
Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse
15
Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse Jean-Michel Itier 1,{ , Pablo Iba ´n ˜ ez 2,{ , Marı ´a Angeles Mena 3 , Nacer Abbas 2,z , Charles Cohen-Salmon 4 , Georg Andrees Bohme 5 , Michel Laville 5 , Jeremy Pratt 5 , Olga Corti 2 , Laurent Pradier 5 , Gwe ´na ¨ elle Ret 1 , Chantal Joubert 4 , Magali Periquet 2 , Francisco Araujo 5 , Julia Negroni 4 , Marı ´a Jose ´ Casarejos 3 , Santiago Canals 3 , Rosa Solano 3 , Alba Serrano 6 , Eva Gallego 6 , Marina Sa ´ nchez 6 , Patrice Dene `fle 1 , Jesu ´ s Benavides 5 , Gu ¨ nter Tremp 1 , Thomas A. Rooney 5, * , Alexis Brice 2,7 and Justo Garcı ´a de Ye ´ benes 6 1 Functional Genomics Department, Aventis Pharma SA, 13 Quai Jules Guesde, F-94400 Vitry-sur-Seine, France, 2 INSERM U289, Groupe Hopitalier Pitie ´ -Salpe ˆ trie `re, 47, Bd de l’Ho ˆpital, 75651 Paris Cedex 13, France, 3 Department of Research, Hospital Ramon y Cajal, Carretera de Colmenar km. 9,100, 28034 Madrid, Spain, 4 CNRS UMR 7593, Hopitalier Pitie ´-Salpe ˆ trie ` re, 47, Bd de l’Ho ˆ pital, 75651 Paris Cedex 13, France, 5 CNS Department, Aventis Pharma SA, 13 Quai Jules Guesde, F-94400 Vitry-sur-Seine, France, 6 Department of Neurology, Fundacio ´n Jime ´nez Dı ´az, Universidad Auto ´noma de Madrid, Avda. de Reyes Cato ´ licos, 2. 28040 Madrid, Spain and 7 De ´partement de Ge ´ne ´ tique, Cytoge ´ne ´ tique et Embryologie, Groupe Hopitalier Pitie ´ -Salpe ˆ trie `re, AP-HP, 47, Bd de l’Ho ˆ pital, 75651 Paris Cedex 13, France Received April 10, 2003; Revised June 13, 2003; Accepted July 10, 2003 Mutations of the parkin gene are the most frequent cause of early onset autosomal recessive parkinsonism (EO-AR). Here we show that inactivation of the parkin gene in mice results in motor and cognitive deficits, inhibition of amphetamine-induced dopamine release and inhibition of glutamate neurotransmission. The levels of dopamine are increased in the limbic brain areas of parkin mutant mice and there is a shift towards increased metabolism of dopamine by MAO. Although there was no evidence for a reduction of nigrostriatal dopamine neurons in the parkin mutant mice, the level of dopamine transporter protein was reduced in these animals, suggesting a decreased density of dopamine terminals, or adaptative changes in the nigrostriatal dopamine system. GSH levels were increased in the striatum and fetal mesencephalic neurons from parkin mutant mice, suggesting that a compensatory mechanism may protect dopamine neurons from neuronal death. These parkin mutant mice provide a valuable tool to better understand the preclinical deficits observed in patients with PD and to characterize the mechanisms leading to the degeneration of dopamine neurons that could provide new strategies for neuroprotection. INTRODUCTION Parkinson’s Disease (PD) is the second most frequent neuro- degenerative disorder with a prevalence of 0.2–0.3% in the population of Europe and North America (1,2). Clinically, PD is best characterized by akinesia or bradykinesia, rigidity, resting tremor and postural abnormalities (3,4). In addition, other neurological deficits, such as peculiar personality traits, cognitive and autonomic dysfunction, depression and sleep disorders often take place (3). The major pathological hallmark of PD is the degeneration of dopaminergic neurons of the substantia nigra, predominantly the ventrolateral portion of the pars compacta that innervates the dorsal striatum. Other monoamine brain nuclei are also damaged, but to a lesser extent than in the substantia nigra. Most PD patients present intracellular eosinophillic inclusions, known as Lewy bodies *To whom correspondence should be addressed. Tel: þ33 158932645; Fax: þ33 158933685; Email: [email protected] { The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors. z Deceased. Human Molecular Genetics, 2003, Vol. 12, No. 18 2277–2291 DOI: 10.1093/hmg/ddg239 Human Molecular Genetics, Vol. 12, No. 18 # Oxford University Press 2003; all rights reserved by guest on January 5, 2016 http://hmg.oxfordjournals.org/ Downloaded from
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse

Parkin gene inactivation alters behaviourand dopamine neurotransmission in the mouse
Jean-Michel Itier1,{, Pablo Ibanez2,{, Marıa Angeles Mena3, Nacer Abbas2,z,
Charles Cohen-Salmon4, Georg Andrees Bohme5, Michel Laville5, Jeremy Pratt5,
Olga Corti2, Laurent Pradier5, Gwenaelle Ret1, Chantal Joubert4, Magali Periquet2,
Francisco Araujo5, Julia Negroni4, Marıa Jose Casarejos3, Santiago Canals3, Rosa Solano3,
Alba Serrano6, Eva Gallego6, Marina Sanchez6, Patrice Denefle1, Jesus Benavides5,
Gunter Tremp1, Thomas A. Rooney5,*, Alexis Brice2,7 and Justo Garcıa de Yebenes6
1Functional Genomics Department, Aventis Pharma SA, 13 Quai Jules Guesde, F-94400 Vitry-sur-Seine,
France, 2INSERM U289, Groupe Hopitalier Pitie-Salpetriere, 47, Bd de l’Hopital, 75651 Paris Cedex 13, France,3Department of Research, Hospital Ramon y Cajal, Carretera de Colmenar km. 9,100, 28034 Madrid, Spain,4CNRS UMR 7593, Hopitalier Pitie-Salpetriere, 47, Bd de l’Hopital, 75651 Paris Cedex 13, France, 5CNS Department,
Aventis Pharma SA, 13 Quai Jules Guesde, F-94400 Vitry-sur-Seine, France, 6Department of Neurology,
Fundacion Jimenez Dıaz, Universidad Autonoma de Madrid, Avda. de Reyes Catolicos, 2. 28040 Madrid,
Spain and 7Departement de Genetique, Cytogenetique et Embryologie, Groupe Hopitalier Pitie-Salpetriere,
AP-HP, 47, Bd de l’Hopital, 75651 Paris Cedex 13, France
Received April 10, 2003; Revised June 13, 2003; Accepted July 10, 2003
Mutations of the parkin gene are the most frequent cause of early onset autosomal recessive parkinsonism(EO-AR). Here we show that inactivation of the parkin gene in mice results in motor and cognitive deficits,inhibition of amphetamine-induced dopamine release and inhibition of glutamate neurotransmission. Thelevels of dopamine are increased in the limbic brain areas of parkin mutant mice and there is a shift towardsincreased metabolism of dopamine by MAO. Although there was no evidence for a reduction of nigrostriataldopamine neurons in the parkin mutant mice, the level of dopamine transporter protein was reduced in theseanimals, suggesting a decreased density of dopamine terminals, or adaptative changes in the nigrostriataldopamine system. GSH levels were increased in the striatum and fetal mesencephalic neurons from parkinmutant mice, suggesting that a compensatory mechanism may protect dopamine neurons from neuronaldeath. These parkin mutant mice provide a valuable tool to better understand the preclinical deficits observedin patients with PD and to characterize the mechanisms leading to the degeneration of dopamine neuronsthat could provide new strategies for neuroprotection.
INTRODUCTION
Parkinson’s Disease (PD) is the second most frequent neuro-degenerative disorder with a prevalence of 0.2–0.3% in thepopulation of Europe and North America (1,2). Clinically, PD isbest characterized by akinesia or bradykinesia, rigidity, restingtremor and postural abnormalities (3,4). In addition, otherneurological deficits, such as peculiar personality traits,
cognitive and autonomic dysfunction, depression and sleepdisorders often take place (3). The major pathological hallmarkof PD is the degeneration of dopaminergic neurons of thesubstantia nigra, predominantly the ventrolateral portion of thepars compacta that innervates the dorsal striatum. Othermonoamine brain nuclei are also damaged, but to a lesser extentthan in the substantia nigra. Most PD patients presentintracellular eosinophillic inclusions, known as Lewy bodies
*To whom correspondence should be addressed. Tel: þ33 158932645; Fax: þ33 158933685; Email: [email protected]{The authors wish it to be known that, in their opinion, the first two authors should be regarded as joint First Authors.zDeceased.
Human Molecular Genetics, 2003, Vol. 12, No. 18 2277–2291DOI: 10.1093/hmg/ddg239
Human Molecular Genetics, Vol. 12, No. 18 # Oxford University Press 2003; all rights reserved
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

(5). These inclusions are immunoreactive for several proteins, suchas a-synuclein and ubiquitin, but are not exclusive to PD since theyhave been observed in other neurological disorders (6,7).
Most cases of PD are sporadic and have been associated witha number of risk factors, including exposure to pesticides,drinking well water, and recent industrialization. This suggeststhat the pathogenesis of the disease may be related to excessiveoxidative stress, lack of free radical scavenging products orabnormalities of mitochondrial energy production (8,9).Indeed, blockade of the mitochondrial respiratory chain orgeneration of oxidative stress by different pharmacologicaltools has been used to generate most of the experimentalmodels of PD in laboratory animals (10–13). In addition to thesporadic forms of PD, familial aggregation is present in up to25% of PD cases. A subset of these families correspond tomonogenic forms of PD; a few families with mutations ingenes encoding for a-synuclein (14) and Nurr1 (15) areassociated with autosomal dominant forms of the disease.However, the majority of familial cases are associated withmutations of the parkin gene (16), which causes the mostcommon form of autosomal recessive parkinsonism (17) andaccounts for approximately 15% of the isolated cases withonset before the age of 45 (18).
In most families with PD related to parkin gene mutations,the disease onset is before age 40 and the clinical response toL-DOPA is good (16,19,20). The majority of patients withhomozygous or combined heterozygous mutations of theparkin gene have severe loss of nigral dopaminergic neurons,but without Lewy bodies (21–24). However, PD patients withparkin mutations can also display atypical phenotypes of lateonset, autosomal dominant transmission and Lewy bodies intheir brain (17,25–27). Age at onset of parkinsonism related toparkin mutations is extremely variable, even within the samefamily or among carriers of the same mutation (20,28). Inaddition, a single parkin mutation is detected in some patients(29), suggesting that this situation could represent a risk factorfor PD. Therefore, the reduction of parkin protein function,and not only total absence of parkin function or a dominantnegative effect of specific mutations, is enough to produceparkinsonism in some individuals. The pathogenic impact ofparkin mutations may also be modulated by other factors.
Parkin is encoded by a gene containing 12 exons. Manydifferent mutations, including deletions and multiplications ofone or several exons, point mutations that alter the openreading frame producing premature stop codons and truncatedproteins or changes of a single amino acid in critical regions ofthe protein have been identified in all exons, including exon 3(17,19). Parkin is a protein of 465 amino acids with a ubiquitinhomology domain at the N terminal, and two ring fingers plusone ‘in between ring’ sequence towards the C terminal. As withmany RING-finger-containing proteins, parkin acts as an E3ubiquitin protein ligase and has been shown to bind to andubiquitinate a number of specific protein substrates, includingCdcrel-1 (30), Pael-R (31), synphilin-1 (32) and glycosylateda-synuclein (33).
