
Kit and PDGFR-α activities are necessary for Notch4/Int3-induced tumorigenesis
11
ORIGINAL ARTICLE Kit and PDGFR-a activities are necessary for Notch4/Int3-induced tumorigenesis A Raafat 1 , A Zoltan-Jones 1 , L Strizzi, S Bargo, K Kimura, D Salomon and R Callahan Oncogenetics Section, Mammary Biology and Tumorigenesis Laboratory, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA Transgenic mice overexpressing Notch4 intracellular domain (Int3) under the control of the whey acidic protein (WAP) or mouse mammary tumor virus-long terminal repeat promoters, develop mammary tumors. Microarray analysis of these tumors revealed high levels of c-Kit expression. Gleevec is a tyrosine kinase inhibitor that targets c-Kit, platelet-derived growth factor receptors (PDGFRs) and c-Abl. This led us to speculate that tyrosine kinase receptor activity might be a driving force in the development of Int3 mammary tumors. WAP-Int3 tumor-bearing mice were treated with continuous release of Gleevec using subcutaneously implanted Alzet pumps. Phoshorylation of c-Kit, PDGFRs and c-Abl is inhibited in Int3 transgenic mammary tumors by Gleevec. Inhibi- tion of these enzymes is associated with a decrease in cell proliferation and angiogenesis, and an induction of apoptosis. To examine the signaling mechanisms under- lying Notch4/Int3 tumorigenesis, we employed small interfering RNA (siRNA) to knock down c-Kit, PDGFRs and c-Abl alone or in combination and observed the effects on soft agar growth of HC11 cells overexpressing Int3. Only siRNA constructs for c-Kit and/or PDGFR-a were able to inhibit HC11-Int3 colony formation in soft agar. Our data demonstrate an inhibitory effect of Gleevec on Int3-induced transformation of HC11 cells and mammary tumors and indicate an oncogenic role for c-Kit and PDGFR-a tyrosine kinases in the context of Int3 signaling. Oncogene (2007) 26, 662–672. doi:10.1038/sj.onc.1209823; published online 31 July 2006 Keywords: Kit; PDGFR-a; Notch4/Int3; Gleevec; phos- phorylation; siRNA Introduction The Notch signaling pathway is involved in cell fate decisions of tissues and organs in several different organisms (Callahan and Raafat, 2001). The Notch gene encodes a transmembrane receptor protein. In mammals, there are four members of the Notch gene family, some of which are targets for mutations that contribute to tumor development (Allenspach et al., 2002). Developmental transitions and malignant trans- formations share many of the same signaling pathways. An emerging view of malignant transformation is that of a developmental program that lacks the precise control and regulation seen during development. Recently, growing evidence supports aberrant Notch signaling in malignant transformation. Notch3 overexpression has been implicated in cases of human lung cancer (Dang et al., 2000) and activation of Notch3 in lung epithelium leads to the inhibition of epithelial differentiation (Dang et al., 2003). In a subset of human T-cell acute lymphoblastic leukemias, there is a disruption in the Notch1 gene locus that leads to activated Notch1 signaling (Pear and Aster, 2004; Chiaramonte et al., 2005). An early indication that Notch4 signaling is involved in mammary gland development and tumor- igenesis stems from the identification of the Notch4 gene as a common insertion site for the mouse mammary tumor virus (MMTV) in a feral strain of mice (Gallahan et al., 1987). MMTV integration into the Notch4 gene results in the transcription of a truncated, constitutively active Notch4 gene product (Int3) that corresponds to the intracellular domain (ICD). Expression of Int3 represents a gain-of-function mutation (Robbins et al., 1992; Gallahan and Callahan, 1997). We have shown that expression of Int3 from either the MMTV long terminal repeat (LTR) or the whey acidic protein (WAP) promoter in transgenic mice blocks normal mammary lobular development and the ability of these females to lactate (Jhappan et al., 1992; Smith et al., 1995; Gallahan et al., 1996). In addition, 100% of the females develop mammary adenocarcinomas (Jhappan et al., 1992; Gallahan et al., 1996; Gallahan and Callahan, 1997). Microarray analysis of MMTV LTR and WAP-Int3 mammary tumor RNAs revealed high steady-state levels of the c-Kit tyrosine kinase receptor, as compared to control FVB mammary tissue (our unpublished data). The c-Kit receptor is a transmembrane tyrosine kinase that belongs to the larger receptor tyrosine kinase type III family (RTK III). This family also includes the c-Abl and platelet-derived growth factor receptors (PDGFR-a Received 4 April 2006; revised 22 May 2006; accepted 23 May 2006; published online 31 July 2006 Correspondence: Dr R Callahan, Mammary Biology and Tumorigen- esis Laboratory, National Cancer Institute, National Institutes of Health, 37 Convent Drive, Bldg 37, Rm 1118A, Bethesda, MD 20892, USA. E-mail: [email protected] 1 These authors contributed equally to this work Oncogene (2007) 26, 662–672 & 2007 Nature Publishing Group All rights reserved 0950-9232/07 $30.00 www.nature.com/onc
