
Neutrophil elastase and syndecan shedding contribute to ...
10
RESEARCH ARTICLE Neutrophil elastase and syndecan shedding contribute to antithrombin depletion in murine anthrax Myung-Chul Chung, Shelley C. Jorgensen, Taissia G. Popova, Charles L. Bailey & Serguei G. Popov National Center for Biodefense and Infectious Diseases, George Mason University, Manassas, VA, USA Correspondence: Serguei G. Popov, National Center for Biodefense and Infectious Diseases, George Mason University, 10900 University Blvd., Manassas, VA 20110, USA. Tel.: 11 703 993 4713; fax: 11 703 993 4288; e-mail: [email protected] Received 29 May 2008; revised 25 August 2008; accepted 26 August 2008. First published online 8 October 2008. DOI:10.1111/j.1574-695X.2008.00480.x Editor: Nicholas Carbonetti Keywords anthrax; disseminated intravascular coagulation; sepsis; antithrombin; syndecan. Abstract Bacillus anthracis infection is associated with severe hemostatic disturbances but their roles and contribution to fatality remain incompletely characterized. We undertook analyses of circulating antithrombin levels during the course of infection using a comparison of lethal and nonlethal murine anthrax models. Plasma samples were obtained from DBA/2 mice challenged intraperitoneally with the spores of either toxigenic B. anthracis Sterne strain or nontoxigenic, avirulent delta Sterne strain. We found that plasma antithrombin levels were rapidly depleted in Sterne spore-challenged mice, concomitant with elevation of neutrophil elastase (NE) and massive syndecan shedding from the liver into circulation. The shed syndecan bound with antithrombin accelerated NE-mediated antithrombin proteolysis. The liver response to infection demonstrated strain-specific compensatory increases of antithrombin and syndecan gene transcription. Both bacterial strains induced changes in blood coagulation parameters consistent with the onset of disseminated intravascular coagulation. We propose that antithrombin depletion proceeding through activation of neutrophils and massive shedding of heparin-like syndecan from the liver into circulation contribute to anthrax coagulopathy. Introduction Bacillus anthracis is a gram-positive, spore-forming bacter- ium. It is the causative agent of anthrax – a highly dangerous disease of humans and several animal species. The disease can be manifested in one of the three distinct forms, namely cutaneous, gastrointestinal, and inhalational, depending on the route of the host exposure to the infectious bacterial spores. If left untreated, anthrax can quickly progress systemically, which is associated with high mortality. Several pathogenic factors of B. anthracis have been identified and characterized. It is commonly accepted that lethal and edema toxins (LT and ET, respectively), along with the antiphagocytic capsule are the major virulence factors of the bacterium (Prince, 2003; Moayeri & Leppla, 2004; Heninger et al., 2006). Recently, other secreted bacterial proteins, including broad-spectrum proteases and hemo- lysins, have been reported as substantial contributors to anthrax pathology and mortality (Popov et al., 2005; Mosser & Rest, 2006; Heffernan et al., 2007). Although studies on the effects of B. anthracis pathogenic factors in vivo and in vitro have yielded important insights into their function, the pathological mechanisms underlying high mortality of systemic anthrax, as well as the immediate cause of death in this disease remains incompletely understood. Hemostatic and hemodynamic abnormalities during an- thrax infection are well known (Smith & Keppie, 1954; Abramova et al., 1993; Guarner et al., 2003; Stearns-Kurosawa et al., 2006). Dilated capillaries are such a common morpho- logic feature of anthrax infection that for many years before the discovery of anthrax toxins, the mechanical obstruction of capillaries by the bacilli was believed to be the cause of death in anthrax septicemia (Albrink, 1961). This theory was dismissed based on the fact that anthrax-infected animals at times die in the absence of severe bacteremia (Eckert & Bonventre, 1963). However, later experiments revealed that widespread pulmonary capillary and kidney glomeruli thrombosis in infected rats, rabbits, and guinea- pigs was not caused solely by the mechanical obstruction of capillaries by a large number of bacilli (Dalldorf & Beal, 1967). The authors suggested thrombosis as the major cause of death and concluded that anthrax pathogenic factors acted directly upon the membranes of capillary endothelium to cause increased permeability and capillary thrombosis. FEMS Immunol Med Microbiol 54 (2008) 309–318 c 2008 George Mason University National Center for Biodefense Journal compilation c 2008 Federation of European Microbiological Societies Published by Blackwell Publishing Ltd. Downloaded from https://academic.oup.com/femspd/article/54/3/309/512730 by guest on 04 July 2022
-
Upload
khangminh22 -
Category
Documents
-
view
2 -
download
0
Transcript of Neutrophil elastase and syndecan shedding contribute to ...

R E S E A R C H A R T I C L E
Neutrophil elastaseand syndecan shedding contribute toantithrombindepletion inmurineanthraxMyung-Chul Chung, Shelley C. Jorgensen, Taissia G. Popova, Charles L. Bailey & Serguei G. Popov
National Center for Biodefense and Infectious Diseases, George Mason University, Manassas, VA, USA
Correspondence: Serguei G. Popov,
National Center for Biodefense and Infectious
Diseases, George Mason University, 10900
University Blvd., Manassas, VA 20110, USA.
Tel.: 11 703 993 4713; fax: 11 703 993
4288; e-mail: [email protected]
Received 29 May 2008; revised 25 August
2008; accepted 26 August 2008.
First published online 8 October 2008.
DOI:10.1111/j.1574-695X.2008.00480.x
Editor: Nicholas Carbonetti
Keywords
anthrax; disseminated intravascular
coagulation; sepsis; antithrombin; syndecan.
Abstract
Bacillus anthracis infection is associated with severe hemostatic disturbances but
their roles and contribution to fatality remain incompletely characterized. We
undertook analyses of circulating antithrombin levels during the course of infection
using a comparison of lethal and nonlethal murine anthrax models. Plasma samples
were obtained from DBA/2 mice challenged intraperitoneally with the spores of
either toxigenic B. anthracis Sterne strain or nontoxigenic, avirulent delta Sterne
strain. We found that plasma antithrombin levels were rapidly depleted in Sterne
spore-challenged mice, concomitant with elevation of neutrophil elastase (NE) and
massive syndecan shedding from the liver into circulation. The shed syndecan
bound with antithrombin accelerated NE-mediated antithrombin proteolysis. The
liver response to infection demonstrated strain-specific compensatory increases of
antithrombin and syndecan gene transcription. Both bacterial strains induced
changes in blood coagulation parameters consistent with the onset of disseminated
intravascular coagulation. We propose that antithrombin depletion proceeding
through activation of neutrophils and massive shedding of heparin-like syndecan
from the liver into circulation contribute to anthrax coagulopathy.