To date, the mechanism(s) by which parkin mutations andderegulation of parkin substrates lead to dopamine neuronal lossand the onset of parkinsonian symptoms is unknown. To furtherunderstand the mechanisms by which mutations in parkin leadto the onset of parkinsonism, we have generated a transgenic
animal model in which the parkin gene has been inactivated.Here we report the construction of a parkin mutant mouse and acharacterization of the phenotype resulting from preventing thefunctional expression of the parkin protein.
RESULTS
Absence of parkin protein in parkinexon 3 deleted mice
A 12 kb fragment containing mouse parkin exon 3 was isolatedfrom the 129SV genomic phage library. A targeting vector wasassembled to replace the 30 end of exon 3 and a substantial partof intron 4 by a neomycin-resistance cassette (Fig. 1A). Thesuccessful deletion was confirmed at the genome level bySouthern blot (Fig. 1B). Northern blot analysis revealed that thetree parkin mRNA disappeared in the mutant mice and isreplaced by a single new band. Using parkin exon 2, 3 or 4 as aprobe, we demonstrated a splice between exon 2 and 4 andskipping of exon 3 in the parkin mRNA present in the mutantmice (Fig. 1C). This result was confirmed by RT–PCRexperiments and sequencing of the amplification products(data not shown). The consequence of such aberrant splicingin this putative coding mRNA is the change of the ORF fromposition codon 57 and the appearance of a stop codon atposition 105. Germ line transmission was obtained withchimeric mice. Mice carrying the homozygous mutation wereviable and fertile. Studies of mortality in the colony showedno excess mortality when compared with wild-type animalsreared in parallel and mutant mice had the same average littersize as wild type mice (7.6� 0.6 versus 8.3� 0.6 young/litter;n¼ 32 and 24, respectively).
Western blots using an antibody raised against amino acids71–92 of the parkin protein indicated that the 48–50 kDa band,corresponding to the parkin protein, had disappeared in theparkin mutant mice (Fig. 1D). These mutants can therefore beconsidered to have lost their ability to produce the parkinprotein.
Clinical phenotype and brain morphology
Transgenic parkin mice appeared to be normal with nomodification of behaviour or handicap. The oldest homozygoustransgenic mice, of both mixed (C57BL6/129SV) and pure(129SV) background, did not show any overt behaviouralchanges up to 24 months of age. No differences in brainmorphology, brain weight and brain size were observed andthere was no evidence of muscle degeneration (data notshown). These findings are in contrast to those found inDrosophila parkin mutants that display a reduced lifespan,male sterility and muscle degeneration (34). Histologicalsections of the brain at the levels of the striatum, hippocampus,brain stem and cerebellum stained with haematoxylin and eosinalso did not reveal any changes (data not shown). However,body weight was reduced from weaning to 500 days of age inboth male and female parkin mutant mice (Fig. 2) and bodytemperature, measured rectally, was reduced in parkin mutantmice at 4 months (37.2� 0.2�C in parkin mutant mice versus38.5� 0.16�C in wild-type mice, P< 0.001, n¼ 14).
2278 Human Molecular Genetics, 2003, Vol. 12, No. 18
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

Protein markers of dopaminergic neurons
Tyrosine hydroxylase (TH) immunoreactivity in the substantianigra and striatum of female mice was similar in wild typeand parkin mutant mice at 15 months of age. Quantitativeanalysis showed no statistically significant difference in thenumber of TH-positive perikaria in the substantia nigra or inTH optical density in the striatum between the two groups(Fig. 3A–F). No differences in TH staining were observed upto 24 months of age (data not shown). This lack of effect onTH staining was confirmed by western blot where nodifferences in the levels of TH protein were observed betweenmale parkin mutant mice and controls at 15 months of age inthe striatum (Fig. 3G and H) or in the limbic region,diencephalon and brain stem (data not shown). In addition,there was no difference in a-synuclein, ubiquitin, synapsin,synaptophysin, Map2 and Neu N staining in mutant mice at24 months of age (data not shown). By contrast, althoughimmunohistochemical examination of dopamine transporter(DAT) staining in wild-type and mutant mice at 15 months ofage showed no significant differences (data not shown), thelevels of DAT and VMAT2, measured by western blot, weresignificantly reduced in the striatum of parkin mutant mice(Fig. 3G and H).
Deficits in learning and exploration inparkin mutant mice
Parkin mutant mice display reduced exploratory behaviour inunfamiliar environments (Fig. 4A) and decreased spontaneousalternation in the exploration of the unknown arm of a T-maze(Fig. 4B), a test used to reveal deficits in working memory. Thisindicates that mutant mice are less willing to explore and/orless able to remember what they had recently explored. In thehanging thread test no differences were observed between wild-type and mutant mice with respect to maximal hanging time(Fig. 4C), or the number of animals refusing the test during thefirst trial (data not shown), which could indicate no differencesin muscular tonus between both groups. However, during re-exposure to the test 3 months later, the number of animalsrejecting the challenge was greater in the wild-type than in themutant group (Fig. 4C). This could be interpreted as abnormallearning by the mutant mice, although other explanations, suchas a more submissive character are also possible.
Spontaneous and amphetamine-induced motor activity
Amphetamine (0.5 mg/kg) reduced locomotor activity in bothmutant and wild type mice, and the percentage reduction with
Figure 1. Inactivation of the parkin gene. (A) The strategy for inactivating the parkin gene in the mouse. The 1097 bp encompassing the 30 end of exon 3 and 50 endof intron 3 were deleted. (B) Southern blot of genomic DNA from wild type and parkin mutant mice. The 6 kb and the 5 kb bands indicate the presence of the wild-type and the mutated parkin allele, respectively. (C) Northern blot: total RNA from wild-type or parkin mutant mice brain were hybridized with a probe corre-sponding to parkin gene exon 2, 3 or 4. (D) Demonstration of the absence of parkin protein by western blot analysis. Brain extract from wild-type or parkin mutantmice was used with an anti-parkin antibody.
Human Molecular Genetics, 2003, Vol. 12, No. 18 2279
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

respect to the baseline was similar for both groups (Fig. 4E).Spontaneous motor activity in familiar environments wasreduced in parkin mutant mice at 6 months of age (Fig. 4D).Amphetamine (1–5 mg/kg) significantly increased motor activa-tion in wild-type, but not in parkin mutant mice (Fig. 4E). Sinceamphetamine is thought to release newly formed, mostlycytoplasmic, catecholamines, rather than vesicular catechol-amines, these findings suggest that deregulation of parkinfunction alters the handling of intracytoplasmic neurotransmit-ters.
Monoamine metabolism in parkin mutant mice
The levels of 5-HT and 5-HIAA in the striatum, limbic system,diencephalon and brainstem were similar in both wild type andmutant animals at 11 months of age (Table 1 and Fig. 5A–D),suggesting that the metabolism of 5-HT is not altered byparkin dysfunction. The endogenous levels of dopamine wereincreased in the limbic regions of parkin mutant mice and theratios of DOPAC/DA and DOPAC/3-MT were also increasedin mutant mice (Table 1 and Fig. 5A–D). Increased dopaminelevels and increased dopamine metabolism to DOPAC viamonoamino oxidase (MAO), an enzyme considered to be mostlyintraneuronal, and to 3-MT via catechol-ortho-methyl-tranferase(COMT), an enzyme considered mostly extraneuronal, suggeststhat parkin dysfunction impairs the release of dopamine andincreases intraneuronal dopamine metabolism via MAO.
To determine which cellular compartment of dopamine isshifted to intraneuronal oxidation, we investigated the turnoverof catecholamines after inhibition of their synthesis by a-methyl-tyrosine (a-MT). The rate of depletion of catechol-amines in a-MT-treated animals was similar in both mutant andwild-type animals (data not shown), suggesting that theturnover is similar in both groups. Since the short term effectsof a-MT on catecholamines are mostly related to the turnoverof the newly synthesized intracytoplasmic pool of catechol-amines, rather than the vesicular pool of catecholamines, ourdata suggest that the increased production of DOPAC in theabsence of parkin is not mediated by changes in dopamine
Figure 2. Parkin mutant mice have decreased body weight. Evolution of bodyweight for wild type (open bars, n¼ 11) and parkin mutant (hatched bars,n¼ 14) mice. *P< 0.05; **P< 0.01; ***P< 0.001 versus wild-type mice,Student’s t-test.
Figure 3. Protein markers of dopamine neurons. (A) Representative photo-micographs of TH immunoreactivity in the striatum of a 15-month-old femalewild-type mouse. (B) Representative photomicographs of TH immunoreactivityin the striatum of a 15-month-old female parkin mutant mouse. (C) TH immuno-reactivity in the substantia nigra of wild-type mouse. (D) TH immunoreactivityin the substantia nigra of parkin mutant mice. (E) Magnification of TH immuno-staining in the substantia nigra in wild-type mice. (F) Magnification of THimmunostaining in the substantia nigra in parkin mutant mice (originalmagnification�50). Bar indicates 500mm for the striatum and 200mm for thesubstantia nigra. TH cell counts and OD-values in wild type and mutant micewere compared using an unpaired t-test. A value of P< 0.05 was considered sta-tistically significant. (G) Representative western blots of tyrosine hydroxylase,dopamine transporter and vesicular monoamine transporter proteins in micestriatum (1–3 from wild type and 4–6 from parkin mutant mice). b-Actin wasused as charge control. (H) Data represent the ratio TH, DAT and VMAT2/b-actin in mice striatum. Values are expressed as the mean� SEM (n¼ 6 animalsin each group). Statistical analysis was performed by ANOVA, followed byStudent’s t-test. *P< 0.05, **P< 0.01 parkin mutant versus wild-type mice.
2280 Human Molecular Genetics, 2003, Vol. 12, No. 18
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

turnover, but due to intracytoplasmatic dopamine beingunprotected against metabolism by MAO.
The increased dopamine metabolism by MAO was associatedwith an increase in the levels of reduced GSH in the striatum(Fig. 5E), the brain area with the highest levels of dopamine,but not in other brain areas (data not shown). There were nosignificant differences in the GSSG levels (data not shown). Tofurther evaluate the changes of GSH in dopamine rich areas, wemeasured GSH levels in neuronal enriched cultured cells fromventral fetal mid brain. We found that there was an increase ofGSH levels in dopamine rich areas of parkin mutant mice fromthe fetal period (Fig. 5F).
[3H]-DA release and [3H]-DA uptake in fetalmidbrain neuronal-enriched cultures
Both basal release of [3H]-DA and Kþ-evoked [3H]-DArelease, which is mainly an index of vesicular dopaminerelease, were similar in neuronal cultures from wild type andparkin mutant mice (Fig. 6A). However, amphetamine-induced[3H]-DA release, which is mainly an index of newly synthe-sized neurotransmitter release, was significantly decreased inneuronal cultures from parkin mutant mice compared to wild-type (Fig. 6A). [3H]-DA uptake was also significantlydecreased in fetal midbrain cultures from parkin mutant micecompared with wild type (Fig. 6B). This data is in agreementwith the reduction of DAT protein levels, suggesting that thereis a decrease in both dopamine transporter protein expressionand function in parkin mutant mice.
