
manual for the - provision of intrauterine devices - WHO ...
Upload
independentCategory
view
0download
0
http://rsx.sagepub.com/Reproductive Sciences
http://rsx.sagepub.com/content/early/2013/11/11/1933719113508820The online version of this article can be found at:
DOI: 10.1177/1933719113508820
published online 12 November 2013Reproductive SciencesKannan, John P. Newnham, Boris W. Kramer, Alan H. Jobe, Jeffrey A. Keelan and Matthew W. Kemp
Masatoshi Saito, Matthew S. Payne, Yuichiro Miura, Demelza J. Ireland, Sarah Stock, Suhas G. Kallapur, Paranthaman S.Model
Polymyxin B Agonist Capture Therapy for Intrauterine Inflammation: Proof-of-Principle in a Fetal Ovine
Published by:
http://www.sagepublications.com
On behalf of:
Society for Gynecologic Investigation
can be found at:Reproductive SciencesAdditional services and information for
http://rsx.sagepub.com/cgi/alertsEmail Alerts:
http://rsx.sagepub.com/subscriptionsSubscriptions:
http://www.sagepub.com/journalsReprints.navReprints:
http://www.sagepub.com/journalsPermissions.navPermissions:
What is This?
- Nov 12, 2013OnlineFirst Version of Record >>
at OhioLink on November 13, 2013rsx.sagepub.comDownloaded from at OhioLink on November 13, 2013rsx.sagepub.comDownloaded from

Original Article
Polymyxin B Agonist Capture Therapyfor Intrauterine Inflammation: Proof-of-Principle in a Fetal Ovine Model
Masatoshi Saito, PhD1,2, Matthew S. Payne, PhD1,Yuichiro Miura, PhD1, Demelza J. Ireland, PhD1, Sarah Stock, PhD1,Suhas G. Kallapur, MD1,3, Paranthaman S. Kannan, PhD3,John P. Newnham, MD1, Boris W. Kramer, PhD1,4, Alan H. Jobe, PhD1,3,Jeffrey A. Keelan, PhD1, and Matthew W. Kemp, PhD1
AbstractIntrauterine infection is a leading cause of preterm birth (PTB), most notably in deliveries occurring before 32 weeks gestation.Preterm infants exposed to intrauterine inflammation are more likely to have a host of neurological, respiratory, gastrointestinal,and visual pathologies. Preventing preterm delivery and protecting the fetus from injury is thus likely to require treatment of bothintrauterine infection and inflammation. Polymyxin B (PMXB) is a cationic peptide antibiotic that binds Escherichia coli lipopoly-saccharides (LPS) and prevents inflammatory activation. We hypothesized that intraamniotic administration of PMXB wouldselectively inhibit LPS-driven inflammation, serving as a proof-of-principle for targeted agonist capture therapy as a treatment forPTB and fetal injury. In vitro studies with primary fetal ovine keratinocytes demonstrated a significant and sustained reduction intumor necrosis factor a and interleukin 8 messenger RNA expression after treatment with PMXB and LPS, relative to cellstreated with LPS alone. In vivo studies with fetal sheep demonstrated a significant reduction in proinflammatory cytokines in theamniotic fluid and fetal lung (but not fetal skin or chorioamnion) in LPS þ PMXB-treated animals, relative to those treated withLPS alone. These data are consistent with a partial resolution of LPS-driven intrauterine inflammation. They suggest the potentialfor agonist capture as a conceptual means of resolving the proparturition inflammation caused by infection of the amniotic cavity.
Keywordspreterm birth, inflammation, sheep, infection, fetus, agonist
Introduction
Preterm birth (PTB) remains a primary cause of neonatal death
and disease.1 The incidence of PTB varies markedly depending
on a host of interacting factors including geographical location,
ethnicity, and socioeconomic status. Of all births worldwide,
14.9 million or 11.1% were estimated to be born preterm in
2010; 2.6% of births in urban China, 7.8% in Hong Kong,
7% in the United Kingdom, 12% in the United States, and
15.5% in Indonesia are classified as preterm.1-4 Globally, PTB
accounts for some 28% of the estimated 4 million neonatal
deaths that occur each year5; in Australia, PTB is responsible
for an estimated 70% of total neonatal deaths.6 Despite marked
improvements in global rates of <5 mortality, to date little
progress has been made in reducing the rates of neonatal
mortality.7
The need for therapies to prevent both preterm delivery and fetal
injury is underscored by the profound societal cost of PTB. Preterm
infants are at an increased risk (in proportion, generally speaking,
to the degree of prematurity) of acute and chronic diseases of the
respiratory (pneumonia, bronchopulmonary dysplasia and chronic
lung disease), ocular (retinopathy of prematurity), neurological
(intra-ventricular hemorrhage, cerebral palsy, executive function/
learning deficit), and gastrointestinal (necrotizing enterocolitis)
systems.7-15 Interestingly, several studies now exist to suggest that
those born late preterm (36-38 weeks gestation) are also at an ele-
vated risk of sudden infant death syndrome and exhibit lower
1 School of Women’s and Infants’ Health, The University of Western Australia,
Perth, Western Australia2 Division of Perinatal Medicine, Tohoku University Hospital, Sendai, Japan3 Division of Pulmonary Biology, Cincinnati Children’s Hospital Medical
Centre, University of Cincinnati School of Medicine, Cincinnati, OH, USA4 Department of Paediatrics, School of Oncology and Developmental Biology,
Maastricht University Medical Centre, Maastricht, the Netherlands
Corresponding Author:
Matthew W. Kemp, School of Women’s and Infants’ Health, The University of
Western Australia, Perth, Western Australia 6009, Australia.
