
Sensory evaluation of ovine milk yoghurt with inulin addition

2002;74:753-760 Ann Thorac SurgNarula, L. Henry Edmunds, Jr and Robert C. Gorman
M. Jackson, Theodore Plappert, Navneet Narula, Martin G. St. John-Sutton, Jagat Sina L. Moainie, Joseph H. Gorman, III, T. Sloane Guy, Frank W. Bowen, III, Benjamin
An ovine model of postinfarction dilated cardiomyopathy
http://ats.ctsnetjournals.org/cgi/content/full/74/3/753on the World Wide Web at:
The online version of this article, along with updated information and services, is located
Print ISSN: 0003-4975; eISSN: 1552-6259. Southern Thoracic Surgical Association. Copyright © 2002 by The Society of Thoracic Surgeons.
is the official journal of The Society of Thoracic Surgeons and theThe Annals of Thoracic Surgery
by on June 11, 2013 ats.ctsnetjournals.orgDownloaded from

An Ovine Model of Postinfarction DilatedCardiomyopathySina L. Moainie, MD, Joseph H. Gorman, III, MD, T. Sloane Guy, MD,Frank W. Bowen, III, MD, Benjamin M. Jackson, MD, Theodore Plappert, CVT,Navneet Narula, MD, Martin G. St. John-Sutton, MBBS, Jagat Narula, MD, PhD,L. Henry Edmunds, Jr, MD, and Robert C. Gorman, MDHarrison Department of Surgical Research, Departments of Medicine and Pathology, University of Pennsylvania School ofMedicine, and the Center for Molecular Cardiology, MCP/Hahnemann University School of Medicine, Philadelphia, Pennsylvania
Background. Coronary arterial disease is the majorcause of congestive heart failure, but suitable animalmodels of postinfarction, dilated cardiomyopathy do notexist. This article describes an ovine model that developsafter an anterobasal infarction.
Methods. The distribution of ovine myocardium sup-plied by the first two diagonal branches of the lefthomonymous artery were determined in 20 slaughter-house hearts and eight live sheep using methylene blueand tetrazolium injections, respectively. Seven addi-tional animals had the infarction and underwent serialhemodynamic, microsphere and echocardiographic stud-ies more than 8 weeks and histologic studies at the eighthweek. Infarcts represented 24.6% � 4.7% and 23.9% �2.2% of the left ventricular mass in slaughterhouse andlive hearts, respectively.
Results. During remodeling, left ventricular end-systolic and end-diastolic volumes increased 115% and73%, respectively, ejection fraction decreased from 41.2%� 6.7% to 29.1% � 5.7%, systolic wall thickening remote
from the infarct decreased by 68%, sphericity indexincreased from 0.465 � 0.088 to 0.524 � 0.038, and leftventricular end-diastolic pressure increased from 1.7 �1.0 to 8.2 � 3.5 mm Hg. Serial microsphere measurementsdocumented normal blood flow (1.34 mL/g per minute) toall uninfarcted myocardium and 22% of normal to theinfarct. Viable myocardium showed mild interstitialfibrosis.
Conclusions. This ovine model meets all criteria forpostinfarction, dilated cardiomyopathy and has the ad-vantages of controlling for variations in coronary arterialanatomy, collateral vascularity, and differences in thenumbers, location, and severity of atherosclerotic lesionsthat confound human studies of the pathogenesis of thisdisease. This simple model contains only infarcted andfully perfused, hypocontractile myocardium producedby a moderate-sized, regional infarction.
(Ann Thorac Surg 2002;74:753–60)© 2002 by The Society of Thoracic Surgeons
Congestive heart failure affects 4.7 million Americansand is fatal for 50% within 5 years of diagnosis [1].
After myocardial infarction 15% to 25% of patients de-velop congestive heart failure within 5 years [2]. On thebasis of an analysis of 13 published reports Gheorghaideand Bonow [3] concluded that coronary artery disease isthe cause of 68% of all patients with congestive heartfailure. In many of these patients postinfarction, dilatedcardiomyopathy is a consequence of ventricular remod-eling and develops slowly over time without furtherinfarction [4]. Most commonly, the dilated, poorly con-tractile ventricle contains a completed segmental infarctand scattered foci of replacement fibrosis throughout theremaining myocardium [5].
The pathogenesis of this process is not well under-stood, but has been attributed to a change in myocardialmaterial properties due to occlusive or nonocclusivecoronary artery disease, myocyte cell loss, acute side-to-
side myocyte slippage, chronic myocyte hypertrophy andlengthening, which produce a marked increase in dia-stolic wall stress [6]. More recent evidence, however,suggests that progressive myocardial cell loss and re-placement fibrosis is independent of further ischemiaand is secondary to postinfarction neurohormonal re-sponses and regional wall stresses and strains [7].