Deficits in glutamate synaptic transmission inparkin mutant mice
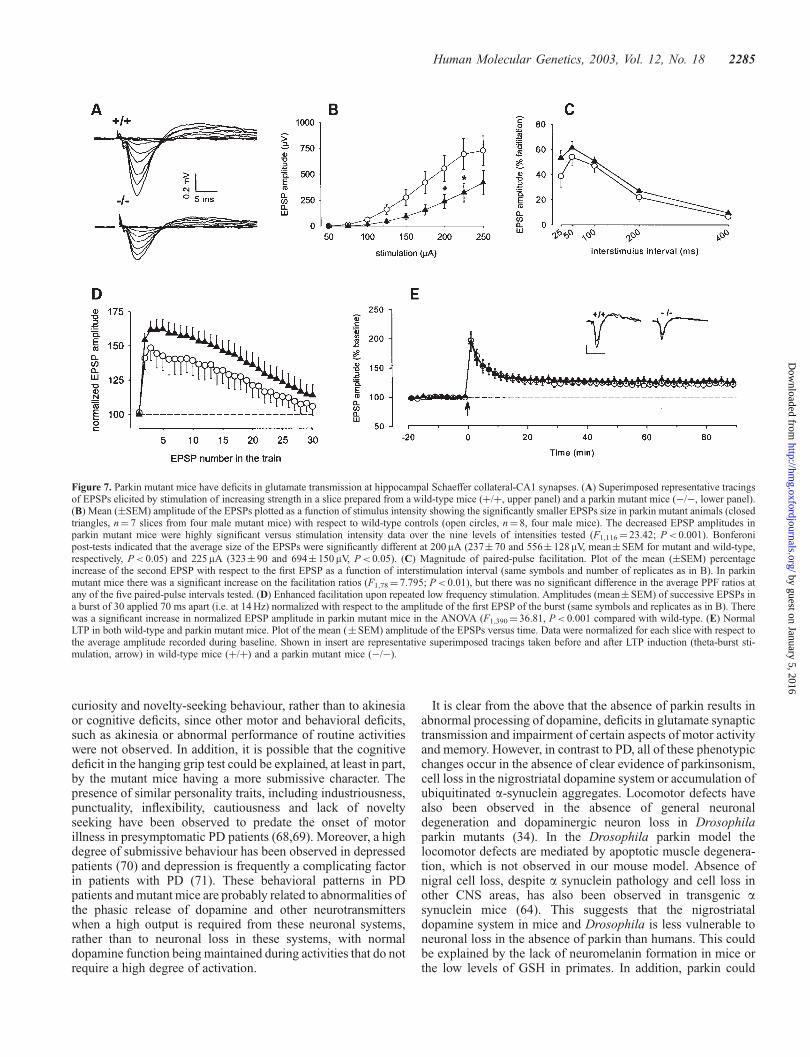
The deficits in dopamine neurotransmission prompted us toexamine the effects of parkin deletion on the pre- andpostsynaptic properties of other synaptic systems. To this end,we examined glutamatergic synaptic transmission in hippocam-pal slices from wild type and parkin mutant mice. First, weanalyzed basal synaptic transmission by applying isolated
stimuli of increasing intensity to the Schaffer collaterals. Theshape of the resulting glutamatergic extracellular field excitatorypost-synaptic potentials (EPSPs) appeared identical in slicesfrom parkin mutant mice and wild-type controls. There was alsono evidence for abnormal excitability, such as traces of repeatedaction-potential firing. However, the amplitude of the EPSPswas smaller in the parkin mutant mice (Fig. 7A and B).
We then examined paired pulse facilitation (PPF), aphenomenon whereby the second pulse of a pair applied inrapid succession elicits increased EPSP when the interval issufficiently short (<500 ms). This phenomenon reflects neuro-transmitter release (35). We observed a slight increase in PPFin parkin mutant animals (Fig. 7C). We therefore activated thepathway in a more sustained manner by applying trains of 30pulses at a fixed frequency of 14 Hz. Under these conditions oflow-frequency stimulation, the size of the EPSPs in the parkinmutant animals were further increased (Fig. 7D). Previousstudies have established that manipulations that decreaseneurotransmitter release result in increased synaptic facilitationin the hippocampal slice (35–37). Therefore, the observation ofa decreased EPSP size in response to single stimulation,together with an enhanced facilitation upon repeated stimula-tion, suggests that deletion of parkin results in decreasedpresynaptic glutamate release. By contrast, there was nodifference in the ability to induce long-term potentiation(LTP) in slices from wild-type and parkin mutant groups(Fig. 7E), suggesting that postsynaptic glutamate-mediatedevents are not altered in this brain region.
DISCUSSION
We have generated and characterized a knock out mouse modelin which exon 3 of the parkin gene has been deleted byhomologous recombination. Deletion of exon 3 has beenshown to be one of the most common parkin mutations foundin patients with autosomal recessive early-onset parkinsonism(17,19). As in parkinsonian patients with parkin mutations,deletion of parkin exon 3 in mice abolishes their capacity to
Table 1. Monoamine metabolism in limbic system, striatum, diencephalon and brain stem of wild-type and knockout mice for parkin
Groups Limbic system Striatum Diencephalon Brain stem
Wild-type Parkin knockout Wild-type Parkin knockout Wild-type Parkin knockout Wild-type Parkin knockout
DA 2339� 132 3350� 390* 8570� 1583 10814� 1123 1031� 218 1147� 117 175� 4.4 164� 8.4143% 126% 111% 94%
3-MT 127.4� 8.4 131� 12 247� 40 245� 14 41.5� 8 40.85� 11 nd nd103% 99% 98%
DOPAC 223� 16 348� 18*** 252� 52 338� 26 140� 30 135� 15 57.5� 3.1 71� 2.5156% 134% 96% 122%
HVA 221� 16 273� 27 374� 61 416� 28 124� 21 134� 16 64.8� 4.13 52� 3.0123% 111% 108% 99%
NA 478� 91 467� 57 441� 44 420� 30 1534� 191 1635� 95 714� 44 776� 2098% 95% 106% 108%
5-HT 435� 85 448� 35 1056� 173 1063� 61 1376� 160 1498� 93 1367� 54 1574� 12593103% 100% 108% 115%
5-HIAA 77.4� 18 79� 19 102� 16 127� 16 222� 11 257� 21 338� 15 424� 24102% 124% 115% 125%
Results are expressed in ng per g of fresh tissue. Values are the mean�SEM (n¼ 5–6). Statistical analysis was performed by one-way analysis of variance followedby the Student’s t-test. *P< 0.05, ***P< 0.001 parkin mutant versus wild-type mice.
Human Molecular Genetics, 2003, Vol. 12, No. 18 2281
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

produce the parkin protein. These mice therefore reproduce thekey protein deficit that is responsible for the pathogenesis ofthe most frequent cause of autosomal recessive parkinsonism.The parkin mutant mice are viable and reproduce successfully,but display behavioural, biochemical and electrophysiologicalchanges that could help explain some aspects of thepathogenesis of PD. In addition, parkin mutant mice displaydecreased body weight and temperature. The significance of the
body weight and temperature changes is unclear, since similarchanges have been observed in other transgenic models.However, it is noteworthy that both parkin and dopaminergicsignalling have been implicated in the control of body weightand thermoregulation (38–41).
Loss of parkin function is associated with deregulation ofdopamine handling. Thus, the endogenous levels of dopamine,but not the turnover of dopamine, are increased in brain areas
Figure 4. Parkin mutant mice have motor and cognitive deficits. (A) Open field activity in 170-day-old wild-type (open bars, n¼ 14) and parkin mutant (striped bars,n¼ 14) male mice over two consecutive periods of 3 min immediately following the introduction of the mouse into the motility cage reveals a reduced exploratorybehaviour during the first period of testing without changes in the motor activity during the second period. **P< 0.01 versus wild-type mice (Student’s t-test).(B) Alternance in a T maze: male 140-day-old wild-type (open bars, n¼ 10) and parkin mutant (hatched bars, n¼ 14) mice were allowed 3 min to explore the mazewhen one of its arms were blocked. The next day animals were returned to the maze with both arms open. The first arm to be visited was noted. Wild-type mice showed ahigher probability of visiting the previously unknown arm than the parkin mutant mice. *P< 0.05 (Fisher’s exact test). (C) Hanging thread test: the data show the longesttime that animals of 12 months of age managed to hang on to the taught string (best of three trials). There was no difference between hanging times achieved by naivewild-type (open bars) and mutant mice (hatched bars) and no difference between the number of animals refusing to try the test (n¼ 3/28 wild-type versus 3/25 mutant).However, when animals were re-exposed to the test 3 months later, a significantly higher proportion of wild-type animals refused to grip the string. *P< 0.05 (Fischer’sexact test). (D) Basal motor activity in control wild-type (open bar) and parkin mutant mice (shaded bar) at 6 months of age. Motor activity was decreased in the parkinmutant mice. ***P< 0.001 (Student’s t-test). (E) Amphetamine-induced motor activity in control wild type (shaded diamond) and parkin mutant mice (open triangle) at6 months of age. The data show the cumulative amount of motor activity at 6 months of age in mice treated with amphetamine (0.5–5 mg/kg, i.p.) over a period of 1 hbefore and after treatment with amphetamine and the mean� SEM of the counts observed during the periods of 5 min expressed as a percentage of baseline for eachgroup. Amphetamine (0.5 mg/kg) reduced motor activity to a similar extent in both mutant and wild-type groups. Amphetamine (1 and 5 mg/kg) increased motor activityin wild-type, but not in mutant mice. Motor activity was evaluated in groups of five animals. ***P< 0.001 (Student’s t-test).
2282 Human Molecular Genetics, 2003, Vol. 12, No. 18
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

rich in dopamine terminals and the ratio of the MAO-deriveddopamine metabolite, DOPAC, over the COMT-deriveddopamine metabolite, 3-MT, was significantly greater in parkinmutant mice. These changes suggest that abolition of parkinfunction interferes with dopamine release. Since MAO-mediated metabolism of dopamine is considered to be mainlyintracellular and COMT activity is mostly extracellular, thesefindings suggest that there is a shift towards increasedintraneuronal dopamine metabolism by MAO in parkin mutantmice. The increase in the oxidative reduction of dopamine byMAO should result in enhanced production of H2O2, sinceMAO generates one molecule of DOPAC and H2O2 permolecule of dopamine metabolized. Increasing the productionof H2O2 could cause neuronal damage, due to excessiveproduction of free radicals, if the normal scavenging systemsare not able to cope with the excess of oxidative stress (42).Moreover, a number of studies have implicated oxidative stressin the neuronal death associated with PD (43) and severereductions in the levels of GSH have been observed in thesubstantia nigra of PD patients (44,45). Decreased GSH levels
have also been reported in human neuroblastoma and humanteratoma cells transfected with mutant parkin proteins througha mechanism that appears to be independent of parkin ubiquitinligase activity (46).
