Determination ofcarbonyl content in oxidatively modified proteins. MethodsEnzymol 186:464–478.
Levine RL, Mosoni L, Berlett BS, Stadtman ER. 1996.Methionine residues as endogenous antioxidants in proteins.Proc Natl Acad Sci USA 93:15036–15040.
Levine RL, Wehr N, Williams JA, Stadtman ER, Shacter E.2000b. Determination of carbonyl groups in oxidizedproteins. Methods Mol Biol 99:15–24.
LimPS,ChengYM,WeiYH. 2002. Increase in oxidative damageto lipids and proteins in skeletal muscle of uremic patients.Free Radic Res 36:295–301.
Lim A, Prokaeva T, McComb ME, Connors LH, Skinner M,Costello CE. 2003. Identification of S-sulfonation and S-thiolation of a novel transthyretin Phe33Cys variant from apatient diagnosed with familial transthyretin amyloidosis.Protein Sci 12:1775–1785.
Lind C, Gerdes R, Hamnell Y, Schuppe-Koistinen I, vonLowenhielm HB, Holmgren A, Cotgreave IA. 2002.Identification of S-glutathionylated cellular proteins duringoxidative stress and constitutive metabolism by affinitypurification and proteomic analysis. Arch BiochemBiophys406:229–240.
Lungarella G, Cavarra E,Martorana PA. 1999.Models of geneticemphysema: The C57 Bl/6J mice and their mutants: Tight-skin, pallid and beige. In: Stockley RA, editor. Molecularbiology of the lung. Vol. I: Emphysema and infection. Basel:Birkhauser Verlag, pp 19–36.
Lusini L, Tripodi SA, Rossi R, Giannerini F, Giustarini D, delVecchio MT, Barbanti G, Cintorino M, Tosi P, Di SimplicioP. 2001. Altered glutathione anti-oxidantmetabolism duringtumor progression in human renal-cell carcinoma. Int JCancer 91:55–59.
Lyras L, Cairns NJ, Jenner A, Jenner P, Halliwell B. 1997. Anassessment of oxidative damage to proteins, lipids, andDNAin brain from patients with Alzheimer’s disease. J Neuro-chem 68:2061–2069.
MacCoss MJ, McDonald WH, Saraf A, Sadygov R, Clark JM,Tasto JJ,GouldKL,WoltersD,WashburnM,WeissA,ClarkJI, Yates JR III. 2002. Shotgun identification of proteinmodifications from protein complexes and lens tissue. ProcNatl Acad Sci USA 99:7900–7905.
REDOX PROTEOMICS AND DISEASE &
93

MacMillan-Crow LA, Crow JP, Thompson JA. 1998. Peroxyni-trite-mediated inactivation of manganese superoxide dis-mutase involves nitration and oxidation of critical tyrosineresidues. Biochemistry 37:1613–1622.
MacMillan-Crow LA, Crow JP, Kerby JD, Beckman JS,Thompson JA. 1996. Nitration and inactivation of manga-nese superoxide dismutase in chronic rejection of humanrenal allografts. Proc Natl Acad Sci USA 93:11853–11858.
MacPherson H, Ralston S. 2000. Nitric oxide and bonedestruction. In: Winyard P, Blake D, Evans C, editors. Freeradicals and inflammation. Basel: Birkhauser.
MacPherson JC, Comhair SA, Erzurum SC, Klein DF, LipscombMF, Kavuru MS, Samoszuk MK, Hazen SL. 2001.Eosinophils are a major source of nitric oxide-derivedoxidants in severe asthma: Characterization of pathwaysavailable to eosinophils for generating reactive nitrogenspecies. J Immunol 166:5763–5772.
Maier KL, Leuschel L, Costabel U. 1992. Increased oxidizedmethionine residues in BAL fluid proteins in acute orchronic bronchitis. Eur Respir J 5:651–658.
Mallozzi C, Di Stasi MA, Minetti M. 2001. Peroxynitrite-dependent activation of src tyrosine kinases lyn and hck inerythrocytes is under mechanistically different pathways ofredox control. Free Radic Biol Med 30:1108–1117.
Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX,Kane LS, Gow AJ, Stamler JS. 1999. Fas-induced caspasedenitrosylation. Science 284:651–654.
Mantle D, FalkousG,Walker D. 1999. Quantification of proteaseactivities in synovial fluid from rheumatoid and osteoar-thritis cases: Comparison with antioxidant and free radicaldamage markers. Clin Chim Acta 284:45–58.
Mapp PI,KlockeR,WalshDA,Chana JK, StevensCR,GallagherPJ, Blake DR. 2001. Localization of 3-nitrotyrosine torheumatoid and normal synovium. Arthritis Rheum 44:1534–1539.
Markesbery WR. 1997. Oxidative stress hypothesis inAlzheimer’s disease. Free Radic Biol Med 23:134–147.
Marotta E, Lapolla A, Fedele D, Senesi A, Reitano R, Witt M,Seraglia R, Traldi P. 2003. Accurate mass measurements byFourier transform mass spectrometry in the study ofadvanced glycation end products/peptides. JMass Spectrom38:196–205.
Marvin LF, Delatour T, Tavazzi I, Fay LB, Cupp C, Guy PA.2003. Quantification of o,o0-dityrosine, o-nitrotyrosine, ando-tyrosine in cat urine samples by LC/ electrosprayionization-MS/MS using isotope dilution. Anal J Chem75:261–267.