Email: [email protected]
Reproductive Sciences00(0) 1-9ª The Author(s) 2013Reprints and permission:sagepub.com/journalsPermissions.navDOI: 10.1177/1933719113508820rs.sagepub.com
at OhioLink on November 13, 2013rsx.sagepub.comDownloaded from

intelligence quotient scores than contemporaries delivered
between 39 and 41 weeks, potentially due to exposure to a sub-
optimal uterine environment.16,17 High on-going rates of PTB
also convey a significant financial cost. The annual societal
economic burden of prematurity in the United States is esti-
mated at US$26.2 billion. A single very early (delivery before
32 weeks) preterm infant born in England or Wales that sur-
vives to adulthood is estimated to cost the public £61,781 more
than a contemporary, term-born survivor.4,18
Uterine infection and inflammation is involved in at least
40% of all cases of prematurity and is the causal pathway that
is best understood from a clinical, molecular, and patho-
physiological perspective (for review see7,8,11,19). Microbial
pathogen-associated molecular patterns (including peptidogly-
can, lipopolysaccharides [LPS], and lipoteichoic acids) are
recognized by innate immune system receptors that trigger a
series of signaling cascades leading to the local production of
proparturition mediators including, proinflammatory cytokines/
chemokines (interleukins [IL] 1b, IL-6, IL-8, tumor necrosis fac-
tor [TNF] a), prostaglandins, and matrix metalloproteinases by
gestational tissues.7,8,11 Within the fetus, systemic inflammation,
or the fetal inflammatory response syndrome, is characterized by
cord blood IL-6 in excess of 11 pg/mL and is indicative of uter-
ine infection and adverse neonatal outcomes.20
An appreciation for the role of inflammation in both preterm
labor and fetal injury has led to increased efforts to develop
PTB prophylaxis that resolve both intrauterine infection and
resultant intrauterine inflammation. The majority of anti-
inflammatory approaches tested to date involve the disruption
of inflammatory signaling (eg, inhibitors of nuclear factor
kB(NF-kB) signaling, and IL-1 antagonists) or administration
of anti-inflammatory agents such as IL-10.21,22 Although a
number of these approaches show some promise, their clinical
application may be hampered by concerns relating to their
nonspecific modulation of fetal inflammatory signaling. In
addition to playing a key role in normal pregnancy, appropri-
ately regulated inflammation (controlled by ubiquitous media-
tors such as NF-kB) also regulate normal fetal immunological
development and organogenesis.23-25
The microorganisms involved in PTB, and the ligand
receptor interactions responsible for the induction of pathologi-
cal inflammation are increasingly well understood.8,19,26,27 We
hypothesized that inhibition of inflammatory signaling by
binding of key PAMP ligands by an inert agent may serve as
a selective, effective, and nontoxic means of resolving intrau-
terine inflammation.
Polymyxin B (PMXB) is a cyclic, cationic peptide antibiotic
that prevents LPS-driven inflammatory activation by binding
the lipid A core of LPS.28,29 A number of earlier studies have
demonstrated the utility of PMXB in resolving LPS-driven
inflammation. Relative to LPS stimulation alone, Stokes and
colleagues demonstrated that PMXB blocked TNF-a produc-
tion by rat alveolar macrophages following costimulation with
LPS in both in vitro experiments and in an in vivo rat model.29
More recently, PMXB, or synthetic derivatives, was used as
LPS binding agents to prevent inflammatory activation in a
number of settings including hemoperfusion and as a treatment
for endotoxemia in horses.30-33 As a proof of principle study,
we hypothesized that administration of PMXB would inhibit
LPS-driven intrauterine inflammation in a well-characterized
ovine model of human pregnancy.
Materials and Methods
Animals
All procedures with animals were performed at The University
of Western Australia (Perth, Western Australia) after approval
by The University of Western Australia Animal Ethics Com-
mittee. Twenty four date-mated Australian merino ewes with
singleton pregnancies were randomized to receive (1) a single
ultrasound-guided intraamniotic injection of 10 mg PMXB
(81271; Sigma Aldrich, St Louis, Missouri; PMXB group;
n¼ 8) or (2) a single ultrasound-guided intraamniotic injection
of 10 mg LPSs from Escherichia coli O55: B5 (LPS; L2880,
Sigma Aldrich; LPS group; n ¼ 8); or (3) a single ultrasound-
guided intraamniotic injection of 10 mg PMXB immediately
followed by 10 mg LPS (PMXBþ LPS group; n¼ 8). Success-
ful placement of intraamniotic injections were confirmed with
electrolyte (Cl�) analysis of amniotic fluid sample using a Sie-
mens Rapidlab 1265 Analyzer (Siemens, Munich, Germany).
Injections were performed at 122 day + 1 day gestational age
(GA). Fetuses were surgically delivered after 2 days and both
ewe and fetus were euthanized with an intravenous injection
of pentobarbitone (100 mg/kg). Fetal tissues for protein and
messenger RNA (mRNA) expression analyses were snap
frozen in liquid nitrogen. No fetal losses occurred during the
experiment. Fetal lung tissues for histological analysis were
inflation fixed in 10% neutral-buffered formalin for 24 hours
before paraffin embedding.
Fetal Epidermal Keratinocyte Isolation and Culture
Primary ovine epidermal keratinocytes were collected from the
skin of 124 d GA fetuses (no in utero exposure to either LPS
or PMXB) following surgical delivery and euthanasia, as previ-
ously described.26,34 Briefly, full-thickness fetal skin was cut
into 3-mm2 pieces and washed 3 times in 70% ethanol and 3
times in sterile phosphate-buffered saline (PBS) containing 5
mg/mL streptomycin and 3.3mg/mL penicillin. Tissue pieces
were immersed in a 3% solution of iodine for 5 minutes then
incubated for 48 hours in sterile PBS containing 1U/mL
Dispase II (17105-014; Life Technologies, Carlsbad, Califor-
nia). Epidermal sheets were removed from the dermis using ster-
ile forceps and placed into 5 mL 0.1% trypsin in EDTA.
Epidermal sheets were disrupted by incubating cells for 20 min-
utes in a shaking water bath at 37�C. Trypsinization was halted
with the addition of saline containing 5% fetal calf serum. Cells
were pelleted by centrifugation (1000 rpm, 10 minutes, 24�C)
and washed 3 times in sterile PBS. Isolated keratinocytes were
cultured in 25 cm2 flasks and cultured at 37�C in a 5% CO2
atmosphere in Keratinocyte-SFM media (17005-042; Life
2 Reproductive Sciences 00(0)
at OhioLink on November 13, 2013rsx.sagepub.comDownloaded from

Technologies) containing 1 � Penicillin-Streptomycin (15140-
122; Life Technologies). All cells used in this study were
passage 3.