In parkin mutant mice we have detected reduced levels of DATin the striatum, which suggests an initial damage to thepresynaptic terminals of nigrostriatal dopamine neurons.However, since there is no obvious loss of nigrostriataldopamine neurons in parkin mutant mice, it is possible thatthese animals develop more powerful free radical protectingsystems than the wild-type. Evidence supporting this possibilityis provided by the observation that the levels of GSH areselectively increased in the striatum, the brain area with thehighest dopamine levels, as well as in neuronal cultures from thedopamine rich ventral midbrain of parkin mutant mice. GSH is acritical factor that regulates the neurotoxic or neurotrophiceffects of catecholamines and other free radical donors on DAneurons (47–49). For example, increasing catecholamine levels,similar to those observed in parkin mutant mice, also increasesGSH levels and produces neuroprotective effects in both
Figure 5. Monoamine metabolism in parkin mutant mice is shifted towards the H2O2 producing MAO pathway, and a compensatory increase in GSH levels. (A)Monoamine and metabolites in limbic system. (B) Monoamine and metabolites in the striatum. (C) Monoamine and metabolites in the diencephalon. (D)Monoamine and metabolites in the brain stem. (E) GSH levels in striatum. (F) GSH levels in fetal ventral mid brain neuronal cultures. Wild-type and parkin mutantmice at 11 months of age. (A–D) Results were calculated as ng/g of fresh tissue and then expressed as a ratio. (E) Results are expressed as mg GSH/g of freshtissue. (F) Results are expresed as ng GSH/mg protein. Values are the mean�SEM (n¼ 6–9). Statistical analysis was performed by one-way analysis of variance,followed by the Newman–Kleus multiple comparision test. *P< 0.05; **P< 0.01, ***P< 0.001.
Human Molecular Genetics, 2003, Vol. 12, No. 18 2283
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

cultured fetal rat neurons and neuroblastoma cells and inhibitionof this increased GSH formation switches the neuroprotectiveeffects of catecholamines to a neurotoxic action (47–49). Sincewe found increased GSH levels in both fetal neurons and brainregions of postnatal animals, this mechanism of neuroprotectionappears to be active from the very early stages of dopamine celldevelopment.
The parkin mutant mice also show a number of deficits indopamine function, such as abnormal baseline motor activity,lack of amphetamine-induced increased locomotor activity,reduced amphetamine-induced dopamine release in fetal neur-onal cultures, as well as decreased levels of VMAT2 and DATprotein and reduced [3H]-DA uptake. Amphetamine mainlyincreases the release of dopamine from the intracytoplasmicrather than the vesicular pool (50–54). Since the release ofdopamine evoked by KCl, which is considered as an index ofvesicular release, was unaffected in fetal neuronal cultures frommutant mice, this indicates that the reduction of amphetamine-induced dopamine release is mediated by alterations in theintracytoplasmic pool of dopamine.
The changes observed in VMAT2 and DAT protein levelsand the reduction of [3H]-DA uptake may be related to thepathogenetic mechanisms of parkin dysfunction, or they could
signal the first indication of initial damage to the dopaminenigrostriatal terminals. However, we cannot exclude a con-tribution of adaptative modifications in response to otherchanges in DA metabolism. Transgenic mice with reducedVMAT2 expression also show reduced amphetamine-inducedDA release (55). Therefore, the abnormalities of DA releaseobserved in the parkin mutant mice may be related tothe reduction of VMAT2 levels. Since VMAT2 controls theintraneuronal storage of DA, preventing its intracytoplasmicoxidation and preparing it for exocytotic release, the reductionof VMAT2 levels could enhance the neurotoxicity of highintracytoplasmic DA concentrations (56). There is a substantialamount of information indicating that VMAT2 and DAT arealtered in patients with PD (57) and in presymptomaticindividuals (58) and that their activity, as measured by modernneuroimaging techniques, can be used to measure the densityof DA terminals while aging (59) and the severity of DA cellpresynaptic terminal loss during the progression of PD (60).
Examination of hippocampal electrophysiology demonstratesthat abnormal neurotransmitter release in parkin mutant mice isnot restricted to the nigrostriatal DA pathway but is also evidentas significant deficits in glutamate neurotransmitter release atthe level of the Schaffer collateral-CA1 synapses. Similardeficits in hippocampal electrophysiology have been observedin a synuclein KO mice (61). The inhibition of neurotransmit-ter release in the absence of parkin is consistent with previousobservations in which parkin has been shown to be localized tothe interface between the synaptic vesicles and the cytoplasm(62) and to control the degradation of protein substrates,e.g. Cdcrel-1, that have been implicated in the inhibition ofsynaptic transmission (30).
Dopamine neurotransmission is also altered in mice withmutations of a-synuclein. For example, similar to parkinmutant mice, mice lacking a-synuclein develop normally, donot display loss of dopaminergic neurons, but have decreasedlocomotor responses to amphetamine and reduced striataldopamine levels (63). By contrast, overexpression of A53Tmutated a-synuclein in mice induces a more pronouncedphenotype characterized by a severe movement disorderleading to paralysis and death (64). Moreover, in contrast tomesencephalic neurons from parkin mutant mice whereKCl-induced dopamine release is unaffected and ampheta-mine-induced dopamine release is decreased, in mesencephalicneurons overexpressing the A53T a-synuclein mutation,KCl-induced dopamine release is decreased and ampheta-mine-induced dopamine release is increased (65). Expressionof the A53T mutation in PC12 cells also produces loss of KCl-induced dopamine release and alterations of the ubiquitin-dependent degradation system (66).
The alterations in dopamine and glutamate neurotransmissioncould be at the origin of some of the motor and cognitive deficitsin parkin mutant mice, which are also present as presymptomaticsymptoms in patients with PD prior to the development of overtclinical symptoms. For example, the decline in spatial workingmemory observed in mutant mice in the spontaneous alternationtask has also been observed in PD patients with mild clinicalsymptoms (67). PD patients with severe clinical symptomsdisplay impairment of spatial, verbal and visual workingmemory (67). The reduced exploration observed in mutantmice in the open field studies is probably related to a lack of
Figure 6. Parkin mutant mice have normal basal spontaneous release of [3H]-dopamine and KCl-induced release of [3H]-dopamine, but reduced ampheta-mine-induced release of [3H]-dopamine as well as reduced [3H]-dopamineuptake. (A) [3H]-DA release in 7 DIV-old cultures from fetal (E13) midbrainmice are the mean�SEM (n¼ 4–6). Statistical analysis was performed byone-way analysis of variance followed by the Newman–Keuls multiple compar-ison test. ***P< 0.001 for amphetamine-induced [3H]-DA release in parkinmutant versus wild-type mice. (B) [3H]-DA uptake in 7 DIV-old cultures fromfetal (E13) midbrain mice are the mean� SEM (n¼ 12). Statistical analysis wasperformed by one-way analysis of variance followed by the Newman–Keulsmultiple comparison test. ***P< 0.001 parkin mutant versus wild-type mice.
2284 Human Molecular Genetics, 2003, Vol. 12, No. 18
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from
curiosity and novelty-seeking behaviour, rather than to akinesiaor cognitive deficits, since other motor and behavioral deficits,such as akinesia or abnormal performance of routine activitieswere not observed. In addition, it is possible that the cognitivedeficit in the hanging grip test could be explained, at least in part,by the mutant mice having a more submissive character. Thepresence of similar personality traits, including industriousness,punctuality, inflexibility, cautiousness and lack of noveltyseeking have been observed to predate the onset of motorillness in presymptomatic PD patients (68,69). Moreover, a highdegree of submissive behaviour has been observed in depressedpatients (70) and depression is frequently a complicating factorin patients with PD (71). These behavioral patterns in PDpatients and mutant mice are probably related to abnormalities ofthe phasic release of dopamine and other neurotransmitterswhen a high output is required from these neuronal systems,rather than to neuronal loss in these systems, with normaldopamine function being maintained during activities that do notrequire a high degree of activation.
It is clear from the above that the absence of parkin results inabnormal processing of dopamine, deficits in glutamate synaptictransmission and impairment of certain aspects of motor activityand memory. However, in contrast to PD, all of these phenotypicchanges occur in the absence of clear evidence of parkinsonism,cell loss in the nigrostriatal dopamine system or accumulation ofubiquitinated a-synuclein aggregates. Locomotor defects havealso been observed in the absence of general neuronaldegeneration and dopaminergic neuron loss in Drosophilaparkin mutants (34). In the Drosophila parkin model thelocomotor defects are mediated by apoptotic muscle degenera-tion, which is not observed in our mouse model. Absence ofnigral cell loss, despite a synuclein pathology and cell loss inother CNS areas, has also been observed in transgenic asynuclein mice (64). This suggests that the nigrostriataldopamine system in mice and Drosophila is less vulnerable toneuronal loss in the absence of parkin than humans. This couldbe explained by the lack of neuromelanin formation in mice orthe low levels of GSH in primates. In addition, parkin could
Figure 7. Parkin mutant mice have deficits in glutamate transmission at hippocampal Schaeffer collateral-CA1 synapses. (A) Superimposed representative tracingsof EPSPs elicited by stimulation of increasing strength in a slice prepared from a wild-type mice (þ/þ, upper panel) and a parkin mutant mice (�/�, lower panel).(B) Mean (�SEM) amplitude of the EPSPs plotted as a function of stimulus intensity showing the significantly smaller EPSPs size in parkin mutant animals (closedtriangles, n¼ 7 slices from four male mutant mice) with respect to wild-type controls (open circles, n¼ 8, four male mice). The decreased EPSP amplitudes inparkin mutant mice were highly significant versus stimulation intensity data over the nine levels of intensities tested (F1,116¼ 23.42; P< 0.001). Bonferonipost-tests indicated that the average size of the EPSPs were significantly different at 200 mA (237� 70 and 556� 128mV, mean�SEM for mutant and wild-type,respectively, P< 0.05) and 225mA (323� 90 and 694� 150mV, P< 0.05). (C) Magnitude of paired-pulse facilitation. Plot of the mean (�SEM) percentageincrease of the second EPSP with respect to the first EPSP as a function of interstimulation interval (same symbols and number of replicates as in B). In parkinmutant mice there was a significant increase on the facilitation ratios (F1,78¼ 7.795; P< 0.01), but there was no significant difference in the average PPF ratios atany of the five paired-pulse intervals tested. (D) Enhanced facilitation upon repeated low frequency stimulation. Amplitudes (mean�SEM) of successive EPSPs ina burst of 30 applied 70 ms apart (i.e. at 14 Hz) normalized with respect to the amplitude of the first EPSP of the burst (same symbols and replicates as in B). Therewas a significant increase in normalized EPSP amplitude in parkin mutant mice in the ANOVA (F1,390¼ 36.81, P< 0.001 compared with wild-type. (E) NormalLTP in both wild-type and parkin mutant mice. Plot of the mean (�SEM) amplitude of the EPSPs versus time. Data were normalized for each slice with respect tothe average amplitude recorded during baseline. Shown in insert are representative superimposed tracings taken before and after LTP induction (theta-burst sti-mulation, arrow) in wild-type mice (þ/þ) and a parkin mutant mice (�/�).