Introduction
Bacillus anthracis is a gram-positive, spore-forming bacter-
ium. It is the causative agent of anthrax – a highly dangerous
disease of humans and several animal species. The disease
can be manifested in one of the three distinct forms, namely
cutaneous, gastrointestinal, and inhalational, depending on
the route of the host exposure to the infectious bacterial
spores. If left untreated, anthrax can quickly progress
systemically, which is associated with high mortality. Several
pathogenic factors of B. anthracis have been identified and
characterized. It is commonly accepted that lethal and
edema toxins (LT and ET, respectively), along with the
antiphagocytic capsule are the major virulence factors of
the bacterium (Prince, 2003; Moayeri & Leppla, 2004;
Heninger et al., 2006). Recently, other secreted bacterial
proteins, including broad-spectrum proteases and hemo-
lysins, have been reported as substantial contributors to
anthrax pathology and mortality (Popov et al., 2005; Mosser
& Rest, 2006; Heffernan et al., 2007). Although studies on
the effects of B. anthracis pathogenic factors in vivo and in
vitro have yielded important insights into their function, the
pathological mechanisms underlying high mortality of
systemic anthrax, as well as the immediate cause of death in
this disease remains incompletely understood.
Hemostatic and hemodynamic abnormalities during an-
thrax infection are well known (Smith & Keppie, 1954;
Abramova et al., 1993; Guarner et al., 2003; Stearns-Kurosawa
et al., 2006). Dilated capillaries are such a common morpho-
logic feature of anthrax infection that for many years before
the discovery of anthrax toxins, the mechanical obstruction
of capillaries by the bacilli was believed to be the cause of
death in anthrax septicemia (Albrink, 1961). This theory
was dismissed based on the fact that anthrax-infected
animals at times die in the absence of severe bacteremia
(Eckert & Bonventre, 1963). However, later experiments
revealed that widespread pulmonary capillary and kidney
glomeruli thrombosis in infected rats, rabbits, and guinea-
pigs was not caused solely by the mechanical obstruction of
capillaries by a large number of bacilli (Dalldorf & Beal,
1967). The authors suggested thrombosis as the major cause
of death and concluded that anthrax pathogenic factors
acted directly upon the membranes of capillary endothelium
to cause increased permeability and capillary thrombosis.
FEMS Immunol Med Microbiol 54 (2008) 309–318 c� 2008 George Mason UniversityNational Center for Biodefense
Journal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd.
Dow
nloaded from https://academ
ic.oup.com/fem
spd/article/54/3/309/512730 by guest on 04 July 2022

Further studies (Grinberg et al., 2001; Culley et al., 2005;
Stearns-Kurosawa et al., 2006) strongly supported these
observations, and it has been recognized recently (Berry
et al., 2003) that vascular pathology in anthrax patients is
indicative of the onset of condition known as disseminated
intravascular coagulation (DIC) or consumptive coagulo-
pathy (Stearns-Kurosawa et al., 2006).
DIC is often associated with microbial sepsis and typically
sets off intravascular thrombosis, tissue hypoxia, activation
of the fibrinolytic pathways, depletion of coagulation fac-
tors, inability to maintain hemostasis, and overt bleeding
(Opal & Esmon, 2003). All these features are present in the
course of anthrax infection, and some of them, such as
thrombocytopenia and vascular leakage, have also been
reported in the cell and animal models of anthrax toxemia
(Gozes et al., 2006; Stearns-Kurosawa et al., 2006). Clinical
manifestations of DIC may be underlined by different
disorders and range from the absence of overt symptoms to
shock and death. The pathogenic mechanisms that trigger
DIC in anthrax are currently unknown, and the contribu-
tion of DIC to anthrax mortality remains to be elucidated.
Available evidence indicates that the coagulopathic effect of
the anthrax infectious process is complex, and likely repre-
sents a concerted action of toxins (Ascenzi et al., 2002;
Moayeri & Leppla, 2004; Popova et al., 2006; Turk, 2007;
Watson et al., 2007) and other pathogenic factors such as
broad-spectrum proteases and hemolytic proteins (Bonventre
& Eckert, 1963; Popov et al., 2005; Chung et al., 2006;
Heffernan et al., 2007). We recently reported that
B. anthracis-challenged mice demonstrate increases in the
amount of circulating soluble proteoglycans, namely synde-
can (SDC)-1, -4, as well as plasma depletion of the func-
tional von Willebrand factor and its natural regulator
ADAMTS13, implicating these pathogenic processes in the
development of hemorrhage (Popova et al., 2006; Chung
et al., 2008). Bacillus anthracis proteases have been shown to
be hemorrhagic in the mouse skin test (Popov et al., 2005),
and able to degrade a range of proteins critically involved in
the regulation of hemostasis in vitro, such as a2-macroglo-
bulin, a2-antiplasmin, and a1-protease inhibitor, which are
the most important serum regulators of plasmin and blood
elastase (Chung et al., 2006). With regard to blood fibrino-
lysis, B. anthracis proteases can also cleave fibrinogen and
therefore may impair the clotting process during anthrax
infection (Chung et al., 2006, 2008).
In sepsis, antithrombin is quickly depleted by virtue of its
consumption during complex formation with activated
clotting factors and after proteolytic degradation by elastase
secreted from polymorphonuclear leukocytes (Jordan et al.,
1987). Antithrombin is a plasma-derived serine protease
inhibitor from a single chain glycoprotein, which controls
the activity of thrombin and the inhibition of several other
proteases of the coagulation cascade. Antithrombin has
potent anticoagulant activity and is significantly enhanced
by heparin and vessel wall-associated glycosaminoglycans
(GAGs). Despite acting as an acceleratory cofactor for
antithrombin, heparin was also found to considerably accel-
erate the in vitro inactivation of antithrombin by neutrophil
elastase (NE) (Jordan et al., 1987). Sharp decreases of
antithrombin in sepsis has been shown previously to cause
a dramatic imbalance in hemostasis and homeostasis result-
ing in an excess of activated factors. In fact, when anti-
thrombin levels decrease to o 30% of normal, DIC patients
show almost absolute lethality with sepsis (Roemisch et al.,
2002). Although B. anthracis infection induces DIC with
severe sepsis, the underlying mechanisms responsible
remain poorly understood.
In the present study, we undertook analyses of circulating
antithrombin levels and several other hemostatic parameters
in the murine model of anthrax. Plasma samples were
obtained from mice challenged with the spores of either
toxigenic B. anthracis Sterne strain (pXO11, pXO2�), which
is virulent in DBA/2 mice, or nontoxigenic, avirulent delta
Sterne strain (pXO1�, pXO2�). Strong antithrombin deple-
tion from circulation at the late stages of toxigenic infection
was observed compared with nontoxigenic one. By bio-
chemical analyses, we demonstrate that activation of neu-
trophils and massive shedding of heparin-like syndecan
from the liver into circulation is one of the pathological
mechanisms of antithrombin depletion.
Materials and methods
Animals
Female, 9-week-old DBA/2 mice were purchased from
Jackson Laboratory (Bar Harbor, ME) and maintained at
Biocon Inc. (Rockville, MD). Food and water were supplied
ad libitum. Animals were allowed to acclimate to their
surroundings for 1 week before experiments. The George
Mason University Institutional Animal Care and Use Com-
mittee and Biocon Animal Care and Use Committee/Insti-
tutional Review Board have approved all protocols before
animal experiments.
Preparation of mouse plasma
Mice (n = 5–10 per treatment group) were challenged in-
traperitoneally (i.p.) with 5� 106 spores of B. anthracis
nonencapsulated Sterne strain 34F2 (pXO11, pXO2�) or
nontoxigenic delta Sterne strain (pXO1�, pXO2�) obtained
from the Colorado Serum Company (Boulder, CO) and
National Center for Biodefense and Infectious Diseases
(Manassas, VA), respectively. Approximately 200–500 mL of
blood were drawn from the survived animals (n = 3–5 per
treatment group at every time point) via the retro-orbital
FEMS Immunol Med Microbiol 54 (2008) 309–318c� 2008 George Mason UniversityNational Center for BiodefenseJournal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd.
