
Therapeutic B cell depletion impairs adaptive and autoreactive CD4+ T cell activation in mice
Upload
independentCategory
view
4download
0
of June 13, 2013.This information is current as
of Rapamycin Signaling PathwayNutrient/Energy-Sensing Mammalian TargetAutoreactive CD4+ T Cells through the Leptin Modulates the Survival of
Giuseppe MatareseFortunata Carbone, Paolo Chieffi, Antonio La Cava and Mario Galgani, Claudio Procaccini, Veronica De Rosa,
http://www.jimmunol.org/content/185/12/7474doi: 10.4049/jimmunol.1001674November 2010;
2010; 185:7474-7479; Prepublished online 15J Immunol
MaterialSupplementary
4.DC1.htmlhttp://www.jimmunol.org/content/suppl/2010/11/15/jimmunol.100167
Referenceshttp://www.jimmunol.org/content/185/12/7474.full#ref-list-1
, 16 of which you can access for free at: cites 46 articlesThis article
Subscriptionshttp://jimmunol.org/subscriptions
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/ji/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/cgi/alerts/etocReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. All rights reserved.9650 Rockville Pike, Bethesda, MD 20814-3994.The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 13, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

The Journal of Immunology
Leptin Modulates the Survival of Autoreactive CD4+ T Cellsthrough the Nutrient/Energy-Sensing Mammalian Target ofRapamycin Signaling Pathway
Mario Galgani,*,† Claudio Procaccini,*,† Veronica De Rosa,*,† Fortunata Carbone,*,†
Paolo Chieffi,‡ Antonio La Cava,x and Giuseppe Matarese*,†
Chronic inflammation can associate with autoreactive immune responses, including CD4+ T cell responses to self-Ags. In this
paper, we show that the adipocyte-derived proinflammatory hormone leptin can affect the survival and proliferation of autore-
active CD4+ T cells in experimental autoimmune encephalomyelitis, an animal model of human multiple sclerosis. We found that
myelin olygodendrocyte glycoprotein peptide 35–55 (MOG35–55)-specific CD4+ T cells from C57BL/6J wild-type mice could not
transfer experimental autoimmune encephalomyelitis into leptin-deficient ob/obmice. Such a finding was associated with a reduced
proliferation of the transferred MOG35–55-reactive CD4+ T cells, which had a reduced degradation of the cyclin-dependent kinase
inhibitor p27kip1 and ERK1/2 phosphorylation. The transferred cells displayed reduced Th1/Th17 responses and reduced delayed-
type hypersensitivity. Moreover, MOG35–55-reactive CD4+ T cells in ob/ob mice underwent apoptosis that associated with a down-
modulation of Bcl-2. Similar results were observed in transgenic AND-TCR- mice carrying a TCR specific for the pigeon cyto-
chrome c 88–104 peptide. These molecular events reveal a reduced activity of the nutrient/energy-sensing AKT/mammalian target
of rapamycin pathway, which can be restored in vivo by exogenous leptin replacement. These results may help to explain a link
between chronic inflammation and autoimmune T cell reactivity. The Journal of Immunology, 2010, 185: 7474–7479.
Several studies have shown that calorie restriction (CR)can modulate metabolic and physiologic changes that canbe beneficial in cancer, metabolic syndrome, inflammation,
and autoimmunity (1–3). Leptin, an adipocyte-derived hormone ofthe long-chain helical cytokine family (4, 5), has been proposed tolink nutritional status and certain immune responses (6, 7). Leptinconcentration is proportional to fat mass and is positively as-sociated with overweight, obesity, and increased susceptibility tochronic inflammation and autoimmunity (6, 7). At the immunelevel, leptin promotes Th1 proinflammatory responses in auto-immunity (8, 9), although it can also induce the expression of IL-1receptor antagonist in human monocytes (10) or IL-4 (11) and
IL-10 (12) and suppress IFN-g (12) or IL-6 production caused byendotoxin (13).Leptin-deficient (ob/ob) mice and leptin receptor-deficient mice
(db/db) mice display numerous immune abnormalities (14, 15),including CD4+ T cell lymphopenia, increased numbers of naturalregulatory T cells, and a resistance to autoimmune disorders in-cluding experimental autoimmune encephalomyelitis (EAE), ex-perimental colitis, Ag-induced arthritis, and type 1 diabetes (16).The cytokines IL-6, IL-7, IL-15, and IL-21 have all been im-
plicated in the homeostatic control of lymphocyte numbers (17–19), and homeostatic proliferation of regulatory T cells has beenobserved during T cell lymphopenia. An uncontrolled expansionof autoreactive T cells could favor autoimmunity (20–24), but inob/ob mice, CD4+ lymphopenia does not associate with homeo-static proliferation of T cells and autoimmunity (14–16).In this paper, we show that leptin can affect the survival and
proliferation of autoreactive T cells by modulating the activity ofthe survival protein Bcl-2, Th1/Th17 cytokines, and the nutrient/energy-sensing AKT-mammalian target of rapamycin (mTOR) path-way. Findings that link body fat mass and autoreactivity alsosuggest the possibility to regard obesity as a disease with auto-immune components (25–29).
Materials and MethodsMice
Eight- to 10-wk-old female C57BL/6J (B6), wild-type (WT), and C57/BL/6J-ob/ob-leptin–deficient mice were purchased from Charles River Labo-ratories (Calco, Italy) and from Harlan (Correzana, Italy). The B10.Cg.Tg(TcrAND)53Hed/J (AND-TCR transgenic [Tg]) pigeon cytochrome c(PCC)88–104-specific transgenic mice were purchased from The JacksonLaboratory (Bar Harbor, ME). B6-WT and ob/ob mice were age-matchedfor individual experiments and were group-housed in the same cagesaccording to different experimental condition, with a 12-h light-dark cycle.All experiments were performed under approved protocol in accordancewith animal use guidelines of the Istituto Superiore di Sanita (Rome, Italy).
