
Ultrafast Dynamics of Adenine Derivatives Studied by Time ...
Upload
independentCategory
view
2download
0
doi:10.1006/jmbi.2000.4127 available online at http://www.idealibrary.com on J. Mol. Biol. (2000) 303, 93±110
Molecular Enzymology of the EcoRV DNA-(Adenine-N 6)-Methyltransferase: Kinetics of DNA Binding andBending, Kinetic Mechanism and Linear Diffusion ofthe Enzyme on DNA
Humaira Gowher and Albert Jeltsch*
Institut fuÈ r BiochemieFachbereich 8, Heinrich-Buff-Ring 58, 35392, GiessenGermany
E-mail address of the [email protected]
Abbreviations used: MTase, DNAAdoMet, S-adenosylmethionine; AdS-adenosylhomocysteine; mA, 6-met2AP, 2-aminopurine.
0022-2836/00/010093±18 $35.00/0
The EcoRV DNA-(adenine-N6)-methyltransferase recognizes GATATCsequences and modi®es the ®rst adenine residue within this site. Weshow here, that the enzyme binds to the DNA and the cofactor S-adeno-sylmethionine (AdoMet) in an ordered bi-bi fashion, with AdoMet beingbound ®rst. M.EcoRV binds DNA in a non-speci®c manner and theenzyme searches for its recognition site by linear diffusion with a rangeof approximately 1800 bp. During linear diffusion the enzyme continu-ously scans the DNA for the presence of recognition sites. Upon speci®cM.EcoRV-DNA complex formation a strong increase in the ¯uorescenceof an oligonucleotide containing a 2-aminopurine base analogue at theGAT-2AP-TC position is observed which, most likely, is correlated withDNA bending. In contrast to the GAT-2AP-TC substrate, a G-2AP-TATCsubstrate in which the target base is replaced by 2-aminopurine does notshow an increase in ¯uorescence upon M.EcoRV binding, demonstratingthat 2-aminopurine is not a general tool to detect base ¯ipping. Stopped-¯ow experiments show that DNA bending is a fast process with rate con-stants >10 sÿ1. In the presence of cofactor, the speci®c complex adopts asecond conformation, in which the target sequence is more tightly con-tacted by the enzyme. M.EcoRV exists in an open and in a closed statethat are in slow equilibrium. Closing the open state is a slow process(rate constant �0.7 minÿ1) that limits the rate of DNA methylation undersingle turnover conditions. Product release requires opening of the closedcomplex which is very slow (rate constant �0.05-0.1 minÿ1) and limitsthe rate of DNA methylation under multiple turnover conditions.M.EcoRV methylates DNA sequences containing more than one recog-nition sites in a distributive manner. Since the dissociation rate from non-speci®c DNA does not depend on the length of the DNA fragment, DNAdissociation does not preferentially occur at the ends of the DNA.
# 2000 Academic Press
Keywords: protein-DNA interaction; enzyme mechanism; facilitateddiffusion; kinetic mechanism; 2-aminopurine
*Corresponding authorIntroduction
In addition to the regular nucleobases adenine,guanine, cytosine and thymine, DNA from prokar-yotes and eukaryotes also contains the methylated
ing author:n.de
methyltransferase;oHyc,hyladenine;
bases N6-methyladenine, N4-methylcytosine andC5-methylcytosine. These modi®cations are intro-duced post-replicatively in a sequence-speci®cmanner by DNA methyltransferases (MTases)using S-adenosylmethionine (AdoMet) as a donorfor an activated methyl group. DNA methylationplays important roles in the control of geneexpression, DNA replication and DNA repair (forreviews, see Ahmad & Rao, 1996; Bestor &Verdine, 1994; Cheng, 1995a,b; Dryden, 1999;Messer & Noyer-Weidner, 1998). In eukaryotes it isalso involved in development and epigenetic pro-
# 2000 Academic Press

94 Molecular Enzymology of M.EcoRV
cesses like imprinting and X-inactivation (forreviews, see Colot & Rossignol, 1999; Wolffe &Matzke, 1999). The establishment and maintenanceof the physiological pattern of DNA methylation isabsolutely essential in mammals, because knock-out of different DNA methyltransferases in miceleads to embryonic lethality (Li et al., 1992; Okanoet al., 1999). The important role of DNA methyl-ation in bacteria is underscored by the recent ®nd-ing, that DNA MTases also are involved inpathogenicity of different human pathogens(Escherichia coli (Hale et al., 1998), Salmonella typhi-murium (Enserink, 1999; Garcia-Del Portillo et al.,1999; Heithoff et al., 1999), Neisseria meningitidis(Bucci et al., 1999)). By far the most bacterial DNAMTases are components of restriction-modi®cationsystems which as a kind of immune system protectbacteria against infection by bacteriophages (forreviews, see Heitman, 1993; Wilson & Murray,1991). These systems are present in almost all bac-terial species. In addition to the MTase, they con-tain a restriction endonuclease that cleavesincoming phage DNA at speci®c recognition sites(for a review, see Pingoud & Jeltsch, 1997). The cel-lular DNA is not attacked, because it is methylatedby the MTase within the recognition site of theendonuclease.
All DNA methyltransferases modify DNA in asequence-speci®c manner, i.e. only nucleotideswithin a certain recognition sequence of theenzyme are methylated. Beyond their importantbiological functions, DNA methyltransferases arealso very interesting from a mechanistic point ofview: in order to methylate the DNA at speci®csites, a DNA MTase must ®rst interact with its twosubstrates, namely DNA and S-adenosylmethio-nine (AdoMet), the cofactor of the reaction. Upon®rst encounter, the enzyme binds the DNA non-speci®cally and immediately starts scanning theDNA by linear diffusion for the presence of therecognition site. During speci®c complex for-mation, DNA MTases induce dramatic confor-mational changes of the DNA, because the targetbase is ¯ipped out of the DNA helix, as shown bystructural (Gong et al., 1997; Klimasauskas et al.,1994; Labahn et al., 1994; Reinisch et al., 1995;Schluckebier et al., 1995; Tran et al., 1998) and bio-chemical analyses with many different MTases(Allan et al., 1998a,b; Allan & Reich, 1996; Cal &Connolly, 1997; Holz et al., 1998; Jeltsch et al., 1998,1999b; Klima-sauskas & Roberts, 1995a,b, 1998;Reddy & Rao, 2000). In M.HhaI it has been shownby an NMR analysis that the ¯ipped base exists inat least two states: one state in which the ¯ippedbase is in a mobile conformation and one in whichthe base is tightly packed into a hydrophobic bind-ing site in the protein (Klimasauskas et al., 1998). Inaddition, many DNA MTases bend their DNA sub-strate considerably (Cal & Connolly, 1996; Dubey& Bhattacharya, 1997; Garcia et al., 1996). There-fore, DNA binding and target site recognition byDNA MTases is a multistep process that involves
several conformational changes of the enzyme-DNA complex.
Here, the adenine-N6 MTase M.EcoRV is investi-gated which recognizes GATATC sequences andmodi®es the ®rst adenine residue within this site(Nwosu et al., 1988). M.EcoRV binds to oligonu-cleotides with binding constants between 5 � 106
Mÿ1 and 1 � 108 Mÿ1 (Cal & Connolly, 1997;Jeltsch et al., 1998; Szczelkun & Connolly, 1995).The af®nity for DNA is increased in the presenceof cofactor (Cal & Connolly, 1997; Szczelkun &Connolly, 1995). The enzyme approaches the DNAfrom the major groove side (Szczelkun et al., 1995)and bends the DNA by approximately 60 � towardsthe major groove (Cal & Connolly, 1996). The stea-dy-state rates of DNA methylation are low(kcat � 4 hÿ1) (Jeltsch et al., 1998; Roth & Jeltsch,2000). It is interesting that longer DNA substratesare modi®ed at a higher rate, suggesting the invol-vement of linear diffusion in target site location(Jeltsch et al., 1998). The enzyme ¯ips out its targetbase and three aromatic amino acid residues havebeen implicated in the stabilization of the ¯ippednucleotide (Friedrich et al., 1998; Jeltsch et al., 1998,1999b; Roth et al., 1998). The recognition of thetarget base is relaxed, because substrates contain-ing cytosine instead of adenine are also modi®ed(Jeltsch et al., 1999a). In this work, we investigatethe complete catalytic cycle of this enzyme, i.e. (i)the order of substrate and cofactor binding; (ii) lin-ear diffusion and target-site location; (iii) the kin-etics of conformational changes of the enzyme andthe DNA; (iv) processivity of DNA methylation;and (v) the pathway of the release of the DNA.
Results
The order of substrate binding to M.EcoRV
The catalytic cycle of DNA MTases is initiatedby binding of the enzyme to the DNA substrateand to AdoMet, the cofactor for methylation. Ingeneral, binding of these substrates could occur ina random or in a sequential order with one ofthem binding ®rst. So far, the kinetic mechanism ofDNA MTases has been studied for some bacterialadenine-N6 MTases (M.EcoP15I (Rao et al., 1989),M.EcoRI (Reich & Mashhoon, 1991; Surby & Reich,1996a), M.EcaI (Szilak et al., 1993), CcrM (Berdiset al., 1998)) and cytosine-C5 MTases (M.HhaI(Lindstrom et al., 2000; Wu & Santi, 1987), M.MspI(Bhattacharya & Dubey, 1999)), and the murineDnmt1 MTase (Flynn & Reich, 1998). It is interest-ing that these studies show that all cytosine-C5
MTases studied so far follow an ordered bi-bimechanism in which the DNA is bound ®rst,whereas the mechanisms of the adenine-N6 MTasesdiffer from each other: the mechanism of the CcrMenzyme appears to be similar to that of the cyto-sine-C5 MTases. M.EcoRI also has an ordered bi-bimechanism but it ®rst binds to the AdoMet,whereas M.EcoP15I and M.EcaI follow a random,rapid equilibrium mechanism. In the light of the
Molecular Enzymology of M.EcoRV 95
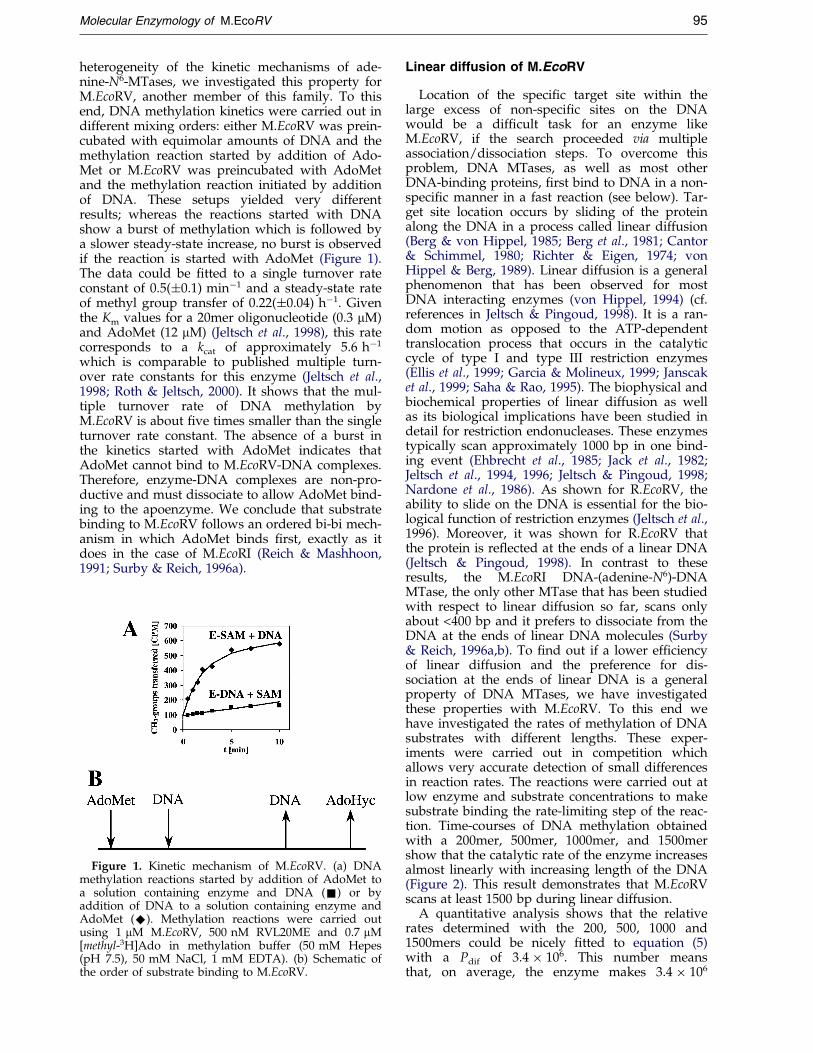
heterogeneity of the kinetic mechanisms of ade-nine-N6-MTases, we investigated this property forM.EcoRV, another member of this family. To thisend, DNA methylation kinetics were carried out indifferent mixing orders: either M.EcoRV was prein-cubated with equimolar amounts of DNA and themethylation reaction started by addition of Ado-Met or M.EcoRV was preincubated with AdoMetand the methylation reaction initiated by additionof DNA. These setups yielded very differentresults; whereas the reactions started with DNAshow a burst of methylation which is followed bya slower steady-state increase, no burst is observedif the reaction is started with AdoMet (Figure 1).The data could be ®tted to a single turnover rateconstant of 0.5(�0.1) minÿ1 and a steady-state rateof methyl group transfer of 0.22(�0.04) hÿ1. Giventhe Km values for a 20mer oligonucleotide (0.3 mM)and AdoMet (12 mM) (Jeltsch et al., 1998), this ratecorresponds to a kcat of approximately 5.6 hÿ1
which is comparable to published multiple turn-over rate constants for this enzyme (Jeltsch et al.,1998; Roth & Jeltsch, 2000). It shows that the mul-tiple turnover rate of DNA methylation byM.EcoRV is about ®ve times smaller than the singleturnover rate constant. The absence of a burst inthe kinetics started with AdoMet indicates thatAdoMet cannot bind to M.EcoRV-DNA complexes.Therefore, enzyme-DNA complexes are non-pro-ductive and must dissociate to allow AdoMet bind-ing to the apoenzyme. We conclude that substratebinding to M.EcoRV follows an ordered bi-bi mech-anism in which AdoMet binds ®rst, exactly as itdoes in the case of M.EcoRI (Reich & Mashhoon,1991; Surby & Reich, 1996a).
