
A heat shock protein70 fusion protein with α1-antitrypsin in plasma of Type 1 diabetic subjects
Upload
independentCategory
view
2download
0
of April 28, 2016.This information is current as
Polymorphonuclear Neutrophils1-Protease Inhibitor at the Surface of Human
αInhibition of Neutrophil Elastase by
Juliano and Francis GauthierBrice Korkmaz, Sylvie Attucci, Marie-Lise Jourdan, Luiz
http://www.jimmunol.org/content/175/5/3329doi: 10.4049/jimmunol.175.5.3329
2005; 175:3329-3338; ;J Immunol
Referenceshttp://www.jimmunol.org/content/175/5/3329.full#ref-list-1
, 21 of which you can access for free at: cites 58 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2005 by The American Association of9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on April 28, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from
by guest on April 28, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

Inhibition of Neutrophil Elastase by �1-Protease Inhibitor atthe Surface of Human Polymorphonuclear Neutrophils1
Brice Korkmaz,* Sylvie Attucci,* Marie-Lise Jourdan,† Luiz Juliano,‡ and Francis Gauthier2*
The uncontrolled proteolytic activity in lung secretions during lung inflammatory diseases might be due to the resistance ofmembrane-bound proteases to inhibition. We have used a new fluorogenic neutrophil elastase substrate to measure the activity offree and membrane-bound human neutrophil elastase (HNE) in the presence of �1-protease inhibitor (�1-Pi), the main physio-logical inhibitor of neutrophil serine proteases in lung secretions. Fixed and unfixed neutrophils bore the same amounts of activeHNE at their surface. However, the HNE bound to the surface of unfixed neutrophils was fully inhibited by stoichiometric amountsof �1-Pi, unlike that of fixed neutrophils. The rate of inhibition of HNE bound to the surface of unfixed neutrophils was the sameas that of free HNE. In the presence of �1-Pi, membrane-bound elastase is almost entirely removed from the unfixed neutrophilmembrane to form soluble irreversible complexes. This was confirmed by flow cytometry using an anti-HNE mAb. HNE activityrapidly reappeared at the surface of HNE-depleted cells when they were triggered with the calcium ionophore A23187, and thisactivity was fully inhibited by stoichiometric amounts of �1-Pi. HNE was not released from the cell surface by oxidized, inactive�1-Pi, showing that active inhibitor is required to interact with active protease from the cell surface. We conclude that HNEactivity at the surface of human neutrophils is fully controlled by �1-Pi when the cells are in suspension. Pericellular proteolysiscould be limited to zones of contact between neutrophils and subjacent protease substrates where natural inhibitors cannotpenetrate. The Journal of Immunology, 2005, 175: 3329–3338.
H uman neutrophil elastase (HNE)3 plays a pivotal role inthe pathophysiology of acute lung injury. Its concentra-tion is increased in clinical and animal models of the
disease, and typical symptoms of lung injury are produced by theadministration of HNE, which are reduced by inhibiting HNE (re-viewed in Refs. 1–3). HNE is released from neutrophils at inflam-matory sites when they are stimulated by a variety of compounds,including cytokines and chemoattractants such as TNF-�, IL-8,and fMLP (4). There is now a growing body of evidence that HNE,like related serine proteases from primary granules, remains inlarge part bound to the external surface of the plasma membrane,where it cleaves biologically relevant and synthetic substrates (5).Released, free proteases become pre-eminent upon further activa-tion and subsequent cell lysis, as in the expectorations of cysticfibrosis patients and those suffering from severe acute lung injurysuch as acute respiratory distress syndrome (6). However, the reg-
ulation of proteolytic activity at inflammatory sites under condi-tions of mild neutrophil-dependent inflammation mainly concernsmembrane-bound serine proteases that are exposed at the cell sur-face after mobilization of their primary granules. The extracellularmilieu contains excess concentrations of high affinity protease in-hibitors under these conditions, and these should normally controlproteolytic activity in the pericellular environment. Several mech-anisms have been proposed to explain the prolonged proteolyticactivity that remains in the presence of inhibitors, including theproteolytic or oxidative inactivation of inhibitors (7), the adher-ence of cells and proteases to physiological substrates (8–11), andthe resistance of membrane-bound proteases to naturally occurringprotein inhibitors (4, 5, 12). Whereas it has been clearly shown thatprotease inhibitors, and especially �1-protease inhibitor (�1-Pi),confine proteolytic activity to the pericellular environment andhave little effect on the breakdown of proteins that are in closecontact with neutrophils (9), the persistence of proteolytic activityat the cell surface, because the access of proteinase inhibitors isrestricted by steric hindrance, is more controversial. Although ithas been shown that large inhibitors inhibit proteolytic activity atthe cell surface less efficiently than do those of lower Mr (4), mem-brane-bound proteases are fully active on protein substrates of highMr (13–15). Most experiments designed to measure proteolyticactivity at the surface of purified neutrophils have used fixed ac-tivated cells to increase the HNE concentration at the cell surface,as this improves the detection of active protease and avoids theleakage of protease from the cell (5). New fluorogenic HNE sub-strates that are more specific and more sensitive than those previ-ously used have been recently prepared (16–18). These are moresuitable for quantifying HNE and studying the regulation of itsactivity by inhibitors at the surface of quiescent and triggered neu-trophils. This is particularly important for developing anti-inflammatory treatment for lung inflammatory diseases using in-hibitors designed to target both free and membrane-bound forms ofactive proteases at inflammatory sites (19–21). Several attempts
*University Francois Rabelais, Institut National de la Sante et de la Recherche Medi-cale Unite 618 Proteases et Vectorisation Pulmonaires, and Institut Federatif de Re-cherche 135 Imagerie Fonctionnelle; †Institut National de la Sante et de la RechercheMedicale Equipe 211 Nutrition et Cancer, Tours, France; and ‡Universidade Federal,Departamento de Biofısica, Escola Paulista de Medicina, Sao Paulo, Brazil
Received for publication March 4, 2005. Accepted for publication May 18, 2005.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported in France by Vaincre la Mucoviscidose and by the Fon-dation pour la Recherche Medicale, and in Brazil by Fundacao de Amparo a Pesquisado Estado de Sao Paulo and Conselho Nacional de Desenvolvimento Cientıfico eTecnologico.2 Address correspondence and reprint requests to Dr. Francis Gauthier, Institut Na-tional de la Sante et de la Recherche Medicale Unite 618, Faculty of Medicine, 10Boulevard Tonnelle, 37032 Tours Cedex, France. E-mail address: [email protected] Abbreviations used in this paper: HNE, human neutrophil elastase; �1-Pi, �1-pro-tease inhibitor; Abz, ortho-aminobenzoic acid; BAL, bronchoalveolar lavage; BALF,BAL fluid; EDDnp, N-(2,4-dinitrophenyl) ethylenediamine; mHNE, membrane-bound HNE; PMN, polymorphonuclear leukocyte; Pr3, protease 3; SLPI, secretoryleukocyte protease inhibitor.
The Journal of Immunology
Copyright © 2005 by The American Association of Immunologists, Inc. 0022-1767/05/$02.00
by guest on April 28, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

have been made to treat patients with cystic fibrosis with aerosol-ized or inhaled inhibitors, but the results have not been conclusivepartly due to their limited access to plugged areas (22–24). Thepartition of free and membrane-associated proteases in lung in-flammatory secretions and their relative sensitivities to inhibitorsappears to be an important factor influencing treatment design. Wehave compared the activities of free HNE and membrane-boundHNE (mHNE); studied the interaction of �1-Pi, the main serineprotease inhibitor in lung fluids, with freshly purified, quiescent,and triggered human neutrophils; and examined the fate of �1-Pi-HNE complexes after cell-inhibitor interaction.