*Laboratorio di Immunologia, Istituto di Endocrinologia e Oncologia Sperimentale,Consiglio Nazionale delle Ricerche; †Laboratorio di Immunologia, Dipartimento diBiologia e Patologia Cellulare e Molecolare, Universita di Napoli “Federico II”;‡Dipartimento di Medicina Sperimentale, II Universita di Napoli, Napoli, Italy;and xDepartment of Medicine, David Geffen School of Medicine, University ofCalifornia, Los Angeles, Los Angeles, CA 90095
Received for publication May 20, 2010. Accepted for publication October 12, 2010.
This work was supported by grants from the European Union Ideas Programme (toG.M.), European Research Council-Starting Independent “LeptinMS” Grant 202579(to G.M.), and Telethon-Juvenile Diabetes Research Foundation International GrantGJT08004 (to G.M.). A.L.C. is supported in part by the National Institutes of HealthGrant AR53239.
Address correspondence and reprint requests to Dr. Giuseppe Matarese or Dr. AntonioLaCava, Laboratorio di Immunologia, Istituto di Endocrinologia eOncologia Sperimen-tale, ConsiglioNazionale delle Ricerche,Via S. Pansini 5, 80131,Napoli, Italy (G.M.), orDepartment of Medicine, David Geffen School of Medicine, University of California,Los Angeles, 1000 Veteran Avenue 32-59, Los Angeles, CA 90095 (A.L.C.). E-mailaddresses: [email protected] (G.M.) and [email protected] (A.L.C.)
The online version of this article contains supplemental material.
Abbreviations used in this paper: B6, C57BL/6J; CR, calorie restriction; DTH,delayed-type hypersensitivity; EAE, experimental autoimmune encephalomyelitis;mTOR, mammalian target of rapamycin; P, phospho; PCC, pigeon cytochrome c;r-leptin, recombinant leptin; S6, S6 ribosomal protein; Tg, transgenic; WT, wild-type.
Copyright� 2010 by TheAmericanAssociation of Immunologists, Inc. 0022-1767/10/$16.00
www.jimmunol.org/cgi/doi/10.4049/jimmunol.1001674
by guest on June 13, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

Peptides
MOG35–55 peptide (MEVGWYRSPFSRVVHLYRNGK) was used for theinduction of EAE (30), and PCC88–104 peptide (KAERADLIAYLKQ-ATAK) was used to stimulate AND-TCR-Tg cells (31).
Induction of adoptive EAE and clinical assessment
The induction of EAE has been described previously (30). Ten female donorB6-WT mice (6–8 wk old) were primed s.c. with 300 mg MOG35–55
peptide in CFA distributed over four sites. Recipient syngeneic naivefemale ob/ob, PBS or recombinant leptin (r-leptin)–treated, and B6-WTcontrol mice were injected i.v. with 53 106 CD4+ T cells in a final volumeof 500 ml PBS. Mice received 200 ng pertussis toxin immediately after celltransfer and 1 d later. Individual mice were monitored daily for clinicalsigns of disease for up to 30 d after adoptive transfer, as described pre-viously (30).
Treatment with leptin and rapamycin
Mouse r-leptin was purchased from R&D Systems (Oxon, U.K.); puritywas .97%. The endotoxin level was ,0.1 ng/mg leptin, as determined bythe Limulus amebocyte lysate method. Mice comprised two groups (n = 6–11/group) of ob/ob-leptin–deficient obese mice and one group (n = 6–10/group) of B6-WT age- and sex-matched control mice. For adoptively-in-duced disease, mice were treated with r-leptin (0.5 mg/g initial body weighttwice daily in 200 ml volume i.p.) starting 3 d before the transfer ofMOG35–55-specific CD4
+ T cells and continuing over a period of 30 d. Ofthe groups of leptin-deficient mice, one was injected with 200 ml PBStwice daily (at 10:00 AM and 6:00 PM); the second group was injected withmurine r-leptin (0.5 mg/g initial body weight twice daily in 200 ml volumei.p.). For the group of B6-WT mice, PBS was injected twice daily ac-cording to the same schedule. All mice were weighed, and food intakewas recorded daily. Rapamycin was dissolved in DMSO and then broughtto volume with PBS. For in vivo treatment with rapamycin (Sigma-Aldrich, St. Louis, MO), B6-WT mice adoptively transferred with CFSE+
MOG35–55-specific CD4+ T cells were injected once (day 0) i.p. with100 mg rapamycin/mouse (final volume, 200 ml). The control group re-ceived the same volume of vehicle once. Leptin-treated mice received onesingle dose of 100 mg r-leptin/mouse once. Mice were sacrificed 7 dposttreatment, and cells were analyzed by flow cytometry.
Delayed-type hypersensitivity
Delayed-type hypersensitivity (DTH) responses to MOG35–55 peptide byadoptively transferred MOG35–55-specific CD4+ T cells were quantifiedusing a time-dependent footpad swelling assay (32). Footpad thickness wasmeasured at 7 d after transfer and at 12, 24, 48, and 72 h after challenge orat 1, 7, or 14 d by a “blinded” investigator, as described previously (30).
Proliferation assays
Spleen cells were obtained from mice at different time points after adop-tive transfer and used for proliferation assays, as described previously (30).For experiments with AND-TCR-Tg mice, DCEK transfectants (murinefibroblasts cells transfected with the Ek mouse MHC class II molecule)(31) were used as APCs to activate in vitro CD4+ AND-TCR-Tg miceT cells. T cells were incubated and harvested as described previously (30).
