
Neuroendocrine Responses to m-Chlorophenylpiperazine and i-Tryptophan in Bulimia
10
Neuroendocrine Responses to m-Chlorophenylpiperazine and i-Tryptophan in Bulimia Timothy D. Brewerton, MD; Edward A. Mueller, MD; Michael D. Lesem, MD; Harry A. Brandt, MD; Bonnie Quearry, MS; D. Ted George, MD; Dennis L. Murphy, MD; David C. Jimerson, MD \s=b\ Preclinical and clinical evidence supports a theory of se- rotonin (5-hydroxytryptamine [5-HT]) dysregulation in bu- limia. We therefore studied the prolactin (PRL) and cortisol responses following challenges with the postsynaptic 5-HT receptor agonist m-chlorophenylpiperazine (m-CPP), 0.5 mg/kg orally, the 5-HT precursor L-tryptophan, 100 mg/kg intravenously, and placebo in a group of 28 normal weight bulimic patients and 16 healthy controls. Patients with bu- limia, regardless of the presence of major depression, had significantly blunted PRL responses following m-CPP ad- ministration compared with those in controls. In contrast, only bulimic patients with concurrent major depression had significantly blunted PRL responses following L-tryptophan administration compared with those in nondepressed bu- limic patients and controls. Cortisol responses following m-CPP were not significantly different for bulimic patients vs controls, although there was a trend toward blunted cor- tisol responses following L-tryptophan administration in the depressed bulimic patients. These differences in neuroen- docrine responses were not related to differences in age, percent of average body weight, medications, time of day, peak plasma drug levels, or baseline estradiol levels. Sea- sonal variations in PRL responses to both agents were iden- tified, although covariation for season did not alter the group differences. The PRL responses following m-CPP ad- ministration were inversely correlated to baseline cortisol levels in the bulimic patients, but not in the controls, sug- gesting a dampening effect by hypothalamic-pituitary- adrenal axis dysfunction on postsynaptic 5-HT receptor sensitivity. The reasons for the differing hormonal responses to these two serotonergic agents may relate to differential involvement of presynaptic and postsynaptic mechanisms, 5-HT receptor subtypes, and anatomical loci of action. The blunted PRL responses to m-CPP administration suggest that postsynaptic 5-HT receptor sensitivity is altered in bulimia nervosa, and that similar alterations in 5-HT receptors at or above the level of the hypothalamus may contribute to binge eating and other behavioral symptoms. (Arch Gen Psychiatry. 1992;49:852-861) A spectrum of behavioral, pharmacological, and clinical studies in animals and humans suggest that altered function of the neurotransmitter serotonin (5-hydroxy¬ tryptamine [5-HT]) may play an important role in bulimia. Symptoms that span alterations in eating behavior, affec¬ tive modulation, and impulse control have all been asso¬ ciated with alterations in 5-HT function. Manipulations of the 5-HT system result in marked changes in appetite, eating behavior, and body weight.1-2 5-Hydroxytryptamine is thought to play an inhibitory role in feeding and to be selectively involved in the mediation of satiety. Generally, treatments and procedures that lead to an increased availability of 5-HT in the synaptic cleft or that di¬ rectly activate 5-HT receptors have been reported to reduce food consumption, particularly carbohydrate consumption. For example, both L-tryptophan (L-TRP) and fenfluramine have been reported to decrease carbohydrate intake selec¬ tively in obese subjects.3"5 Conversely, interventions that ei¬ ther directly or indirectly decrease 5-HT receptor activation are reported to decrease satiety and increase food consump¬ tion and weight gain. In laboratory animals, blockade of 5-HT function in the "satiety center" of the ventromedial hy¬ pothalamus results in marked increases in food ingestion and weight gain.2 A relevant clinical example is the increased rate of weight gain that was reported in a subgroup of pa¬ tients with anorexia nervosa who were treated with the 5-HT antagonist cyproheptadine.6 Further indirect evidence for deficient serotonin activity in bulimic patients derives from several striking similari¬ ties between bulimia and depression. The hypothesis that major depression involves a reduction in serotonin fune- Accepted for publication August 26, 1991. From the Section on Biomedical Psychiatry, Laboratory of Clinical Science, National Institute of Mental Health, Bethesda, Md. Dr Brewer- ton is now with the Department of Psychiatry and Behavioral Sciences, Medical University of South Carolina, Charleston. Dr Mueller is now with the Rutland (Vt) Mental Health Center. Dr Lesem is now with the Department of Psychiatry, University of Texas Medical School Hospital, Houston. Dr Brandt is now with the Department of Psychiatry, Univer- sity of Maryland School of Medicine, Baltimore. Dr George is now with the Laboratory of Clinical Studies, National Institute of Alcoholism and Alcohol Abuse, Bethesda, Md. Dr Jimerson is now with the Department of Psychiatry, Beth Israel Hospital and Harvard Medical School, Boston, Mass. Reprint requests to Department of Psychiatry and Behavioral Sciences, Medical University of South Carolina, 171 Ashley Ave, Charleston, SC 29425-0742 (Dr Brewerton). DownloadedFrom:http://archpsyc.jamanetwork.com/byTimBrewertonon03/28/2015
Transcript of Neuroendocrine Responses to m-Chlorophenylpiperazine and i-Tryptophan in Bulimia

Neuroendocrine Responses to m-Chlorophenylpiperazineand i-Tryptophan in Bulimia
Timothy D. Brewerton, MD; Edward A. Mueller, MD; Michael D. Lesem, MD; Harry A. Brandt, MD;Bonnie Quearry, MS; D. Ted George, MD; Dennis L. Murphy, MD; David C. Jimerson, MD
\s=b\Preclinical and clinical evidence supports a theory of se-rotonin (5-hydroxytryptamine [5-HT]) dysregulation in bu-limia. We therefore studied the prolactin (PRL) and cortisolresponses following challenges with the postsynaptic 5-HTreceptor agonist m-chlorophenylpiperazine (m-CPP), 0.5mg/kg orally, the 5-HT precursor L-tryptophan, 100 mg/kgintravenously, and placebo in a group of 28 normal weightbulimic patients and 16 healthy controls. Patients with bu-limia, regardless of the presence of major depression, hadsignificantly blunted PRL responses following m-CPP ad-ministration compared with those in controls. In contrast,only bulimic patients with concurrent major depression hadsignificantly blunted PRL responses following L-tryptophanadministration compared with those in nondepressed bu-limic patients and controls. Cortisol responses followingm-CPP were not significantly different for bulimic patientsvs controls, although there was a trend toward blunted cor-tisol responses following L-tryptophan administration in thedepressed bulimic patients. These differences in neuroen-docrine responses were not related to differences in age,
percent of average body weight, medications, time of day,peak plasma drug levels, or baseline estradiol levels. Sea-sonal variations in PRL responses to both agents were iden-tified, although covariation for season did not alter thegroup differences. The PRL responses following m-CPP ad-ministration were inversely correlated to baseline cortisollevels in the bulimic patients, but not in the controls, sug-gesting a dampening effect by hypothalamic-pituitary-adrenal axis dysfunction on postsynaptic 5-HT receptorsensitivity. The reasons for the differing hormonal responsesto these two serotonergic agents may relate to differentialinvolvement of presynaptic and postsynaptic mechanisms,5-HT receptor subtypes, and anatomical loci of action. Theblunted PRL responses to m-CPP administration suggest thatpostsynaptic 5-HT receptor sensitivity is altered in bulimianervosa, and that similar alterations in 5-HT receptors at or
above the level of the hypothalamus may contribute tobinge eating and other behavioral symptoms.
(Arch Gen Psychiatry. 1992;49:852-861)
A spectrum of behavioral, pharmacological, and clinicalstudies in animals and humans suggest that altered
function of the neurotransmitter serotonin (5-hydroxy¬tryptamine [5-HT]) may play an important role in bulimia.Symptoms that span alterations in eating behavior, affec¬tive modulation, and impulse control have all been asso¬ciated with alterations in 5-HT function.
Manipulations of the 5-HT system result in marked
changes in appetite, eating behavior, and body weight.1-25-Hydroxytryptamine is thought to play an inhibitory rolein feeding and to be selectively involved in the mediation ofsatiety. Generally, treatments and procedures that lead to anincreased availability of 5-HT in the synaptic cleft or that di¬rectly activate 5-HT receptors have been reported to reducefood consumption, particularly carbohydrate consumption.For example, both L-tryptophan (L-TRP) and fenfluraminehave been reported to decrease carbohydrate intake selec¬tively in obese subjects.3"5 Conversely, interventions that ei¬ther directly or indirectly decrease 5-HT receptor activationare reported to decrease satiety and increase food consump¬tion and weight gain. In laboratory animals, blockade of5-HT function in the "satiety center" of the ventromedial hy¬pothalamus results in marked increases in food ingestionand weight gain.2 A relevant clinical example is the increasedrate of weight gain that was reported in a subgroup of pa¬tients with anorexia nervosa who were treated with the 5-HTantagonist cyproheptadine.6
Further indirect evidence for deficient serotonin activityin bulimic patients derives from several striking similari¬ties between bulimia and depression. The hypothesis thatmajor depression involves a reduction in serotonin fune-
Accepted for publication August 26, 1991.From the Section on Biomedical Psychiatry, Laboratory of Clinical
Science, National Institute of Mental Health, Bethesda, Md. Dr Brewer-ton is now with the Department of Psychiatry and Behavioral Sciences,Medical University of South Carolina, Charleston. Dr Mueller is nowwith the Rutland (Vt) Mental Health Center. Dr Lesem is now with theDepartment of Psychiatry, University of Texas Medical School Hospital,Houston. Dr Brandt is now with the Department of Psychiatry, Univer-sity of Maryland School of Medicine, Baltimore. Dr George is now withthe Laboratory of Clinical Studies, National Institute of Alcoholism andAlcohol Abuse, Bethesda, Md. Dr Jimerson is now with the Departmentof Psychiatry, Beth Israel Hospital and Harvard Medical School, Boston,Mass.
Reprint requests to Department of Psychiatry and Behavioral Sciences,Medical University of South Carolina, 171 Ashley Ave, Charleston, SC29425-0742 (Dr Brewerton).
Downloaded From: http://archpsyc.jamanetwork.com/ by Tim Brewerton on 03/28/2015

Normal Weight Normal WeightBulimics Controls
-1m-CPP L-TRP m-CPP L-TRP(n=26) (n=23) (n=15) (n=16)
Age, y 24.7±4.8 24.1 ±4.8 26.4±5.3 26.8±5.3
Weight, kg 54.7±4.9 55.3±5.2 57.5±6.3 57.5±6.2
ABW, % 92.7±7.9 92.4±7.7 95.6±7.7 95.5±7.8Illness
duration, y 5.5±3.5 5.7±3.7
Binge frequency,No./wk 13.8+9.0 12.1 ±8.5
*m-CPP indicates m-chlorophenylpiperazine; L-TRP, L-tryptophan;and ABW, average body weight. Data are given as mean±SD.
tion has been reviewed elsewhere.7"9 Bulimic patients havehigh frequencies of depressed mood,10"14 lifetime12"18 andfamily histories of affective illness,1819 and therapeutic re¬
sponsiveness to antidepressant medications.2029 Imipra-mine,20"23 desipramine,24·25 phenelzine,26 isocarboxazid,27fluoxetine,28"30 and other antidepressants that are clinicallyeffective in treating both depression and bulimia areknown to potentiate central 5-HT function31"41 among otherneurochemical effects.