Materials and MethodsMaterials
Human neutrophil elastase (EC 3.4.21.37) and �1-Pi were obtained fromAthens Research & Technology. Trypsin (EC 3.4.21.4), A23187, and per-oxidase-conjugated goat polyclonal anti-rabbit IgG Abs and anti-mouseF(ab�)2 FITC goat Abs and fMLP were from Sigma-Aldrich. Secretoryleukocyte protease inhibitor (SLPI) and TNF-� were from R&D Systems.The specific elastase inhibitor EPI-hNE4 was a kind gift of F. Saudubray(Debiopharm, Lausanne, Switzerland). N,N-dimethylformamide was fromMerck; N-chlorosuccinimide was from Valeant Pharmaceuticals. Polymor-phprep and Lymphoprep were from Nycomed. Fluorescein-5-maleimidewas from Interchim. Polyclonal anti-�1-Pi Abs were purchased from DadeBehring. Mouse IgG1 FITC, CD16b FITC, and CD63 PE mAbs were fromBeckman Coulter. Anti-HNE mAbs were from Biogenesis. All other re-agents were of analytical grade.
Isolation of blood polymorphonuclear leukocytes (PMNs) andbronchoalveolar lavage (BAL) fluid (BALF) collection
Human PMNs were purified from 8-ml samples of peripheral blood col-lected from healthy volunteers into EDTA-containing tubes essentially aspreviously reported (25). The PMN pellet recovered after lysing the eryth-rocytes was washed twice in PBS containing 4 mM EGTA and 1% (w/v)BSA. Purified PMNs were kept at room temperature in this buffer withgentle shaking and washed with PBS containing 4 mM EGTA just beforeuse. Cell viability was checked by trypan blue exclusion.
PMN were activated by suspending �3 � 106 cells/ml in PBS contain-ing 1 mM CaCl2 and 1 mM MgCl2 and incubating them with A23187 (1�M final) for 15 min at 37°C. Alternatively, cells were primed with TNF-�(10 ng/ml final) for 15 min at 37°C and then activated by fMLP (10�8 Mfinal) for 30 min at 37°C. Quiescent or triggered PMNs were fixed byincubating them for 3 min on ice with 3% (w/v) paraformaldehyde and0.25% (v/v) glutaraldehyde, as reported by Owen et al. (5). The PMNswere analyzed by flow cytometry and scanning electron microscopy,washed twice with PBS-EGTA, suspended at �3 � 106 cells/ml in thesame buffer, and used for enzyme assays. BALF from patients with acuterespiratory distress syndrome that contained �80% PMNs were centri-fuged at 1000 � g for 10 min, and cell fraction was analyzed in the sameway as purified blood PMNs for their mHNE activity.
Patients were referred to the Department of Pneumology or to the Med-ical Intensive Care Unit of Bretonneau University Hospital. They fulfilledthe diagnostic criteria for acute respiratory distress syndrome, and under-went BAL for diagnostic or therapeutic investigation. The decision to un-dertake BAL was taken by the clinicians. The protocol was approved by thelocal institution Ethics Committee. BAL was performed, as previouslyreported (26). BALF samples were collected under sterile conditions,and the fifth fraction, which was not contaminated with epithelial orbronchial cells, was filtered and retained for biochemical and enzymaticanalysis (26).
Flow cytometry analyses
Flow cytometry was performed on a Corixa Epics Elite ESP flow cytom-eter equipped with a 488-nm argon laser. The forward and side scatteringof each sample were measured for at least 10,000 events. Samples con-tained �99% PMNs, no monocytes, and �1% lymphocytes. The percent-age of activated PMNs after purification was checked by double labelingand incubating 5 � 105 PMNs in 200 �l of final with 20 �l of mAbsanti-CD16b FITC and 20 �l of mAbs anti-CD63 PE for 20 min at roomtemperature. Controls were treated under the same conditions by incubat-ing cells with 20 �l of mAbs anti-IgG1 FITC. Mixtures were centrifugedat 500 � g for 5 min; the resulting pellets were washed twice in PBS andsuspended for flow cytometry. Any mHNE Ag at the neutrophil surface
was detected before and after �1-Pi (10�9-10�8 M final) treatment, incu-bating 5 � 105 quiescent or activated, unfixed and fixed PMNs with anti-HNE mAbs (Biogenesis clone 39A) diluted 1/50, for 30 min at 4°C, fol-lowed by two washes in PBS and incubation with FITC-conjugated F(ab�)2
from goat anti-mouse IgG (diluted 1/100) for 30 min at 4°C. The experi-mental conditions for using the anti-HNE Ab were adapted from (27).
Membrane-binding analysis of �1-Pi labeled with fluorescein-5-maleimide
A solution of �1-Pi (10�4 M final) was incubated for 2 h at room temper-ature with 1 mM fluorescein-5-maleimide prepared as a 15 mM stock so-lution in dimethylformamide (28). Excess fluorescein-5-maleimide was re-moved by ultrafiltration, and the inhibitory capacity of the labeled �1-Piwas compared with that of the unlabeled inhibitor. Freshly prepared PMNs(7.5 to 9 � 105 cells), corresponding to �5 � 10�9 M HNE, were incu-bated with 5 � 10�9 M-labeled �1-Pi in 150 ml of PBS and 4 mM EGTAfor 10 min at room temperature. The PMNs were collected by centrifuga-tion at 500 � g for 5 min, and the supernatant was removed. The remainingcells were suspended in the same volume of buffer and fixed, as describedabove. Labeled cells were analyzed by flow cytometry.
Scanning electron microscopy
Approximately 1.5 � 106 cells were washed in PBS, deposited on poly-lysine-coated glass slides, and left to adhere for a few minutes. They werefixed with 1% glutaraldehyde (v/v) and 4% (w/v) paraformaldehyde in 0.1M phosphate buffer, pH 7.4, postfixed in 2% osmium tetroxide, dehydratedin a graded acetone series, dried to the critical point using carbon dioxide,and sputter coated with platinum. Fixed cells were examined in a Gemini982 Leo scanning electron microscope.
Enzyme assays
The activities of free HNE and mHNE were measured in PBS. Free HNEwas titrated with �1-Pi, the titer of which had been determined using bo-vine trypsin titrated with p-nitrophenyl-p�-guanidinobenzoate (29). Trypsinwas prepared as a 2 � 10�4 M stock solution in 100 mM Tris-HCl buffer,pH 8, 50 mM CaCl2, then used in the same buffer as HNE. Because thereis no significant �1-Pi inactivation via the substrate pathway upon inter-action with HNE (30), a 1:1 stoichiometry was assumed. mHNE activitywas quantified by comparing the rate of hydrolysis of its specific substrate(ortho-aminobenzoic acid (Abz)-APEEIMRRQ-N-(2,4-dinitrophenyl) eth-ylenediamine (EDDnp) (18)) with that of titrated HNE under the sameexperimental conditions. The concentration of Abz-APEEIMRRQ-EDDnpwas determined by measuring the absorbance at 365 nm, using E365 nm �17,300 M�1 cm�1 for EDDnp.
Unactivated and activated, unfixed or fixed PMNs (1 � 105 to 5 � 105
cells), or purified proteases used as controls, were incubated with 20 �MAbz-APEEIMRRQ-EDDnp in polypropylene microplate wells selected fortheir low binding properties (Hard-Shell Thin-Wall Microplates; MJ Re-search) at room temperature in activity buffer (10 mM PBS, pH 7.4). Thefluorescence was recorded at lex � 320 nm and lem � 420 nm using amicroplate fluorescence reader (Spectra Max Gemini; Molecular Devices)under continuous stirring.
Release of membrane-bound proteases
The release of HNE into PBS/EGTA was checked by incubating unfixedand fixed cells (3 � 106 cells/ml) in buffer for up to 1 h and measuring thepeptidase activities in supernatants cleared of cells by centrifugation for 5min at 500 � g. Cell pellets were suspended in the same buffer, and mem-brane-bound activity was measured using the same procedure. The proce-dure was repeated using the same buffer supplemented with 1.5 M NaCl.PMNs were kept for 1 h at room temperature or 24 h at 37°C under gentlestirring, collected by centrifugation at 500 � g for 15 min, and resuspendedin PBS containing 1.5 M NaCl (final). The HNE activity in the supernatantsof unfixed and fixed cells and on suspended fixed cells was measured andcompared with the activity on unfractionated treated cells.