Massy ZA, Borderie D, Nguyen-Khoa T, Drueke TB, EkindjianOG, Lacour B. 2003. Increased plasma S-nitrosothiol levelsin chronic haemodialysis patients. Nephrol Dial Transplant18:153–157.
Matsumoto A, Comatas KE, Liu L, Stamler JS. 2003. Screeningfor nitric oxide-dependent protein–protein interactions.Science 301:657–661.
MauriceMM, Nakamura H, Gringhuis S, Okamoto T, Yoshida S,Kullmann F, Lechner S, van-der-Voort EA, Leow A,
Versendaal J, Muller-Ladner U, Yodoi J, Tak PP, BreedveldFC, Verweij CL. 1999. Expression of the thioredoxin–thioredoxin-reductase system in inflamed joints of patientswith rheumatoid arthritis. Arthritis Rheum 42:2430–2439.
Maziak W, Loukides S, Culpitt S, Sullivan P, Kharitonov SA,Barnes PJ. 1998. Exhaled nitric oxide in chronic obstructivepulmonary disease. Am J Respir Crit Care Med 157:998–1002.
McKillop AM,Meade A, Flatt PR, O’Harte FP. 2003. Evaluationof the site(s) of glycation in human proinsulin by ion-trapLCQ electrospray ionization mass spectrometry. Regul Pept113:1–8.
Metz TO, Alderson NL, Thorpe SR, Baynes JW. 2003.Pyridoxamine, an inhibitor of advanced glycation andlipoxidation reactions: A novel therapy for treatment ofdiabetic complications. Arch BiochemBiophys 419:41–49.
Mikkelsen RB, Wardman P. 2003. Biological chemistry ofreactive oxygen and nitrogen and radiation-induced signaltransduction mechanisms. Oncogene 22:5734–5754.
Milhavet O, Lehmann S. 2002. Oxidative stress and the prionprotein in transmissible spongiform encephalopathies.Brain Res Rev 38:328–339.
Milhavet O, McMahon HE, Rachidi W, Nishida N, Katamine S,Mange A, Arlotto M, Casanova D, Riondel J, Favier A,Lehmann S. 2000. Prion infection impairs the cellularresponse to oxidative stress. Proc Natl Acad Sci USA97:13937–13942.
Milzani A, Rossi R, Di Simplicio P, Giustarini D, Colombo R,Dalle-Donne I. 2000. The oxidation produced by hydrogenperoxide on Ca-ATP-G-actin. Protein Sci 9:1774–1782.
Minetti M, Mallozzi C, Di Stasi AM. 2002. Peroxynitriteactivates kinases of the src family and upregulates tyrosinephosphorylation signaling. Free Radic Biol Med 33:744–754.
Minetti M, Pietraforte D, Carbone V, Salzano AM, Scorza G,Marino G. 2000. Scavenging of peroxynitrite by oxyhemo-globin and identification of modified globin residues.Biochemistry 39:6689–6697.
Miyata T, Inagi R, Wada Y, Ueda Y, Iida Y, Takahashi M,Taniguchi N, Maeda K. 1994. Glycation of human beta 2-microglobulin in patients with hemodialysis-associatedamyloidosis: Identification of the glycated sites. Biochem-istry 33:12215–12221.
Miyata T, Inagi R, Asahi K, Yamada Y, Horie K, Sakai H, UchidaK, Kurokawa K. 1998. Generation of protein carbonyls byglycoxidation and lipoxidation reactions with autoxidationproducts of ascorbic acid and polyunsaturated fatty acids.FEBS Lett 437:24–28.
Mollace V, Nottet HSLM, Clayette P, Turco MC, Muscoli C,Salvemini D, Perno CF. 2001. Oxidative stress andneuroAIDS: Triggers, modulators and novel antioxidants.Trends Neurosci 24:411–416.
Moriel P, Pereira IR, Bertolami MC, Abdalla DS. 2001. Isceruloplasmin an important catalyst for S-nitrosothiolgeneration in hypercholesterolemia? Free Radic Biol Med30:318–326.
& DALLE-DONNE ET AL.
94

Morrison D, Rahman I, Lannan S, MacNee W. 1999. Epithelialpermeability, inflammation, and oxidant stress in the airspaces of smokers. Am J Respir Crit Care Med 159:473–479.
Moskovitz J, Flescher E, Berlett BS, Azare J, Poston JM,Stadtman ER. 1998. Overexpression of peptide-methioninesulfoxide reductase in Saccharomyces cerevisiae and humanT cells provides them with high resistance to oxidativestress. Proc Natl Acad Sci USA 95:14071–14075.
Moskovitz J, Bar-Noy S, Williams WM, Requena J, Berlett BS,StadtmanER. 2001.Methionine sulfoxide reductase (MsrA)is a regulator of antioxidant defense and lifespan inmammals. Proc Natl Acad Sci USA 98:12920–12925.
Moskovitz J, Singh VK, Requena J, Wilkinson BJ, Jayaswal RK,Stadtman ER. 2002. Purification and characterization ofmethionine sulfoxide reductases from mouse and Staphylo-coccus aureus and their substrate stereospecificity. BiochemBiophys Res Commun 290:62–65.