Cytokine measurement
Leptin, IL-7, IL-15, and IL-21 were measured using ELISA kits from R&DSystems (Minneapolis, MN), BioScientific (Austin, TX), and BioLegend(San Diego, CA), respectively. Measurements were performed according to
FIGURE 1. Impaired capacity of autoreactive CD4+ MOG35–55-reactive
T cells to transfer EAE, DTH, and Th1/Th17 immunity in ob/ob mice. A,
Mean clinical score of adoptively induced EAE in B6-WT, ob/ob-PBS–
treated, and ob/ob-leptin–treated mice. Only B6-WT and ob/ob-leptin–
treated mice groups develop clinical signs of disease, with a similar disease
score. Ob/ob mice treated with PBS are resistant to EAE induction when
adoptively transferred with 5 3 106 encephalitogenic MOG35–55-specific
CD4+ T lymphocytes. Ob/ob-leptin–treated group were injected with
r-leptin starting 3 d before transfer until day 25 (1 mg/g body weight). Data
are representative of three independent experiments with similar results (n =
5 mice/group). B, Kinetics of DTH responses at day 7 posttransfer in B6-
WT, ob/ob-PBS–treated, and ob/ob mice treated with leptin. Seven days
after adoptive transfer of MOG35–55-specific CD4+ T cells, mice were
challenged with 50 mg MOG35–55 peptide into the footpad, and DTH re-
sponse was measured in time kinetics (0–72 h); data are representative of
three independent experiments with similar results (n = 5 mice/group;
means 6 SD of footpad-swelling responses); **p , 0.05 of ob/ob PBS
versus B6-WT and ob/ob leptin treated. C, DTH response over time at 1, 7,
and 14 d after adoptive transfer of MOG35–55-specific CD4+ T cells. Plotted
data refer to 24-h DTH footpad swelling. Data are representative of two
independent experiments with similar results, showing means 6 SD of
footpad-swelling responses; *p, 0.01 and **p, 0.05 of ob/ob PBS versus
B6-WT and ob/ob leptin treated. D, Dose-dependent MOG35–55 specific
in vitro proliferation of splenocytes pulsed with MOG35–55 from the three
groups of mice previously transferred with MOG35–55-specific CD4+
T cells. Data are representative of two independent experiments showing
means 6 SD of [3H]thymidine incorporation; *p , 0.01 of ob/ob PBS
versus B6-WT and ob/ob leptin treated. E, In vitro cytokine release in
supernatants from in vitro proliferation of splenocytes to MOG35–55 from
the three groups of mice previously transferred with B6-WT MOG35–55-
specific CD4+ T cells; *p , 0.01 and **p , 0.05 of ob/ob PBS versus B6-
WT and ob/ob leptin treated.
The Journal of Immunology 7475
by guest on June 13, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

the manufacturer’s instructions. Soluble IL-1a, IL-2, IL-4, IL-5, IL-10, IL-17A, IFN-g GM-CSF, and TNF-a mouse cytokines were measured usingbeads-based Analyte Detection Assay (Th1/Th2 FlowCytomix Kit; BenderMedSystems, Vienna, Austria), according to the manufacturer’s instruc-tions.
Flow cytometry, cell sorting, and biochemical analyses
Untouched CD4+ T cells (isolated by negative selection using the MiltenyiBiotec T Cell Isolation kit and an AutoMACS cell separator) from donormice were stained with the fluorescent dye CFSE from Molecular Probes(Eugene, OR) used at 1 mg/ml. A total of 5 3 106 CD4+CFSE+-labeledT cells were injected into the tail vein of B6-WT, ob/ob, and leptin-treatedob/ob mice. For flow cytometric analyses of CFSE+CD4+ T cells 7 and14 d after transfer, spleens of three mice groups were harvested, and 1 3106 cells were analyzed using a FACSCalibur (BD Biosciences, San Jose,CA) and CellQuest software (BD Biosciences). For biochemical analyses,0.5–1 3 106 CD4+CFSE+ cells were obtained from the spleen of eachgroup of WT, ob/ob, and ob/ob-leptin–replaced mice using High-SpeedCells Sorting (MoFlo; DakoCytomation, Glostrup, Denmark); cells were.99% pure. For Western blotting, sorted cells were lysed as describedpreviously (33). The Abs used were the following: anti-p27Kip-1, anti-phospho (P)-AKT, anti-AKT and anti–Bcl-2, and anti-S6 ribosomal pro-tein (S6) and anti–P-S6 (all from Cell Signaling Technology, Beverly,MA). All filters were quantified by densitometric analysis of the bandsusing the program ScionImage 1.63 for Macintosh (Scion, Frederick, MD).FACS analyses for intracellular signaling on ex vivo CFSE+ cells fixed andpermeabilized were performed by intracellular staining of P-S6 usingPE-conjugated P-S6 Ab (Cell Signaling Technology).
Statistical analyses
Analyses were performed using Mann-Whitney U test (for unpaired twogroup analyses) and Kruskal-Wallis ANOVA test (for three or more groupanalyses). Results are expressed as mean 6 SD; p , 0.05 was consideredstatistically significant.