310 M.-C. Chung et al.
Dow
nloaded from https://academ
ic.oup.com/fem
spd/article/54/3/309/512730 by guest on 04 July 2022

sinus into 50 mL of 0.5 M EDTA and centrifuged at 900 g for
10 min to obtain platelet poor plasma (PPP).
Bacterial load and course of infection
Mice were euthanized at 24, 48, and 72 h post-spore
challenge and the livers were collected to measure bacterial
dissemination. Liver tissues were homogenized in
phosphate-buffered saline (PBS), serially diluted, and plated
on Luria–Bertani agar to determine numbers of CFU. The
highest bacterial burden was 2.0� 105 CFUg�1 (48 h) in liver
of Sterne-challenged mice and 5.2� 104 CFUg�1 (72 h) in
delta Sterne-challenged mice. The 50% mortality took place
at 72 h post-Sterne challenge; there was 100% survival in all
delta Sterne-spore challenged mice (Supporting Informa-
tion, Fig. S1).
Heparin cofactor antithrombin assay
For the quantitative determination of antithrombin in
mouse plasma, a heparin cofactor assay kit (Chromogenix,
Coamatic Antithrombin Kit) was used according to the
manufacturer’s recommendation. Briefly, 50mL of 1 : 20
diluted plasma were mixed with 50 mL of human factor Xa
in the presence of heparin and incubated at 37 1C for 90 s in
a 96-well microtiter plate. To measure the residual factor Xa
activity, 50 mL of substrate (N-a-Cbo-Arg-Gly-Arg-p-
nitroanilide) were added to the reaction for 90 s. Then 50mL
of 20% acetic acid were added to each microplate well and
the absorbance was read at 405 nm. Relative antithrombin
activity (%) was evaluated based on a standard curve using
pooled control plasma.
Western blot
Plasma samples (corresponding to 20 mg of total protein)
from normal control and spore-challenged mice were boiled
in sodium dodecyl sulfate (SDS) sample buffer containing
30 mM dithiothreitol. The samples were loaded onto a
NuPAGE 4–12% Bis Tris Gel (Invitrogen) and transferred
onto a polyvinylidene difluoride membrane using the iBlot
Gel Transfer System (Invitrogen). The membrane was
blocked with a solution of 5% milk powder in PBS,
incubated with primary sheep a-mouse antithrombin anti-
body (Abcam) or mouse a-human NE antibody (Santa Cruz
Biotechnology), and finally incubated with an horse radish
peroxidase (HRP)-conjugated secondary antibody (rabbit
a-sheep IgG from Abcam, or sheep a-mouse IgG from GE
Healthcare, correspondingly). The membranes were devel-
oped using an ECL Western blotting detection kit (Amer-
sham Biosciences) and Kodak BioMax light film or a
Molecular Imager ChemiDoc XRS System (Bio-Rad).
Ligand blot
Two micrograms of human antithrombin (Sigma) were
loaded onto a NuPAGE 4–12% Bis-Tris Gel (Invitrogen)
and transferred onto a nitrocellulose membrane using the
iBlot Gel Transfer System (Invitrogen). The membrane was
incubated overnight at 4 1C in PBS containing 0.05%
Tween-20 (PBST), washed three times with PBST, incubated
with the protein ligand solution in PBST for 1.5 h at room
temperature and then incubated with a corresponding
primary antibody (1 : 3000 dilution) for 2 h. Primary anti-
bodies were sheep anti-mouse antithrombin (Abcam),
mouse anti-mouse NE (Santa Cruz Biotechnology), mouse
anti-mouse SDC-1 (BD Pharmingen), goat anti-mouse
SDC-4 (Santa Cruz Biotechnology) and chicken anti-
human a1-protease inhibitor (Abcam). Culture supernatants
of anthrolysin O (ALO)-treated normal murine mammary
gland cells (NMuMGCs) (150mL) were prepared by treating
the cultured cells with 1 mg mL�1 of ALO for 24 h to induce
syndecan shedding as described previously (Popova et al.,
2006). Finally, the membranes were washed and detected
with corresponding secondary antibodies described above.
Immunoprecipitation
Pooled plasma (30mL) from 48-h Sterne spore-challenged
mice was diluted in PBS (1 : 10) and precleared by adding
20 mL of suspended (25% v/v) protein A/G-agarose beads
(Santa Cruz Biotechnology). Primary antibodies (2mg)
against antithrombin, NE, or SDC-1 were added after
incubation for 1 h at 4 1C. Then, 20 mL of protein A/G-
agarose beads were added followed by incubation at 4 1C on
a platform rocker overnight. The immunoprecipitates were
collected by centrifugation at 900 g for 30 s at 4 1C and
washed four times with 1.0 mL of PBS. After the final wash,
the proteins were eluted by directly adding 40mL of 2�electrophoresis sample buffer. After boiling for 3 min, the
samples were subjected to electrophoresis and Western blot
analysis with antithrombin antibody as described above.
Enzyme-linked immunosorbent assays (ELISAs)
Wells of a 96-mictotiter (NUNC MaxiSorp) plate were
coated with 50 mL of plasma samples from five mice
(0.01–20mg of total protein) in 50 mM carbonate buffer
(pH 9.0) and incubated overnight at 4 1C. Coated wells were
blocked with 1% bovine serum albumin in PBS, and
primary antibodies were added in a 1 : 3000 dilution. The
antibodies were against the following antigens: mouse
a-human D-dimer antibody (Abcam, ab-10050); goat
a-mouse PAI-1 (Santa Cruz Biotechnology, sc-6644);
goat a-mouse PAI-2 (Santa Cruz Biotechnology, sc-6649);
rabbit a-mouse a2-antiplasmin (Abcam, ab-28326); rabbit
a-mouse plasminogen (Abcam, ab-28319); mouse a-human
FEMS Immunol Med Microbiol 54 (2008) 309–318 c� 2008 George Mason UniversityNational Center for Biodefense
Journal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd.
311Antithrombin depletion in a murine anthrax model
Dow
nloaded from https://academ
ic.oup.com/fem
spd/article/54/3/309/512730 by guest on 04 July 2022

thrombin (Abcam, ab-17199); sheep a-human antithrom-
bin (Abcam, ab-8776); and chicken a-human a1-antitrypsin
(Abcam, ab-14226). All antibodies against human proteins
cross-reacted with corresponding mouse proteins. The
pooled plasma samples from five mice were incubated in
triplicates in the coated wells at room temperature for 1 h,
and washed three times with PBST. Fifty microliters of
corresponding HRP-conjugated secondary antitotal IgG
antibodies were added in a 1 : 10 000 dilution followed by
1-h incubation at room temperature. After washing five
times with PBST, the tetramethylbenzidine (TMB) substrate
(100mL) was added. The reaction was stopped with 1 M
H2SO4 (50 mL) and the absorbance was read at 450 nm.
