
The role of neutrophil extracellular traps in the pathogenesis ...
Upload
independentCategory
view
5download
0
Research Paper
J Vasc Res 2001;38:47–58
NFÎB Signaling in Posthypoxic EndothelialCells: Relevance to E-Selectin Expression andNeutrophil Adhesion
Satoshi Kokuraa Carol Ann Rhoadsa Robert E. Wolfb
Toshikazu Yoshikawac D. Neil Grangera Tak Yee Awa
aDepartment of Molecular and Cellular Physiology and bCenter of Excellence in Arthritis and Rheumatism,Louisiana State University Health Sciences Center, Shreveport, La., USA; cFirst Department of Internal Medicine,Kyoto Prefectural University of Medicine, Kyoto, Japan
Received: February 22, 2000Accepted after revision: August 15, 2000
Tak Yee Aw, PhDDepartment of Molecular and Cellular Physiology, LSU Health Sciences Center1501 Kings HighwayShreveport, LA 71130-3932 (USA)Tel. +1 318 675 6032, Fax +1 318 675 4217, E-Mail [email protected]
ABCFax + 41 61 306 12 34E-Mail [email protected]
© 2001 S. Karger AG, Basel
Accessible online at:www.karger.com/journals/jvr
Key WordsAnoxia-reoxygenation W Redox imbalance W Proteintyrosine kinase W Protein kinase C W Protein tyrosinephosphatase W NFÎB W E-selectin expression
AbstractOur previous studies have implicated the nuclear tran-scription factor ÎB (NFÎB) in the regulation of adhesionmolecule expression in endothelial cells exposed toanoxia-reoxygenation (A/R) or a redox imbalance. Theobjectives of this study were (1) to define the kinetics ofNFÎB activation by examining IÎB· degradation and thenuclear translocation of p65 in response to A/R or redoximbalance (induced by treatment of cells with diamideand buthionine sulfoximine) and (2) to determine wheth-er the signal for IÎB· degradation, nuclear translocationof p65, and E-selectin-mediated neutrophil adhesion isrelated to the activity of protein tyrosine kinase (PTK),protein tyrosine phosphatase (PTPase) and/or proteinkinase C (PKC). The results demonstrate that both A/Rand redox imbalance led to IÎB· degradation within30 min and the concomitant appearance of p65 in thenucleus, consistent with rapid cytosolic activation ofNFÎB and subsequent nuclear translocation of the acti-vated p65 subunit. Inhibition of PKC blocked IÎB· degra-
dation and p65 translocation in A/R-challenged, but notredox-altered, endothelial cells. However, both A/R- andredox-induced NFÎB activation was blocked by inhibitionof PTK. Similarly, A/R-induced E-selectin expression andneutrophil-endothelial cell adhesion were blocked byinhibition of PKC or PTK, while only PTK inhibited theredox-induced adhesion response. Pretreatment of cellswith N-acetyl cysteine effectively blocked A/R- or redox-induced IÎB degradation and significantly attenuated therespective neutrophil adhesion responses. Collectively,these findings indicate that A/R-induced E-selectin ex-pression and neutrophil-endothelial cell adhesion aremediated by both PKC and PTK, which signal rapid acti-vation of NFÎB. This A/R-induced NFÎB signaling re-sponse appears to be mediated, at least in part, by intra-cellular redox imbalance.
Copyright © 2001 S. Karger AG, Basel
Introduction
Exposure of endothelial cells to anoxia/reoxygenation(A/R) induces leukocyte adhesion by increasing the sur-face expression of various endothelial cell adhesion mole-cules (ECAMs) [1, 2]. We have previously reported thatexposure of human umbilical vein endothelial cells
Dow
nloa
ded
by:
L.S
.U. H
ealth
Sci
ence
Cen
ter
20
6.17
6.17
5.20
0 -
7/11
/201
3 8:
55:1
2 P
M

48 J Vasc Res 2001;38:47–58 Kokura/Rhoads/Wolf/Yoshikawa/Granger/Aw
(HUVECs) to 60 min of anoxia, followed by up to 10 hreoxygenation elicits a biphasic neutrophil adhesion re-sponse that is related to endothelial oxidant productionand differentially modulated by transcription-indepen-dent (30 min, phase 1) and transcription-dependent (4 h,phase 2) upregulation of ECAMs [1]. The multisubunitnuclear factor ÎB (NFÎB) is a member of transcriptionfactors involved in regulating the biosynthesis of E-selectin [3–5], intercellular adhesion molecule 1 [5], andvascular cell adhesion molecule 1 [6, 7]. In the phase 2adhesion response elicited by A/R, de novo synthesis ofE-selectin was mediated by NFÎB which was activated byreactive oxygen species [1]. In other studies, we and othershave shown that changes in cellular thiol status contributeto the NFÎB activation mediated by A/R, as well as tumornecrosis factor (TNF), interleukin 1, lipopolysaccharideor H2O2 [8–16]. The results of our study indicated thatA/R induced an intracellular redox imbalance and sus-tained increase in oxidized glutathione (GSSG) relative toreduced glutathione (GSH), which leads to the activationof NFÎB and the consequent biosynthesis of differentECAMs that mediate neutrophil-endothelial adhesion[16]. These findings suggest that an intracellular redoximbalance may be a critical early event in the A/R signal-ing processes that ultimately result in neutrophil-endothe-lial cell adhesion.
In the cytoplasm, inactive NFÎB exists as a heterodi-meric complex of the subunits p50 and p65 that bind to acytoplasmic retention protein, IÎB·. Upon activation,IÎB· is rapidly degraded, and the p50/p65 heterodimer istranslocated from the cytoplasm into the nucleus where itbinds with high affinity to a ÎB motif. Phosphorylationand subsequent proteolysis of IÎB· have been reported inTNF- and H2O2-mediated inflammatory responses [17,18]. In human endothelial cells, inhibition of protein tyro-sine kinase (PTK) appears to block the signal transductionsteps that are essential for the release of NFÎB in responseto TNF [19]. Furthermore, it has been shown that TNF-dependent NFÎB activation was abrogated by inhibitorsof protein tyrosine phosphatase (PTPase) in monocyticand endothelial cells [20]. It has also been reported thatthe selective inhibitor of protein kinase C (PKC) partiallyprevents the activation of NFÎB in response to H2O2 inporcine endothelial cells [21]. Thus, it appears that differ-ential signaling pathways may be involved in NFÎB sig-naling by different inflammatory stimuli.
The signaling mechanisms that activate NFÎB duringA/R are poorly understood. The objectives of this studywere to (1) define the kinetics of NFÎB activation byexamining IÎB· degradation and the nuclear transloca-
tion of p65 in response to A/R or redox imbalance and (2)determine whether the signal for IÎB· degradation, nu-clear translocation of p65 and E-selectin expression aremediated by PTK, PTPase and/or PKC.
Materials and Methods
SubjectsThe procedures used to obtain human neutrophils and human
umbilical cords were approved by the Institutional Review Board forHuman Research at the Louisiana State University Health SciencesCenter. Freshly discarded human umbilical cords were obtainedfrom the delivery suite of the Louisiana State University HealthSciences Center. Each blood donor provided written consent and wascompensated for participating in the study.
HUVEC Culture and Isolation of NeutrophilsHUVECs were harvested and cultured as previously described
[22, 23]. Human neutrophils were isolated as described [16].