Figure 1. Kinetic mechanism of M.EcoRV. (a) DNAmethylation reactions started by addition of AdoMet toa solution containing enzyme and DNA (&) or byaddition of DNA to a solution containing enzyme andAdoMet (^). Methylation reactions were carried outusing 1 mM M.EcoRV, 500 nM RVL20ME and 0.7 mM[methyl-3H]Ado in methylation buffer (50 mM Hepes(pH 7.5), 50 mM NaCl, 1 mM EDTA). (b) Schematic ofthe order of substrate binding to M.EcoRV.
Linear diffusion of M.EcoRV
Location of the speci®c target site within thelarge excess of non-speci®c sites on the DNAwould be a dif®cult task for an enzyme likeM.EcoRV, if the search proceeded via multipleassociation/dissociation steps. To overcome thisproblem, DNA MTases, as well as most otherDNA-binding proteins, ®rst bind to DNA in a non-speci®c manner in a fast reaction (see below). Tar-get site location occurs by sliding of the proteinalong the DNA in a process called linear diffusion(Berg & von Hippel, 1985; Berg et al., 1981; Cantor& Schimmel, 1980; Richter & Eigen, 1974; vonHippel & Berg, 1989). Linear diffusion is a generalphenomenon that has been observed for mostDNA interacting enzymes (von Hippel, 1994) (cf.references in Jeltsch & Pingoud, 1998). It is a ran-dom motion as opposed to the ATP-dependenttranslocation process that occurs in the catalyticcycle of type I and type III restriction enzymes(Ellis et al., 1999; Garcia & Molineux, 1999; Janscaket al., 1999; Saha & Rao, 1995). The biophysical andbiochemical properties of linear diffusion as wellas its biological implications have been studied indetail for restriction endonucleases. These enzymestypically scan approximately 1000 bp in one bind-ing event (Ehbrecht et al., 1985; Jack et al., 1982;Jeltsch et al., 1994, 1996; Jeltsch & Pingoud, 1998;Nardone et al., 1986). As shown for R.EcoRV, theability to slide on the DNA is essential for the bio-logical function of restriction enzymes (Jeltsch et al.,1996). Moreover, it was shown for R.EcoRV thatthe protein is re¯ected at the ends of a linear DNA(Jeltsch & Pingoud, 1998). In contrast to theseresults, the M.EcoRI DNA-(adenine-N6)-DNAMTase, the only other MTase that has been studiedwith respect to linear diffusion so far, scans onlyabout <400 bp and it prefers to dissociate from theDNA at the ends of linear DNA molecules (Surby& Reich, 1996a,b). To ®nd out if a lower ef®ciencyof linear diffusion and the preference for dis-sociation at the ends of linear DNA is a generalproperty of DNA MTases, we have investigatedthese properties with M.EcoRV. To this end wehave investigated the rates of methylation of DNAsubstrates with different lengths. These exper-iments were carried out in competition whichallows very accurate detection of small differencesin reaction rates. The reactions were carried out atlow enzyme and substrate concentrations to makesubstrate binding the rate-limiting step of the reac-tion. Time-courses of DNA methylation obtainedwith a 200mer, 500mer, 1000mer, and 1500mershow that the catalytic rate of the enzyme increasesalmost linearly with increasing length of the DNA(Figure 2). This result demonstrates that M.EcoRVscans at least 1500 bp during linear diffusion.
A quantitative analysis shows that the relativerates determined with the 200, 500, 1000 and1500mers could be nicely ®tted to equation (5)with a Pdif of 3.4 � 106. This number meansthat, on average, the enzyme makes 3.4 � 106

Figure 2. Linear diffusion of M.EcoRV. (a) Competitive methylation assay: 10-20 nM of the 200mer, 500mer and1000mer were incubated for de®ned times at ambient temperature with 10 nM M.EcoRV in methylation buffer(50 mM Hepes (pH 7.5), 50 mM NaCl, 1 mM EDTA) containing 1 mM AdoMet (Sigma). MgCl2 and R.EcoRV wereadded to the reaction mixture to ®nal concentrations of 10 mM and 100 nM, respectively, and the samples incubatedfor 60 minutes at 37 �C to cleave all unmodi®ed EcoRV sites. Unmodi®ed 200mer is converted to a 133mer, 500meryields a 333mer and 1000mer yields a 673mer. The cleavage pattern was analyzed on a 6 % TPE/6 % (w/v) polyacryl-amide gel and the radioactivity visualized using a Phosphorimager (Canberra Packard). (b) Relative rates of methyl-ation of substrates of different lengths by M.EcoRV. The data were determined by competitive methylationexperiments of the substrates in 50 mM Hepes (pH 7.5), 50 mM NaCl, 1 mM EDTA, 1 mM AdoMet. The line rep-resents a ®t to equation (5) with a Pdif of 3.4 � 106.
96 Molecular Enzymology of M.EcoRV
diffusional steps on the DNA before it dissociates.Since linear diffusion can be described by a one-dimensional random walk, the average number ofbasepairs scanned during linear diffusion is givenby the square-root of the number of steps. There-fore, the range of linear diffusion of M.EcoRV isapprox. 1800 bp. Since the analysis relies on theslight curvature of the rate of methylation versuslength of substrate curve, the value of 1800 bpshould be regarded as a lower limit of the range oflinear diffusion. This means that the ef®ciency oflinear diffusion of M.EcoRV is very high, whencompared with results obtained with otherenzymes (R.EcoRI: 850 bp (Ehbrecht et al., 1985);R.EcoRV: 700 bp (Jeltsch et al., 1996) and M.EcoRI:400 bp (Surby & Reich, 1996a)).
Steady-state binding of M.EcoRV tooligonucleotides containing 2-aminopurine
2-Aminopurine (2AP) is a base analogue that is¯uorescent in solution. However, when it is locatedin the DNA helix, the stacking interactions withneighboring base-pairs strongly quench the ¯uor-escence (Ward et al., 1969). 2AP ¯uorescenceincreases after proteins, like DNA MTases (Allanet al., 1998a; Allan & Reich, 1996; Holz et al., 1998;Reddy & Rao, 2000), RNA polymerases (Jia et al.,1996), DNA polymerases (Frey et al., 1995), heli-cases (Raney et al., 1994) or glycosylases (Stiverset al., 1999) bind to the DNA. In general, changesin 2AP ¯uorescence could be correlated with localunstacking caused by conformational changes ofthe DNA, like partial melting, bending or unwind-
ing. In the case of some DNA MTases, a strongincrease in ¯uorescence is observed if the targetbase for methylation is replaced by 2AP. It hasbeen suggested that this effect is caused by the ¯ip-ping of the base analogue out of the DNA (Allanet al., 1998a; Allan & Reich, 1996; Holz et al., 1998).The kinetics of this process were investigated withM.EcoRI showing that DNA binding, DNA bend-ing, base ¯ipping and DNA methylation occuralmost concerted in one fast step (Allan et al.,1998a).
We have used three different 20mer oligonucleo-tide substrates containing 2AP: AP1 contains thebase analogue at the site of DNA modi®cation (G-2AP-TATC/GmATATC) and AP2 contains a GAT-2AP-TC/GmATATC site. AP3 was used as control,which contains the modi®cation immediatelybefore the GATATC site. The changes in the 2AP¯uorescence upon mixing of the substrates withM.EcoRV are given in Table 1, examples of theemission ¯uorescence spectra are shown inFigure 3(a) and (b). It is surprising that AP1 didnot show any signi®cant ¯uorescence change,whereas AP2 shows a strong signal. The ¯uor-escence change in AP2 is observed in the presenceof AdoHyc and AdoMet as well as in the absenceof cofactor (data not shown).
The result that the AP1 substrate (G-2AP-TATC)does not show a signi®cant ¯uorescence changeupon M.EcoRV binding, differs from resultsobtained with other MTases in analogous exper-iments where binding to substrates in which thetarget base is replaced by 2AP led to ¯uorescencechanges which have been interpreted to re¯ect

Table 1. Fluorescence changes, DNA-binding constants and rates of DNA methylation of the 2AP-containingsubstrates
Substrate AP1 AP2 AP3
Change in fluorescence (fold)a 1.4 11.2 1.2KAss/107 (Mÿ1)b 1.8(�0.5) 2.7(�0.7) 2.2(�0.5)kmet (fmol/minute)c 0.04 2.6 2.4
a The relative changes of the 2-aminopurine ¯uorescence were reproducible within �20 %.b Binding experiments were carried out in the presence of 1 mM AdoHyc.c DNA methylation experiments were carried out using 0.5 mM oligonucleotide, 1 mM M.EcoRV and 0.73 mM AdoMet in
methylation buffer. Rates were reproducible within �20 %.
Molecular Enzymology of M.EcoRV 97
base ¯ipping (Allan et al., 1998a; Allan & Reich,1996; Holz et al., 1998; Pues et al., 1999; Reddy &Rao, 2000). DNA-binding experiments showed thatthe lack of effect in the case of M.EcoRV is not dueto a lack of binding of M.EcoRV to the AP1-sub-strate (Table 1). Therefore, we have investigatedthe rate of DNA methylation of the various sub-strates: AP2 is modi®ed at a high rate, whereas therate of methylation of AP1 is very low, as expectedbecause in this substrate the target base is replacedby the analogue (Table 1). This result con®rms thatthe AP1 substrate carries a modi®ed base at thetarget position and the AP2 substrate does not, rul-ing out that the substrates were interchangedduring synthesis or handling.