Inhibition of mHNE by �1-Pi, SLPI, and EPI-hNE4
Unactivated and activated, unfixed or fixed PMNs (105 to 5 � 105 cells),corresponding to 0.5–1.5 � 10�9 M active purified HNE, were incubatedwith equimolar concentrations and up to a 1000-fold molar excess of �1-Pifor 15–40 min under stirring. Residual HNE activity was then measured byadding Abz-APEEIMRRQ-EDDnp (20 �M final). Progress curves for in-hibition were recorded using equimolar amounts of �1-Pi and mHNE(�10�9 M final). Inhibitor and substrate were added simultaneously toquiescent or activated PMNs. Fluorescence was recorded with the Gemini
3330 INHIBITION OF MEMBRANE-BOUND ELASTASE BY �1-Pi
by guest on April 28, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

fluorometer at room temperature. These experiments were repeated usingequimolar concentrations of mHNE and the potent, specific low Mr elastaseinhibitor EPI-hNE4, whereas a molar excess of SLPI was used to take intoaccount its 0.1 nM Ki toward HNE (31), and its putative inhibition ofcathepsin G.
Oxidation of �1-Pi
The �1-Pi (40 �M) was oxidized by a 20-fold molar excess of N-chloro-succinimide (32), and excess oxidant was removed by ultrafiltration. Theinhibitory capacity of the oxidized �1-Pi was compared with that of nativeinhibitor.
Activation of PMNs by the calcium ionophore during enzymeassay
The HNE activity at the surface of quiescent, unfixed cells was totallyinhibited by �1-Pi. The cells were resuspended in PBS containing 1 mMCaCl2 and 1 mM MgCl2 in the microplate wells, then activated with thecalcium ionophore A23187. The reappearance of HNE activity was mon-itored continuously at room temperature, and further inhibition experi-ments with �1-Pi were conducted as above.
Electrophoresis and Western blotting
PMNs (�70,000 cells/40 �l) corresponding to an HNE concentration of�5 � 10�8 M and similar concentrations of free HNE were incubated with�1-Pi (5 � 10�8 M final) in PBS and 4 mM EGTA, for 2 min with gentlestirring, at room temperature. Cell mixtures were then centrifuged at 500 �g for 5 min. The supernatants were recovered, and the formation of HNE-�1-Pi complexes was analyzed by SDS-PAGE on 12% acrylamide/bisac-rylamide gels. The resolved proteins were transferred to nitrocellulosemembranes, and the complexes were detected by incubation with rabbitpolyclonal anti-�1-Pi Abs (diluted 1/1,000), followed by peroxidase-cou-pled goat polyclonal anti-rabbit IgG Abs (diluted 1/15,000), using the Re-naissance Plus kit (Valeant Pharmaceuticals).
ResultsElastase activity in suspensions of unfixed and fixed PMNs
HNE activity has been detected at the surface of activated neutro-phils that were fixed to improve stability and avoid intracellular
proteases leaking out during the time cells were reacted with sub-strates and/or inhibitors (5). Analysis of unactivated and activatedneutrophils by flow cytometry before and after fixation indicatedthat anti-CD16b Abs reacted with their surface Ag at the surface ofunfixed cells, but not at that of fixed cells (Fig. 1), while anti-CD63Abs, used as a marker of neutrophil activation, reacted less effi-ciently with fixed activated neutrophils (Fig. 1). This suggests thatthe accessibility of epitopes at the cell surface is altered by fixa-tion. The morphology of the surface of fixed neutrophils is signif-icantly different from that of unfixed neutrophils (Fig. 1). Wetherefore measured the activities of HNE at the surface of fixedand unfixed neutrophils (2–4 � 105 cells in 150 �l final) beforeand after activation by the calcium ionophore A23187 or by fMLPusing a specific, sensitive elastase substrate (Abz-APEEIMRRQ-EDDnp) that allows the measurement of HNE at the cell surfaceeven in the presence of the closely related protease 3 (18). Fixationdid not alter the HNE activity of either quiescent or activated neu-trophils. Unfixed and fixed neutrophils activated with the iono-phore had 3–5 times greater activity than unactivated cells, whilethe activity of fMLP-activated cells was 1.5–2 times greater. Therecovery of fluorescence after the total hydrolysis of the substrateby free HNE and mHNE was the same. Because an oxidized Metin the substrate sequence prevents further cleavage by HNE (16),there can have been no significant oxidation of the Met residue inthe substrate by oxidants released from activated neutrophils. Inagreement with these data, flow cytometry indicated that 10–30%of unactivated, unfixed PMNs bore HNE Ag at their surface,whereas all of the activated PMNs were labeled with the anti-HNEmAbs (Fig. 2B). This correlates well with the 3- to 5-fold higherHNE activity using activated cells. The fluorescence at the surfaceof fixed cells differed from that of unfixed cells, but the fluores-cence with activated cells was still greater (Fig. 2C).
FIGURE 1. Flow cytometry analy-sis of quiescent (A and B) and triggered(C and D) purified human PMNs before(A and C) and after (B and D) fixationwith glutaraldehyde/formaldehyde, asrevealed with anti-CD16b and anti-CD63 Abs. The CD16b on quiescent,unfixed PMNs are labeled (A), but notthose on fixed cells (B). Triggered, un-fixed PMNs are labeled with both anti-CD16b and anti-CD63 Abs (C),whereas only some triggered, fixedcells are labeled by anti-CD63 Abs (D).The typical morphologies of quiescentand triggered PMNs before and afterfixation by scanning electron micros-copy are shown in insets for eachanalysis.
3331The Journal of Immunology
by guest on April 28, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

HNE stability at the surface of unfixed neutrophils
We checked the stability of the HNE at the surface of unfixedneutrophils by recording activities in supernatants and suspendedcells. Approximately 20% of the mHNE activity was found insupernatants of quiescent PMNs in PBS/EGTA. This fraction re-mained stable for at least 1 h (Fig. 3A), suggesting that mHNE andfree HNE are in equilibrium under these conditions. This is sup-ported by our finding that there was no increase in the percentageof free HNE using unfixed, activated PMNs despite the increasedHNE. Less than 10% of total HNE was found in the supernatant offixed, quiescent, or triggered PMNs treated in the same way. In-cubating unfixed cells in the PBS/EGTA buffer supplemented with
1.5 M NaCl for 60 min released all of the active mHNE (Fig. 3A),whereas only 50% of the HNE was released from the surface offixed neutrophils under the same conditions (Fig. 3B). The totalHNE activities in supernatants and suspended fixed cells (unfixedcells cannot be resuspended because they form aggregates aftersalt treatment and centrifugation) were the same as that initiallyfound at the cell surface (Fig. 3B). Thus, enzymatically activeHNE is bound mostly via electrostatic bonds to the surface ofunfixed cells, and its activity toward synthetic substrates is notaltered by its binding to the cell membrane. The percentage ofHNE released from fixed PMNs by salt treatment remained un-changed after incubation overnight at 37°C (Fig. 3B), suggestingdifferent modes of protease binding to cell surfaces that do not altercatalytic activity. We used the rate of hydrolysis of the fluorogenicsubstrate by free, titrated HNE to calculate that the concentrationof active HNE on quiescent, fixed, or unfixed purified PMNs was�1 nM when 106 cells were incubated in 1 ml of final.
Inhibition of mHNE activity of unfixed and fixed neutrophils
Identical amounts of active HNE were found at the surfaces ofunfixed and fixed neutrophils, suggesting that cell fixation does not
FIGURE 2. Flow cytometry analysis of mHNE in unfixed and fixedblood neutrophils. Neutrophils were labeled with an anti-HNE mAb re-vealed by FITC-conjugated anti-mouse IgG to visualize cell surface HNE.Twenty percent of quiescent unfixed neutrophils and 100% of activatedunfixed neutrophils are labeled with the mAb anti-HNE before and afteractivation with the A23187 calcium ionophore in this representative ex-periment (B). All fixed, activated cells were also labeled, although the basalfluorescence at the surface of fixed quiescent cells (C) differs from that at thesurface of unfixed cells (B). Control unfixed and fixed cells are shown in A.