ResultsAutoreactive CD4+ T cells do not transfer EAE in ob/ob mice
We adoptively transferred 5 3 106 encephalitogenic MOG35–55-specific CD4+ T lymphocytes into leptin-deficient ob/ob micetreated or not with mouse r-leptin. As shown in Fig. 1A, MOG35–55-specific CD4+ T cells did not transfer EAE when injected into ob/ob recipients. In contrast, T cells did induce EAE when transferredinto B6-WT and ob/ob mice treated with r-leptin with a similarfrequency of disease and clinical score in the groups.Transferred MOG35–55-specific CD4+ T cells were studied
in vivo and in vitro. Seven days after transfer of MOG35–55-reactive CD4+ T cells, mice were challenged with 50 mg MOG35–55
peptide in the footpad, and DTH response was measured. Ob/obmice had delayed DTH (12–72 h) as compared with B6-WT andr-leptin–treated ob/ob mice (Fig. 1B). The monitoring of DTHafter adoptive transfer (1, 7, and 14 d) indicated that the DTH waslost when MOG35–55-specific CD4+ T cells were transferred intoob/ob mice (Fig. 1C).To also definewhether leptin deficiency affected the proliferation
of transferred MOG35–55-specific CD4+ T cells, we performeddose-dependent in vitro stimulation with MOG35–55 in autologousserum (to preserve leptin deficiency). Reduced cell proliferation toMOG35–55 peptide of B6-WT cells from ob/ob mice was restoredby r-leptin treatment (Fig. 1D). Also, secretion of proinflammatorycytokines IL-1a, IL-2, IL-6, IL-17A, GM-CSF, IFN-g, and TNF-afrom B6-WT MOG35–55-specific CD4+ T cells from ob/ob micewas reduced and restored by r-leptin administration (Fig. 1E).Leptin deficiency did not alter the production of IL-4 and IL-10from B6-WT T cells in response to MOG35–55, and IL-5 was re-duced similarly to Th1 cytokines. Finally, in adoptively transferredCD4+ B6-WT T cells stimulated in vitro with MOG35–55, the levelsof IL-15 and IL-21 were significantly reduced (Fig. 1E), and IL-7was undetectable (data not shown). To exclude that the observed
FIGURE 2. Leptin deficiency impairs survival of autoreactive CD4+
T cells in vivo. A, Representative flow cytometry of CFSE+ CD4+
T cells recovered from B6-WT (upper black panel), ob/ob-PBS (middle
blue panel), and ob/ob-leptin (lower red panel) treated mice 7 d after
adoptive transfer of in vitro-activated MOG35–55-specific CFSE-labeled
CD4+ T cells from B6-WT (see schematic diagram of experimental
procedure). One experiment representative of three; *p , 0.05, ob/ob
PBS versus B6-WT and ob/ob leptin treated. B, Histograms repre-
sent the percentage (upper panel) and absolute number (lower panel) of
CFSE+ T cells detected at 7 and 14 d postadoptive transfer in spleen and
B6-WT (black bars), ob/ob PBS-treated (blue bars), and ob/ob-rleptin–
treated mice (red bars). Data are representative of three independent
experiments. Data are shown as mean 6 SD; *p , 0.05 and **p , 0.01,
respectively. C, Representative flow cytometry of annexin V staining in
CFSE+ CD4+ T cells recovered from B6-WT (upper panel), ob/ob-PBS
(middle panel), and ob/ob-leptin (lower panel)-treated mice 7 d after
adoptive transfer of in vitro-activated MOG35–55-specific CFSE-labeled
CD4+ T cells from B6-WT. Data are representative of three independent
experiments and indicate the percentage of Annexin V+ cells. Data are
shown as mean 6 SD; *p , 0.01. D, Histograms show averaged value
of annexin V staining in CFSE+CD4+ T cells from three independent
experiments. Data are shown as mean 6 SD; *p , 0.01 and **p , 0.05,
respectively.
7476 LEPTIN MODULATES CD4+ T CELL SURVIVAL VIA THE mTOR PATHWAY
by guest on June 13, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
differences were due to quantitative effects of cells recovery, wenormalized the in vitro experiments for the same number of CFSE+
CD4+ T cells in each well and confirmed that autoreactive CD4+
T cells exposed to a leptin-free microenvironment secrete less Th1/Th17 inflammatory and prosurvival cytokines.
Leptin controls the survival of autoreactive CD4+ T cells
To define whether leptin deficiency influences in vivo survival andproliferation of autoreactive MOG35–55-reactive T cells, CD4+
T cells purified from MOG35–55-immunized B6-WT mice 7 d afterimmunization were activated in vitro for 3 d with MOG35–55
peptide, CFSE labeled, and adoptively transferred into B6-WT,ob/ob, and r-leptin–treated ob/ob recipient mice (Fig. 2). Cellswere monitored longitudinally as CFSE+ MOG35–55-reactiveCD4+ T cells. Seven days posttransfer, the percentage of CFSE+
CD4+ T cells in ob/ob mice was dramatically reduced whencompared with the other groups (Fig. 2A, 2B). At later time points(day 14 posttransfer), CD4+ T cells were still reduced in numbersin ob/ob mice as compared with the other two groups of mice (Fig.2B). To confirm the findings using clonal T cell populations, weused CD4+ T cells from AND-TCR Tg mice that carry a TCRspecific for PCC88–104. We adoptively transferred into B6-WT, ob/ob treated or not with r-leptin, the CFSE+ PCC88–104-specificCD4+ T cells after 3 d in vitro activation with PCC88–104 peptideloaded onto MHC class II-(Ek)-expressing DCEK transfectants(31, 32). It was confirmed in this system that leptin deficiencyassociated with a reduced recovery of CFSE+ PCC88–104-specificCD4+ transgenic T cells in ob/ob mice (Supplemental Fig. 1A, 1B)at 7 and 14 d after transfer (Supplemental Fig. 1B). To addresswhether these findings associated with an increased apoptosis ofthe transferred cells, we evaluated the expression of annexin V inCFSE+ MOG35–55-specific CD4+ T cells recovered from thespleens of B6-WT and ob/ob treated or not with r-leptin. AnnexinV levels were significantly increased in B6-WT CD4+ T cells from
ob/ob mice (Fig. 2C, 2D), and a similar extent of apoptosis wasobserved from T cells from transgenic mice (data not shown).