Given the ®nding of a strong ¯uorescencechange of the AP2 substrate (GAT-2AP-TC), weinvestigated if M.EcoRV might be able to modifyalso the second adenine within the GATATCsequence. Therefore, we investigated if fully meth-ylated oligonucleotides containing GmATATC/GmATATC site are modi®ed by M.EcoRV using avery sensitive methylation assay (Roth & Jeltsch,2000). The results were compared with substratescontaining GmATmATC/GmATmATC sites. How-ever, in agreement with earlier results (Nwosuet al., 1988), none of these substrates was signi®-cantly modi®ed (data not shown). The strong ¯uor-escence change of the AP2 substrate cannot becorrelated with base ¯ipping but must be due to adifferent structural change of the DNA helix. Sincethe 2AP ¯uorescence is altered by local unstackingof the DNA and M.EcoRV is known to bend theDNA by 60 � (Cal & Connolly, 1996), a process thatdisrupts or at least weakens stacking interactionsof neighboring base-pairs, DNA bending is themost likely cause of the ¯uorescence change ofAP2. Recently, a strong change of 2AP ¯uorescencein a substrate which does not carry the 2AP substi-tution at the position of modi®cation was alsoreported for the EcoP15I adenine-N6 MTase (Reddy& Rao, 2000).
{ A variant of model 1 which uses reduced enzymeconcentrations to account for the possibility that not allenzyme molecules are active did not improve the ®ts.Similarly, another modi®ed version of model 1 in whichthe speci®c M.EcoRV-DNA complex can directly releasethe DNA did not allow a ®t to the data.
Fast-kinetic analyses of M.EcoRV binding tothe GAT-2AP-TC substrate
Since only the AP2 substrate shows a ¯uor-escence change, this effect obviously must be dueto speci®c binding of the MTase to the DNA.Therefore, the AP2 substrate (GAT-2AP-TC) offersa unique tool to study the kinetics of speci®c DNAbinding and bending by M.EcoRV. The time-courses of DNA binding were determined at con-centrations of M.EcoRV and DNA varying between100 and 700 nM in stopped-¯ow experiments. Inaddition, the time-course of enzyme-DNA complexdissociation was measured. These experimentswere carried out in the absence of cofactor as wellas in the presence of AdoHyc or AdoMet.Examples of the time-courses obtained are shownin Figures 4 and 5. All data that were obtainedunder the same conditions (i.e. in the absence ofcofactor, with AdoHyc or with AdoMet) were sim-ultaneously ®tted to kinetic models. As the enzyme®rst binds non-speci®cally to DNA, model 1 is thesimplest model to analyze these data. In thismodel (shown below), only the speci®c complex(EDsp) has a ¯uorescence signal:
E�D�k1
kÿ1
EDnsp�k2
kÿ2
EDsp
where EDnsp is the non-speci®c enzyme-DNA com-plex; EDsp is the speci®c enzyme-DNA complex.
However, only the data obtained in the absenceof cofactor could be ®t to this model (Table 2).Trials to ®t the data obtained in the presence ofAdoHyc or AdoMet to model 1 resulted in largesystematic errors (Figure 4). Therefore, model 2was used to ®t these data which is extended by anadditional conformational change of the speci®cenzyme-DNA complex and assigns a different ¯u-orescence signal to the two speci®c enzyme-DNAcomplexes. As shown in Figure 5, the data aredescribed well by this modi®ed model, demon-strating that the speci®c M.EcoRV-DNA complexisomerizes into a different conformation in the pre-sence of cofactor{. The rate constants resultingfrom the ®ts are given in Table 2. In both ®ts, the¯uorescence of the ESsp
0complex is 0.7-0.9 of that
of the ESsp complex. This indicates that the ¯uor-escence of the 2AP probe in both of these confor-mations is much higher than in the usual B-form
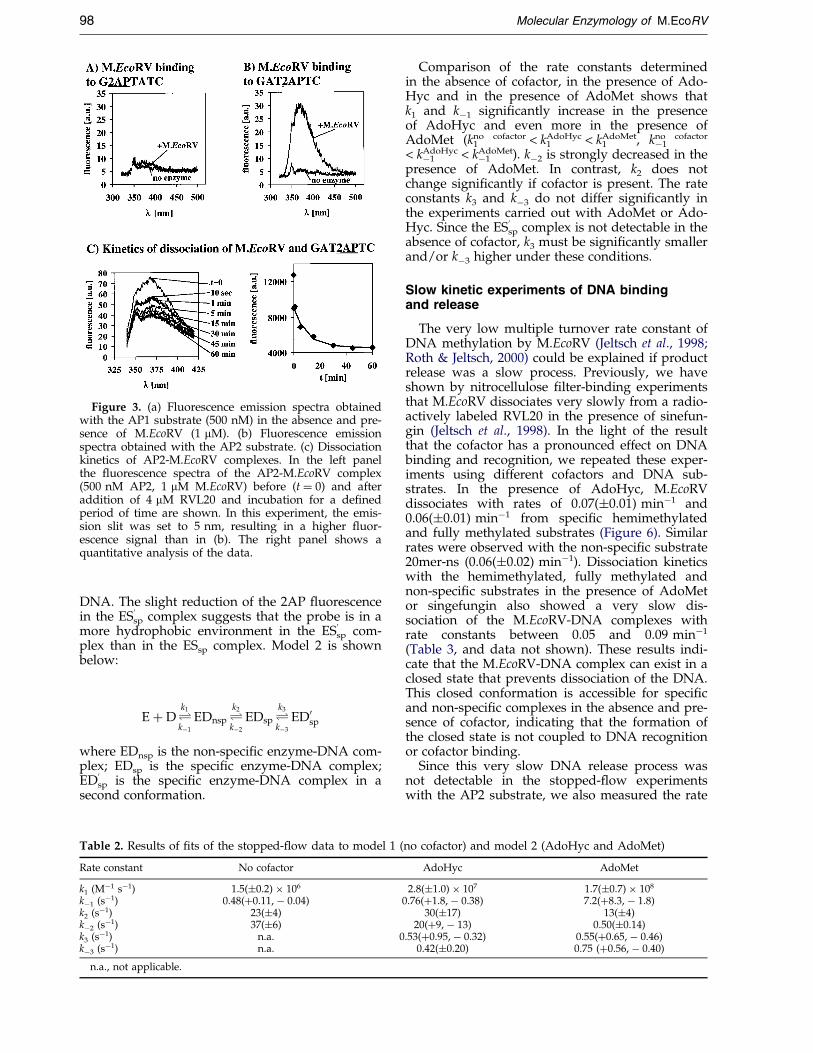
Figure 3. (a) Fluorescence emission spectra obtainedwith the AP1 substrate (500 nM) in the absence and pre-sence of M.EcoRV (1 mM). (b) Fluorescence emissionspectra obtained with the AP2 substrate. (c) Dissociationkinetics of AP2-M.EcoRV complexes. In the left panelthe ¯uorescence spectra of the AP2-M.EcoRV complex(500 nM AP2, 1 mM M.EcoRV) before (t � 0) and afteraddition of 4 mM RVL20 and incubation for a de®nedperiod of time are shown. In this experiment, the emis-sion slit was set to 5 nm, resulting in a higher ¯uor-escence signal than in (b). The right panel shows aquantitative analysis of the data.
98 Molecular Enzymology of M.EcoRV
DNA. The slight reduction of the 2AP ¯uorescencein the ESsp
0complex suggests that the probe is in a
more hydrophobic environment in the ESsp
0com-
plex than in the ESsp complex. Model 2 is shownbelow:
E�D�k1
kÿ1
EDnsp�k2
kÿ2
EDsp�k3
kÿ3
ED0sp
where EDnsp is the non-speci®c enzyme-DNA com-plex; EDsp is the speci®c enzyme-DNA complex;EDsp
0is the speci®c enzyme-DNA complex in a
second conformation.
Table 2. Results of ®ts of the stopped-¯ow data to model 1 (
Rate constant No cofactor
k1 (Mÿ1 sÿ1) 1.5(�0.2) � 106
kÿ1 (sÿ1) 0.48(�0.11, ÿ 0.04)k2 (sÿ1) 23(�4)kÿ2 (sÿ1) 37(�6)k3 (sÿ1) n.a. 0kÿ3 (sÿ1) n.a.
n.a., not applicable.
Comparison of the rate constants determinedin the absence of cofactor, in the presence of Ado-Hyc and in the presence of AdoMet shows thatk1 and kÿ1 signi®cantly increase in the presenceof AdoHyc and even more in the presence ofAdoMet (k1
no cofactor < k1AdoHyc < k1
AdoMet, kÿ1no cofactor
< kÿ1AdoHyc < kÿ1
AdoMet). kÿ2 is strongly decreased in thepresence of AdoMet. In contrast, k2 does notchange signi®cantly if cofactor is present. The rateconstants k3 and kÿ3 do not differ signi®cantly inthe experiments carried out with AdoMet or Ado-Hyc. Since the ESsp
0complex is not detectable in the
absence of cofactor, k3 must be signi®cantly smallerand/or kÿ3 higher under these conditions.
Slow kinetic experiments of DNA bindingand release
The very low multiple turnover rate constant ofDNA methylation by M.EcoRV (Jeltsch et al., 1998;Roth & Jeltsch, 2000) could be explained if productrelease was a slow process. Previously, we haveshown by nitrocellulose ®lter-binding experimentsthat M.EcoRV dissociates very slowly from a radio-actively labeled RVL20 in the presence of sinefun-gin (Jeltsch et al., 1998). In the light of the resultthat the cofactor has a pronounced effect on DNAbinding and recognition, we repeated these exper-iments using different cofactors and DNA sub-strates. In the presence of AdoHyc, M.EcoRVdissociates with rates of 0.07(�0.01) minÿ1 and0.06(�0.01) minÿ1 from speci®c hemimethylatedand fully methylated substrates (Figure 6). Similarrates were observed with the non-speci®c substrate20mer-ns (0.06(�0.02) minÿ1). Dissociation kineticswith the hemimethylated, fully methylated andnon-speci®c substrates in the presence of AdoMetor singefungin also showed a very slow dis-sociation of the M.EcoRV-DNA complexes withrate constants between 0.05 and 0.09 minÿ1
(Table 3, and data not shown). These results indi-cate that the M.EcoRV-DNA complex can exist in aclosed state that prevents dissociation of the DNA.This closed conformation is accessible for speci®cand non-speci®c complexes in the absence and pre-sence of cofactor, indicating that the formation ofthe closed state is not coupled to DNA recognitionor cofactor binding.