Animal models of postinfarction congestive heart fail-ure do not faithfully replicate the clinical course ofpostinfarction, dilated cardiomyopathy. In rats, verylarge infarctions (50%) are required to produce myocytehypertrophy and delayed cell loss [5]. Multiple micro-sphere injections or sequential infarctions can producecardiac failure in dogs, sheep, and calves [8, 9], but thereproducibility and relevance of these models to thepathogenesis of the human disease is questionable.Other methods of inducing congestive heart failure byrapid pacing, heart block, adriamycin, and pressureoverload are not relevant to postinfarction dilatedcardiomyopathy.
During a study of the role of papillary muscles in thedevelopment of chronic ischemic mitral regurgitation in
Accepted for publication May 28, 2002.
Address reprint requests to Dr Gorman, Department of Surgery, 6Silverstein, Hospital of the University of Pennsylvania, Philadelphia,PA 19104; e-mail: [email protected].
© 2002 by The Society of Thoracic Surgeons 0003-4975/02/$22.00Published by Elsevier Science Inc PII S0003-4975(02)03827-4
by on June 11, 2013 ats.ctsnetjournals.orgDownloaded from
sheep, Gorman and colleagues [10] observed that ligationof the first two diagonal arteries of the left homony-mous (equivalent to the human left anterior descending)artery produced massive ventricular dilatation of theentire ventricle within 8 weeks. This infarct involvedonly 24% of the left ventricular (LV) mass and did notimmediately produce heart failure. The present studyconfirms and expands these findings and establishes anew, reproducible, ovine model of postinfarction, dilatedcardiomyopathy.
Material and Methods
Infarct SizingIN VITRO STUDIES. Coronary arterial anatomy was deter-mined in 20 excised Dorsett hybrid sheep hearts thatwere obtained from a local slaughterhouse. None of thesehearts and none of the in vivo studies described wereincluded in the previous report [10]. All hearts wereflushed with heparinized saline solution (10 U/mL) andstored in refrigerated saline solution containing 1g ofchloramphenicol. Because sheep lack preformed collat-eral vessels [11, 12], coronary injection of methylene bluedye effectively stains all myocardium supplied by theinjected artery or group of arteries and none of theadjacent myocardium supplied by uninjected arteries.Ten milliliters of methylene blue dye was injected byhand simultaneously into the first (D1) and second (D2)diagonal branches of the left homonymous artery,termed the left anterior descending coronary artery inthis article. After injection, stained hearts were slicedorthogonal to the long axis at 5-mm intervals from apex
to base. Each slice was traced onto acetate paper andcarefully marked to differentiate stained and unstainedmyocardium. Tracings were digitized (Truegrid, HoustonInstruments, Houston, TX) and the percentage of myo-cardium served by the combination of D1 and D2 wascalculated with customized software (Asyst SoftwareTechnologies, Rochester, NY).
IN VIVO STUDIES. In compliance with the Guide for the Careand Use of Laboratory Animals published by the Na-tional Institutes of Health (National Institutes of Healthpublication 85-23, revised 1985) 8 Dorsett hybrid sheepbetween 35 and 45 kg and bred for laboratory use(Animal Biotech Industries, Doylestown, PA) were in-duced with sodium thiopental (12.5 mg/kg intravenously)and intubated. Anesthesia was maintained with 1% to 2%isoflurane in oxygen. All animals received glycopyrrate(0.4 mg intravenously) 1 hour before incision. All animalsunderwent left thoracotomy with clean technique andligation of D1 and D2. Animals received magnesium (1gintravenously) and lidocaine (3 mg/kg intravenously)before infarction and an infusion of lidocaine (2 mg/min)for 60 minutes afterward. The percentage of infarctedmyocardium was determined after infusion of triphenyltetrazolium chloride for 30 minutes as described previ-ously [13].