Standard curves were made from a dilution of normal
mouse plasma and the results were expressed relative to
controls. For TAT analysis, wells of a 96-well plate (NUNC
MaxiSorp) were coated with 100 mL of anti-antithrombin
antibody (1 mg mL�1 in coating buffer). Plates were incu-
bated overnight at 4 1C. The plates were washed three times
with PBST, and 200mL of PBS/10% fetal calf serum (FCS)
was added to each well as a blocking step for 1 h followed by
addition of 50 mL of standard or fourfold-diluted plasma
samples in PBST/1% FCS for 1 h on a shaker. The plate was
washed three times with PBST and then incubated with
100mL of antithrombin antibody (0.5mg mL�1) in PBST/1%
FCS for 1 h. The wells were washed three times with PBST
and incubated with 100 mL of HRP-antibody in PBST
followed by washes and TMB solution as described above.
Reverse-transcriptase (RT)-PCR
For determination of SDC-1, -4, and antithrombin mRNA
expression, total RNA was prepared from frozen liver
sections stored at � 80 1C. Random-primed cDNA was
prepared from 5mg of total RNA using the Platinum PCR
Supermix (Invitrogen). Specific primers for antithrombin,
50-ATG GTA CAT TCC TCA CCC TGC G (sense) and 50-
AGC ACC ACT GCT CCC ACA ATG A (antisense); SDC-1,
50-ATG AGA CGC GCG GCG CTC TGG CTT (sense) and
50-GGCGTA GAA CTC CTC CTG CTT GGT (antisense);
SDC-4, 50-AAC CAC ATC CCT GAG AAT GC (sense) and
50-AGG AAA ACG GCA AAG AGG AT (antisense) were
used during RT-PCR. After amplification, PCR products
were separated on a 1.7% agarose gel and stained with
ethidium bromide. The gel photographs were analyzed by
densitometry and the expression levels were normalized by
the amount of b-actin mRNA.
Statistical analyses
A paired two-tailed Student’s t-test was used for all statistical
analyses. The results are presented as means� SD. Values of
Po 0.05 were considered statistically significant.
Results
Depletion of antithrombin in plasma isassociated with its proteolytic degradation
Reduced antithrombin activity in the blood is often con-
sidered one of the most important diagnostic indicators of
nonsurvival in sepsis (Spero et al., 1980; Bick et al., 1996). In
order to examine circulating antithrombin levels, DBA/2
mice were challenged intraperitoneally with 5� 106 spores
of toxigenic Sterne or nontoxigenic delta Sterne strains.
Plasma samples from randomly chosen individual mice were
prepared using EDTA, loaded onto the SDS-polyacrylamide
gel electrophoresis (PAGE) and analyzed using Western blot.
Sterne-challenged mice demonstrated a strong reduction in
the overall intensity of bands recognized by the antithrom-
bin-specific antibody (Fig. 1a and c). The reduction was
most intense between 24 and 48 h postchallenge, while
animals sacrificed for analysis at 72 h postchallenge demon-
strated less pronounced antithrombin depletion. For inter-
pretation of these results, it has to be taken into account that
before 48 h postchallenge the population of animals is
characteristic of the disease onset and is not yet skewed by
high mortality, which takes place around 72 h (Popov et al.,
2005). In contrast, at 72 h postchallenge the live animals
represent mainly resistant survivors. In comparison, plas-
mas from delta Sterne-challenged mice showed only subtle
changes in the intensities of the bands immunoreacting with
the anti-antithrombin antibody (Fig. 1b and c). At 72 h
post-delta Sterne-challenge all mice survived, and the
amount of antithrombin in plasma returned to its level in
healthy mice (Fig. 1c) indicating convalescence after disease.
Next, we used the heparin cofactor assay to test the
amount of functional antithrombin in the plasma of
Sterne-challenged mice. This assay is based on the ability of
antithrombin to inhibit the activity of factor Xa in the
presence of heparin. The assay is able to discriminate
between antithrombin-deficient and antithrombin nondefi-
cient individuals better than thrombin-based assays
(Demers et al., 1993) and gives accurate results in patients
receiving heparin therapy (Bohner et al., 1994). We found
that the heparin cofactor activity gradually decreased after
Sterne spore challenge (Fig. 1d). At 72 h postinfection, the
heparin cofactor activity remained low, indicating a func-
tional deficiency of circulating antithrombin in spite of the
fact that the animals survived up to this time showed total
antithrombin protein levels close to normal (Fig. 1a and c).
In order to determine whether the depletion of antith-
rombin from circulation could be explained by decreased
expression of the antithrombin gene, total RNA was pre-
pared from the liver of B. anthracis spore-challenged mice
was analyzed using RT-PCR. The results showed that during
the course of toxigenic infection, the levels of antithrombin
FEMS Immunol Med Microbiol 54 (2008) 309–318c� 2008 George Mason UniversityNational Center for BiodefenseJournal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd.
312 M.-C. Chung et al.
Dow
nloaded from https://academ
ic.oup.com/fem
spd/article/54/3/309/512730 by guest on 04 July 2022

gene expression steadily increased, while the nontoxigenic
infection did not induce a statistically significant response
(Fig. 2). This suggested that depletion of antithrombin in
plasma was associated with its proteolytic degradation, but
not suppression of the antithrombin gene expression.
Shed SDC-1 and -4 interact with antithrombinand accelerate neutrophil elastase-mediatedantithrombin proteolysis in vitro
NE expression significantly correlates with DIC severity and
has a good predictive value for mortality in patients
suspected of having DIC (Song et al., 2007). During DIC,
NE is able to inactivate antithrombin by a specific and
limited proteolytic cleavage (Jordan et al., 1987). We, there-
fore, suggested that antithrombin depletion found in toxi-
genic B. anthracis-challenged mice could be relevant to the
induction of NE during infection and tested with NE
antibodies the plasma samples previously used for the
analysis of antithrombin. NE levels were significantly in-
creased at 24 and 48 h post-Sterne spore challenge (Fig. 3).
Western blot analysis of antithrombin and NE from plasma
of each mouse demonstrated that high levels of NE corre-
lated with the reduced levels of antithrombin (Figs 1 and 3).
To prove a possible causal connection between enzymatic
activity of NE and antithrombin levels, we carried out
experiments with purified NE. Inactivation of antithrombin
by NE is known to be considerably accelerated by heparan
sulfate proteoglycans derived from endothelial cells (Jordan
et al., 1987). On the surface of endothelium, antithrombin
associates with proteoglycans such as SDC-1 and -4 posses-
sing sulfated carbohydrate chains structurally similar to
heparin, and the anticoagulant heparin subfraction also
demonstrates high-affinity binding for antithrombin
(Jordan et al., 1987; San Antonio et al., 1994; Woods et al.,
1998, 2000; Ueno et al., 2001). Bacillus anthracis accelerates
SDC-1 shedding due to the activity of proteolytic and
hemolytic proteins (Popova et al., 2006). Therefore, we
tested whether shed syndecans accelerate NE-dependent
antithrombin proteolysis. First, we investigated the interac-
tion of isolated antithrombin and NE proteins with shed
syndecans using the ligand blot assay. As a source of
syndecans, we used culture supernatant of NMuMGCs
treated with B. anthracis hemolysin ALO (Popova et al.,
Fig. 1. Antithrombin protein level and functional activity are reduced in plasma of spore-challenged mice. Western blots of antithrombin in plasma from
Sterne (a) and delta Sterne (b) spore-challenged mice. Numbers indicate individual mice for comparison with NE analysis in Fig. 3. Arrows indicate the positions
on gels corresponding to molecular mass of antithrombin. (c) Densitometric analysis of antithrombin bands in (a) and (b). Open and gray bars indicate relative
antithrombin levels in plasmas from delta Sterne- and Sterne-challenged mice, correspondingly. �P = 0.001 and ��P = 0.025 relative to control mice. (d)
Heparin cofactor activity of antithrombin in plasma of Sterne spore-challenged mice relative to unchallenged control. �P = 0.02 and ��Po 0.01 compared
with control unchallenged mice.