Murray J, Taylor SW, Zhang B, Ghosh SS, Capaldi RA. 2003.Oxidative damage to mitochondrial complex I due toperoxynitrite: Identification of reactive tyrosines by massspectrometry. J Biol Chem 278:37223–37230.
Myatt L, Rosenfield RB, Eis AL, Brockman DE, Greer I, Lyall F.1996. Nitrotyrosine residues in placenta. Evidence ofperoxynitrite formation and action. Hypertension 28:488–493.
Nadeem A, Chhabra SK, Masood A, Raj HG. 2003. Increasedoxidative stress and altered levels of antioxidants in asthma.J Allergy Clin Immunol 111:72–78.
Naito C, Niwa T. 2000. Analysis of glutathionyl hemoglobinlevels in diabetic patients by electrospray ionizationliquid chromatography-mass spectrometry: Effect of vita-min E administration. J Chromatogr B Biomed Sci Appl746:91–94.
Nakamura H, Masutani H, Yodoi J. 2002. Redox imbalanceand its control in HIV infection. Antiox Redox Signaling4:455–464.
Naslund J, Schierhorn A, Hellman U, Lannfelt L, Roses AD,Tjernberg LO, Silberring J, Gandy SE, Winblad B, Green-gard P, Nordstedt C, Terenius L. 1994. Relative abundanceof Alzheimer A beta amyloid peptide variants in Alzheimerdisease and normal aging. Proc Natl Acad Sci USA 91:8378–8382.
Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T,Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP,Giardino I, Brownlee M. 2000. Normalizing mitochondrialsuperoxide production blocks three pathways of hypergly-caemic damage. Nature 404:787–790.
Niwa T, Naito C, Mawjood AHM, Imai K. 2000. Increasedglutathionyl hemoglobin in diabetes mellitus and hyperli-pidemia demonstrated by liquid chromatography/electro-spray ionization-mass spectrometry. Clin Chem 46:82–88.
Okamoto T, Akaike T, Sawa T, Miyamoto Y, van der Vliet A,Maeda H. 2001. Activation of matrix metalloproteinases byperoxynitrite-induced protein S-glutathiolation via disulfideS-oxide formation. J Biol Chem 276:29596–29602.
Page NM, Woods RJ, Gardiner SM, Lomthaisong K, GladwellRT, Butlin DJ, Manyonda IT, Lowry PJ. 2000. Excessiveplacental secretion of neurokininBduring the third trimestercauses pre-eclampsia. Nature 405:797–800.
Pandey A, Mann M. 2000. Proteomics to study genes andgenomes. Nature 405:837–846.
Panek J, Vohradsky J. 1999. Point pattern matching in theanalysis of twodimensional gel electropherograms. Electro-phoresis 20:3483–3491.
Paron I, D’Elia A, D’Ambrosio C, Scaloni A, D’Aurizio F,Prescott A, Damante G, Tell G. 2003. A proteomic approachto identify early molecular targets of oxidative stress inhuman epithelial lens cells. Biochem J 2003 Dec 16 [Epubahead of print; DOI 10.1042/BJ2003/190].
Pasinelli P, Houseweart MK, Brown RH, Jr., Cleveland DW.2000. Caspase-1 and -3 are sequentially activated in motorneuron death in Cu,Zn superoxide dismutase-mediatedfamilial amyotrophic lateral sclerosis. Proc Natl Acad SciUSA 97:13901–13906.
PastoreA,TozziG,GaetaLM,GiannottiA,Bertini E, FedericiG,Digilio MC, Piemonte F. 2003a. Glutathione metabolismand antioxidant enzymes in children with Down’s syn-drome. J Pediatr 142:583–585.
Pastore A, Tozzi G, Gaeta LM, Bertini E, Serafini V, DiCesare S, Bonetto V, Casoni F, Carrozzo R, Federici G,Piemonte F. 2003b. Actin glutathionylation increases infibroblasts of patients with Friedreich’s ataxia: A potentialrole in the pathogenesis of the disease. J Biol Chem 43:42588–42595.
Pennathur S, Jackson-Lewis V, Przedborski S, Heinecke JW.1999. Mass spectrometric quantification of 3-nitrotyrosine,ortho-tyrosine, and o,o0-dityrosine in brain tissue of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice, amodel of oxidative stress in Parkinson’s disease. J BiolChem 274:34621–34628.
Peterson KP, Pavlovich JG, Goldstein D, Little R, England J,Peterson CM. 1998. What is hemoglobin A1c? An analysisof glycated hemoglobins by electrospray ionization massspectrometry. Clin Chem 44:1951–1958.
Petersson AS, Steen H, Kalume DE, Caidahl K, Roepstorff P.2001. Investigation of tyrosine nitration in proteins by massspectrometry. J Mass Spectrom 36:616–625.
Piemonte F, Pastore A, Tozzi G, Tagliacozzi D, Santarelli FM,Carrozzo R, Casali C, Damiano M, Federici G, Bestini E.2001. Glutathione in blood of patients with Friedreich’sataxia. Eur J Clin Invest 31:1007–1011.
Pietraforte D, Salzano AM, Scorza G, Marino G, Minetti M.2002. Mechanism of peroxynitrite interaction with ferrichemoglobin and identification of nitrated tyrosine residues.CO(2) inhibits heme-catalyzed scavenging and isomeriza-tion. Biochemistry 40:15300–15309.