Transcript of Kit and PDGFR-α activities are necessary for Notch4/Int3-induced tumorigenesis

ORIGINAL ARTICLE
Kit and PDGFR-a activities are necessary for Notch4/Int3-induced
tumorigenesis
A Raafat1, A Zoltan-Jones1, L Strizzi, S Bargo, K Kimura, D Salomon and R Callahan
Oncogenetics Section, Mammary Biology and Tumorigenesis Laboratory, National Cancer Institute, National Institutes of Health,Bethesda, MD, USA
Transgenic mice overexpressing Notch4 intracellulardomain (Int3) under the control of the whey acidic protein(WAP) or mouse mammary tumor virus-long terminalrepeat promoters, develop mammary tumors. Microarrayanalysis of these tumors revealed high levels of c-Kitexpression. Gleevec is a tyrosine kinase inhibitor thattargets c-Kit, platelet-derived growth factor receptors(PDGFRs) and c-Abl. This led us to speculate thattyrosine kinase receptor activity might be a driving forcein the development of Int3 mammary tumors. WAP-Int3tumor-bearing mice were treated with continuous releaseof Gleevec using subcutaneously implanted Alzet pumps.Phoshorylation of c-Kit, PDGFRs and c-Abl is inhibitedin Int3 transgenic mammary tumors by Gleevec. Inhibi-tion of these enzymes is associated with a decrease in cellproliferation and angiogenesis, and an induction ofapoptosis. To examine the signaling mechanisms under-lying Notch4/Int3 tumorigenesis, we employed smallinterfering RNA (siRNA) to knock down c-Kit, PDGFRsand c-Abl alone or in combination and observed the effectson soft agar growth of HC11 cells overexpressing Int3.Only siRNA constructs for c-Kit and/or PDGFR-a wereable to inhibit HC11-Int3 colony formation in soft agar.Our data demonstrate an inhibitory effect of Gleevec onInt3-induced transformation of HC11 cells and mammarytumors and indicate an oncogenic role for c-Kit andPDGFR-a tyrosine kinases in the context of Int3 signaling.Oncogene (2007) 26, 662–672. doi:10.1038/sj.onc.1209823;published online 31 July 2006
Keywords: Kit; PDGFR-a; Notch4/Int3; Gleevec; phos-phorylation; siRNA
Introduction
The Notch signaling pathway is involved in cell fatedecisions of tissues and organs in several different
organisms (Callahan and Raafat, 2001). The Notchgene encodes a transmembrane receptor protein. Inmammals, there are four members of the Notch genefamily, some of which are targets for mutations thatcontribute to tumor development (Allenspach et al.,2002). Developmental transitions and malignant trans-formations share many of the same signaling pathways.An emerging view of malignant transformation is that ofa developmental program that lacks the precise controland regulation seen during development. Recently,growing evidence supports aberrant Notch signaling inmalignant transformation. Notch3 overexpression hasbeen implicated in cases of human lung cancer (Danget al., 2000) and activation of Notch3 in lung epitheliumleads to the inhibition of epithelial differentiation (Danget al., 2003). In a subset of human T-cell acutelymphoblastic leukemias, there is a disruption in theNotch1 gene locus that leads to activated Notch1signaling (Pear and Aster, 2004; Chiaramonte et al.,2005). An early indication that Notch4 signaling isinvolved in mammary gland development and tumor-igenesis stems from the identification of the Notch4 geneas a common insertion site for the mouse mammarytumor virus (MMTV) in a feral strain of mice (Gallahanet al., 1987). MMTV integration into the Notch4 generesults in the transcription of a truncated, constitutivelyactive Notch4 gene product (Int3) that corresponds tothe intracellular domain (ICD). Expression of Int3represents a gain-of-function mutation (Robbins et al.,1992; Gallahan and Callahan, 1997). We have shownthat expression of Int3 from either the MMTV longterminal repeat (LTR) or the whey acidic protein (WAP)promoter in transgenic mice blocks normal mammarylobular development and the ability of these females tolactate (Jhappan et al., 1992; Smith et al., 1995;Gallahan et al., 1996). In addition, 100% of the femalesdevelop mammary adenocarcinomas (Jhappan et al.,1992; Gallahan et al., 1996; Gallahan and Callahan,1997).