Human Molecular Genetics, 2003, Vol. 12, No. 18 2285
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

work in concert with other factors to induce the degeneration ofdopaminergic neurons and/or compensatory changes couldprotect against parkin loss of function. A key role for suchregulatory factors has been observed in dopaminergic neuronswhere L-DOPA and catecholamines can produce neurotoxic(72,73) or neurotrophic (47,74–76) effects, depending on theconcentration of GSH and other free radical scavengers (77–79).The presence of such compensatory mechanisms could explain,at least in part, the lack of a clear parkinsonian phenotype inparkin mutant mice and the large variability in age of onset andclinical severity of parkinsonism related to mutations of parkin,even in members of the same family (19,20).
In conclusion, this parkin mutant mouse model displays anumber of behavioural and biochemical changes whichreproduce some of the presymptomatic aspects of PD. However,these mice do not display a clear parkinsonian phenotype.This model therefore provides a valuable tool to betterunderstand some of the preclinical deficits observed inpatients with PD and to further examine the potential com-pensatory mechanisms that prevent the onset of a parkinsonianphenotype and which could provide new strategies forneuroprotection.
MATERIALS AND METHODS
Inactivation of the parkin gene: cloning of the mouseparkin gene and creation of the targeting vector
A genomic phage library from the 129SvJ mouse strain(Stratagene, lambda FIX II ref. 946313) was screened with a ratparkin cDNA probe. A clone containing a 12kb genomicfragment was identified and shown to contain exon 3 of themouse parkin gene, plus about 2.4 kb of intron 2 at its 50 end.
Two oligonucleotides were synthesized to amplify a sequenceof 1092 bp containing 1030 bp at the 30 end of intron 2 and62 bp at the 50 end of exon 3. The oligonucleotide used at the 50
end carries an EcoRI cloning site (ccg gaa ttc gtg tcc ttg gtg gtagat tca), whereas that used at the 30 end carries a BamHI (cgcgga tcc tga ctt ctc ctc cgt ggt ctc). The XhoI site on the PCRfragment was destroyed and then the fragment was cloned atsites EcoRI and BamHI in the pPN2T plasmid, provided byDr E. Rubin’s laboratory (Berkeley, CA, USA). This plasmidcontains a positive selection cassette (neomycin gene), plus twonegative selection cassettes (TK gene).
The long arm of the homologous recombination vector wasobtained by cloning a 7 kb EcoRI–SalI fragment from the 12 kbgenomic fragment in a Bluescript plasmid. The EcoRI site is179 bp from the 50 extremity of the exon 3. The NotI site of thisDNA fragment was then destroyed. New sites of NotI and XhoIcloning were then introduced into the SalI site and the EcoRIsite of the polylinker, respectively. Sequencing revealed that theEcoRI site was removed. The 7 kb genomic fragment wascloned in the previous plasmid containing the BamHI–EcoRIfragment plus the Neo and TK genes by using NotI–XhoI sites.In this construction, the parkin gene was interrupted by theneomycin resistance cassette within exon 3. The first 62 nucleicacids of exon 3 were preserved. The introduced deletion coversan area of 1097 bp, encompassing the last 179 nucleic acids ofexon 3 and the first 918 nucleic acids of intron 3.
Tools for homologous recombination
PCRs. Three oligonucleotide pairs were used to select therecombinant ES cells; pair A, tttccaaatgtgtcagtttc/tttgagtaagagc-cactaagg; pair B, tgttccacatacacttcattc/tttgagtaagagccactaagg;pair C, cagtattgttttgccaagttc/tttgagtaagagccactaagg.
The PCR conditions were as follows: reaction mixturecontaining 50 fg of DNA, 1.5 ml of four dNPTs 4� 25 mM, 4 mlof each oligonucleotide at 100 ng/ml, 10 ml of MgCl2 1 mM,10 ml of buffer Taq 10x, 0.5 ml of Taq polymerase in 5 U/ml(Perkin Elmer) and 100 ml of water QSP. Amplification cycleswere 8 min at 95�C, then 50 cycles at 95�C (0.5 min), 53�C(0.5 min), 72�C (1 min) and finally 10 min at 72�C.
Southern blots. A 1.3 kb probe, external to the recombinationvector and located in the 50 of this, makes it possible to discri-minate a wild allele of the Parkin gene from a mutated alleleafter having performed homologous recombination. After enzy-matic digestion by the endonuclease BamHI, the marked proberevealed a fragment of 6 kb for the wild allele and a fragment of5 kb for the mutated allele. The methods for Southern blotswere as described by Sambrook et al. (80)
Cell culture. The homologous recombination vector was elec-troporated into 129SV ES CK35 cells obtained from thePasteur Institute. 288 clones resistant to double selection(Neo/TK) were plated. Analysis of the genetic content of thesecells by PCR and then by Southern blot made it possible toselect 41 clones showing evidence of homologous recombina-tion in the parkin gene.
Mutant mice. Two clones were microinjected into blasto-cystes from C57BL6 mice. Germ line transmission wasobtained from the chimera mice of the two microinjectedclones. The parkin mutation was maintained in a pure 129SVand in a mixed 129SV/C57BL6 (50/50) genetic background.
Analysis of parkin protein expression
Antibody production. An antibody to mouse parkin wasraised in rabbits following the methods described by Stichelet al. (81). Briefly, a peptide was synthesized correspondingto residues 71–92 of the mouse parkin protein and this wascoupled to GFAP. New Zealand White rabbits were injecteds.c. with 150–250 mg of protein emulsified in KeyholeLimpet Haemacyonin and then boosted nine times. Rabbitswere bled 2 weeks after the last boost and serum purified byaffinity column and used for immunoblotting studies.
Western blots. Tissue samples from parkin mutant and wild-type control mice were homogenized with a tissue mincer in alysis buffer composed of 50 mM HEPES pH 7.5, 150 mM NaCl,1 mM EGTA, 10% (w/v) glycerol, 1% Triton X-100 (v/v),1.5 mM MgCl2, and a protease inhibitor cocktail. The proteinconcentration in the crude preparation was measured accordingto the standard Bradford assay. Blots were performed on homo-genates from mouse brain on 10% acrylamide gels with MOPSrunning buffer (NuPAGE, Invitrogen). Parkin antibody, as above,
2286 Human Molecular Genetics, 2003, Vol. 12, No. 18
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

was used at 1/500 and revealed with peroxidase-conjugated goatanti-rabbit (Jackson ImmunoResearch) at 1/8000. Protein wasrevealed with an ECL kit (Amersham-Pharmacia).
Phenotype
Breeding colonies were established on the 129SV backgroundor on a mixed 50/50% 129SV/C57BL6 background and theiroffspring used for experimentation. Litter size and mortalitywere observed and recorded in the breeding colony. Afterweaning, mice were maintained in same-sex groups of four tosix per cage under normal animal house conditions withconstant temperature (21� 1�C) and a 12 h light/dark cycle.Unless otherwise stated, animals of mixed background wereused as these animals reproduced more prolifically.
Behaviour
Behavioural studies were carried out by operators blinded to thegenotype of the animals being tested:
(1) Long-term behavioural observations—both male andfemale transgenic mice and wild-type animals wereobserved at monthly intervals, up to the age of 24 months.
(2) Growth rate and body temperature—groups of mice weremonitored for changes in body weight at regular intervalsusing a Mettlar Toledo balance. Body temperature wasmeasured via a rectal probe lubricated with saline.
(3) Open field activity—animals were placed in a Perspex box(41� 28 cm) with a cross dividing it into four equalcompartments. The blinded operator sat at 2 m distancefrom the cage and counted each time the animal traversedthe lines of the cross over two consecutive periods of 3 min.
(4) Spontaneous alternation—animals were placed in a T mazewith one arm blocked and allowed 3 min to explore themaze. Fifteen minutes later they were placed in the samemaze, this time with both arms open, and allowed to enterinto the arm of their choice and the latency and arm choicerecorded. Where the animal had been allowed access to onearm in the first trial and they chose the other arm in thesecond trial, alternation was considered to have takenplace. This is a valid cue for good working memoryperformance and normally, in almost 100% of cases,animals alternate, following a strong exploratory motiva-tion. However, alternation is only possible if the subjectsremember the exploration of the first arm (first trial). Thisis a test classically used to estimate spatial workingmemory in rodents using a motor task without fooddeprivation (no food motivation and reinforcement) (82),the only reinforcement being the strength of the animals’novel environment exploration.
(5) Grip strength—animals were measured using the Colum-bus grip strength meter. Animals were allowed threeattempts to hold on while being pulled backwards acrossthe grid attached to the force-meter. The highest forceobtained, normally the first reading, was recorded.
(6) Hanging thread—animals were placed on a 50 cm cottonthread (in the middle of the thread, 25 cm from eachplatform) suspended between two platforms 25 cm fromthe ground. The animal’s forelegs were placed on the
thread, immediately inducing a grasping behaviour. Toreach any of the two platforms, the mice had to place theirback legs on the thread. Both the time on the wire and thetime until a back leg was placed on the wire were recorded.Refusal to grip was also noted. Animals underwent threetests per session and were re-tested at 3 monthly intervals.
(7) Exploratory behaviour, social interaction and motoractivity were measured in drug-naive animals and afterpharmacological treatment with amphetamine. Sponta-neous motor activity was measured over a 30 min period,after 30 min of adaptation, using automated activity cageswith photoelectric cells without drug treatment and afteramphetamine (0.5, 1 and 5 mg/kg i.p.). In amphetaminetreated mice, the animals were returned to the activitycages 40 min after the treatment and then recorded.
Histology and western blots
Male and female mice were used at different ages (2–24 months)for these experiments. Animals were anaesthetized with pento-barbital (130 mg/kg; Sigma, St Quentin, France) and transcar-dially perfused with 4% paraformaldehyde in 0.1 M phosphatebuffer saline (PBS). Brains were removed, postfixed, andcryoprotected. Routine histological sections at the level of thestriatum, hippocampus, brain stem and cerebellum were stainedwith haematoxylin and eosin. Immunohistochemistry wasperformed, as described previously (83), on free-floatingcryomicrotome-cut sections (20mm in thickness) from the wholebrain. After incubation in 1% H2O2, followed by 0.2% Triton X-100 and then 2% normal goat serum in 0.1 PBS–0.2% gelatin, thesections were stained overnight at 4�C using a polyclonalantibody against tyrosine hydroxylase (TH) (1/1000; rabbitpolyclonal; Pel Freez, Rogers, AR, USA), dopamine transporter(DAT) (1/10000; kindly provided by B. Giros), a-synuclein (1/2000; affinity-purified rabbit polyclonal AB5038P; ChemiconInternational), ubiquitin (1/3000; rabbit polyclonal; Dako),synapsin (1/2000; polyclonal rabbit; Sigma), synaptophysin (1/1000; monoclonal mouse; Sigma), Map2 (1/500; monoclonalmouse; Sigma) or NeuN (1/2000; monoclonal mouse; ChemiconInternational). Sections were then treated with biotinylated anti-rabbit (1/200) secondary antibodies (Vectastain; VectorLaboratory, Burlingame, CA, USA), and subsequently incubatedwith avidinbiotinylated horseradish peroxidase complex. Theperoxidase was revealed by incubation with 0.05% 3,30-diaminobenzidine tetrahydrochloride containing 0.015% hydro-gen peroxide. All sections were stained simultaneously foranimals of the same age using the same solutions. The totalnumber of TH-stained cells with clearly visible nuclear borderswas estimated, using a previously described method and an imageanalysis system (84) (Visioscan 2000, Biocom, Les Ulis, France).All sections were coded and examined blind. In the striatum,measurement of optical density of TH immunostaining, as anindex of the density of dopaminergic axons and nerve terminals,was performed using the same image analysis system.