Pignatelli B, Li CQ, Boffetta P, Chen Q, Ahrens W, Nyberg F,Mukeria A, Bruske-Hohlfeld I, Fortes C, Constantinescu V,Ischiropoulos H, Ohshima H. 2001. Nitrated and oxidizedplasma proteins in smokers and lung cancer patients. CancerRes 61:778–784.
REDOX PROTEOMICS AND DISEASE &
95

Pinamonti S, Muzzoli M, Chicca MC, Papi A, Ravenna F, FabbriLM, Ciaccia A. 1996. Xanthine oxidase activity inbronchoalveolar lavage fluid from patients with chronicobstructive pulmonary disease. Free Radic Biol Med 21:147–155.
Podrez EA, Abu-Soud HM, Hazen SL. 2000. Myeloperoxidase-generated oxidants and atherosclerosis. Free Radic BiolMed 28:1717–1725.
Poggioli S, Bakala H, Friguet B. 2002. Age-related increase ofprotein glycation in peripheral blood lymphocytes isrestricted to preferential target proteins. Exp Gerontol37:1207–1215.
Powell SA, Gurzenda EM, Wahezi SE. 2001. Actin is oxidizedduringmyocardial ischemia. FreeRadicBiolMed 30:1171–1176.
Pratico D, Basili S, Vieri M, Cordova C, Violi F, Fitzgerald GA.1998a. Chronic obstructive pulmonary disease is associatedwith an increase in urinary levels of isoprostane F2alpha-III,an index of oxidant stress. Am J Respir Crit Care Med158:1709–1714.
Pratico D, Lee VM-Y, Trojanowski JQ, Rokach J, Fitzgerald GA.1998b. Increased F2-isoprostanes in Alzheimer’s disease:Evidence for enhanced lipid peroxidation in vivo. FASEB J12:1777–1783.
Previero A, Coletti-Previero MA, Jolles P. 1967. Localization ofnon-essential tryptophan residues for the biological activityof lysozyme. J Mol Biol 24:261–268.
Prusiner SB, Scott MR, Dearmond SJ, Cohen FE. 1998. Prionprotein biology. Cell 93:337–348.
Pryor WA. 2001. Bio-assays for oxidative stress status (BOSS).Amsterdam: Elsevier Science B.V, 286 p.
Rabek JP, Boylston WH III, Papaconstantinou J. 2003.Carbonylation of ER chaperone proteins in aged mouseliver. Biochem Biophys Res Commun 305:566–572.
Rabilloud T, Heller M, Gasnier F, Luche S, Rey C, Aebersold R,Benahmed M, Louisot P, Lunardi J. 2002. Proteomicsanalysis of cellular response to oxidative stress. Evidencefor in vivo overoxidation of peroxiredoxins at their activesite. J Biol Chem 277:19396–19401.
Rahman I, Skwarska E, MacNee W. 1997. Attenuation ofoxidant/antioxidant imbalance during treatment of exacer-bations of chronic obstructive pulmonary disease. Thorax52:565–568.
Rahman I, MacNee W. 1996. Role of oxidants/antioxidants insmoking-induced lung diseases. Free Radic Biol Med21:669–681.
Rahman I, MacNee W. 1998. Role of transcription factors ininflammatory lung diseases. Thorax 53:601–612.
Rahman I, Morrison D, Donaldson K, MacNee W. 1996.Systemic oxidative stress in asthma, COPD, and smokers.Am J Respir Crit Care Med 154:1055–1060.
Rahman I, van Schadewijk AA, Crowther AJ, Hiemstra PS, StolkJ, MacNee W, De Boer WI. 2002. 4-Hydroxy-2-nonenal, aspecific lipid peroxidation product, is elevated in lungs ofpatients with chronic obstructive pulmonary disease. Am JRespir Crit Care Med 166:490–495.
Ramsay PL, Demayo FJ, Hegemier SE,WeardenME, Smith CV,Welty SE. 2001. Clara cell secretory protein oxidation andexpression in premature infants who develop bronchopul-monary dysplasia. Am J Respir Crit Care Med 164:155–161.
Renke J, Popadiuk S, Korzon M, Bugajczyk B, Wozniak M.2000. Protein carbonyl groups content as a useful clinicalmarker of antioxidant barrier impairment in plasma ofchildrenwith juvenile chronic arthritis. FreeRadic BiolMed29:101–104.
Requena JR, FuMX, AhmedMU, Jenkins AJ, Lyons TJ, BaynesJW, Thorpe SR. 1997. Quantification of malondialdehydeand 4-hydroxynonenal adducts to lysine residues in nativeand oxidized human low-density lipoprotein. Biochem J322:317–325.
Requena JR, Chao CC, Levine RL, Stadtman ER. 2001a.Glutamic and aminoadipic semialdehydes are the maincarbonyl products of metal-catalyzed oxidation of proteins.Proc Natl Acad Sci USA 98:69–74.
Requena JR, Groth D, Legname G, Stadtman ER, Prusiner SB,Levine RL. 2001b. Copper-catalyzed oxidation of therecombinant SHa(29-231) prion protein. Proc Natl AcadSci USA 98:7170–7175.
Rijcken B, Britton J. 1998. Epidemiology of chronic obstructivepulmonary disease. In: Postma DS, Siafakas NM, editors.Management of chronic obstructive pulmonary disease.Sheffield, UK: ERS Journals Ltd. pp 41–73.