Assessment of RemodelingINITIAL INSTRUMENTATION. Seven additional Dorsett hybridsheep were intubated and anesthetized as describedpreviously. Surface electrocardiogram and arterial bloodpressure were continuously monitored. A sterile left
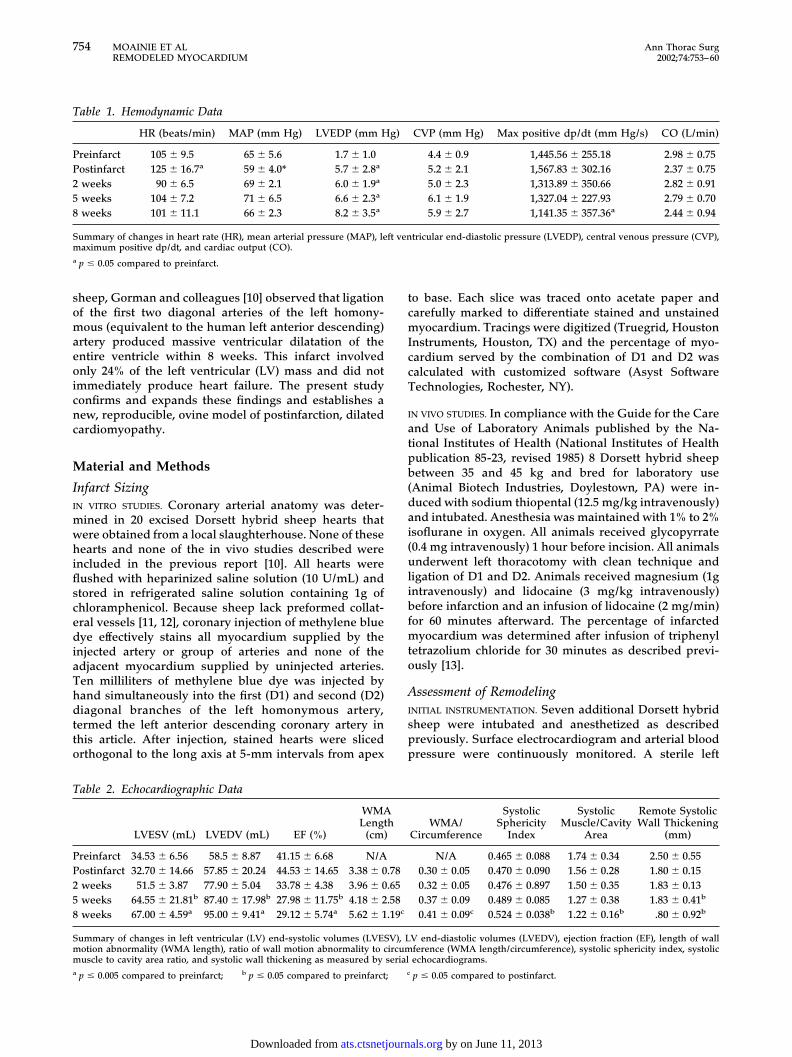
Table 1. Hemodynamic Data
HR (beats/min) MAP (mm Hg) LVEDP (mm Hg) CVP (mm Hg) Max positive dp/dt (mm Hg/s) CO (L/min)
Preinfarct 105 � 9.5 65 � 5.6 1.7 � 1.0 4.4 � 0.9 1,445.56 � 255.18 2.98 � 0.75Postinfarct 125 � 16.7a 59 � 4.0* 5.7 � 2.8a 5.2 � 2.1 1,567.83 � 302.16 2.37 � 0.752 weeks 90 � 6.5 69 � 2.1 6.0 � 1.9a 5.0 � 2.3 1,313.89 � 350.66 2.82 � 0.915 weeks 104 � 7.2 71 � 6.5 6.6 � 2.3a 6.1 � 1.9 1,327.04 � 227.93 2.79 � 0.708 weeks 101 � 11.1 66 � 2.3 8.2 � 3.5a 5.9 � 2.7 1,141.35 � 357.36a 2.44 � 0.94
Summary of changes in heart rate (HR), mean arterial pressure (MAP), left ventricular end-diastolic pressure (LVEDP), central venous pressure (CVP),maximum positive dp/dt, and cardiac output (CO).a p � 0.05 compared to preinfarct.
Table 2. Echocardiographic Data
LVESV (mL) LVEDV (mL) EF (%)
WMALength
(cm)WMA/
Circumference
SystolicSphericity
Index
SystolicMuscle/Cavity
Area
Remote SystolicWall Thickening
(mm)
Preinfarct 34.53 � 6.56 58.5 � 8.87 41.15 � 6.68 N/A N/A 0.465 � 0.088 1.74 � 0.34 2.50 � 0.55Postinfarct 32.70 � 14.66 57.85 � 20.24 44.53 � 14.65 3.38 � 0.78 0.30 � 0.05 0.470 � 0.090 1.56 � 0.28 1.80 � 0.152 weeks 51.5 � 3.87 77.90 � 5.04 33.78 � 4.38 3.96 � 0.65 0.32 � 0.05 0.476 � 0.897 1.50 � 0.35 1.83 � 0.135 weeks 64.55 � 21.81b 87.40 � 17.98b 27.98 � 11.75b 4.18 � 2.58 0.37 � 0.09 0.489 � 0.085 1.27 � 0.38 1.83 � 0.41b
8 weeks 67.00 � 4.59a 95.00 � 9.41a 29.12 � 5.74a 5.62 � 1.19c 0.41 � 0.09c 0.524 � 0.038b 1.22 � 0.16b .80 � 0.92b
Summary of changes in left ventricular (LV) end-systolic volumes (LVESV), LV end-diastolic volumes (LVEDV), ejection fraction (EF), length of wallmotion abnormality (WMA length), ratio of wall motion abnormality to circumference (WMA length/circumference), systolic sphericity index, systolicmuscle to cavity area ratio, and systolic wall thickening as measured by serial echocardiograms.a p � 0.005 compared to preinfarct; b p � 0.05 compared to preinfarct; c p � 0.05 compared to postinfarct.