Muscle samples from five transgenic and five wild-type malemice at 20 months of age were embebbed in paraffin, cut on amicrotome in 5 mm sections and mounted on 2� gelatinizedslides. A Masson Trichrome staining was performed, and slideswere examined by optic microscopy.
Human Molecular Genetics, 2003, Vol. 12, No. 18 2287
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

Samples (25 mg) of striatum, limbic region, diencephalon andbrain stem were analyzed for TH and DAT proteins by westernblots, according to Mena et al. (85). Samples (40 mg) of striatumwere analysed for VMAT2 protein by western blots, accordingto Handler et al. (86). The membranes were incubated withmouse anti-TH from Chemicon (1:10 000), mouse anti-DATfrom Chemicon (1:5000), rabbit anti-VMAT2 from Chemicon(1:500) and mouse anti-b-actin from Sigma (1:10 000), as acontrol of charge. For VMAT2 detection, an enchancedchemiluminescence (SuperSignal West Dura ExtendedDuration Substrate) kit from Pierce Chemical was used.
Pharmacology
Monoamines and their metabolites. Endogenous monoamineand metabolite levels in brain regions: After decapitation, brainparts were dissected, according to Carlsson and Lindqvist (87)into the dopamine (DA)-rich limbic portion, the corpora striata,the rest of the cerebral hemispheres, the diencephalon and thelower brain stem. The brain parts were frozen on dry ice and thelevels of DA and its metabolites, 3-methoxy-tyramine (3-MT),3,4-dihydroxyphenylacetic acid (DOPAC) and homovanillicacid (HVA), noradrenaline (NA) and its metabolite, 4-hydroxy-3-methoxy-phenyl-glycol (MHPG), serotonin (5-HT)and its metabolite, 5-hydroxy-indole-acetic acid (5-HIAA),were measured by HPLC with an ESA coulochem detector,according to Mena et al. (85). Briefly, the tissue was sonicatedin 8 vols (w/v) of 0.4 N perchloric acid with 0.5 mM Na2S2O5
and 2% EDTA and then centrifuged for 10 min. Monoaminelevels were determined from 20 ml of the resulting supernatant.
Catecholamine turnover. Endogenous whole brain levelsof NA and DA, with and without catecholamine synthesisinhibition by alpha-methyltyrosine (a-MT), were measured.Mice were sacrificed 1 h after being injected with a-MT(250 mg/kg, i.p.) (88).
Measurement of glutathione levels. Total glutathione (GSH)levels were measured by the method of Tietze (89). Briefly,cells from fetal midbrain cultures or brain samples were soni-cated in 0.4 N PCA and centrifuged, and the supernatants wereneutralized with 4 vols of NaH2PO4, 0.1 M (5 mM EDTA), pH7.5. GSH content was measured in a P96 automatic reader bythe addition of DTNB (0.6 mM), NADPH (0.2 mM), GSHreductase (1 U) and the reaction monitored at 412 nM for6 min. Oxidized glutathione (GSSG) was measured in the cellsby the method of Griffith (90). Reduced GSH, after PCAextraction and pH neutralization, was derivatized with 2-vinyl-pyridine at room temperature for 1 h and the reaction carriedout as above. GSH was obtained by subtracting GSSG levelsfrom total GSH levels (47).
DA uptake and release in fetal midbrain neuronalcultures. Neuronal-enriched cultures from embryonic wildtype and parkin mutant mice midbrain E-13 (crown–rumplength 10–12 mm) were obtained and prepared, according toMena et al. (91). The cells were seeded in B27/Neurobasal
TM medium with 15% fetal calf serum (B27/NBL-FCS) at adensity of 2� 105 cells/cm2 in multiwells or glass coverslides coated with poly-D-lysine (4.5 mg/cm2) in 0.1 M boratebuffer, pH 8.4. The cultures were kept in a humidifiedchamber at 37�C in a 5% CO2 atmosphere for 6–7 days in vitro.Twenty-four hours after plating, the cells were changedto serum-free defined medium (B27/NBL), as previouslyreported (91).
Basal and Kþ- and amphetamine-induced [3H]-DA releasewas investigated in cells after 6–7 days in culture. Each culturewas washed once with 0.5 ml of Krebs–Ringer–HEPES buffer(KRH), and then loaded with [3H]-DA by incubating them for20 min at 37�C with KRH containing [3H]-DA (5� 10�8
M)and desipramine (5� 10�5
M). After loading, the cells wererinsed four times with KRH. The basal release of [3H]-DA wasmeasured in cultures incubated for 5 min at 23�C with 0.5 ml ofKRB. Kþ- or amphetamine-induced [3H]-DA release wasmeasured in similarly treated cultures, but with added KCl(50 mM) or amphetamine (10 mM). The radioactivity in themedia was measured by liquid scintillation spectrometry. At theend of the experiment, residual intracellular radioactivity wasextracted from the cells by incubating them for 30 min with0.2 ml of NaOH (0.4 N), and then counted. [3H]-DA release isexpressed as a percentage of the total intracellular content of[3H]-DA present in each culture at the time of the releaseincubation, to correct for variations in the amount of [3H]-DAtaken up by different cultures.
High affinity [3H]-DA uptake was measured after incubationof the cells with 10�8
M [3H]-DA (70 Ci/mmol), in the presenceof pargyline 10�5
M and ascorbic acid 10�3M, at 37�C for
20 min. Non-specific uptake/binding was calculated in thepresence of 10�5
M mazindol (77). Proteins were measuredaccording to the BCA assay.
Hippocampal electrophysiology. Hippocampal slices(�500mM thick) were prepared from male parkin mutant miceof 64–68 weeks. Age- and sex-matched wild-type micewere used as controls. Slices were incubated in a submersion-typerecording chamber through which artificial cerebrospinal fluid(ACSF: 124 mM NaCl, 3 mM KCl, 1.25 mM NaH2PO4, 1.3 mM
MgSO4, 2 mM CaCl2, 26 mM NaHCO3 and 10 mM glucose)was continuously perfused at 2.5–3 ml/min. ACSF was bubbledwith a mixture of 95% O2/5% CO2 and maintained at31.5� 0.5�C. Test stimulations (0.1 ms duration) were deliveredat constant intensity through bipolar stainless steel electrodesplaced in the stratum radiatum of the CA1 area. Extracellular fieldexcitatory post-synaptic potentials (EPSPs) were recorded with amonopolar tungsten electrode placed in the same region on thetrajectory of the Schaffer collaterals. The following features ofsynaptic physiology were assessed: basal transmission, paired-pulse facilitation, repetitive low-frequency stimulation and long-term potentiation. The latter three properties were evaluated at astimulation strength eliciting EPSPs of approximately half themaximal size. Analysis of paired-pulse facilitation was performedby delivering five pairs of stimuli at decreasing intervals (400,200, 100, 50 and 25 ms). Response to low-frequency stimulationwas explored by delivering two sets of 30 stimuli at 70 msintervals repeated 30 s apart and averaged. To induce long-termpotentiation, repeated brief trains of high frequency stimulation
2288 Human Molecular Genetics, 2003, Vol. 12, No. 18
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

were delivered following a protocol consisting of five bursts offive pulses, each burst at 100 Hz, with 200 ms between bursts(theta-burst stimulation).
Data analysis
All data presented are expressed as the mean� SEM. Theresults were statistically evaluated for significance by usingone-way analysis of variance, followed by Student’s t-test, theNewman–Keuls multiple comparison test or Bonferoni posthoc test. Means among groups were considered significantlydifferent when P< 0.05.
ACKNOWLEDGEMENTS
This work was supported by grants from the ComunidadAutonoma de Madrid, grant CAM 8.5/49/2001 and Fondo deInvestigaciones Sanitarias, FIS 2002/PI20265 to J.G.Y. andM.A.M., and from the Association France Parkinson to A.B.The authors thank Mrs Rosario Villaverde and Elodie Martınfor technical help and Thomas Debeir, Rita Raisman, PatrickMichel and Etienne Hirsch for helpful discussions. This paperis dedicated to the memory of Nacer Abbas.
REFERENCES
1. de Rijk, M.C., Launer, L.J., Berger, K., Breteler, M.M., Dartigues, J.F.,Baldereschi, M., Fratiglioni, L., Lobo, A., Martinez-Lage, J.,Trenkwalder, C. and Hofman, A. (2000) Prevalence of Parkinson’sdisease in Europe: A collaborative study of population-based cohorts.Neurologic Diseases in the Elderly Research Group, Neurology, 54,S21–23.
2. Elbaz, A., Bower, J.H., Maraganore, D.M., McDonnell, S.K., Peterson, B.J.,Ahlskog, J.E., Schaid, D.J. and Rocca, W.A. (2002) Risk tables forparkinsonism and Parkinson’s disease. J. Clin. Epidemiol., 55, 25–31.
3. Jankovic, J. (1992) Pathophysiology and clinical assessment of motorsymptoms in Parkinson’s disease. In Koller, W.C. (ed.), Handbook ofParkinson’s Disease. Marcel Dekker, New York, pp. 129–157.
4. Louis, E.D., Levy, G., Cote, L.J., Mejia, H., Fahn, S. and Marder, K.(2002) Diagnosing Parkinson’s disease using videotaped neurologicalexaminations: validity and factors that contribute to incorrect diagnoses.Mov. Disord., 17, 513–517.
5. Forno, L.S. (1996) Neuropathology of Parkinson’s disease. J. Neuropathol.Exp. Neurol., 55, 259–272.
6. Dickson, D.W. (2001) Alpha-synuclein and the Lewy body disorders. Curr.Opin. Neurol., 14, 423–432.
7. Schneider, J.A., Bienias, J.L., Gilley, D.W., Kvarnberg, D.E., Mufson, E.J.and Bennett, D.A. (2002) Improved detection of substantia nigra pathologyin Alzheimer’s disease. J. Histochem. Cytochem., 50, 99–106.
8. Beal, M.F. (2002) Oxidatively modified proteins in aging and disease. FreeRadic. Biol. Med., 32, 797–803.
9. Schapira, A.H., Gu, M., Taanman, J.W., Tabrizi, S.J., Seaton, T., Cleeter, M.and Cooper, J.M. (1998) Mitochondria in the etiology and pathogenesis ofParkinson’s disease. Ann. Neurol., 44, S89–98.
10. Betarbet, R., Sherer, T.B. and Greenamyre, J.T. (2002) Animal models ofParkinson’s disease. Bioessays, 24, 308–318.
11. Feger, J., Pessiglione, M., Francois, C., Tremblay, L. and Hirsch, E. (2002)Experimental models of Parkinson’s disease. Ann. Pharm. Fr., 60, 3–21.
12. Gerlach, M. and Riederer, P. (1996) Animal models of Parkinson’s disease:an empirical comparison with the phenomenology of the disease in man.J. Neural Transm., 103, 987–1041.