FIGURE 3. HNE activity at the surface and in the supernatant of un-fixed (A) and fixed (B) quiescent PMNs. Approximately 20% of the totalHNE is released spontaneously when unfixed cells are suspended in PBS/EGTA for at least 60 min, whereas 100% HNE is found in supernatant afterNaCl treatment (A). Fixed cells release �10% HNE into the supernatantwhen incubated in PBS/EGTA, and �50% when incubated with 1.5 MNaCl, even after 24 h (B).
3332 INHIBITION OF MEMBRANE-BOUND ELASTASE BY �1-Pi
by guest on April 28, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from
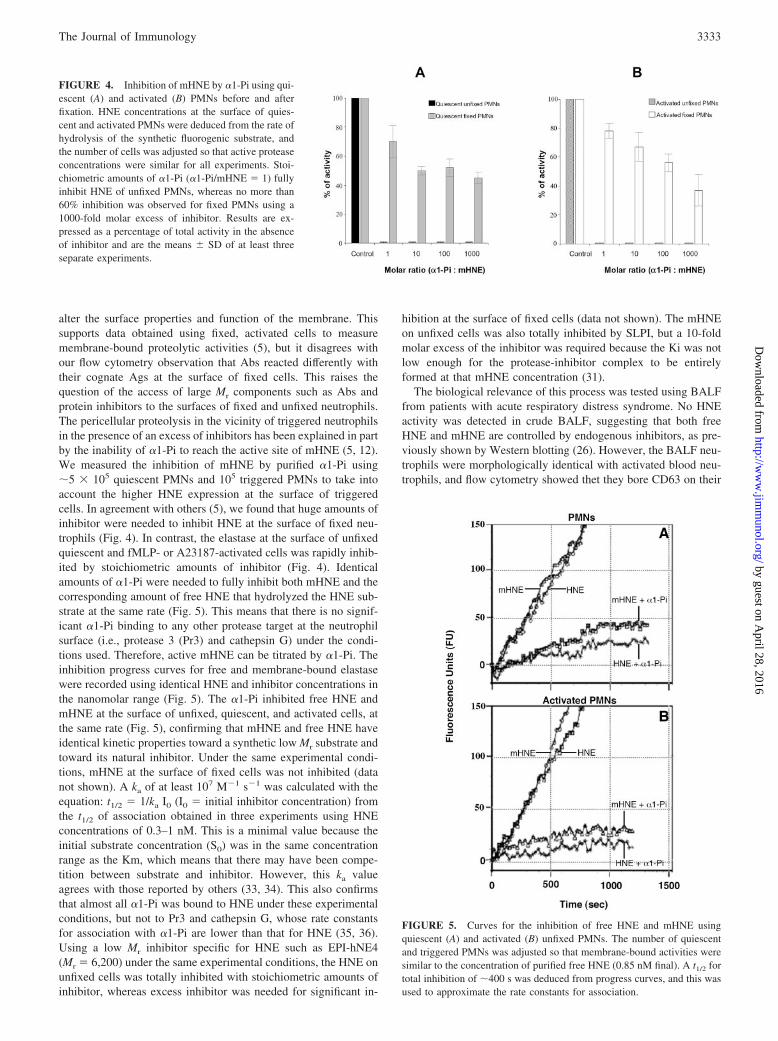
alter the surface properties and function of the membrane. Thissupports data obtained using fixed, activated cells to measuremembrane-bound proteolytic activities (5), but it disagrees withour flow cytometry observation that Abs reacted differently withtheir cognate Ags at the surface of fixed cells. This raises thequestion of the access of large Mr components such as Abs andprotein inhibitors to the surfaces of fixed and unfixed neutrophils.The pericellular proteolysis in the vicinity of triggered neutrophilsin the presence of an excess of inhibitors has been explained in partby the inability of �1-Pi to reach the active site of mHNE (5, 12).We measured the inhibition of mHNE by purified �1-Pi using�5 � 105 quiescent PMNs and 105 triggered PMNs to take intoaccount the higher HNE expression at the surface of triggeredcells. In agreement with others (5), we found that huge amounts ofinhibitor were needed to inhibit HNE at the surface of fixed neu-trophils (Fig. 4). In contrast, the elastase at the surface of unfixedquiescent and fMLP- or A23187-activated cells was rapidly inhib-ited by stoichiometric amounts of inhibitor (Fig. 4). Identicalamounts of �1-Pi were needed to fully inhibit both mHNE and thecorresponding amount of free HNE that hydrolyzed the HNE sub-strate at the same rate (Fig. 5). This means that there is no signif-icant �1-Pi binding to any other protease target at the neutrophilsurface (i.e., protease 3 (Pr3) and cathepsin G) under the condi-tions used. Therefore, active mHNE can be titrated by �1-Pi. Theinhibition progress curves for free and membrane-bound elastasewere recorded using identical HNE and inhibitor concentrations inthe nanomolar range (Fig. 5). The �1-Pi inhibited free HNE andmHNE at the surface of unfixed, quiescent, and activated cells, atthe same rate (Fig. 5), confirming that mHNE and free HNE haveidentical kinetic properties toward a synthetic low Mr substrate andtoward its natural inhibitor. Under the same experimental condi-tions, mHNE at the surface of fixed cells was not inhibited (datanot shown). A ka of at least 107 M�1 s�1 was calculated with theequation: t1/2 � 1/ka I0 (I0 � initial inhibitor concentration) fromthe t1/2 of association obtained in three experiments using HNEconcentrations of 0.3–1 nM. This is a minimal value because theinitial substrate concentration (S0) was in the same concentrationrange as the Km, which means that there may have been compe-tition between substrate and inhibitor. However, this ka valueagrees with those reported by others (33, 34). This also confirmsthat almost all �1-Pi was bound to HNE under these experimentalconditions, but not to Pr3 and cathepsin G, whose rate constantsfor association with �1-Pi are lower than that for HNE (35, 36).Using a low Mr inhibitor specific for HNE such as EPI-hNE4(Mr � 6,200) under the same experimental conditions, the HNE onunfixed cells was totally inhibited with stoichiometric amounts ofinhibitor, whereas excess inhibitor was needed for significant in-
hibition at the surface of fixed cells (data not shown). The mHNEon unfixed cells was also totally inhibited by SLPI, but a 10-foldmolar excess of the inhibitor was required because the Ki was notlow enough for the protease-inhibitor complex to be entirelyformed at that mHNE concentration (31).
The biological relevance of this process was tested using BALFfrom patients with acute respiratory distress syndrome. No HNEactivity was detected in crude BALF, suggesting that both freeHNE and mHNE are controlled by endogenous inhibitors, as pre-viously shown by Western blotting (26). However, the BALF neu-trophils were morphologically identical with activated blood neu-trophils, and flow cytometry showed thet they bore CD63 on their
FIGURE 4. Inhibition of mHNE by �1-Pi using qui-escent (A) and activated (B) PMNs before and afterfixation. HNE concentrations at the surface of quies-cent and activated PMNs were deduced from the rate ofhydrolysis of the synthetic fluorogenic substrate, andthe number of cells was adjusted so that active proteaseconcentrations were similar for all experiments. Stoi-chiometric amounts of �1-Pi (�1-Pi/mHNE � 1) fullyinhibit HNE of unfixed PMNs, whereas no more than60% inhibition was observed for fixed PMNs using a1000-fold molar excess of inhibitor. Results are ex-pressed as a percentage of total activity in the absenceof inhibitor and are the means � SD of at least threeseparate experiments.
FIGURE 5. Curves for the inhibition of free HNE and mHNE usingquiescent (A) and activated (B) unfixed PMNs. The number of quiescentand triggered PMNs was adjusted so that membrane-bound activities weresimilar to the concentration of purified free HNE (0.85 nM final). A t1/2 fortotal inhibition of �400 s was deduced from progress curves, and this wasused to approximate the rate constants for association.