754 MOAINIE ET AL Ann Thorac SurgREMODELED MYOCARDIUM 2002;74:753–60
by on June 11, 2013 ats.ctsnetjournals.orgDownloaded from

thoracotomy was then performed and the heart exposed.Monofilament nylon snares were loosely passed aroundthe first and second diagonal coronary arteries at theirorigin from the left anterior descending coronary artery.Coronary snares were briefly tightened. This transientischemia outlined the sharp border between ischemicand perfused myocardium and identified the location ofthe future infarct. After marking the infarct borders, thecoronary snares were loosened and six myocardial mark-ers (2-mm nonabsorbable polyvinyl chloride beads) weresutured along the mid-ventricular short axis at the leftanterior descending coronary artery, at the center of theinfarct, at the lateral edges of the infarct, at the posteriordescending artery, and at the midpoint between theposterior descending artery and the infarct edge. A leftatrial port catheter (Access Technologies, Skokie, IL) wasinserted for future injection of fluorescent microspheresand placed in a subcutaneous pocket. Coronary snareswere passed through pressure tubing and added to the
subcutaneous pocket. The thoracotomy was closed inlayers. The left chest was drained with a single chest tubeintroduced through a separate stab incision. After emer-gence from anesthesia the chest tube and endotrachealtube were removed. Animals received buphrenorphine(5 mg/kg) just before extubation and flunixin meglumine(1 mg/kg) for postoperative pain relief. Enrofloxacin(10 mg/kg intramuscularly) was administered on postop-erative days 1 and 2.
INFARCTION. Animals were allowed 7 to 10 days to recover.General anesthesia was again induced and the animalwas again intubated. Subcutaneous coronary snares wereexposed and tightened to produce myocardial infarction.Each animal received magnesium sulfate (1 g intrave-nously), bretylium (10 mg/kg intravenously), and lido-caine (3 mg/kg intravenous bolus followed by 2 mg/mininfusion) before infarction.
ECHOCARDIOGRAPHY. Quantitative two-dimensional subdi-aphragmatic echocardiograms were obtained before in-farction and at 30 minutes, 2, 5, and 8 weeks afterinfarction [10]. A sterile midline laparotomy or right orleft subcostal incisions were made and subdiaphragmatictwo-dimensional echocardiographic images were ob-tained using a 5-MHz probe (Hewlett Packard 77020A,Palo Alto, CA). Images were recorded on 1⁄2-inch video-tape at 30 Hz. (Panasonic AG-6300 VHS Recorder, Mat-sushita Electrical Ind. Co. Ltd, Osaka, Japan). Left ven-tricular short-axis images at three levels (at the tips of thepapillary muscles, at the bases of the papillary muscles,and at the apex) and two orthogonal long-axis views wereobtained. Previous reports have validated the reproduc-ibility and effectiveness of this technique for evaluatingLV remodeling in sheep [14].
Left ventricular volumes at end-systole and end-diastole were calculated using Simpson’s rule [15]. Ejec-tion fraction was calculated as the ratio of the differenceof end-diastolic and end-systolic volumes to the end-
Fig 1. (A) Serial transdiaphragmatic echocardiograms in short axisat the high papillary muscle level recorded from a representativeanimal at baseline, 30 minutes, 2, 5, and 8 weeks after infarction,illustrating progressive left ventricular dilation over time. (B) Serialtransdiaphragmatic echocardiograms recorded in the left ventricularlong axis view from a representative animal at baseline, 30 minutes,2, 5, and 8 weeks after infarction demonstrating progressive left ven-tricular dilation over time.
Fig 2. Left ventricular end-systolic volume (mL) with error bars rep-resenting standard error. p � 0.05, preinfarct versus 5 weeks; p �
0.005 preinfarct versus 8 weeks. Line represents the left ventricularend-systolic volume.
755Ann Thorac Surg MOAINIE ET AL2002;74:753–60 REMODELED MYOCARDIUM
by on June 11, 2013 ats.ctsnetjournals.orgDownloaded from

diastolic volume. Systolic wall thickening was measuredin short-axis views of two regions remote from theinfarct, the mid-septum and lateral wall between thepapillary muscles, using an offline analysis of echocar-diographic images (Tomtec Corp, Chicago, IL). Remotewall thickening is reported as an average of these twovalues.
Left ventricular myocardial area was calculated in thehigh papillary muscle short-axis echocardiographic im-ages by subtracting the cavity area from the total epicar-dial area at end-systole.
Left ventricular sphericity index, as an indicator of LVshape change, was calculated as the ratio of the diastolicshort-axis LV internal diameter to the diastolic long-axisLV length (a ratio of unity would represent a sphere) [4].The circumferential length of the regional wall motion
abnormality and total internal ventricular circumferencewere also measured at each postinfarction time point.
Assessment of Myocardial PerfusionSerial microsphere injections were performed at baselineand 2, 5, and 8 weeks after infarction to determineregional myocardial perfusion at each time point. Fifteenmillion, color-coded, 15.5-�m diameter NuFlow Fluores-cent microspheres (IMT Laboratories, Irvine, CA) wereinjected at each time interval through the left atrial port.A reference blood sample was withdrawn using a con-stant withdrawal syringe (Harvard Apparatus Co, Cam-bridge, MA) from the right femoral artery beginning 5seconds before microsphere injection and continuing for80 seconds to allow measurements of regional perfusion.