Roberts JM,Cooper DW. 2001. Pathogenesis and genetics of pre-eclampsia. Lancet 357:53–56.
Roberts JM, Hubel CA. 1999. Is oxidative stress the link in thetwo-stage model of pre-eclampsia? Lancet 354:788–789.
Roberts JM, Redman CW. 1993. Pre-eclampsia: More thanpregnancy-induced hypertension. Lancet 341:1447–1451.
Rosen P, Nawroth PP, King G, Moller W, Tritschler H-J, PackerL. 2001. The role of oxidative stress in the onset andprogression of diabetes and its complications: A summary ofa Congress Series sponsored by UNESCO-MCBN, theAmerican Diabetes Association and the German DiabetesSociety. Diabetes Metab Res Rev 17:189–212.
Ruan H, Tang XD, Chen ML, Joiner ML, Sun G, Brot N,Weissbach H, Heinemann SH, Iverson L, Wu CF, Hoshi T,Chen ML, Joiner MA, Heinemann SH. 2002. High-qualitylife extension by the enzyme peptide methionine sulfoxidereductase. Proc Natl Acad Sci USA 99:2748–2753.
Saetta M, Turato G, Maestrelli P, Mapp CE, Fabbri LM. 2001.Cellular and structural bases of chronic obstructivepulmonary disease. Am J Respir Crit Care Med 163:1304–1309.
Sandhu JK, Robertson S, Birnboim HC, Goldstein R. 2003.Distribution of protein nitrotyrosine in synovial tissues ofpatients with rheumatoid arthritis and osteoarthritis. JRheumatol 30:1173–1181.
Sano M, Ernesto C, Thomas RG, Klauber MR, Schafer K,Grundman M, Woodbury P, Growdon J, Cotman CW,Pfeiffer E, Schneider LS, Thal LJ. 1997. A controlled trial ofselegiline, alpha-tocopherol, or both as treatment for
& DALLE-DONNE ET AL.
96

Alzheimer’s disease. The Alzheimer’s disease cooperativestudy. N Engl J Med 336:1216–1222.
Sanz-Cameno P, Medina J, Garcia-Buey L, Garcia-Sanchez A,Borque MJ, Martin-Vilchez S, Gamallo C, Jones EA,Moreno-Otero R. 2002. Enhanced intrahepatic induciblenitric oxide synthase expression and nitrotyrosine accumu-lation in primary biliary cirrhosis and autoimmune hepatitis.J Hepatol 37:723–729.
Saraswathi M, Nakanishi T, Shimizu A. 1999. Relativequantification of glycated Cu–Zn superoxide dismutase inerythrocytes by electrospray ionization mass spectrometry.Biochim Biophys Acta 1426:483–490.
Sarver A, Scheffler NK, ShetlarMD,Gibson BW. 2001. Analysisof peptides and proteins containing nitrotyrosine by matrix-assisted laser desorption/ionization mass spectrometry. JAm Soc Mass Spectrom 12:439–448.
Scaloni A, Perillo V, Franco P, Fedele E, Froio R, Ferrara L,Bergamo P. 2002. Characterization of heat-induced lacto-sylation products in caseins by immunoenzymatic and massspectrometric methodologies. Biochim Biophys Acta 1598:30–39.
Schafer FQ,BuettnerGR. 2001. Redox environment of the cell asviewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med 30:1191–1212.
Schett G, Tohidast-Akrad M, Steiner G, Smolen J. 2001. Thestressed synovium. Arthritis Res 3:80–86.
Schey KL, Finley EL. 2000. Identification of peptide oxidationby tandem mass spectrometry. Acc Chem Res 33:299–306.
Schock BC, Sweet DG, Halliday HL, Young IS, Ennis M. 2001.Oxidative stress in lavage fluid of preterm infants at risk ofchronic lung disease. Am J Physiol Lung Cell Mol Physiol281:L1386–L1391.
Schoneich C, Williams TD. 2002. Cu(II)-catalyzed oxidation ofbeta-amyloid peptide targets His13 and His14 over His6:Detection of 2-oxo-histidine by HPLC-MS/MS. Chem ResToxicol 15:717–722.
Schwaller M, Wilkinson B, Gilbert HF. 2003. Reduction–reoxidation cycles contribute to catalysis of disulfideisomerization by protein-disulfide isomerase. J Biol Chem278:7154–7159.
Shacter E, Williams JA, Lim M, Levine RL. 1994. Differentialsusceptibility of plasma proteins to oxidative modification.Examination byWestern blot immunoassay. FreeRadic BiolMed 17:429–437.
ShigenagaMK, LeeHH,Blount BC, Christen S, Shigeno ET,YipH, Ames BN. 1997. Inflammation and NO(X)-inducednitration: Assay for 3-nitrotyrosine by HPLC with electro-chemical detection. Proc Natl Acad Sci USA 94:3211–3216.
Shishehbor MH, Aviles RJ, Brennan ML, Fu X, Goormastic M,Pearce GL, Gokce N, Keaney JF, Penn MS, Sprecher DL,Vita JA, Hazen SL. 2003. Association of nitrotyrosine levelswith cardiovascular disease and modulation by statintherapy. JAMA 289:1675–1680.
Singer II, Kawka DW, Scott S, Weidner JR, Mumford RA, RiehlTE, Stenson WF. 1996. Expression of inducible nitric oxide
synthase and nitrotyrosine in colonic epithelium in inflam-matory bowel disease. Gastroenterology 111:871–885.
