
Perturbed skeletal muscle insulin signaling in the adult female intrauterine growth-restricted rat
Upload
independentCategory
view
1download
0
Chronic Intrauterine Pulmonary Hypertension Increases Endothelial Cell Rho-
Kinase Activity and Impairs Angiogenesis in vitro.
Jason Gien MD 1
Gregory J Seedorf, BS 2
Vivek Balasubramaniam MD 2
Nancy Tseng, BS, MS
Neil Markham, BS 2
Steven H Abman MD 2
From the Pediatric Heart Lung Center, Sections of Neonatology1 and Pulmonary Medicine2, Department of Pediatrics, University of Colorado School of Medicine, Denver, CO
Correspondence:Dr Jason Gien P18-4402K Mail Stop 831712800 East 19TH Ave.PO Box 6511Aurora, CO 80045 Phone: 303 724 4065Fax: 303 724 4072Email: [email protected]
Page 1 of 41Articles in PresS. Am J Physiol Lung Cell Mol Physiol (July 11, 2008). doi:10.1152/ajplung.00516.2007
Copyright © 2008 by the American Physiological Society.

Abstract
Persistent pulmonary hypertension of the newborn (PPHN) is characterized
by endothelial dysfunction and decreased vascular growth. The role of rho-kinase
activity in modulating endothelial function and regulating angiogenesis during
normal lung development and in PPHN are unknown. We hypothesized that PPHN
increases rho-kinase activity in fetal pulmonary artery endothelial cells (PAECs)
and impairs angiogenesis in vitro. Proximal PAECs were harvested from fetal
sheep with partial ligation of the ductus arteriosus in utero (PPHN) and age-
matched controls. Rho-kinase activity, was measured by rhoA, RhoGTP and P-
MYPT-1 protein content. The effects of rho-kinase activity on angiogenesis, eNOS
protein expression and NO production were determined in normal and PPHN
PAECs. Angiogenesis was assessed by tube formation in vitro with/without Y-
27632, (rho-kinase inhibitor), and calpeptin, (rho-kinase activator), in
presence/absence of L-NA (NOS inhibitor). RhoA, rho-GTP and P-MYPT-1 protein
were increased in PPHN PAECs. Tube formation was reduced by 29% in PPHN
PAECs (p<0.001) and increased with Y-27632 treatment in normal and PPHN
PAECs with PPHN PAECs achieving similar values to normal PAECs. L-NA
inhibited the Y-27632-induced increase in tube formation in normal but not PPHN
PAECs. Calpeptin reduced tube formation in normal and PPHN PAECs. eNOS
expression, was reduced by 42% in PPHN PAECs (p<0.01). Y-27632 increased
eNOS protein and NO production in normal and PPHN PAECs. Calpeptin
decreased eNOS protein only in normal PAECs, but reduced NO production in
Page 2 of 41

normal and PPHN PAECs. We conclude that rho-kinase activity is increased in
PPHN PAECs, which down-regulates eNOS protein and NO production and
impairs angiogenesis in vitro.
Key terms: Persistent pulmonary hypertension of the newborn, pulmonary
hypertension, angiogenesis, vasculogenesis, rho-kinase, nitric oxide, endothelial
nitric oxide synthase, endothelial cells, lung vascular development.
Page 3 of 41

Introduction
Persistent pulmonary hypertension of the newborn (PPHN) is a clinical
syndrome characterized by elevated pulmonary vascular resistance (PVR) that
persists after birth, leading to extrapulmonary right to left shunting and profound
hypoxemia. Mechanisms responsible for elevated PVR in PPHN include increased
vascular tone, hypertensive remodeling and in the most severe cases, impaired
angiogenesis or vascular growth (12). Impaired angiogenesis is usually seen in the
setting of PPHN with lung hypoplasia, such as with congenital diaphragmatic hernia
(12,14). In the presence of lung hypoplasia, decreased arterial number plays an
especially prominent role in maintaining high PVR, resulting in disease that is often
refractory to vasodilator therapies, such as inhaled nitric oxide (14). In this setting,
novel strategies that can stimulate vascular growth and increase arterial number
may improve outcomes of neonates with severe PPHN and lung hypoplasia.
However, mechanisms that impair angiogenesis and enhance lung vascular growth
in severe PPHN are poorly understood.
Past studies have shown that partial ligation of the ductus arteriosus (DA) in
late gestation fetal sheep provides a useful animal model for studying the
pathogenesis and treatment of PPHN (3,29,54). In this model, partial DA ligation
increases pulmonary artery pressure without causing sustained elevations of
pulmonary blood flow or hypoxemia (1). At delivery, PVR remains elevated and
causes hypoxemia due to extrapulmonary shunting despite mechanical ventilation
with supplemental oxygen (1). Physiologically, this model of PPHN is characterized
Page 4 of 41

by marked endothelial dysfunction, as reflected by the early loss of endothelium-
dependent vasodilation, with down-regulation of lung endothelial NO synthase
(eNOS) expression, impaired NO production, increased superoxide generation and
increased ET-1 expression (16,17,22,41,48,49). Overall, these and other findings
suggest that disruption of normal endothelial function in the fetal lung increases
pulmonary vasoconstriction and causes abnormal vasoreactivity in PPHN. In
addition to its role in the regulation of vascular tone, the endothelial cell also
modulates vascular structure and growth. Recent studies in this experimental
model have shown that chronic intrauterine pulmonary hypertension impairs lung
angiogenesis and cause lung hypoplasia (11). We recently demonstrated that
endothelial cells from PPHN fetal sheep maintain an abnormal phenotype in vitro,
which is characterized by decreased growth and impaired tube formation (9).
However, how hemodynamic stress induced by hypertension alters endothelial cell
function and impairs vascular growth in PPHN is unknown.
Rho-kinase signaling is a complex pathway responsible for cellular
proliferation, migration, differentiation and gene expression in diverse vascular beds
(26). Rho-kinase activity has been shown to regulate smooth muscle cell
contraction and vascular tone in systemic and pulmonary circulations (4,5,32,53).
During lung development, rho-kinase activity maintains high PVR in the fetal lung
(35) and may contribute to increased vascular tone in neonatal pulmonary
hypertension. In adult animal models of pulmonary hypertension, rho-kinase
activity is increased (21,26,32,52), however these studies have largely focused on
the effects of rho-kinase activation on smooth muscle cell function, demonstrating
Page 5 of 41

that increased rho-kinase activity elevates vascular tone, mediates calcium
sensitization and contributes to hypertensive remodeling (5,32,42,51,53). In adult
models of pulmonary hypertension, acute treatment with rho-kinase inhibitors
causes potent pulmonary vasodilation and chronic therapy prevents vascular
remodeling and improves survival (20,21,26,30,34).
In addition to its effects in smooth muscle cells, rho-kinase activity also
modulates endothelial cell function. RhoGTPases are key regulators of endothelial
permeability (2,50,55) and rhoA activation increases vascular permeability
(10,39,47). Whether rho-kinase activity regulates angiogenesis, especially in the
lung circulation, is controversial. In adult models of pulmonary hypertension due to
chronic hypoxia, inhibition of rho-kinase activity prevents pulmonary hypertension
and inhibits angiogenesis (13). Unlike these findings in the adult lung, experimental
pulmonary hypertension in fetal sheep is associated with reduced vascular growth
and impaired angiogenesis (11). Whether increased rho-kinase activity impairs
endothelial cell function and reduces vascular growth in severe pulmonary
hypertension, especially in the developing lung or in neonatal pulmonary
hypertension, remains unknown.
Since eNOS protein expression is decreased in PPHN PAECs (9) and rho-
kinase activity regulates eNOS protein expression and NO production (27,33,36),
we proposed to test whether reduced eNOS protein expression and tube formation
in PPHN PAECS is due to increased rho-kinase activity. Specifically, we
hypothesized that chronic intrauterine pulmonary hypertension would increase rho-
kinase activity in pulmonary artery endothelial cells, resulting in endothelial cell
Page 6 of 41

dysfunction, decreased eNOS expression and impaired angiogenesis. We further
hypothesized that increased rho-kinase activity would impair angiogenesis in fetal
and hypertensive PAECs due to reduced NO production. In this study, we report
increased rho-kinase activity in PAECs from PPHN lambs, and that rho-kinase
inhibition increases eNOS expression and NO production and enhances tube
formation in vitro. Overall, these findings support the hypothesis that rho-kinase
activation contributes to endothelial cell dysfunction and impaired angiogenesis in
PPHN.
Page 7 of 41