Microarray analysis of MMTV LTR and WAP-Int3mammary tumor RNAs revealed high steady-state levelsof the c-Kit tyrosine kinase receptor, as compared tocontrol FVB mammary tissue (our unpublished data).The c-Kit receptor is a transmembrane tyrosine kinasethat belongs to the larger receptor tyrosine kinase typeIII family (RTK III). This family also includes the c-Abland platelet-derived growth factor receptors (PDGFR-a
Received 4 April 2006; revised 22 May 2006; accepted 23 May 2006;published online 31 July 2006
Correspondence: Dr R Callahan, Mammary Biology and Tumorigen-esis Laboratory, National Cancer Institute, National Institutes ofHealth, 37 Convent Drive, Bldg 37, Rm 1118A, Bethesda, MD 20892,USA.E-mail: [email protected] authors contributed equally to this work
Oncogene (2007) 26, 662–672& 2007 Nature Publishing Group All rights reserved 0950-9232/07 $30.00
www.nature.com/onc

and PDGFR-b) (Besmer et al., 1986). The role of theRTK III family in human malignancies is well known.In chronic myeloid leukemia (CML), c-Abl is activatedby a translocation to create a Bcr/Abl fusion protein(Marley and Gordon, 2005). Human gastrointestinalstromal tumors (GIST) harbor activating mutations ofc-Kit (Lasota et al., 1999; Taniguchi et al., 1999; Kimet al., 2000) or, in c-Kit-negative subsets, activatingmutations of PDGFR-a (Heinrich et al., 2003). The aimof the present study was to investigate the significance ofelevated levels of c-Kit in MMTV LTR and WAP-Int3mammary tumors and to determine whether theexpression of c-Kit and/or other RTK III familymembers, in the context of Int3 signaling, contributesto malignant growth.
Results
Expression of Gleevec targets in WAP-Int3 mammarytumorsTo investigate the effects of Int3 expression on altera-tions in gene expression, we performed a gene arraystudy comparing normal mammary tissue from FVB/Nfemales and WAP-Int3 or MMTV LTR-Int3 tumors(data not shown). Of particular interest to us was theincreased steady-state levels of c-Kit RNA in theWAP-Int3 and MMTV LTR-Int3 tumors as compared tomammary tissue from normal virgin or pregnant FVB/N mice (Figure 1a). To determine whether other relatedRTK III including c-Abl, PDGFR-a and PDGFR-bwere also expressed in these tumors, total RNA extractsfrom three independent primary WAP-Int3 mammarytumors were analysed for c-Kit, c-Abl, PDGFR-a andPDGFR-b expression by PCR with reverse transcription(RT–PCR). All three tumors were positive for theseother RTK III RNAs (Figure 1b). Additionally,immunohistochemical analysis demonstrated that allthree primary tumors express the respective proteins(Figure 1c). The c-Kit and PDGFR-a proteins(Figure 1c, panels 5 and 6, respectively) are expressedin most of the tumor epithelium, whereas PDGFR-b(Figure 1c, panel 7) was detected primarily in the stromaof the tumors. Only a minor fraction of the tumorepithelium was stained with the c-Abl antibody(Figure 1c, panel 8).
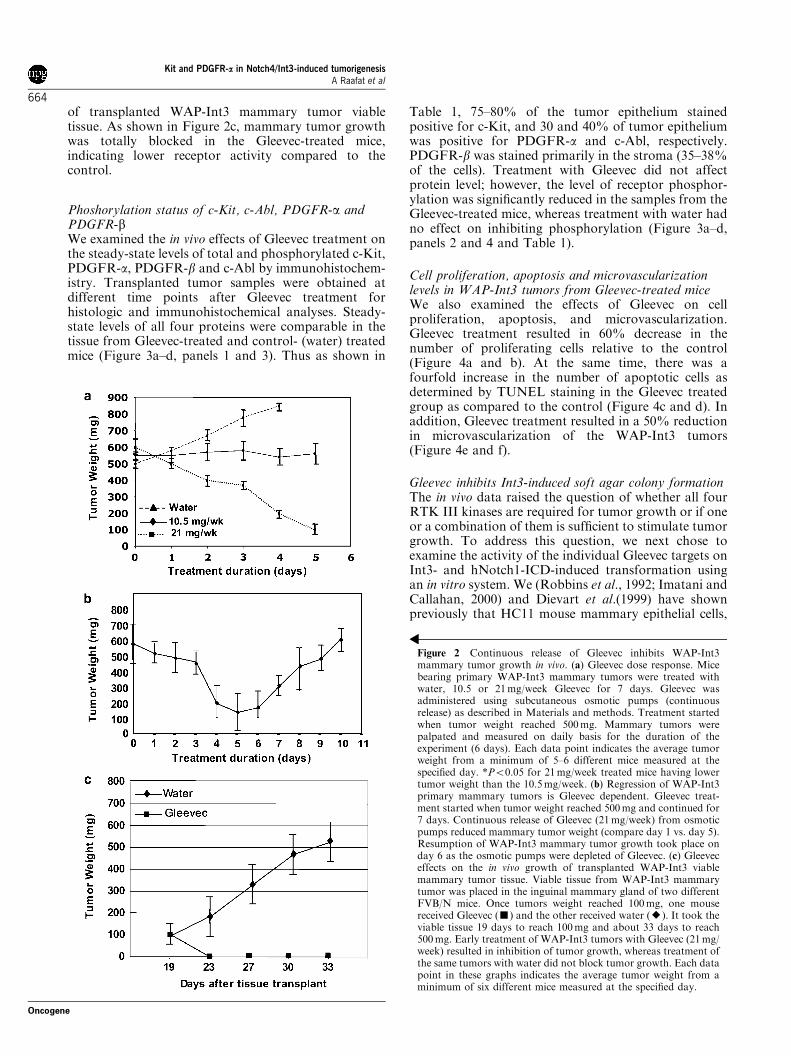