3333The Journal of Immunology
by guest on April 28, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

cell surface (Fig. 6). HNE activity was detected in cells that hadbeen centrifuged and equilibrated in PBS, which indicates that newHNE molecules were exposed at the cell surface of BALF PMNs.The concentration of newly exposed HNE was similar (�1 nMusing 1–1.5 � 106 cells/ml) to that of the mHNE at the surface ofpurified blood PMNs. The mHNE in the buffered cell fraction ofBALF was totally inhibited by similar or slightly higher concen-trations of �1-Pi, confirming the biological relevance of thisprocess.
The fate of �1-Pi-HNE complexes
The formation of the serpin-protease complex is complex, involv-ing pole-to-pole displacement of the protease in the complex dueto the cleavage of the reactive-center loop and its full insertion intothe A-sheet of the serpin (37, 38). Thus, �1-Pi might remain or notat the cell surface after complex formation. We used Western blot-ting to look for �1-Pi-HNE complexes in the supernatants of cellsincubated with �1-Pi, and flow cytometry to detect fluorescein-labeled �1-Pi at the neutrophil surface. Mixtures of neutrophilsand �1-Pi were prepared containing the amount of �1-Pi requiredto fully inhibit mHNE activity. They were centrifuged, and thesupernatants were assayed for �1-Pi-protease complexes by SDS-PAGE, followed by Western blotting using anti-�1-Pi Abs. Mostof the �1-Pi initially present in the neutrophil suspension hadformed soluble complexes (Fig. 7A), which suggests that �1-Piremoved the bound protease from the cell surface during the for-mation of the irreversible complex. Oxidized �1-Pi, which inhibitsfree HNE very poorly, did not release HNE from the cell surface.In keeping with these data, flow cytometry showed no significantfluorescence on unfixed neutrophils incubated with fluorescentlylabeled �1-Pi, which demonstrates that no active protease remainsat the cell surface (Fig. 7B). We also observed a nonspecific bind-ing of fluorescent �1-Pi at the surface of fixed cells that does notresult in any significant inhibition of mHNE and confirms that nosignificant amount of HNE is released by �1-Pi from the surface offixed cells (Fig. 7B).
Because fluorescent �1-Pi only reacts with active HNE, we usedanti-HNE mAbs to investigate the fate of total HNE Ag at the cellsurface by flow cytometry after treating cells with �1-Pi, followedby anti-HNE mAbs. After treatment with �1-Pi, most of fluores-cence disappeared from the surface of unfixed, but not of fixed,activated neutrophils, indicating that mHNE is present essentially(�80%) as an active protease at the surface of unfixed activatedPMNs (Fig. 8).
As incubation with �1-Pi removed all active HNE from the cellsurface, we looked at the time-dependent recovery of elastase ac-tivity on suspended HNE-depleted cells. No significant HNE ac-tivity was recovered at the surface of unstimulated cells incubatedwith the specific HNE substrate for 1 h at 25°C (Fig. 9A). How-ever, activity reappeared at the surface of HNE-depleted cells thathad been activated with the calcium ionophore A23187 (Fig. 9Bb);it was comparable to that at the surface of control, triggered cells
FIGURE 6. Flow cytometry analysis of BALF PMNs using anti-CD16band anti-CD63 Abs. Anti-CD63 labeling demonstrates that almost all cellsare activated. Inset, Shows BALF PMN morphology by scanning electronmicroscopy.
FIGURE 7. A, Immunoblot analysis of �1-Pi (5 � 10�8 M final) afterinteraction with stoichiometric amount of free HNE (b), supernatants ofquiescent PMNs whose concentration was first adjusted so that their initial,membrane-bound HNE concentration was 5 � 10�8 M final, then incu-bated for 2 min with a stoichiometric amount of �1-Pi (c), supernatants ofthe same number of fixed PMNs incubated as in b (d). Purified �1-Pi (Mr �52 kDa) is shown in a. The band at �80 kDa indicates the irreversible�1-Pi-HNE complex, and that of lower Mr in c corresponds to a degradedform of the inhibitor. Most of the �1-Pi is found as a soluble complex afterincubation with unfixed PMNs (c), but not after incubation with fixedPMNs (d). B, Flow cytometry analysis of unfixed PMNs after they havebeen incubated as in c with fluorescently labeled �1-Pi (�1-Pi�) (blackline), and of fixed PMNs incubated as in d (dashed line). The gray linecorresponds to control PMNs. No fluorescence remains on unfixed PMNsthat have been first incubated with �1-Pi�, which further demonstrates thatall active protease has been removed from the cell surface. Unspecificbinding of �1-Pi� was observed at the surface of fixed cells, resulting in aminimal release of HNE, as observed in d, and no inactivation of themembrane-bound protease.
3334 INHIBITION OF MEMBRANE-BOUND ELASTASE BY �1-Pi
by guest on April 28, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

that had not been incubated with �1-Pi (Fig. 9B, a and b). The newactivity at the surface of depleted then activated cells was stoi-chiometrically inhibited by adding further �1-Pi (Fig. 9B, c and d).
There was a substrate-independent increase in fluorescence (Fig.9Be), probably due to the in situ activation of neutrophils thatrelease significant amounts of fluorescent NADPH, as previouslyreported when cells were activated by phorbol myristate or by thechemotactic peptide fMLP (39).
DiscussionInflammatory lung diseases are due, at least in part, to the impairedregulation of protease activities by endogenous inhibitors at in-flammatory sites. The local binding capacity of these inhibitorsmay be overwhelmed by the proteases brought by infiltrating neu-trophils and other inflammatory cells including bacteria (40)and/or by the oxidative inactivation of critical Met residues byhalogenated oxidants (41) and by proteolysis (42). However, un-controlled proteolytic activity is not due solely to the overwhelm-ing of the inhibitory capacity at inflammatory sites. Neutrophilproteases preserve their catalytic activity in the presence of pro-tease inhibitors in several ways (reviewed in Ref. 4), such as thetight binding of neutrophil proteases to their target substrate, ad-herence of neutrophils to the extracellular matrix with the forma-tion of an area of preserved proteolytic activity (43), and the re-sistance of membrane-bound proteases to inhibition byendogenous inhibitors (5). A more complete understanding ofthese mechanisms for preserving protease activity in the pericel-lular environment is necessary before protease inhibitors can beused as therapeutic tools to modulate the tissue destruction asso-ciated with acute inflammatory diseases. Whereas it has beenclearly demonstrated that the access of protease inhibitors to thezone of contact between adherent neutrophils and subjacent sub-strates is impaired (43), their access to membrane-bound proteasesin an environment replete with biologically active inhibitors ismore controversial. A negative correlation was reported betweenthe size of the inhibitor and its effectiveness against membrane-bound serine proteases, but these results were obtained with fixed,activated neutrophils (5, 44) or with purified membranes (12).Fixed and unfixed activated neutrophils retain their cell surfaceprotease activity on synthetic low Mr substrates and on proteinsubstrates (5, 13, 15), but fixation can alter their accessibilityand/or conformation. We have shown a difference in the surfacemorphologies of fixed and unfixed neutrophils by scanning elec-tron microscopy, and different reactivities of Abs to membraneAgs by flow cytometry. This agrees with findings demonstrating achange in surface membrane and protein charge by chemicalfixation (45).
FIGURE 9. A, Hydrolysis of Abz-APEEIMRRQ-EDDnp by quiescent PMNs (0.7 � 105 cells/well cor-responding to an HNE concentration of �0.3 nM) be-fore (a) and after (b) adding �1-Pi. The amount ofinhibitor was deduced from the titration curve (inset).B, Progress curves for the inhibition of mHNE aftercells were cleared of their surface HNE by incubationwith stoichiometric amounts of �1-Pi and then acti-vated in situ by the calcium ionophore A23187. Curve(b) shows the kinetics of hydrolysis of the fluorogenicsubstrate by newly expressed HNE on triggered cells;c, by triggered cells in the presence of 0.6 nM �1-Pi;and d, 1.5 nM �1-Pi. e, Shows the spontaneous releaseof fluorescence by triggered cells in the absence of sub-strate, demonstrating the almost total inhibition of newHNE at the surface of triggered cells by stoichiometricamounts of inhibitor. Curve (a) shows the hydrolysisby activated PMNs that have intact mHNE.