A/R Protocol and Modulation of Cellular GSH and GSSGThe in vitro model of A/R used in this study is similar to that
previously reported [2]. Confluent HUVEC monolayers were ex-posed to anoxia for 60 min by incubating in a Plexiglas chamber thatwas continuously purged (1 l/min) with an anoxic gas mixture (93%N2-5% CO2-2% H2). Reoxygenation was initiated by exposingHUVECs to a 100% humidified atmosphere with 5% CO2, withreoxygenation periods ranging between 0 and 4 h. In a separate seriesof experiments, GSH and GSSG levels in HUVECs were modifiedusing a combination of diamide (Sigma) and buthionine sulfoximine(BSO, Sigma) as previously described [16]. To effect changes in GSHand GSSG, the HUVEC culture medium was removed and cells wereincubated with diamide (0.2 mmol/l) in fresh EGM (Clonetics). After15 min of diamide treatment, the medium was removed and the cellswere incubated with BSO (1 mmol/l) in fresh EGM to prevent resyn-thesis of GSH for periods ranging between 0 and 4 h. HUVEC mono-layers thus treated will be referred to as redox-altered endothelialcells.
Treatment Protocols and Neutrophil Adhesion AssayHUVEC monolayers were preincubated with the following inhib-
itors: (1) a PTK inhibitor, genistein (100 Ìmol/l; Calbiochem), (2) aPKC inhibitor, chelerythrine chloride (5 Ìmol/l; Calbiochem), (3) aPTPase inhibitor, phenylarsine oxide (PAO, 0.1 Ìmol/l; Sigma) andstimulated with A/R or diamide + BSO as described in the figurelegends. After 4 h of stimulation, these reagents were removed, andthe assays for neutrophil adhesion or E-selectin expression were per-formed. In some experiments, HUVEC monolayers were pretreatedwith 5 mmol/l N-acetyl cysteine (NAC), a thiol reductant, for 60 minprior to exposure of cells to A/R or diamide + BSO.
E-Selectin Expression ELISAAn E-selectin expression ELISA was performed as previously
described [1, 16]. Briefly, HUVECs were incubated for 30 min at37°C with the anti-E-selectin monoclonal antibody (mAb) CL3 [mu-rine IgG1 F(ab))2 antihuman E-selectin], a gift from Dr. DonaldAnderson (Pharmacia-Upjohn Laboratories, Kalamazoo, Mich.,USA) diluted 1:1,000 in HBSS/PBS with 5% fetal bovine serum. The
Dow
nloa
ded
by:
L.S
.U. H
ealth
Sci
ence
Cen
ter
20
6.17
6.17
5.20
0 -
7/11
/201
3 8:
55:1
2 P
M
NFÎB Signaling in Posthypoxic EndothelialCells
J Vasc Res 2001;38:47–58 49
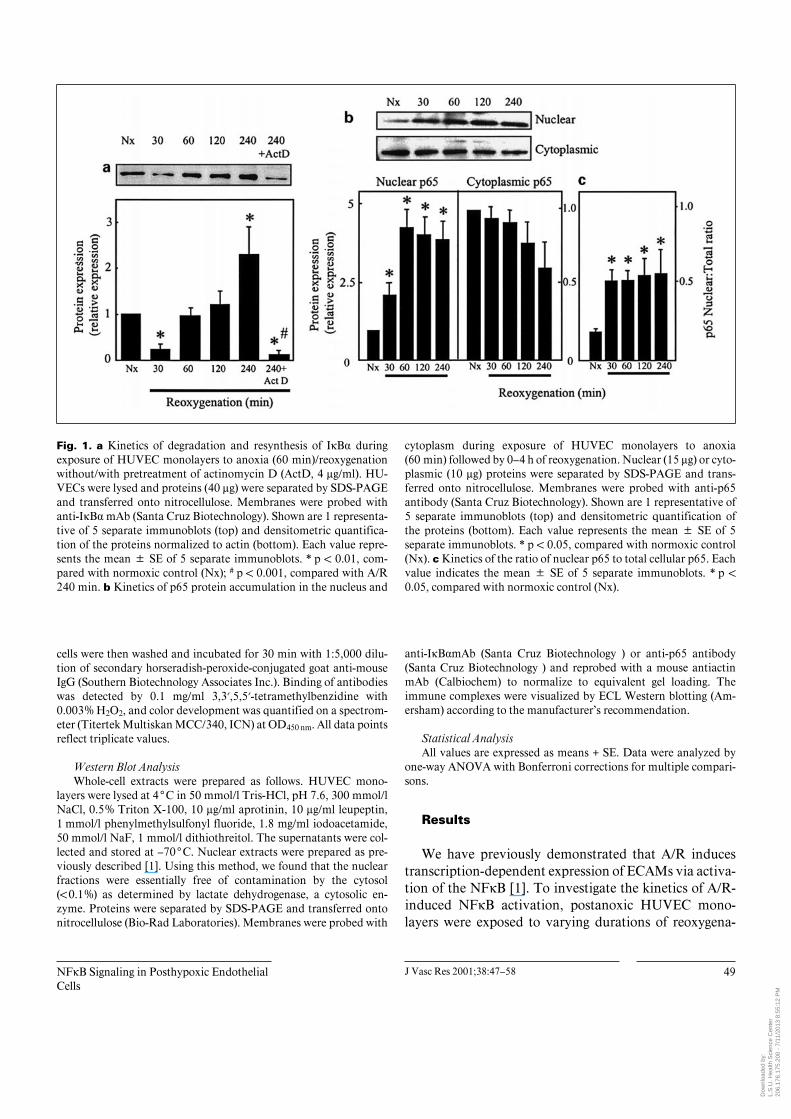
Fig. 1. a Kinetics of degradation and resynthesis of IÎB· duringexposure of HUVEC monolayers to anoxia (60 min)/reoxygenationwithout/with pretreatment of actinomycin D (ActD, 4 Ìg/ml). HU-VECs were lysed and proteins (40 Ìg) were separated by SDS-PAGEand transferred onto nitrocellulose. Membranes were probed withanti-IÎB· mAb (Santa Cruz Biotechnology). Shown are 1 representa-tive of 5 separate immunoblots (top) and densitometric quantifica-tion of the proteins normalized to actin (bottom). Each value repre-sents the mean B SE of 5 separate immunoblots. * p ! 0.01, com-pared with normoxic control (Nx); # p ! 0.001, compared with A/R240 min. b Kinetics of p65 protein accumulation in the nucleus and
cytoplasm during exposure of HUVEC monolayers to anoxia(60 min) followed by 0–4 h of reoxygenation. Nuclear (15 Ìg) or cyto-plasmic (10 Ìg) proteins were separated by SDS-PAGE and trans-ferred onto nitrocellulose. Membranes were probed with anti-p65antibody (Santa Cruz Biotechnology). Shown are 1 representative of5 separate immunoblots (top) and densitometric quantification ofthe proteins (bottom). Each value represents the mean B SE of 5separate immunoblots. * p ! 0.05, compared with normoxic control(Nx). c Kinetics of the ratio of nuclear p65 to total cellular p65. Eachvalue indicates the mean B SE of 5 separate immunoblots. * p !0.05, compared with normoxic control (Nx).
cells were then washed and incubated for 30 min with 1:5,000 dilu-tion of secondary horseradish-peroxide-conjugated goat anti-mouseIgG (Southern Biotechnology Associates Inc.). Binding of antibodieswas detected by 0.1 mg/ml 3,3),5,5)-tetramethylbenzidine with0.003% H2O2, and color development was quantified on a spectrom-eter (Titertek Multiskan MCC/340, ICN) at OD450 nm. All data pointsreflect triplicate values.