Gleevec dose responseTo ascertain whether members of the RTK III familyare involved in Int3-induced mammary tumorigenesis,we undertook a dose–response study to determine theeffects of Gleevec on WAP-Int3 tumor-bearing mice.We used 21 or 10.5mg/week of Gleevec per mouse. Micetreated with a 10.5mg/week dose showed no significantchange in the tumor weight. In contrast, there was asignificant reduction in mammary tumor weight (TW) inmice treated with 21mg/week (Figure 2a). Therefore, adose of 21mg/week was used in all subsequent experi-ments. Mice treated with water showed a significantcontinuous increase in tumor weight. We had to
euthanize these mice at day 4 of water treatmentbecause of the tumor weight. Histological analysis ofhematoxylin and eosin (H&E)-stained liver and kidneysections from Gleevec-treated mice did not show anyevident morphological alterations such as necrosis orinflammatory changes due to toxicity (data not shown).These results demonstrate that WAP-Int3 mammarytumors are sensitive to Gleevec and further provide astrong rationale that the activation of one or more of theRTK III targets of Gleevec, in the context of Int3signaling, likely contributes to tumor growth. Todetermine the range of Gleevec effects on tumor growth,we treated five WAP-Int3 tumor-bearing mice with21mg/week Gleevec for 1 week and monitored TWdaily. At the end of the treatment period, TW wassignificantly reduced (Figure 2b) in all mice. When thepumps were depleted of Gleevec after day 6, there was aresumption of tumor growth, suggesting that contin-uous inhibition of Gleevec targets is needed toeffectively inhibit tumor growth. In a separate experi-ment, we examined the effect of Gleevec on the growth
Figure 1 Expression of Gleevec-sensitive kinases in mouse Int3transgenic mammary tumors. (a) Evaluation of c-Kit expression inFVB/N and Int3 transgenic mammary glands. Northern blotanalysis of FVB/N tissue of virgin and 15 days pregnant (P15)mammary glands, also mammary tissue of 15 days pregnant WAP-Int3 (Wap-P15), MMTV LTR-Int3 (LTR-V) and WAP-Int3(WAP-V) tumors from virgin mice. The membrane was strippedand re-probed with GAPDH. (b) Qualitative RT–PCR analysis ofGleevec targets in three independent WAP-Int3 primary mammarytumors shows detectable levels of c-Kit, PDGFR-a , PDGFR-band c-Abl. (c) Photomicrographs of immunohistochemical stainingof c-Kit (panels 1and 5), PDGFR-a (panels 2 and 6), PDGFR-b(panels 3 and 7) and c-Abl (panels 4 and 8) in WAP-Int3 mammarygland tumors; negative controls for c-Kit, PDGFR-a, PDGFR-band c-Abl are panels 5, 6, 7 and 8, respectively. Exhibited sectionswere counterstained with hematoxylin. Magnification, � 40.
Kit and PDGFR-a in Notch4/Int3-induced tumorigenesisA Raafat et al
663
Oncogene
of transplanted WAP-Int3 mammary tumor viabletissue. As shown in Figure 2c, mammary tumor growthwas totally blocked in the Gleevec-treated mice,indicating lower receptor activity compared to thecontrol.