FIGURE 8. Flow cytometry analysis of quiescent unfixed PMNs (A)and of activated unfixed human PMNs before (B) and after (C) treatmentwith �1-Pi, as revealed with anti-HNE mAbs. In keeping with kinetic data,�1-Pi treatment results in a large decrease of fluorescence from the surfaceof unfixed, activated (C) cells, but not of fixed activated cells (inset of C).PMN activation was controlled by cell surface CD63 expression.
3335The Journal of Immunology
by guest on April 28, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

We therefore compared the HNE activity at the surface of qui-escent, unfixed neutrophils with that on fixed neutrophils before orafter activation by a calcium ionophore or by fMLP. This waspossible because we used the specific fluorogenic HNE substrateAbz-APEEIMRRQ-EDDnp that is not cleaved by Pr3 and is sen-sitive enough to detect rapidly subnanomolar concentrations offree HNE and mHNE (18). HNE activity is resistant to inhibitionat the surface of fixed cells, as reported (5), but mHNE is fullyinhibited by stoichiometric amounts of �1-Pi when quiescent oractivated unfixed cells are kept in suspension. The same result wasobtained using activated neutrophils isolated from BALF from pa-tients with acute respiratory distress syndrome that were morpho-logically similar to the activated blood neutrophils.
We also found that the inhibition rate of mHNE is similar to thatmeasured with free HNE used at the same concentration, and thatthe HNE-�1-Pi complexes are all found in the soluble fraction.This probably favors their rapid elimination via the receptors toserpin-protease complexes (see Ref. 46 for review). But the mech-anism of interaction between �1-Pi and mHNE is not yet under-stood. The �1-Pi could either bind to HNE at the cell surface andextract the membrane-bound protease to form the irreversible com-plex, or it could displace an equilibrium between free and mem-brane-bound protease by reacting only with the free enzyme. Thepeculiar way that serpins react with their target proteases (38)might explain that complexes do not remain at the cell surface andare found in the soluble fraction, which gives support to the formerhypothesis, but the spontaneous release of mHNE from purifiedneutrophils after they have been centrifuged and equilibrated inbuffer agrees with the latter.
Our finding that stoichiometric amounts of �1-Pi almost com-pletely inhibit mHNE agrees with our recent finding that the in-hibitor binds essentially to HNE, and not to Pr3 or cathepsin G,although these potential targets for this inhibitor are also present atthe cell surface (26). This is explained, at least in part, by thedifferent rate constants for association and by the two-step mech-anism of Pr3 inhibition that could favor HNE binding when com-petition occurs (47).
Incubating purified neutrophils with 1.5 M NaCl releases allmHNE activity from the surface of quiescent and triggered, un-fixed neutrophils. This agrees with the results obtained by Kolk-enbrock et al. (14) using membranes of activated neutrophils. Butonly part of the mHNE can be released from fixed neutrophilsunder the same conditions, further demonstrating that fixation al-ters cell surface properties. Exactly how HNE binds to the neu-trophil membrane is not yet elucidated, but previously reporteddata suggest that it differs from that of related serine proteasesfrom neutrophil primary granules (48). Nevertheless, all mHNEthat is enzymatically active on peptide substrates can be inhibitedby �1-Pi; however, it is bound. Our flow cytometry studies indi-cate that a major part of the fluorescence revealed by anti-HNEmAbs is removed from the cell surface after �1-Pi treatment. Theminor part of HNE that remains at the cell surface after �1-Pitreatment is inactive or has an inaccessible active site so that itcannot be removed by its inhibitor. At least part of mHNE couldbe bound to CR3, the major adhesion protein of PMNs, through amechanism that is dependent on its enzymatic activity (49) andcould result in the inactivation of the protease (50).
Oxidation of inhibitors by reactive oxygen species released fromactivated neutrophils may also preserve membrane-bound proteo-lytic activity by inactivating lung protease inhibitors that have anoxidation-sensitive Met residue at their inhibitory site. We show inthis study that �1-Pi inhibits free HNE and mHNE stoichiometri-cally at the same rate. This suggests that the inhibitor is not inac-tivated, even in the immediate cell environment, because oxidized
�1-Pi inhibits HNE with a 2000-fold lower Ka (51). Similarly, thehydrolysis of the HNE-specific substrate Abz-APEEIMDRQ-EDDnp, which also contains a critical Met residue at its P1� siteand is no longer hydrolyzed once it is oxidized, remains unaltered(16). This agrees with the results of Chamba et al. (11), showingthat oxidants have little influence on extracellular proteolysis byneutrophils. The absence of Met oxidation from both the substrateand inhibitor may be because the neutrophils were transferred tothe reaction mixture after they had been activated and washed. Theneutrophils were fluorescent when they were activated directly inthe reaction mixture (Fig. 9), indicating the release of NADPH, anessential factor for the production of reactive oxygen species (52).However, there was no significant inactivation of �1-Pi or theHNE substrate, under our experimental conditions, during the timeneeded for total inhibition of mHNE.
The HNE activity of neutrophils whose mHNE has been com-pletely removed by �1-Pi may rapidly reappear after they are trig-gered. Newly exposed HNE molecules that remain bound to thecells as well as secreted HNE can be immediately inhibited by any�1-Pi in the cell suspension. Hence, the proteolytic activity ofneutrophil serine proteases is essentially due to adherent neutro-phils that generate a protected pericellular environment, wherethey contact the underlying physiological substrate, as long as ac-tive inhibitors, especially �1-Pi, are present in the pericellular en-vironment. In contrast, the activity at the surface of nonadherentcells is controlled by the inhibitors in the fluid phase. This is con-firmed by our observation that there is no HNE activity in BALFsupernatants of patients with moderate inflammation, but that thisactivity appears with time after cells have been centrifuged andsuspended in an inhibitor-free medium (our unpublished data).Therefore, any HNE activity at the surface of neutrophils circu-lating in blood must be immediately inhibited by circulating �1-Pi.This also means that the low concentrations of HNE and relatedproteases that we and others find at the surface of unstimulatedhuman PMNs by measuring their enzymatic activity or by immu-nofluorescence and immunogold staining (5, 18, 48, 53) probablyappeared during their purification, once the cells were separatedfrom plasma replete with inhibitors. HNE may be permanentlypresent at the surface of activated neutrophils and its activitytightly controlled by endogenous inhibitors, especially �1-Pi,which is the most abundant HNE inhibitor in plasma and in BALF(54–56). But this control may be hampered once triggered neu-trophils adhere and define a zone of restricted accessibility forinhibitors at the cell-subjacent substrate interface. The recent dem-onstration that neutrophil-bound HNE cleaves vascular endothe-lium cadherin, involved in the maintenance of endothelium integ-rity, during the process of transmigration supports this hypothesis(57). HNE activity should thus be limited to the zone of contactbetween adherent neutrophil and vascular endothelium, as sug-gested by previous studies, because of the rapid inhibition ofmHNE by �1-Pi at the surface of nonadherent cells (9, 58). TheHNE activity at the surface of nonadherent PMNs is therefore fullycontrolled by �1-Pi and related physiological inhibitors as long asthey are present in excess in the microenvironment, and activatedcells have not interacted with opsonized surfaces either duringtransendothelial migration or when they bind to insoluble matrixcomponents. The permanent clearance of newly exposed HNEmolecules from the cell surface therefore participates in the rapidconsumption of active inhibitor molecules present locally, thuscontributing to the disruption of the protease-inhibitor balance,helping to make inflammatory diseases involving PMN recruit-ment chronic.