Western Blot AnalysisWhole-cell extracts were prepared as follows. HUVEC mono-
layers were lysed at 4°C in 50 mmol/l Tris-HCl, pH 7.6, 300 mmol/lNaCl, 0.5% Triton X-100, 10 Ìg/ml aprotinin, 10 Ìg/ml leupeptin,1 mmol/l phenylmethylsulfonyl fluoride, 1.8 mg/ml iodoacetamide,50 mmol/l NaF, 1 mmol/l dithiothreitol. The supernatants were col-lected and stored at –70°C. Nuclear extracts were prepared as pre-viously described [1]. Using this method, we found that the nuclearfractions were essentially free of contamination by the cytosol(!0.1%) as determined by lactate dehydrogenase, a cytosolic en-zyme. Proteins were separated by SDS-PAGE and transferred ontonitrocellulose (Bio-Rad Laboratories). Membranes were probed with
anti-IÎB·mAb (Santa Cruz Biotechnology ) or anti-p65 antibody(Santa Cruz Biotechnology ) and reprobed with a mouse antiactinmAb (Calbiochem) to normalize to equivalent gel loading. Theimmune complexes were visualized by ECL Western blotting (Am-ersham) according to the manufacturer’s recommendation.
Statistical AnalysisAll values are expressed as means + SE. Data were analyzed by
one-way ANOVA with Bonferroni corrections for multiple compari-sons.
Results
We have previously demonstrated that A/R inducestranscription-dependent expression of ECAMs via activa-tion of the NFÎB [1]. To investigate the kinetics of A/R-induced NFÎB activation, postanoxic HUVEC mono-layers were exposed to varying durations of reoxygena-
Dow
nloa
ded
by:
L.S
.U. H
ealth
Sci
ence
Cen
ter
20
6.17
6.17
5.20
0 -
7/11
/201
3 8:
55:1
2 P
M

50 J Vasc Res 2001;38:47–58 Kokura/Rhoads/Wolf/Yoshikawa/Granger/Aw
Fig. 2. Inhibitors of PTK and PKC dose-dependently block IÎB·degradation. HUVECs were incubated without or with 2 concentra-tions of genistein (50 or 100 Ìmol/l) or chelerythrine chloride (2.5 or5 Ìmol/l) and exposed to 60 min of anoxia followed by 30 min ofreoxygenation. HUVECs exposed to normoxia served as controls.Cell lysates (40 Ìg protein) were separated by SDS-PAGE, and nitro-cellulose membranes were probed with anti-IÎB· mAb. Shown is 1representative of 2 separate immunoblots. a Genistein. b Chelery-thrine.
tion, and cell extracts were analyzed for IÎB· degradationand nuclear translocation of p65 by Western blots. Fig-ure 1a shows that IÎB· disappeared within 30 min afterreoxygenation, suggesting rapid degradation of the pro-tein. IÎB· expression reappeared after 1 h and remainedelevated for 4 h. The increased IÎB· protein expressionwas blocked by actinomycin D, demonstrating that newprotein synthesis is responsible for the reappearance ofIÎB·. To confirm that the dissociation of IÎB· results intranslocation of the activated NFÎB subunits from thecytoplasm to the nucleus, nuclear extracts were assayedfor an increase in p65 protein levels. The kinetics of p65appearance in the nucleus (fig. 1b) revealed an increasednuclear p65 level after 30 min of reoxygenation andreached a plateau at 3.5- to 4-fold the control value at60 min. Over the same time period, cytoplasmic p65 pro-tein levels remained relatively unchanged although therewas a tendency towards a decrease at 4 h after anoxia, butthe values are highly variable and not significant (fig. 1b).Figure 1c shows that the ratio of nuclear p65 to total p65was low during normoxia, consistent with a primary cyto-plasmic localization of NFÎB in the unstimulated state.Upon A/R stimulation, this ratio increased significantlywithin 30 min and remained constant over 4 h (fig. 1c),indicating that activated p65 is rapidly and maximallytranslocated into the nucleus within 30–60 min followingits activation in the face of an unchanging total NFÎBpool.
Cytokines, such as TNF, and oxidants have been dem-onstrated to activate NFÎB through activation of PTK[17, 19, 24–26] or PKC [21]. Therefore, inhibitors of eith-er PTK (genistein), PKC (chelerythrine) or PTPase (PAO)
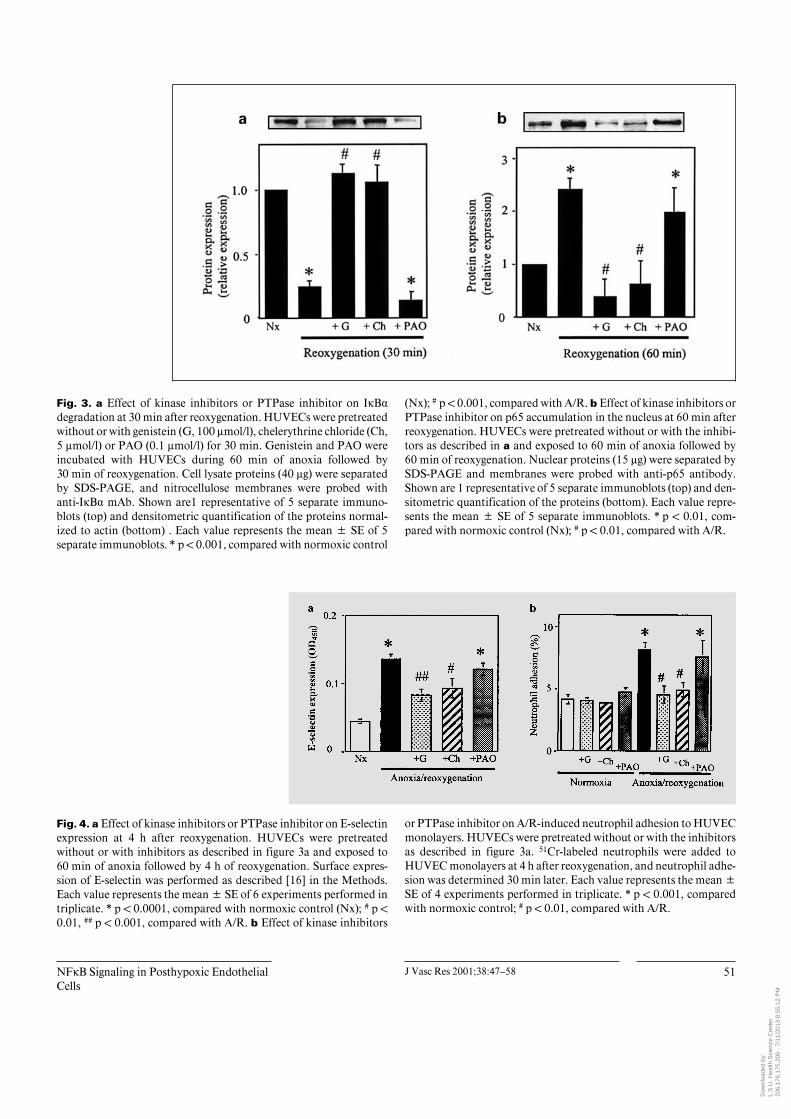
were examined for their ability to block NFÎB activationin A/R-exposed endothelial cells. In our studies, we usedconcentrations of genistein (100 Ìmol/l), chelerythrinechloride (5 Ìmol/l) and PAO (0.1 Ìmol/l) that had pre-viously been shown to completely and selectively inhibitthe respective enzyme activities in various cells includinghuman endothelial cells [19, 20, 26, 27]. We found thatgenistein and chelerythrine dose-dependently inhibitedIÎB degradation, and maximal inhibitions were achievedwith 100 Ìmol/l genistein and 5 Ìmol/l chelerythrine(fig. 2a and b, respectively). Figure 3 shows the effects ofthe inhibitors against PTK, PKC or PTPase on IÎB· deg-radation and p65 nuclear translocation induced by A/R.The results in figure 3a show that A/R-induced IÎB· deg-radation was significantly blocked by pretreatment ofHUVEC monolayers with the PTK inhibitor genistein orthe PKC inhibitor chelerythrine chloride, but not by thePTPase inhibitor PAO. Similarly, p65 nuclear transloca-tion induced by A/R is inhibited by pretreatment withgenistein or chelerythrine chloride but not by PAO(fig. 3b). Because previous studies have implicated NFÎBin A/R-induced transcription of E-selectin, which me-diates neutrophil-HUVEC adhesion, we tested the effectsof inhibitors against PTK, PKC or PTPase on E-selectinexpression (fig. 4a) and neutrophil adhesion (fig. 4b) in-duced by A/R. The results show that the A/R-induced E-selectin expression and neutrophil adhesion responseswere significantly attenuated by genistein or chelerythrinechloride, but not by PAO. Collectively, these results areconsistent with a role for both PTK and PKC in signalingNFÎB activation during A/R by stimulating IÎB· degra-dation, p65 translocation from the cytoplasm to the nu-cleus and upregulation of E-selectin expression, whichpromotes neutrophil-endothelial cell adhesion.