Leptin deficiency associates with a decrease of Bcl-2expression, P-ERK1/2, and reduced degradation of the cellcycle inhibitor p27kip1 in CD4+ T cells
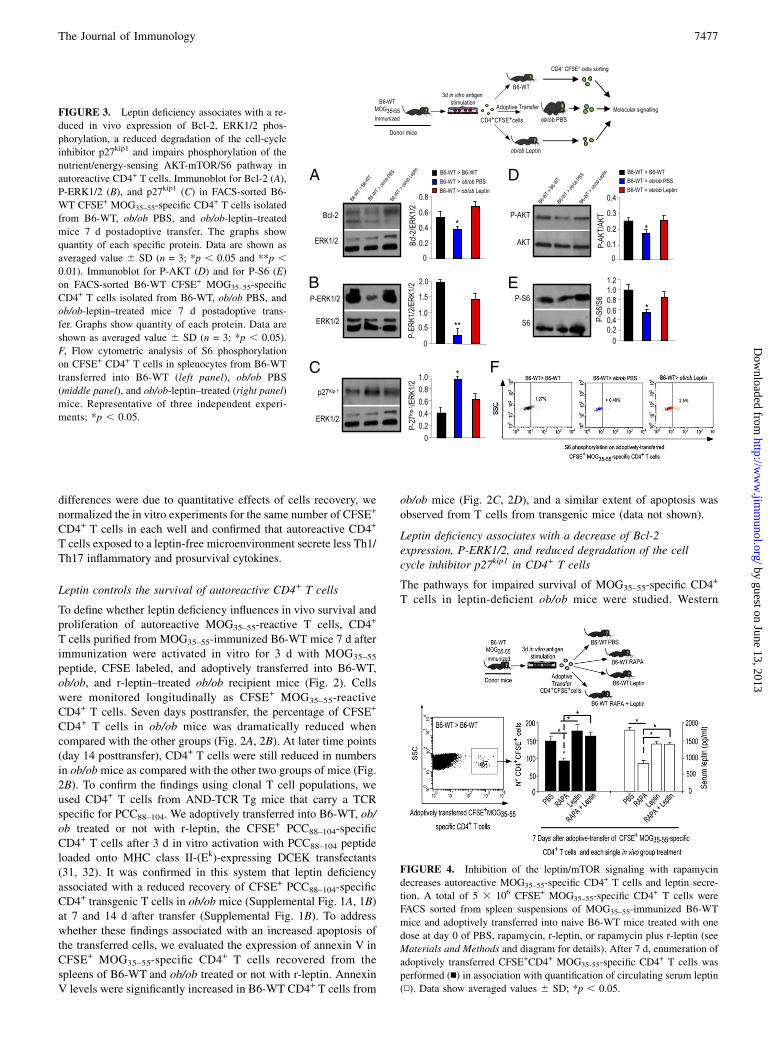
The pathways for impaired survival of MOG35–55-specific CD4+
T cells in leptin-deficient ob/ob mice were studied. Western
FIGURE 3. Leptin deficiency associates with a re-
duced in vivo expression of Bcl-2, ERK1/2 phos-
phorylation, a reduced degradation of the cell-cycle
inhibitor p27kip1 and impairs phosphorylation of the
nutrient/energy-sensing AKT-mTOR/S6 pathway in
autoreactive CD4+ T cells. Immunoblot for Bcl-2 (A),
P-ERK1/2 (B), and p27kip1 (C) in FACS-sorted B6-
WT CFSE+ MOG35–55-specific CD4+ T cells isolated
from B6-WT, ob/ob PBS, and ob/ob-leptin–treated
mice 7 d postadoptive transfer. The graphs show
quantity of each specific protein. Data are shown as
averaged value 6 SD (n = 3; *p , 0.05 and **p ,0.01). Immunoblot for P-AKT (D) and for P-S6 (E)
on FACS-sorted B6-WT CFSE+ MOG35–55-specific
CD4+ T cells isolated from B6-WT, ob/ob PBS, and
ob/ob-leptin–treated mice 7 d postadoptive trans-
fer. Graphs show quantity of each protein. Data are
shown as averaged value 6 SD (n = 3; *p , 0.05).
F, Flow cytometric analysis of S6 phosphorylation
on CFSE+ CD4+ T cells in splenocytes from B6-WT
transferred into B6-WT (left panel), ob/ob PBS
(middle panel), and ob/ob-leptin–treated (right panel)
mice. Representative of three independent experi-
ments; *p , 0.05.
FIGURE 4. Inhibition of the leptin/mTOR signaling with rapamycin
decreases autoreactive MOG35–55-specific CD4+ T cells and leptin secre-
tion. A total of 5 3 106 CFSE+ MOG35–55-specific CD4+ T cells were
FACS sorted from spleen suspensions of MOG35–55-immunized B6-WT
mice and adoptively transferred into naive B6-WT mice treated with one
dose at day 0 of PBS, rapamycin, r-leptin, or rapamycin plus r-leptin (see
Materials and Methods and diagram for details). After 7 d, enumeration of
adoptively transferred CFSE+CD4+ MOG35-55-specific CD4+ T cells was
performed (n) in association with quantification of circulating serum leptin
(N). Data show averaged values 6 SD; *p , 0.05.
The Journal of Immunology 7477
by guest on June 13, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

blot analysis showed that FACS-sorted B6-WT MOG35–55-specificCD4+ T cells, isolated from ob/ob mice, had lower levels of Bcl-2and P-ERK1/2; these data were associated with a reduced degra-dation of the cell cycle inhibitor p27Kip1 as compared with B6-WTCD4+ T cells from B6-WTand leptin-treated ob/ob mice (Fig. 3A–C). These findings suggest an increased apoptosis, reduced pro-liferation, and cell cycle arrest in the absence of leptin.