3336 INHIBITION OF MEMBRANE-BOUND ELASTASE BY �1-Pi
by guest on April 28, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

AcknowledgmentsWe thank Catherine Girardin for technical assistance, Eric Hazouard andMartine Ferrandiere for providing blood samples and BALF,Pierre-Yves Sizaret for performing scanning electron microscopy,Francois Saudubray (Debiopharm) for providing EPI-hNE4 samples,Owen Parkes for editing the English text, and Joseph Bieth (Institut Na-tional de la Sante et de la Recherche Medicale Unite 392) for criticallyreading the manuscript.
DisclosuresThe authors have no financial conflict of interest.
References1. Lee, W. L., and G. P. Downey. 2001. Leukocyte elastase: physiological functions
and role in acute lung injury. Am. J. Respir. Crit. Care Med. 164: 896–904.2. Kawabata, K., T. Hagio, and S. Matsuoka. 2002. The role of neutrophil elastase
in acute lung injury. Eur. J. Pharmacol. 451: 1–10.3. Moraes, T. J., C. W. Chow, and G. P. Downey. 2003. Proteases and lung injury.
Crit. Care Med. 31: S189–S194.4. Owen, C. A., and E. J. Campbell. 1999. The cell biology of leukocyte-mediated
proteolysis. J. Leukocyte Biol. 65: 137–150.5. Owen, C. A., M. A. Campbell, P. L. Sannes, S. S. Boukedes, and E. J. Campbell.
1995. Cell surface-bound elastase and cathepsin G on human neutrophils: a novel,non-oxidative mechanism by which neutrophils focus and preserve catalytic ac-tivity of serine proteinases. J. Cell Biol. 131: 775–789.
6. Lee, C. T., A. M. Fein, M. Lippmann, H. Holtzman, P. Kimbel, andG. Weinbaum. 1981. Elastolytic activity in pulmonary lavage fluid from patientswith adult respiratory-distress syndrome. N. Engl. J. Med. 304: 192–196.
7. Matheson, N. R., P. S. Wong, and J. Travis. 1979. Enzymatic inactivation ofhuman �-1-proteinase inhibitor by neutrophil myeloperoxidase. Biochem. Bio-phys. Res. Commun. 88: 402–409.
8. Hornebeck, W., and H. P. Schnebli. 1982. Effect of different elastase inhibitors onleukocyte elastase pre-adsorbed to elastin. Hoppe-Seyler’s Z. Physiol. Chem. 363:455–458.
9. Campbell, E. J., and M. A. Campbell. 1988. Pericellular proteolysis by neutro-phils in the presence of proteinase inhibitors: effects of substrate opsonization.J. Cell Biol. 106: 667–676.
10. Morrison, H. M., H. G. Welgus, R. A. Stockley, D. Burnett, and E. J. Campbell.1990. Inhibition of human leukocyte elastase bound to elastin: relative ineffec-tiveness and two mechanisms of inhibitory activity. Am. J. Respir. Cell Mol. Biol.2: 263–269.
11. Chamba, A., S. C. Afford, R. A. Stockley, and D. Burnett. 1991. Extracellularproteolysis of fibronectin by neutrophils: characterization and the effects of re-combinant cytokines. Am. J. Respir. Cell Mol. Biol. 4: 330–337.
12. Bangalore, N., and J. Travis. 1994. Comparison of properties of membrane boundversus soluble forms of human leukocytic elastase and cathepsin G. Biol. Chem.Hoppe-Seyler 375: 659–666.
13. Salcedo, R., K. Wasserman, and M. Patarroyo. 1997. Endogenous fibronectin ofblood polymorphonuclear leukocytes: stimulus-induced secretion and proteolysisby cell surface-bound elastase. Exp. Cell Res. 233: 33–40.
14. Kolkenbrock, H., J. Zimmermann, G. R. Burmester, and N. Ulbrich. 2000. Ac-tivation of progelatinase B by membranes of human polymorphonuclear granu-locytes. Biol. Chem. 381: 49–55.
15. DiMartino, S. J., A. B. Shah, G. Trujillo, and R. R. Kew. 2001. Elastase controlsthe binding of the vitamin D-binding protein (Gc-globulin) to neutrophils: a po-tential role in the regulation of C5a co-chemotactic activity. J. Immunol. 166:2688–2694.
16. Korkmaz, B., S. Attucci, E. Hazouard, M. Ferrandiere, M. L. Jourdan,M. Brillard-Bourdet, L. Juliano, and F. Gauthier. 2002. Discriminating betweenthe activities of human neutrophil elastase and proteinase 3 using serpin-derivedfluorogenic substrates. J. Biol. Chem. 277: 39074–39081.
17. Koehl, C., C. G. Knight, and J. G. Bieth. 2003. Compared action of neutrophilproteinase 3 and elastase on model substrates: favorable effect of S�-P� interac-tions on proteinase 3 catalysis. J. Biol. Chem. 278: 12609–12612.
18. Korkmaz, B., S. Attucci, T. Moreau, E. Godat, L. Juliano, and F. Gauthier. 2004.Design and use of highly specific substrates of neutrophil elastase and proteinase3. Am. J. Respir. Cell Mol. Biol. 30: 801–807.
19. Hubbard, R. C., M. L. Brantly, S. E. Sellers, M. E. Mitchell, and R. G. Crystal.1989. Anti-neutrophil-elastase defenses of the lower respiratory tract in �1-an-titrypsin deficiency directly augmented with an aerosol of �1-antitrypsin. Ann.Intern. Med. 111: 206–212.
20. Hubbard, R. C., and R. G. Crystal. 1990. Strategies for aerosol therapy of �1-antitrypsin deficiency by the aerosol route. Lung 168: 565–578.
21. Vogelmeier, C., R. Buhl, R. F. Hoyt, E. Wilson, G. A. Fells, R. C. Hubbard,H. P. Schnebli, R. C. Thompson, and R. G. Crystal. 1990. Aerosolization ofrecombinant SLPI to augment antineutrophil elastase protection of pulmonaryepithelium. J. Appl. Physiol. 69: 1843–1848.
22. McElvaney, N. G., R. C. Hubbard, P. Birrer, M. S. Chernick, D. B. Caplan,M. M. Frank, and R. G. Crystal. 1991. Aerosol �1-antitrypsin treatment for cysticfibrosis. Lancet 337: 392–394.
23. McElvaney, N. G., B. Doujaiji, M. J. Moan, M. R. Burnham, M. C. Wu, andR. G. Crystal. 1993. Pharmacokinetics of recombinant secretory leukoprotease
inhibitor aerosolized to normals and individuals with cystic fibrosis. Am. Rev.Respir. Dis. 148: 1056–1060.
24. Doring, G. 1999. Serine proteinase inhibitor therapy in �(1)-antitrypsin inhibitordeficiency and cystic fibrosis. Pediatr. Pulmonol. 28: 363–375.
25. Attucci, S., B. Korkmaz, L. Juliano, E. Hazouard, C. Girardin,M. Brillard-Bourdet, S. Rehault, P. Anthonioz, and F. Gauthier. 2002. Measure-ment of free and membrane-bound cathepsin G in human neutrophils using newsensitive fluorogenic substrates. Biochem. J. 366: 965–970.
26. Korkmaz, B., P. Poutrain, E. Hazouard, M. de Monte, S. Attucci, andF. L. Gauthier. 2005. Competition between elastase and related proteases fromhuman neutrophil for binding to �1-Pi. Am. J. Respir. Cell Mol. Biol. 32: 553–559.
27. Durant, S., M. Pederzoli, Y. Lepelletier, S. Canteloup, P. Nusbaum, P. Lesavre,and V. Witko-Sarsat. 2004. Apoptosis-induced proteinase 3 membrane expres-sion is independent from degranulation. J. Leukocyte Biol. 75: 87–98.
28. Mellet, P., C. Boudier, Y. Mely, and J. G. Bieth. 1998. Stopped flow fluorescenceenergy transfer measurement of the rate constants describing the reversible for-mation and the irreversible rearrangement of the elastase-�1-proteinase inhibitorcomplex. J. Biol. Chem. 273: 9119–9123.