We have recently documented that the leukocyte-endothelial cell interactions elicited by chemical induc-tion of redox imbalance closely mimicked those inducedby A/R [1, 16]. Figure 5a shows the effect of A/R on theintracellular GSSG/GSH status in HUVECs after 60 minof anoxia followed by 0–4 h of reoxygenation. A/R expo-sure caused a significant increase in GSSG with respect toGSH within 30 min that remained elevated for 4 h. Incomparison, treatment of HUVECs with diamide (0.2mmol/l, 15 min) + BSO (1 mmol/l) to chemically induce aredox change [16] that is independent of A/R similarlycaused an imbalance in the GSSG-to-GSH ratio (fig. 5b)that kinetically resembled that induced by A/R. This cor-respondence in redox shifts suggests a role for redox regu-lation of the cellular and molecular signaling mechanismsin A/R-induced leukocyte-endothelial cell interactions.
Dow
nloa
ded
by:
L.S
.U. H
ealth
Sci
ence
Cen
ter
20
6.17
6.17
5.20
0 -
7/11
/201
3 8:
55:1
2 P
M
NFÎB Signaling in Posthypoxic EndothelialCells
J Vasc Res 2001;38:47–58 51
Fig. 3. a Effect of kinase inhibitors or PTPase inhibitor on IÎB·degradation at 30 min after reoxygenation. HUVECs were pretreatedwithout or with genistein (G, 100 Ìmol/l), chelerythrine chloride (Ch,5 Ìmol/l) or PAO (0.1 Ìmol/l) for 30 min. Genistein and PAO wereincubated with HUVECs during 60 min of anoxia followed by30 min of reoxygenation. Cell lysate proteins (40 Ìg) were separatedby SDS-PAGE, and nitrocellulose membranes were probed withanti-IÎB· mAb. Shown are1 representative of 5 separate immuno-blots (top) and densitometric quantification of the proteins normal-ized to actin (bottom) . Each value represents the mean B SE of 5separate immunoblots. * p ! 0.001, compared with normoxic control
(Nx); # p ! 0.001, compared with A/R. b Effect of kinase inhibitors orPTPase inhibitor on p65 accumulation in the nucleus at 60 min afterreoxygenation. HUVECs were pretreated without or with the inhibi-tors as described in a and exposed to 60 min of anoxia followed by60 min of reoxygenation. Nuclear proteins (15 Ìg) were separated bySDS-PAGE and membranes were probed with anti-p65 antibody.Shown are 1 representative of 5 separate immunoblots (top) and den-sitometric quantification of the proteins (bottom). Each value repre-sents the mean B SE of 5 separate immunoblots. * p ! 0.01, com-pared with normoxic control (Nx); # p ! 0.01, compared with A/R.
Fig. 4. a Effect of kinase inhibitors or PTPase inhibitor on E-selectinexpression at 4 h after reoxygenation. HUVECs were pretreatedwithout or with inhibitors as described in figure 3a and exposed to60 min of anoxia followed by 4 h of reoxygenation. Surface expres-sion of E-selectin was performed as described [16] in the Methods.Each value represents the mean B SE of 6 experiments performed intriplicate. * p ! 0.0001, compared with normoxic control (Nx); # p !0.01, ## p ! 0.001, compared with A/R. b Effect of kinase inhibitors
or PTPase inhibitor on A/R-induced neutrophil adhesion to HUVECmonolayers. HUVECs were pretreated without or with the inhibitorsas described in figure 3a. 51Cr-labeled neutrophils were added toHUVEC monolayers at 4 h after reoxygenation, and neutrophil adhe-sion was determined 30 min later. Each value represents the mean BSE of 4 experiments performed in triplicate. * p ! 0.001, comparedwith normoxic control; # p ! 0.01, compared with A/R.
Dow
nloa
ded
by:
L.S
.U. H
ealth
Sci
ence
Cen
ter
20
6.17
6.17
5.20
0 -
7/11
/201
3 8:
55:1
2 P
M

52 J Vasc Res 2001;38:47–58 Kokura/Rhoads/Wolf/Yoshikawa/Granger/Aw
Fig. 5. Time course of change in GSSG-to-GSH ratio in HUVECs exposed to 60 min ofanoxia followed by 4 h of reoxygenation (a)or to diamide + BSO for 0–4 h (b). Each val-ue indicates the mean B SEM of 4 experi-ments. * p ! 0.01 versus control.
Fig. 6. a Kinetics of degradation and resynthesis of IÎB· duringexposure of HUVECs to diamide + BSO without or with pretreat-ment with actinomycin D (ActD, 4 Ìg/ml). Shown are 1 representa-tive of 5 separate immunoblots (top) and densitometric quantifica-tion of the proteins normalized to actin (bottom). Each value repre-sents the mean B SE of 5 separate immunoblots. * p ! 0.001, com-pared with control (Ctl); # p ! 0.001, compared with the grouptreated for 240 min. b Kinetics of p65 protein accumulation in thenucleus during exposure of HUVECs to diamide + BSO. Nuclear
(15 Ìg) or cytoplasmic (10 Ìg) proteins were separated by SDS-PAGE, and nitrocellulose membranes were probed with anti-p65antibody. Shown are 1 representative of 5 separate immunoblots(top) and densitometric quantification of the proteins (bottom). Eachvalue represents the mean B SE of 5 separate immunoblots. * p !0.05, compared with control (Ctl). c Kinetics of the ratio of nuclearp65 to total cellular p65. Each value indicates the mean B SE of 5separate immunoblots. * p ! 0.05, compared with untreated control(Ctl).