In Vitro PMXB Dose–Response Study
To confirm dose–response studies performed previously,28,29
keratinocytes were subcultured into 12-well culture plates at
a density of 2 � 105 cells/well, 16 hours prior to commencing
the study. The >95% viability of cells added to each well was
confirmed with trypan blue staining. Keratinocytes were
exposed to 2 mL keratinocyte-SFM media containing (1) 1
mg LPS; (2) 1 mg LPS and 1 mg PMXB (1:1 w/w); (3) 1 mg LPS
and 10 mg PMXB (1:10, w/w); or (4) 1 mg LPS and 100 mg
PMXB (1:100 w/w). Each exposure was performed once in
triplicate for 0, 1, 2, 3, 6, and 12 hours.
Assessment of In Vivo LPS Concentration
LPS concentrations in amniotic fluid were determined using a
Pierce LAL Chromogenic Endotoxin Quantitation Kit (88282;
Thermo Scientific, Rockford, Illinois) incorporating a 3-point
standard curve.
Hematology
Complete blood counts and differential analyses were
performed by VetPath Laboratory Services (Perth, Western
Australia) using an automated Coulter counter adapted for
ovine specimens.
Fetal Lung Histology
Tissues from 5, randomly selected animals from each group
were analyzed for histological changes. Sections of 5 mm thick-
ness from formalin-fixed lung (right upper lobe) tissues
embedded in paraffin blocks were stained with hematoxylin
and eosin. Immunofluorescent staining of fetal lung for CD3
(A0452, Dako, Glostrup, Denmark, working concentration
1:100) was performed as previously published.35 CD3 cells
in fetal lung were quantified by counting positively stained
cells in 5 randomly selected, nonoverlapping fields at 20�objective magnification.
Isolation and Purification of RNA
Total RNA was extracted from keratinocyte monolayers or
fetal tissue homogenized in liquid nitrogen using TRIzol
(15596-018; Life Technologies). Extracted RNA was treated
with Turbo DNase (AM2238; Life Technologies) to remove
any residual DNA, following manufacturer’s instructions.
RNA yields from fetal tissues were normalized to 100 ng/mL
using nuclease-free water (AM9937; Life Technologies).
Relative Quantification of mRNA Expression
Polymerase chain reaction (PCR) primers specific for ovine IL-8
and TNF-a were used to perform quantitative PCR (qPCR) reac-
tions using Power SYBR Green PCR Master Mix (4367659;
Life Technologies) on complementary DNA generated from
600 ng DNase-treated RNA isolated from cultured epidermal
keratinocytes. Power SYBR Green qPCR cycling conditions
were: 1 � 5 minute initial denaturation (95�C), 35 � 30-
second denaturation (95�C), 30-second annealing (60�C), 30-
second extension (60�, 1 � final 0.5�C stepped temperature
ramp 60�C-95�C). Reactions were performed on a Qiagen
(Venlo, the Netherlands) Rotorgene in a final volume of 20
mL as described previously.26,34 The Cq values were normalized
to ovine-specific glyceraldehyde 3-phosphate dehydrogenase to
calculate difference in Cq (dCq) and fold-change values. Reac-
tion efficiencies and specificities were within limits proposed in
the Minimum Information for Publication of Quantitative Real-
Time PCR Experiments (MIQE) guidelines.36
The PCR primers and hydrolysis probes specific for ovine
IL-1b, IL-6, IL-8, TNF-a, MCP-2, Hepcidin (a regulator of iron
homeostasis that is increased in response to inflammation),
serum amalyoid A3 (SAA-3) protein (a marker of inflamma-
tion), and C-reactive protein (CRP, Custom Targets, Life Tech-
nologies) sequences were used to perform quantitative PCRs
on DNase-treated RNA isolated from fetal tissue. Reactions
were performed using an EXPRESS One-Step SuperScript
qRT-PCR Kit (1178101K; Life Technologies) with 400 ng
total RNA template in a total volume of 20 mL in accordance
with the manufacturer’s instructions. Reaction cycling condi-
tions consisted of 15 minutes reverse transcription at 50�C and
an initial denaturation at 95�C for 20 seconds, followed by 40
cycles of 95�C for 3 seconds and 60�C for 30 seconds (acqui-
sition phase). All reactions were performed in fast 96-well
plates on a ViiA7 real-time PCR thermocycler (Life Technolo-
gies). The Cq values were normalized to 18s rRNA and
expressed as fold changes relative to pooled control values
+ standard deviation (SD). Reaction efficiencies were within
limits proposed in the MIQE guidelines.36 Six randomly
selected animals from each group were analyzed for changes
in cytokine/chemokine mRNA expression. The dCq values
were used for statistical analyses as described subsequently.
Enzyme-Linked Immunosorbent Assays
Six randomly selected animals from each group were analyzed
for changes in cytokine/chemokine protein expression. Quanti-
fication of IL-1b and IL-8 protein concentrations in amniotic
fluid and alveolar wash samples was performed as previously
described.37 All samples were diluted 1:5 in assay buffer and
thoroughly mixed by vortexing prior to analysis. Quantification
of IL-6 protein concentration in ovine amniotic fluid was
performed using an identical protocol with the following
modifications: Plate wells were coated overnight at 4�C with
5 mg/mL capture antibody (MCA1659; ABD Serotech,
Kidlington, United Kingdom). Recombinant sheep IL-6 protein
Saito et al 3
at OhioLink on November 13, 2013rsx.sagepub.comDownloaded from

standards (Protein Express, Cincinnati, Ohio) and amniotic
fluid samples were diluted 1:2 in assay buffer and incubated
overnight at 4�C. The detection antibody (AHP424; ABD
Serotech) was diluted 1:750 in assay buffer. All samples and
standards were assayed in duplicate.
Statistical Analyses
All values are expressed as mean + 1 SD. All statistical anal-
yses were performed using IBM SPSS Statistics for Windows,
Version 20.0 (IBM Corp, Armonk, New York). Data were
assessed for normality with Shapiro-Wilk Tests. Normally dis-
tributed (parametric) data were screened for outliers with
Dixon Q-Parameter and differences tested for significance with
1-way analysis of variance (ANOVA) employing an a value of
.05. Multiple post hoc comparisons were performed using
Tukey test. Apparent differences in nonparametric data were
tested for significance with Kruskal-Wallis 1-way ANOVA
employing an a value of .05. Multiple post hoc comparisons
were performed using Mann-Whitney tests with an a value cor-
rected for n multiple comparisons.