Methods
Isolation and culture of fetal ovine pulmonary arterial endothelial cells.
Isolation and culture of fetal ovine pulmonary arterial endothelial cells. All
procedures and protocols were reviewed and approved by the Animal Care and
Use Committee at the University of Colorado Health Sciences Center, Denver, CO.
The left and right pulmonary arteries were isolated from late-gestation normal fetal
sheep (mixed-breed Columbia-Rambouillet pregnant ewes at 135 days gestation
(n=4), term = 147 days) and from fetal sheep that had undergone partial ligation of
the ductus arteriosus in utero 7-10 days prior to euthanasia (PPHN)(n=4)(as
previously described, (3,29,54). Proximal PAECs were isolated as previously
described (9,22) and endothelial cell phenotype confirmed by positive
immunostaining for von Willebrands Factor (vWF), eNOS, vascular endothelial
(VE)-cadherin. VEGF-R2 (KDR), positive uptake of ac-LDL and negative staining
for desmin. Cells from passage 4 and 5 were used for each of the study
experiments and cells from each animal were kept separate throughout all
passages and for all experiments.
ELISA: ELISA was performed using the G-LISA RhoA Activation Assay
(Cytoskeleton Inc, Denver, CO #BK124) and the assay was performed according
to the manufacturers instructions. Briefly PAECs from control and PPHN lambs
were grown to 50-70% confluence in 150mm dishes and cell lysate collected by
Page 8 of 41

scraping the dishes. Lysates were snap frozen in liquid nitrogen and stored at -
80°C. After thawing protein concentrations were determined and samples were
prepared with identical protein concentrations. The rhoA activation assay was
performed in triplicate and rho GTP signal was determined by measuring
absorbance at 490nm using a microplate spectrophotometer. Differences in
absorbance between normal and PPHN PAECs were measured and quantified.
Membrane-cytosolic separation: Membrane fraction separation was
performed using the ProteoExtract Native Membrane Protein Extraction Kit
(Calbiochem, Cat#444810 San Diego, CA). Briefly, PAECs from normal and PPHN
fetal sheep were grown to 95% confluence in 150mm dishes and cells were
detached from the dishes using 0.25% trypsin. Membrane protein was extracted
from whole cell lysates per manufacturer instructions.
The cytosolic fraction was extracted using the Mem-PER Eukaryotic
membrane extraction kit (Pierce Biotechnology Inc (catalog # 89826) Rockford, IL).
PAECs from normal and PPHN fetal sheep were grown to 95% confluence in
150mm dishes and cells were detached from the dishes using 0.25% trypsin.
Cytosolic protein was extracted from whole cell lysates per manufacturer
instructions.
Protein content in the membrane and cytosolic samples was determined
by the Bicinchoninic acid assay (BCA) (Pierce Biotechnology Inc (catalog #
23225) Rockford, IL), using bovine serum albumin as the standard. Twenty µg of
protein sample per lane was resolved by SDS polyacrylamide gel
Page 9 of 41

electrophoresis. Proteins from the gel were transferred to nitrocellulose
membrane and Rho A protein was detected by western blot analysis (See
protocol below).
Tube Formation Assay: The ability of fetal PAECs to form vascular
structures in vitro was assayed by plating PAEC’s on type 1 collagen. Collagen
was pipetted into 24 well tissue culture dishes (250 µl/well) and allowed to
polymerize at 37°C for one hour. PAEC from normal and PPHN fetal sheep were
seeded at a density of 5 x 104 cells/well in serum free DMEM supplemented with
and without Y-27632 (1µM; rho-kinase inhibitor) ,calpeptin (100µg/ml; rho-kinase
activator) and Y-27632 (1µM) with nitro-L-arginine (LNA; a NOS inhibitor; 4mM).
Doses for each drug were determined by preliminary experiments and published
studies (40). The lowest dose for which an effect was seen was used for all drugs.
PAECs were incubated in 3% oxygen conditions in order to simulate the low
oxygen environment in the normal fetus (9). After 6 hours, branch point counting
was performed in blinded fashion under 10X magnification from each of 4 wells, as
previously described (38).
Western Blot Analysis: PAECs from normal and PPHN animals were
grown on 150mm cloning dishes in DMEM supplemented with 5% serum. At 70%
confluence, PAECs were treated with Y-27632 (1µM) for 24 hours and calpeptin
(100µg/ml) were for 30 minutes per the manufacturer’s recommendations. Cells
were washed with ice cold PBS x 2 and lysed in radioimmunoprecipitation (RIPA)
buffer (PBS, 1% Nonidet P-40, 0.5%, sodium deoxycholate, 0.1% SDS, PMSF
Page 10 of 41

[10mg/ml], aprotinin [16 µl/ml], and sodium orthovanadate [1mM]). Cell lysates
were scraped off the dishes, sonicated, and centrifuged at 10,000 x g for 30 min
at 4°C. The supernatant was removed and protein content in the supernatant
was determined by the BCA assay (Pierce Biotechnology Inc (catalog # 23225)
Rockford, IL), using bovine serum albumin as the standard. 20 µg of protein
sample per lane was resolved by SDS polyacrylamide gel electrophoresis, and
proteins from the gel were transferred to nitrocellulose membrane.
Rho A: Blots were blocked for 30 minutes in 5% nonfat dry milk dissolved in
buffer 1 (10mM tris-hcl, 150mM NaCl, 0.05% tween-20, PH 8.0) Blots were
incubated for 2 hours at room temperature with anti ROCK-II/ROK∝ (BD610624
BD Biosciences, San Jose, CA) (1:500) diluted in 5% nonfat dry milk in buffer 1.
After washing, blots were incubated for 1 hour at room temperature with goat
anti-mouse HRP conjugated secondary (Chemicon, Billerica, MA)(1:10000).
Bands of interest were visualized by enhanced chemiluminenscence (ECL+ kit;
Amersham Pharmacia Biotech, Buckinghamshire, UK), identified by molecular
weight as identified by the manufacturer for the protein of interest.
Phospho-MYPT-1: Blots were blocked for 30 minutes with 5% nonfat dry
milk dissolved in buffer 1 (10mM tris-hcl, 150mM NaCl, 0.05% tween-20, PH 8.0).
Blots were then incubated overnight with Phospho-MYPT1 (Thr853) Antibody
(#4563 Cell Signaling, Danvers, MA) (1:500). After washing, blots were incubated
for 1 hour at room temperature with goat anti-rabbit HRP conjugated secondary
Page 11 of 41