Impulsivity, a related clinical feature that is common toboth bulimia and some subtypes of depression, is modu¬lated by 5-HT neurotransmission.42'43 Some forms of ag¬gression toward self and others can be conceptualized asdisinhibited behavior.43 Muricidal activity in rats is in¬duced by a diet low in L-TRP, as well as administration ofp-chlorophenylalanine, which is an inhibitor of 5-HT syn¬thesis, and is reversed by oral L-TRP administration.44Lower levels of cerebrospinal fluid 5-hydroxyindoleaceticacid in humans have been associated with suicide at¬tempts, especially by violent means.45"47 Low cerebrospinalfluid 5-hydroxyindoleacetic acid levels after probenecidtreatment have been reported in weight-recovered bulimicanorectic patients as compared with those in weight-recovered nonbulimic anorectic patients.48 Bulimic anorec¬tic patients share clinical features with normal weight bu¬limic patients besides binge eating that are often indicativeof impulsivity, such as suicidality, alcohol/drug abuse,stealing, and impulsive sexual behavior.4951 Interestingly,several authors have noted the association between dis¬turbed eating behaviors and self-mutilation.52"55
Given such data, we investigated neuroendocrine re¬
sponses to two serotonergic agents as a means of evaluat¬ing central serotonin function in bulimia. Neuroendocrineresponses to the 5-HT precursor L-TRP) are used to eval¬uate 5-HT function in humans. L-Tryptophan administra¬tion increases plasma prolactin (PRL)5661 levels in humansby a pathway that is antagonized by the 5-HT antagonistsmethysergide59 and metergoline.60 Prolactin responses toboth intravenous L-TRP56<M.,a and oral fenfluramine63 areblunted in major depression, presumably reflecting the se¬rotonin deficiency that has been hypothesized in this pa¬tient group,7"9 although one group implicates differences inthe pharmacokinetics of L-TRP.56 We also used m-chlor¬ophenylpiperazine (m-CPP), a triazolopyridine metaboliteof the antidepressant drug trazodone, and a selectiveserotonin (5-HT) receptor agonist that produces neuroen¬
docrine, behavioral, food intake, and other physiological
alterations, presumably as a result of postsynaptic stimu¬lation of 5-HT receptors.6471 m-Chlorophenylpiperazinehas equipotent affinity at all 5-HT receptor subtypes, as
well as similar affinity for a2-adrenergic receptors.72 Pre-clinical studies suggest that m-CPP acts particularly at5-HTlc and 5-HT1B receptors to produce hypophagia,73-74although 5-HTib receptors have not been identified in hu¬mans.75 The induced release of PRL and cortisol (CORT) isblocked by metergoline76 and appears to be mediated by5-HT, receptors.77 The drug acts as an agonist at 5-HTlc re¬
ceptors, but as a pure antagonist at 5-HT2 receptors.78SUBJECTS AND METHODS
The demographic features of the normal weight bulimicpatients and the age- and weight-matched female normal controlsare seen in the Table. There was not complete overlap between them-CPP- and L-TRP-treated patient and control groups. All sub¬jects were studied after at least 4 weeks off all medications,including birth control pills. In addition, patients were free ofmonoamine oxidase inhibitors for at least 8 weeks before study.At admission, all patients met DSM-III criteria for bulimia79; ret¬rospectively, all patients were also found to meet DSM-III-R cri¬teria for bulimia nervosa.8" Other past and present diagnoses weredetermined by means of a Schedule for Affective Disorders andSchizophrenia-Lifetime version (SADS-L)81 interview that wasconducted by a trained social worker. DSM-III Axis I diagnoses,as determined by the patient's treating psychiatrist, were con¬firmed by a second member of the treatment team. None of thepatients with bulimia met present or past DSM-III criteria for an¬orexia nervosa. Normal controls were selected on the basis of anabsence of present or past major psychiatric illness, as determinedby the SADS-L. Weights of patients ranged between 80% to 120%of average body weight (% ABW), whereas the weights of controlsranged between 85% to 120 %ABW, as determined by standard¬ized weight tables.82 All subjects were in good general health, notpregnant, and free of significant medical illness on the basis of a
thorough medical evaluation, which included a physical exami¬nation, electrocardiogram, and appropriate laboratory tests. Inaddition, controls were eliminated on the basis of a lifetime fam¬ily history of any psychiatric disorder in a first-degree relative.Furthermore, controls were also eliminated if any first-degreerelative had a history of obesity. Each subject received an oral andwritten explanation of the purposes, procedures, and potentialhazards of the project and gave informed consent.
All subjects were studied at the National Institute of MentalHealth Clinical Center Eating Disorders Unit, Bethesda, Md, af¬ter overnight bed rest and a 14-hour fast (except water). In addi¬tion, 20 of the 26 bulimic subjects were inpatients at the time ofthe study and had been abstinent from binge eating and vomit¬ing for at least 4 weeks. Inpatients were treated on a highly struc¬tured, locked inpatient unit with supervised meals and use of thebathroom. The daily caloric intake for all inpatients was pre¬scribed and consisted of approximately 126 kj/kg (30 kcal). AUmeals were prepared and checked for an appropriate balance ofmacronutrients, minerals, and vitamins by experienced dietitians.Patients were not allowed to exercise and were checked period¬ically by nursing staff to prevent excessive activity. All subjectsalso observed limited activity levels and a low-monoamine,alcohol-free, low-caffeine diet for at least 72 hours before study.Subjects who smoked were not allowed to do so on the days ofthe study. A 20-gauge intravenous catheter that was attached toa heparinized double stopcock was inserted by 9 AM on each ofthe procedure days. Double-blind administration of m-CPP (0.5mg/kg orally), L-TRP (100 mg/kg intravenously), or placebo was
performed on separate days in random order at least 48 hoursapart. Each subject had an infusion of saline solution or L-TRP andthe same number of capsules (placebo or m-CPP), except for sixpatients with indwelling venous lines who received only capsuleson each day of 2 days (placebo or m-CPP). Vital signs wereobtained at least twice before the infusion and then every 30 min-
Downloaded From: http://archpsyc.jamanetwork.com/ by Tim Brewerton on 03/28/2015
utes throughout the procedure. Two baseline blood samples wereobtained at approximately 9:45 AM and 9:55 AM. L-Tryptophan(100 mg/kg) in a 1-mg/mL solution of 0.45% saline solution or anidentical volume of saline solution alone (placebo) was infused ata constant rate during 20 minutes, starting at 10 AM. Blood sam¬
ples were obtained at the following times after drug or placeboadministration: 30, 45, 60, 75, 90, 120, 150, 180, and 210 minutes.The 30-, 45-, and 75-minute samples were not used in the m-CPPanalyses. Samples were kept on ice and centrifuged. Plasma was
aliquoted and frozen for subsequent assay. Prolactin, CORT, andestradiol (E2) levels were determined by radioimmunoassay. TheL-TRP and m-CPP levels were measured by high-pressure liquidchromatography.8385 The Hamilton Depression Rating Scale(HAM-D)86 was administered by one of the authors (T.D.B.), andsubjects completed the Beck Depression Inventory (BDI)87 on themorning of each study day. A side effects checklist was obtainedat regular intervals throughout the course of the study. Subjectsrated the severity of physical side effects (headache, nausea,weakness, dizziness, etc) on a four-point scale (0, not at all; 1, mild;2, moderate; and 3, severe).
Data AnalysisBefore analysis, all data were tested for normal distribution and
homogeneity of variance with the use of SAS PROC UNVARI-ATE. Parametric data were analyzed over time via repeated mea¬sures analysis of variance (RAÑOVA). Bonferroni's t tests werethen computed, when appropriate, for each time point in theanalysis. Parametric data were analyzed by group with the use ofthe unpaired t test. Nonparametric data were analyzed by groupwith the use of the Mann-Whitney LI test. All variances are
reported as SDs.
RESULTSBaseline Measures
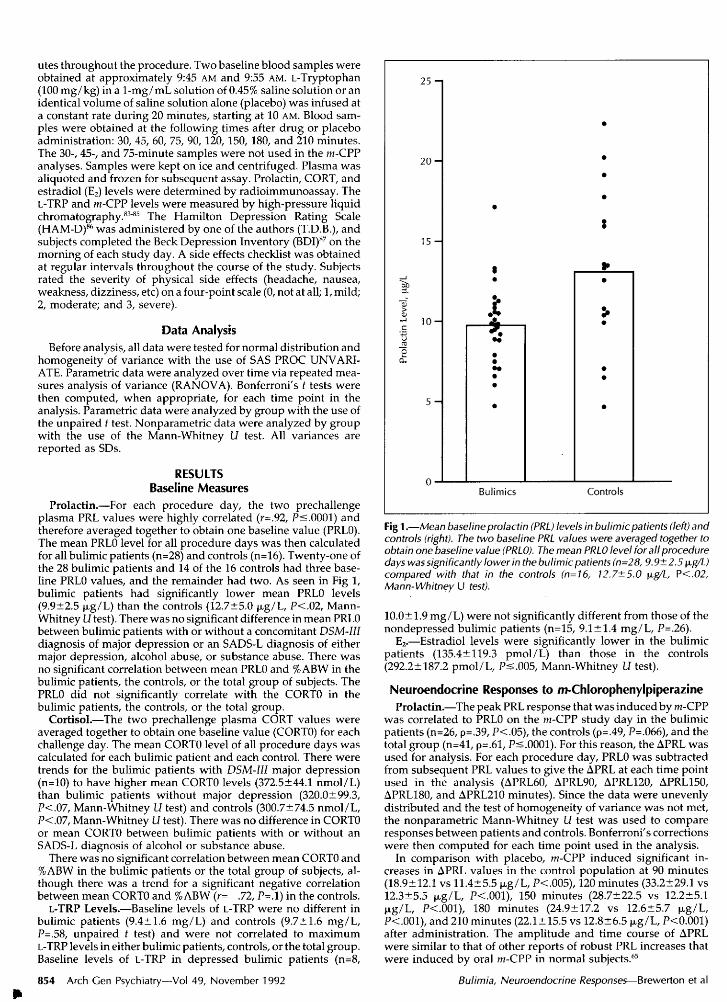
Prolactin.—For each procedure day, the two prechallengeplasma PRL values were highly correlated (r=.92, Ps.0001) andtherefore averaged together to obtain one baseline value (PRL0).The mean PRL0 level for all procedure days was then calculatedfor all bulimic patients (n=28) and controls (n=16). Twenty-one ofthe 28 bulimic patients and 14 of the 16 controls had three base¬line PRL0 values, and the remainder had two. As seen in Fig 1,bulimic patients had significantly lower mean PRL0 levels(9.9±2.5 µg/L) than the controls (12.7±5.0 µg/L, P<.02, Mann-Whitney U test). There was no significant difference in mean PRL0between bulimic patients with or without a concomitant DSM-IIIdiagnosis of major depression or an SADS-L diagnosis of eithermajor depression, alcohol abuse, or substance abuse. There wasno significant correlation between mean PRL0 and %ABW in thebulimic patients, the controls, or the total group of subjects. ThePRL0 did not significantly correlate with the CORTO in thebulimic patients, the controls, or the total group.
Cortisol.—The two prechallenge plasma CORT values were
averaged together to obtain one baseline value (CORTO) for eachchallenge day. The mean CORTO level of all procedure days wascalculated for each bulimic patient and each control. There weretrends for the bulimic patients with DSM-III major depression(n=10) to have higher mean CORTO levels (372.5±44.1 nmol/L)than bulimic patients without major depression (320.0±99.3,P<.07, Mann-Whitney U test) and controls (300.7±74.5 nmol/L,P<.07, Mann-Whitney U test). There was no difference in CORTOor mean CORTO between bulimic patients with or without anSADS-L diagnosis of alcohol or substance abuse.