Sittipunt C, Steinberg KP, Ruzinski JT, Myles C, Zhu S,Goodman RB, Hudson LD, Matalon S, Martin TR. 2001.Nitric oxide and nitrotyrosine in the lungs of patients withacute respiratory distress syndrome. Am J Respir Crit CareMed 163:503–510.
Smith MA, Perry G, Richey PL, Sayre LM, Anderson UE, BealMF, Kowall N. 1996. Oxidative damage in Alzheimer’s.Nature 382:120–121.
Smith MA, Richey Harris PL, Sayre LM, Beckman G, Perry GJ.1997. Widespread peroxynitrite-mediated damage in Alz-heimer’s disease. Neuroscience 17:2653–2657.
Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G.2000. Oxidative stress in Alzheimer’s disease. BiochimBiophys Acta 1502:139–144.
Smith MA, Casadesus G, Joseph JA, Perry G. 2002. Amyloid-band t serve antioxidant functions in the aging andAlzheimerbrain. Free Radic Biol Med 33:1194–1199.
Sochaski MA, Jenkins AJ, Lyons TJ, Thorpe SR, Baynes JW.2001. Isotope dilution gas chromatography/mass spectro-metrymethod for the determination of methionine sulfoxidein protein. Anal Chem 73:4662–4667.
Song EJ, Kim YS, Chung JY, Kim E, Chae SK, Lee KJ. 2000.Oxidative modification of nucleoside diphosphate kinaseand its identification by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Biochemistry39:10090–10097.
Stadtman ER, Berlett BS. 1998. Reactive oxygen-mediatedprotein oxidation in aging and disease. Drug Metab Rev30:225–243.
Stadtman ER, Moskovitz J, Berlett BS, Levine RL. 2002. Cyclicoxidation and reduction of protein methionine residues is animportant antioxidant mechanism. Mol Cell Biochem 234-235:3–9.
Stamler JS, Lamas S, Fang FC. 2001. Nitrosylation. Theprototypic redox-based signaling mechanism. Cell 106:675–683.
Su JH, Deng GM, Cotman CW. 1997. Neuronal DNA damageprecedes tangle formation and is associated with upregula-tion of nitrotyrosine in Alzheimer’s brain. Brain Res 774:193–199.
Sullivan DM, Wehr NB, Fergusson MM, Levine RL, Finkel T.2000. Identification of oxidant-sensitive proteins: TNF-ainduces protein glutathiolation. Biochemistry 39:11121–11128.
Suzuki D, Miyata T, Saotome N, Horie K, Inagi R,Yasuda Y, Uchida K, Izuhara Y, Yagame M, Sakai H,Kurokawa K. 1999. Immunohistochemical evidence for anincreased oxidative stress and carbonyl modification ofproteins in diabetic glomerular lesions. J Am Soc Nephrol10:822–832.
Synek M, Beasley R, Frew AJ, Goulding D, Holloway L, LampeFC,RocheWR,Holgate ST. 1996. Cellular infiltration of theairways in asthma of varying severity. Am JRespir Crit CareMed 154:224–230.
REDOX PROTEOMICS AND DISEASE &
97

Taggart C, Cervantes-Laurean D, Kim G, McElvaney NG, WehrN,Moss J, Levine RL. 2000. Oxidation of either methionine351 or methionine 358 in alpha 1-antitrypsin causes loss ofanti-neutrophil elastase activity. J Biol Chem 275:27258–27265.
Takahashi M, Lu YB, Myint T, Fujii J, Wada Y, Taniguchi N.1995. In vivo glycation of aldehyde reductase, a major 3-deoxyglucosone reducing enzyme: Identification of glyca-tion sites. Biochemistry 34:1433–1438.
Takayama F, Tsutsui S, Horie M, Shimokata K, Niwa T. 2001.Glutathionyl hemoglobin in uremic patients undergoinghemodialysis and continuous ambulatory peritoneal dialy-sis. Kidney Int Suppl 78:S155–158.
Tarpey M, Fridovich I. 2001. Methods of detection of vascularreactive species. Nitric oxide, superoxide, hydrogen per-oxide, and peroxynitrite. Circ Res 89:224–236.
Taylor SW, Fahy E, Murray J, Capaldi RA, Ghosh SS. 2003.Oxidative post-translational modification of tryptophanresidues in cardiac mitochondrial proteins. J Biol Chem278:19587–19590.
Telci A, Cakatay U, Kayali R, Erdogan C, Orhan Y, Sivas A,Akcay T. 2000a.Oxidative protein damage in plasma of type2 diabetic patients. Horm Metab Res 32:40–43.
Telci A, Cakatay U, Salman S, Satman I, Sivas A. 2000b.Oxidative protein damage in early stage Type 1 diabeticpatients. Diabetes Res Clin Pract 50:213–223.
Tell G, Scaloni A, Pellizzari L, Formisano S, Pucillo C, DamanteG. 1998. Redox potential controls the structure and DNAbinding activity of the paired domain. J Biol Chem 273:25062–25072.
Thornalley PJ. 2002. Glycation in diabetic neuropathy: Char-acteristics, consequences, causes, and therapeutic options.Int Rev Neurobiol 50:37–57.