(Santa Cruz Biotech, SC2054). Bands of interest were visualized by enhanced
chemiluminenscence (ECL+ kit; Amersham Pharmacia Biotech,
Buckinghamshire, UK), identified by molecular weight as identified by the
manufacturer for the protein of interest.
eNOS: Blots were blocked for 30 minutes in 2% ECL advance
(Amersham Pharmacia Biotech, Buckinghamshire, UK) dissolved in PBS with
0.05% Tween 20 after which time the blots were incubated for 1 hour with
BD610297 (eNOS/NOS III)(1:1000) diluted in 2% ECL advance. After washing,
blots were incubated for 1 hour at room temperature with goat anti-mouse HRP
conjugated secondary (Chemicon)(1:20000). Bands of interest were visualized
using the ECL advance kit, identified by molecular weight as identified by the
manufacturer for the protein of interest.
All blots were then stripped and reprobed with an antibody to β-actin
(Sigma,St. Louis, A5316). Densitometry was performed using NIH Image (v1.61).
Changes in protein expression were analyzed after normalizing for β-actin
expression.
Nitric Oxide Assay: NO production was determined with the DAF-FM
Nitric Oxide indicator (Molecular Probes, Eugene, OR #D-23844). 5 x 103 normal
and PPHN PAECs were plated in 96 well plates in DMEM with 5% FBS under
3% oxygen conditions. Cells were allowed to adhere overnight after which normal
and PPHN PAECs were incubated with DAF-FM with and without Y-27632 (1µM)
and calpeptin (100µg/ml) in PBS for 1 hour. PBS containing DAF-FM fluorescent
Page 12 of 41

probe was transferred to a black 96 well plate and NO production measured in
response rho-kinase activation and inhibition using a microplate reader with
fluorescence excitation and emission maxima of 495 and 515 nm, respectively.
Comparisons were made between normal and PPHN cells with respect to NO
production.
Statistical analysis. Data are presented as means ± SEM. Statistical analysis
was performed with the Prism 4 software package (GraphPad Software, San Diego,
CA). Statistical comparisons were made using analysis of variance for tube formation
assays with Bonferroni post test analysis. Unpaired t test was used for western blot
and ELISA analysis and NO production studies. P < 0.05 was considered significant.
Page 13 of 41

Results
Increased Rho-kinase activity in fetal PAECS from PPHN Lambs. In
comparison with controls, PAECs from PPHN lambs exhibited increased rho-
kinase activity. Western blot analysis on whole cell lysates from normal and PPHN
PAECs demonstrated a 93% increase in total Rho A protein in PAECs from PPHN
lambs (p<0.05)(fig 1a). As determined by ELISA, rho-GTP, the active form of rho,
was increased by 53% in PPHN PAECs (p<0.001) (fig 1b.) In addition to increased
rhoA and rho-GTP protein, phosphorylation of MYPT-1, another measure of rho-
kinase activity, was increased 65% in PPHN PAECs (p<0.01) (fig 1c). When
separated into membrane and cytosolic protein fractions, cell lysates from PPHN
PAECs demonstrate increased rho A protein in both membrane and cystosolic
fractions when compared with normal controls. In comparison with control PAECs,
rho A membrane and cytosolic protein contents were increased by 34% and 52%,
respectively (p<0.01) (fig 2).
Activation of rho-kinase with calpeptin. Western blot analysis on whole cell
lystates from normal and PPHN PAECs demonstrated that rho-kinase activation
with calpeptin increased phosphorylation of MYPT-1 by 85% in normal PAECs
(p<0.01) (fig 3a). With calpeptin treatment there was no further increase in
phosphorylation of MYPT-1 in PPHN PAECs (p=NS)(fig3b.).
Effect of Rho-kinase inhibition and stimulation on tube formation in vitro.
Treatment with Y-27632, a rho-kinase inhibitor, increased tube formation in both
Page 14 of 41

normal and PPHN PAECs. Tube formation was increased by 13% (p<0.01) and
31% (p<0.001) in normal and PPHN PAECs, respectively (fig 4a). Rho-kinase
inhibition increased tube formation by PPHN PAECs to values achieved in normal
PAECs. Treatment with calpeptin, decreased tube formation in both normal and
PPHN PAECs by 29% (p<0.001) and 21% (p<0.01), respectively. (fig 4b). The
addition of S-nitroso-N-acetylpenicillamine (SNAP) as an NO donor did not prevent
the decrease in tube formation due to rho-kinase activation. Tube formation
remained decreased by 25% (p<0.001) in normal and by 17% (p<0.001) in PPHN
PAECs (fig not shown).
Effect of Rho-kinase inhibition and stimulation on eNOS expression. Rho-kinase
inhibition with Y-27632 increased eNOS protein content in both normal and PPHN
PAECs. eNOS protein expression was increased by 30% (p<0.01) and 58% (p<0.05)
in normal and PPHN PAECs, respectively (fig 5a). Rho-kinase activation with
calpeptin decreased eNOS protein expression by 28% (p<0.01) in normal PAECs,
however, calpeptin did not cause a further decrease in eNOS protein expression in
PPHN PAECs ((fig 5b), which was decreased by 42% (p<0.01) at baseline.
Effect of Rho-kinase inhibition and activation on nitric oxide (NO) production
in normal and PPHN PAECs. Rho-kinase inhibition with Y-27632 increased NO
production in both normal and PPHN PAECs (fig 6a.). With rho-kinase inhibition
NO production increased 63% (p<0.001) in normal and 64% (p<0.001) in PPHN
Page 15 of 41

PAECs. Rho-kinase activation decreased NO production by 31% (p<0.05) and
25% (p>0.05) in normal and PPHN PAECs respectively (fig 6b).
Increase in tube formation with rho-kinase inhibition is nitric oxide dependent in
normal but not PPHN PAECs. The increase in tube formation seen with rho-kinase
inhibition was reversed with nitric oxide synthase inhibition using L-NA in normal
PAECs. Tube formation decreased by 30% (p<0.001) with the addition of L-NA to
Y-27632 (fig 7). Values achieved were 22% lower than that achieved by normal
controls. In PPHN PAECs, L-NA had no effect on the increase in tube formation
with rho-kinase inhibition (fig 7).
Page 16 of 41

Discussion
In addition to increased pulmonary vascular tone and hypertensive remodeling,
impaired angiogenesis also contributes to high PVR in severe PPHN, especially in
the setting of lung hypoplasia (12). Previous studies have shown that pulmonary
hypertension during late gestation impairs fetal lung vascular growth in vivo (11),
and causes abnormalities in endothelial cell phenotype that persist in vitro (9).
However, mechanisms through which sustained elevations of pulmonary arterial
pressure inhibit lung angiogenesis during development are unknown. Since rho-
kinase activity modulates eNOS protein expression and activity, we hypothesized
that increased rho-kinase activity may account for the change in endothelial cell
phenotype seen in PPHN. We found that rho kinase activity, as assessed by rhoA,
and rhoGTP protein expression and phosphorylation of MYPT-1, was increased in
PAECs harvested from PPHN lambs. We also found that treatment with Y27632, a
rho kinase inhibitor, increased eNOS protein expression and NO production and
rescued the abnormal in vitro phenotype, restoring tube formation by PPHN
PAECs to normal levels. In addition, treatment with calpeptin, a rho kinase
activator, increased phosphorylation of MYPT-1, decreased eNOS protein
expression and NO production, and decreased tube formation in vitro in normal
PAECs. These findings demonstrate that chronic intrauterine pulmonary
hypertension increases rho kinase activity in lung vascular endothelium, which
Page 17 of 41

contributes to impaired angiogenesis, reduced eNOS protein content and
decreased NO production in PPHN.
This is the first study of rho-kinase activity in fetal PAECs and these findings
demonstrate increased rho-kinase activity in PPHN PAECS, suggesting a role for
the rho-kinase pathway in regulating angiogenesis in the developing lung. Prior
studies in experimental pulmonary hypertension have demonstrated increased rho-
kinase activity in the adult, but these reports have primarily focused on rho-kinase
activity in the smooth muscle cell and its effect on vascular tone and hypertensive
remodeling in pulmonary hypertension. With exposure to acute hypoxia inhibition
of rho-kinase activity attenuates the constrictor response in adult rats (37,52), while
after chronic hypoxia, rho-kinase inhibition decreases mean pulmonary artery
pressure (30,32). Pulmonary hypertension induced by chronic hypoxia has
previously been attributed to structural changes in the pulmonary vasculature
including hypertensive remodeling which produces a fixed increase in resistance
(13). Sustained inhibition of rho-kinase throughout the period of hypoxic exposure
attenuates pulmonary hypertension and prevents vascular remodeling (20). Adult
rats when treated with a single dose of monocrotaline develop severe pulmonary
hypertension and vascular remodeling (8,46). Chronic rho-kinase inhibition in this
setting prevents pulmonary vascular remodeling, by suppressing vascular smooth
muscle cell proliferation and macrophage infiltration (19). These studies indicate
that rho-kinase–mediated pathways are substantially involved in the pathogenesis
of pulmonary hypertension contributing significantly to vascular tone and
hypertensive remodeling in PPHN.
Page 18 of 41