Dow
nloa
ded
by:
L.S
.U. H
ealth
Sci
ence
Cen
ter
20
6.17
6.17
5.20
0 -
7/11
/201
3 8:
55:1
2 P
M
NFÎB Signaling in Posthypoxic EndothelialCells
J Vasc Res 2001;38:47–58 53
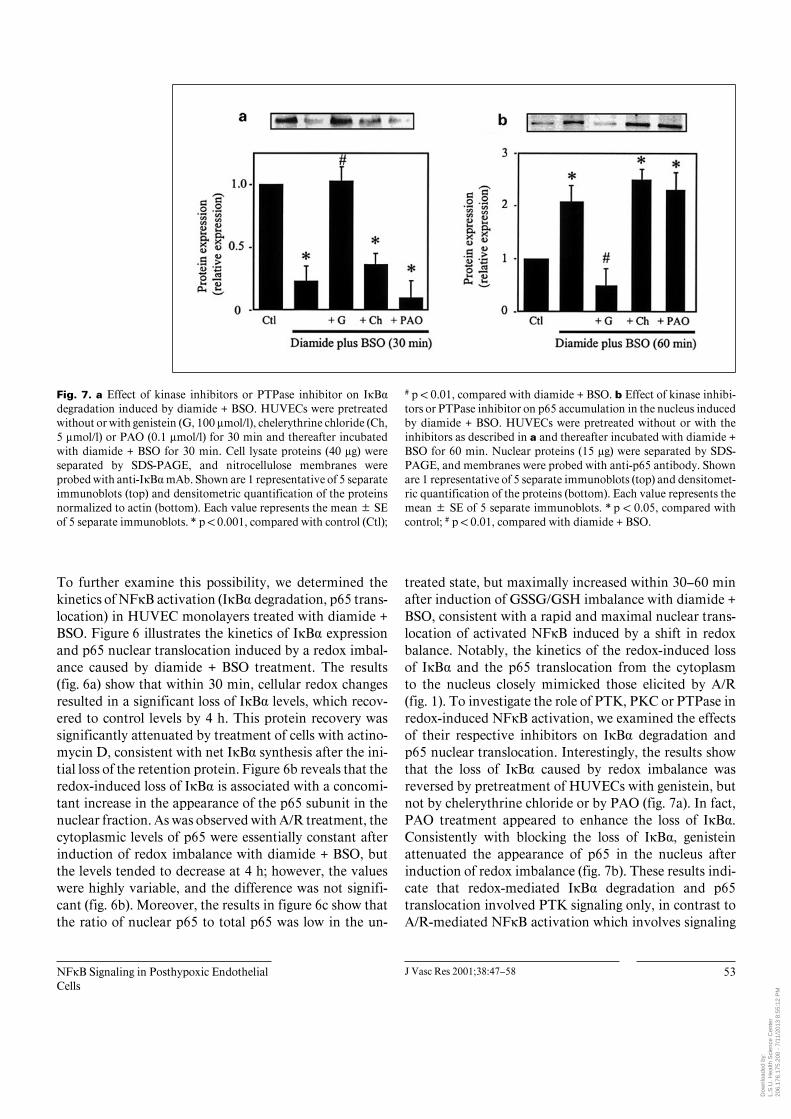
Fig. 7. a Effect of kinase inhibitors or PTPase inhibitor on IÎB·degradation induced by diamide + BSO. HUVECs were pretreatedwithout or with genistein (G, 100 Ìmol/l), chelerythrine chloride (Ch,5 Ìmol/l) or PAO (0.1 Ìmol/l) for 30 min and thereafter incubatedwith diamide + BSO for 30 min. Cell lysate proteins (40 Ìg) wereseparated by SDS-PAGE, and nitrocellulose membranes wereprobed with anti-IÎB· mAb. Shown are 1 representative of 5 separateimmunoblots (top) and densitometric quantification of the proteinsnormalized to actin (bottom). Each value represents the mean B SEof 5 separate immunoblots. * p ! 0.001, compared with control (Ctl);
# p ! 0.01, compared with diamide + BSO. b Effect of kinase inhibi-tors or PTPase inhibitor on p65 accumulation in the nucleus inducedby diamide + BSO. HUVECs were pretreated without or with theinhibitors as described in a and thereafter incubated with diamide +BSO for 60 min. Nuclear proteins (15 Ìg) were separated by SDS-PAGE, and membranes were probed with anti-p65 antibody. Shownare 1 representative of 5 separate immunoblots (top) and densitomet-ric quantification of the proteins (bottom). Each value represents themean B SE of 5 separate immunoblots. * p ! 0.05, compared withcontrol; # p ! 0.01, compared with diamide + BSO.
To further examine this possibility, we determined thekinetics of NFÎB activation (IÎB· degradation, p65 trans-location) in HUVEC monolayers treated with diamide +BSO. Figure 6 illustrates the kinetics of IÎB· expressionand p65 nuclear translocation induced by a redox imbal-ance caused by diamide + BSO treatment. The results(fig. 6a) show that within 30 min, cellular redox changesresulted in a significant loss of IÎB· levels, which recov-ered to control levels by 4 h. This protein recovery wassignificantly attenuated by treatment of cells with actino-mycin D, consistent with net IÎB· synthesis after the ini-tial loss of the retention protein. Figure 6b reveals that theredox-induced loss of IÎB· is associated with a concomi-tant increase in the appearance of the p65 subunit in thenuclear fraction. As was observed with A/R treatment, thecytoplasmic levels of p65 were essentially constant afterinduction of redox imbalance with diamide + BSO, butthe levels tended to decrease at 4 h; however, the valueswere highly variable, and the difference was not signifi-cant (fig. 6b). Moreover, the results in figure 6c show thatthe ratio of nuclear p65 to total p65 was low in the un-
treated state, but maximally increased within 30–60 minafter induction of GSSG/GSH imbalance with diamide +BSO, consistent with a rapid and maximal nuclear trans-location of activated NFÎB induced by a shift in redoxbalance. Notably, the kinetics of the redox-induced lossof IÎB· and the p65 translocation from the cytoplasmto the nucleus closely mimicked those elicited by A/R(fig. 1). To investigate the role of PTK, PKC or PTPase inredox-induced NFÎB activation, we examined the effectsof their respective inhibitors on IÎB· degradation andp65 nuclear translocation. Interestingly, the results showthat the loss of IÎB· caused by redox imbalance wasreversed by pretreatment of HUVECs with genistein, butnot by chelerythrine chloride or by PAO (fig. 7a). In fact,PAO treatment appeared to enhance the loss of IÎB·.Consistently with blocking the loss of IÎB·, genisteinattenuated the appearance of p65 in the nucleus afterinduction of redox imbalance (fig. 7b). These results indi-cate that redox-mediated IÎB· degradation and p65translocation involved PTK signaling only, in contrast toA/R-mediated NFÎB activation which involves signaling
Dow
nloa
ded
by:
L.S
.U. H
ealth
Sci
ence
Cen
ter
20
6.17
6.17
5.20
0 -
7/11
/201
3 8:
55:1
2 P
M

54 J Vasc Res 2001;38:47–58 Kokura/Rhoads/Wolf/Yoshikawa/Granger/Aw
Fig. 8. a Effect of kinase inhibitors or PTPase inhibitor on E-selectinexpression at 4 h after the addition of diamide + BSO. HUVECs werepretreated without or with the inhibitors for 30 min and thereafterwere incubated with diamide + BSO for 4 h. Each value representsthe mean B SE of 6 experiments performed in triplicate. * p ! 0.001,compared with normoxic control (Ctl); # p ! 0.01, compared withA/R. b Effects of kinase inhibitors or PTPase inhibitor on neutrophiladhesion to HUVEC monolayers induced by diamide + BSO. Pre-
treatment of HUVEC monolayers with inhibitors and incubationwith diamide + BSO were as described in a. 51Cr-labeled neutrophilswere added to HUVEC monolayers at 4 h after the induction ofredox imbalance, and neutrophil adhesion was determined 30 minlater. Each value represents the mean B SE of 4 experiments per-formed in triplicate. * p ! 0.001, compared with normoxic control;# p ! 0.001, compared with A/R.