Results
In Vitro PMXB Dose Response for LPS
Relative to treatment with culture media containing LPS only,
expression of IL-8 and TNF-a mRNA was significantly reduced
(P < .05) in ovine keratinocytes exposed to culture media supple-
mented with LPS and PMXB (Figure 1). A 1:1 or 1:10 ratio of
LPS:PMXB resulted in the most consistent suppression of IL-8
and TNF-a mRNA expression. As no significant difference
(P > .05) in IL-8 or TNF-a expression was identified between
a 1:1 and 1:10 LPS–PMXB dose, the lower 1:1 LPS–PMXB
ratio was selected for subsequent in vivo studies.
Fetal Weight, Hematology, and Amniotic Fluid EndotoxinConcentrations
No differences (P > .05) were observed in fetal birth weight (kg),
arterial cord blood pH, pO2 (mm Hg), pCO2 (mm Hg), or
hemoglobin (g/L) between PMXB-, PMXB þ LPS-, LPS-
exposed animals, respectively (Table 1). No differences (P >
.05) were observed in fetal arterial cord total white blood cells
(1011/L), neutrophil (109/L), lymphocyte (109/L), monocyte
(109/L), or eosinophil counts (109/L) between PMXB-, PMXB
þ LPS-, and LPS-exposed animals, respectively (Table 1). No
significant difference (P ¼ .345) in amniotic fluid LPS concen-
tration was observed between PMXB þ LPS- and LPS-exposed
animals. Amniotic fluid LPS concentrations from PMXB þLPS- and LPS-exposed animals were significantly increased
(P < .000) relative to amniotic fluid from PMXB-exposed ani-
mals (Figure 2).
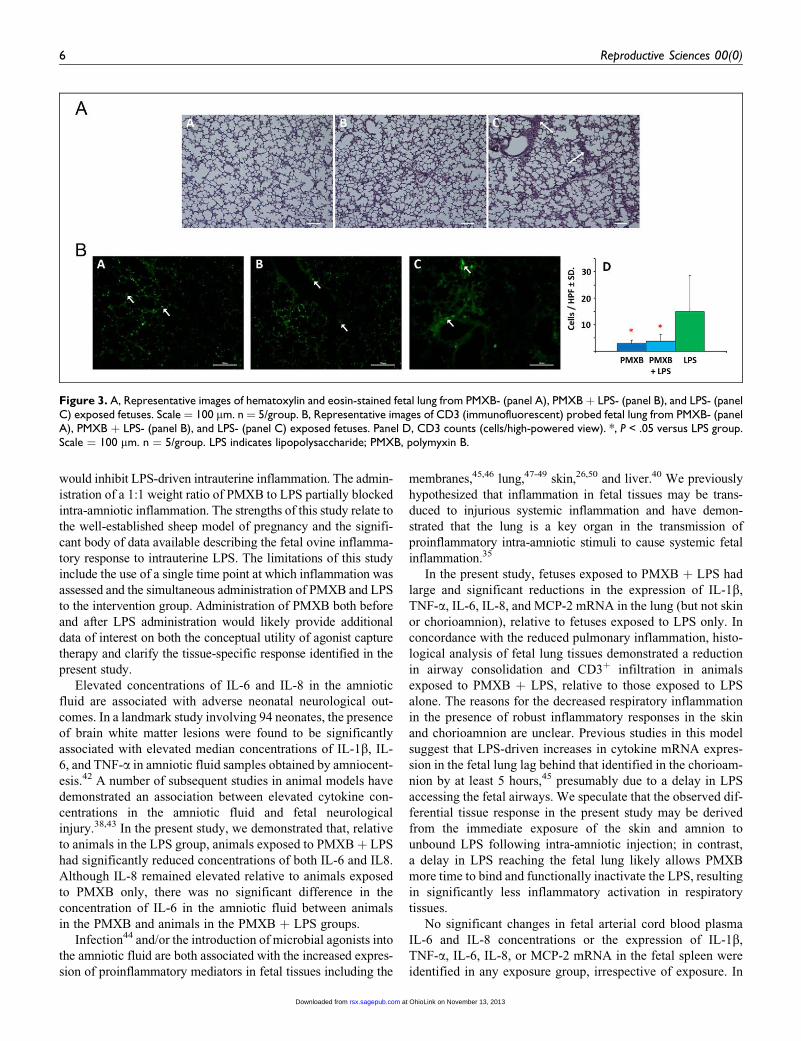
Fetal Lung Histology
Analysis of hematoxylin and eosin-stained lung (right upper
lobe) sections from LPS-exposed animals demonstrated
numerous focal areas of consolidation and extensive airway
inflammatory cells (Figure 3A; panel C, arrows), consistent
with alveolar inflammation. Similar consolidation/airway
involvement was absent from sections taken from PMXB- and
PMXB þ LPS-exposed animals (Figure 3A; panels A and B,
respectively). The number of CD3-positive cells was signifi-
cantly increased in lung tissue of LPS-exposed animals (15.0
Figure 1. Expression of interleukin (IL) 8 (panel A) and tumor necrosis factor (TNF) a (panel B) mRNA (fold change) in primary fetal ovinekeratinocytes exposed to LPS þ polymyxin B (PMXB), relative to LPS-exposure alone. *, P < .05. Note logarithmic scale: values <1 representreduced mRNA expression relative to LPS stimulation alone; values >1 represent increased mRNA expression relative to LPS stimulation alone.mRNA indicates messenger RNA; LPS, lipopolysaccharide
Table 1. Fetal Arterial Cord Blood Measurements at Delivery.
PMXB PMXB þ LPS LPS
Fetal birth weight, kg 2.8 + 0.4 3.0 + 0.2 2.6 + 0.4Arterial cord blood, pH 7.2 + 0.05 7.2 + 0.05 7.2 + 0.1pO2, mm Hg 83.0 + 9 77 + 6 83.0 + 11pCO2, mm Hg 4.60 + 1.3 5.2 + 2.4 5.3 + 3.7Hemoglobin, g/L 130.0 + 15.5 122.0 + 9.4 122.0 + 15.0Total white blood cells,
1011/L3.7 + 1.9 3.5 + 1.0 3.1 + 1.7
Neutrophils, 109/L 0.4 + 0.2 0.4 + 0.3 0.3 + 0.3Lymphocytes, 109/L 2.6 + 1.6 2.3 + 0.7 2.1 + 1.0Monocytes, 109/L 0.2 + 0.1 0.2 + 0.2 0.2 + 0.1Eosinophils, 109/L 0.06 + 0.05 0.04 + 0.04 0.02 + 0.04
Abbreviations: LPS, lipopolysaccharide; PMXB, polymyxin B.