There was no significant correlation between mean CORTO and%ABW in the bulimic patients or the total group of subjects, al¬though there was a trend for a significant negative correlationbetween mean CORTO and %ABW (r=-.72, P=.l) in the controls.
L-TRP Levels.—Baseline levels of L-TRP were no different inbulimic patients (9.4±1.6 mg/L) and controls (9.7±1.6 mg/L,P=.58, unpaired t test) and were not correlated to maximumL-TRP levels in either bulimic patients, controls, or the total group.Baseline levels of L-TRP in depressed bulimic patients (n=8,
Fig 1.—Mean baseline prolactin (PRL) levels in bulimic patients (left) andcontrols (right). The two baseline PRL values were averaged together toobtain one baseline value (PRLO). The mean PRLO level forali proceduredays was significantly lower in the bulimic patients (n=28, 9.9± 2.5 µ / )compared with that in the controls (n=16, 12.7±5.0 µ /L, P<.02,Mann-Whitney U fesfj.
10.0±1.9 mg/L) were not significantly different from those of thenondepressed bulimic patients (n=15, 9.1±1.4 mg/L, P=.26).
E2.—Estradiol levels were significantly lower in the bulimicpatients (135.4± 119.3 pmol/L) than those in the controls(292.2±187.2 pmol/L, Ps.005, Mann-Whitney U test).Neuroendocrine Responses to m-ChlorophenylpiperazineProlactin.—The peak PRL response that was induced by m-CPP
was correlated to PRLO on the m-CPP study day in the bulimicpatients (n=26, p=.39, P<.05), the controls (p=.49, P=.066), and thetotal group (n=41, p=.61, P<.0001). For this reason, the APRL was
used for analysis. For each procedure day, PRLO was subtractedfrom subsequent PRL values to give the APRL at each time pointused in the analysis (APRL60, APRL90, APRL120, APRL150,APRL180, and APRL210 minutes). Since the data were unevenlydistributed and the test of homogeneity of variance was not met,the nonparametric Mann-Whitney ¡J test was used to compareresponses between patients and controls. Bonferroni's correctionswere then computed for each time point used in the analysis.
In comparison with placebo, m-CPP induced significant in¬creases in APRL values in the control population at 90 minutes(18.9 + 12.1 vs 11.4±5.5 µg/L, P-C005), 120 minutes (33.2±29.1 vs12.3±5.5 µg/L, P<.001), 150 minutes (28.7±22.5 vs 12.2±5.1µg/L, P<.001), 180 minutes (24.9±17.2 vs 12.6±5.7 µg/L,P<.001), and 210 minutes (22.1 ±15.5 vs 12.8±6.5 µg/L, P<0.001)after administration. The amplitude and time course of APRLwere similar to that of other reports of robust PRL increases thatwere induced by oral m-CPP in normal subjects.65
Downloaded From: http://archpsyc.jamanetwork.com/ by Tim Brewerton on 03/28/2015
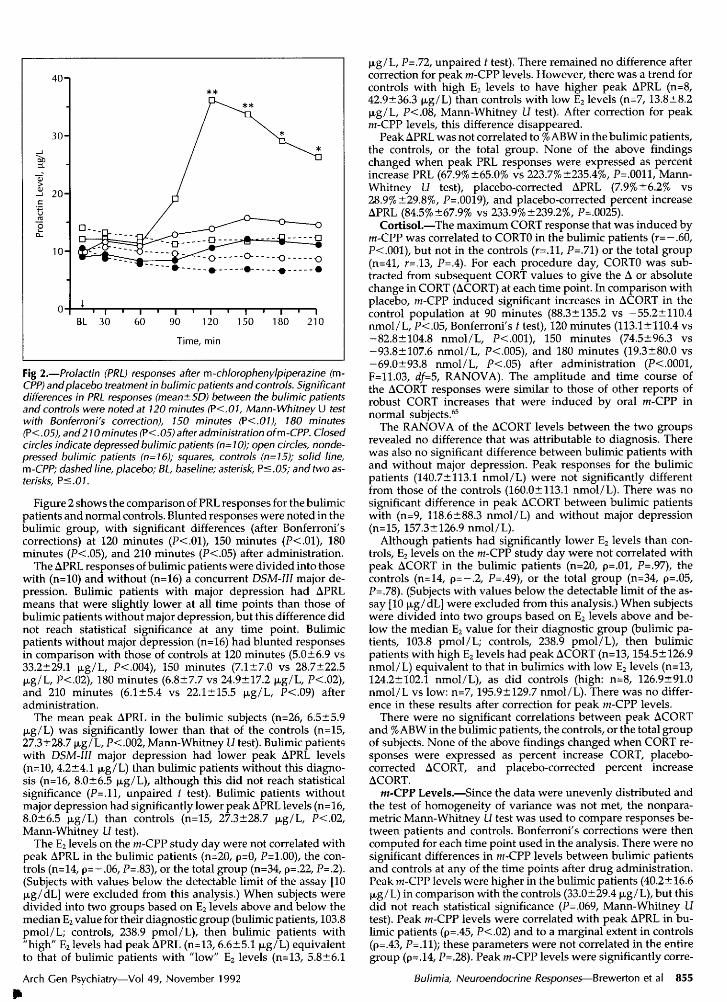
Fig 2.—Prolactin (PRL) responses after m-chlorophenylpiperazine (m-CPP) andplacebo treatment in bulimic patients and controls. Significantdifferences in PRL responses (mean±SD) between the bulimic patientsand controls were noted at 120 minutes (P<.01, Mann-Whitney U testwith Bonferroni's correction), 150 minutes (?<.0l), 180 minutes(P<.05), and210 minutes (P<.05) after administration ofm-CPP. Closedcircles indicate depressed bulimic patients (n=10); open circles, nonde-pressed bulimic patients (n=16); squares, controls (n=15); solid line,m-CPP; dashed line, placebo; BL, baseline; asterisk, Ps.05; and two as¬terisks, Ps.01.
Figure 2 shows the comparison of PRL responses for the bulimicpatients and normal controls. Blunted responses were noted in thebulimic group, with significant differences (after Bonferroni'scorrections) at 120 minutes (P<.01), 150 minutes (P<.01), 180minutes (P<.05), and 210 minutes (P<.05) after administration.
The APRL responses of bulimic patients were divided into thosewith (n=10) and without (n=16) a concurrent DSM-III major de¬pression. Bulimic patients with major depression had APRLmeans that were slightly lower at all time points than those ofbulimic patients without major depression, but this difference didnot reach statistical significance at any time point. Bulimicpatients without major depression (n=16) had blunted responsesin comparison with those of controls at 120 minutes (5.0±6.9 vs33.2±29.1 µg/L, P<.004), 150 minutes (7.1±7.0 vs 28.7±22.5µg/L, P<.02), 180 minutes (6.8±7.7 vs 24.9±17.2 µg/L, P<.02),and 210 minutes (6.1±5.4 vs 22.1±15.5 µg/L, P<.09) afteradministration.
The mean peak APRL in the bulimic subjects (n=26, 6.5±5.9µg/L) was significantly lower than that of the controls (n=15,27.3±28.7 µg/L, P<.002, Mann-Whitney Li test). Bulimic patientswith DSM-III major depression had lower peak APRL levels(n=10, 4.2±4.1 µg/L) than bulimic patients without this diagno¬sis (n=16, 8.0+6.5 µg/L), although this did not reach statisticalsignificance (P=.ll, unpaired t test). Bulimic patients withoutmajor depression had significantly lower peak APRL levels (n=16,8.0±6.5 µg/L) than controls (n=15, 27.3±28.7 µg/L, P<.02,Mann-Whitney U test).
The E2 levels on the m-CPP study day were not correlated withpeak APRL in the bulimic patients (n=20, p=0, P=1.00), the con¬trols (n=14, p=-.06, P=.83), or the total group (n=34, p=.22, P=.2).(Subjects with values below the detectable limit of the assay [10µg/dL] were excluded from this analysis.) When subjects weredivided into two groups based on E2 levels above and below themedian E2 value for their diagnostic group (bulimic patients, 103.8pmol/L; controls, 238.9 pmol/L), then bulimic patients with"high" E2 levels had peak APRL (n=13, 6.6±5.1 µg/L) equivalentto that of bulimic patients with "low" E2 levels (n=13, 5.8±6.1
µg/L, P=.72, unpaired t test). There remained no difference aftercorrection for peak m-CPP levels. However, there was a trend forcontrols with high E2 levels to have higher peak APRL (n=8,42.9±36.3 µg/L) than controls with low E2 levels (n=7,13.8±8.2µg/L, P<.08, Mann-Whitney U test). After correction for peakm-CPP levels, this difference disappeared.
Peak APRL was not correlated to % ABW in the bulimic patients,the controls, or the total group. None of the above findingschanged when peak PRL responses were expressed as percentincrease PRL (67.9%±65.0% vs 223.7%±235.4%, P=.0011, Mann-Whitney L? test), placebo-corrected APRL (7.9% ±6.2% vs
28.9% ±29.8%, P=.0019), and placebo-corrected percent increaseAPRL (84.5% ±67.9% vs 233.9% ±239.2%, P=.0025).
Cortisol.—The maximum CORT response that was induced bym-CPP was correlated to CORTO in the bulimic patients (r=-.60,P<.001), but not in the controls (r=.ll, P=.71) or the total group(n=41, r=.13, P=.4). For each procedure day, CORTO was sub¬tracted from subsequent CORT values to give the A or absolutechange in CORT (ACORT) at each time point. In comparison withplacebo, m-CPP induced significant increases in ACORT in thecontrol population at 90 minutes (88.3±135.2 vs -55.2±110.4nmol/L, P<.05, Bonferroni's f test), 120 minutes (113.1±110.4 vs-82.8±104.8 nmol/L, P<.001), 150 minutes (74.5±96.3 vs-93.8±107.6 nmol/L, P<.005), and 180 minutes (19.3+80.0 vs-69.0±93.8 nmol/L, P<.05) after administration (P<.0001,F=11.03, df=5, RANOVA). The amplitude and time course ofthe ACORT responses were similar to those of other reports ofrobust CORT increases that were induced by oral m-CPP innormal subjects.65
The RANOVA of the ACORT levels between the two groupsrevealed no difference that was attributable to diagnosis. Therewas also no significant difference between bulimic patients withand without major depression. Peak responses for the bulimicpatients (140.7±113.1 nmol/L) were not significantly differentfrom those of the controls (160.0±113.1 nmol/L). There was no
significant difference in peak ACORT between bulimic patientswith (n=9, 118.6±88.3 nmol/L) and without major depression(n=15, 157.3±126.9 nmol/L).
Although patients had significantly lower E2 levels than con¬trols, E2 levels on the m-CPP study day were not correlated withpeak ACORT in the bulimic patients (n=20, p=.01, P=.97), thecontrols (n=14, p=-.2, P=.49), or the total group (n=34, p=.05,P=.78). (Subjects with values below the detectable limit of the as¬
say [10 µg/dL] were excluded from this analysis.) When subjectswere divided into two groups based on E2 levels above and be¬low the median E2 value for their diagnostic group (bulimic pa¬tients, 103.8 pmol/L; controls, 238.9 pmol/L), then bulimicpatients with high E2 levels had peak ACORT (n=13,154.5±126.9nmol/L) equivalent to that in bulimics with low E2 levels (n=13,124.2±102.1 nmol/L), as did controls (high: n=8, 126.9±91.0nmol/L vs low: n=7, 195.9+129.7 nmol/L). There was no differ¬ence in these results after correction for peak m-CPP levels.