After the end of the 8-week study animals were eutha-nized and hearts explanted. Myocardial markers placedat the time of initial instrumentation were used to definethe infarct borders. Samples were obtained from myocar-dium between adjacent myocardial markers yieldingsamples from within the infarct, the myocardium within1 cm of the infarct (border zone myocardium), andremote LV myocardium immediately adjacent to theposterior descending artery. Myocardial samples andreference blood samples were analyzed using flow cy-tometry for microsphere content by IMT Laboratories.Regional perfusion was calculated using the followingformula: Qm � (Cm x Qr)/Cr, where Qm � myocardialblood flow per gram (mL/min per gram) of sample, Cm �microsphere count per gram of tissue in sample, Qr �withdrawal rate of the reference blood sample (mL/min),and Cr � microsphere count in the reference bloodsample.
Hemodynamic MeasurementsThe surface electrocardiogram and arterial blood pres-sure were continuously monitored (Sonometrics Inc,London, Ontario, Canada) and recorded during all datacollection procedures. A high-fidelity pressure trans-
Fig 3. Left ventricular sphericity index calculated as the ratio of leftventricular internal diameter (LVID) in short axis compared to leftventricular length (measured as distance from mitral annulus to api-cal endocardium in left ventricular long axis view). Error bars repre-sent standard error. p � 0.005 for 8 week data compared to prein-farct. Line represents the systolic (Sys) sphericity index.
Fig 4. Remote systolic wall thickening (mm) of remote myocardiummeasured as mean difference of wall thickness at end-diastole and atend-systole in two different regions remote from the infarct, the mid-septum and posterior wall between the papillary muscles; error barsrepresent standard error. Line represents the wall thickening.
Fig 5. Regional perfusion of myocardial samples (mL/g/min) ob-tained from infarct, border zone, and remote myocardium as deter-mined by serial microsphere injections. f � baseline; � � perfu-sion at 2 weeks; z � perfusion at 5 weeks; p � perfusion at 8weeks. Error bars represent standard error. p � 0.005, infarct per-fusion versus border zone and remote myocardial perfusion.
756 MOAINIE ET AL Ann Thorac SurgREMODELED MYOCARDIUM 2002;74:753–60
by on June 11, 2013 ats.ctsnetjournals.orgDownloaded from

ducer (Spc-350, Millar Instruments Inc, Houston, TX) wasinserted from the femoral artery into the left ventricle forcontinuous LV pressure monitoring (Hewlett-Packard78534c monitor). A pulmonary artery catheter was alsoplaced for each data collection procedure. Thermodilu-tion cardiac output was measured in triplicate at eachserial time point for each animal.
HistologySections of the myocardium from the infarct, borderzone, and remote myocardium were fixed in 5% bufferedformalin, paraffin embedded, cut at 3 to 4 �m, andstained with either hematoxylin and eosin or Masson’strichrome. Light microscopic analysis of these specimenspermitted qualitative assessment of fibrosis, myocytehypertrophy, and myocyte degeneration in the infarct,border zone, and remote myocardium.
StatisticsFor all measurements at the same time points, values arereported as means and standard deviations. Differencesbetween baseline measurements and measurements atsubsequent times were compared using analysis of vari-ance with repeated measures.
Results
Infarct SizingTogether D1 and D2 supplied 24.6% � 4.7% of the LVmass at the anterior base in 20 slaughterhouse hearts and23.9% � 2.2% in 8 sheep studied with tetrazolium chlo-ride after in vivo ligation of both vessels. The territoryserved by the two vessels varied reciprocally betweeneach other, but the mass supplied by D1 and D2 wasconstant and agreed closely with previous measurements[10].
HemodynamicsHemodynamic data are summarized in Table 1. Leftventricular end-diastolic pressure increased from 1.7 �1.0 mm Hg at baseline to 8.2 � 3.5 mm Hg 8 weeks afterinfarction (p � 0.05). Maximum LV dp/dt decreased from1,445 � 255 mm Hg/s at baseline to 1,141 � 357 mm Hg/s8 weeks after infarction (p � 0.05).
EchocardiographyEchocardiographic data are summarized in Table 2. Adramatic increase in LV size was observed during 8weeks after infarction (Fig 1). Left ventricular end-
Fig 6. Sections of myocardium from border zone and remote myocardium stained with Masson’s trichrome. (A) Border zone low power (�2.5),arrow indicates the junction between infarct scar and border zone myocardium. (B) Border zone high power (�20), blue staining around myo-cytes indicates increased fibrosis. (C) Remote low power (�2.5). (D) Remote high power (�20), blue staining around myocytes indicates in-creased fibrosis.
757Ann Thorac Surg MOAINIE ET AL2002;74:753–60 REMODELED MYOCARDIUM
by on June 11, 2013 ats.ctsnetjournals.orgDownloaded from

diastolic volume increased from 58.6 � 8.9 mL at baselineto 95 � 9.4 mL (p � 0.005) or 73.2% at 8 weeks afterinfarction. Similar increases were seen in LV end-systolicvolume, which increased from 34.5 � 6.6 mL at baselineto 67 � 4.6 mL (115.3%) at 8 weeks (p � 0.005) (Fig 2). Theincrease in LV volumes involved the entire ventricle andno regional or asymmetrical areas of dilatation wereobserved.