Leptin modulates the survival of autoreactive CD4+ T cellsthrough the nutrient/energy-sensing AKT-mTOR pathway
We tested whether leptin affects the expression of the protein ki-nase B (AKT) and its downstream energy-sensing mTOR pathway(34–36). We found that FACS-sorted MOG35–55-specific CD4+
T cells from ob/ob mice had lower levels of phosphorylation ofboth AKT and S6, which is downstream of the mTOR pathway(Fig. 3D, 3E). These results were confirmed by FACS for P-S6 inB6-WT CFSE+ MOG35–55-specific CD4+ T cells (Fig. 3F).Because P-S6 appeared reduced by leptin deficiency in B6-WT
autoreactive T cells, we performed in vivo experiments usingrapamycin, an mTOR inhibitor. Interestingly, acute/short-term ra-pamycin treatment in vivo resembled the effects of leptin defi-ciency in terms of recovery of MOG35–55-specific CD4+ T in B6-WT mice. B6-WT CFSE+ MOG35–55 pathogenic CD4+ T cellswere analyzed 7 d after adoptive transfer into B6-WT mice treatedwith a single dose of PBS, rapamycin, r-leptin, or r-leptin plusrapamycin (Fig. 4, n) on day 0. Rapamycin treatment significantlyreduced the number of B6-WT CFSE+ MOG35–55 T cells; theseeffects were prevented by coinjection of r-leptin with rapamycin(Fig. 4, n). In this last condition, r-leptin could partially sustainmTOR activation, as demonstrated by P-S6 levels in CD4+ T cellsfrom rapamycin plus r-leptin–treated mice. These results also cor-related with the capacity of rapamycin to reduce serum leptinlevels in B6-WT mice (Fig. 4, N), and these effects were notsecondary to weight loss (data not shown).The serum leptin levels were unchanged in leptin-treated mice as
compared with controls, likely because they were analyzed after7 d of a single dose of r-leptin whose half-life is ∼180 min (37).
DiscussionSeveral reports have suggested that leptin can affect survival ofimmune cells in vitro (38, 39). To our knowledge, this study is thefirst to report that leptin has a key role in the survival and pro-liferation of autoreactive CD4+ T cells in vivo. Leptin-deficientob/ob mice were protected from adoptively transferred EAE, and theprotection was associated with a progressive decline in the survivalof adoptively transferred autoreactive MOG35–55-specific CD4+
T cells and reduced secretion of Th1/Th17 proinflammatory cyto-kines. Also, CD4+ autoreactive T cells had a significant downregu-lation of the survival protein Bcl-2, reduction in P-ERK1/2, andcell cycle arrest associated with reduced degradation of the cellcycle inhibitor p27kip1. Importantly, a significant impairment oc-curred at the level of the nutrient-energy–sensing AKT-mTOR/S6signaling pathway. These phenomena were all reversed in vivoby r-leptin administration, suggesting that leptin can activate themTORpathway in autoreactive CD4+ T cells (Supplemental Fig. 2).Studies published by Piccio et al. (3) and by our group (40) have
shown that nutritional deprivation or CR can reduce the magnitudeand disease score of EAE. The so-called “frugal phenotype,” inwhich the survival of chronically food-restricted mice is higherthan that of ad libitum-fed mice, fits well into our current findingsof reduced T cell autoreactivity in the absence of leptin. Also,chronic rapamycin treatment is known to increase significantly ro-dent survival (41). Although the precise mechanisms for the ef-fects of rapamycin need to be elucidated, through mTOR inhibi-
tion, rapamycin can reduce the absorption of amino acids and glu-cose and dampen proinflammatory cytokines (including leptin)(42). This is interesting because rapamycin can improve diseasecurse and progression in EAE and type 1 diabetes by increasingregulatory T cell responses and dampening Th1/Th17 responses(43–45).Leptin represents a link with the environment by informing
about available energy reserves as stored fat (5). Microenviron-mental requirements for immune cells, including autoreactiveCD4+ T cells, are not well known, although many cytokines, che-mokines, and other mediators have been described as capable ofsignificantly affecting survival and homeostasis of the T cells. Inthis paper, we describe a mechanism of leptin-based modulation ofautoreactive CD4+ T cell survival through the nutrient energy-sensing mTOR pathway and the expression of Bcl-2. The re-duced secretion of cytokines such as IL-6, IL-15, IL-21, andGM-CSF also appeared important. B6-WT myelin-specific CD4+
T cells in the leptin-free microenvironment showed a reducedsecretion of IL-6, IL-15, IL-21, and GM-CSF when restimulatedin vitro with MOG35–55. Other cytokines such as IL-1a, IL-2, IL-17A, IFN-g, and TNF-a were also downmodulated in autoreactiveMOG35–55-specific CD4+ B6-WT cells when transferred intoleptin-deficient ob/ob mice. Incidentally, the fact that IL-1a andIL-17A were reduced by leptin deficiency is in line with a recentreport of a key role for mTOR in the maintenance of Th17 cellproliferation and cytokine secretion by IL-1 in EAE (46).In summary, this study shows a previously unappreciated role of
leptin in the promotion of the survival of CD4+ autoreactive T cells.The implications of these findings in relation to nutritional dep-rivation or CR can suggest targeting of leptin-based pathways ofintervention for the modulation of autoreactive T cell activity.
AcknowledgmentsWe thank Salvatore De Simone and Francesco D’Agnello for technical help
in cell sorting at the MoFlo Core Facility. This work is dedicated to the
memory of Eugenia Papa and Serafino Zappacosta.
DisclosuresThe authors have no financial conflicts of interest.
References1. Duan, W., Z. Guo, H. Jiang, M. Ware, and M. P. Mattson. 2003. Reversal of
behavioral and metabolic abnormalities, and insulin resistance syndrome, bydietary restriction in mice deficient in brain-derived neurotrophic factor. Endo-crinology 144: 2446–2453.
2. Maswood, N., J. Young, E. Tilmont, Z. Zhang, D. M. Gash, G. A. Gerhardt,R. Grondin, G. S. Roth, J. Mattison, M. A. Lane, et al. 2004. Caloric restrictionincreases neurotrophic factor levels and attenuates neurochemical and behavioraldeficits in a primate model of Parkinson’s disease. Proc. Natl. Acad. Sci. USA101: 18171–18176.
3. Piccio, L., J. L. Stark, and A. H. Cross. 2008. Chronic calorie restriction at-tenuates experimental autoimmune encephalomyelitis. J. Leukoc. Biol. 84: 940–948.