There were no significant correlations between peak ACORTand % ABW in the bulimic patients, the controls, or the total groupof subjects. None of the above findings changed when CORT re¬
sponses were expressed as percent increase CORT, placebo-corrected ACORT, and placebo-corrected percent increaseACORT.
m-CPP Levels.—Since the data were unevenly distributed andthe test of homogeneity of variance was not met, the nonpara-metric Mann-Whitney U test was used to compare responses be¬tween patients and controls. Bonferroni's corrections were thencomputed for each time point used in the analysis. There were no
significant differences in m-CPP levels between bulimic patientsand controls at any of the time points after drug administration.Peak m-CPP levels were higher in the bulimic patients (40.2±16.6µg/L) in comparison with the controls (33.0±29.4 µg/L), but thisdid not reach statistical significance (P=.069, Mann-Whitney Litest). Peak m-CPP levels were correlated with peak APRL in bu¬limic patients (p=.45, P<.02) and to a marginal extent in controls(p=.43, P=.ll); these parameters were not correlated in the entiregroup (p=.14, P=.28). Peak m-CPP levels were significantly corre-
Downloaded From: http://archpsyc.jamanetwork.com/ by Tim Brewerton on 03/28/2015

lated with peak ACORT in bulimic patients (p=.77, P-C.0001) andthe combined group (p=.43, P<.0005), but not in the controls alone(p=.25, P=.36).
By using peak m-CPP levels as the independent variable andpeak APRL as the dependent variable in an analysis of cova-
riance, an even greater difference in APRL response to m-CPPbetween bulimic patients (5.0±12.4 µg/L) and controls(30.0±12.5 µg/L, P<.0001, F=42.8, df=l, analysis of covariance)was discerned. No significant difference in peak ACORT be¬tween bulimic patients and controls was observed after correc¬
tion for peak m-CPP levels.Depression Ratings.—On the m-CPP study day, bulimic pa¬
tients had significantly higher HAM-D scores (16.4±8.2) and BDIscores (7.8±8.0) than normal controls (1.1 ±1.6, 1.0+1.3, P<.001,Mann-Whitney Li test). In the bulimic patients, neither HAM-Dscores nor BDI scores were significantly correlated to PRLO, peakAPRL, CORTO, or peak ACORT.
Side Effects Checklist.—The m-CPP challenge procedure was
generally well tolerated by all subjects. The most common sideeffect that was reported by both bulimic patients and controlsduring the procedure was mild nausea. There were no differencesin maximum nausea ratings during the m-CPP procedure daybetween bulimic patients (0.73±0.92; range, 0 to 3) and controls(0.87±0.94; range, 0 to 3). Maximum ratings of nausea were notsignificantly correlated with peak APRL or peak ACORT in thebulimic patients. However, there were trends for maximum nau¬sea ratings to be significantly correlated with peak APRL (p=.47,P<.08), but not with peak ACORT, in the controls. Maximumnausea ratings were correlated with maximum m-CPP levels inthe controls (p=.61, P<.02), but not in the bulimic patients or thetotal group.
Migrainelike headaches were a late-occurring side effect in 54%of subjects 8 to 12 hours after receiving m-CPP. These results havebeen discussed elsewhere.88·89
Behavioral Variables.—In the bulimic patients, there was a
trend for the frequency of binge eating during the month beforeadmission to be negatively correlated with peak APRL (n=26,p=-.37, P=.06), but not with peak ACORT. No significant corre¬lations were found between peak APRL or peak ACORT and thenumber of past suicide attempts, the number of years of bingeeating or vomiting, or the presence of stealing (items from theDiagnostic Survey of the Eating Disorders).'"1
Seasonal Variation.—Because season (seasons were defined inthe traditional manner by their respective solstice and equinox)is known to influence serotonin function significantly,91 aKruskal-Wallis test was performed with the use of season as a
grouping variable. There were statistically significant differencesin peak APRL when analyzed by season in the total group( 2=12.6, df=3, P=.0056) and in the patients alone (x2=8.16, d/=3,P=.043, Fig 3), but not in the controls alone. Post hoc Mann-Whitney Li tests with Bonferroni corrections disclosed a signifi¬cant difference between winter (n=10,28.4±26.5 µg/L) and sum¬mer (n=14, 5.9+4.4 µg/L, P<.01) in the total group, and betweenwinter (n=5,14.0±7.9 µg/L) and summer (n=9,4.0±2.7 µg/L), aswell as winter and fall (n=6, 3.7±4.4 µg/L) in the patients. Theseresults have been discussed in detail elsewhere.92 Peak ACORTwas also found to vary significantly by season in the total group(P=.034, Kruskal-Wallis test), although there were no significantpost hoc differences. There were no significant seasonal variationsin peak m-CPP levels in either the patients, the controls, or the to¬tal group. None of these results changed when depressed patientswere removed from the analysis. The seasonal variations do notappear to be relevant to the peak APRL differences between bu¬limic patients and controls. Both groups were randomly dis¬persed across the seasons (P=.73, 2).
Neuroendocrine Responses to L-TRPProlactin.—The peak PRL response that was induced by L-TRP
was not correlated to PRLO on the L-TRP study day in either thebulimic patients, the controls, or the total group. To adjust fordifferences in baseline PRL values between patients and controls,
Fig 3.—Peak Aprolactin (APRL) following m-chlorophenylpiperazine(m-CPP) administration by season. There were statistically significantdifferences in peak APRL (+SD) when analyzed by season (P=.0056,Kruskal-Wallis test). Post-hoc Mann-Whitney U fesfs and Bonferronicorrections disclosed a significant difference between winter and sum¬mer (P<.01) in the total group. Hatched squares indicate bulimicpatients; open squares, controls; closed circles, depressedpatients; opencircles, nondepressed; and star, Ps.O/.
the APRL was used for analysis. For each procedure day, PRLOwas subtracted from subsequent PRL values to give the APRL ateach time point used in the analysis (APRL30, APRL45, APRL60,APRL75, APRL90, APRL120, APRL180, and APRL210 minutes).
In comparison with placebo, L-TRP induced significant in¬creases in plasma PRL concentrations over time in the controlpopulation (timeXtreatment, P<.0001, RANOVA) (Fig 4). Bon¬ferroni's t tests were significant for APRL values at 30 minutes(32.2±18.4 vs 13.9±6.4 µg/L, P<.001), 45 minutes (32.6±18.6 vs13.2±6.2 µg/L, P<.001), 60 minutes (26.2±14.8 vs 13.1±5.8 µg/L,P<.001), 75 minutes (23.3 + 14.2 vs 11.7+5.2 µg/L, P-COOl), and90 minutes (22.3±18.4 vs 11.4+5.1 µg/L, P<.01) after adminis¬tration of L-TRP. The amplitude and time course of PRL responseswere similar to those of other reports of robust PRL increases thatwere induced by intravenous L-TRP in normal subjects.56-57-59"62
No significant differences were noted in APRL responses for thebulimic patients and normal controls (timeXdiagnosisXtreat-ment, P=,4, RANOVA). As seen in Fig 4, bulimic patients withmajor depression (n=8) had PRL levels that were significantlylower than those of bulimic patients without major depression(n=15, P<.001, timeXdiagnosisxtreatment, RANOVA). AfterBonferroni's corrections, bulimic patients with major depressionhad blunted responses in comparison with those of bulimic pa¬tients without major depression at 30 minutes (P<.05), 75 minutes(P<.05), 150 minutes (P<.05), 180 minutes (P<.05), and 210 min¬utes (P<.05) after administration of L-TRP. Prolactin levels in thedepressed bulimic patients were not significantly lower thanthose in controls (n=16, P=.23, RANOVA).
The mean peak APRL in the bulimic patients (17.6±10.1 µg/L)was not significantly lower than that of the controls (25.4±21.0µg/L, P=.21, unpaired t test). However, bulimic patients withDSM-III major depression (n=8) had lower peak APRL levels(9.9±7.4 µg/L) than bulimic patients without this diagnosis(n=15, 21.7±8.9 µg/L, P<.005, unpaired t test). Bulimic patientswith major depression also had significantly lower peak APRLlevels than controls (P<.02, Mann-Whitney Li test).
The E2 levels on the L-TRP study day were not correlated withpeak APRL in the bulimic patients (n=22, r=-.22, P=.31), the con¬trols (n=16, r=-.23, P=.4), or the total group (n=38, r=-.16, P=.34).(Subjects with values below the detectable limit of the assay [10µg/dL] were excluded from this analysis.) When subjects weredivided into two groups based on E2 levels above and below themedian E2 value for their diagnostic group (bulimic patients: 92.3pmol/L; controls: 196.7 pmol/L), then bulimic patients with highE2 levels had peak APRL (n=ll, 15.0±10.7 µg/L) equivalent tothat in bulimic patients with low E2 levels (n=ll, 20.9±9.0 µg/L,P=.18). Likewise, peak APRL in the controls with high E2 levels(n=8, 20.1 + 16.6 µg/L) was equivalent to the peak APRL in thecontrols with low E2 levels (n=8, 30.7±24.6 µg/L, P=.33). The E2levels were not significantly different between the depressed
Downloaded From: http://archpsyc.jamanetwork.com/ by Tim Brewerton on 03/28/2015
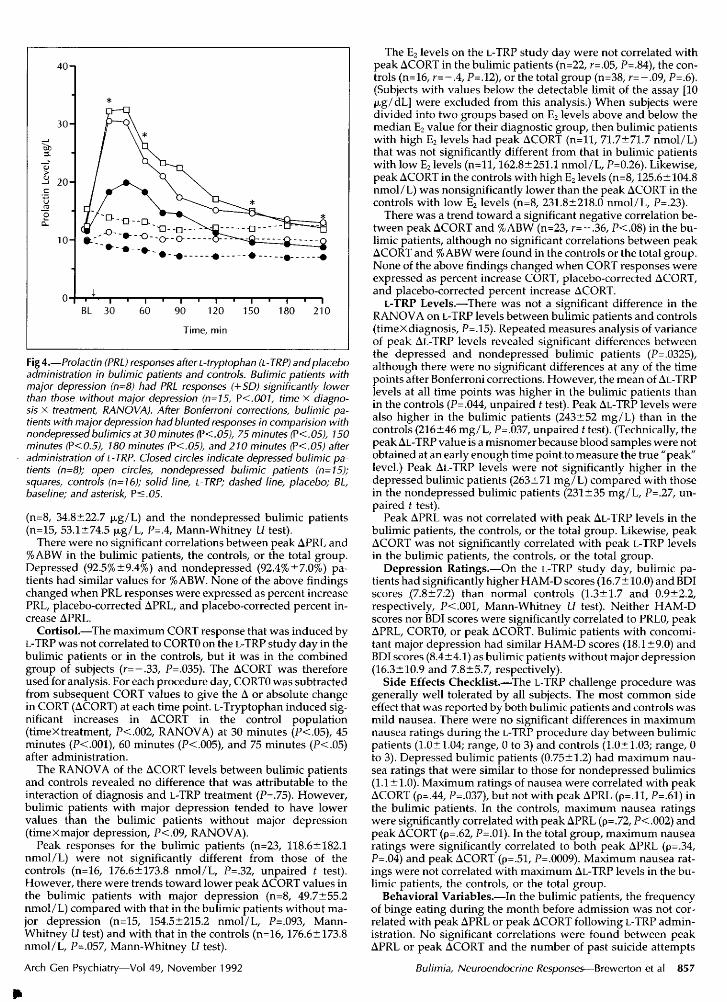
Fig 4.—Prolactin (PRL) responses after L-tryptophan (L-TRP) and placeboadministration in bulimic patients and controls. Bulimic patients withmajor depression (n=8) had PRL responses (+SD) significantly lowerthan those without major depression (n=l5, P<.001, time x diagno¬sis x treatment, RANOVA). After Bonferroni corrections, bulimic pa¬tients with major depression had blunted responses in comparision withnondepressedbulimics at 30 minutes (P<.05), 75 minutes (P<.05), 150minutes (P<0.5), 180 minutes (P<.05), and 210 minutes (P<.05) afteradministration of L-TRP. Closed circles indicate depressed bulimic pa¬tients (n=8); open circles, nondepressed bulimic patients (n=l5);squares, controls (n=16); solid line, L-TRP; dashed line, placebo; BL,baseline; and asterisk, P<.05.