Fig. 2. Gene expression for antithrombin measured using RT-PCR rela-
tive to actin in spore-challenged mouse plasma. Three mice from each
group were subjected to RT-PCR. Expression was calculated as a fold
change relative to control (mean� SD). P-values were evaluated by
paired Student’s t-test.
FEMS Immunol Med Microbiol 54 (2008) 309–318 c� 2008 George Mason UniversityNational Center for Biodefense
Journal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd.
313Antithrombin depletion in a murine anthrax model
Dow
nloaded from https://academ
ic.oup.com/fem
spd/article/54/3/309/512730 by guest on 04 July 2022

2006). Antithrombin blotted onto a nitrocellulose mem-
brane was incubated with NE, culture supernatant, or
a1-protease inhibitor as a negative control. After intensive
washing, interaction of antithrombin with the overlaid
proteins was detected using specific antibodies. The results
demonstrated that antithrombin interacted with NE, shed
SDC-1 and -4 from culture supernatant, but not with
a1-protease inhibitor (Fig. 4a). The interaction between
antithrombin and NE or syndecan was further confirmed
using immunoprecipitation analysis (Fig. 4b).
Bacillus anthracis -challenged mice showsyndecan shedding in liver blood vessels
Damage to the liver plays a central role in abnormal
hemostasis and the development of DIC (Mammen, 1992).
Syndecan shedding can be stimulated by different cell stress
factors including B. anthracis-secreted proteins, and the liver
has been suggested as one of the primary targets (Watchorn
et al., 2002; Kovalszky et al., 2004; Popova et al., 2006). Using
immunofluorescence microscopy with a SDC-1-specific
primary antibody and a fluorescein-labeled secondary anti-
body, we evaluated the amount of syndecan in the liver of
spore-challenged mice. Control samples demonstrated that
SDC-1 was readily detectable as green fluorescence in the
lining of normal liver sinusoids and the lumen of liver blood
vessel was stained negative (Fig. 5a and c). Conversely,
Sterne spore-challenged mice demonstrated a considerable
increase in the amount of expressed SDC-1 and shortening
of hepatocyte cords indicating a physiological response of
the liver to infection (Fig. 5b). Concomitantly, a massive
amount of SDC-1-positive aggregates was detected in the
lumen of blood vessels (Fig. 5d) in agreement with our
previous ELISA data demonstrating the appearance of shed
SDC-1 in the blood of infected mice (Popova et al., 2006).
SDC-1 gene expression was increased in both Sterne and
delta Sterne-challenged mice (Fig. 6a), which has been
shown previously to correlate with changes in cytoskeleton
organization (Lebakken & Rapraeger, 1996). SDC-4-
deficient mice have impaired repair to tissue injury and are
more susceptible to thrombus formation and endotoxin
shock (Ishiguro et al., 2001). In our study, SDC-4 gene
expression was upregulated only by the toxigenic strain
(Fig. 6b).
Markers for coagulopathy in plasma ofB. anthracis spore-challenged mice
In order to further characterize hemostatic abnormalities
during anthrax infection we analyzed plasmas of challenged
mice using a conventional ELISA with specific antibodies
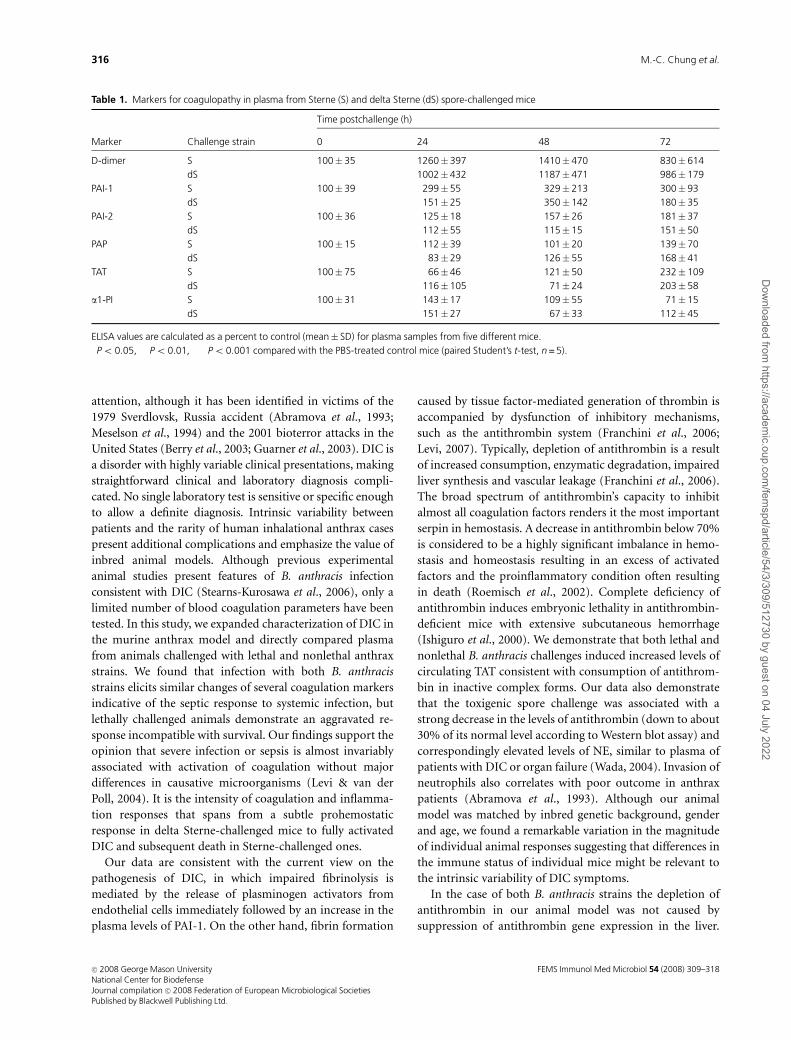
against several key proteins associated with DIC (Table 1).
According to the current understanding of DIC, the deter-
mination of fibrin degradation products in plasma is critical
for diagnosis (Levi & ten Cate, 1999; Schrecengost et al.,
2003). Analysis of D-dimers, the breakdown products of the
fibrin mesh, helps to differentiate degradation of cross-
linked fibrin formed in the process of coagulation from
fibrinogen degradation products. Bacteremia typically re-
sults in a rapidly occurring increase in fibrinolytic activity,
which becomes impaired due to a sustained level of
Fig. 4. Antithrombin interacts with human NE, shed SDC-1 and SDC-4
in vitro and in vivo. (a) Ligand blot analysis. Antithrombin was immobi-
lized on the nitrocellulose membrane, incubated with indicated analytes,
and immunodetected using specific antibodies. a1-PI is shown as a
negative control. Culture supernatant of ALO-treated NMuMGCs served
as a source of SDC-1. (b) Immunoprecipitation (IP) of proteins from
plasma of Sterne spore-challenged mice at 24 and 48 h postinfection
with antithrombin-, NE-, and SDC-1-specific antibodies demonstrate the
presence of bound antithrombin detected using immunoblotting (IB)
with antithrombin-specific antibody.