We recently reported that prolonged intrauterine pulmonary hypertension
impairs angiogenesis and decreases alveolarization and lung weight in fetal lambs
(11). Intrauterine pulmonary hypertension also directly alters endothelial cell
function and impairs growth and tube formation by isolated PAECs in vitro. (9).
Thus, pulmonary hypertension itself can impair endothelial cell function, reduce
vascular growth and cause lung hypoplasia. How pulmonary hypertension alters
endothelial cell function and contributes to impaired angiogenesis in PPHN
remains unknown, but our results implicate the rho-kinase signal transduction
pathway as contributing to endothelial cell dysfunction and impaired vascular
growth in PPHN.
Our studies demonstrate a role for the rho-kinase pathway in regulating
these important endothelial cell functions and contributing to normal blood vessel
formation in the developing lung as well as impaired angiogenesis in PPHN.
Whether decreased alveolarization in PPHN is mediated by rho-kinase is unknown.
Prior studies have implicated rho-kinase signaling in regulating alveolarization
during development (26,28), but mechanisms underlying these findings were not
explored. Past studies have suggested that inhibition of vascular growth impairs
alveolarization (18,44,45). We speculate that high rho-kinase activity in PPHN
PAECs decreases vascular growth and subsequent alveolarization. Fetal lung
explants incubated for 48 hours in 3% oxygen show increased branching as well
as membrane associated rhoA when compared with room air controls (7,26).
Page 19 of 41

Recent studies have demonstrated that under hypoxic condition rho-kinase is
activated (26,37,43,52), suggesting that the increase in lung branching seen under
hypoxic conditions may be mediated by rho-kinase. The fawn hooded rat (FHR) is
a genetic model of pulmonary hypertension and is characterized by increased rho-
kinase activity (31). FHRs when exposed to mild hypoxia seen at Denver’s altitude
develop alveolar simplification and pulmonary hypertension (23,24). In this model
chronic rho-kinase inhibition improves alveolarization and vascular growth. These
studies support our findings that while rho-kinase activity during fetal life may
regulate lung growth, inappropriate rho-kinase activation during fetal life or
persistence of rho-kinase activation after birth, may impair angiogenesis and lung
growth.
Prior studies have demonstrated that inhibition of rho-kinase upregulates
and activates eNOS, increasing the production of NO (27,33,36). Long-term
inhibition of rho-kinase activity is protective against pulmonary hypertension and
right ventricular hypertrophy in hypoxia exposed adult mice (20), but was less
effective in eNOS (-/-) mice (20), which suggests that eNOS activation after rho-
kinase inhibition is responsible for these protective effects. We report that rho-
kinase inhibition increases eNOS protein expression and activity in both normal
and PPHN PAECs, which supports the concept that eNOS activation and
increased NO production may be responsible for enhanced angiogenesis in vitro
by normal and PPHN PAECs. However, the effect of rho-kinase inhibition on
angiogenesis was lost in normal but not PPHN PAECS with NOS inhibition using
Page 20 of 41

L-NA. This finding suggests an NO independent effect of rho-kinase inhibition
and, raises the possibility that in conditions associated with dysfunctional eNOS,
and impaired NO production, rho-kinase inhibitors may have even greater
therapeutic benefit. Interestingly, Lohn et al demonstrated that in the models of
genetically reduced endothelial NO production (eNOS-/- mice and spontaneous
hypertensive rats) and in models of pharmacologically reduced endogenous NO
production (LNAME treatment), rho-kinase inhibition produced a strong
vasodilator response, which also suggests that inhibition of rho kinase has NO-
independent effects as well (25).
Potential limitations of this study include the use of fetal PAECs harvested
from relatively large vessels, and that differences may exist in the behavior of
these cells as compared to microvascular PAECs. Since microvascular PAECs
may primarily be involved in lung angiogenesis during development in vivo, future
studies are needed to compare and contrast rho-kinase activity in microvascular
PAECs. While calpeptin markedly increased phosphorylation of MYPT-1 in normal
PAECs, there was no further increase in phosphorylation of MYPT-1 in PPHN
PAECs. NO production, however, was significantly reduced with calpeptin
treatment in both normal and PPHN PAECs. While the effects of calpeptin
treatment may not be mediated through rho-kinase, the decrease in NO production
with calpeptin treatment may support the concept that the detrimental effects of
rho-kinase activation are the result of decreased eNOS activity. Another potential
limitation is the fact that angiogenesis was only measured in vitro, and whether
Page 21 of 41

rho-kinase inhibition enhances angiogenesis in vivo remains unknown.
In conclusion, we found that chronic intrauterine pulmonary hypertension
increases rho-kinase activity in PAECs harvested from PPHN lambs and that this
increase in rho-kinase activity directly contributes to downregulated eNOS,
decreased NO production and impaired angiogenesis in vitro. Rho-kinase inhibition
reversed the abnormal in vitro phenotype previously described in PPHN PAECs.
This effect however was found to be NO independent. These findings suggest that
treatment strategies that down regulate or inhibit rho-kinase activation in PPHN
may enhance angiogenesis in vivo, and may be especially important in treating
pulmonary hypertension in the presence of endothelial dysfunction and lung
hypoplasia.
Page 22 of 41

Acknowledgements:
This work was supported in part by grants from iNO Therapeutics (Gien) and the
NIH R01 HL068702 (Abman).
Page 23 of 41

1. Abman SH SP, Accurso FJ. Failure of postnatal adaptation of the
pulmonary circulation after chronic intrauterine pulmonary hypertension in fetal
lambs. J Clin Invest 83: 1849-1858, 1989.
2. Arbajal JM SRJ. RhoA inactivation enhances endothelial cell barrier
function. RhoA inactivation enhances endothelial cell barrier function. Am J
Physiol. 1999, 277:C955-C964.
3. Belik J KF, Baldwin F, Rabinovitch M. Pulmonary hypertension and
vascular remodeling in fetal sheep. Am J Physiol 266: H2303-2309, 1994.
4. Bi D NJ, Niiro N, Hirano K, Kanaide H. Contractile properties of the
cultured vascular smooth muscle cells: the crucial role played by RhoA in the
regulation of contractility. Circ Res 96: 890-897, 2005.
5. Bussemaker E PF, Forrster S, Herbrig K, Gross P, Passauer J, Brandes
RP. Rho kinase contributes to basal vascular tone in humans: role of
endothelium-derived nitric oxide. Am J Physiol Heart Circ Physiol 293: H541-547,
2007.
6. Chapados R, Khotaro A; Ihida-Stansbury, K; McKean, D; Gates, AT.;
Kern, M; Merklinger, S; Elliott, J; Plant, A; Shimokawa, H; Jones, PL. ROCK
Controls Matrix Synthesis in Vascular Smooth Muscle Cells: Coupling
Vasoconstriction to Vascular Remodeling. Circulation Research 99: 837-844,
2006.
7. Gebb SA JP. Hypoxa and Lung Branching Morphogenesis. Adv Exp Med
Biol 543: 117-125, 2003.
8. Ghodsi F WJ. Changes in pulmonary structure and function induced by
monocrotaline intoxication. Am J Physiol 240: H149-155., 1981.
9. Gien J SG, Balasubramaniam V, Markham N, Abman SH. Chronic
Page 24 of 41