(n=8, 34.8±22.7 µg/L) and the nondepressed bulimic patients(n=15, 53.1±74.5 µg/L, P=.4, Mann-Whitney Li test).
There were no significant correlations between peak APRL and%ABW in the bulimic patients, the controls, or the total group.Depressed (92.5%±9.4%) and nondepressed (92.4%±7.0%) pa¬tients had similar values for %ABW. None of the above findingschanged when PRL responses were expressed as percent increasePRL, placebo-corrected APRL, and placebo-corrected percent in¬crease APRL.
Cortisol.—The maximum CORT response that was induced byL-TRP was not correlated to CORTO on the L-TRP study day in thebulimic patients or in the controls, but it was in the combinedgroup of subjects (r=—.33, P=.035). The ACORT was thereforeused for analysis. For each procedure day, CORTO was subtractedfrom subsequent CORT values to give the A or absolute changein CORT (ACORT) at each time point. L-Tryptophan induced sig¬nificant increases in ACORT in the control population(timeXtreatment, P<.002, RANOVA) at 30 minutes (P<.05), 45minutes (P<.001), 60 minutes (P<.005), and 75 minutes (P<.05)after administration.
The RANOVA of the ACORT levels between bulimic patientsand controls revealed no difference that was attributable to theinteraction of diagnosis and L-TRP treatment (P=.75). However,bulimic patients with major depression tended to have lowervalues than the bulimic patients without major depression(timeXmajor depression, P<,09, RANOVA).
Peak responses for the bulimic patients (n=23, 118.6± 182.1nmol/L) were not significantly different from those of thecontrols (n=16, 176.6±173.8 nmol/L, P=.32, unpaired t test).However, there were trends toward lower peak ACORT values inthe bulimic patients with major depression (n=8, 49.7±55.2nmol/L) compared with that in the bulimic patients without ma¬
jor depression (n=15, 154.5+215.2 nmol/L, P=.093, Mann-Whitney Li test) and with that in the controls (n=16, 176.6±173.8nmol/L, P=.057, Mann-Whitney Li test).
The E2 levels on the L-TRP study day were not correlated withpeak ACORT in the bulimic patients (n=22, r=.05, P=.84), the con¬trols (n=16, r=-.4, P=.12), or the total group (n=38, r=-.09, P=.6).(Subjects with values below the detectable limit of the assay [10µg/dL] were excluded from this analysis.) When subjects weredivided into two groups based on E2 levels above and below themedian E2 value for their diagnostic group, then bulimic patientswith high E2 levels had peak ACORT (n=ll, 71.7±71.7 nmol/L)that was not significantly different from that in bulimic patientswith low E2 levels (n=ll, 162.8±251.1 nmol/L, P=0.26). Likewise,peak ACORT in the controls with high E2 levels (n=8,125.6± 104.8nmol/L) was nonsignificantly lower than the peak ACORT in thecontrols with low E2 levels (n=8, 231.8±218.0 nmol/L, P=.23).
There was a trend toward a significant negative correlation be¬tween peak ACORT and %ABW (n=23, r=-.36, P<.08) in the bu¬limic patients, although no significant correlations between peakACORT and % ABW were found in the controls or the total group.None of the above findings changed when CORT responses were
expressed as percent increase CORT, placebo-corrected ACORT,and placebo-corrected percent increase ACORT.
L-TRP Levels.—There was not a significant difference in theRANOVA on L-TRP levels between bulimic patients and controls(timeX diagnosis, P=.15). Repeated measures analysis of varianceof peak Al-TRP levels revealed significant differences betweenthe depressed and nondepressed bulimic patients (P=.0325),although there were no significant differences at any of the timepoints after Bonferroni corrections. However, the mean of Al-TRPlevels at all time points was higher in the bulimic patients thanin the controls (P=.044, unpaired í test). Peak AL-TRP levels werealso higher in the bulimic patients (243±52 mg/L) than in thecontrols (216±46 mg/L, P=.037, unpaired t test). (Technically, thepeak Al-TRP value is a misnomer because blood samples were notobtained at an early enough time point to measure the true "peak"level.) Peak Al-TRP levels were not significantly higher in thedepressed bulimic patients (263±71 mg/L) compared with thosein the nondepressed bulimic patients (231 ±35 mg/L, P=.27, un¬
paired f test).Peak APRL was not correlated with peak Al-TRP levels in the
bulimic patients, the controls, or the total group. Likewise, peakACORT was not significantly correlated with peak L-TRP levelsin the bulimic patients, the controls, or the total group.
Depression Ratings.—On the L-TRP study day, bulimic pa¬tients had significantly higher HAM-D scores (16.7± 10.0) and BDIscores (7.8±7.2) than normal controls (1.3±1.7 and 0.9±2.2,respectively, P<.001, Mann-Whitney Li test). Neither HAM-Dscores nor BDI scores were significantly correlated to PRLO, peakAPRL, CORTO, or peak ACORT. Bulimic patients with concomi¬tant major depression had similar HAM-D scores (18.1 ±9.0) andBDI scores (8.4±4.1) as bulimic patients without major depression(16.3±10.9 and 7.8±5.7, respectively).
Side Effects Checklist.—The L-TRP challenge procedure was
generally well tolerated by all subjects. The most common sideeffect that was reported by both bulimic patients and controls wasmild nausea. There were no significant differences in maximumnausea ratings during the L-TRP procedure day between bulimicpatients (1.0±1.04; range, 0 to 3) and controls (1.0±1.03; range, 0to 3). Depressed bulimic patients (0.75±1.2) had maximum nau¬sea ratings that were similar to those for nondepressed bulimics(1.1 ±1.0). Maximum ratings of nausea were correlated with peakACORT (p=.44, P=.037), but not with peak APRL (p=.ll, P=.61) inthe bulimic patients. In the controls, maximum nausea ratingswere significantly correlated with peak APRL (p=.72, P<.002) andpeak ACORT (p=.62, P=.01). In the total group, maximum nausea
ratings were significantly correlated to both peak APRL (p=.34,P=.04) and peak ACORT (p=.51, P=.0009). Maximum nausea rat¬ings were not correlated with maximum AL-TRP levels in the bu¬limic patients, the controls, or the total group.
Behavioral Variables.—In the bulimic patients, the frequencyof binge eating during the month before admission was not cor¬related with peak APRL or peak ACORT following L-TRP admin¬istration. No significant correlations were found between peakAPRL or peak ACORT and the number of past suicide attempts
Downloaded From: http://archpsyc.jamanetwork.com/ by Tim Brewerton on 03/28/2015

or the number of years of binge eating or vomiting. Peak APRLand peak ACORT were not significantly different betweenpatients with and without a history of stealing or a past SADS-Ldiagnosis of alcohol or drug abuse.
Seasonal Variation.—Given the seasonal variation in APRLand ACORT responses following m-CPP administration, as notedabove, an analysis of variance was performed with the use ofseason as a grouping variable for ApRL and ACORT responsesfollowing L-TRP treatment. However, because the depressed pa¬tients had lower peak APRL and peak ACORT responses, thesewere eliminated from this analysis. Significant seasonal differ¬ences in peak APRL were identified in the nondepressed patients(P=.0064), the controls (P=.02), and the combined group (P=.0003).Post hoc f tests with Bonferroni corrections revealed significantdifferences between fall (n=4,50.7±22.2 µg/L) and winter (n=10,24.1±15.4 µg/L), fall and spring (n=6, 21.2+6.1 µg/L), and falland summer (n=ll, 14.6±3.9 µg/L) in the combined group. Sig¬nificant post hoc differences were found between fall (n=3,55.1±25.0 µg/L) and summer (n=7, 14.0±4.2 µg/L) in thecontrols, although no such differences were found in the nonde¬pressed patients.
Peak ACORT was not found to vary significantly by season inthe patients, the controls, or the total group, regardless of whetherdepressed patients were included in the analysis.
Interrelationships Between Responses tom-Chlorophenylpiperazine and L-TRP
m-CPP (PRL and CORT).—Peak APRL was correlated to peakACORT in the bulimic patients (p=.40, P=.042) and in the entiregroup (p=.33, P=.036), but not in the controls alone (p=.08, P=.77).Peak APRL was inversely correlated to CORTO in the bulimic pa¬tients (p= —.40, P=.043) and, to a lesser extent, in the entire group(p=-.26, P=.l), but not in the controls alone (p=.36, P=.18). PeakACORT was correlated to PRLO in the bulimic patients (p=.41,P=.038), but not in the controls or in the entire group.
L-TRP (PRL and CORT).—Peak APRL was correlated to peakACORT in the controls (n=16, r=.67, P-C005) and in the entiregroup (n=39, r=.35, P=.042), but not in the bulimic patients alone(n=23, r=-.04, P=.88). Peak APRL was not correlated to CORTOin the bulimic patients, the controls alone, or the entire group.Peak ACORT was not correlated to PRLO in the bulimic patients,the controls, or the entire group.
PRL (m-CPP and L-TRP).—Peak APRL following m-CPP treat¬ment was not correlated to peak APRL following L-TRP treatmentin the bulimic patients (p=.25, P=.27), in the controls alone (p=.18,P=.54), or in the entire group (p=.26, P=.13).
CORT (m-CPP and L-TRP).—Peak ACORT following m-CPPwas not correlated to peak ACORT following L-TRP administra¬tion in the bulimic patients (p=.02, P=.93), in the controls alone(p=-.17, P=.55), or in the entire group (p=-.09, P=.63).
Order Effects.—There were no significant effects or order ofdrug administration for peak APRL or peak ACORT in either thepatients or the controls.
COMMENTOur finding of lower baseline PRL levels in the bulimic
patients as compared with that in the healthy controls issimilar to that previously reported.93 There is evidence tosupport94 and refute95-96 a role for 5-HT in determiningbaseline PRL levels, although other neurochemical sys¬tems may be involved, such as dopamine and the opiates.
The reduced APRL responses to m-CPP, as demon¬strated in this study of bulimic females, is, to our knowl¬edge, the first neuroendocrine evidence of serotonergicdysfunction in normal weight bulimic patients. The alteredresponses cannot be attributed to the controlled variablesof age, weight, percent of average body weight, activity,medications, or time of day. Nor are they likely to be dueto a defect in the pituitary lactotroph since we saw normal
PRL responses following L-TRP administration, and thisoccurs in bulimic patients following treatment with pro-tirelin (thyrotropin-releasing hormone).93 Whether our
finding of an alteration in 5-HT functioning represents acausative factor in the pathogenesis of bulimia nervosa, isa sequela of the disorder, or a combination of both cannotbe stated with certainty.