Fig. 3. NE level is increased in plasma of Sterne spore-challenged mice.
(a) Western blot of NE in plasma of Sterne spore-challenged mice. The
major band with a molecular mass around 60 kDa in unchallenged
plasma corresponds to NE. Open triangles indicate complexes of NE with
unidentified proteins detected with anti-NE antibody during infection.
Numbers indicate individual mice for comparison with antithrombin
analysis in Fig. 1a. (b) Densitometric analysis of NE bands in (a).
FEMS Immunol Med Microbiol 54 (2008) 309–318c� 2008 George Mason UniversityNational Center for BiodefenseJournal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd.
314 M.-C. Chung et al.
Dow
nloaded from https://academ
ic.oup.com/fem
spd/article/54/3/309/512730 by guest on 04 July 2022

plasminogen activator inhibitor-1 (PAI-1), the main regu-
lator of endogenous fibrinolysis (Paul et al., 2005). In
agreement with this, Table 1 shows that progression of the
infectious process in the case of both B. anthracis strains
resulted in a sharp increase in D-dimer activity as early as
24 h postinfection followed by a slow decline at 72 h. The D-
dimer formation was accompanied by the generation of PAI-
1, and to the lesser extent, of PAI-2. This process took place
faster in the case of the virulent infection (Po 0.05 at 24 h).
In agreement with moderate elevation of plasmin–antiplasmin
complex (PAP) in DIC patients (Mavrommatis et al., 2001),
the levels of PAP in our experiments were slightly increased
above normal indicating that the generation of plasmin and
the fibrinolytic process were impaired.
The dynamics of DIC can be judged by measuring plasma
concentration of thrombin–antithrombin (TAT) complexes.
This marker is highly sensitive to low-grade activation of
coagulation but may have a limited specificity in complex
pathological conditions because it reflects the net effect of
generation and consumption of both thrombin and antith-
rombin. Our results demonstrate that TAT levels gradually
increased and became significantly elevated (Po 0.05) at
72 h postinfection in both Sterne and delta Sterne spore-
challenged mice. Taken together with the depletion of the
levels of circulating antithrombin (Fig. 1), these data in-
dicate a quick onset of a prothrombotic condition during
anthrax infection.
Discussion
The host septic response plays a key role in anthrax
pathogenesis (Smith & Keppie, 1954; Stearns-Kurosawa
et al., 2006). Bacterial infections, in particular septicemia,
are the most common causes of DIC (Kreger et al., 1980;
Mavrommatis et al., 2001). However, in anthrax, DIC as an
underlying pathophysiological condition attracted little
Fig. 5. Immunofluorescent staining of liver
using SDC-1-specific primary antibody and
fluorescein-labeled secondary antibody. SDC-1
(green) in the lining of sinusoids (a, b) and in the
lumen of liver blood vessel of control (a, c) and
Sterne spore-challenged mice at 72 h postinfection
(p.i.) (b, d). Brightness of images in (c) and (d) has
been increased to demonstrate accumulation
of SDC-1 aggregates in vessel lumen (d).
Fig. 6. Gene expression for SDC-1 (a), and SDC-4 (b) measured using
RT-PCR relative to actin in spore-challenged mouse plasma. Three mice
from each group were subjected to RT-PCR. Expression was calculated as
a fold change relative to control. P-values were calculated using paired
Student’s t-test.
FEMS Immunol Med Microbiol 54 (2008) 309–318 c� 2008 George Mason UniversityNational Center for Biodefense
Journal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd.
315Antithrombin depletion in a murine anthrax model
Dow
nloaded from https://academ
ic.oup.com/fem
spd/article/54/3/309/512730 by guest on 04 July 2022
attention, although it has been identified in victims of the
1979 Sverdlovsk, Russia accident (Abramova et al., 1993;
Meselson et al., 1994) and the 2001 bioterror attacks in the
United States (Berry et al., 2003; Guarner et al., 2003). DIC is
a disorder with highly variable clinical presentations, making
straightforward clinical and laboratory diagnosis compli-
cated. No single laboratory test is sensitive or specific enough
to allow a definite diagnosis. Intrinsic variability between
patients and the rarity of human inhalational anthrax cases
present additional complications and emphasize the value of
inbred animal models. Although previous experimental
animal studies present features of B. anthracis infection
consistent with DIC (Stearns-Kurosawa et al., 2006), only a
limited number of blood coagulation parameters have been
tested. In this study, we expanded characterization of DIC in
the murine anthrax model and directly compared plasma
from animals challenged with lethal and nonlethal anthrax
strains. We found that infection with both B. anthracis
strains elicits similar changes of several coagulation markers
indicative of the septic response to systemic infection, but
lethally challenged animals demonstrate an aggravated re-
sponse incompatible with survival. Our findings support the
opinion that severe infection or sepsis is almost invariably
associated with activation of coagulation without major
differences in causative microorganisms (Levi & van der
Poll, 2004). It is the intensity of coagulation and inflamma-
tion responses that spans from a subtle prohemostatic
response in delta Sterne-challenged mice to fully activated
DIC and subsequent death in Sterne-challenged ones.
Our data are consistent with the current view on the
pathogenesis of DIC, in which impaired fibrinolysis is
mediated by the release of plasminogen activators from
endothelial cells immediately followed by an increase in the
plasma levels of PAI-1. On the other hand, fibrin formation
caused by tissue factor-mediated generation of thrombin is
accompanied by dysfunction of inhibitory mechanisms,
such as the antithrombin system (Franchini et al., 2006;
Levi, 2007). Typically, depletion of antithrombin is a result
of increased consumption, enzymatic degradation, impaired
liver synthesis and vascular leakage (Franchini et al., 2006).
The broad spectrum of antithrombin’s capacity to inhibit
almost all coagulation factors renders it the most important
serpin in hemostasis. A decrease in antithrombin below 70%
is considered to be a highly significant imbalance in hemo-
stasis and homeostasis resulting in an excess of activated
factors and the proinflammatory condition often resulting
in death (Roemisch et al., 2002). Complete deficiency of
antithrombin induces embryonic lethality in antithrombin-
deficient mice with extensive subcutaneous hemorrhage
(Ishiguro et al., 2000). We demonstrate that both lethal and
nonlethal B. anthracis challenges induced increased levels of
circulating TAT consistent with consumption of antithrom-
bin in inactive complex forms. Our data also demonstrate
that the toxigenic spore challenge was associated with a
strong decrease in the levels of antithrombin (down to about
30% of its normal level according to Western blot assay) and
correspondingly elevated levels of NE, similar to plasma of
patients with DIC or organ failure (Wada, 2004). Invasion of
neutrophils also correlates with poor outcome in anthrax
patients (Abramova et al., 1993). Although our animal
model was matched by inbred genetic background, gender
and age, we found a remarkable variation in the magnitude
of individual animal responses suggesting that differences in
the immune status of individual mice might be relevant to
the intrinsic variability of DIC symptoms.