Intrauterine Pulmonary Hypertension Impairs Angiogenesis and Growth of Ovine
Fetal Pulmonary Artery Endothelial Cells in vitro. Am J Respir Crit Care Med
176: 1146-1153, 2007.
10. Gorovoy M NR, Niu J, Vogel S, Predescu D, Miyoshi J, Takai Y, Kini V,
Mehta D, Malik AB, Voyno-Yasenetskaya T. Rho GDI-1 Modulation of the Activity
of Monomeric RhoGTPase Rho A Regulates Endothelial Barrier Function in
Mouse Lungs. Circ Res 101: 50-58, 2007.
11. Grover TR PT, Balasubramaniam V, Markham NE, Abman SH. Pulmonary
hypertension impairs alveolarization and reduces lung growth in the ovine fetus.
Am J Physiol Lung Cell Mol Physiol 288: L648-L654, 2005.
12. Hopkins N MP. The structural basis of pulmonary hypertension in chronic
lung disease: remodelling, rarefaction or angiogenesis? J Anat 201: 335-348,
2002.
13. Howell K PR, McLoughlin P. Chronic hypoxia causes angiogenesis in
addition to remodelling in the adult rat pulmonary circulation. J Physiol 547: 133–
145, 2003.
14. Hwang SJ LK, Lee KH,Hwang JH, Choi CW, Shim JW, Chang YS, Park
WS. Factors affecting the response to inhaled nitric oxide therapy in persistent
pulmonary hypertension of the newborn infants. Yonsei Med J 45: 49-55, 2004.
15. Hyvelin JM HK, Nichol A, Costello CM, Preston RJ, McLoughlin P.
Inhibition of Rho-Kinase Attenuates Hypoxia-Induced Angiogenesis in the
Pulmonary Circulation. Circ Res 97: 185 – 191, 2005.
16. Ivy DD Z, JW.; Dubus, MF.; Fox, JJ.; Kinsella, JP.; Abman, SH. Chronic
Intrauterine Pulmonary Hypertension Alters Endothelin Receptor Activity in the
Ovine Fetal Lung. Pediatric Research 39: 435-442, 1996.
Page 25 of 41

17. Ivy DD P, TA.; Ziegler, JW.; Galan, HL.; Kinsella, JP.; Tuder, RM.; Abman,
SH. Prolonged Endothelin A Receptor Blockade Attenuates Chronic Pulmonary
Hypertension in the Ovine Fetus. Journal of Clinical Investigation 99: 1179-1186,
1997.
18. Jakkula M LCT, Gebb S, Hirth KP, Tuder RM, Voelkel NF, Abman SH.
Inhibition of angiogenesis decreases alveolarization in the developing rat lung.
Am J Physiol Lung Cell Mol Physiol 279: L600-607., 2000.
19. Kohtaro A HS, Keiko M, Toyokazu U, Keiji O, Yasuharu M, Tsuyoshi H,
Yutaka N, Kozo K, Katsuo S, Akira T. Long-Term Treatment With a Rho-Kinase
Inhibitor Improves Monocrotaline-Induced Fatal Pulmonary Hypertension in Rats.
Circ Res 94: 385 – 393, 2004.
20. Kohtaro A MD PSTMKOM, PhD; Takatoshi H MD; Toyokazu U MD, PhD;
Yoshihiro F MD, PhD; Kozo K MD, PhD, Hiroaki S MD, PhD. Long-Term
Inhibition of Rho-kinase Ameliorates Hypoxia-Induced Pulmonary Hypertension
in Mice. J Cardiovasc Pharmacol 48: 280-285, 2006.
21. Kohtaro AM, PhD; Shunsuke T MS; Keiji, Oi MD, PhD; Takatoshi H MD;
Toyokazu U MD, PhD;Yoshihiro F MD, PhD; Kozo K MD, PhD, Hiroaki S MD,
PhD. Inhibition of Rho-kinase ameliorates hypoxia-induced PH in mice, eNOS
activation may be involved. J Cardiovasc Pharmacol 48: 280-285, 2006.
22. Konduri GG OJ, Shi Y, Kirkwood AP, Jr. Decreased association of HSP90
impairs endothelial nitric oxide synthase in fetal lambs with persistent pulmonary
hypertension. Am J Physiol Heart Circ Physiol 285: H204–H211, 2003.
23. Le Cras TD KD, Gebb S, Markham NE, Shannon JM, Tuder RM, Abman
SH. Abnormal lung growth and the development of pulmonary hypertension in
the Fawn-Hooded rat. Am J Physiol 277: L709-718,1999.
Page 26 of 41

24. Le Cras TD KD, Markham NE, Abman SA. Early abnormalities of
pulmonary vascular development in the Fawn-Hooded rat raised at Denver's
altitude. Am J Physiol Lung Cell Mol Physiol 279: L283-291, 2000.
25. Lohn M SK, Bleich M, Busch AE, Ivashchenko Y. Inhibition of Rho-kinase
stimulates nitric oxide-independent vasorelaxation. European Journal of
Pharmacology 507: 179 – 186, 2005.
26. McMurtry IF BN, Fagan KA, Nagaoka T, Gebb SA Oka M. Hypoxia and
Rho/Rho-kinase Signalling: Lung Development Versus Hypoxic Pulmonary
Hypertension. Adv Exp Med Biol 543: 127-137, 2003.
27. Ming XF VH, Barandier C, Ruffieux J, Kaibuchi K, Rusconi S, Yang Z. Rho
GTPase/Rho Kinase Negatively Regulates Endothelial Nitric Oxide Synthase
Phosphorylation through the Inhibition of Protein Kinase B/Akt in Human
Endothelial Cells. Mol Cell Biol 22: 8467-8477, 2002.
28. Moore KA MDHS, M.D., Ph.D., Kong Y, M.D., Ph.D., Sunday ME, M.D.,
Ph.D. Ingber DE, M.D., Ph.D. Control of Embryonic Lung Branching
Morphogenesis by the Rho Activator, Cytotoxic Necrotizing Factor 1. Journal of
Surgical Research 104: 95–100, 2002.
29. Morin FC. Ligating the ductus arteriosus before birth causes persistent
pulmonary hypertension in the newborn lamb. Pediatr Res 25: 245-250, 1989.
30. Nagaoka T FK, Gebb SA, Morris KG, Suzuki T, Shimokawa H, McMurtry
IF, Oka M. Inhaled Rho Kinase Inhibitors Are Potent and Selective Vasodilators
in Rat Pulmonary Hypertension. Am J Respir Crit Care Med 171: 494–499, 2005.
31. Nagaoka T GS, Karoor V, Homma N, Morris KG, McMurtry IF, Oka M.
Involvement of RhoA/Rho kinase signaling in pulmonary hypertension of the
Page 27 of 41