Left ventricular sphericity index increased from0.465 � 0.088 at baseline to 0.524 � 0.038 at 8 weeks (p �
0.05) (Fig 3). A decrease in LV systolic muscle-to-cavityarea ratio was also observed; the ratio decreased from1.74 � 0.34 before infarction to 1.22 � 0.16 at 8 weeks. Thecircumferential length of the wall motion abnormalityincreased from 3.38 � 0.78 cm immediately after infarc-tion to 5.62 � 1.19 cm at 8 weeks (p � 0.05). The ratio ofthe wall motion abnormality-to-ventricular circumfer-ence also increased significantly by 8 weeks (Table 2).
Ejection fraction decreased from 41.2% � 6.7% atbaseline to 29.1% � 5.7% at 8 weeks after infarction (p �
0.05). Remote systolic wall thickening decreased progres-sively from 2.50 � 0.5 to 0.80 � 0.8 mm (p � 0.05) (Fig 4).
Myocardial PerfusionPerfusion of myocardium not involved in the infarct(border zone and remote regions) remained constantthroughout the study and did not vary significantlybetween the two regions. Mean myocardial blood flow asdetermined by microsphere injection in the uninfarctedregions was 1.320 � 0.199, 1.326 � 0.393, 1.381 � 0.175mL/g per minute at 2, 5, and 8 weeks after infarct,respectively. Myocardial samples collected from the in-farct territory had a mean measured perfusion of 0.306 �0.045, 0.184 � 0.050, and 0.399 � 0.049 mL/g per minute at2, 5, and 8 weeks after infarct, respectively (p � 0.005infarct versus uninfarcted myocardium at each timepoint). Figure 5 summarizes the regional blood flow ofthe myocardial samples.
HistologyAnalysis of the histologic sections from the region of theinfarct demonstrated fibrosis consistent with healed in-farction. Specimens from the border zone showed patchyinterstitial and replacement fibrosis (Fig 6). Myocytevacuolization (myofibrillarlytic cells) and mild hypertro-phy was evident in the border zone sections (Fig 7).Sections of the remote myocardium demonstrated mildpatchy interstitial fibrosis in some of the animals.
Comment
The global increase in LV diastolic and systolic volumesand sphericity index and the reductions in systolic wallthickening, ejection fraction, and ratio of systolic muscle-to-cavity area 8 weeks after anterobasal infarction of 24%of the LV mass meet all criteria for the diagnosis ofpostinfarction, dilated cardiomyopathy. The dilatationinvolves the entire ventricle and not just the infarct.These remodeling changes are associated with a progres-sive increase in LV end-diastolic pressure and a signifi-cant reduction in LV maximum dp/dt index. The model isboth reproducible and robust and mimics a subset ofpatients who develop postinfarction dilated cardiomyop-athy after a single, moderate-sized infarct [4, 16].
In sheep similar-sized infarcts in other locations re-model to produce chronic ischemic mitral regurgitationand anteroapical LV aneurysm [13, 17]. These models andthe present model are unique in that the only interven-tion is a moderate-sized infarction of 21% to 24% of theLV mass [13, 17]. The only difference between models isthe location of the infarction. Together these three ovinemodels reproduce the three major clinical causes ofpostinfarction congestive heart failure. The controlledvariables of all three models are the lack of collateralblood flow to the infarcted area, maintenance of normalblood flow to uninfarcted areas, and the moderate size ofthe infarcted myocardium. These variables are uncon-trolled and confounding variables in patients and inother animal models.
Coronary artery disease is now the most commoncause of heart failure. Cardiomyopathy due to coronaryarterial disease may develop from multiple infarctions of
Fig 7. Sections of myocardium from border zone (A) and remote (B)regions stained with Masson’s trichrome (magnification, �20). Thesection from the border zone (A) shows extensive myocyte vacuoliza-tion (myofibrillarlytic cells). Myofibrillarlytic cells are not present inthe remote region.
758 MOAINIE ET AL Ann Thorac SurgREMODELED MYOCARDIUM 2002;74:753–60
by on June 11, 2013 ats.ctsnetjournals.orgDownloaded from

varying size in different locations throughout the myo-cardium [3, 18] but often results after a single moderate-sized infarct that is initially well tolerated hemodynami-cally [4]. The heterogeneity of human coronary anatomy,variable collateral vascularity, and differences in thelocation, number, and degree of atherosclerotic obstruc-tions produce the chronic ischemic syndromes of hiber-nating and stunned myocardium that contribute to globalmyocardial dysfunction. Stunned myocardium is definedas perfused, hypocontractile myocardium produced by aprior episode of ischemia and is reversible [19]. Hiber-nating myocardium is defined as chronically poorly per-fused, hypocontractile myocardium, which is also revers-ible [20]. Techniques have been developed thateffectively identify and treat chronically ischemic myo-cardium, but these strategies have not produced consis-tent results in reversing or arresting the progressive heartfailure associated with postinfarction cardiomyopathy[21].