Since this very slow DNA release process wasnot detectable in the stopped-¯ow experimentswith the AP2 substrate, we also measured the rate
no cofactor) and model 2 (AdoHyc and AdoMet)
AdoHyc AdoMet
2.8(�1.0) � 107 1.7(�0.7) � 108
0.76(�1.8, ÿ 0.38) 7.2(�8.3, ÿ 1.8)30(�17) 13(�4)
20(�9, ÿ 13) 0.50(�0.14).53(�0.95, ÿ 0.32) 0.55(�0.65, ÿ 0.46)
0.42(�0.20) 0.75 (�0.56, ÿ 0.40)
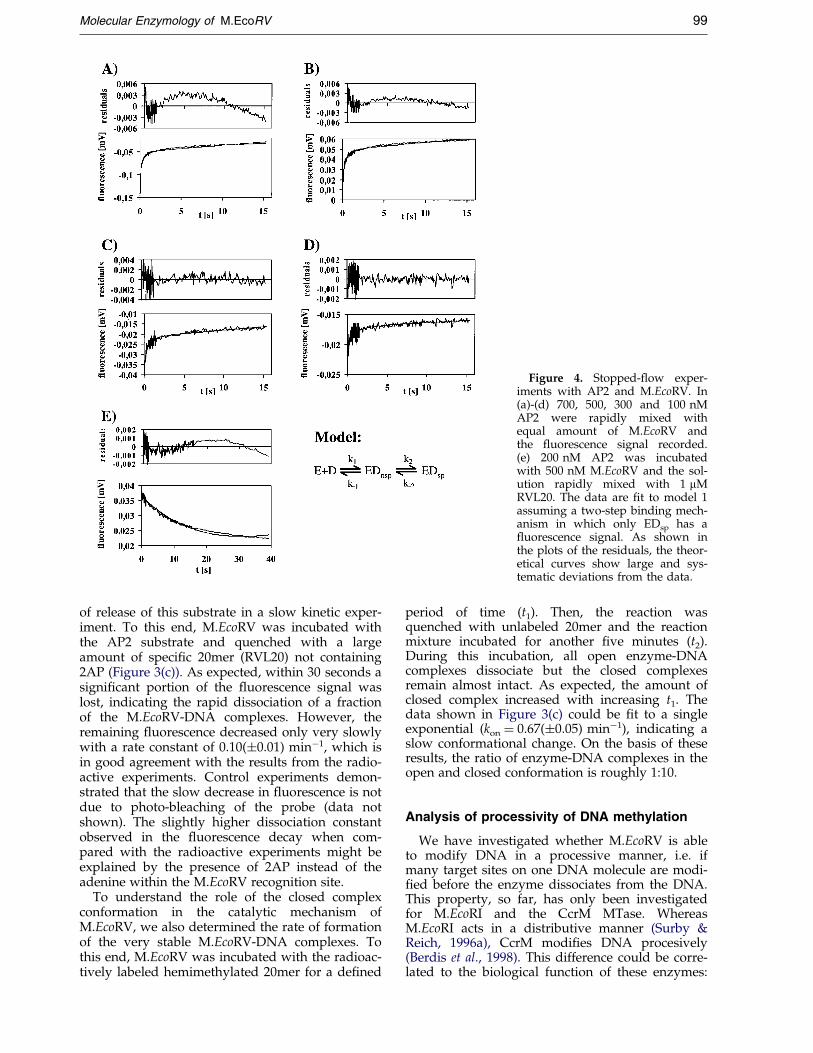
Figure 4. Stopped-¯ow exper-iments with AP2 and M.EcoRV. In(a)-(d) 700, 500, 300 and 100 nMAP2 were rapidly mixed withequal amount of M.EcoRV andthe ¯uorescence signal recorded.(e) 200 nM AP2 was incubatedwith 500 nM M.EcoRV and the sol-ution rapidly mixed with 1 mMRVL20. The data are ®t to model 1assuming a two-step binding mech-anism in which only EDsp has a¯uorescence signal. As shown inthe plots of the residuals, the theor-etical curves show large and sys-tematic deviations from the data.
Molecular Enzymology of M.EcoRV 99
of release of this substrate in a slow kinetic exper-iment. To this end, M.EcoRV was incubated withthe AP2 substrate and quenched with a largeamount of speci®c 20mer (RVL20) not containing2AP (Figure 3(c)). As expected, within 30 seconds asigni®cant portion of the ¯uorescence signal waslost, indicating the rapid dissociation of a fractionof the M.EcoRV-DNA complexes. However, theremaining ¯uorescence decreased only very slowlywith a rate constant of 0.10(�0.01) minÿ1, which isin good agreement with the results from the radio-active experiments. Control experiments demon-strated that the slow decrease in ¯uorescence is notdue to photo-bleaching of the probe (data notshown). The slightly higher dissociation constantobserved in the ¯uorescence decay when com-pared with the radioactive experiments might beexplained by the presence of 2AP instead of theadenine within the M.EcoRV recognition site.
To understand the role of the closed complexconformation in the catalytic mechanism ofM.EcoRV, we also determined the rate of formationof the very stable M.EcoRV-DNA complexes. Tothis end, M.EcoRV was incubated with the radioac-tively labeled hemimethylated 20mer for a de®ned
period of time (t1). Then, the reaction wasquenched with unlabeled 20mer and the reactionmixture incubated for another ®ve minutes (t2).During this incubation, all open enzyme-DNAcomplexes dissociate but the closed complexesremain almost intact. As expected, the amount ofclosed complex increased with increasing t1. Thedata shown in Figure 3(c) could be ®t to a singleexponential (kon � 0.67(�0.05) minÿ1), indicating aslow conformational change. On the basis of theseresults, the ratio of enzyme-DNA complexes in theopen and closed conformation is roughly 1:10.
Analysis of processivity of DNA methylation
We have investigated whether M.EcoRV is ableto modify DNA in a processive manner, i.e. ifmany target sites on one DNA molecule are modi-®ed before the enzyme dissociates from the DNA.This property, so far, has only been investigatedfor M.EcoRI and the CcrM MTase. WhereasM.EcoRI acts in a distributive manner (Surby &Reich, 1996a), CcrM modi®es DNA procesively(Berdis et al., 1998). This difference could be corre-lated to the biological function of these enzymes:

Figure 5. Stopped-¯ow exper-iments with AP2 and M.EcoRV. In(a)-(e) the same data are shown asin Figure 2. The data are ®t tomodel 2 assuming a three-stepbinding mechanism in which EDsp
and EDsp0 have different ¯uor-
escence signals. As shown in theplots of the residuals, the theoreti-cal curves nicely ®t the data.
100 Molecular Enzymology of M.EcoRV
whereas M.EcoRI, like M.EcoRV, is part of a restric-tion/modi®cation system, CcrM is an orphanMTase, that is involved in the cell-cycle regulationof Caulobacter cresentus (Stephens et al., 1996). To®nd out if M.EcoRV methylates the DNA in a pro-cessive manner, the rates of methylation of twoEcoRV sites on a single oligonucleotide substratewere studied. The amount of DNA in which onlyone of the two EcoRV sites is methylated duringthe time-course of DNA methylation was observed.As shown in Figure 7(a), this intermediate shouldnot occur at all or decay quickly if the enzyme fol-lowed a processive mechanism. In contrast, someamount of intermediate is formed if DNA methyl-ation is non-processive and this fraction remainsalmost constant for several turnovers. We havechosen the restriction protection method to moni-tor this reaction, because it allows detection of themethylation status of individual recognition sites.To this end, the substrate was either labeled on the50 end of the top strand or bottom strand,incubated with M.EcoRV for increasing periods oftime and the methylation analyzed by digestionwith R.EcoRV. As shown in Figure 7(b), a fast
burst of methylation is observed in which about15 % of site 1 and 30 % of site 2 are modi®ed. Thisburst fraction of 45 % nicely corresponds to theratio of enzyme (20 nM) to DNA (50 nM) in thisexperiment demonstrating that the enzyme prep-aration used has a very high speci®c activity.During this burst, a single methyl group is trans-ferred to the DNA, so corresponding amounts ofintermediates are formed. As clearly shown inFigure 7(b), these amounts do not decrease duringthe following turnovers, although the total amountof methylation increases, as indicated by theincreasing amounts of ®nal product in which bothEcoRV sites are modi®ed. Therefore, we tried to ®tthe data to a distributive mechanism of DNAmethylation. In this model, the time-course ofDNA methylation at each site is independent ofthe methylation status of the second site on eachsubstrate. The amount of DNA methylation at eachsite is given by:
cimet � bursti � ki
mt � t
where cimet is the concentration of DNA modi®ed
at site i; bursti is the burst of methylation at site i;

Figure 6. Kinetics of the transition of M.EcoRVbetween the open and closed states. (a) Dissociation kin-etics of radioactively labeled hemimethylated and fullymethylated oligonucleotides from M.EcoRV. The kineticswere assayed in 50 mM Hepes (pH 7.5), 50 mM NaCl,1 mM EDTA in the presence of 1 mM AdoHyc. (b) Kin-etics of the formation of the closed complex. In theseexperiments, labeled hemimethylated oligonucleotides(10 nM) were incubated for increasing times withM.EcoRV (200 nM). Then, the unlabeled substrateRVL20 was added to a ®nal concentration of 0.5 mMand the sample incubated for ®ve minutes. During thisincubation, the open M.EcoRV-DNA complexes almostcomplete dissociate whereas the closed complexes arestill intact. Therefore, the fraction of closed complexescan be determined.
Figure 7. Processivity of M.EcoRV. (a) Schematic rep-resentation of the processive and distributive methyl-ation of substrates with two recognition sites.Unmethylated EcoRV sites are represented by a &,methylated by a &. (b) Experimental data obtained withsubstrates labeled at the 50 end of the upper strand(upper panel) and at the 50 end of the lower strand(lower panel), respectively. Kinetic studies were carriedout using 50 nM of the oligonucleotide and 10 nMM.EcoRV in methylation buffer (50 mM Hepes (pH 7.5),50 mM NaCl, 1 mM EDTA) containing 1 mM AdoMet(Sigma). Note, that in either case the completelyunmethylated and one of the hemimethylated intermedi-ates cannot be distinguished. The lines show a global ®tof both data sets to a distributive mechanism of DNAmethylation (equation (3)) with k1
mt � 0.041 minÿ1 andk2
mt � 0.079 minÿ1.
Molecular Enzymology of M.EcoRV 101
kimt is the multiple turnover rate of DNA methyl-
ation at site i.As shown in Figure 7(b), a global ®t of both data
sets to one set of rate constants leads to an excel-lent agreement between experimental and theoreti-cal data. We conclude that M.EcoRV does notmethylate the DNA in a processive manner. Thisresult is in agreement with the kinetic mechanismderived from the methylation experiments initiatedby addition of AdoMet or DNA, because anordered bi-bi mechanism of substrate binding inwhich AdoMet binds ®rst precludes a processivemechanism of DNA methylation.
It is interesting that the sizes of the bursts and therates of modi®cation of both recognition sites devi-ate from each other (burst1 � 15 %, burst2 � 30 %,ki
mt � 0.041 minÿ1, k2mt �0.079 minÿ1). This differ-
ence can be understood if one considers theprobability of each site to be located by linear diffu-sion: site 1 is located at position 6-11, site 2 at pos-ition 12-17 of the 33 mer. Therefore, if M.EcoRVassociates to the substrate at position 1-11 it will
Table 3. Rates of dissociation (kÿ1) of M.EcoRV fromsubstrates of different lengths
Length of the DNA (bp) kÿ1 (minÿ1)
20 0.057(�0.013)200 0.050(�0.012)500 0.048(�0.025)1000 0.060(�0.005)
To study dissociation from non-speci®c DNA, all experimentswere carried out in the presence of 0.1 mM AdoMet.
®rst meet site 1 during linear diffusion and if associ-ation occurs at position 12-33, site 2 will be encoun-tered ®rst. Therefore, if the association is randomand each base-pair constitutes a non-speci®c bind-ing site, the probabilities of M.EcoRV to reach site 1and site 2 are 11/33 and 22/33. This means that site2 should be modi®ed twofold faster than site 1,exactly as found in the experiment. This result con-®rms that linear diffusion indeed is the usual path-way for target site location. Moreover, it shows,that the enzyme recognizes each target site duringlinear diffusion with high ®delity, because ifM.EcoRV would tend to overlook recognition sites,both sites had an equal chance to be reached bylinear diffusion.
The pathway of dissociation from the DNA
DNA binding kinetics with M.EcoRI provide evi-dence that this enzyme prefers to dissociate from alinear piece of DNA at the ends (Surby & Reich,1996b). We were interested to ®nd out if M.EcoRVfollows the same pathway to dissociate from DNA.To this end, dissociation kinetics of M.EcoRV weredetermined with substrates of different lengths, i.e.a 20mer, a 200mer, a 500mer and a 1000mer. These

102 Molecular Enzymology of M.EcoRV
experiments were carried out in the presence ofAdoMet which results in modi®cation of the targetsite. Since M.EcoRV does not bind to methylatedsubstrates with any preference under these con-ditions (data not shown), basically the kinetics ofdissociation from non-speci®c DNA are observedin these experiments. Our data show that the rateconstants of M.EcoRV dissociation from all sub-strates are virtually identical although the length ofthe substrates is varied 50-fold (Table 3). Since avariation of substrate length by a factor of 50results in a 50-fold change of the ratio of internalbinding sites to end-sites, these data preclude anyimportant contribution of end dissociation to theregular pathway of dissociation.
Discussion
In this work the key steps in the catalytic mech-anism of the M.EcoRV DNA-(adenine-N6)-methyl-transferase, i.e. the order and kinetics of substrateand cofactor binding, target site location by lineardiffusion, kinetics of conformational changes of theenzyme, kinetics of DNA bending, processivity ofDNA methylation and the pathway of product dis-sociation is investigated.