4 Reproductive Sciences 00(0)
at OhioLink on November 13, 2013rsx.sagepub.comDownloaded from

+ 14.0). Relative to LPS-exposed fetuses, significantly fewer
CD3-positive cells were present in the lung of PMXB- (3 +1.3; P ¼ .004) and PMXB þ LPS- (3.7 + 2.6; P ¼ .009)
exposed fetuses. No difference (P¼ 1.0) was detected between
CD3-positive cells in PMXB- and PMXB þ LPS-exposed ani-
mals. Representative images are shown in Figure 3B.
Cytokine and Acute-Phase Protein mRNA Expression
Expression of IL-1b, IL-6, IL-8, and MCP-2 mRNA was signif-
icantly increased (P < .010) in chorioamnion tissue from LPS-
and LPS þ PMXB-exposed animals, relative to chorioamnion
tissue from PMXB-control animals. No significant difference
in TNF-a mRNA expression (P¼ .270) was identified between
treatment groups. No significant difference in expression of IL-
1b, TNF-a, IL-6, IL-8, or MCP-2 mRNA was identified
between LPS- and LPS þ PMXB-exposed animals (data not
shown). Expression of IL-1b, TNF-a, IL-6, IL-8, and MCP-2
mRNA was significantly increased (P < .015) in lung tissue
(right lower lobe) from LPS-exposed animals, relative to both
PMXB- and PMXB þ LPS-exposed animals. No significant
difference (P > .05) in IL-1b or IL-6 mRNA expression was
observed in PMXB þ LPS-exposed lung, relative to lung from
fetuses exposed to PMXB (Figure 4; panel A). No significant
difference (P > .05) in IL-1b, TNF-a, or IL-6 mRNA expres-
sion was identified in PMXB þ LPS- or LPS-exposed skin,
relative to skin from fetuses exposed to PMXB. The IL-8 and
MCP-2 mRNA expression was equivalently increased (P <
.05) in fetal skin exposed to both PMXB þ LPS and LPS,
relative to skin from fetuses exposed to PMXB (Figure 4; panel
B). No significant differences in assessed cytokine/chemokine
mRNA expression were identified in the fetal spleen PMXB-,
PMXB þ LPS-, or LPS-exposed fetuses (Figure 4; panel C).
Hepcidin and SAA-3 mRNA expression were significantly
(P < .02) increased in liver from fetuses exposed to LPS,
relative to fetuses exposed to PMXB. No differences in liver
hepcidin or SAA-3 mRNA were detected between PMXB- and
PMXB þ LPS-exposed animals (Figure 5). An apparent
increase in both liver CRP mRNA in LPS-exposed fetuses,
relative to PMXB-exposed fetuses, and apparent reduction in
liver CRP mRNA in PMXB þ LPS-exposed fetuses (relative
to LPS-exposed fetuses) approached statistical significance
(P ¼ .02 and .08 [Pcrit ¼ .02]).
Amniotic Fluid and Alveolar Wash CytokineConcentrations
The concentration of IL-6 in amniotic fluid was significantly
increased in LPS-exposed animals, relative to animals exposed
to either PMXB (P ¼ .007) or PMXB þ LPS (P ¼ .018). The
concentration of amniotic fluid IL-8 was significantly
increased in LPS-exposed animals, relative to animals exposed
to either PMXB (P¼ .000) or PMXBþ LPS (P¼ .014), and in
PMXB þ LPS-exposed animals, relative to PMXB-exposed
animals (P ¼ .011; Figure 6). The concentration of IL-8 in
alveolar wash samples from LPS-exposed fetuses (140 + 10
ng/mL) was significantly increased relative to PMXB (52 +54 pg/mL; P ¼ .001) and PMXB þ LPS (0.19 + 0.13 ng/
mL; P¼ .002). In contrast, there was no statistically significant
difference in the concentration of IL-8 in the alveolar wash
between animals exposed to PMXB and PMXB þ LPS (P ¼.999).
Discussion
Intrauterine inflammation (frequently caused by microbial
colonization of the amniotic environment7) is hypothesized to
be a primary cause of PTB, most notably in deliveries prior
to 32-week gestation.7,19,20 Much remains to be understood
with regard to the organ-specific short- and long-term out-
comes of exposure to aberrant antenatal inflammation, as high-
lighted by research into the impact of antenatal inflammation
on fetal lung development.15 However, pathological intrauter-
ine inflammation is hypothesized to alter the developmental
trajectory of several fetal organs including the lung,15 brain,38
and gastrointestinal tract.39 Antenatal exposure to pathological
inflammation also programs alterations in glucose and lipid
metabolism that persist into neonatal life.40 In pregnancy,
inflammation is involved both in normal fetal and in gestational
development as well as the pathological processes underlying
PTB and changes in fetal organogenesis.23-25,41 Developing
anti-inflammatory treatments that resolve pathogenic intrauter-
ine inflammation without affecting fetal or gestational develop-
ment is thus likely to constitute an important step in preventing
PTB and fetal injury.
The cationic peptide antibiotic PMXB binds the lipid A core
of LPS, inhibiting proinflammatory activation of the innate arm
of the immune system.28-30,32 Rather than attempting to use
potent pharmacological agents to block intrauterine inflamma-
tion, we hypothesized that adapting the concept of inhibiting
inflammation by binding and functionally inactivating micro-
bial agonist (which we term agonist capture therapy) may be
a strategy for PTB prevention. As a proof-of-principle experi-
ment, we asked whether intra-amniotic administration of PMXB
Figure 2. Amniotic fluid endotoxin concentration at delivery. *, P < .05versus polymyxin B (PMXB) group. n ¼ 5/group.
Saito et al 5
at OhioLink on November 13, 2013rsx.sagepub.comDownloaded from
would inhibit LPS-driven intrauterine inflammation. The admin-
istration of a 1:1 weight ratio of PMXB to LPS partially blocked
intra-amniotic inflammation. The strengths of this study relate to
the well-established sheep model of pregnancy and the signifi-
cant body of data available describing the fetal ovine inflamma-
tory response to intrauterine LPS. The limitations of this study
include the use of a single time point at which inflammation was
assessed and the simultaneous administration of PMXB and LPS
to the intervention group. Administration of PMXB both before
and after LPS administration would likely provide additional
data of interest on both the conceptual utility of agonist capture
therapy and clarify the tissue-specific response identified in the
present study.