29. Serveau, C., T. Moreau, G. X. Zhou, A. ElMoujahed, J. Chao, and F. Gauthier.1992. Inhibition of rat tissue kallikrein gene family members by rat kallikrein-binding protein and �1-proteinase inhibitor. FEBS Lett. 309: 405–408.
30. Silverman, G. A., P. I. Bird, R. W. Carrell, F. C. Church, P. B. Coughlin,P. G. Gettins, J. A. Irving, D. A. Lomas, C. J. Luke, R. W. Moyer, et al. 2001.The serpins are an expanding superfamily of structurally similar but functionallydiverse proteins: evolution, mechanism of inhibition, novel functions, and a re-vised nomenclature. J. Biol. Chem. 276: 33293–33296.
31. Boudier, C., and J. G. Bieth. 1989. Mucus proteinase inhibitor: a fast-actinginhibitor of leukocyte elastase. Biochim. Biophys. Acta 995: 36–41.
32. Padrines, M., and J. G. Bieth. 1993. Oxidized and Met3583Leu mutated �1-proteinase inhibitor as substrates of Pseudomonas aeruginosa elastase. Biochim.Biophys. Acta 1163: 61–66.
33. Beatty, K., J. Bieth, and J. Travis. 1980. Kinetics of association of serine pro-teinases with native and oxidized �-1-proteinase inhibitor and �-1-antichymo-trypsin. J. Biol. Chem. 255: 3931–3934.
34. Frommherz, K. J., B. Faller, and J. G. Bieth. 1991. Heparin strongly decreases therate of inhibition of neutrophil elastase by �1-proteinase inhibitor. J. Biol. Chem.266: 15356–15362.
35. Rao, N. V., N. G. Wehner, B. C. Marshall, W. R. Gray, B. H. Gray, andJ. R. Hoidal. 1991. Characterization of proteinase-3 (PR-3), a neutrophil serineproteinase: structural and functional properties. J. Biol. Chem. 266: 9540–9548.
36. Duranton, J., C. Adam, and J. G. Bieth. 1998. Kinetic mechanism of the inhibi-tion of cathepsin G by �1-antichymotrypsin and �1-proteinase inhibitor. Bio-chemistry 37: 11239–11245.
37. Wright, H. T., and J. N. Scarsdale. 1995. Structural basis for serpin inhibitoractivity. Proteins 22: 210–225.
38. Huntington, J. A., R. J. Read, and R. W. Carrell. 2000. Structure of a serpin-protease complex shows inhibition by deformation. Nature 407: 923–926.
39. Heintzelman, D. L., R. Lotan, and R. R. Richards-Kortum. 2000. Characterizationof the autofluorescence of polymorphonuclear leukocytes, mononuclear leuko-cytes and cervical epithelial cancer cells for improved spectroscopic discrimina-tion of inflammation from dysplasia. Photochem. Photobiol. 71: 327–332.
40. Sponer, M., H. P. Nick, and H. P. Schnebli. 1991. Different susceptibility ofelastase inhibitors to inactivation by proteinases from Staphylococcus aureus andPseudomonas aeruginosa. Biol. Chem. Hoppe-Seyler 372: 963–970.
41. Carp, H., and A. Janoff. 1980. Potential mediator of inflammation: phagocyte-derived oxidants suppress the elastase-inhibitory capacity of �1-proteinase in-hibitor in vitro. J. Clin. Invest. 66: 987–995.
42. Desrochers, P. E., and S. J. Weiss. 1988. Proteolytic inactivation of �-1-protein-ase inhibitor by a neutrophil metalloproteinase. J. Clin. Invest. 81: 1646–1650.
43. Rice, W. G., and S. J. Weiss. 1990. Regulation of proteolysis at the neutro-phil-substrate interface by secretory leukoprotease inhibitor. Science 249:178 –181.
44. Owen, C. A., and E. J. Campbell. 1998. Angiotensin II generation at the cellsurface of activated neutrophils: novel cathepsin G-mediated catalytic activitythat is resistant to inhibition. J. Immunol. 160: 1436–1443.
45. Burry, R. W., and J. G. Wood. 1979. Contributions of lipids and proteins to thesurface charge of membranes: an electron microscopy study with cationized andanionized ferritin. J. Cell Biol. 82: 726–741.
46. Janciauskiene, S. 2001. Conformational properties of serine proteinase inhibitors(serpins) confer multiple pathophysiological roles. Biochim. Biophys. Acta 1535:221–235.
47. Duranton, J., and J. G. Bieth. 2003. Inhibition of proteinase 3 by �1-antitrypsinin vitro predicts very fast inhibition in vivo. Am. J. Respir. Cell Mol. Biol. 29:57–61.
48. Witko-Sarsat, V., P. Lesavre, S. Lopez, G. Bessou, C. Hieblot, B. Prum,L. H. Noel, L. Guillevin, P. Ravaud, I. Sermet-Gaudelus, et al. 1999. A largesubset of neutrophils expressing membrane proteinase 3 is a risk factor for vas-culitis and rheumatoid arthritis. J. Am. Soc. Nephrol. 10: 1224–1233.
49. Zimmermann, F., K. Lautenschlager, V. Heppert, A. Wentzensen, G. M. Hansch,and C. Wagner. 2005. Expression of elastase on polymorphonuclear neutrophilsin vitro and in vivo: identification of CD11b as ligand for the surface-boundelastase. Shock 23: 216–223.
3337The Journal of Immunology
by guest on April 28, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from

50. Cai, T. Q., and S. D. Wright. 1996. Human leukocyte elastase is an endogenousligand for the integrin CR3 (CD11b/CD18, Mac-1, �M�2) and modulates poly-morphonuclear leukocyte adhesion. J. Exp. Med. 184: 1213–1223.
51. Padrines, M., M. Schneider-Pozzer, and J. G. Bieth. 1989. Inhibition of neutro-phil elastase by �-1-proteinase inhibitor oxidized by activated neutrophils. Am.Rev. Respir. Dis. 139: 783–790.
52. Vogt, W. 1995. Oxidation of methionyl residues in proteins: tools, targets, andreversal. Free Radical Biol. Med. 18: 93–105.
53. Campbell, E. J., M. A. Campbell, and C. A. Owen. 2000. Bioactive proteinase 3on the cell surface of human neutrophils: quantification, catalytic activity, andsusceptibility to inhibition. J. Immunol. 165: 3366–3374.
54. Boudier, C., A. Pelletier, A. Gast, J. M. Tournier, G. Pauli, and J. G. Bieth. 1987.The elastase inhibitory capacity and the �1-proteinase inhibitor and bronchialinhibitor content of bronchoalveolar lavage fluids from healthy subjects. Biol.Chem. Hoppe-Seyler 368: 981–990.
55. Nadziejko, C., I. Finkelstein, and J. R. Balmes. 1995. Contribution of secretoryleukocyte proteinase inhibitor to the antiprotease defense system of the peripherallung: effect of ozone-induced acute inflammation. Am. J. Respir. Crit. Care Med.152: 1592–1598.
56. Frass, O. M., F. Buhling, M. Tager, H. Frass, S. Ansorge, C. Huth, and T. Welte.2001. Antioxidant and antiprotease status in peripheral blood and BAL fluid aftercardiopulmonary bypass. Chest 120: 1599–1608.
57. Hermant, B., S. Bibert, E. Concord, B. Dublet, M. Weidenhaupt, T. Vernet, andD. Gulino-Debrac. 2003. Identification of proteases involved in the proteolysis ofvascular endothelium cadherin during neutrophil transmigration. J. Biol. Chem.278: 14002–14012.
58. Cepinskas, G., M. Sandig, and P. R. Kvietys. 1999. PAF-induced elastase-de-pendent neutrophil transendothelial migration is associated with the mobilizationof elastase to the neutrophil surface and localization to the migrating front. J. CellSci. 112: 1937–1945.
3338 INHIBITION OF MEMBRANE-BOUND ELASTASE BY �1-Pi
by guest on April 28, 2016
http://ww
w.jim
munol.org/
Dow
nloaded from
Copyright © 2022 FDOKUMEN




