Kinetic mechanism of M.EcoRV
Like M.EcoRI, M.EcoRV follows an ordered bi-bimechanism with AdoMet binding before DNA.Enzyme-DNA complexes are non-catalytic com-plexes and must dissociate prior to subsequent cat-alytic rounds. If enzyme-AdoMet complexes aremixed with DNA a burst of methylation isobserved, because the single turnover rate constantof DNA methylation is about ®vefold higher thanthe multiple turnover constant. This is an interest-ing result because now the order of substrate bind-ing is known for three cytosine-C5 and ®veadenine-N6 MTases. Whereas, all cytosine-C5
MTases follow a sequential bi-bi mechanism withthe DNA binding ®rst, the adenine-N6 MTasesappear rather heterogeneous in this behavior: twoof them follow an ordered bi-bi mechanism inwhich AdoMet binds ®rst (M.EcoRI and M.EcoRV),two have a random equilibrium mechanism(EcoP15I and M.EcaI) and one has an orderedmechanism in which the DNA binds ®rst (CcrM).It would be interesting to see how these mechanis-tic differences are re¯ected in the structures of thecorresponding enzyme-DNA complexes.
Linear diffusion
After binding AdoMet, the EcoRV MTaseencounters the DNA non-speci®cally followed bylocation of its target site on the DNA. This processinvolves linear diffusion of the enzyme on theDNA. With a range of approximately 1800 bp line-ar diffusion is performed very ef®ciently byM.EcoRV when compared to results obtained forother enzymes (R.EcoRI, 850 bp (Ehbrecht et al.,
1985); R.EcoRV, 700 bp (Jeltsch et al., 1996); andM.EcoRI, 400 bp (Surby & Reich, 1996a)). The highef®ciency of linear diffusion of M.EcoRV could bedue to the formation of a very stable enzyme-DNAcomplex in which the protein is in a closed confor-mation, which prevents DNA dissociation. Duringlinear diffusion, the enzyme accurately scans theDNA for the presence of recognition sites. Withrespect to the ef®ciency of target site location,M.EcoRV resembles the EcoRI restriction enzyme(Jeltsch et al., 1994) but differs from the R.EcoRVenzyme, which tends to ``overlook'' recognitionsites during linear diffusion (Jeltsch & Pingoud,1998). This result also suggests that linear diffusionof M.EcoRV is a sliding rather than hopping move-ment. In order to explain high ef®ciency of targetsite recognition, sliding most likely follows thehelical pitch of the DNA (Jeltsch et al., 1994). It hasbeen argued that the short range of linear diffusionof M.EcoRI might be a special adaptation to thebiological function of an MTase which is part of arestriction/modi®cation system (Surby & Reich,1996a). However, our data show that a short rangeof linear diffusion is not a general property ofMTases which are one part of restriction/modi®-cation systems.
Fluorescence changes of oligonucleotidescontaining the 2AP base analogue
The ¯uorescence changes of oligonucleotidescontaining the 2AP base analogue upon binding ofthe M.EcoRV DNA adenine methyltransferase havebeen investigated. This base analogue is ¯uorescentwith excitation and emission maxima at 310 and360 nm, respectively. Its ¯uorescence is stronglyquenched within the DNA helix, but becomes vis-ible if base stacking is reduced. The ¯uorescentprobes were situated at different positions in oligo-nucleotide substrates containing the EcoRV recog-nition site (GATATC): at the target position thatbecomes modi®ed by the MTase (G-2AP-TATC), atthe center of the recognition site (GAT-2AP-TC)and immediately 30 to the recognition site. The G-2AP-TATC substrate was used because MTasesmove their target bases out of the DNA helix andit has been shown with the M.EcoRI, M.TaqI,M.HhaI and M.EcoP15I DNA MTases that sub-strates containing 2AP at the target site of theMTase show a strong increase in ¯uorescenceupon binding of the MTase (Allan et al., 1998a;Allan & Reich, 1996; Holz et al., 1998; Reddy &Rao, 2000). However, here we show that a similareffect is not observed with M.EcoRV. This ®ndingsuggests some differences in the interaction withthe extrahelical base between M.EcoRV the otherMTases: either the equilibrium between the ¯ippedand the not ¯ipped target base is on the side of thenon-¯ipped state in the case of M.EcoRV, or thebinding pocket for the ¯ipped base might be morehydrophobic in M.EcoRV than in the other MTases.The latter model is supported by the presence ofleast three aromatic amino acid residues near the

Molecular Enzymology of M.EcoRV 103
adenine-binding site in M.EcoRV which appear tocontribute to the binding of the ¯ipped target basein M.EcoRV, one tryptophan and two tyrosine resi-dues (Friedrich et al., 1998; Jeltsch et al., 1999b;Roth et al., 1998).
In contrast to the G-2AP-TATC substrate, theGAT-2AP-TC substrate shows a strong ¯uor-escence change upon M.EcoRV binding. This signaldepends on speci®c complex formation whichleads to a 60 � bend of the DNA. Large changes in2AP ¯uorescence in substrates which do not carrythe substitution at the site of methylation recentlywere also observed with M.EcoP15I (Reddy & Rao,2000). Therefore, changes in 2AP ¯uorescenceshould not be regarded as proof of base ¯ippingbut as evidence for conformational changes of theDNA which lead to unstacking of the 2AP base.Base ¯ipping might be one example of suchchanges.
Fast kinetics of DNA binding and bending
The ¯uorescence change of the GAT-2AP-TCsubstrate was used to study the kinetics of DNAbinding and release, and DNA bending byM.EcoRV in stopped-¯ow experiments. Three con-formations of the enzyme-DNA complex could beidenti®ed that are in rapid equilibrium: First,M.EcoRV binds non-speci®cally to DNA with abimolecular rate constant of 1.5 � 106 sÿ1 Mÿ1 inthe absence of cofactor, 2.8 � 107 sÿ1 Mÿ1 in thepresence of AdoHyc and 1.7 � 108 sÿ1 Mÿ1 in thepresence of AdoMet. This increase in the bimolecu-lar rate constant of association in the presence ofAdoMet can only be explained if AdoMet bindsto the enzyme prior to the DNA. It is interestingthat the rate of decay of this complex (kÿ1)also increases in the presence of AdoMet(kÿ1
no cofactor < kÿ1AdoHyc < kÿ1
AdoMet) suggesting that bind-ing of the cofactor induces an open state of theenzyme that facilitates DNA binding and release.This conclusion is in agreement with the kineticevidence in favor of an ordered bi-bi mechanism ofM.EcoRV in which AdoMet binds before the DNA.
The non-speci®c M.EcoRV-DNA complex iso-merizes to a speci®c complex conformation inwhich the DNA is bent in a fast step with rate con-stants (k2) between 10 and 30 sÿ1. This result is inagreement with the ®ndings that DNA has a highconformational ¯exibility (Jeltsch, 1998) and veryfast rates of DNA bending have been observed forother proteins, like the TATA box binding protein(Parkhurst et al., 1996). Although the rate of DNAbending by M.EcoRV is almost independent of thepresence and nature of the cofactor, the stability ofthe bent enzyme-DNA complex is much higher inthe presence of AdoMet as indicated by the 50-foldlower rate constant of decay of this complex (kÿ2).This result is in agreement with the previous ®nd-ing that AdoMet contributes to DNA recognitionby M.EcoRV (Cal & Connolly, 1997; Szczelkun &Connolly, 1995) and other DNA MTases, like EcoRI(Reich & Mashhoon, 1991), EcaI (Szilak et al., 1993)
or E. coli dam (Bergerat & Guschlbauer, 1990). Inthe presence of cofactor, the speci®c enzyme-DNAcomplex isomerizes into a second conformationESsp
0. According to the slight decrease in the ¯uor-
escence intensity of the 2AP probe, it is in a morehydrophobic environment in ESsp
0. This result
could mean that the recognition site is more tightlycontacted in the ESsp
0complex. It is in agreement
with previous results, which demonstrated thathydrophobic interactions strongly contribute to theinteraction energy of the speci®c M.EcoRV-DNAcomplex (Jeltsch et al., 1998).
The identi®cation of two different speci®cEcoRV-DNA complexes is in good agreement withdata obtained for M.HhaI, showing that the ¯ippedtarget base exists in two different conformations,one loose conformation and one in which the tar-get base is tightly packed into its binding pocket ofthe enzyme (Klimasauskas et al., 1998). It is inter-esting that in these experiments, addition of Ado-Hyc was shown to greatly increase the fraction oftightly bound cytosine (experiments with AdoMetwere not carried out, because under these con-ditions turnover would take place). This resultshows that in M.HhaI the cofactor also contributesto DNA recognition. Moreover, stopped-¯owexperiments with M.EcoRI demonstrated that thespeci®c M.EcoRI-DNA complex isomerizes with arate constant of 0.6 sÿ1 into a different confor-mation (Allan et al., 1998a). The rate constants forthese isomerizations are very similar for M.EcoRI(0.6 sÿ1) and M.EcoRV (k3 0.55 sÿ1). These datasuggest that upon speci®c complex formation,DNA MTases and the target DNA undergo amutual induced ®t which has been suggested to bea general mechanism of sequence-speci®c DNAinteraction (Rhodes et al., 1996; Spolar & Record,1994).
Finally, we have shown that in addition to theopen M.EcoRV-DNA complex which allows forrapid binding and release of DNA, the enzymealso exists in a closed state in which DNAexchange is not possible. In enzyme-DNA com-plexes the equilibrium between the open andclosed states is on the side of the closed state.However, in the apoenzyme it is shifted towardsthe open state in the presence of cofactor. If anenzyme-DNA complex adopts a closed state, DNArelease is prevented. The closed state is accessiblefor speci®c and non-speci®c complexes indepen-dent of the presence of cofactor. The existence of aclosed M.EcoRV-DNA complex explains the largerange of linear diffusion by this enzyme. The rateof formation of the closed state (0.67 minÿ1) issimilar to the single turnover rate constant of DNAmethylation by M.EcoRV (0.55 minÿ1). Theseresults suggest that this conformational changecould be the rate-limiting step of DNA methylationby this enzyme. This could explain why the rate ofmethyl group transfer catalyzed by M.EcoRV evenunder single turnover conditions is much smallerthan methylation rates determined with M.EcoRI(41 sÿ1) (Reich & Mashhoon, 1993). Multiple turn-

Figure 8. Mechanistic model of the interaction ofM.EcoRV with DNA. A, AdoMet; H, AdoHyc; D, DNA;Dmet, modi®ed DNA; E, M.EcoRV. The enzyme exists inan open and a closed state. The AdoMet-enzyme-DNAcomplex has at least three different conformations: anon-speci®c one, AEDnsp, and two speci®c ones, AEDsp
and AEDsp0. For details, see the text.
104 Molecular Enzymology of M.EcoRV
over appears to be limited by the release of DNAfrom the closed complex, because the rate constantof dissociation of closed M.EcoRV-DNA complexes(0.05-0.1 minÿ1) is very similar to the kcat value ofM.EcoRV (4 hÿ1) (Jeltsch et al., 1998; Roth & Jeltsch,2000). Most likely, the transition from the open tothe closed complex encompasses closing of theDNA-binding cleft of the enzyme and leads to acomplex in which the enzyme encircles the DNA.A similar conformational change is observed withM.HhaI, where a loop which does not adopt anordered structure in the apoenzyme encircles theDNA in the M.HhaI-DNA complex (Cheng et al.,1993; Klimasauskas et al., 1994).
Processivity of DNA methylation and pathwayof DNA release
After catalysis, M.EcoRV must dissociate fromthe DNA to allow the next turnover because thisenzyme is not able to methylate DNA processively.With respect to this property, M.EcoRV againresembles M.EcoRI. It should be noticed that thedistributive mechanism of DNA methylation ofthese two enzymes is a direct consequence of theirorder of substrate binding because in both casesAdoHcy cannot leave ternary enzyme-DNA-AdoHcy complexes and AdoMet cannot enterenzyme-DNA complexes. It would be interestingto see if more of the MTases with a different kineticmechanism might be able to modify DNA proces-sively.