Tohgi H, Abe T, Yamazaki K, Murata T, Ishizaki E, Isobe C.1999. Remarkable increase in cerebrospinal fluid 3-nitrotyrosine in patients with sporadic amyotrophic lateralsclerosis. Ann Neurol 46:129–131.
Torre D, Pugliese A, Speranza F. 2002. Role of nitric oxide inHIV-1 infection: Friend or foe? Lancet Infect Dis 2:273–280.
Tsikas D, Schwedhelm E, Stutzer FK, Gutzki FM, Rode I, MehlsC, Frolich JC. 2003. Accurate quantification of basal plasmalevels of 3-nitrotyrosine and 3-nitrotyrosinoalbumin by gaschromatography-tandem mass spectrometry. J ChromatogrB Analyt Technol Biomed Life Sci 784:77–90.
Turchan J, Pocernich CB, Gairola C, Chauhan A, Schifitto G,Butterfield DA, Buch S, Narayan O, Sinai A, Geiger J,Berger JR, Elford H, Nath A. 2003. Oxidative stress in HIVdemented patients and protection ex vivo with novelantioxidants. Neurology 60:307–314.
Tyurin VA, Liu SX, Tyurina YY, Sussman NB, Hubel CA,Roberts JM, Taylor RN, Kagan VE. 2001. Elevated levels ofS-nitrosoalbumin in preeclampsia plasma. Circ Res 88:1210–1215.
Upston JM, Niu X, Brown AJ, Mashima R, Wang H,Senthilmohan R, Kettle AJ, Dean RT, Stocker R. 2002.
Disease stage-dependent accumulation of lipid and proteinoxidation products in human atherosclerosis. Am J Pathol160:701–710.
Valentine JS. 2002.Do oxidativelymodified proteins causeALS?Free Radic Biol Med 33:1314–1320.
Van der Vliet A, Eiserich JP, Shigenaga MK, Cross CE. 1999.Reactive nitrogen species and tyrosine nitration in therespiratory tract: Epiphenomena or a pathobiologicmechan-ism of disease? Am J Respir Crit Care Med 160:1–9.
Van der Vliet A, Nguyen MN, Shigenaga MK, Eiserich JP,Marelich GP, Cross CE. 2000.Myeloperoxidase and proteinoxidation in cystic fibrosis. Am J Physiol Lung Cell MolPhysiol 279:L537–546.
van Montfort RL, Congreve M, Tisi D, Carr R, Jhoti H. 2003.Oxidation state of the active-site cysteine in protein tyrosinephosphatase 1B. Nature 423:773–777.
Varadarajan S, Yatin SM, Aksenova M, Butterfield DA. 2000.Alzheimer’s amyloid b peptide-associated free radicaloxidative stress and neurotoxicity. J Struct Biol 130:184–208.
Varsila E, Pesonen E, Andersson S. 1995. Early protein oxidationin the neonatal lung is related to development of chroniclung disease. Acta Paediatr 84:1296–1299.
Veale DJ,Maple C. 1996. Cell adhesionmolecules in rheumatoidarthritis. Drugs Aging 9:87–92.
Vilardo PG, Scaloni A, Amodeo P, Barsotti C, Cecconi I,Cappiello M, Lopez Mendez B, Rullo R, Dal Monte M, DelCorso A, Mura U. 2001. Thiol/disulfide interconversion inbovine lens aldose reductase induced by intermediates ofglutathione turnover. Biochemistry 40:11985–11994.
Viner RI, Williams TD, Schoneich C. 1999. Peroxynitritemodification of protein thiols: Oxidation, nitrosylation,and S-glutathiolation of functionally important cysteineresidue(s) in the sarcoplasmic reticulum Ca-ATPase.Biochemistry 38:12408–12415.
VogtW. 1995.Oxidation ofmethionyl residues in proteins: Tools,targets, and reversal. Free Radic Biol Med 18:93–106.
Weissbach H, Etienne F, Hoshi T, Heinemann SH, Lowther WT,Matthews B, St John G, Nathan C, Brot N. 2002. Peptidemethionine sulfoxide reductase: Structure, mechanism ofaction, and biological function. Arch Biochem Biophys397:172–178.
Wildhirt SM,WeisM, Schulze C, Conrad N, Pehlivanli S, RiederG, Enders G, von Scheidt W, Reichart B. 2001. Expressionof endomyocardial nitric oxide synthase and coronaryendothelial function in human cardiac allografts. Circula-tion 104:1336–1343.
Winterbourn CC, Vissers MC, Kettle AJ. 2000a. Myeloperox-idase. Curr Opin Hematol 7:53–58.
Winterbourn CC, Kettle AJ. 2000. Biomarkers of myeloperox-idase-derived hypochlorous acid. Free Radic Biol Med 29:403–409.
Winterbourn CC, Buss IH, Chan TP, Plank LD, Clark MA,Windsor JA. 2000. Protein carbonyl measurements showevidence of early oxidative stress in critically ill patients.Crit Care Med 28:143–149.
& DALLE-DONNE ET AL.
98

Winterbourn CC, BonhamMJ, Buss H, Abu-Zidan FM,WindsorJA. 2003. Elevated protein carbonyls as plasma markers ofoxidative stress in acute pancreatitis. Pancreatology 3:375–382.