Elevated concentrations of IL-6 and IL-8 in the amniotic
fluid are associated with adverse neonatal neurological out-
comes. In a landmark study involving 94 neonates, the presence
of brain white matter lesions were found to be significantly
associated with elevated median concentrations of IL-1b, IL-
6, and TNF-a in amniotic fluid samples obtained by amniocent-
esis.42 A number of subsequent studies in animal models have
demonstrated an association between elevated cytokine con-
centrations in the amniotic fluid and fetal neurological
injury.38,43 In the present study, we demonstrated that, relative
to animals in the LPS group, animals exposed to PMXBþ LPS
had significantly reduced concentrations of both IL-6 and IL8.
Although IL-8 remained elevated relative to animals exposed
to PMXB only, there was no significant difference in the
concentration of IL-6 in the amniotic fluid between animals
in the PMXB and animals in the PMXB þ LPS groups.
Infection44 and/or the introduction of microbial agonists into
the amniotic fluid are both associated with the increased expres-
sion of proinflammatory mediators in fetal tissues including the
membranes,45,46 lung,47-49 skin,26,50 and liver.40 We previously
hypothesized that inflammation in fetal tissues may be trans-
duced to injurious systemic inflammation and have demon-
strated that the lung is a key organ in the transmission of
proinflammatory intra-amniotic stimuli to cause systemic fetal
inflammation.35
In the present study, fetuses exposed to PMXB þ LPS had
large and significant reductions in the expression of IL-1b,
TNF-a, IL-6, IL-8, and MCP-2 mRNA in the lung (but not skin
or chorioamnion), relative to fetuses exposed to LPS only. In
concordance with the reduced pulmonary inflammation, histo-
logical analysis of fetal lung tissues demonstrated a reduction
in airway consolidation and CD3þ infiltration in animals
exposed to PMXB þ LPS, relative to those exposed to LPS
alone. The reasons for the decreased respiratory inflammation
in the presence of robust inflammatory responses in the skin
and chorioamnion are unclear. Previous studies in this model
suggest that LPS-driven increases in cytokine mRNA expres-
sion in the fetal lung lag behind that identified in the chorioam-
nion by at least 5 hours,45 presumably due to a delay in LPS
accessing the fetal airways. We speculate that the observed dif-
ferential tissue response in the present study may be derived
from the immediate exposure of the skin and amnion to
unbound LPS following intra-amniotic injection; in contrast,
a delay in LPS reaching the fetal lung likely allows PMXB
more time to bind and functionally inactivate the LPS, resulting
in significantly less inflammatory activation in respiratory
tissues.
No significant changes in fetal arterial cord blood plasma
IL-6 and IL-8 concentrations or the expression of IL-1b,
TNF-a, IL-6, IL-8, or MCP-2 mRNA in the fetal spleen were
identified in any exposure group, irrespective of exposure. In
Figure 3. A, Representative images of hematoxylin and eosin-stained fetal lung from PMXB- (panel A), PMXB þ LPS- (panel B), and LPS- (panelC) exposed fetuses. Scale ¼ 100 mm. n ¼ 5/group. B, Representative images of CD3 (immunofluorescent) probed fetal lung from PMXB- (panelA), PMXB þ LPS- (panel B), and LPS- (panel C) exposed fetuses. Panel D, CD3 counts (cells/high-powered view). *, P < .05 versus LPS group.Scale ¼ 100 mm. n ¼ 5/group. LPS indicates lipopolysaccharide; PMXB, polymyxin B.
6 Reproductive Sciences 00(0)
at OhioLink on November 13, 2013rsx.sagepub.comDownloaded from

contrast, LPS exposure yielded variable, but statistically signif-
icant increases in liver CRP, hepcidin, and SAA-3 mRNA
expression in the fetal liver relative to animals in both PMXB
and PMXB þ LPS groups. The apparent reductions identified
in hepatic acute-phase protein expression in the PMXB þ LPS
group, relative to the LPS group, were not statistically signifi-
cant (P > .05).
Conclusions
The administration of PMXB decreased LPS-driven inflamma-
tion in the fetal lung but not the fetal skin, chorioamnion, or
liver. Taken together, these data suggest that the concept of
in utero agonist capture therapy offers some promise as a
means of resolving intrauterine inflammation in a targeted
fashion. Two points of particular interest may be drawn from
these data. First, a substantial decrease in pulmonary inflamma-
tion alone was associated with a large reduction in amniotic
fluid IL-6 and IL-8 concentrations. It is tempting to speculate
that the inflammatory state of the fetal lung is thus a major
determinant of amniotic fluid inflammation, although further
work is required to confirm this hypothesis. Second, although
the use of PMXB substantially decreased pulmonary and
amniotic fluid inflammation, this alone was insufficient to
prevent the induction of systemic fetal inflammation, as
demonstrated by increased acute-phase protein expression in
the liver of PMXB þ LPS-treated animals. These data suggest
that successful dampening of fetal inflammation likely requires
inhibiting the production of proinflammatory mediators in
most and perhaps all the fetal tissues that may come into
contact with proinflammatory agonist. This observation is per-
tinent to both our agonist capture strategy and alternative
approaches employing pharmacological methods to downregu-
lated inflammatory signaling pathways. Moreover, given data
Figure 4. Cytokine/chemokine messenger RNA (mRNA) expressionin fetal lung (panel A), skin (panel B), and spleen (panel C). *, P < .05versus PMXB group. �, P < .05 versus PMXB þ lipopolysaccharide(LPS) group. n ¼ 6/group. PMXB indicates polymyxin B.
Figure 6. Concentration of amniotic fluid IL-6 and IL-8 protein. *, P <.05 versus PMXB group. �, P < .05 versus PMXB þ lipopolysaccharide(LPS) group. n¼ 6/group. IL indicates interleukin; PMXB, polymyxin B.
Figure 5. Acute-phase protein messenger RNA (mRNA) expressionin the fetal liver. *, P < .05 versus polymyxin B (PMXB) group. n ¼ 6/group.