We found that the rate constant of dissociationof M.EcoRV from non-speci®c DNA does notdepend on the length of substrate between 20 and1000 bp. Since no difference in the dissociation kin-etics was observed although the ratio of end tointernal binding positions changes over 50-fold, weconclude that dissociation of M.EcoRV from DNAdoes not occur at the ends of linear DNA. There-fore, dissociation must proceed via a conformation-al change of the enzyme from the closed to theopen state. Taken together, M.EcoRV behaves verysimilarly to M.EcoRI with respect to the kineticmechanism, linear diffusion and processivity withthe exception that the range of linear diffusion ismuch larger in M.EcoRV and that end dissociationis not involved in product dissociation inM.EcoRV.
Biological implications
How do these properties relate to the biologicalfunction of M.EcoRV? It is clear that the ability forlinear diffusion is required for each protein thatspeci®cally interacts with DNA to locate its targetsite within a reasonable time. However, there areadditional adaptations to the particular biologicalrole by M.EcoRV. The enzyme is one component ofa restriction/modi®cation system whose functionis to protect the bacterial cell from infection by bac-teriophages. In order to be able to cleave the phageDNA, it is necessary that the endonuclease binds
to the phage DNA before it is completely modi®edby the methyltransferase. Since M.EcoRV, likemany other prokaryotic MTases, does not differen-tiate between unmethylated and hemimethylatedrecognition sites (Jeltsch et al., 1998), this conditioncan only be met if the MTase is a slow enzyme.M.EcoRV indeed is a slow enzyme if the multipleturnover is considered. In particular, the inabilityto modify DNA processively is important becauseit prevents methylation of a whole phage DNAwith just one MTase molecule bound. The distribu-tive mechanism of DNA methylation mechanismin turn is based on the ordered bi-bi mechanism ofsubstrate binding in which AdoMet must bind®rst. Therefore, it seems that basic enzymological

Molecular Enzymology of M.EcoRV 105
properties directly contribute to the biological func-tion of M.EcoRV.
Conclusions
The results of this study are summarized inFigure 8. M.EcoRV binds to the cofactor AdoMetand the DNA in an ordered bi-bi reaction in whichthe cofactor has to be bound ®rst. The enzymeexists in an open state that allows exchange of theDNA substrate and a closed state in which theDNA is tightly bound by the enzyme. The slowrelease of DNA from the closed state explains thehigh ef®ciency of linear diffusion of this enzymeon DNA. The enzyme binds the DNA substrateand the cofactor AdoMet in an ordered bi-bi mech-anism with AdoMet being bound ®rst. Therefore,the enzyme is unable to modify DNA in a proces-sive manner. Cofactor binding to the apoenzymeshifts the conformational equilibrium to the openstate thereby promoting DNA binding. After DNAbinding, a non-speci®c enzyme-DNA complex isformed that isomerizes into a speci®c complexafter the enzyme has reached a speci®c site. Uponspeci®c M.EcoRV-DNA complex formation, theDNA is bent by 60 � in a fast process. The cofactorof the methylation reaction, AdoMet, but not theproduct of the reaction, AdoHyc, promotes speci®ccomplex formation. In the presence of cofactor, asecond conformation is accessible for speci®cenzyme-DNA complexes. Like the apoenzyme, thedifferent enzyme-DNA complexes exist in an openand closed complex conformation. DNA methyl-ation only takes place in the closed complex con-formation whereas DNA exchange occurs in theopen state. Product release does not preferentiallytake place at the ends of linear DNA. Since theconformational change between the open andclosed complexes is a slow process, formation ofthe closed enzyme-substrate complex and openingof the enzyme-product complex are the rate limit-ing steps of the methylation reaction under singleand multiple turnover conditions, respectively.
Experimental Procedures
Oligonucleotides and enzyme preparations
All oligonucleotides used in this study were pur-chased from Interactiva (Ulm, Germany) or PRIMM(Milano, Italy) in an HPLC puri®ed form. The followingsubstrates were used:
20ME: d(GATCGTAGATATCGCATCGA)/d(TCGATGCGmATATCTACGATC)
20MM: d(GATCGTAGATATCGCATCGA)/d(TCGATGCGmATATCTACGATC)
AP1: d(GATCGTAG-2AP-TATCGCATCGA)/d(TCGATGCGmATATCTACGATC)
AP2: d(GATCGTAGAT-2AP-TCGCATCGA)/d(TCGATGCGmATATCTACGATC)
AP3: d(GATCGT-2AP-GATATCGCATCGA)/d(TCGATGCGmATATCTACGATC)
20ME: d(GATCGTAGATATCGCATCGA)/d(TCGATGCGmATATCTACGATC)
RV2sites: d(GCGCC GATATCGATATC ATCACCGATGGGGAA)/d(TTCCCCATCGGTGAT GATATCGATATC GGCGC)
In addition, substrates containing more mA residuesand a palindromic non-speci®c substrate 20mer-nsd(GATCGACGATATCGTCGATC)2 were employed. Apalindromic 20mer (RVL20: d(GATCGACGATATCGTC-GATC)2) was used as competitor in the dissociationkinetics.
M.EcoRV was puri®ed as an N-terminal His6-fusionprotein as described (Jeltsch et al., 1999a). The activity ofthe M.EcoRV preparations was >80 % in active-sitetitrations carried out as described by Friedrich et al.(2000).
DNA-binding experiments
Equilibrium DNA-binding constants and dissociationrate constants of enzyme-DNA complexes were analyzedby nitrocellulose ®lter-binding experiments as described(Jeltsch et al., 1998, 1999b). Unless otherwise stated,all experiments were carried out in the presence of1 mM S-adenosylhomocysteine (AdoHyc) to preventDNA methylation. To determine equilibrium DNA-bind-ing constants, 5-10 nM labeled oligonucleotide was incu-bated with 50-1000 nM M.EcoRV in binding buffer(50 mM Hepes (pH 7.5), 50 mM NaCl, 1 mM EDTA,50 mg/ml bovine serum albumin (BSA) supplementedwith 1 mM AdoMet, AdoHyc or sinefungin as indicated)for 30 minutes and sucked through a nitrocellulose mem-brane (Protran BA 85, Schleicher & SchuÈ ll, Dassel). Afterwashing the membrane with buffer (50 mM Hepes(pH 7.5), 50 mM NaCl, 1 mM EDTA), the bound radio-activity was analyzed using an Instant Imager (CanberraPackard) and the experimental CPM-values ®tted toequation (1), which directly follows from the de®nitionof a bimolecular binding equilibrium:
CPMtheo �A� B
2� cE;tot � cD;tot � 1
KAss
�
ÿ��������������������������������������������������������������������������������������
cE;tot � cD;tot � 1
KAss
� �2
ÿ4� cE;tot � cD;tot
s�
�1�where A is the background correction; B is the factoraccounting for ®lter binding ef®ciency and speci®cactivity of the DNA substrate; KAss is the equilibriumbinding constant; cE,tot is the total enzyme concentration;cD,tot is the total DNA concentration.
For the dissociation kinetics, 10 nM labeled oligonu-cleotide was incubated with 200 nM M.EcoRV for30 minutes in binding buffer to allow complexformation. Then, the sample was diluted 20-fold in buf-fer containing unlabelled RVL20 at a ®nal concentrationof 0.5 mM. After increasing times, samples were suckedthrough a nitrocellulose membrane and washed immedi-ately with buffer (50 mM Hepes (pH 7.5), 50 mM NaCl,1 mM EDTA). The bound radioactivity was analyzed

106 Molecular Enzymology of M.EcoRV
using an Instant Imager (Canberra Packard) and the data®tted to equation (2), which is the integrated form of therate equation of a monomolecular decay process:
CPMtheo � A� B� eÿkdis�t �2�where A is the background correction; B is the factoraccounting for ®lter binding ef®ciency and speci®cactivity of the DNA substrate; kdis is the rate constant ofcomplex dissociation.
To determine the rate of closed complex formation,radiolabeled hemimethylated oligonucleotides (10 nM)were incubated for increasing times (t1) with M.EcoRV(200 nM). Then, unlabeled RVL20 was added to a ®nalconcentration of 0.5 mM and the reaction mixture incu-bated for ®ve minutes (t2). After the second incubation,the samples were sucked through a nitrocellulose mem-brane and the bound radioactivity analyzed. The datawere ®tted to equation (3), which describes the kineticsof a conformational change. With this experimentalsetup, the fraction of closed complexes can be deter-mined, because during the incubation with RVL20 (t2)the open M.EcoRV-DNA complexes dissociate almostcompletely whereas the closed complexes remain almostintact:
CPMtheo � A� B� �1ÿ eÿk�t� �3�where A is the background correction; B is the ®lterbinding ef®ciency; k is the rate constant of closedcomplex formation.
Steady-state fluorescence experiments
To measure the ¯uorescence change of oligonucleo-tides containing 2AP upon M.EcoRV binding, 500 nMdouble-stranded oligonucleotide containing 2AP was dis-solved in buffer (50 mM Hepes (pH 7.5), 50 mM NaCl,1 mM EDTA, 0.001 % Lubrol) containing 0.1 mM Ado-Met and the ¯uorescence signal of the 2AP probe deter-mined. A second sample of the oligonucleotide wasmixed with 1 mM M.EcoRV and analyzed after 15 min-utes incubation. 2AP-¯uorescence was excited at 313 nmin a F4500 spectro¯uorimeter (Hitachi). Uncorrectedemission spectra were recorded between 320 and500 nm. Emission and excitation slits were set to 2.5 nm.The data were analyzed by integration of the ¯uor-escence peak after subtraction of a constant backgroundlevel.
For the enzyme-DNA complex dissociation exper-iments, 500 nM AP2 oligonucleotide was incubated with1 mM M.EcoRV in 50 mM Hepes (pH 7.5), 50 mM NaCl,1 mM EDTA, 0.001 % (v/v) Lubrol, 0.1 mM AdoMet for30 minutes to allow complex formation. After determin-ing the ¯uorescence of the sample, 20mer oligonucleotide(RVL20) was added to a concentration of 4 mM, mixedand the ¯uorescence spectra analyzed after increasingtimes of incubation. The data were analyzed by inte-gration of the ¯uorescence peaks after subtraction of aconstant background level and ®t to equation (2).
Stopped-flow experiments
Stopped-¯ow experiments were carried out in an SF3-QF device (BioLogic, Claix). To study the time-course ofDNA binding, syringe 1 and syringe 2 were ®lled with100-700 nM AP2 oligonucleotide in SF-buffer (50 mMHepes (pH 7.5), 50 mM NaCl, 1 mM EDTA, 0.001 %Lubrol in some experiments supplemented with 0.1 mM
AdoHyc or 0.1 mM AdoMet as indicated). Prior to theexperiments SF-buffer was passed through a 2 mm mem-brane ®lter and degassed. Syringe 3 was ®lled with 100-700 nM M.EcoRV in SF-buffer. The samples were rapidlymixed and the ¯uorescence of the 2AP probe was excitedat 313 nm. Emission was observed using a 340 nm cut-off ®lter. To study dissociation of enzyme-DNA com-plexes, syringe 3 was ®lled with enzyme and DNA inSF-buffer. The enzyme-DNA complexes were rapidlymixed with RVL20 dissolved in SF-buffer (syringe 1 and2) (®nal concentrations: 500 nM M.EcoRV, 200 nM DNA,1 mM RVL20). Under the experimental conditions, thedead time of the experiment was 3.1 ms in both setups:10-20 ¯uorescence traces were averaged for each exper-iment. The results were globally ®t to different modelsby least-squares ®ts. To this end, the behavior of the sys-tems was calculated on the basis of the correspondingrate equations by numerical integration using an Excelworksheet. For error analysis, all rate constants wereindividually minimized and maximized upon variationof all other constants until the mean error of the resulting®t was signi®cantly higher than in the best ®t as judgedby a t-test.