fawn-hooded rat. J Appl Physiol 100: 996-1002, 2006.
32. Nagaoka T MY, Casanova N, Bauer N, Gebb S, Mcmurtry I, Oka M.
Rho/Rho kinase signaling mediates increased basal pulmonary vascular tone in
chronically hypoxic rats. Am J Physiol Lung Cell Mol Physiol 287: L665-667,
2004.
33. Noma K ON, Liao JK. Physiological role of ROCKs in the cardiovascular
system. Am J Physiol Cell Physiol 290: C661-C668, 2006.
34. Oka M HN, Taraseviciene-Stewart L, Morris KG< Kraskauskas D, Burns
N, Voelkel NF, McMurtry IF. Rho kinase-mediated vasoconstriction is important
in severe occlusive pulmonary arterial hypertension in rats. Circ Res 100: 923-
929, 2007.
35. Parker TA RG, Grover TR, Abman SH. Rho kinase activation maintains
high pulmonary vascular resistance in the ovine fetal lung. Am J Physiol Lung
Cell Mol Physiol 291: L976-982, 2006.
36. Rikitake YL, JK. Rho GTPases, Statins, and Nitric Oxide. Circ Res 97:
1232-1235, 2005.
37. Robertson TP DM, Ward JP, Aaronson, PI, Evans AM. Inhibition of
Sustained Hypoxic Vasoconstriction by Y-27632 in Isolated Intrapulmonary
Arteries and Perfused Lung of the Rat. Br J Pharmacol 131: 5-9, 2000.
38. Salani D TG, Rosano L, Di Castro V, Borsotti P, Giavazzi R, and Bagnato
A. Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells
and stimulates neovascularization in vivo. Am J Pathol 157: 1703-1711., 2000.
39. Sawafuji M IA, Kohno M, Koh H, Tasaka S, Ishii Y, Kobayashi K. Role of
Rho-kinase in reexpansion pulmonary edema in rabbits. Am J Physiol Lung Cell
Page 28 of 41

Mol Physiol 289: L946-953, 2005.
40. Schoenwaelder SM BK. Evidence for a Calpeptin-sensitive Protein-
tyrosine Phosphatase Upstream of the Small GTPase Rho. A Novel Role For
The Calpain Inhibitor Calpeptin in the inhibition of protein-tyrosine phosphotases.
J Biol Chem 274: 14359-14367, 1999.
41. Shaul PW Y-UI, German Z, Chen Z, Steinhorn RH, Morin FC. Pulmonary
endothelial NO synthase gene expression is decreased in fetal lambs with
pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 16: L1005-L1012,
1997.
42. Stenmark KF, KA.; Frid, MG. Hypoxia-Induced Pulmonary Vascular
Remodeling: Cellular and Molecular Mechanisms. Circulation Research 99: 675-
691, 2006.
43. Takemoto MM, PhD; Sun, J MD, PhD; Hiroki, J MD; Shimokawa, H MD,
PhD; Liao, JK. MD. Rho-Kinase Mediates Hypoxia-Induced Downregulation of
Endothelial Nitric Oxide Synthase. Circulation 106: 57-62, 2002.
44. Thebaud B. Angiogenesis in lung development, injury and repair:
implications for chronic lung disease of prematurity. Neonatology 91: 291-297,
2007.
45. Thebaud B AS. Bronchopulmonary dysplasia: where have all the vessels
gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir
Crit Care Med 175: 978-985, 2007.
46. Valdivia E LJ, Hayashi Y, Sonnad J. Alterations in pulmonary alveoli after
a single injection of monocrotaline. Arch Pathol 84: 64-76, 1967.
47. van Nieuw AG vHV. Endogenous RhoA Inhibitor Protects Endothelial
Barrier. Circ Res 101: 7-9, 2007.
48. Villamor E, Le Cras TD, Horan MP, Halbower AC,Tuder RM, Abman SH.
Page 29 of 41

Chronic intrauterine pulmonary hypertension impairs endothelial nitric oxide
synthase in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 16: L1013-
L1020, 1997.
49. Villanueva ME ZF, Svinarich DM, Konduri, G. Decreased Gene
Expression of Endothelial Nitric Oxide Synthase in Newborns with Persistent
Pulmonary Hypertension. Pediatric Research: 44: 338-343, 1998.
50. Vouret-Craviari V BC, Boulter E, van Obberghen-Schilling E. Distinct
signals via Rho GTPases and Src drive shape changes by thrombin sphingosine-
1-phosphate in endothelial cells. J Cell Sci 115: 2475-2484, 2002.
51. Wang J WL, Foxson J, Shimoda LA, Sylvester JT. Ca2+ signaling in
hypoxic pulmonary vasoconstriction: effects of myosin light chain and Rho kinase
antagonists. Am J Physiol Lung Cell Mol Physiol 293: L674-685, 2007.
52. Wang Z JN, Ganguli S, Swartz DR, Li L, Rhoades RA. Rho-kinase
Activation is Involved in Hypoxia-induced Pulmonary Vasoconstriction. Am J
Respir Cell Mol Biol 25: 628-635, 2001.
53. Ward JP KG, Snetkov VA, Aaronson PI. Protein kinases in vascular
smooth muscle tone--role in the pulmonary vasculature and hypoxic pulmonary
vasoconstriction. Pharmacol Ther 104: 207-231, 2004.
54. Wild LM, Morin FC III. Ligating the ductus arteriosus before birth remodels
the pulmonary vasculature of the lamb. Pediatr Res 25: 251-257, 1989.
55. Wojciak-Stothard B PS, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc2
regulate endothelial cell permeability. J Cell Sci 114: 1343-1355, 2001.
Page 30 of 41

Figure Legends
Figure 1. Increased Rho-kinase activity in PAECs from PPHN Sheep.
RhoA, rhoGTP and phosphorylated MYPT-1 protein expression was assessed in
fetal ovine PAECs from normal and PPHN lambs by western blot analysis and
ELISA. In comparison with normal PAECs total rhoA (fig 1a), rhoGTP (fig1b) and
phosphorylated MYPT-1( fig 1c.) protein was increased in PPHN PAECs.
Error bars represent SD from mean.
Figure 2. Increased Membrane and Cystosolic Rho A Protein Expression in
PAECs from PPHN Sheep. Whole cell lysates were separated into membrane
and cytosolic fractions and both membrane and cytosolic RhoA protein was
increased in PPHN PAECs when compared with normal controls.
Error bars represent SD from mean.
Figure 3. Effects of Calpeptin on Rho-Kinase Activation in Normal and PPHN
PAECs. Phosphorylated MYPT-1 protein expression was assessed in normal
and PPHN PAECs in response to calpeptin treatment (rho-kinase activator). In
response to calppeptin treatmet phosphorylation of MYPT-1 was increased in
normal (fig 3a.) but not PPHN PAECs (fig 3b).
Page 31 of 41

Figure 4. Effect of Rho-kinase Inhibition and Stimulation on Tube Formation in vitro.
Fetal ovine PAECs from normal and PPHN sheep were plated on collagen in serum
free media under 3% oxygen conditions with and without Y-27632 (1µM) (rho-kinase
inhibitor) and calpeptin (100µg/ml)(rho-kinase activator). Y27632 treatment increased
the number of branch points in PPHN and normal PAECs (fig 4a), increasing the
number of branch points by PPHN PAECs to similar values seen in normal PAECs.
Calpeptin decreased tube formation in both normal and PPHN PAECs (fig 4b). Error
bars represent SD from mean.
Figure 5. Effect of Rho-kinase Inhibition and Activation on eNOS Protein Expression.
Cell lysates were collected from normal and PPHN PAECs with and without Y-27632
(1µM) (rho-kinase inhibitor) and calpeptin (100µg/ml)(rho-kinase activator). Rho-kinase
inhibition increased eNOS protein expression in both normal and PPHN PAECs (fig
5a.). Rho-kinase activation decreased eNOS protein expression in normal but not
PPHN PAECs (fig 5b.). Error bars represent SD from mean.
Figure 6. Effect of Rho-kinase Inhibition and Activation on Nitric Oxide (NO)
production in normal and PPHN PAECs. Rho-kinase inhibition with Y-27632
increased NO production in both normal and PPHN PAECs (fig 6a). Rho-kinase
activation decreased NO production in both normal and PPHN PAECs (fig 6b).
Page 32 of 41