In the case of both B. anthracis strains the depletion of
antithrombin in our animal model was not caused by
suppression of antithrombin gene expression in the liver.
Table 1. Markers for coagulopathy in plasma from Sterne (S) and delta Sterne (dS) spore-challenged mice
Marker Challenge strain
Time postchallenge (h)
0 24 48 72
D-dimer S 100� 35 1260�397�� 1410� 470�� 830� 614�
dS 1002�432�� 1187� 471�� 986� 179���
PAI-1 S 100� 39 299�55� 329� 213� 300� 93�
dS 151�25��� 350� 142� 180� 35�
PAI-2 S 100� 36 125�18 157� 26 181� 37
dS 112�55 115� 15 151� 50
PAP S 100� 15 112�39 101� 20 139� 70
dS 83�29 126� 55 168� 41�
TAT S 100� 75 66�46 121� 50 232� 109
dS 116�105 71� 24 203� 58�
a1-PI S 100� 31 143�17 109� 55 71� 15
dS 151�27 67� 33 112� 45
ELISA values are calculated as a percent to control (mean� SD) for plasma samples from five different mice.�Po 0.05, ��Po 0.01, ���Po 0.001 compared with the PBS-treated control mice (paired Student’s t-test, n = 5).
FEMS Immunol Med Microbiol 54 (2008) 309–318c� 2008 George Mason UniversityNational Center for BiodefenseJournal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd.
316 M.-C. Chung et al.
Dow
nloaded from https://academ
ic.oup.com/fem
spd/article/54/3/309/512730 by guest on 04 July 2022

Antithrombin is known to be susceptible to NE-mediated
cleavage in the presence of blood proteoglycans, such as
SDC-1 (Jordan et al., 1987). We reported previously in-
creased shedding of syndecans into the blood of B. anthracis
spore-challenged mice (Popova et al., 2006), and our present
results implicate syndecan shedding as the cause of acceler-
ated antithrombin degradation by NE. However, activation
of NE may not be the only mechanism of antithrombin
depletion during infection. Relevant to this, vascular leakage
and hemorrhage have been demonstrated previously in mice
administered LT and other secreted proteases (Cui et al.,
2005; Culley et al., 2005).
In summary, our findings reveal early aggressive hemo-
static changes in the murine anthrax model consistent with
DIC, acceleration of the coagulation cascade, and subse-
quent host septic response, which need to be taken into
account in future therapeutic interventions.
Acknowledgement
This work was supported by the United States Department
of Defense grant DAMD 17-03-C-01220.
Authors’contribution
M.-C.C. and S.C.J. contributed equally to this study.
References
Abramova F, Grinberg L, Yampolskaya O & Walker D (1993)
Pathology of inhalational anthrax in 42 cases from the
Sverdlovsk outbreak of 1979. Proc Natl Acad Sci USA 90:
2291–2294.
Albrink W (1961) Pathogenesis of inhalation anthrax. Bacteriol
Rev 25: 268–273.
Ascenzi P, Visca P, Ippolito G, Spallarossa A, Bolognesi M &
Montecucco C (2002) Anthrax toxin: a tripartite lethal
combination. FEBS Lett 531: 384–388.
Berry K, Colvin S, Blythe D et al. (2003) Follow-up of deaths
among U.S. postal service workers potentially exposed to
Bacillus anthracis – district of Columbia, 2001–2002. JAMA
290: 2119–2120.
Bick R, Strauss J & Frenkel E (1996) Thrombosis and hemorrhage
in oncology patients. Hematol Oncol Clin North Am 10:
875–907.
Bohner J, von Pape K & Blaurock M (1994) Thrombin-based
antithrombin assays show overestimation of antithrombin III
activity in patients on heparin therapy due to heparin cofactor
II influence. Thromb Haemost 71: 280–283.
Bonventre P & Eckert N (1963) The biologic activities of Bacillus
anthracis and Bacillus cereus culture filtrates. Am J Pathol 43:
201–211.
Chung MC, Popova TG, Millis BA, Mukherjee DV, Zhou W,
Liotta LA, Petricoin EF, Chandhoke V, Bailey C & Popov SG
(2006) Secreted neutral metalloproteases of Bacillus anthracis
as candidate pathogenic factors. J Biol Chem 281:
31408–31418.
Chung MC, Popova TG, Jorgensen SC, Dong L, Chandhoke V,
Bailey CL & Popov SG (2008) Degradation of circulating von
Willebrand factor and its regulator ADAMTS13 implicates
secreted Bacillus anthracis metalloproteases in anthrax
consumptive coagulopathy. J Biol Chem 283: 9531–9542.
Cui X, Li Y, Moayeri M et al. (2005) Late treatment with a
protective antigen-directed monoclonal antibody improves
hemodynamic function and survival in a lethal toxin-infused
rat model of anthrax sepsis. J Infect Dis 191: 422–434.
Culley N, Pinson D, Chakrabarty A, Mayo M & Levine S (2005)
Pathophysiological manifestations in mice exposed to anthrax
lethal toxin. Infect Immun 73: 7006–7010.
Dalldorf F & Beal F (1967) Capillary thrombosis as a cause of
death in experimental anthrax. Arch Path 83: 154–161.
Demers C, Henderson P, Blajchman M et al. (1993) An
antithrombin III assay based on factor Xa inhibition provides a
more reliable test to identify congenital antithrombin III
deficiency than an assay based on thrombin inhibition.
Thromb Haemost 69: 231–235.
Eckert N & Bonventre P (1963) In vivo effects of Bacillus anthracis
culture filtrates. J Infect Dis 112: 226–232.
Franchini M, Lippi G & Manzato F (2006) Recent acquisitions in
the pathophysiology, diagnosis and treatment of disseminated
intravascular coagulation. Thrombosis J 4: 4.
Gozes Y, Moayeri M, Wiggins J & Leppla S (2006) Anthrax lethal
toxin induces ketotifen-sensitive intradermal vascular leakage
in certain inbred mice. Infect Immun 74: 1266–1272.
Grinberg R, Abramova F, Yampolskaya O, Walker D & Smith J
(2001) Quantitative pathology of inhalational anthrax I:
quantitative microscopic findings. Mod Pathol 5: 482–495.
Guarner J, Jernigan J, Shieh W, Tatti K, Flannagan L & Stephens D
(2003) Pathology and pathogenesis of bioterrorism-related
inhalational anthrax. Am J Path 163: 701–709.
Heffernan B, Thomason B, Herring-Palmer A & Hanna P (2007)
Bacillus anthracis anthrolysin O and three phospholipases C
are functionally redundant in a murine model of inhalation
anthrax. FEMS Microbiol Lett 271: 98–105.
Heninger S, Drysdale M, Lovchik J, Hutt J, Lipscomb M &
Koehler T (2006) Toxin-deficient mutants of B. anthracis are
lethal in a murine model for pulmonary anthrax. Infect Immun
74: 6067–6074.
Ishiguro K, Kojima T, Kadomatsu K et al. (2000) Complete
antithrombin deficiency in mice results in embryonic lethality.
J Clin Invest 106: 873–878.
Ishiguro K, Kadomatsu K, Kojima T et al. (2001) Syndecan-4
deficiency leads to high mortality of lipopolysaccharide-
injected mice. J Biol Chem 276: 47483–47488.