Recent, short-term dieting, which can cause 5-HT recep¬tor upregulation,97 is an unlikely cause of the observedchanges. Twenty of the 26 patients were studied after a 3-to 4-week period of careful weight maintenance and absti¬nence from binge eating and purging. Furthermore, the sixoutpatients studied who were actively engaged in bulimicbehaviors before study had APRL responses equivalent tothose of the 20 abstinent inpatients. Nevertheless, thesefindings could represent persistent effects of long-term di¬eting. Binge eating and vomiting, which may enhancecentral 5-HT synthesis,98 could conceivably result in down-regulation of postsynaptic 5-HT receptors.31-33-35-39-99 Al¬though the 3- to 4-week period of abstinence might restoreat least a partial return to premorbid conditions, long-termalterations in 5-HT receptor functioning as a result ofchronic binge eating and vomiting are certainly possible.However, it may be that the blunted responses reflect a
premorbid state that predisposes these patients towardbulimia nervosa and perhaps affective illness. The strongtrend for an inverse correlation between weekly binge fre¬quency during the month before admission and peakAPRL is compatible with either of the above interpreta¬tions.
Drug levels of m-CPP do not account for the blunted PRLresponses in the bulimic patients, since m-CPP levels were
essentially equivalent in the two groups of subjects.Furthermore, when peak APRL was corrected for m-CPPlevels, the differences between bulimic patients and con¬trols was actually magnified.
Although PRL responses following m-CPP in the bulimicpatients with major depression (38% of the sample) were
lower than those in the bulimic patients without major de¬pression (62%), this difference did not reach statistical sig¬nificance. The PRL responses following m-CPP treatmentfor both groups were significantly lower than those forcontrols. The presence of major depression cannot there¬fore fully account for the variance between bulimic pa¬tients and controls. Moreover, neither peak APRL nor peakACORT correlated with HAM-D or BDI scores.Peak APRLfollowing m-CPP administration was inversely correlatedto baseline CORT in the bulimic patients and, to a lesserextent, in the entire group. This finding is similar to the re¬
port in animals of blunted PRL responses following m-CPPtreatment after long-term CORT treatment.100 Other re¬
ports also have suggested an inverse relationship between5-HT function and the hypothalamic-pituitary-adrenalaxis.101102 In brief, these findings suggest that chronic stressor other poorly understood factors may further compro¬mise postsynaptic 5-HT receptor sensitivity.
Cortisol responses, following m-CPP administration, didnot differ between the bulimic patients and the controls.The differences between PRL and CORT responsivity maybe because different anatomical pathways,103106 as well as
different 5-HT receptor subtypes,107"109 are involved in theirstimulation. Cortisol secretion results from corticotropin-releasing hormone stimulation at the level of the me-diobasal hypothalamus, as well as a direct action by 5-HTon the adrenal gland.105106
Downloaded From: http://archpsyc.jamanetwork.com/ by Tim Brewerton on 03/28/2015

Our findings of blunted PRL responses following L-TRPin bulimic patients with major depression are consistentwith findings from previous reports.61,62 The blunting of thePRL responses in the depressed group of bulimic patientscould not be accounted for by differences in age, % ABW,recent activity or dieting, medications, time of day, orL-TRP plasma levels, which was a major variable in a priorstudy.56 Since depressed bulimic patients did not have sig¬nificantly lower E2 levels than nondepressed bulimicpatients, the blunting also does not appear to be due todecreased E2 levels. In addition to blunted PRL responsesfollowing L-TRP in the depressed bulimic patients, therewas also a trend toward blunted CORT responses in thedepressed bulimic patients in comparison with those of thenondepressed bulimic patients and the controls. This iscompatible with a more generalized blunting of 5-HT re¬
sponsiveness in the depressed vs the nondepressed patientgroup, although alterations in the hypothalamic-pituitary-adrenal axis may contribute to this finding.
Although the 17ß-estradiol (E2) level was lower inbulimic patients in comparison with that in controls, wecould not establish a direct link between E2 levels and PRLor CORT responses to m-CPP or L-TRP. Estradiol has beenshown to stimulate PRL synthesis and release,110"114 in partmediated by serotonergic neurons and receptors.115"119 Ourdata analyses were limited by the fact that a number ofpatient E2 values were below the sensitivity of the assay.Thus, our PRL abnormalities could possibly be secondaryto dysfunction in the reproductive system.
Although the exact locus of the 5-HT abnormality thatis responsible for a blunted PRL response cannot be knownwith certainty, current evidence from studies in humansindicates that PRL release is stimulated by 5-HTj receptorsand inhibited by 5-HT2 receptors.108 In contrast, animaldata indicate that CORT stimulation is mediated by both5-HT, and 5-HT2 receptors.109 Normal or enhanced sensi¬tivity of 5-HT2 receptors, together with decreased sensitiv¬ity of 5-HT] receptors, would be a possible interpretationof these data.89
Why was there significant blunting of PRL in both thedepressed and nondepressed bulimic patients followingm-CPP treatment and only in the depressed bulimicpatients following L-TRP treatment? One possible reasonis that we simply did not study enough patients. Anotherrests on the different mechanisms of action between thesetwo agents. To the extent that L-TRP stimulation of PRLinvolves both presynaptic and postsynaptic serotonergicmechanisms, and m-CPP stimulation involves onlypostsynaptic receptor mechanisms, it is suggested thatmajor depression, as a state, may preferentially involve a
disruption of presynaptic 5-HT mechanisms.120'126 Our datasuggest a possible defect in L-TRP transport or metabolismassociated with major depression, since the depressed bu¬limic patients had higher L-TRP levels over time than boththe nondepressed bulimic patients and controls. Interest¬ingly, the ratio of peak Al-TRP to peak APRL was signif¬icantly lower over time in bulimic patients compared withthat in controls (P<.03, RANOVA). This ratio was espe¬cially lower in depressed bulimic patients compared withthat in controls (P<.01) and would be compatible with im¬paired transport of L-TRP across the blood-brain barrier ora decreased rate of 5-HT synthesis or release.
Another possible explanation for the normal responsesin the nondepressed bulimic patients is that the decreasein postsynaptic sensitivity may be compensated for by an
increase in presynaptic output of 5-HT. This adaptivecountermeasure may fail during the development of thestate of major depression, possibly because of the effects ofCORT on 5-HT receptor sensitivity.
There remains the possibility that L-TRP administrationmay stimulate or influence PRL and/or CORT release vianonserotonergic mechanisms, either through the action ofone of its metabolites, eg, tryptamine, kynurenine, or me-latonin, or through stimulation of another neurotransmit¬ter or peptide system.
In summary, our findings indicate that postsynapticresponsiveness in hypothalamic-pituitary-serotonergicpathways that mediate PRL release is impaired in bulimianervosa, regardless of the presence of major depression.Similar alterations in other 5-HT pathways at or above thelevel of the hypothalamus may contribute to binge eatingand other behavioral symptoms in bulimic patients.89127Further studies that will explore the role of altered 5-HTreceptor function in bulimia nervosa are warranted.
The authors gratefully acknowledge the assistance of Joy Kassett,MSW, MPH, with the Schedule for Affective Disorders andSchizophrenia-Lifetime version interviews; John Bartko, PhD, withstatistical consultation; the support of the National Institute of Men¬tal Health 3-East nursing staff; and Naomi Bloom, MD, with dataanalysis.
References1. Blundell JE. Serotonin and appetite. Neuropharmacology. 1984;23:
1537-1551.2. Blundell JE. Is there a role for serotonin (5-hydroxy-tryptamine) in feed-
ing? Int J Obes. 1977;1:15-42.3. Stunkard AJ, Craighead LW, O'Brien R. Controlled trial of behavior
therapy, pharmacotherapy, and their combination in the treatment of obesity.Lancet. 1980;2:1045-1047.
4. Wurtman JJ, Wurtman RJ. Suppression of carbohydrate consumption assnacks and at mealtime by dl-fenfluramine or tryptophan. In: Garrattini S, ed.Anorectic Agents: Mechanism of Actions and of Tolerance. New York, NY:Raven Press; 1981:169-181.
5. Wurtman JJ, Wurtman RJ, Growdon JH, Henry P, Lipscomb A, ZeiselSH. Carbohydrate craving in obese people: suppression by treatments affect-ing serotonergic transmission. Int J Eat Disord. 1981;1:2-15.
6. Halmi KA, Eckert ED, LaDu TJ, Cohen J. Anorexia nervosa treatment ef-ficacy of cyproheptadine and amitriptyline. Arch Gen Psychiatry. 1986;43:177-181.
7. Murphy DL, Campell I, Costa JL. Current status of the indoleamine hy-pothesis of affective disorders. In: Lipton MA, DiMascio A, Killam KF, eds.Psychopharmacology: A Generation ofProgress. New York, NY: Raven Press;1978:1234-1248.
8. Coppen A, Wood K. 5-Hydroxytryptamine in the pathogenesis of affec-tive disorders. In: Ho BT, Schoolar JC, Usdin E, eds. Serotonin in BiologicalPsychiatry. New York, NY: Raven Press; 1982:249-258.
9. Meltzer HY, Lowy MT. The serotonin hypothesis of depression. In:Meltzer HY, ed. Psychopharmacology: The Third Generation of Progress.New York, NY: Raven Press; 1987:513-526.
10. Weiss SR, Ebert MH. Psychological and behavioral characteristics ofnormal-weight bulimics and normal-weight controls. Psychosom Med.1983;45:293-303.
11. Abraham SF, Beumont PJV. How patients describe bulimia or bingeeating. Psychol Med. 1982;12:625-635.
12. Johnson C, Larson R. Bulimia: an analysis of moods and behavior.Psychosom Med. 1982;44:341-351.
13. Hatsukami DK, Mitchell JE, Eckert ED. Eating disorders: a variant ofmood disorders? Psychiatr Clin North Am. 1984;7:349-365.
14. Brewerton TD, Heffernan MM, Rosenthal NE. Psychiatric aspects ofthe relationship between eating and mood. Nutr Rev. 1986;44(suppl):78-88.
15. Brewerton TD, Lyriard RB, Laraia M, O'Neil P, Ballenger JC. Comor-bidity of psychiatric disorders in bulimia nervosa. In: Program and abstractsof the Fourth International Conference on Eating Disorders; April 27-29,1990; New York, NY. Abstract 236.
16. Hudson JI, Pope HG, Jonas JM, Yurgelun-Todd D. Phenomenologicrelationship of eating disorders to major affective disorder. Psychiatry Res.1983;9:345-354.
17. Walsh BT, Roose SP, Glassman AH, Savik C. Bulimia and depression.Psychosom Med. 1985;47:123-131.
18. Hudson JI, Pope HG, Yurgelun-Todd D, Jonas JM, Frandenburg FR. Acontrolled study of lifetime prevalence of affective and other psychiatric dis-orders in bulimic outpatients. Am J Psychiatry. 1987;144:1283-1287.
Downloaded From: http://archpsyc.jamanetwork.com/ by Tim Brewerton on 03/28/2015

19. Kassett JA, Gershon ES, Maxwell ME, Guroff JJ, Kazuba DM, Smith AL,Brandt HA, Jimerson DC. Psychiatric disorders in the first-degree relatives ofprobands with bulimia nervosa. Am J Psychiatry. 1989;146:1468-1471.
20. Hudson JI, Pope HG, Jonas JM. Treatment of bulimia with antidepres-sants: theoretical considerations and clinical findings. In: Stunkard AJ, StellarE, eds. Eating and Its Disorders. New York, NY: Raven Press; 1984:259-273.
21. Pope HG, Hudson JI, Jonas JM, Yurgelum-Todd D. Bulimia treatedwith imipramine: a placebo-controlled, double-blind study. Am J Psychiatry.1983;140:554-558.
22. Agras WS, Dorian B, Kirkley BG, Arnow B, Bachman J. Imipramine inthe treatment of bulimia: a double-blind controlled study. Int J Eat Disord.1987;6:29-38.