13. Grunblatt, E., Mandel, S. and Youdim, M.B. (2000) Neuroprotectivestrategies in Parkinson’s disease using the models of 6- hydroxydopamineand MPTP. Ann. NY Acad. Sci., 899, 262–273.
14. Polymeropoulos, M.H., Lavedan, C., Leroy, E., Ide, S.E., Dehejia, A.,Dutra, A., Pike, B., Root, H., Rubenstein, J., Boyer, R. et al. (1997)
Mutation in the alpha-synuclein gene identified in families with Parkinson’sdisease. Science, 276, 2045–2047.
15. Le, W.D., Xu, P., Jankovic, J., Jiang, H., Appel, S.H., Smith, R.G. andVassilatis, D.K. (2003) Mutations in NR4A2 associated with familialParkinson disease. Nat. Genet., 33, 85–89.
16. Kitada, T., Asakawa, S., Hattori, N., Matsumine, H., Yamamura, Y.,Minoshima, S., Yokochi, M., Mizuno, Y. and Shimizu, N. (1998) Mutationsin the parkin gene cause autosomal recessive juvenile parkinsonism.Nature, 392, 605–608.
17. Lucking, C.B., Durr, A., Bonifati, V., Vaughan, J., De Michele, G.,Gasser, T., Harhangi, B.S., Meco, G., Denefle, P., Wood, N.W. et al. (2000)Association between early-onset Parkinson’s disease and mutations in theparkin gene. French Parkinson’s Disease Genetics Study Group. New Engl.J. Med., 342, 1560–1567.
18. Periquet, M., Latouche, M., Lohmann, E., Rawal, N., De Michele, G.,Ricard, S., Teive, H., Fraix, V., Vidailhet, M., Nicholl, D. et al. (2003)Parkin mutations are frequent in patients with isolated early-onsetparkinsonism. Brain, 126, 1271–1278.
19. Abbas, N., Lucking, C.B., Ricard, S., Durr, A., Bonifati, V., De Michele, G.,Bouley, S., Vaughan, J.R., Gasser, T., Marconi, R. et al. (1999) A widevariety of mutations in the parkin gene are responsible for autosomalrecessive parkinsonism in Europe. French Parkinson’s Disease GeneticsStudy Group and the European Consortium on Genetic Susceptibility inParkinson’s Disease. Hum. Mol. Genet., 8, 567–574.
20. Hoenicka, J., Vidal, L., Morales, B., Ampuero, I., Jimenez-Jimenez, F.J.,Berciano, J., del Ser, T., Jimenez, A., Ruiz, P.G. and de Yebenes, J.G. (2002)Molecular findings in familial Parkinson disease in Spain. Arch. Neurol.,59, 966–970.
21. Hayashi, S., Wakabayashi, K., Ishikawa, A., Nagai, H., Saito, M.,Maruyama, M., Takahashi, T., Ozawa, T., Tsuji, S. and Takahashi, H.(2000) An autopsy case of autosomal-recessive juvenile parkinsonismwith a homozygous exon 4 deletion in the parkin gene. Mov. Disord.,15, 884–888.
22. Mori, H., Kondo, T., Yokochi, M., Matsumine, H., Nakagawa-Hattori, Y.,Miyake, T., Suda, K. and Mizuno, Y. (1998) Pathologic and biochemicalstudies of juvenile parkinsonism linked to chromosome 6q. Neurology,51, 890–892.
23. Takahashi, H., Ohama, E., Suzuki, S., Horikawa, Y., Ishikawa, A.,Morita, T., Tsuji, S. and Ikuta, F. (1994) Familial juvenile parkinsonism:clinical and pathologic study in a family. Neurology, 44, 437–441.
24. Van de Warrenburg, B.P., Lammens, M., Lucking, C.B. Denefle, P.,Wesseling, P., Booij, J., Praamstra, P., Quinn, N., Brice, A. andHorstink, M.W. (2001) Clinical and pathologic abnormalities in a familywith parkinsonism and parkin gene mutations. Neurology, 56, 555–557.
25. Farrer, M., Chan, P., Chen, R., Tan, L., Lincoln, S., Hernandez, D.,Forno, L., Gwinn-Hardy, K., Petrucelli, L. et al. (2001) Lewy bodies andparkinsonism in families with parkin mutations. Ann. Neurol., 50, 293–300.
26. Klein, C., Pramstaller, P.P., Kis, B., Page, C.C., Kann, M., Leung, J.,Woodward, H., Castellan, C.C., Scherer, M., Vieregge, P. et al. (2000)Parkin deletions in a family with adult-onset, tremor-dominantparkinsonism: expanding the phenotype. Ann. Neurol., 48, 65–71.
27. Morales, B., Martınez, A., Gonzalo I. Vidal, L., Gomez-Tortosa, E.,Rabano, A., Ampuero, I., Sanchez, M., Hoenicka, J. and Garcia deYebenes, J. (2002) Steele–Richardson–Olzewsky syndrome in a patientwith a single C212Y mutation in the parkin protein. Mov. Disord.,17, 1374–1380.
28. Lohmann, E., Periquet, M., Bonifati, V., Wood, N.W., De Michele, G.,Bonnet, A.M., Fraix, V., Broussolle, E., Horstink, M.W., Vidailhet, M.,Verpillat, P. et al. (2003) How much phenotypic variation can be attributedto parkin genotype? The French Parkinson’s Disease Genetics Study Groupand the European Consortium on Genetic Susceptibility in Parkinson’sDisease. Ann. Neurol., 54, 176–185.
29. West, A., Periquet, M., Lincoln, S., Lucking, C.B., Nicholl, D., Bonifati, V.,Rawal, N., Gasser, T., Lohmann, E., Deleuze J.F. et al. (2002) Complexrelationship between Parkin mutations and Parkinson disease. Am. J. Med.Genet., 114, 584–591.
30. Zhang, Y., Gao, J., Chung, K.K., Huang, H., Dawson, V.L. and Dawson, T.M.(2000) Parkin functions as an E2-dependent ubiquitin-protein ligase andpromotes the degradation of the synaptic vesicle-associated protein,CDCrel-1. Proc. Natl Acad. Sci. USA, 97, 13354–13359.
31. Imai, Y., Soda, M., Inoue, H., Hattori, N., Mizuno, Y. and Takahashi, R.(2001) An unfolded putative transmembrane polypeptide, which can leadto endoplasmic reticulum stress, is a substrate of Parkin. Cell, 105, 891–902.
Human Molecular Genetics, 2003, Vol. 12, No. 18 2289
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

32. Chung, K.K., Zhang, Y., Lim, K.L., Tanaka, Y., Huang, H., Gao, J.,Ross, C.A., Dawson, V.L. and Dawson, T.M. (2001) Parkin ubiquitinatesthe alpha-synuclein-interacting protein, synphilin-1: implications forLewy-body formation in Parkinson disease. Nat. Med., 7, 1144–1150.
33. Shimura, H., Schlossmacher, M.G., Hattori, N., Frosch, M.P.,Trockenbacher, A., Schneider, R., Mizuno, Y., Kosik, K.S. and Selkoe, D.J.(2001) Ubiquitination of a new form of alpha-synuclein by parkin fromhuman brain: implications for Parkinson’s disease. Science, 293, 263–269.
34. Greene, J.C., Whitworth, A.J., Kuo, I., Andrews, L.A., Feany, M.B. andPollanck, L.J. (2003) Mitochondrial pathology and apoptotic muscledegeneration in Drosophila parkin mutants. Proc. Natl Acad. Sci. USA,100, 4078–4083.
35. Zucker, R.S. (1989). Short-term synaptic plasticity. A. Rev. Neurosci.,12, 13–31.
36. Creager, R., Dunwiddie, T. and Lynch, G. (1980) Paired-pulse andfrequency facilitation in the CA1 region of the in vitro rat hippocampus.J. Physiol., 299, 409–424.
37. Dunwiddie, T.V. and Haas, H.L. (1985) Adenosine increases synapticfacilitation in the in vitro rat hippocampus: evidence for a presynapticsite of action. J. Physiol., 369, 365–377.
38. Beites, C.L., Xie, H., Bowser, R. and Trimble, W.S. (1999) The septinCDCrel-1 binds syntaxin and inhibits exocytosis. Nat. Neurosci., 2,434–439.
39. Diaz-Torga, G., Feierstein, C., Libertun, C., Gelman, D., Kelly, M.A.,Low, M.J., Rubinstein, M. and Becu-Villalobos, D. (2002) Disruption ofthe D2 dopamine receptor alters GH and IGF-I secretion and causesdwarfism in male mice. Endocrinology, 143, 1270–1279.
40. Lipton, J.M. and Clark, W.G. (1986) Neurotransmitters in temperaturecontrol. A. Rev. Physiol., 48, 613–623.
41. Boulay, D., Depoortere, R., Perrault, G., Borrelli, E. and Sanger, D.J.(1999) Dopamine D2 receptor knock-out mice are insensitive to thehypolocomotor and hypothermic effects of dopamine D2/D3 receptoragonists. Neuropharmacology, 38, 1389–1396.
42. Bharath, S., Hsu, M., Kaur, D., Rajagopalan, S. and Andersen, J.K. (2002)Glutathione, iron and Parkinson’s disease. Biochem. Pharmac., 64,1037–1048.
43. Adams, J.D., Chang, M.L. and Klaidman, L.K. (2001) ParkinsonsDisease-redox mechanisms. Curr. Med. Chem., 8, 809–814.
44. Riederer, P., Sofic, E., Rausch, W.D., Schmidt, B., Reynolds, G.P.,Jellinger, K. and Youdim, M.B. (1989) Transition metals, ferritin,glutathione, and ascorbic acid in parkinsonian brains. J. Neurochem.,52, 515–520.
45. Sofic, E., Lange, K.W., Jellinger, K. and Riederer, P. (1992) Reduced andoxidized glutathione in the substantia nigra of patients with Parkinson’sdisease. Neurosci. Lett., 142, 128–130.
46. Hyun, D-H., Lee, M.H., Hattori, N., Kubo, S. I., Mizuno, Y., Halliwell, B.and Jenner, P. (2002) Effect of wild-type or mutant parkin on oxidativedamage, nitric oxide, antioxidant defenses, and the proteasome. J. Biol.Chem., 277, 28572–28577.
47. Mena, M.A., Davila, V., Bogaluvsky, J. and Sulzer, D. (1998) A synergisticneurotrophic response to l-dihydroxyphenylalanine and nerve growthfactor. Mol. Pharmac., 54, 678–686.
48. Rodriguez-Marın, E., Canals, S., Casarejos, M.J., de Bernardo, S.,Handler, A. and Mena, M.A. (2001) L-DOPA and glia-conditioned mediumhave additive effects on tyrosine hydroxylase expression in humancatecholamine-rich neuroblastoma NB69 cells. J. Neurochem., 78, 535–545.
49. Canals, S., Casarejos, M.J., de Bernardo, S., Rodriguez-Marın, E. andMena, M.A. (2001) Glutathione depletion switches nitric oxideneurotrophic effects to cell death in midbrain cultures: implications forParkinson’s disease. J. Neurochem., 79, 1183–1195.