Figure 7. Increase in Tube Formation with Rho-kinase inhibition is Nitric Oxide
Dependent in Normal but not PPHN PAECs. Fetal ovine PAECs from normal and
PPHN sheep were plated on collagen in serum free media under 3% oxygen
conditions with and without Y-27632 (1µM) (rho-kinase inhibitor) in the presence and
absence of L-NA(4mM). The addition of L-NA to Y-27632 decreased tube formation in
normal but not PPHN PAECs (fig 7.). Error bars represent SD from mean.
Page 33 of 41
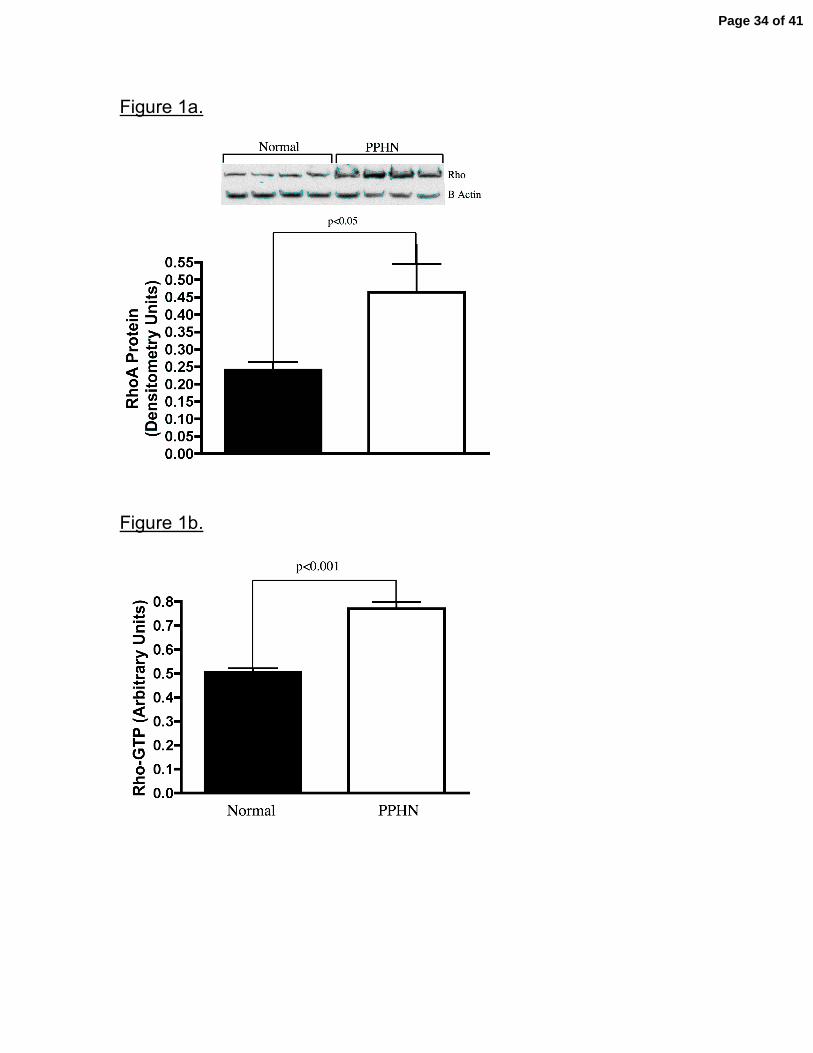
Figure 1a.
Figure 1b.
Page 34 of 41

Figure 1c.
Page 35 of 41
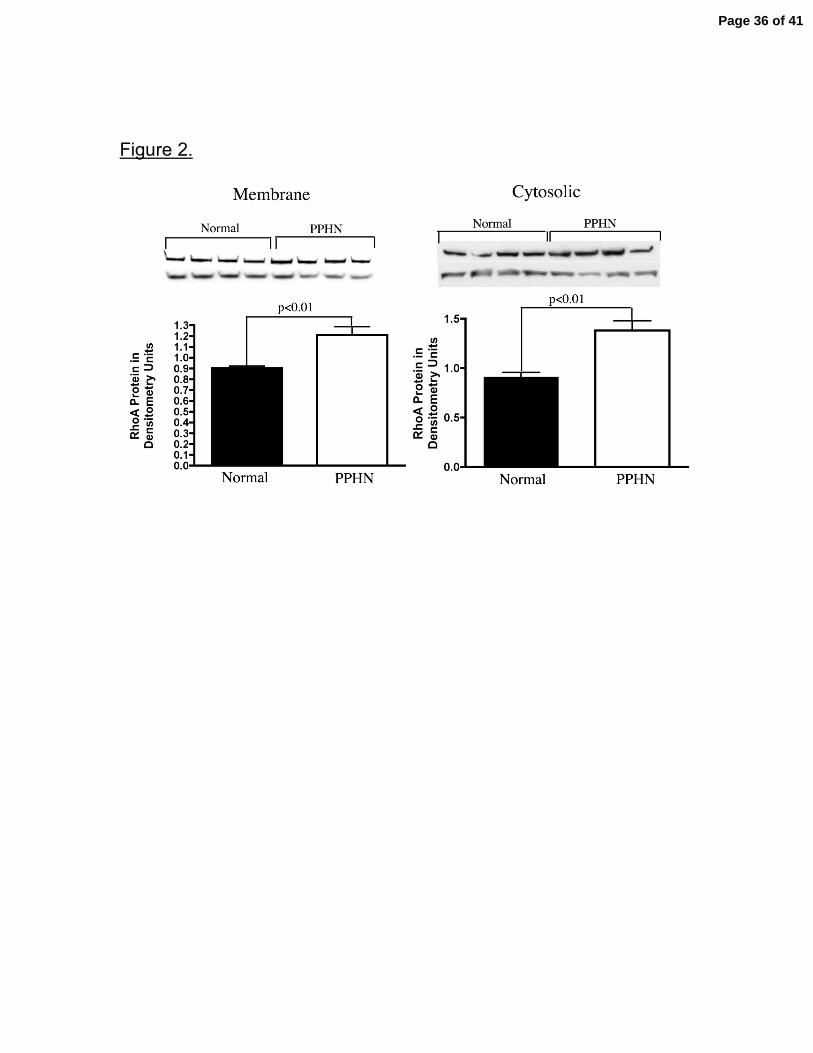
Figure 2.
Page 36 of 41