23. Pope HG, Hudson JI. Antidepressant drug therapy for bulimia: currentstatus. J Clin Psychiatry. 1986;47:339-345.
24. Hughes PL, Wells LA, Cunningham CJ, Ilstrup DM. Treating bulimiawith desipramine: a double-blind, placebo-controlled study. Arch Gen Psy-chiatry. 1986;43:182-186.
25. Barlow J, Blouin J, Blouin A, Perez E. Treatment of bulimia with desi-pramine: a double-blind crossover study. Can J Psychiatry. 1988;33:129-133.
26. Walsh BT, Stewart JW, Rose SP, Gladis M, Glassman AH. Treatmentof bulimia with phenelzine: a double-blind, placebo-controlled study. ArchGen Psychiatry. 1984;41:1105-1109.
27. Kennedy SH, Piran N, Warsh JJ, Prendergast P, Mainprize E, WhynotC, Garfinkel PE. A trial of isocarboxazid in the treatment of bulimia nervosa.
J Clin Psychopharmacol. 1988;8:391-396.28. Enas GG, Pope HG, Levine LR. Fluoxetine in bulimia nervosa:
double-blind study. In: Program and abstracts of the 142nd meeting of theAmerican Psychiatric Association; May 4-6, 1989; San Francisco, Calif. Ab-stract NR386.
29. Freeman CPL, Hampson M. Fluoxetine as a treatment for bulimia ner-vosa. Int JObes1987;11(suppl 3):171-177.
30. Benfield P, Heel RC, Lewis SP. Fluoxetine: a review of its pharmaco-dynamic and pharmacokinetic properties, and therapeutic efficacy in de-pressive illness. Drugs. 1986;32:481-508.
31. Fuxe K, Ogren S-O, Agnati LF, et al. Chronic antidepressant treatmentand central 5-HT synapses. Neuropharmacology. 1983;22:389-400.
32. Charney DC, Menkes DB, Heninger GR. Receptor sensitivity and themechanism of action of antidepressant treatment: implications for the etiol-ogy and therapy of depression. Arch Gen Psychiatry. 1981;38:1160-1180.
33. Gastpar M, Wakelin JS. Selective 5-HT Reuptake Inhibitors: Novel or
Commonplace Agents. New York, NY: S Karger AG; 1988.34. Charney DS, Heninger DR, Sternberg DE. Serotonin function and
mechanism of action of antidepressant treatment: effects of amitriptyline anddesipramine. Arch Gen Psychiatry. 1983;41:359-365.
35. Willner P. Antidepressants and serotonergic transmission: an integra-tive review. Psychopharmacology (Berl). 1985;85:387-404.
36. Cowen PJ, Geaney DP, Sch\l=a"\chterM, Green AR, Elliott JM. Desi-pramine treatment in normal subjects: effects on neuroendocrine responsesto tryptophan and on platelet serotonin (5-HT)-related receptors. Arch GenPsychiatry. 1986;43:61-67.
37. deMontigny C, Aghajanian GK. Tricyclic antidepressants: long-termtreatment increases responsivity of rat forebrain neurons to serotonin.Science. 1978;202:1303-1306.
38. deMontigny C, Blier P. Electrophysiological laspects of serotoninneuropharmacology: implications for antidepressant treatments. In: GreenAR, ed. Neuropharmacology ofSerotonin. New York, NY: Oxford UniversityPress Inc; 1985:181-195.
39. \l=O"\grenS-O, Fuxe K. Effects of antidepressant drugs on serotonergic re-
ceptors. In: Green AR, ed. Neuropharmacology ot Serotonin. New York, NY:Oxford University Press Inc; 1985:131-180.
40. Blier P, deMontigny C, Chaput Y. Modification of the serotonergicsystem by antidepressant treatments: implications for the therapeutic re-
sponse in major depression. J Clin Psychopharmacol. 1987;7:245-355.41. Whitaker-Azmitia, PM, Perovtka SV, eds. The neuropharmacology of
serotonin. Ann N Y Acad Sci. 1990;600:.42. Linnoila M, Virkhunen M, Scheinin M, Nuutila A, Rimon R, Goodwin
FK. Low cerebrospinal fluid 5-hydroxyindoleacetic acid concentrationdifferentiates impulsive from nonimpulsive violent behavior. Life Sci. 1983;33:2609-2614.
43. Asberg M, Schalling D, Traskman-Bendz L, Wagner A. Psychobiologyof suicide, impulsivity, and related phenomena. In: Meltzer H, ed. Psychop-harmacology: The Third Generation of Progress. New York, NY: Raven Press;1987:655-668.
44. Valzelli L, Bernasconi S, Dalessandro M. Effect of tryptophan admin-istration on spontaneous and p-PCA-induced muricidal aggression in labo-ratory rats. Pharmacol Res. 1981;13:891-897.
45. Asberg M, Thoren P, Traskman L. 'Serotonin depression': a biochem-ical subgroup within the affective disorders? Science. 1976;191:478.
46. Asberg M, Traskman L, Thoren P. 5-HIAA in the cerebrospinal fluid:a biochemical suicide predictor? Arch Gen Psychiatry. 1976;33:1193.
47. Brown GL, Ebert MH, Boyer PF, et al. Aggression, suicide, and sero-tonin: relationships to csf amine metabolites. Am J Psychiatry. 1982;139:741-746.
48. Kaye WH, Ebert MH, Gwirtsman HE, Weiss SR. Differences in brainserotonergic metabolism between nonbulimic and bulimic patients with an-
orexia nervosa. Am J Psychiatry. 1984;141:1598-1601.49. Garfinkel PE, Garner DM. Subtypes of anorexia nervosa. In: Garfinkel
PE, Garner DM, eds. Anorexa Nervosa: A Multidimensional Perspective. NewYork, NY: Brunner/Mazel Inc; 1982:40-57.
50. Crisp AH, Hsu LKG, Harding B. The starving hoarder and voraciousspender: stealing in anorexia nervosa. J Psychosom Res. 1980;24:225-231.
51. Russell GFM. Bulimia nervosa: an ominous variant of anorexianervosa. Psychol Med. 1979;9:429-448.
52. French AP, Nelson HL. Genital self-mutilation in women. Arch GenPsychiatry. 1972;27:618-620.
53. Goldney RD, Simpson IG. Female genital self-mutilation, dysorexiaand the hysterical personality: the Caenis syndrome. Can J Psychiatry. 1975;20:435-441.
54. Simpson MA. The phenomenology of self-mutilation in a general hos-pital setting. Can J Psychiatry. 1975;20:429-434.
55. Rosenthal RJ, Rinzler C, Wallsh R, Klausner E. Wrist-cutting syndrome:the meaning of a gesture. Am J Psychiatry. 1972;11:1363-1368.
56. Koyama T, Meltzer HY. A biochemical and neuroendocrine study ofthe serotonergic system in depression. In: Hippius H, et al, eds. New Resultsin Depression. New York, NY: Springer-Verlag NY Inc; 1986:169-188.
57. Charney DS, Heninger GR, Reinhard JF, Sternberg DE, Hafstead KM.The effect of IV I-tryptophan on prolactin and growth hormone and mood inhealthy subjects. Psychopharmacology (Berl). 1982;7:212-222.
58. Van Pragg HM. In search of the mode of action of antidepressants:5-HTP/tyrosine mixtures in depressions. Neuropharmacology. 1983;22:433\p=m-\440.
59. Maclntoe JH, Turkington RW. Stimulation of human prolactin secre-tion by intravenous infusion of I-tryptophan. J Clin Invest. 1973;52:1972\p=m-\1978.
60. Deakin JFW, Pennell I. 5-HT receptor subtypes and depression. psy-chopharmacology. 1986;89:S24.
61. Heninger GR, Charney DS, Sternberg DE. Serotonergic function indepression: prolactin response to intravenous tryptophan in depressedpatients and healthy subjects. Arch Gen Psychiatry. 1984;41:398-402.
62. Cowen PJ, Charig EM. Neuroendocrine responses to intravenous tryp-tophan in major depression. Arch Gen Psychiatry. 1987;44:958-966.
63. Siever LJ, Murphy DL, Slater S, de la Vega E, Lipper S. Plasma prolac-tin changes following fenfluramine in depressed serotonergic responsivity indepression. Life Sci. 1984;34:1029-1039.
64. Caccia S, Fong MH, Garattini S, Zanini MG. Plasma concentrations oftrazodone and 1-(3-chlorophenyl) piperazine in man after a single oral doseof trazodone. J Pharm Pharmacol. 1982;34:605-606.
65. Mueller EA, Murphy DL, Sunderland T. Neuroendocrine effects ofm-chlorophenylpiperazine, a serotonin agonist, in humans. JClin EndocrinolMetab. 1985;61:1179-1184.
66. Invernizzi R, Cotecchia S, De Biasi A, Mennini T, Pataccini R, Sama-nin R. Effects of m-chlorophenylpiperazine on receptor binding and brainmetabolism of monoamine in rats. Neurochem Int. 1981;3:239-244.
67. Fuller RW, Snoddy HD, Mason NR, Owen JE. Disposition and phar-macological effects of m-chlorophenylpiperazine in rats. Neuropharmacol-ogy. 1981;20:155-162.
68. Aloi JA, Insel TR, Mueller EA, Murphy DL. Neuroendocrine and be-havioral effects of m-chlorophenylpiperazine administration in monkeys. LifeSci. 1984;34:1325-1331.
69. Samanin R, Mennini T, Ferraris A. m-Chlorophenylpiperazine: a cen-tral serotonin agonist causing powerful anorexia in rats. Naunyn Schmiede-bergs Arch Pharmacol. 1982;308:605.
70. Martin LL, Sanders-Bush E. Comparison of the pharmacological char-acteristics of t-HT, and 5-HT2 binding sites with those of serotonin autore-ceptors which modulate serotonin release. Naunyn Schmiedebergs ArchPharmacol. 1982;321:165-170.
71. Murphy DL, Mueller EA, Hill JL, Tolliver TJ, Jacobsen FM. Compara-tive anxiogenic, neuroendocrine, and other physiologic effects ofm-chlorophenylpiperazine given intravenously or orally to healthy volun-teers. Psychopharmacology (Berl). 1989;98:275-282.
72. Hamik A, Peroutka SJ. 1-(m-Chlorophenyl) piperazine (m-CPP) inter-actions with neurotransmitter receptors in the human brain. Biol Psychiatry.1989;25:569-575.
73. Kennett GA, Curzon G. Evidence that hypophagia induced by m-CPPand TFMPP requires 5-HT1c and 5-HT1B receptors: hypophagia induced byRU 24969 only requires 5-HT1B receptors. Psychopharmacology (Berl).1988;96:93-100.
74. Kennett GA, Curzon G. Evidence that m-CPP may have behaviouraleffects mediated by central 5-HT1c receptors. Br J Pharmacol. 1988;94:137\p=m-\147.
75. Hoyer D, Pagos A, Probst A, Palacios JM. Serotonin receptors in thehuman brain, I: characterizations and autoradiographic localization of5-HT1A recognition sites: apparent absence of 5-HT1B recognition sites. BrainRes. 1986;376:85-96.
76. Mueller EA, Murphy DL, Sunderland T. Further studies of the putativeserotonin agonist, m-chlorophenylpiperazine: evidence for a serotoninreceptor mediated mechanism of action in humans. Psychopharmacology(Berl). 1986;89:388-391.
77. Bagby G, Szemeredi K, Kanyicska B, Murphy DL. Different serotoninreceptors mediate blood pressure, heart rate, plasma catecholamine and
Downloaded From: http://archpsyc.jamanetwork.com/ by Tim Brewerton on 03/28/2015

prolactin responses to m-chlorophenylpiperazine in conscious rats. J Phar-macol Exp Ther. 1989;250:72-78.