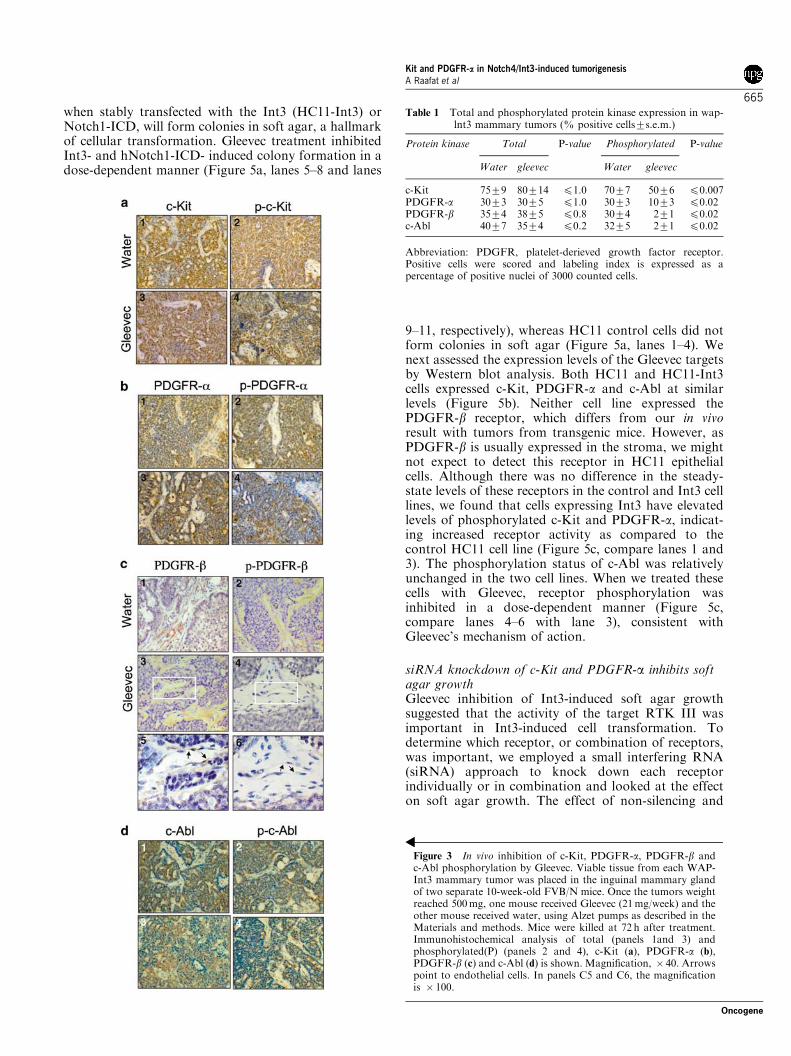
Phoshorylation status of c-Kit, c-Abl, PDGFR-a andPDGFR-bWe examined the in vivo effects of Gleevec treatment onthe steady-state levels of total and phosphorylated c-Kit,PDGFR-a, PDGFR-b and c-Abl by immunohistochem-istry. Transplanted tumor samples were obtained atdifferent time points after Gleevec treatment forhistologic and immunohistochemical analyses. Steady-state levels of all four proteins were comparable in thetissue from Gleevec-treated and control- (water) treatedmice (Figure 3a–d, panels 1 and 3). Thus as shown in
Table 1, 75–80% of the tumor epithelium stainedpositive for c-Kit, and 30 and 40% of tumor epitheliumwas positive for PDGFR-a and c-Abl, respectively.PDGFR-b was stained primarily in the stroma (35–38%of the cells). Treatment with Gleevec did not affectprotein level; however, the level of receptor phosphor-ylation was significantly reduced in the samples from theGleevec-treated mice, whereas treatment with water hadno effect on inhibiting phosphorylation (Figure 3a–d,panels 2 and 4 and Table 1).
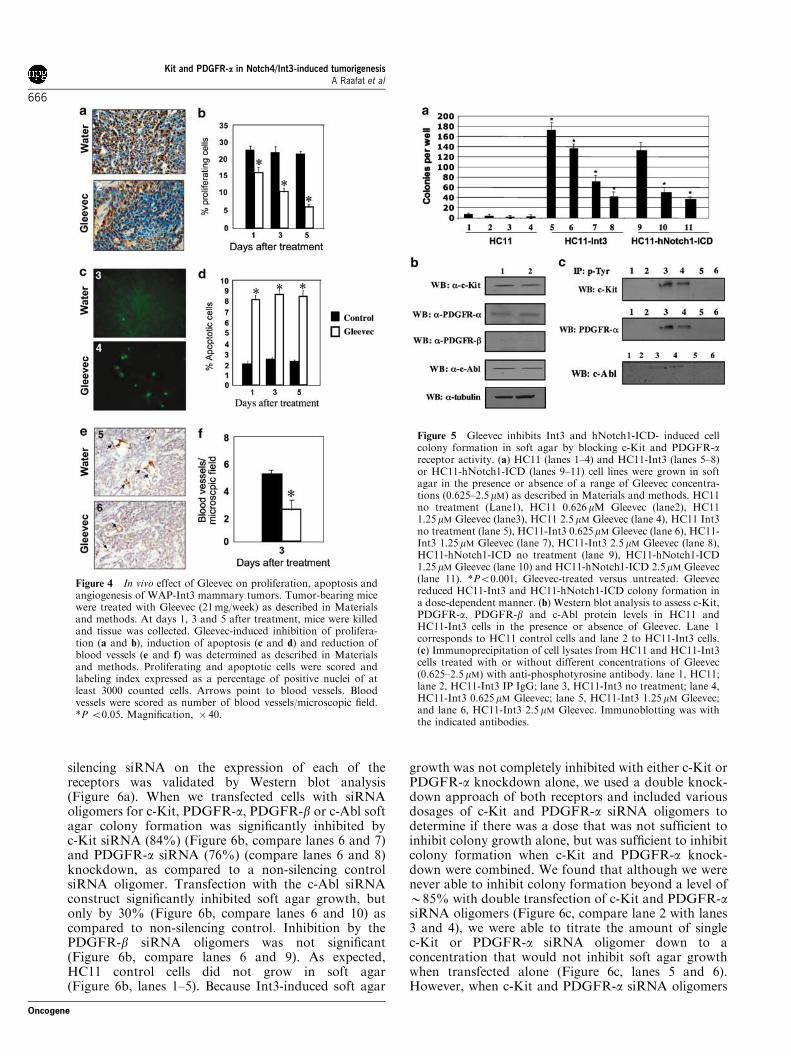
Cell proliferation, apoptosis and microvascularizationlevels in WAP-Int3 tumors from Gleevec-treated miceWe also examined the effects of Gleevec on cellproliferation, apoptosis, and microvascularization.Gleevec treatment resulted in 60% decrease in thenumber of proliferating cells relative to the control(Figure 4a and b). At the same time, there was afourfold increase in the number of apoptotic cells asdetermined by TUNEL staining in the Gleevec treatedgroup as compared to the control (Figure 4c and d). Inaddition, Gleevec treatment resulted in a 50% reductionin microvascularization of the WAP-Int3 tumors(Figure 4e and f).