Recently, clinical and experimental evidence has de-fined a third type of viable but impaired myocardium,termed remodeled myocardium. Remodeled myocar-dium, previously termed border zone myocardium, isdefined as fully perfused, hypocontractile myocardiumadjacent to an infarct [22]. Narula and colleagues [23],using a novel nuclear imaging protocol, have docu-mented that approximately 30% of hypocontractile seg-ments in ischemic cardiomyopathic patients could beclassified as remodeled myocardium. Jackson and col-leagues (personal communication) have demonstratedstretched, hypocontractile fully perfused myocardium,“remodeled myocardium,” in an experimental model ofanteroapical LV aneurysm. This new sheep model ofischemic cardiomyopathy further advances the conceptof remodeled myocardium and suggests that a regionalinfarct can initiate a myopathic process that can spreadbeyond the border zone to involve the entire ventricle.Whether the myopathic mechanism responsible for re-modeled myocardium is reversible or not is unknown.The inconsistent results of salvage surgical proceduresdesigned to restore normal ventricular geometry andreduce ventricular wall stress suggest that the myopathicprocess may not be reversible [24–27].
In the absence of measurable ischemia outside theinfarct region, this sheep model of postinfarction, dilatedcardiomyopathy does not include either stunned or hi-bernating myocardium and, therefore, is a pure model ofremodeled myocardium. The simplicity of the model—myocardium is either infarcted or normally perfused—and the absence of confounding variables provide anopportunity to elucidate the mechanism of the progres-sive myopathic process whereby fully perfused, normallycontractile myocardium becomes hypocontractile afterregional transmural infarction. The presence of vacuol-ized myocytes in normally perfused myocardium at theinfarct border suggests a mechanism for the contractiledysfunction that develops in this model. Narula andcolleagues (personal communication) have shown thatthe majority of vacuolized myocytes, also termed myofi-brillarlytic cells, are apoptotic and are associated with an
upregulation cytoplasmic caspase-3. Both myofibrillar-lytic [28] and apoptotic processes [29] in myocytes havebeen proposed to be reversible phenomena. The associ-ation between myocyte apoptosis and remodeled myo-cardium is unique and provides a new therapeutic targetfor the treatment or prevention of heart failure secondaryto postinfarction LV remodeling.
This work was supported by HL36308 and HL63594 from theNational Heart Lung Blood Institute, National Institutes ofHealth, Bethesda, MD, and a grant from the Mary L. SmithCharitable Trust, Philadelphia, PA.
The editor thanks Dr Bartley P. Griffith for managing the blindedpeer review.
References
1. 1998. Heart and stroke statistical update. www.americanheart.org.
2. Kannel WB, Sorlie P, McNamara PM. Prognosis after theinitial myocardial infarction: the Framingham study. Am JCardiol 1979;44:53–9.
3. Gheorghiade M, Bonow RO. Chronic heart failure in theunited states: a manifestation of coronary artery disease.Circulation 1998;97:282–9.
4. St. John-Sutton M, Pfeffer MA, Moye L, et al. Cardiovasculardeath and left ventricular remodeling two years after myo-cardial infarction: baseline predictors and impact of long-term use of captopril. Information from the Survival andVentricular Enlargement (SAVE) trial. Circulation 1997;96:3294–9.
5. Schuster EH, Bulkley BH. Ischemic cardiomyopathy: a clin-icopathologic study of fourteen patients. Am Heart J 1980;100:506–12.
6. Anversa P, Li P, Zhang X, et al. Ischemic myocardial injuryand ventricular remodelling. Cardiovasc Res 1993;27:145–57.
7. Narula J, Arbustini, Chandrashekhar Y, et al. Apoptosis andthe systolic dysfunction in congestive heart failure. CardiolClin 2001;19:113–26.
8. Sabbah HN, Stein PD, Kono T, et al. A canine model ofchronic heart failure produced by multiple sequential coro-nary microembolizations. Am J Physiol 1991;290:H1379–84.
9. Huang Y, Kawaguchi O, Zeng B, et al. A stable ovinecongestive heart failure model. ASAIO J 1997;43:M0037–42.
10. Gorman JH III, Gorman RC, Plappert T, et al. Infarct size andlocation determine development of mitral regurgitation inthe sheep model. J Thorac Cardiovasc Surg 1998;115:615–22.
11. Vastesaeger MM, Van Der Straeten PP, Friart J, et al. Lesanastomoses intercoronariennes telles qu’elles apparaissenta la coronarographie post morten. Acta Cardiologica 1957;12:365–401.
12. Schaper W. Collateral circulation of the heart. Amsterdam:North Holland, 1971.
13. Markovitz LJ, Savage EB, Ratcliffe MB, et al. Large animalmodel of left ventricular aneurysm. Ann Thorac Surg 1989;48:838–45.