78. Conn PJ, Sanders-Bush E. Relative efficacious of piperazines at thephosphoinositide hydrolysis-linked serotonergic (5-HT2 and 5-HT1C) recep-tors. J Pharmacol Exp Ther. 1987;242:552-557.
79. American Psychiatric Association. Diagnostic and Statistical Manualof Mental Disorders. 3rd ed. Washington, DC: American Psychiatric PressInc; 1980.
80. American Psychiatric Association. Diagnostic and Statistical Manualof Mental Disorders. revised 3rd ed. Washington, DC: American PsychiatricPress Inc; 1986.
81. Spitzer RL, Endicott J. Schedule for Affective Disorders andSchizophrenia-Lifetime version. Biometrics Research. New York, NY: NewYork State Psychiatric Institute; 1975.
82. Society of Actuaries and Association of Life Insurance Medical Direc-tors of America. Build Study, 1979. Chicago, Ill: Society of Actuaries; 1980.
83. Miller RL, Devane CL. Analysis of trazodone and m-chloro-phenylpiperazine in plasma and brain tissue by high-performance liquid chro-matography. J Chromatogr. 1986;374:388-393.
84. Narang PK, Courtney J, Garrick NA, Blumhardt T, Murphy DL. In:Program and abstracts of the 133rd annual meeting of the AmericanPharmaceutical Association; 1986; San Francisco, Calif. Abstract P-61.
85. Larsson M, Forsman A, Hallgren J. HPLC assays of 5-HIAA and tryp-tophan in cerebrospinal fluid and 5-HT and tryptophan in blood: a method-ological study with clinical applications. Methods Find Exp Clin Pharmacol.1988;10:453-460.
86. Hamilton M. A rating scale for depression. J Neurol NeurosurgPsychiatry. 1960;6:278-296.
87. Beck AT, Ward CH, Mendelson M, Mock J, Erbaugh J. An inventoryfor measuring depression. Arch Gen Psychiatry. 1961;4:561-571.
88. Brewerton TD, Murphy D, Mueller EA, Jimerson DC. The induction ofmigraine-like headaches by the serotonin agonist, m-chlorophenyl-piperazine. Clin Pharmacol Ther. 1988;43:605-609.
89. Brewerton TD, Murphy DL, Lesem DT, Brandt HA, Jimerson DC. Se-rotonin dysregulation in bulimia nervosa: neuroendocrine and headache re-
sponses. In: Van Praag H, Brown S, eds. Serotonin in Psychiatric Disorders.Einstein Monograph Series. New York, NY: Brunner/Mazel Inc; 1990:239\x=req-\259.
90. Johnson C. Initial consultation for patients with bulimia and anorexianervosa. In: Garner DM, Garfinkel PE, eds. Handbook of Psychotherapy forAnorexia Nervosa and Bulimia. New York, NY: Guilford Press; 1985:19-51.
91. Brewerton TD, Berrettini W, Nurnberger J, Linnoila M. An analysis ofseasonal fluctuations of CSF monoamines and neuropeptides in normal con-trols: findings with 5-HIAA and HVA. Psychiatry Res. 1988;23:257-265.
92. Brewerton TD. Seasonal variation of serotonin function in humans:research and clinical implications. Ann Clin Psychiatry. 1989;1:1-12.
93. Levy AB, Dixon KN, Malarkey WB. Pituitary response to TRH in bu-limia. Biol Psychiatry. 1988;23:476-484.
94. Benkelfat C, Murphy DL, Zohar J, Hill JL, Grover G, Insel TR. Clomi-pramine in obsessive-compulsive disorder: further evidence for a serotoner-gic mechanism of action. Arch Gen Psychiatry. 1990;46:23-28.
95. Willoughby JO, Menadue M, Jervois P. Function of serotonin in phys-iologic secretion of growth hormone and prolactin: action of 5,7\x=req-\dihydroxytryptamine, fenfluramine and p-chlorophenylalanine. Brain Res.1982;249:291-299.
96. Richards GE, Holland FJ, Aubert ML, Ganong WF, Kaplan SL, Grum-bach MM. Regulation of prolactin and growth hormone secretion. Neuroen-docrinology. 1980;30:139-143.
97. Goodwin GM, Fraser S, Stump K, Fairburn CG, Elliott JM, Cowen PJ.Dieting and weight loss in volunteers increases the number of alpha2-adrenoceptors and 5-HT receptors on blood platelets without effects on
[3H]imipramine binding. J Affective Disord. 1987;12:267-274.98. Kaye WH, Gwirtsman HE, Brewerton TD, George DT, Wurtman RJ.
Bingeing behavior and plasma amino acids: a possible involvement of brainserotonin in bulimia. Psychiatry Res. 1988;23:31-43.
99. Mennini T. Poggesi E, Caccia S, Bendotti C, Borsini F, Samanin R.Adaptive changes in central serotonin receptors after long-term drug treat-ment in rats. In: Littauer UZ, Dudai Y, Silman I, eds. Neurotransmitter andTheir Receptors. New York, NY: John Wiley & Sons Inc; 1980:101-118.
100. Bagby G, Calogero AE, Aulakh CS, Szemeredi K, Murphy DL. Long-term cortisol treatment impairs behavioral and neuroendocrine responses to5-HT1 agonists in the rat. Neuroendocrinology. 1989;50:241-247.
101. Stokes PE, Maas JW, Davis JM, Koslow SH, Casper RC, Stoll PM.Biogenic amine and metabolite levels in depressed patients with high versusnormal hypothalamic-pituitary-adrenocortical activity. Am J Psychiatry.1987;144:868-872.
102. Roy A, Everett D, Pickar D, Paul SM. Platelet tritiated imipraminebinding and serotonin uptake in depressed patients and controls. Arch GenPsychiatry. 1987;44:320-327.
103. Van de Kar LD, Bethea CL. Pharmacological evidence that seroton-ergic stimulation of prolactin secretion is mediated via the dorsal raphe nu-cleus. Neuroendocrinology. 1982;35:225-230.
104. Van de Kar LD, Wilkinson CW, Skrobik Y, Brownfield MS, GanongWF. Evidence that serotonergic neurons in the dorsal raphe nucleus exert a
stimulatory effect on the secretion of renin but not of corticosterone. BrainRes. 1982;235:233-243.
105. Van de Kar LD, Karteszi M, Bethea CL, Banong WF. Serotonergicstimulation of prolactin and corticosterone secretion is mediated by differentpathways from the mediobasal hypothalamus. Neuroendocrinology. 1985;41:380-384.
106. Verdesca AS, Westermann CD, Crampton RS, Black WC, Nedeljk-ovic RI, Hilton JG. Direct adrenocortical stimulatory effect of serotonin. AmJ Physiol. 1961;201:1065-1067.
107. Bero LA, Kurn CM. Differential ontogeny of opioid, dopamingericand serotonergic regulation of prolactin secretion. J Pharmacol Exp Ther.1987;240:825-830.
108. Charig EM, Anderson IM, Robinson JM, Nutt DJ, Cowen PJ.Tryptophan and prolactin release: evidence for interaction between 5-HT1and 5-HT2 receptors. Hum Psychopharmacol. 1986;1:93-97.
109. Koenig JI, Gudelsky GA, Meltzer HY. Stimulation of corticosteroneand beta-endorphin secretion in the rat by selective 5-HT receptor subtypeactivation. Eur J Pharmacol. 1987;137:1-8.
110. Yamamoto N, Seo H, Suganuma N, Matsui N, Nakane T, KuwayamaA, Kageyama N. Effect of estrogen on prolactin mRNA in the rat pituitary.Neuroendocrinology. 1986;42:494-497.
111. Kaplan SE, DeNicola AF. Protein and RNA synthesis in pituitary tu-mors from F344 rats given implants of estrogen. J Natl Cancer Inst. 1976;56:37-42.
112. Lieberman LE, Szjan I, Brudman JA. Cell proliferation in the rat pi-tuitary gland: a mechanism of control in prolactin rats. Experientia. 1979;35:689-691.
113. MacLeod RM, Abad A, Edison LL. In vivo effect of sex hormone onthe in vitro synthesis of prolactin and growth hormone in normal and pitu-itary tumour-bearing rats. Endocrinology. 1969;84:1475-1483.
114. Maurer AR, Gorski J. Effect of estradiol-17\g=b\and pimozide on
prolactin synthesis in male and female rats. Endocrinology. 1977;101:76-84.115. Caligaris L, Taleisnik S. Involvement of neurones containing
5-hydroxytryptamine in the mechanism of prolactin release induced by oe-strogen. J Endocrinol. 1974;62:25-33.
116. Biegon A, Samuel D. Modulation of serotonin receptors by steroid sexhormones in the rat brain. In: Littauer UZ, Dudai Y, Silman I, Teichberg VI,Vogel Z, eds. Neurotransmitters and Their Receptors. New York, NY: JohnWiley & Sons Inc; 1976:119.
117. Pan J-T, Gala RR. The serotonin 5-HT2 receptor system, but not thealpha1-adrenergic receptor system, is involved in the estrogen-induced after-noon prolactin surge in the rat. Life Sci. 1988;42:1869-1874.
118. Pan J-T, Gala RR. The influence of raphe lesions,p-chlorophenylalanine and ketanserin on the estrogen-induced afternoonprolactin surge. Endocrinology. 1987;120:2070-2077.
119. Jahn GA, Deis RP. A possible dual regulation of prolactin by the se-
rotoninergic system in rats at pro-oestrus and during late pregnancy: role ofovarian hormones. J Endocrinol. 1987;112:367-374.
120. Healy D, Leonard BE. Monoamine transport in depression: kineticsand dynamics. J Affective Disord. 1987;12:91-103.
121. Roy A, Everett D, Pickar D, Paul SM. Platelet tritiated imipraminebinding and serotonin uptake in depressed patients and controls. Arch GenPsychiatry. 1987;44:320-327.
122. Tanimoto K, Maeda K, Terada T. Alteration of platelet [3H]imi-pramine binding in mildly depressed patients correlates with disease sever-
ity. Biol Psychiatry. 1985;20:329-352.123. Kanof PD, Coccaro EF, Johns CA, Siever LJ, Davis KL. Platelet
[3H]imipramine binding in psychiatric disorders. Biol Psychiatry. 1987;22:278-286.
124. Suranyi-Cadotte BE, Wood PL, Nair NPV, Schwartz G. Normaliza-tion of platelet [3H]imipramine binding in depressed patients during remis-sion. Eur J Pharmacol. 1982;85:357.
125. Langer SZ, Sechter D, Loo H, Raisman R, Zarifian E. Electroconvul-sive shock therapy and maximum binding of platelet tritiated imipraminebinding in depression. Arch Gen Psychiatry. 1986;43:949-952.
126. Delgado PL, Charney DS, Price LH, Aghajanian GK, Landis H,Heninger GR. Serotonin function and mechanism of antidepressant action:reversal of antidepressant-induced remission by rapid depletion of plasmatryptophan. Arch Gen Psychiatry. 1990;47:411-418.
127. Brewerton TD, Brandt HA, Lesem MD, Murphy DL, Jimerson DC.Serotonin in eating disorders. In: Coccaro EF, Murphy DL, eds. Serotonin inMajor Psychiatric Disorders. Progress in Psychiatry Monograph Series.Washington, DC: American Psychiatric Press Inc; 1990:153\x=req-\184.
Downloaded From: http://archpsyc.jamanetwork.com/ by Tim Brewerton on 03/28/2015




