50. Di Chiara, G. and Imperato, A. (1988) Drugs abused by humanspreferentially increase synaptic dopamine concentrations in the mesolimbicsystem of freely moving rats. Proc. Natl Acad. Sci. USA, 85, 5274–5278.
51. Rudnick, G. and Wall, S.C. (1992) The molecular mechanism of ‘ecstasy’[3,4-methylenedioxy-methamphetamine (MDMA)]: serotonin transportersare targets for MDMA-induced serotonin release. Proc. Natl Acad. Sci.USA, 89, 1817–1821.
52. Sulzer, D., Maidment, N.T. and Rayport, S. (1993) Amphetamine and otherweak bases act to promote reverse transport of dopamine in ventralmidbrain neurons. J. Neurochem., 60, 527–535.
53. Sulzer, D., Chen, T.K., Lau, Y.Y., Kristensen, H., Rayport, S. and Ewing,A. (1995) Amphetamine redistributes dopamine from synaptic vesicles to thecytosol and promotes reverse transport. J. Neurosci., 15, 4102–4108.
54. Giros, B., Jaber, M., Jones, S.R., Wightman, R.M. and Caron, M.G. (1996)Hyperlocomotion and indifference to cocaine and amphetamine in micelacking the dopamine transporter. Nature, 379, 606–612.
55. Patel, J., Mooslehner, K.A., Chan, P.M. Emson, P.C. and Stamford, J.A.(2003) Presynaptic control of striatal dopamine neurotransmission in adultvesicular monoamine transporter 2 (VMAT2) mutant mice. J. Neurochem.,85, 898–910.
56. Shuldinger, S. (1994) A molecular glimpse of vesicular monoaminetransporters. J. Neurochem., 62, 2067–2078.
57. Miller, G.W., Erickson, J.D., Perez, J.T., Penland, S.N., Mash, D.C.,Rye, D.B. and Levey, A.I. (1999) Immunochemical analysis of vesicularmonoamine transporter (VMAT2) protein in Parkinson’s disease. Exp.Neurol., 156, 138–148.
58. Fernandez, H.H., Friedman, J.H., Fischman, A.J., Noto, R.B. andLannon, M.C. (2001) Is altropane SPECT more sensitive to fluoroDOPA PETfor detecting early Parkinson’s disease? Med. Sci. Monit., 7, 1339–1343.
59. Moll, G.H., Mehnert, C., Wicker, M., Bock, N., Rothenberger, A.,Ruther, E. and Huether, G. (2000) Age-associated changes in the densitiesof presynaptic monoamine transporters in different regions of the rat brainfrom early juvenile life to late adulthood. Brain Res. Dev. Brain Res.,7, 251–257.
60. Parkinson Study Group (2002) Dopamine transporter brain imaging toassess the effects of pramipexole vs levodopa on Parkinson diseaseprogression. JAMA, 3, 1653–1661.
61. Cabin, D.E., Shimazu, K., Murphy, D., Cole, N.B., Gottschalk, W.,McIlwain, K.L., Orrison, B., Chen, A., Ellis, C.E., Paylor, R. et al. (2002)Synaptic vesicle depletion correlates with attenuated synaptic responsesto prolonged repetitive stimulation in mice lacking alpha-synuclein.J. Neurosci., 22, 8797–8807.
62. Kubo, S.I., Kitami, T., Noda, S., Shimura, H., Uchiyama, Y., Asakawa, S.,Minoshima, S., Shimizu, N., Mizuno, Y. and Hattori, N. (2001) Parkin isassociated with cellular vesicles. J. Neurochem., 78, 42–54.
63. Abeliovich, A., Schmitz, Y., Farinas, I., Choi-Lundberg, D., Ho, W.H.,Castillo, P.E., Shinsky, N., Verdugo, J.M., Armanini, M., Ryan, A. et al.(2000) Mice lacking alpha-synuclein display functional deficits in thenigrostriatal dopamine system. Neuron, 25, 239–252.
64. Giasson, B.I., Duda, J.E., Quinn, S.M., Zhang, B., Trojanowski, J.Q. andLee, V.M. (2002) Neuronal alpha-synucleinopathy with severe movementdisorder in mice expressing A53T human alpha-synuclein. Neuron,34, 521–533.
65. Lotharius, J., Barg, S., Wiekop, P., Lundberg, C., Raymon, H.K. andBrundin, P. (2002) Effect of mutant alpha-synuclein on dopaminehomeostasis in a new human mesencephalic cell line. J. Biol. Chem.,277, 38884–38894.
66. Stefanis, L., Larsen, K.E., Rideout, H.J., Sulzer, D. and Greene, L.A. (2001)Expression of A53T mutant but not wild-type alpha-synuclein in PC12cells induces alterations of the ubiquitin-dependent degradation system,loss of dopamine release, and autophagic cell death. J. Neurosci.,21, 9549–9560.
67. Owen, A.M., Iddon, J.L., Hodges, J.R., Summers, B.A. and Robbins, T.W.(1997) Spatial and non-spatial working memory at different stages ofParkinson’s disease. Neuropsychologia, 35, 519–532.
68. Heberlein, I., Ludin, H.P., Scholz, J. and Vieregge, P. (1998) Personality,depression, and premorbid lifestyle in twin pairs discordant for Parkinson’sdisease. J. Neurol. Neurosurg. Psychiat., 64, 262–266.
69. Menza, M. (2000) The personality associated with Parkinson’s disease.Curr. Psychiatry Rep., 2, 421–426.
70. O’Connor, L., Berry, J., Weiss, J. and Gilbert, P. (2002) Guilt, fear,submission, and empathy in depression. J. Affect. Disord., 71, 13–19.
71. Cummings, J.L. (1992) Depression and Parkinson’s disease. Am. J.Psychiat., 149, 443–454.
72. Michel, P.P. and Hefti, F. (1990) Toxicity of 6-hydroxydopamine anddopamine for dopaminergic neurons in culture. J. Neurosci. Res., 26,428–435.
73. Mena, M.A., Pardo, B., Casarejos, M.J., Fahn, S. and Garcia de Yebenes, J.(1992) Neurotoxicity of levodopa on catecholamine-rich neurons. Mov.Disord., 7, 23–31.
74. Murer, M.G., Dziewczapolski, G., Menalled, L.B., Garcia, M.C., Agid, Y.,Gershanik, O. and Raisman-Vozari, R. (1998) Chronic levodopa is not toxicfor remaining dopamine neurons, but instead promotes their recovery, inrats with moderate nigrostriatal lesions. Ann. Neurol., 43, 561–575.
75. Troadec, J.D., Marien, M., Darios, F., Hartmann, A., Ruberg, M.,Colpaert, F. and Michel, P.P. (2001) Noradrenaline provides long-term
2290 Human Molecular Genetics, 2003, Vol. 12, No. 18
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from

protection to dopaminergic neurons by reducing oxidative stress.J. Neurochem., 79, 200–210.
76. Troadec, J.D., Marien, M., Mourlevat, S., Debeir, T., Ruberg, M.,Colpaert, F. and Michel, P.P. (2002) Activation of the mitogen-activatedprotein kinase (ERK1/2) signaling pathway by cyclic AMP potentiatesthe neuroprotective effect of the neurotransmitter noradrenaline ondopaminergic neurons. Mol. Pharmac., 62, 1043–1052.
77. Mena, M.A., Casarejos, M.J., Carazo, A., Paino, C.L. and Garciade Yebenes, J. (1996) Glia conditioned medium protects fetal ratmidbrain neurones in culture from L-DOPA toxicity. Neuroreport, 7,441–445.
78. Mena, M.A., Casarejos, M.J. and Garcia de Yebenes, J. (1999) The effectof glia-conditioned medium on dopamine neurons in culture. Modulationof apoptosis, tyrosine hydroxylase expression and 1-methyl-4-phenylpyridinium toxicity. J. Neural Transm., 106, 1105–1123.
79. Pardo, B., Mena, M.A., Casarejos, M.J., Paino, C.L. and De Yebenes, J.G.(1995). Toxic effects of L-DOPA on mesencephalic cell cultures: protectionwith antioxidants. Brain Res., 682, 133–143.
80. Sambrook, J., Fritsch, E.F. and Maniatis, T. (1989) Molecular Cloning.A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold SpringHarbor, NY.
81. Stichel, C., Augustin, M., Kuhn, K., Zhu, X., Engels, P., Ullmer, C. andLubbert, H. (2000) Parkin expression in adult mouse brain. Eur. J.Neurosci., 12, 1–15.
82. Jaffard, R., Dubois, M. and Galey, D. (1981) Memory of a choice directionin a T maze as measured by spontaneous alternation in mice: effects ofintertribal interval and reward. Behav. Proc., 6, 11–21.
83. Hirsch, M.D. and Helke, C.J. (1988) Bulbospinal thyrotropin-releasinghormone projections to the intermediolateral cell column: a double
fluorescence immunohistochemical-retrograde tracing study in the rat.Neuroscience, 25, 625–637.
84. Hunot, S., Brugg, B., Ricard, D., Michel, P.P., Muriel, M.P., Ruberg, M.,Faucheux, B.A., Agid, Y. and Hirsch, E.C. (1997) Nuclear translocationof NF-kappaB is increased in dopaminergic neurons of patients withparkinson disease. Proc. Natl Acad. Sci. USA, 94, 7531–7536.
85. Mena, M.A., Casarejos, M.J., Bonin, A., Ramos, J.A. and Garcia deYebenes, J. (1995) Effects of dibutyryl cyclic AMP and retinoic acid on thedifferentiation of dopamine neurons: prevention of cell death by dibutyrylcyclic AMP. J. Neurochem., 65, 2612–2620.
86. Handler, A., Lobo, M.V.T., Alonso, F.J., Paino, C.L. and Mena, M.A. (2000)Functional implications of the noradrenergic-cholinergic switch induced byretinoic acid in NB69 neuroblastoma cells. J. Neurosci. Res., 60, 311–320.
87. Carlsson, A. and Lindqvist, M. (1973) Effect of ethanol on thehydroxylation of tyrosine and tryptophan in rat brain in vivo. J. Pharm.Pharmac., 25, 437–440.
88. Garcia de Yebenes, J., Carlsson, A. and Mena, M.A. (1978) The effect oftaurine on motor behavior, body temperature and monoamine metabolismin rat brain. Naunyn-Smiedeberg Archv. Pharmac., 304, 95–99.
89. Tietze, F. (1969) Enzymic method for quantitative determination ofnanogram amounts of total and oxidized glutathione: applications tomammalian blood and other tissues. Anal. Biochem., 27, 502–522.
90. Griffith, O.W. (1980) Determination of glutathione and glutathionedisulfide using glutathione reductase and 2-vinylpyridine. Anal. Biochem.,106, 207–212.
91. Mena, M.A., Casarejos, M.J., Carazo, A., Paino, C.L. and Garcia deYebenes, J. (1997) Glia protect fetal midbrain dopamine neurons in culturefrom L-DOPA toxicity through multiple mechanisms. J. Neural.Transm.,104, 317–328.
Human Molecular Genetics, 2003, Vol. 12, No. 18 2291
by guest on January 5, 2016http://hm
g.oxfordjournals.org/D
ownloaded from




