Fig. 9. Effect of NAC on IÎB· degradation and neutrophil adhesioninduced by A/R (a) or treatment with diamide B BSO (redox, b).HUVECs were pretreated with 5 mmol/l NAC for 60 min prior toexposure of cells to A/R or diamide + BSO. In studies on IÎB degra-dation (a), NAC-pretreated endothelial cells were exposed to either60 min of anoxia followed by 30 min of reoxygenation or incubatedfor 30 min with diamide + BSO; thereafter Western analyses were
performed for IÎB· expression. In studies on neutrophil adhesion(b), NAC-pretreated endothelial cells were either exposed to 60 minof anoxia followed by 4 h of reoxygenation or incubated for 4 h withdiamide + BSO; thereafter the neutrophil adhesion assay was per-formed as described in figure 8. * p ! 0.05 as compared to control;# p ! 0.05 as compared to A/R- or redox-treated groups.
Dow
nloa
ded
by:
L.S
.U. H
ealth
Sci
ence
Cen
ter
20
6.17
6.17
5.20
0 -
7/11
/201
3 8:
55:1
2 P
M

NFÎB Signaling in Posthypoxic EndothelialCells
J Vasc Res 2001;38:47–58 55
by both PTK and PKC. To determine the contributions ofthese signaling pathways to the E-selectin expression andneutrophil adhesion mediated by a redox imbalance,HUVEC monolayers exposed to diamide + BSO weretreated with inhibitors against either PTK, PKC orPTPase. Figure 8 shows that redox-induced E-selectin ex-pression (fig. 8a) and neutrophil adhesion (fig. 8b) weresignificantly attenuated by genistein, but not by chelery-thrine chloride or PAO, which supports the contentionthat the inflammatory response induced by a redox imbal-ance is specifically and predominately mediated by acti-vation of PTK.
To address our suggestion that an A/R-induced redoxchange mediates NFÎB-dependent neutrophil-endothelialinteractions, we pretreated HUVEC monolayers with athiol reductant, NAC (5 mmol/l), prior to exposure of cellsto A/R or diamide + BSO. Figure 9 illustrates the effect ofNAC on IÎB degradation and polymorphonuclear celladhesion. The results show that NAC effectively blockedthe degradation of IÎB induced by A/R or redox imbal-ance induced by diamide + BSO (fig. 9a), consistent withinhibition of NFÎB activation with restoration of redoxstatus. Accordingly, NAC significantly attenuated theneutrophil adhesion responses caused by A/R or redoximbalance (fig. 9b). Collectively, these results provide evi-dence that the A/R-induced loss of endothelial redox bal-ance is an important contributor to A/R-mediated NFÎB-dependent neutrophil adhesion.
Discussion
We have previously demonstrated that A/R stimulatesleukocyte adhesion to HUVECs by inducing adhesionmolecule expression [1, 2] that is mediated in part byA/R-induced intracellular redox imbalance and the conse-quent activation of NFÎB and AP-1 [16]. It is well docu-mented that activation of NFÎB involves its release fromIÎB· and the subsequent translocation of active NFÎBinto the nucleus [28]. Beg et al. [29] have reported that thepresence of IÎB is sufficient to retain both the p50 andp65 subunits of NFÎB in the cytoplasm, while the absenceof IÎB allows these subunits to be transported into thenucleus. In the present study, cytoplasmic levels of IÎB·were monitored to assess the time course of activation ofNFÎB after stimulation of endothelial cells with eitherA/R or redox imbalance. Our results clearly demonstratethat exposure of HUVECs to A/R leads to rapid degrada-tion of IÎB· and the concomitant appearance of p65 inthe nucleus. These findings are consistent with our pre-
vious observation that A/R elicits the activation of NFÎBas determined by the electrophoretic mobility gel shiftassay [1].
A notable observation in the present study is that A/Rinduces a loss of IÎB· within 30 min after reoxygenationand an equally rapid translocation of the p65 subunit intothe nucleus (between 30 and 60 min following cytoplas-mic activation), indicating that activation of NFÎB per seis a rapid process. Moreover, the observation that thekinetics of IÎB· degradation closely parallels the nuclearappearance of p65 indicates that the rate-limiting step inthe activation of NFÎB is degradation of IÎB·. Theseresults are consistent with previous reports by Beg et al.[17] and Koong et al. [30]. Interestingly, our results showthat postanoxic recovery of IÎB· is also a rapid process,which occurs within 30 min after loss of the protein. Thefinding that the reappearance of IÎB· is inhibited bytreatment with actinomycin D indicates that de novo syn-thesis of IÎB· is responsible for recovery of the protein.Similar results were also obtained with stimulation byTNF or PMA, which raises the interesting possibility thatactivation of NFÎB may be involved in the transcrip-tional induction of IÎB· itself [17, 31]. Paradoxically,NFÎB appeared to localize to the nucleus over 4 h even asthe cytoplasmic levels of IÎB· rise at 60 min. The reasonfor this interesting observation is unclear. There are sever-al possible explanations. Conceivably, the 30-min timedelay between the observed maximal loss of IÎB· at30 min after anoxia (fig. 1a) and the maximal appearancein nuclear p65 at 60 min (fig. 1b) may reflect the actualtime it takes for the movement of p65 into the nucleusfrom the cytoplasm following NFÎB activation. The sus-tained levels of accumulated p65 over 4 h within thenucleus suggest a slow turnover of nuclear p65. Anotherpossibility may be that newly synthesized IÎB· has aweaker affinity for NFÎB or that newly synthesized IÎB·and perinuclear NFÎB pools are compartmentalized indifferent regions of the cytoplasm. Significantly, thismeans that, in the face of a large NFÎB cytoplasmic pool,newly synthesized IÎB· may have minimal overall impacton inactivating the perinuclear NFÎB that is expected tobe involved in activation and nuclear translocation. In-deed, our data support the notion that the average pool ofNFÎB in the cytoplasm is large and essentially unchang-ing as evidenced by the relatively constant and stable p65levels in the cytoplasmic compartment even as the nuclearp65 levels increased (fig. 1b, c). Moreover, this interpreta-tion is further consistent with the idea that only a local-ized fraction of NFÎB (i.e. perinuclear NFÎB) activelyparticipates in signaling in response to an inflammatory
Dow
nloa
ded
by:
L.S
.U. H
ealth
Sci
ence
Cen
ter
20
6.17
6.17
5.20
0 -
7/11
/201
3 8:
55:1
2 P
M

56 J Vasc Res 2001;38:47–58 Kokura/Rhoads/Wolf/Yoshikawa/Granger/Aw
Fig. 10. Schematic representation of pro-posed intracellular mechanisms involved inNFÎB-mediated E-selectin expression.
stimulus such as A/R. These are intriguing considerationsthat warrant further investigation.
Previous studies have implicated a role for proteinkinases in the induction of endothelial cell adhesion mole-cules by TNF, and the activation of NFÎB by TNF orH2O2 [19, 21, 30]. Our results invoke a role for PKC andPTK in A/R-induced degradation of IÎB· and the nucleartranslocation of p65, consistently with the function ofthese two protein kinases in NFÎB activation. This inhibi-tion of transcription factor activation directly correlatedwith the suppression of E-selectin expression and theadhesion of neutrophils to HUVEC monolayers inducedby A/R, thus implicating protein kinase-mediated NFÎBsignaling in the A/R-induced inflammatory response. Ourfinding that the kinase inhibitors blocked IÎB· degrada-tion and prevented nuclear accumulation of p65 levelssuggests that NFÎB activation represents a major mecha-nism by which A/R increases trans-acting factors. How-ever, our findings do not allow us to exclude the possibili-ty that A/R also increases the binding affinity of the fac-tors to DNA; indeed, the affinity of these trans-acting fac-tors to DNA may be influenced by the intracellular thiolstatus [32, 33] which we found to be altered during A/R(fig. 5), in agreement with previous findings [16].