DNA methylation experiments with labeled AdoMet
DNA methylation experiments were carried out inbuffer (50 mM Hepes (pH 7.5), 50 mM NaCl, 1 mMEDTA) either by premixing M.EcoRV and AdoMet andstarting the reaction with DNA or by premixingM.EcoRV and DNA and starting the methylation reactionby addition of AdoMet. In both sets of reactions,1 mM M.EcoRV, 500 nM oligonucleotide and 0.7 mM[methyl-3H]AdoMet (2.7 TBq/mmol; NEN) were used. Atincreasing time-points, 2 ml aliquots were withdrawnand analyzed using the biotin-avidin microplate assay asdescribed by Roth & Jeltsch (2000).
Analysis of processivity of M.EcoRV
To analyze processivity of DNA methylation catalyzedby M.EcoRV, the rate of methylation of the RV2sites sub-strate which harbors two M.EcoRV sites was analyzed bythe restriction protection method. This technique directlyallows to visualize the individual methylation status ofboth target sites. RV2sites was radioactively labeled inone strand using phage T4 polynucleotide kinase (MBI):50 nM oligonucleotide was incubated with 10 nMM.EcoRV in methylation buffer (50 mM Hepes (pH 7.5),50 mM NaCl, 1 mM EDTA) containing 1 mM AdoMet(Sigma). After de®ned times, 10 ml aliquots were with-drawn and the reaction stopped by freezing in liquidnitrogen. After addition of MgCl2 to a ®nal concentrationof 10 mM, the samples were incubated with 100 nMR.EcoRV for 60 minutes. The cleavage pattern of the oli-gonucleotide was analyzed on a denaturing 20 % (w/v)polyacrylamide gel and the radioactivity visualizedusing a Phosphorimager (Canberra Packard). The rela-tive amounts of unmethylated and methylated DNAwere ®t to distributive or processive models of DNAmethylation.
Analysis of DNA methylation of several substratesin competition
To be able to measure the rates of DNA methylationof up to three substrates in competition, i.e. in one reac-tion tube, a restriction-protection assay was employed.

Molecular Enzymology of M.EcoRV 107
In these experiments, PCR products of 200, 500, 1000and 1500 bp length were used. In each case one of thePCR primers was labeled with T4 polynucleotide kinaseand [g-P32]ATP such that all substrates are labeled onone end. Since all substrates were ampli®ed frompAT153, all of them contain the EcoRV recognition site inthe same sequence context. The PCR fragments werepuri®ed using Qiaquick Spin puri®cation kits (Qiagen).Gel analyses con®rmed the presence of only one singleband at the expected molecular mass in each case. Forone set of methylation experiments, 10-20 nM 200mer,500mer and 1000mer and for a second set of experimentssame amounts of the 1000mer and 1500mer were incu-bated for de®ned times at ambient temperature with10 nM M.EcoRV in methylation buffer containing 1 mMAdoMet (Sigma). After de®ned times, 10 ml aliquotswere withdrawn and the reaction was quenched byfreezing in liquid nitrogen. Finally, MgCl2 and R.EcoRVwere added to the reaction mixture to ®nal concen-trations of 10 mM and 100 nM, respectively, and thesamples incubated for 60 minutes at 37 �C. The sampleswere analyzed on TPE/6 % polyacrylamide gels and theradioactivity visualized using a Phosphorimager (Can-berra Packard). The relative amounts of unmethylatedand methylated DNA were ®t to a single exponentialcurve to determine the rate constants of DNA methyl-ation.
Analysis of linear diffusion
The shape of the curve showing the rate of methyl-ation of M.EcoRV versus substrate length can be used fora quantitative analysis of the ef®ciency of linear diffu-sion. According to the theory of linear diffusion the over-all rate of association to a DNA fragment (kon) is givenby (Ehbrecht et al., 1985):
kon=k�1 �XL
n�1
eÿ�nÿs�2=Pdif �4�
where kon is the overall rate of association to the sub-strate; k�1 is the association rate constant to a non-speci®c site; L is the length of the DNA fragment; s isthe position of the EcoRV site; Pdif is the probability todiffuse one step along the DNA rather than to dissociatefrom the DNA.
Then, the relative rate of association to the speci®c siteon two substrates, A and B, is given by the ratio of thecorresponding sums of equation (4). If the catalytic rateis limited by the rate of target site location, the rate ofmethylation of one substrate relative to the other isgiven by:
VA=VB � kAon
kBon
�XLA
n�1
eÿ�nÿsA�2=Pdif
XLB
n�1
eÿ�nÿsB�2=Pdif �5�
where vi is the rate of modi®cation of site i; kion is the
association rate constant to site i; Li is the length of sub-strate i; si is the position of the EcoRV site in substrate i.
Acknowledgments
Expert technical assistance by H. BuÈ ngen is gratefullyacknowledged. Thanks are due to A. Pingoud for stimu-
lating discussion, encouragement and support. Thiswork has been supported by the Deutsche Forschungsge-meinschaft (JE 252/2-3 and JE 252/1-1).
References
Ahmad, I. & Rao, D. N. (1996). Chemistry and biologyof DNA methyltransferases. Crit. Rev. Biochem. Mol.Biol. 31, 361-380.
Allan, B. W. & Reich, N. O. (1996). Targeted base stack-ing disruption by the EcoRI DNA methyltransferase.Biochemistry, 35, 14757-14762.
Allan, B. W., Beechem, J. M., Lindstrom, W. M. & Reich,N. O. (1998a). Direct real time observation of base¯ipping by the EcoRI DNA methyltransferase. J. Biol.Chem. 273, 2368-2373.
Allan, B. W., Reich, N. O. & Beechem, J. M. (1998b).Simultaneous real-time measurement of binding:base-¯ipping, and dissociation: ``base un¯ipping'',in EcoRI DNA methyltransferase. Biophys. J. 74,A69.
Berdis, A. J., Lee, I., Coward, J. K., Stephens, C., Wright,R., Shapiro, L. & Benkovic, S. J. (1998). A cell cycle-regulated adenine DNA methyltransferase fromCaulobacter crescentus processively methylatesGANTC sites on hemimethylated DNA. Proc. NatlAcad. Sci. USA, 95, 2874-2879.
Berg, O. G. & von Hippel, P. H. (1985). Diffusion-con-trolled macromolecular interactions. Annu. Rev. Bio-phys. Biophys. Chem. 14, 131-60.
Berg, O. G., Winter, R. B. & von Hippel, P. H. (1981).Diffusion-driven mechanisms of protein transloca-tion on nucleic acids. 1. Models and theory. Bio-chemistry, 20, 6929-6948.
Bergerat, A. & Guschlbauer, W. (1990). The double roleof methyl donor and allosteric effector of S-adeno-syl-methionine for Dam methylase of E. coli. Nucl.Acids Res. 18, 4369-4375.
Bestor, T. H. & Verdine, G. L. (1994). DNA methyltrans-ferases. Curr. Opin. Cell Biol. 6, 380-389.
Bhattacharya, S. K. & Dubey, A. K. (1999). Kinetic mech-anism of cytosine DNA methyltransferase MspI.J. Biol. Chem. 274, 14743-14749.
Bucci, C., Lavitola, A., Salvatore, P., Del, Giudice L.,Massardo, D. R., Bruni, C. B. & Alifano, P. (1999).Hypermutation in pathogenic bacteria: frequentphase variation in meningococci is a phenotypictrait of a specialized mutator biotype. Mol. Cell, 3,435-445.
Cal, S. & Connolly, B. A. (1996). The EcoRV modi®cationmethylase causes considerable bending of DNAupon binding to its recognition sequence GATATC.J. Biol. Chem. 271, 1008-1015.
Cal, S. & Connolly, B. A. (1997). DNA distortion andbase ¯ipping by the EcoRV DNA methyltransferase.J. Biol. Chem. 272, 490-496.
Cantor, C. R. & Schimmel, P. R. (1980). Biophysical Chem-istry, W.H. Freeman and Company, San Francisco.
Cheng, X. (1995a). DNA modi®cation by methyltrans-ferases. Curr. Opin. Struct. Biol. 5, 4-10.
Cheng, X. (1995b). Structure and function of DNAmethyltransferases. Annu. Rev. Biophys. Biomol.Struct. 24, 293-318.
Cheng, X., Kumar, S., Posfai, J., P¯ugrath, J. W. &Roberts, R. J. (1993). Crystal structure of the HhaIDNA methyltransferase complexed with S-adeno-syl-L-methionine. Cell, 74, 299-307.

108 Molecular Enzymology of M.EcoRV
Colot, V. & Rossignol, J. L. (1999). Eukaryotic DNAmethylation as an evolutionary device. Bioessays, 21,402-411.
Dryden, D. T. F. (1999). Bacterial DNA methyltransfer-ases. In S-Adenosyl-dependent Methyltransferases:Structures and Functions (Cheng, X. & Blumenthal,R. M., eds), pp. 283-340, World Scienti®c PublishingCo. Pte. Ltd., Singapore.
Dubey, A. K. & Bhattacharya, S. K. (1997). Angle andlocus of the bend induced by the MspI DNAmethyltransferase in a sequence-speci®c complexwith DNA. Nucl. Acids Res. 25, 2025-2029.
Ehbrecht, H.-J., Pingoud, A., Urbanke, C., Maass, G. &Gualerzi, C. (1985). Linear diffusion of restrictionendonucleases on DNA. J. Biol. Chem. 260, 6160-6166.
Ellis, D. J., Dryden, D. T. F., Berge, T., Edwardson, J. M.& Henderson, R. M. (1999). Direct observation ofDNA translocation and cleavage by the EcoKI endo-nuclease using atomic force microscopy. NatureStruct. Biol. 6, 15-17.
Enserink, M. (1999). Gene may promise new route topotent vaccines. Science, 284, 883.
Flynn, J. & Reich, N. (1998). Murine DNA (cytosine-5-)-methyltransferase: steady-state and substrate trap-ping analyses of the kinetic mechanism. Biochemis-try, 37, 15162-15169.
Frey, M. W., Sowers, L. C., Millar, D. P. & Benkovic, S. J.(1995). The nucleotide analog 2-aminopurine as aspectroscopic probe of nucleotide incorporation bythe Klenow fragment of Escherichia coli polymerase Iand bacteriophage T4 DNA polymerase. Biochemis-try, 34, 9185-9192.
Friedrich, T., Roth, M., Helm-Kruse, S. & Jeltsch, A.(1998). Functional mapping of the EcoRV DNAmethyltransferase by random mutagenesis andscreening for catalytically inactive mutants. Biol.Chem. 379, 475-480.
Friedrich, T., Fatemi, M., Gowhar, H., Leismann, O. &Jeltsch, A. (2000). Speci®city of DNA binding andmethylation by the M. FokI DNA methyltransferase.Biochim. Biophys. Acta, 1480, 145-159.
Garcia, L. R. & Molineux, I. J. (1999). Translocation andspeci®c cleavage of bacteriophage T7 DNA in vivoby EcoKI. Proc. Natl Acad. Sci. USA, 96, 12430-12435.
Garcia, R. A., Bustamante, C. J. & Reich, N. O. (1996).Sequence-speci®c recognition by cytosine C5 andadenine N6 DNA methyltransferases requires differ-ent deformations of DNA. Proc. Natl Acad. Sci. USA,93, 7618-7622.
Garcia-Del Portillo, F., Pucciarelli, M. G. & Casadesus, J.(1999). DNA adenine methylase mutants of Salmo-nella typhimurium show defects in protein secretion,cell invasion, and M cell cytotoxicity. Proc. NatlAcad. Sci. USA, 96, 11578-11583.
Gong, W., O'Gara, M., Blumenthal, R. M. & Cheng, X.(1997). Structure of PvuII DNA-(cytosine N4) meth-yltransferase, an example of domain permutationand protein fold assignment. Nucl. Acids Res. 25,2702-2715.
Hale, W. B., van der Woude, M. W., Braaten, B. A. &Low, D. A. (1998). Regulation of uropathogenicEscherichia coli adhesin expression by DNA methyl-ation. Mol. Genet. Metab. 65, 191-196.