Gleevec inhibits Int3-induced soft agar colony formationThe in vivo data raised the question of whether all fourRTK III kinases are required for tumor growth or if oneor a combination of them is sufficient to stimulate tumorgrowth. To address this question, we next chose toexamine the activity of the individual Gleevec targets onInt3- and hNotch1-ICD-induced transformation usingan in vitro system. We (Robbins et al., 1992; Imatani andCallahan, 2000) and Dievart et al.(1999) have shownpreviously that HC11 mouse mammary epithelial cells,
Figure 2 Continuous release of Gleevec inhibits WAP-Int3mammary tumor growth in vivo. (a) Gleevec dose response. Micebearing primary WAP-Int3 mammary tumors were treated withwater, 10.5 or 21mg/week Gleevec for 7 days. Gleevec wasadministered using subcutaneous osmotic pumps (continuousrelease) as described in Materials and methods. Treatment startedwhen tumor weight reached 500mg. Mammary tumors werepalpated and measured on daily basis for the duration of theexperiment (6 days). Each data point indicates the average tumorweight from a minimum of 5–6 different mice measured at thespecified day. *Po0.05 for 21mg/week treated mice having lowertumor weight than the 10.5mg/week. (b) Regression of WAP-Int3primary mammary tumors is Gleevec dependent. Gleevec treat-ment started when tumor weight reached 500mg and continued for7 days. Continuous release of Gleevec (21mg/week) from osmoticpumps reduced mammary tumor weight (compare day 1 vs. day 5).Resumption of WAP-Int3 mammary tumor growth took place onday 6 as the osmotic pumps were depleted of Gleevec. (c) Gleeveceffects on the in vivo growth of transplanted WAP-Int3 viablemammary tumor tissue. Viable tissue from WAP-Int3 mammarytumor was placed in the inguinal mammary gland of two differentFVB/N mice. Once tumors weight reached 100mg, one mousereceived Gleevec (’) and the other received water (E). It took theviable tissue 19 days to reach 100mg and about 33 days to reach500mg. Early treatment of WAP-Int3 tumors with Gleevec (21mg/week) resulted in inhibition of tumor growth, whereas treatment ofthe same tumors with water did not block tumor growth. Each datapoint in these graphs indicates the average tumor weight from aminimum of six different mice measured at the specified day.
Kit and PDGFR-a in Notch4/Int3-induced tumorigenesisA Raafat et al
664
Oncogene
when stably transfected with the Int3 (HC11-Int3) orNotch1-ICD, will form colonies in soft agar, a hallmarkof cellular transformation. Gleevec treatment inhibitedInt3- and hNotch1-ICD- induced colony formation in adose-dependent manner (Figure 5a, lanes 5–8 and lanes
9–11, respectively), whereas HC11 control cells did notform colonies in soft agar (Figure 5a, lanes 1–4). Wenext assessed the expression levels of the Gleevec targetsby Western blot analysis. Both HC11 and HC11-Int3cells expressed c-Kit, PDGFR-a and c-Abl at similarlevels (Figure 5b). Neither cell line expressed thePDGFR-b receptor, which differs from our in vivoresult with tumors from transgenic mice. However, asPDGFR-b is usually expressed in the stroma, we mightnot expect to detect this receptor in HC11 epithelialcells. Although there was no difference in the steady-state levels of these receptors in the control and Int3 celllines, we found that cells expressing Int3 have elevatedlevels of phosphorylated c-Kit and PDGFR-a, indicat-ing increased receptor activity as compared to thecontrol HC11 cell line (Figure 5c, compare lanes 1 and3). The phosphorylation status of c-Abl was relativelyunchanged in the two cell lines. When we treated thesecells with Gleevec, receptor phosphorylation wasinhibited in a dose-dependent manner (Figure 5c,compare lanes 4–6 with lane 3), consistent withGleevec’s mechanism of action.