14. Gorman JH III, Gorman RC, Jackson BM, et al. Distortions ofthe mitral valve in acute ischemic mitral regurgitation. AnnThorac Surg 1997;64:1026–31.
15. Folland ED, Parisi AF, Moynihan, et al. Assessment of leftventricular ejection fraction and volumes by real-time, two-dimensional echocardiography. A comparison of cineangio-graphic and radionuclide techniques. Circulation 1979;60:760–6.
16. Cigarroa RG, Lange RA, Hillis LD. Prognosis after acutemyocardial infarction in patients with and without residualanterograde coronary blood flow. Am J Cardiol 1989;64:155–60.
17. Llaneras MR, Nance ML, Streicher JT, et al. Large animal
759Ann Thorac Surg MOAINIE ET AL2002;74:753–60 REMODELED MYOCARDIUM
by on June 11, 2013 ats.ctsnetjournals.orgDownloaded from

model of ischemic mitral regurgitation. Ann Thorac Surg1994;57:432–9.
18. Beltrami CA, Finato N, Rocco M, et al. Structural basis ofend-stage failure in ischemic cardiomyopathy in humans.Circulation 1994;89:151–63.
19. Braunwald E, Kloner RA. The stunned myocardium: pro-longed, postischemic ventricular dysfunction. Circulation1982;66:1146–9.
20. Rahimtoola SH. The hibernating myocardium. Am Heart J1989;117:211–21.
21. Pagley PR, Beller GA, Watson DD, Gimple LW, Ragosta M.Improved outcome after coronary bypass surgery in patientswith ischemic cardiomyopathy and residual myocardial vi-ability. Circulation 1997;96:793–800.
22. Pfeffer MA, Braunwald E. Ventricular remodeling after myo-cardial infarction. Experimental observations and clinicalimplications. Circulation 1990;81:1161–72.
23. Narula J, Dawson MS, Singh BK, et al. Noninvasive charac-terization of stunned, hibernating, remodeled, and nonvia-ble myocardium in ischemic cardiomyopathy. J Am CollCardiol 2000;36:1913–9.
24. Couper GS, Bunton RW, Birjiniuk V, et al. Relative risks of
left ventricular aneurysmectomy in patients with akineticscars versus true dyskinetic aneurysms. Circulation. 82(5suppl):248–56.
25. Grossi EA, Chinitz LA, Galloway AC, et al. Endoventricularremodeling of left ventricular aneurysm. Functional, clinical,and electrophysiological results. Circulation 1995;92(9 suppl):II98–100.
26. Hayward MP. Dynamic cardiomyoplasty: time to wrap it up?Heart 1999;82:263–4.
27. Franco-Cereceda A, McCarthy PM, Blackstone EH, et al.Partial left ventriculectomy for dilated cardiomyopathy: isthis an alternative to transplantation? J Thorac CardiovascSurg 2001;121: 879–93.
28. Narula J, et al. J Am Coll Cardiol 2002 (submitted).29. Edwalds GM, Said JW, Block MI, Herscher LL, Siegel RJ,
Fishbein MC. Myocytolysis (vacuolar degeneration) of myo-cardium: immunohistochemical evidence of viability. HumPathol 1984;15:753–6.
30. Narula J, Aurbustini E, Chandrashekhar Y, Schwaiger M.Apoptosis and systolic dysfunction in congestive heart fail-ure: story of apoptosis interruptus and zombie myocytes.Cardiol Clin 2001;19:113–26.
760 MOAINIE ET AL Ann Thorac SurgREMODELED MYOCARDIUM 2002;74:753–60
by on June 11, 2013 ats.ctsnetjournals.orgDownloaded from

2002;74:753-760 Ann Thorac SurgNarula, L. Henry Edmunds, Jr and Robert C. Gorman
M. Jackson, Theodore Plappert, Navneet Narula, Martin G. St. John-Sutton, Jagat Sina L. Moainie, Joseph H. Gorman, III, T. Sloane Guy, Frank W. Bowen, III, Benjamin
An ovine model of postinfarction dilated cardiomyopathy
& ServicesUpdated Information
http://ats.ctsnetjournals.org/cgi/content/full/74/3/753including high-resolution figures, can be found at:
References http://ats.ctsnetjournals.org/cgi/content/full/74/3/753#BIBL
This article cites 26 articles, 14 of which you can access for free at:
Citations http://ats.ctsnetjournals.org/cgi/content/full/74/3/753#otherarticles
This article has been cited by 21 HighWire-hosted articles:
Subspecialty Collections
http://ats.ctsnetjournals.org/cgi/collection/myocardial_infarction Myocardial infarction
following collection(s): This article, along with others on similar topics, appears in the
Permissions & Licensing
[email protected]: orhttp://www.us.elsevierhealth.com/Licensing/permissions.jsp
in its entirety should be submitted to: Requests about reproducing this article in parts (figures, tables) or
Reprints [email protected]
For information about ordering reprints, please email:
by on June 11, 2013 ats.ctsnetjournals.orgDownloaded from
Copyright © 2022 FDOKUMEN




