Figure 3a
Figure 3b
Page 37 of 41
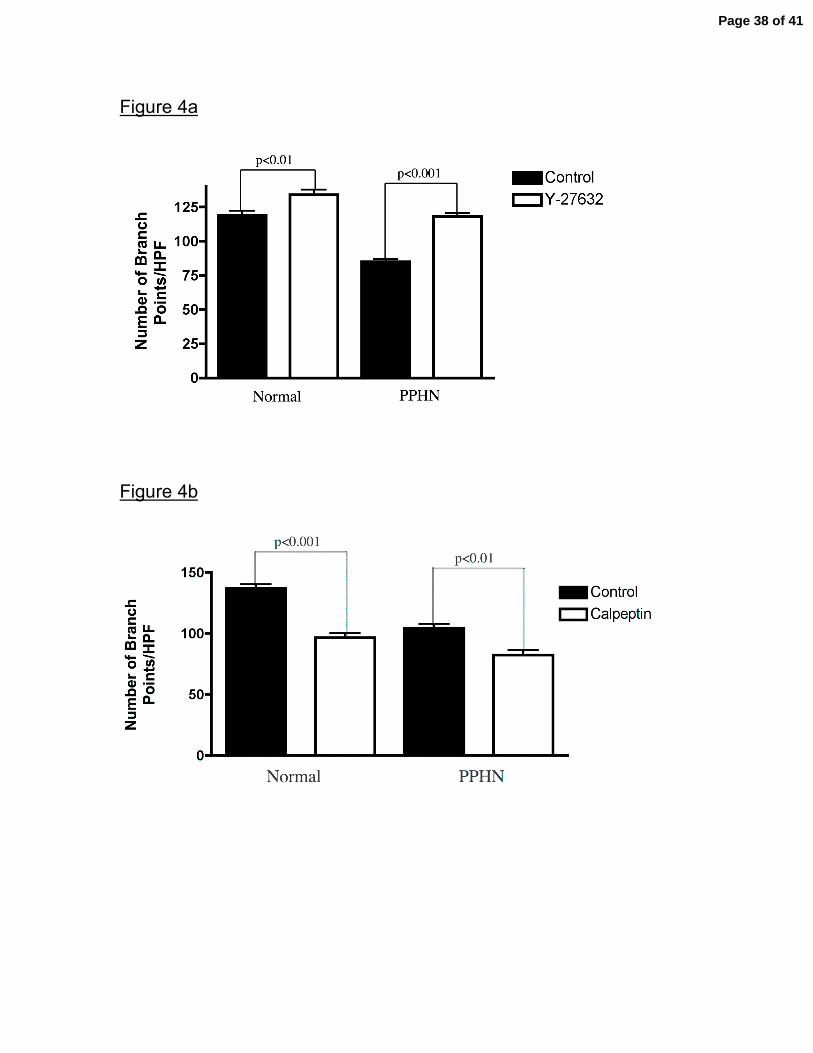
Figure 4a
Figure 4b
Page 38 of 41
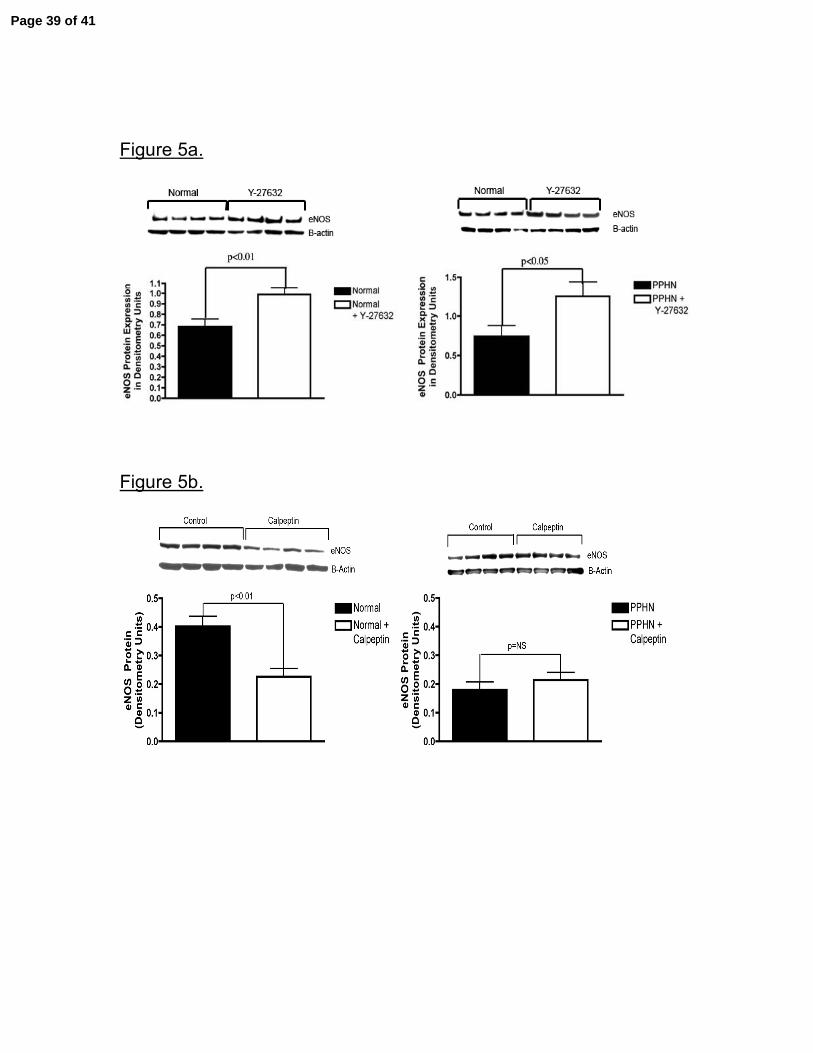
Figure 5a.
Figure 5b.
Page 39 of 41
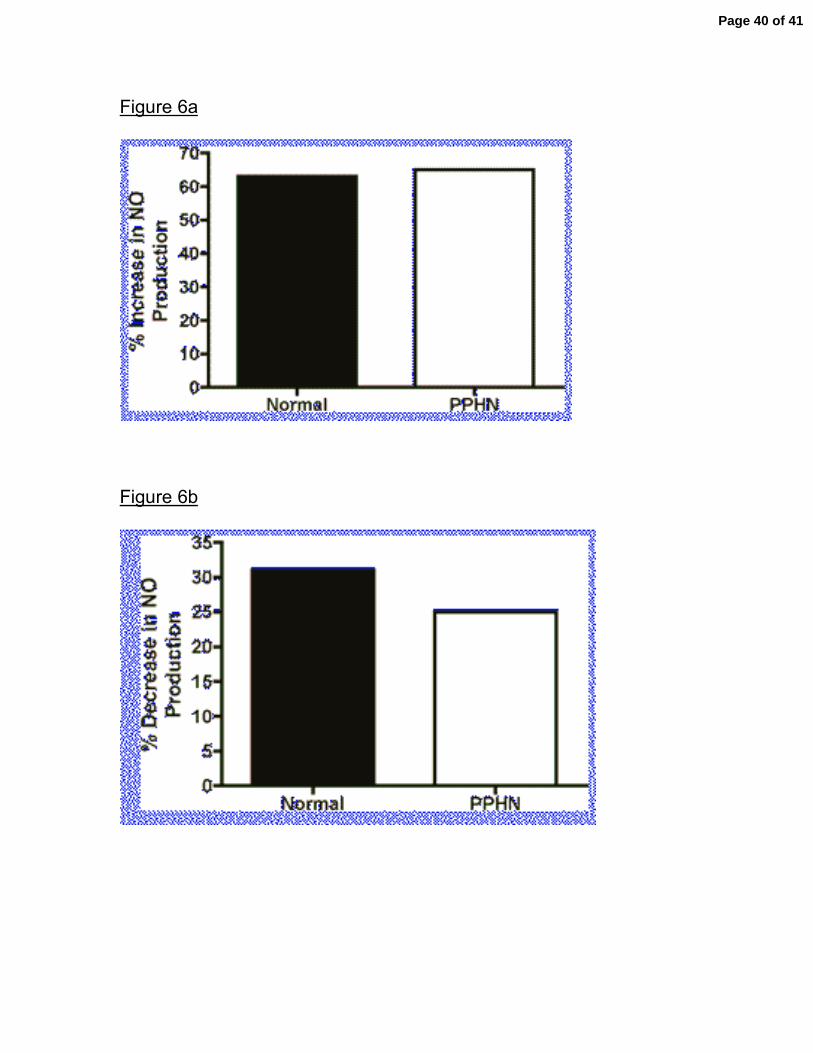
Figure 6a
Figure 6b
Page 40 of 41

Figure 7.
Page 41 of 41
Copyright © 2022 FDOKUMEN




