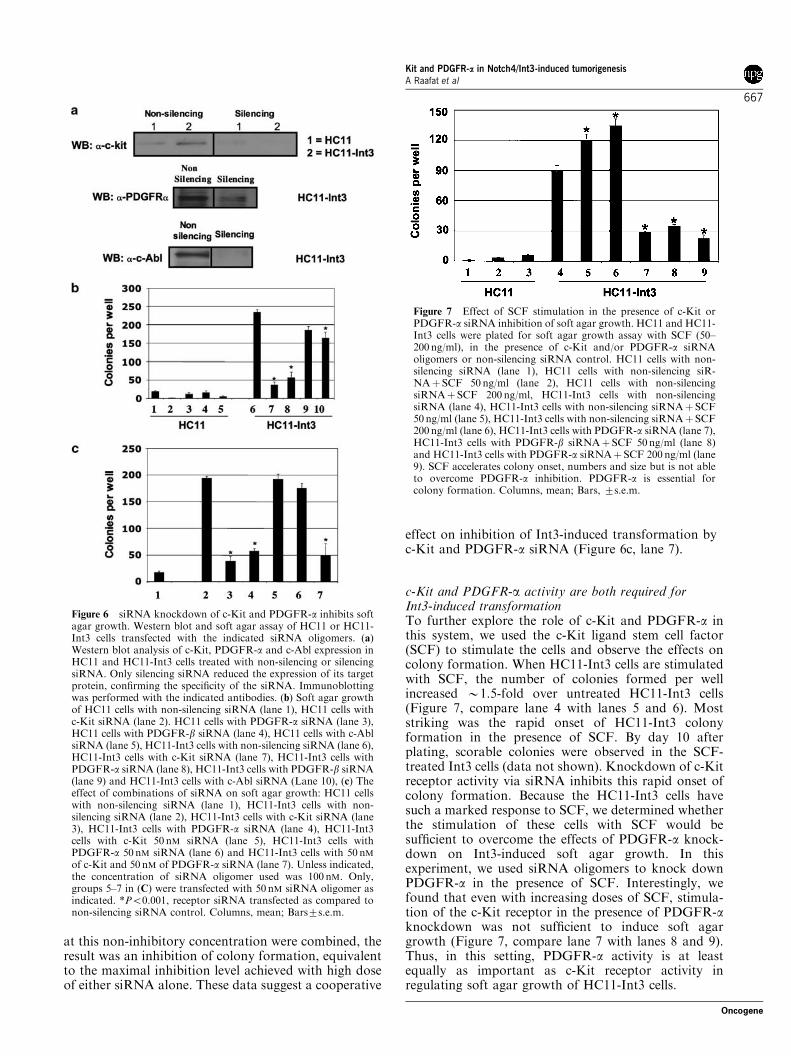
siRNA knockdown of c-Kit and PDGFR-a inhibits softagar growthGleevec inhibition of Int3-induced soft agar growthsuggested that the activity of the target RTK III wasimportant in Int3-induced cell transformation. Todetermine which receptor, or combination of receptors,was important, we employed a small interfering RNA(siRNA) approach to knock down each receptorindividually or in combination and looked at the effecton soft agar growth. The effect of non-silencing and
Figure 3 In vivo inhibition of c-Kit, PDGFR-a, PDGFR-b andc-Abl phosphorylation by Gleevec. Viable tissue from each WAP-Int3 mammary tumor was placed in the inguinal mammary glandof two separate 10-week-old FVB/N mice. Once the tumors weightreached 500mg, one mouse received Gleevec (21mg/week) and theother mouse received water, using Alzet pumps as described in theMaterials and methods. Mice were killed at 72 h after treatment.Immunohistochemical analysis of total (panels 1and 3) andphosphorylated(P) (panels 2 and 4), c-Kit (a), PDGFR-a (b),PDGFR-b (c) and c-Abl (d) is shown. Magnification, � 40. Arrowspoint to endothelial cells. In panels C5 and C6, the magnificationis � 100.
Table 1 Total and phosphorylated protein kinase expression in wap-lnt3 mammary tumors (% positive cells7s.e.m.)
Protein kinase Total P-value Phosphorylated P-value
Water gleevec Water gleevec
c-Kit 7579 80714 p1.0 7077 5076 p0.007PDGFR-a 3073 3075 p1.0 3073 1073 p0.02PDGFR-b 3574 3875 p0.8 3074 271 p0.02c-Abl 4077 3574 p0.2 3275 271 p0.02
Abbreviation: PDGFR, platelet-derieved growth factor receptor.Positive cells were scored and labeling index is expressed as apercentage of positive nuclei of 3000 counted cells.