It is significant that chemically induced redox imbal-ance, independently of A/R, resulted in similar kinetics ofGSSG/GSH shifts, IÎB· degradation and nuclear translo-cation of p65, E-selectin expression and neutrophil adhe-sion. These results, taken together, suggest that an earlyloss of cellular redox homeostasis is an important contri-
butor to the initiating event in A/R-mediated NFÎB acti-vation, consistent with previous observations [1, 16]. Ourfindings that NAC effectively blocked A/R- and redox-induced IÎB· degradation and attenuated the respectiveneutrophil adhesion responses provide strong support forthis hypothesis. Although the kinetics of NFÎB activation(i.e., IÎB· degradation and p65 nuclear translocation)induced by redox imbalance closely mimicked those elic-ited by A/R, it is notable that redox-induced NFÎB activa-tion, E-selectin expression and neutrophil adhesion werespecifically attenuated by inhibition of PTK activity,which contrasts with the participation of both tyrosinekinase and PKC in A/R signaling. These results suggestthat, mechanistically, the inflammatory response elicitedby A/R-induced redox shifts appears to be mediated spe-cifically by the redox-sensitive PTK (fig. 10), while PKClikely mediates an inflammatory response elicited by oth-er mediators released during A/R, such as reactive oxygenradicals. These differences underscore a fundamental dis-tinction between signaling mechanisms that are mediateddirectly by reactive oxygen species and those mediatedthrough redox changes induced by reactive oxygen radi-cals. A participation of oxyradicals in NFÎB signalingduring TNF-·-induced inflammatory responses has beensuggested [17]; however, the role of NFÎB signaling inA/R is unclear at present. Interestingly, previous studiesby Anderson et al. [34] demonstrated cross-talk betweenthe two signaling pathways as evidenced by the findingthat NFÎB activation by a PKC stimulator (PMA) wasinhibited by PTK inhibitors. Our data is consistent with
Dow
nloa
ded
by:
L.S
.U. H
ealth
Sci
ence
Cen
ter
20
6.17
6.17
5.20
0 -
7/11
/201
3 8:
55:1
2 P
M

NFÎB Signaling in Posthypoxic EndothelialCells
J Vasc Res 2001;38:47–58 57
an essential role for tyrosine phosphorylation in NFÎB-dependent expression of ECAMs like E-selectin.
The mechanism by which the A/R-induced loss ofGSH homeostasis modulates tyrosine phosphorylation isunexplained. It is interesting that the studies of Dhawanet al. [20] suggest that diamide may act as a protein tyro-sine phosphatase inhibitor. In this regard, diamide caninhibit tyrosine-phosphatase-catalyzed dephosphoryla-tion via formation of a protein disulfide cross-link in thephosphate transfer domain of the enzyme. Inhibition ofphosphatase activity would be expected to result in hyper-phosphorylation of IÎB· and activation of NFÎB, andsubsequent promotion of neutrophil adhesion. This sug-gestion is supported by our current findings. However, thelack of a potentiation effect of PAO on A/R- or redox-induced E-selectin expression and neutrophil-endothelialcell adhesion is somewhat surprising given (a) that tyro-sine phosphatase and tyrosine kinase function as a regula-tory pair in the phosphorylation/dephosphorylation ofprotein tyrosine residues and (b) that tyrosine phospha-tase activity is inhibited by oxidants and protected byantioxidants [35–37]. Our data show that while inhibitionof tyrosine phosphatase by PAO slightly enhanced thedegradation of IÎB· after challenge by A/R (fig. 3a) orredox imbalance (fig. 7a), the extent of phosphatase inhi-bition did not significantly enhance E-selectin expressionor neutrophil adhesion, suggesting a possible threshold
requirement for IÎB· degradation and transcription-dependent E-selectin expression. Elucidation of the spe-cific contribution of tyrosine phosphorylation in A/R-induced, NFÎB-dependent neutrophil-endothelial cell in-teractions is an ongoing avenue of investigation in ourlaboratory. In this study, we have taken the approach offocusing initially on what we have ascertained as the twokey kinases (i.e. PTK and PKC) at the nodal point to coor-dinate A/R or redox signaling that promotes neutrophiladhesion to postanoxic endothelial cells. We intend infuture studies to systematically evaluate the phosphoryla-tion cascade associated with each of the kinase pathwayswhich clearly show responsiveness to redox influence.
In summary, the present study provides evidence thatA/R-induced E-selectin expression and neutrophil-endo-thelial cell adhesion are mediated by both PKC and PTK,which signal the rapid activation of NFÎB (fig. 10) bystimulating IÎB· degradation and nuclear translocationof p65. This NFÎB signaling in A/R-exposed endothelialcells appears to be mediated, at least in part, by a redoximbalance.
Acknowledgment
This study was supported by a grant from the National Institutesof Health (P01-DK43785).
References
1 Ichikawa H, Flores S, Kvietys PR, Wolf RE,Yoshikawa T, Granger DN, Aw TY: Molecularmechanisms of anoxia/reoxygenation-inducedneutrophil adherence to cultured endothelialcells. Circ Res 1997;81:922–931.
2 Kokura S, Wolf RE , Yoshikawa T, IchikawaH, Granger DN, Aw TY: Endothelial cells ex-posed to anoxia/reoxygenation are hyperadhe-sive to T lymphocytes: Kinetics and molecularmechanisms. Microcirculation, in press.
3 Montgomery KF, Osborn L, Hession C, TizerdR, Goff D, Vassallo C, Tarr PI, Bomsztyk K,Lobb R, Harlan JM, Pohlman TH: Activationof endothelial-leukocyte adhesion molecule 1(ELAM-1) gene transcription. Proc Natl AcadSci USA 1991;88:6523–6527.
4 Read MA, Whitley MZ, Williams AJ, CollinsT: NF-ÎB and IÎB: An inducible regulatory sys-tem in endothelial activation. J Exp Med 1994;179:503–512.
5 Lockyer JM, Colladay JS, Alperin-Lea WL,Hammond T, Buda AJ: Inhibition of nuclearfactor-kB-mediated adhesion molecule expres-sion in human endothelial cells. Circ Res 1998;82:314–320.
6 Iademarco MF, McQuillan JJ, Rosen GD,Dean DC: Characterization of the promotor forvascular cell adhesion molecule-1 (VCAM-1). JBiol Chem 1992;267:16323–16329.
7 7. Neish S, Williams AJ, Palmer HJ, WhitleyMZ, Collins T: Functional analysis of the hu-man vascular cell adhesion molecule-1 promot-er. J Exp Med 1992;176:1583–1593.
8 Staal FJT, Roederer M, Herzenberg LA, Her-zenberg LA: Intracellular thiols regulate activa-tion of nuclear factor ÎB and transcription ofhuman immunodeficiency virus. Proc NatlAcad Sci USA 1990;87:9943–9947.
9 Jabbar SAB, Hoffbrand AV, WickremasingheRG: Redox reagents and staurosporine inhibitstimulation of the transcription regulator NF-ÎB following tumor necrosis factor treatmentof chronic B-leukemia cells. Leukemia Res1994;18:523–530.