Heithoff, D. M., Sinsheimer, R. L., Low, D. A. & Mahan,M. J. (1999). An essential role for DNA adeninemethylation in bacterial virulence. Science, 284, 967-970.
Heitman, J. (1993). On the origins, structures and func-tions of restriction-modi®cation enzymes. In GeneticEngineering (Setlow, J. K., ed.), vol. 15, pp. 57-108,Plenum Press, New York.
Holz, B., Klimasauskas, S., Serva, S. & Weinhold, E.(1998). 2-Aminopurine as a ¯uorescent probe forDNA base ¯ipping by methyltransferases. Nucl.Acids Res. 26, 1076-1083.
Jack, W. E., Terry, B. J. & Modrich, P. (1982). Involve-ment of outside DNA sequences in the majorkinetic path by which EcoRI endonuclease locatesand leaves its recognition sequence. Proc. Natl Acad.Sci. USA, 79, 4010-4014.
Janscak, P., Sandmeier, U. & Bickle, T. A. (1999). Singleamino acid substitutions in the HsdR subunit of thetype IB restriction enzyme EcoAI uncouple theDNA translocation and DNA cleavage activities ofthe enzyme. Nucl. Acids Res. 27, 2638-2643.
Jeltsch, A. (1998). Flexibility of DNA in complex withproteins deduced from the distribution of bendingangles observed by scanning force microscopy. Bio-phys. Chem. 74, 53-57.
Jeltsch, A. & Pingoud, A. (1998). Kinetic characterizationof linear diffusion of the restriction endonucleaseEcoRV on DNA. Biochemistry, 37, 2160-2169.
Jeltsch, A., Alves, J., Wolfes, H., Maass, G. & Pingoud,A. (1994). Pausing of the restriction endonucleaseEcoRI during linear diffusion on DNA. Biochemistry,33, 10215-10219.
Jeltsch, A., Wenz, C., Stahl, F. & Pingoud, A. (1996).Linear diffusion of the restriction endonucleaseEcoRV on DNA is essential for the in vivo functionof the enzyme. EMBO J. 15, 5104-5111.
Jeltsch, A., Friedrich, T. & Roth, M. (1998). Kinetics ofmethylation and binding of DNA by the EcoRVadenine-N6 methyltransferase. J. Mol. Biol. 275, 747-758.
Jeltsch, A., Christ, F., Fatemi, M. & Roth, M. (1999a). Onthe substrate speci®city of DNA methyltransferases:adenine-N6 DNA methyltransferases also modifycytosine residues at position N4. J. Biol. Chem. 274,19538-19544.
Jeltsch, A., Roth, M. & Friedrich, T. (1999b). Mutationalanalysis of the methylation of target base ¯ippingby the EcoRV adenine-N6 DNA methyltransferase.J. Mol. Biol. 285, 1121-1130.
Jia, Y., Kumar, A. & Patel, S. S. (1996). Equilibrium andstopped-¯ow kinetic studies of interaction betweenT7 RNA polymerase and its promoters measuredby protein and 2-aminopurine ¯uorescence changes.J. Biol. Chem. 271, 30451-30458.
Klimasauskas, S. & Roberts, R. J. (1995a). Disruption ofthe target G-C base-pair by the HhaI methyltransfer-ase. Gene, 157, 163-164.
Klimasauskas, S. & Roberts, R. J. (1995b). M. HhaI bindstightly to substrates containing mismatches at thetarget base. Nucl. Acids Res. 23, 1388-1395.
Klimasauskas, S., Kumar, S., Roberts, R. J. & Cheng, X.(1994). Hhal methyltransferase ¯ips its target baseout of the DNA helix. Cell, 76, 357-369.
Klimasauskas, S., Szyperski, T., Serva, S. & Wuthrich, K.(1998). Dynamic modes of the ¯ipped-out cytosineduring HhaI methyltransferase-DNA interactions insolution. EMBO J. 17, 317-324.
Labahn, J., Granzin, J., Schluckebier, G., Robinson, D. P.,Jack, W. E., Schildkraut, I. & Saenger, W. (1994).Three-dimensional structure of the adenine-speci®cDNA methyltransferase M. TaqI in complex with

Molecular Enzymology of M.EcoRV 109
the cofactor S-adenosylmethionine. Proc. Natl Acad.Sci. USA, 91, 10957-10961.
Li, E., Bestor, T. H. & Jaenisch, R. (1992). Targetedmutation of the DNA methyltransferase gene resultsin embryonic lethality. Cell, 69, 915-926.
Lindstrom, W. M., Jr, Flynn, J. & Reich, N. O. (1999).Reconciling structure and function in HhaI DNAcytosine-C-5 methyltransferase. J. Biol. Chem. 275,4912-4919.
Messer, W. & Noyer-Weidner, M. (1998). Timing andtargeting: the biological functions of Dam methyl-ation in E. coli. Cell, 54, 735-737.
Nardone, G., George, J. & Chirikjian, J. G. (1986). Differ-ences in the kinetic properties of BamHI endonu-clease and methylase with linear DNA substrates.J. Biol. Chem. 261, 12128-12133.
Nwosu, V. U., Connolly, B. A., Halford, S. E. & Garnett,J. (1988). The cloning, puri®cation and characteriz-ation of the EcoRV modi®cation methylase. Nucl.Acids Res. 16, 3705-3720.
Okano, M., Bell, D. W., Haber, D. A. & Li, E. (1999).DNA methyltransferases Dnmt3a and Dnmt3b areessential for de novo methylation and mammaliandevelopment. Cell, 99, 247-257.
Parkhurst, K. M., Brenowitz, M. & Parkhurst, L. J.(1996). Simultaneous binding and bending of pro-moter DNA by the TATA binding protein: real timekinetic measurements. Biochemistry, 35, 7459-7465.
Pingoud, A. & Jeltsch, A. (1997). Recognition and clea-vage of DNA by type-II restriction endonucleases.Eur. J. Biochem. 246, 1-22.
Pues, H., Bleimling, N., Holz, B., Wolcke, J. &Weinhold, E. (1999). Functional roles of the con-served aromatic amino acid residues at position 108(motif IV) and position 196 (motif VIII) in base ¯ip-ping and catalysis by the N6-adenine DNA methyl-transferase from Thermus aquaticus. Biochemistry, 38,1426-1434.
Raney, K. D., Sowers, L. C., Millar, D. P. & Benkovic,S. J. (1994). A ¯uorescence-based assay for monitor-ing helicase activity. Proc. Natl Acad. Sci. USA, 91,6644-6648.
Rao, D. N., Page, M. G. P. & Bickle, T. A. (1989). Clon-ing, over-expression and the catalytic properties ofthe EcoP15 modi®cation methylase from Escherichiacoli. J. Mol. Biol. 209, 599-606.
Reddy, Y. V. & Rao, D. N. (2000). Binding of EcoP15IDNA methyltransferase to DNA reveals a largestructural distortion within the recognitionsequence. J. Mol. Biol. 298, 597-610.
Reich, N. O. & Mashhoon, N. (1991). Kinetic mechanismof the EcoRI DNA methyltransferase. Biochemistry,30, 2933-2939.
Reich, N. O. & Mashhoon, N. (1993). Presteady statekinetics of an S-adenosylmethionine-dependentenzyme. J. Biol. Chem. 268, 9191-9193.
Reinisch, K. M., Chen, L., Verdine, G. L. & Lipscomb,W. N. (1995). The crystal structure of HaeIII methyl-transferase covalently complexed to DNA: an extra-helical cytosine and rearranged base-pairing. Cell,82, 143-153.
Rhodes, D., Schwabe, J. W., Chapman, L. & Fairall, L.(1996). Towards an understanding of protein-DNArecognition. Phil. Trans. Roy. Soc. ser. B, 351, 501-509.
Richter, P. H. & Eigen, M. (1974). Diffusion con-trolled reaction rates in spheroidal geometry.Application to repressor-operator association and
membrane bound enzymes. Biophys. Chem. 2, 255-263.
Roth, M. & Jeltsch, A. (2000). Biotin-avidin microplateassay for the quantitative analysis of enzymaticmethylation of DNA by DNA methyltransferases.Biol. Chem. 381, 269-272.
Roth, M., Helm-Kruse, S., Friedrich, T. & Jeltsch, A.(1998). Functional roles of conserved aminoacid residues in DNA methyltransferases investi-gated by site-directed mutagenesis of the EcoRVadenine-M6-methyltransferase. J. Biol. Chem. 273,17333-17342.
Saha, S. & Rao, D. N. (1995). ATP hydrolysis is requiredfor DNA cleavage by EcoPI restriction enzyme.J. Mol. Biol. 247, 559-567.
Schluckebier, G., Labahn, J., Granzin, J., Schildkraut, I.& Saenger, W. (1995). A model for DNA bindingand enzyme action derived from crystallographicstudies of the TaqI N6-adenine-methyltransferase.Gene, 157, 131-134.
Spolar, R. S. & Record, M. T., Jr (1994). Coupling oflocal folding to site-speci®c binding of proteins toDNA. Science, 263, 777-784.
Stephens, C., Reisenauer, A., Wright, R. & Shapiro, L.(1996). A cell cycle-regulated bacterial DNA methyl-transferase is essential for viability. Proc. Natl Acad.Sci. USA, 93, 1210-1214.
Stivers, J. T., Pankiewicz, K. W. & Watanabe, K. A.(1999). Kinetic mechanism of damage site recog-nition and uracil ¯ipping by Escherichia coli uracilDNA glycosylase. Biochemistry, 38, 952-963.
Surby, M. A. & Reich, N. O. (1996a). Contribution offacilitated diffusion and processive catalysis toenzyme ef®ciency: implications for the EcoRI restric-tion-modi®cation system. Biochemistry, 35, 2201-2208.
Surby, M. A. & Reich, N. O. (1996b). Facilitated diffu-sion of the EcoRI DNA methyltransferase isdescribed by a novel mechanism. Biochemistry, 35,2209-2217.
Szczelkun, M. D. & Connolly, B. A. (1995). Sequence-speci®c binding of DNA by the EcoRV restrictionand modi®cation enzymes with nucleic acidand cofactor analogues. Biochemistry, 34, 10724-10733.
Szczelkun, M. D., Jones, H. & Connolly, B. A. (1995).Probing the protein-DNA interface of the EcoRVmodi®cation methyltransferase bound to its recog-nition sequence, GATATC. Biochemistry, 34, 10734-10743.
Szilak, L., Der, A., Deak, F. & Venetianer, P. (1993). Kin-etic characterization of the EcaI methyltransferase.Eur. J. Biochem. 218, 727-733.
Tran, P. H., Korszun, Z. R., Cerritelli, S., Springhorn,S. S. & Lacks, S. A. (1998). Crystal structure of theDpnM DNA adenine methyltransferase from theDpnII restriction system of Streptococcus pneumoniaebound to S-adenosylmethionine. Structure, 6, 1563-1575.
von Hippel, P. H. (1994). Protein-DNA recognition: newperspectives and underlying themes. Science, 263,769-770.
von Hippel, P. H. & Berg, O. G. (1989). Facilitated targetlocation in biological systems. J. Biol. Chem. 264,675-678.
Ward, D. C., Reich, E. & Stryer, L. (1969). Fluorescencestudies of nucleotides and polynucleotides. I. For-mycin, 2- aminopurine riboside, 2,6-diaminopurine

110 Molecular Enzymology of M.EcoRV
riboside, and their derivatives. J. Biol. Chem. 244,1228-1237.
Wilson, G. G. & Murray, N. E. (1991). Restrictionand modi®cation systems. Annu. Rev. Genet. 25,585-627.
Wolffe, A. P. & Matzke, M. A. (1999). Epigenetics: regu-lation through repression. Science, 286, 481-486.
Wu, J. C. & Santi, D. V. (1987). Kinetic and catalyticmechanism of HhaI methyltransferase. J. Biol. Chem.262, 4778-4786.
Edited by J. Karn
(Received 12 June 2000; received in revised form 18 August 2000; accepted 25 August 2000)
Copyright © 2022 FDOKUMEN




