Jordan R, Kilpatrick J & Nelson R (1987) Heparin promotes the
inactivation of AT by neutrophil elastase. Science 14: 777–779.
Kovalszky I, Dudas J, Gallai M, Hollosi P, Tatrai P, Tatrai E &
Schaff Z (2004) Proteoglycans in the liver. Magy Onkol 48:
207–213.
FEMS Immunol Med Microbiol 54 (2008) 309–318 c� 2008 George Mason UniversityNational Center for Biodefense
Journal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd.
317Antithrombin depletion in a murine anthrax model
Dow
nloaded from https://academ
ic.oup.com/fem
spd/article/54/3/309/512730 by guest on 04 July 2022

Kreger B, Craven D & McCabe W (1980) Gram-negative
bacteremia. IV. Re-evaluation of clinical features and
treatment in 612 patients. Am J Med 68: 344–355.
Lebakken C & Rapraeger A (1996) Syndecan-1 mediates cell
spreading in transfected human lymphoblastoid (Raji) cells.
J Cell Biol 132: 1209–1221.
Levi M (2007) Disseminated intravascular coagulation. Crit Care
Med 35: 2191–2195.
Levi M & ten Cate H (1999) Disseminated intravascular
coagulation. N Engl J Med 341: 586–592.
Levi M & van der Poll T (2004) Coagulation in sepsis: all bugs bite
equally. Crit Care Clin 8: 99–100.
Mammen E (1992) Coagulation abnormalities in liver disease.
Hematol Oncol Clin North Am 6: 1247–1257.
Mavrommatis AC, Theodoridis T, Economou M, Kotanidou A, El
Ali M, Christopoulou-Kokkinou V & Zakynthinos SG (2001)
Activation of the fibrinolytic system and utilization of the
coagulation inhibitors in sepsis: comparison with severe sepsis
and septic shock. Intensive Care Med 27: 1853–1859.
Meselson M, Guillemin J, Hugh-Jones M, Langmuir A, Popova I,
Shelokov A & Yampolskaya O (1994) The Sverdlovsk anthrax
outbreak of 1979. Science 266: 1202–1208.
Moayeri M & Leppla S (2004) The roles of anthrax toxin in
pathogenesis. Curr Opin Microbiol 7: 19–24.
Mosser E & Rest R (2006) The Bacillus anthracis cholesterol-
dependent cytolysin, Anthrolysin O, kills human neutrophils,
monocytes and macrophages. BMC Microbiol 6: 56.
Opal S & Esmon C (2003) Bench-to-bedside review: functional
relationships between coagulation and the innate immune
response and their respective roles in the pathogenesis of
sepsis. Crit Care 7: 23–38.
Paul R, Winkler F, Bayerlein I, Popp B, Pfister H & Koedel U
(2005) Urokinase-type plasminogen activator receptor
regulates leukocyte recruitment during experimental
pneumococcal meningitis. J Infect Dis 191: 776–782.
Popov SG, Popova TG, Hopkins S, Weinstein RS, MacAfee R,
Fryxell KJ, Chandhoke V, Bailey C & Alibek K (2005) Effective
antiprotease-antibiotic treatment of experimental anthrax.
BMC Infect Dis 5: 25–39.
Popova TG, Millis B, Bradburne C, Nazarenko S, Bailey C,
Chandhoke V & Popov SG (2006) Acceleration of epithelial
cell syndecan-1 shedding by anthrax hemolytic virulence
factors. BMC Microbiol 6: 8.
Prince A (2003) The host response to anthrax lethal toxin:
unexpected observations. J Clin Invest 112: 656–658.
Roemisch J, Gray E, Hoffmann JN & Wiedermann CJ (2002)
Antithrombin: a new look at the actions of a serine protease
inhibitor. Blood Coagul Fibrinolysis 13: 657–670.
San Antonio JD, Karnovsky MJ, Gay S, Sanderson RD & Lander
AD (1994) Interactions of syndecan-1 and heparin with
human collagens. Glycobiology 4: 327–332.
Schrecengost JE, LeGallo RD, Boyd JC, Moons KG, Gonias SL,
Rose CE Jr & Bruns DE (2003) Comparison of diagnostic
accuracies in outpatients and hospitalized patients of D-dimer
testing for the evaluation of suspected pulmonary embolism.
Clin Chem 49: 1483–1490.
Smith H & Keppie J (1954) Observations on experimental
anthrax: demonstration of a specific lethal factor produced in
vivo by B. anthracis. Nature 173: 869–870.
Song S, Kim H, Park M & Cho H (2007) Neutrophil CD64
expression is associated with severity and prognosis of
disseminated intravascular coagulation. Thromb Res 121:
499–507.
Spero J, Lewis J & Hasiba U (1980) Disseminated intravascular
coagulation. Findings in 346 patients. Thromb Haemost 43:
28–33.
Stearns-Kurosawa D, Lupu F, Taylor F, Kinasewitz G & Kurosawa S
(2006) Sepsis and pathophysiology of anthrax in a nonhuman
primate model. Am J Pathol 169: 433–444.
Turk B (2007) Manipulation of host signalling pathways by
anthrax toxins. Biochem J 402: 405–417.
Ueno M, Yamada S, Zako M, Bernfield M & Sugahara K (2001)
Structural characterization of heparin sulfate and chondroitin
sulfate of syndecan-1 purified from normal murine mammary
gland epithelial cells. J Biol Chem 276: 29134–29140.
Wada H (2004) Disseminated intravascular coagulation. Clin
Chim Acta 344: 13–21.
Watchorn TM, Waddell I & Ross JA (2002) Proteolysis-inducing
factor differentially influences transcriptional regulation in
endothelial subtypes. Am J Physiol Endocrinol Metab 282:
E763–E769.
Watson L, Mock J, Lal H et al. (2007) Lethal and edema toxins of
anthrax induce distinct hemodynamic dysfunction. Front
Biosci 1: 4670–4675.
Woods A & Couchman J (1998) Syndecans: synergistic activators
of cell adhesion. Trends Cell Biol 8: 189–192.
Woods A, Longely R, Tumova S & Couchman J (2000) Syndecan-
4 binding to the high affinity heparin-binding domain of
fibronectin drives focal adhesion formation in fibroblasts. Arch
Biochem Biophys 374: 66–72.
Supporting Information
Additional Supporting Information may be found in the
online version of this article:
Fig. S1. Survival curve of DBA/2 mice (n = 10) challenged
intraperitoneally (i.p.) with 5� 106 spores of Bacillus an-
thracis Sterne strain 34F2 (open circles) or nontoxigenic
delta Sterne strain (closed circles).
Please note: Wiley-Blackwell is not responsible for the
content or functionality of any supporting materials sup-
plied by the authors. Any queries (other than missing
material) should be directed to the corresponding author
for the article.
FEMS Immunol Med Microbiol 54 (2008) 309–318c� 2008 George Mason UniversityNational Center for BiodefenseJournal compilation c� 2008 Federation of European Microbiological SocietiesPublished by Blackwell Publishing Ltd.
318 M.-C. Chung et al.
Dow
nloaded from https://academ
ic.oup.com/fem
spd/article/54/3/309/512730 by guest on 04 July 2022




