4. Zhang, Y., R. Proenca, M. Maffei, M. Barone, L. Leopold, and J. M. Friedman.1994. Positional cloning of the mouse obese gene and its human homologue.Nature 372: 425–432.
5. Friedman, J. M., and J. L. Halaas. 1998. Leptin and the regulation of bodyweight in mammals. Nature 395: 763–770.
6. Matarese, G., A. La Cava, V. Sanna, G. M. Lord, R. I. Lechler, S. Fontana, andS. Zappacosta. 2002. Balancing susceptibility to infection and autoimmunity:a role for leptin? Trends Immunol. 23: 182–187.
7. Matarese, G., S. Moschos, and C. S. Mantzoros. 2005. Leptin in immunology. J.Immunol. 174: 3137–3142.
8. Lord, G. M., G. Matarese, J. K. Howard, R. J. Baker, S. R. Bloom, andR. I. Lechler. 1998. Leptin modulates the T-cell immune response and reversesstarvation-induced immunosuppression. Nature 394: 897–901.
9. Howard, J. K., G. M. Lord, G. Matarese, S. Vendetti, M. A. Ghatei, M. A. Ritter,R. I. Lechler, and S. R. Bloom. 1999. Leptin protects mice from starvation-induced lymphoid atrophy and increases thymic cellularity in ob/ob mice. J.Clin. Invest. 104: 1051–1059.
7478 LEPTIN MODULATES CD4+ T CELL SURVIVAL VIA THE mTOR PATHWAY
by guest on June 13, 2013http://w
ww
.jimm
unol.org/D
ownloaded from

10. Gabay, C., M. Dreyer, N. Pellegrinelli, R. Chicheportiche, and C. A. Meier.2001. Leptin directly induces the secretion of interleukin 1 receptor antagonist inhuman monocytes. J. Clin. Endocrinol. Metab. 86: 783–791.
11. Jaworek, J., J. Bonior, P. Pierzchalski, R. Tomaszewska, J. Stachura, R. Sendur,A. Leja, B. Jachimczak, P. C. Konturek, W. Bielanski, et al. 2002. Leptin protectsthe pancreas from damage induced by caerulein overstimulation by modulatingcytokine production. Pancreatology 2: 89–99.
12. Chatzantoni, K., P. Papathanassopoulos, E. Gourzoulidou, and A. Mouzaki.2004. Leptin and its soluble receptor in plasma of patients suffering fromremitting-relapsing multiple sclerosis (MS) in vitro effects of leptin on type-1and type-2 cytokine secretion by peripheral blood mononuclear cells, T-cells andmonocytes of MS patients. J. Autoimmun. 23: 169–177.
13. Xiao, E., L. Xia-Zhang, N. R. Vulliemoz, M. Ferin, and S. L. Wardlaw. 2003.Leptin modulates inflammatory cytokine and neuroendocrine responses to en-dotoxin in the primate. Endocrinology 144: 4350–4353.
14. Montgomery, L. B., and R. M. Loria. 1986. Humoral immune response in he-reditary and overt diabetes mellitus. J. Med. Virol. 19: 255–268.
15. Fantuzzi, G., and R. Faggioni. 2000. Leptin in the regulation of immunity, in-flammation, and hematopoiesis. J. Leukoc. Biol. 68: 437–446.
16. La Cava, A., and G. Matarese. 2004. The weight of leptin in immunity. Nat. Rev.Immunol. 4: 371–379.
17. Li, L., and V. A. Boussiotis. 2006. Physiologic regulation of central and pe-ripheral T cell tolerance: lessons for therapeutic applications. J. Mol. Med. 84:887–899.
18. Murray, R., T. Suda, N. Wrighton, F. Lee, and A. Zlotnik. 1989. IL-7 is a growthand maintenance factor for mature and immature thymocyte subsets. Int.Immunol. 1: 526–531.
19. Asao, H., C. Okuyama, S. Kumaki, N. Ishii, S. Tsuchiya, D. Foster, andK. Sugamura. 2001. Cutting edge: the common g-chain is an indispensable subunitof the IL-21 receptor complex. J. Immunol. 167: 1–5.
20. King, C., A. Ilic, K. Koelsch, and N. Sarvetnick. 2004. Homeostatic expansion ofT cells during immune insufficiency generates autoimmunity. Cell 117: 265–277.
21. Khoruts, A., and J. M. Fraser. 2005. A causal link between lymphopenia andautoimmunity. Immunol. Lett. 98: 23–31.
22. Marleau, A. M., and N. Sarvetnick. 2005. T cell homeostasis in tolerance andimmunity. J. Leukoc. Biol. 78: 575–584.
23. Teague, T. K., P. Marrack, J. W. Kappler, and A. T. Vella. 1997. IL-6 rescuesresting mouse T cells from apoptosis. J. Immunol. 158: 5791–5796.
24. Schulze-Koops, H. 2004. Lymphopenia and autoimmune diseases. Arthritis Res.Ther. 6: 178–180.
25. Munger, K. L., T. Chitnis, and A. Ascherio. 2009. Body size and risk of MS intwo cohorts of US women. Neurology 73: 1543–1550.
26. Bruining, G. J. 2000. Association between infant growth before onset of juveniletype-1 diabetes and autoantibodies to IA-2: Netherlands Kolibrie study group ofchildhood diabetes. Lancet 356: 655–656.
27. Feuerer, M., L. Herrero, D. Cipolletta, A. Naaz, J. Wong, A. Nayer, J. Lee,A. B. Goldfine, C. Benoist, S. Shoelson, and D. Mathis. 2009. Lean, but notobese, fat is enriched for a unique population of regulatory T cells that affectmetabolic parameters. Nat. Med. 15: 930–939.
28. Winer, S., Y. Chan, G. Paltser, D. Truong, H. Tsui, J. Bahrami, R. Dorfman,Y. Wang, J. Zielenski, F. Mastronardi, et al. 2009. Normalization of obesity-associated insulin resistance through immunotherapy. Nat. Med. 15: 921–929.