Saito et al 7
at OhioLink on November 13, 2013rsx.sagepub.comDownloaded from

demonstrating that bacterial agonist remains in utero for an
extended period,51 the approach used to control inflammation
will likely need to be effective for several weeks at a time
(or able to be repeatedly delivered) and nontoxic to the fetus.
Acknowledgments
The authors wish to acknowledge the generous support received from
Siemens Australia for their donation of Rapidlab 1265 consumables
used in this study. The authors wish to thank Sara and Andrew Ritchie
(Icon Agriculture, Darkan, Western Australia) for the expert provision
of date-mated sheep used in this study.
Authors’ Note
Findings were presented at the 61st Annual Meeting, Society for
Gynaecologic Investigation, Miami, FL, March 16-19, 2011 and the
8th World Congress on Developmental Origins of Health and Disease,
Singapore, November 17-20, 2013.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to
the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for
the research, authorship, and/or publication of this article: This work
was funded by a grant from the Ramaciotti Foundations, Australia, to
MWK and JAK.
References
1. March of Dimes P, Save the Children, WHO. Born Too Soon: The
Gobal Action Report on Preterm Birth. Geneva: World Health
Organization; 2012.
2. Blencowe H, Cousens S, Oestergaard MZ, et al. National,
regional, and worldwide estimates of preterm birth rates in the
year 2010 with time trends since 1990 for selected countries: a
systematic analysis and implications. Lancet. 2012;379(9832):
2162-2172.
3. Newnham JP, Sahota DS, Zhang CY, et al. Preterm birth rates in
Chinese women in China, Hong Kong and Australia—the price of
Westernisation. Aust N Z J Obstet Gynaecol. 2011;51(5):426-431.
4. Petrou S, Eddama O, Mangham L. A structured review of the
recent literature on the economic consequences of preterm birth.
Arch Dis Child Fetal Neonatal Ed. 2011;96(3):F225-F232.
5. Lawn JE, Cousens S, Zupan J, Lancet Neonatal Survival Steering
Team. 4 Million neonatal deaths: when? Where? Why? Lancet.
2005;365(9462):891-900.
6. Tracy SK, Tracy MB, Dean J, Laws P, Sullivan E. Spontaneous
preterm birth of liveborn infants in women at low risk in Australia
over 10 years: a population-based study. BJOG. 2007;114(6):
731-735.
7. Goldenberg RL, Culhane JF, Iams JD, Romero R. Epidemiology
and causes of preterm birth. Lancet. 2008;371(9606):75-84.
8. Bastek JA, Gomez LM, Elovitz MA. The role of inflammation
and infection in preterm birth. Clin Perinatol. 2011;38(3):
385-406.
9. Fawke J. Neurological outcomes following preterm birth. Semin
Fetal Neonatal Med. 2007;12(5):374-382.
10. McAdams RM, Juul SE. Cerebral palsy: prevalence, predictabil-
ity, and parental counseling. Neo Rev. 2011;12(10):e564-e574.
11. Romero R, Espinoza J, Kusanovic JP, et al. The preterm parturi-
tion syndrome. BJOG. 2006;113(suppl 3):17-42.
12. Shatrov JG, Birch SCM, Lam LT, Quinlivan JA, McIntyre S,
Mendz GL. Chorioamnionitis and cerebral palsy: a meta-analysis.
Obstet Gynecol. 2010;116(2 pt 1):387-392.
13. Vermeulen GM, Bruinse HW, de Vries LS. Perinatal risk factors
for adverse neurodevelopmental outcome after spontaneous
preterm birth. Eur J Obstet Gynecol Reprod Biol. 2001;99(2):
207-212.
14. Wood NS, Costeloe K, Gibson AT, et al. The EPICure study:
associations and entecedents of neurological and developmental
disability at the 30 months of age following extremely preterm
birth. Arch Dis Childh Fetal Neonatal Ed. 2005;90(2):F134-F140.
15. Jobe AH. Effects of chorioamnionitis on the fetal lung. Clin
Perinatol. 2012;39(3):441-457.
16. Smith GCS, Pell JP, Dobbie R. Risk of sudden infant death syn-
drome and week of gestation of term birth. Pediatrics. 2003;
111(6 pt 1):1367-1371.
17. Yang S, Platt RW, Kramer MS. Variation in child cognitive
ability by week of gestation among healthy term births. Am J
Epidemiol. 2010;171(4):399-406.
18. Mangham LJ, Petrou S, Doyle LW, Draper ES, Marlow N. The
cost of preterm birth throughout childhood in England and Wales.
Pediatrics. 2009;123(2):e312-e327.
19. Agrawal V, Hirsch E. Intrauterine infection and preterm labor.
Semin Fetal Neonatal Med. 2012;17(1):12-19.
20. Gotsch F, Romero R, Kusanovic JP, et al. The fetal inflammatory
response syndrome. Clin Obstet Gynecol. 2007;50(3):652-683.
21. Keelan JA. Pharmacological inhibition of inflammatory pathways
for the prevention of preterm birth. J Reprod Immunol. 2011;
88(2):176-184.
22. Rinaldi SF, Hutchinson JL, Rossi AG, Norman JE. Anti-
inflammatory mediators as physiological and pharmacological
regulators of parturition. Expert Rev Clin Immunol. 2011; 7(5):
675-696.
23. Hayden MS, West AP, Ghosh S. NF-kB and the immune
response. Oncogene. 2006;25(51):6758-6780.
24. Jiang YJ, Lu B, Crumrine D, Man MQ, Elias PM, Feingold KR.
IL-1a accelerates stratum corneum formation and improves per-
meability barrier homeostasis during murine fetal development.
J Dermatol Sci. 2009;54(2):88-98.
25. Jiang YJ, Lu B, Crumrine D, Elias PM, Feingold KR. IL-6 Stimu-
lates but is not essential for stratum corneum formation and
permeability barrier development during gestation. Exp Derma-
tol. 2010;19(8):e31-e36.
26. Kemp MW, Saito M, Nitsos I, Jobe AH, Kallapur SG, Newnham
JP. Exposure to in utero lipopolysaccharide induces inflammation
in the fetal ovine skin. Reprod Sci. 2010;18(1):88-98.
27. Jones HE, Harris KA, Azizia M, et al. Differing prevalence and
diversity of bacterial species in fetal membranes from very
preterm and term labor. PLoS One. 2009;4(12):e8205.