Woo HA, Chae HZ, Hwang SC, Yang KS, Kang SW, Kim K,Rhee SG. 2003. Reversing the inactivation of peroxiredox-ins caused by cysteine sulfinic acid formation. Science300:653–656.
WoodZA,Schroder E,RobinHarris J, Poole LB. 2003. Structure,mechanism and regulation of peroxiredoxins. TrendsBiochem Sci 28:32–40.
Woods AA, Linton SM, Davies MJ. 2003. Detection of HOCl-mediated protein oxidation products in the extracellularmatrix of human atherosclerotic plaques. Biochem J 370:729–735.
Wu SM, Pizzo SV. 2001. Alpha(2)-Macroglobulin fromrheumatoid arthritis synovial fluid: Functional analysisdefines a role for oxidation in inflammation. Arch BiochemBiophys 391:119–126.
Wu W, Chen BK, d’Avignon A, Hazen SL. 1999. 3-Bromotyr-osine and 3,5-dibromotyrosine aremajor products of proteinoxidation by eosinophil peroxidase: Potential markers foreosinophil-dependent tissue injury in vivo. Biochemistry38:3538–3548.
Wu W, Samoszuk MK, Comhair SAA, Thomassen MJ, FarverCF, Dweik RA, Kavuru MS, Erzurum SC, Hazen SL. 2000.Eosinophils generate brominating oxidants in allergen-induced asthma. J Clin Invest 105:1455–1463.
Wu CT, Eiserich JP, Ansari AA, Coppel RL, Balasubramanian S,Bowlus CL, Gershwin ME, Van De Water J. 2003.Myeloperoxidase-positive inflammatory cells participatein bile duct damage in primary biliary cirrhosis throughnitric oxide-mediated reactions. Hepatology 38:1018–1025.
Yan LJ, Sohal RS. 1998. Gel electrophoretic quantitation ofprotein carbonyls derivatized with tritiated sodium borohy-dride. Anal Biochem 265:176–182.
Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS,Pinsky D, Stern D. 1994. Enhanced cellular oxidative stressby the interaction of advanced glycation end-products withtheir receptors/binding protein. J Biol Chem 269:9889–9897.
Yang KS, Kang SW, Woo HA, Hwang SC, Chae HZ, Kim K,Rhee SG. 2002. Inactivation of human peroxiredoxin Iduring catalysis as the result of the oxidation of the catalyticsite cysteine to cysteine-sulfinic acid. J Biol Chem 277:38029–38036.
YeYZ, StrongM,Huang ZQ, Beckman JS. 1996. Antibodies thatrecognize nitrotyrosine. Methods Enzymol 269:201–209.
Yi D, Ingelse BA, Duncan MW, Smythe GA. 2000. Quantifica-tion of 3-nitrotyrosine in biological tissues and fluids:Generating valid results by eliminating artifactual forma-tion. J Am Soc Mass Spectrom 11:578–586.
YorekMA. 2003. The role of oxidative stress in diabetic vascularand neural disease. Free Radic Res 37:471–480.
Zech B, Wilm M, van Eldik R, Brune B. 1999. Mass spectro-metric analysis of nitric oxide-modified caspase-3. J BiolChem 274:20931–20936.
Zelko IN, Mariani TJ, Folz RJ. 2002. Superoxide dismutasemultigene family:A comparison of theCu,Zn-SOD (SOD1),Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures,evolution, and expression. Free Radic Biol Med 33:337–349.
Zhang H, He S, Mauk AG. 2002. Radical formation at Tyr39 andTyr153 following reaction of yeast cytochrome c peroxidasewith hydrogen peroxide. Biochemistry 41:13507–13513.
Zheng M, Aslund F, Storz G. 1998. Activation of the OxyRtranscription factor by reversible disulfide bond formation.Science 279:1718–1721.
ZhouY,DamskyCH, Fisher SJ. 1997. Preeclampsia is associatedwith failure of human cytotrophoblasts to mimic a vascularadhesion phenotype. One cause of defective endovascularinvasion in this sindrome? J Clin Invest 99:2152–2164.
Zhu S, Basiouny KF, Crow JP, Matalon S. 2000. Carbon dioxideenhances nitration of surfactant protein A by activatedalveolar macrophages. Am J Physiol 278:L1025–L1031.
Zhu S, Ware LB, Geiser T, Matthay MA, Matalon S. 2001.Increased levels of nitrate and surfactant protein A nitrationin the pulmonary edema fluid of patients with acute lunginjury. Am J Respir Crit Care Med 163:166–172.
Ziouzenkova O, Asatryan L, Akmal M, Tetta C, Wratten ML,Loseto-Wich G, Jurgens G, Heinecke J, Sevanian A. 1999.Oxidative cross-linking ofApoB100 and hemoglobin resultsin low density modification in blood. Relevance toatherogenesis caused by hemodialysis. J Biol Chem 274:18916–18924.
Zusterzeel PL, Mulder TP, Peters WH, Wiseman SA, SteegersEA. 2000. Plasma protein carbonyls in nonpregnant, healthypregnant and preeclamptic women. Free Radic Res 33:471–476.
Zusterzeel PL, Rutten H, Roelofs HM, Peters WH, Steegers EA.2001. Protein carbonyls in decidua and placenta of pre-eclamptic women as markers for oxidative stress. Placenta22:213–219.
REDOX PROTEOMICS AND DISEASE &
99




