10 Schreck R, Rieber P, Baeuerle PA: Reactiveoxygen intermediates as apparently widelyused messengers in the activation of the NF-ÎBtranscription factor and HIV-1. EMBO J 1991;10:2247–2258.
11 Sen CK, Khanna S, Reznick AZ, Roy S, PackerL: Glutathione regulation of tumor necrosisfactor-·-induced NF-ÎB activation in skeletalmuscle-derived L6 cells. Biochem Biophys ResCommun 1997;237:645–649.
12 Menon SD, Qin S, Guy GR, Tan YH: Differ-ential induction of NF-ÎB by protein phospha-tase inhibitors in primary and transformed hu-man cells. J Biol Chem 1993;268:26805–26812.
13 Khachigian LM, Collins T, Fries JWU: N-ace-tyl cysteine blocks mesangial VCAM-1 andNF-ÎB expression in vivo. Am J Pathol 1997;151:1225–1229.
14 Tanaka C, Kamata H, Takeshita H, YagisawaH, Hirata H: Redox regulation of lipopolysac-charide (LPS)-induced interleukin-8 (IL-8)gene expression mediated by NF-ÎB and AP-1in human astrocytoma U373 cells. BiochemBiophys Res Commun 1997;232:568–573.
Dow
nloa
ded
by:
L.S
.U. H
ealth
Sci
ence
Cen
ter
20
6.17
6.17
5.20
0 -
7/11
/201
3 8:
55:1
2 P
M

58 J Vasc Res 2001;38:47–58 Kokura/Rhoads/Wolf/Yoshikawa/Granger/Aw
15 Meyer M, Schreck R, Baeuerle PA: H2O2 andantioxidants have opposite effects on activa-tion of NF-ÎB and AP-1 in intact cells: AP-1as secondary antioxidant-responsive factor.EMBO J 1993;12:2005–2015.
16 Kokura S, Wolf RE, Yoshikawa T, GrangerDN, Aw TY: Molecular mechanisms of neutro-phil-endothelial cell adhesion induced by redoximbalance. Circ Res 1999;84:516–524.
17 Beg AA, Finco TS, Nantermet PV, Baldwin ASJr: Tumor necrosis factor and interleukin-1lead to phosphorylation and loss of IÎB·: Amechanism for NF-ÎB activation. Mol CellBiol 1993;13:3301–3310.
18 Traenckner EB-M, Pahl HL, Henkel T,Schmidt KN, Wilk S, Baeuerle PA: Phosphory-lation of human IÎB· on serines 323 and 36controls IÎB· proteolysis and NF-ÎB activa-tion in response to diverse stimuli. EMBO J1995;14:2876–2883.
19 Weber C, Negrescu E, Erl W, Pietsch A, Fran-kenberger M, Löms Ziegler-Heitbrock HW,Siess W, Weber PC: Inhibitors of protein tyro-sine kinase suppress TNF-stimulated inductionof endothelial cell adhesion molecules. J Im-munol 1995;155:445–451.
20 Dhawan S, Singh S, Aggarwal BB: Induction ofendothelial cell surface adhesion molecules bytumor necrosis factor is blocked by proteintyrosine phosphatase inhibitors: Role of the nu-clear transcription factor NF-kappa B. Eur JImmunol 1997;27:2172–2179.
21 Barchowsky A, Munro SR, Morana SJ, Vincen-ti MP, Treadwell M: Oxidant-sensitive andphosphorylation-dependent activation of NF-ÎB and AP-1 in endothelial cells. Am J Physiol1995;269:L829–L836.
22 Yoshida N, Granger DN, Anderson DC, Roth-lein R, Lane C, Kvietys PR: Anoxia/reoxygena-tion-induced neutrophil adherence to culturedendothelial cells. Am J Physiol 1992;262:H1891–H1898.
23 Jaffe EA, Nachman RL, Becker CG, MinckCR: Culture of human endothelial cells derivedfrom umbilical cord veins. J Clin Invest 1973;52:2745–2756.
24 Natarajan K, Manna SK, Chaturvedi MM, Ag-garwal BB: Protein tyrosine kinase inhibitorsblock tumor necrosis factor-induced activationof nuclear factor-kappaB, degradation of I kap-paB alpha, nuclear translocation of p65, andsubsequent gene expression. Arch BiochemBiophys 1998;352:59–70.
25 Lee BS, Kang HS, Pyun KH, Choi I: Roles oftyrosine kinases in the regulation of nitric oxidesynthesis in murine liver cells: Modulation ofNF-ÎB activity by tyrosine kinases. Hepatology1997;25:913–919.
26 Zhu Y, Lin JH-C, Liao H-L, Verna L, Stemer-man MB: Activation of ICAM-1 promoter bylysophosphatidylcholine: Possible involvementof protein tyrosine kinases. Biochim BiophysActa 1997;1345:93–98.
27 McGregor PE, Agrawal DK, Edwards JD: At-tenuation of human leukocyte adherence to en-dothelial cell monolayers by tyrosine kinaseinhibitors. Biochem Biophys Res Commun1994;198:359–365.
28 Baeuerle PA, Baltimore D: IÎB: A specificinhibitor of the NFÎB transcription factor.Science 1988;242:540–546.
29 Beg AA, Ruben SM, Scheinman RI, Haskill S,Rosen CA, Baldwin AS Jr: IÎB interacts withthe nuclear localization sequences of the sub-units of NF-ÎB: A mechanism for cytoplasmicretention. Genes Dev. 1992;6:1899–1913.
30 Koong AC, Chen EY, Giaccia AJ: Hypoxiacauses the activation of nuclear factor ÎBthrough the phosphorylation of IÎB· on tyro-sine residues. Cancer Res 1994;54:1425–1430.
31 Sun SC, Ganchi PA, Ballard DW, Greene WC:NF-ÎB controls expression of inhibitor IÎB·:Evidence for an inducible autoregulatory path-way. Science 1993;259:1912–1915.
32 Israel N, Gougerot-Pocidalo MA, Aillet F, Vi-relizier JL: Redox status of cells influences con-stitutive or induced NF-ÎB translocation andHIV long terminal repeat activity in human Tand monocytic cell lines. J Immunol 1992;149:3386–3393.
33 Mihm S, Galter D, Dröge W: Modulation oftranscription factor NF-ÎB activity by intracel-lular glutathione levels and by variations of theextracellular cysteine supply. FASEB J 1995;9:246–252.
34 Anderson MT, Staal FJT, Gitler C, HerzenbergLA, Herzenberg LA: Separation of oxidant-ini-tiated and redox-regulated steps in the NF-ÎBsignal transduction pathway. Proc Natl AcadSci USA1994;91:11527–11531.
35 Devary Y, Gottlieb RA, Smeal T, Karin M:The mammalian ultraviolet response is trig-gered by activation of Src tyrosine kinases. Cell1992;71:1081–1091.
36 Bauskin A, Alkalay I, Ben-Neriah Y: Redoxregulation of a protein tyrosine kinase in theendoplasmic reticulum. Cell 1991;66:685–696.
37 Fischer EH, Charbonneau H, Tonks NK: Pro-tein tyrosine phosphatases: A diverse family ofintracellular and transmembrane enzymes.Science 1991;253:401–406.
Dow
nloa
ded
by:
L.S
.U. H
ealth
Sci
ence
Cen
ter
20
6.17
6.17
5.20
0 -
7/11
/201
3 8:
55:1
2 P
M
Copyright © 2022 FDOKUMEN




