28. Coyne CP, Fenwick BW. Inhibition of lipopolysaccharide-induced
macrophage tumor necrosis factor-a synthesis by polymyxin B
sulfate. Am J Veterinary Res. 1993;54(2):305-314.
8 Reproductive Sciences 00(0)
at OhioLink on November 13, 2013rsx.sagepub.comDownloaded from

29. Stokes DC, Shenep JL, Fishman M, Hildner WK, Bysani GK,
Rufus K. Polymyxin B prevents lipopolysaccharide-induced
release of tumor necrosis factor-a from alveolar macrophages.
J Infect Dis. 1989;160(1):52-57.
30. Coyne CP, Moritz JT, Fenwick BW. Inhibition of lipopolysacchar-
ide-induced TNF-a production by semisynthetic polymyxin-B
conjugated dextran. Biotechnol Ther. 1994;5(3-4):137-162.
31. Jaber BL, Barrett TW, Cendoroglo Neto M, Sundaram S, King
AJ, Pereira BJ. Removal of cytokine inducing substances by
polymyxin-B immobilized polystyrene-derivative fibers during
in vitro hemoperfusion of 10% human plasma containing Staphy-
lococcus aureus challenge. ASAIO J. 1998;44(1):48-53.
32. Barton MH. Use of Polymyxin B for treatment of endotoxemia in
horses. Comp Cont Educ Pract 2000;22(11):1056.
33. Iwagaki A, Porro M, Pollack M. Influence of synthetic antiendo-
toxin peptides on lipopolysaccharide (LPS) recognition and LPS-
induced proinflammatory cytokine responses by cells expressing
membrane-bound CD14. Infect Immun. 2000;68(3):1655-1663.
34. Kemp MW, Saito M, Kallapur SG, et al. Inflammation of the fetal
ovine skin following in utero exposure to ureaplasma parvum.
Reprod Sci. 2011;18(11):1128-1137.
35. Kemp MW, Senthamarai Kannan P, Saito M, et al. Selective
exposure of the fetal lung and skin/amnion (but not gastro-
intestinal tract) to LPS elicits acute systemic inflammation in fetal
sheep. PLoS One. 2013;8(5):e63355.
36. Bustin SA, Benes V, Garson JA, et al. The MIQE guidelines: min-
imum information for publication of quantitative real-time PCR
experiments. Clin Chem. 2009;55(4):611-622.
37. Zhang L, Saito M, Jobe A, et al. Intra-amniotic administration of
E. coli lipopolysaccharides causes sustained inflammation of the
fetal skin in sheep. Reprod Sci. 2012;19(11):1181-1189.
38. Burd I, Balakrishnan B, Kannan S. Models of fetal brain injury,
intrauterine inflammation, and preterm birth. Am J Reprod Immu-
nol. 2012;67(4):287-294.
39. Wolfs TGAM, Buurman WA, Zoer B, et al. Endotoxin induced
chorioamnionitis prevents intestinal development during gesta-
tion in fetal sheep. PLoS One. 2009;4(6):e5837.
40. Vlassaks E, Gavilanes AWD, et al. Antenatal exposure to
chorioamnionitis affects lipid metabolism in 7-week-old sheep.
J Dev Orig Health Dis. 2012;3(2):103.
41. Liggins GC. Cervical ripening as an inflammatory reaction. In:
Ellwood DA, Anderson ABM, eds. The Cervix in Pregnancy and
Labour, Clinical and Biochemical Investigations. Edinburgh:
Churchill Livingstone; 1981:1.
42. Bo Hyun Y, Jong Kwan J, Romero R, et al. Amniotic fluid inflam-
matory cytokines (interleukin-6, interleukin-1b, and tumor necro-
sis factor-a), neonatal brain white matter lesions, and cerebral
palsy. Am J Obstet Gynecol. 1997;177(1):19-26.
43. Burd I, Bentz AI, Chai J, et al. Inflammation-induced preterm
birth alters neuronal morphology in the mouse fetal brain. J Neu-
rosci Res. 2010;88(9):1872-1881.
44. Gravett MG, Witkin SS, Haluska GJ, Edwards JL, Cook MJ,
Novy MJ. An experimental model for intraamniotic infection and
preterm labor in rhesus monkeys. Am J Obstet Gynecol. 1994;
171(6):1660-1667.
45. Kramer BW, Moss TJ, Willet KE, et al. Dose and time response
after intraamniotic endotoxin in preterm lambs. Am J Respir Crit
Care Med. 2001;164(6):982-988.
46. Grigsby PL, Novy MJ, Adams Waldorf KM, Sadowsky DW,
Gravett MG. Choriodecidual inflammation: a harbinger of the
preterm labor syndrome. Reprod Sci. 2010;17(1):85-94.
47. Kramer BW, Kramer S, Ikegami M, Jobe AH. Injury, inflamma-
tion, and remodeling in fetal sheep lung after intra-amniotic endo-
toxin. Am J Physiol Lung Cell Mol Physiol. 2002;283(2):
L452-L459.
48. Kallapur SG, Willet KE, Jobe AH, Ikegami M, Bachurski CJ.
Intra-amniotic endotoxin: Chorioamnionitis precedes lung
maturation in preterm lambs. Am J Physiol Lung Cell Mol Phy-
siol. 2001;280(3):L527-L536.
49. Davies JK, Shikes RH, Sze CI, et al. Histologic inflammation in
the maternal and fetal compartments in a rabbit model of acute
intra-amniotic infection. Am J Obstet Gynecol. 2000;183(5):
1088-1093.
50. Kim YM, Romero R, Chaiworapongsa T, Espinoza J, Mor G, Kim
CJ. Dermatitis as a component of the fetal inflammatory response
syndrome is associated with activation of Toll-like receptors in
epidermal keratinocytes. Histopathology. 2006;49(5):506-514.
51. Newnham JP, Kallapur SG, Kramer BW, et al. Betamethasone
effects on chorioamnionitis induced by intra-amniotic endotoxin
in sheep. Am J Obstet Gynecol. 2003;189(5):1458-1466.
Saito et al 9
at OhioLink on November 13, 2013rsx.sagepub.comDownloaded from
Copyright © 2022 FDOKUMEN




