29. Lumeng, C. N., I. Maillard, and A. R. Saltiel. 2009. T-ing up inflammation in fat.Nat. Med. 15: 846–847.
30. Matarese, G., A. Di Giacomo, V. Sanna, G. M. Lord, J. K. Howard, A. Di Tuoro,S. R. Bloom, R. I. Lechler, S. Zappacosta, and S. Fontana. 2001. Requirementfor leptin in the induction and progression of autoimmune encephalomyelitis. J.Immunol. 166: 5909–5916.
31. Racioppi, L., G. Matarese, U. D’Oro, M. De Pascale, A. M. Masci, S. Fontana,and S. Zappacosta. 1996. The role of CD4-Lck in T-cell receptor antagonism:evidence for negative signaling. Proc. Natl. Acad. Sci. USA 93: 10360–10365.
32. Zinkernagel, R. M. 1976. H-2 restriction of virus-specific T-cell–mediated ef-fector functions in vivo. II. Adoptive transfer of delayed-type hypersensitivity tomurine lymphocytic choriomeningits virus is restricted by the K and D region ofH-2. J. Exp. Med. 144: 776–787.
33. De Rosa, V., C. Procaccini, G. Calı, G. Pirozzi, S. Fontana, S. Zappacosta, A. LaCava, and G. Matarese. 2007. A key role of leptin in the control of regulatoryT cell proliferation. Immunity 26: 241–255.
34. Delgoffe, G. M., T. P. Kole, Y. Zheng, P. E. Zarek, K. L. Matthews, B. Xiao,P. F. Worley, S. C. Kozma, and J. D. Powell. 2009. The mTOR kinase differ-entially regulates effector and regulatory T cell lineage commitment. Immunity30: 832–844.
35. Araki, K., A. P. Turner, V. O. Shaffer, S. Gangappa, S. A. Keller, M. F. Bachmann,C. P. Larsen, and R. Ahmed. 2009. mTOR regulates memory CD8 T cell-differ-entation. Nature 46: 108–112.
36. Wiederrecht, G. J., C. J. Sabers, G. J. Brunn, M. M. Martin, F. J. Dumont, andR. T. Abraham. 1995. Mechanism of action of rapamycin: new insights into theregulation of G1-phase progression in eukaryotic cells. Prog. Cell Cycle Res. 1:53–71.
37. Ahima, R. S., D. Prabakaran, C. Mantzoros, D. Qu, B. Lowell, E. Maratos-Flier,and J. S. Flier. 1996. Role of leptin in the neuroendocrine response to fasting.Nature 382: 250–252.
38. Fernandez-Riejos, P., R. Goberna, and V. Sanchez-Margalet. 2008. Leptinpromotes cell survival and activates Jurkat T lymphocytes by stimulation ofmitogen-activated protein kinase. Clin. Exp. Immunol. 151: 505–518.
39. Papathanassoglou, E., K. El-Haschimi, X. C. Li, G. Matarese, T. Strom, andC. Mantzoros. 2006. Leptin receptor expression and signaling in lymphocytes:kinetics during lymphocyte activation, role in lymphocyte survival, and responseto high fat diet in mice. J. Immunol. 176: 7745–7752.
40. Sanna, V., A. Di Giacomo, A. La Cava, R. I. Lechler, S. Fontana, S. Zappacosta,and G. Matarese. 2003. Leptin surge precedes onset of autoimmune encepha-lomyelitis and correlates with development of pathogenic T cell responses. J.Clin. Invest. 111: 241–250.
41. Harrison, D. E., R. Strong, Z. D. Sharp, J. F. Nelson, C. M. Astle, K. Flurkey,N. L. Nadon, J. E. Wilkinson, K. Frenkel, C. S. Carter, et al. 2009. Rapamycinfed late in life extends lifespan in genetically heterogeneous mice. Nature 460:392–395.
42. Harle, P., and R. H. Straub. 2006. Leptin is a link between adipose tissue andinflammation. Ann. N. Y. Acad. Sci. 1069: 454–462.
43. Donia, M., K. Mangano, A. Amoroso, M. C. Mazzarino, R. Imbesi,P. Castrogiovanni, M. Coco, P. Meroni, and F. Nicoletti. 2009. Treatment withrapamycin ameliorates clinical and histological signs of protracted relapsingexperimental allergic encephalomyelitis in Dark Agouti rats and induces ex-pansion of peripheral CD4+CD25+Foxp3+ regulatory T cells. J. Autoimmun. 33:135–140.
44. Esposito, M., F. Ruffini, M. Bellone, N. Gagliani, M. Battaglia, G. Martino, andR. Furlan. 2010. Rapamycin inhibits relapsing experimental autoimmune en-cephalomyelitis by both effector and regulatory T cells modulation. J. Neuro-immunol. 220: 52–63.
45. Jiang, G. X., Y. F. Cui, X. Y. Zhong, S. Tai, W. Liu, Z. D. Wang, and Y. G. Shi.2009. Use of a cocktail regimen consisting of soluble galectin-1, rapamycin andhistone deacetylase inhibitor may effectively prevent type 1 diabetes. Arch. Med.Res. 40: 424–426.
46. Gulen, M. F., Z. Kang, K. Bulek, W. Youzhong, T. W. Kim, Y. Chen,C. Z. Altuntas, K. Sass Bak-Jensen, M. J. McGeachy, J. S. Do, et al. 2010.The receptor SIGIRR suppresses Th17 cell proliferation via inhibition ofthe interleukin-1 receptor pathway and mTOR kinase activation. Immunity 32:54–66.
The Journal of Immunology 7479
by guest on June 13, 2013http://w
ww
.jimm
unol.org/D
ownloaded from
Copyright © 2022 FDOKUMEN




