Kit and PDGFR-a in Notch4/Int3-induced tumorigenesisA Raafat et al
665
Oncogene
silencing siRNA on the expression of each of thereceptors was validated by Western blot analysis(Figure 6a). When we transfected cells with siRNAoligomers for c-Kit, PDGFR-a, PDGFR-b or c-Abl softagar colony formation was significantly inhibited byc-Kit siRNA (84%) (Figure 6b, compare lanes 6 and 7)and PDGFR-a siRNA (76%) (compare lanes 6 and 8)knockdown, as compared to a non-silencing controlsiRNA oligomer. Transfection with the c-Abl siRNAconstruct significantly inhibited soft agar growth, butonly by 30% (Figure 6b, compare lanes 6 and 10) ascompared to non-silencing control. Inhibition by thePDGFR-b siRNA oligomers was not significant(Figure 6b, compare lanes 6 and 9). As expected,HC11 control cells did not grow in soft agar(Figure 6b, lanes 1–5). Because Int3-induced soft agar
growth was not completely inhibited with either c-Kit orPDGFR-a knockdown alone, we used a double knock-down approach of both receptors and included variousdosages of c-Kit and PDGFR-a siRNA oligomers todetermine if there was a dose that was not sufficient toinhibit colony growth alone, but was sufficient to inhibitcolony formation when c-Kit and PDGFR-a knock-down were combined. We found that although we werenever able to inhibit colony formation beyond a level ofB85% with double transfection of c-Kit and PDGFR-asiRNA oligomers (Figure 6c, compare lane 2 with lanes3 and 4), we were able to titrate the amount of singlec-Kit or PDGFR-a siRNA oligomer down to aconcentration that would not inhibit soft agar growthwhen transfected alone (Figure 6c, lanes 5 and 6).However, when c-Kit and PDGFR-a siRNA oligomers
Figure 4 In vivo effect of Gleevec on proliferation, apoptosis andangiogenesis of WAP-Int3 mammary tumors. Tumor-bearing micewere treated with Gleevec (21mg/week) as described in Materialsand methods. At days 1, 3 and 5 after treatment, mice were killedand tissue was collected. Gleevec-induced inhibition of prolifera-tion (a and b), induction of apoptosis (c and d) and reduction ofblood vessels (e and f) was determined as described in Materialsand methods. Proliferating and apoptotic cells were scored andlabeling index expressed as a percentage of positive nuclei of atleast 3000 counted cells. Arrows point to blood vessels. Bloodvessels were scored as number of blood vessels/microscopic field.*P o0.05. Magnification, � 40.
Figure 5 Gleevec inhibits Int3 and hNotch1-ICD- induced cellcolony formation in soft agar by blocking c-Kit and PDGFR-areceptor activity. (a) HC11 (lanes 1–4) and HC11-Int3 (lanes 5–8)or HC11-hNotch1-ICD (lanes 9–11) cell lines were grown in softagar in the presence or absence of a range of Gleevec concentra-tions (0.625–2.5mM) as described in Materials and methods. HC11no treatment (Lane1), HC11 0.626mM Gleevec (lane2), HC111.25mM Gleevec (lane3), HC11 2.5mM Gleevec (lane 4), HC11 Int3no treatment (lane 5), HC11-Int3 0.625mM Gleevec (lane 6), HC11-Int3 1.25mM Gleevec (lane 7), HC11-Int3 2.5 mM Gleevec (lane 8),HC11-hNotch1-ICD no treatment (lane 9), HC11-hNotch1-ICD1.25mM Gleevec (lane 10) and HC11-hNotch1-ICD 2.5mM Gleevec(lane 11). *Po0.001, Gleevec-treated versus untreated. Gleevecreduced HC11-Int3 and HC11-hNotch1-ICD colony formation ina dose-dependent manner. (b) Western blot analysis to assess c-Kit,PDGFR-a, PDGFR-b and c-Abl protein levels in HC11 andHC11-Int3 cells in the presence or absence of Gleevec. Lane 1corresponds to HC11 control cells and lane 2 to HC11-Int3 cells.(c) Immunoprecipitation of cell lysates from HC11 and HC11-Int3cells treated with or without different concentrations of Gleevec(0.625–2.5mM) with anti-phosphotyrosine antibody. lane 1, HC11;lane 2, HC11-Int3 IP IgG; lane 3, HC11-Int3 no treatment; lane 4,HC11-Int3 0.625mM Gleevec; lane 5, HC11-Int3 1.25mM Gleevec;and lane 6, HC11-Int3 2.5mM Gleevec. Immunoblotting was withthe indicated antibodies.
Kit and PDGFR-a in Notch4/Int3-induced tumorigenesisA Raafat et al
666
Oncogene
at this non-inhibitory concentration were combined, theresult was an inhibition of colony formation, equivalentto the maximal inhibition level achieved with high doseof either siRNA alone. These data suggest a cooperative
effect on inhibition of Int3-induced transformation byc-Kit and PDGFR-a siRNA (Figure 6c, lane 7).
c-Kit and PDGFR-a activity are both required forInt3-induced transformationTo further explore the role of c-Kit and PDGFR-a inthis system, we used the c-Kit ligand stem cell factor(SCF) to stimulate the cells and observe the effects oncolony formation. When HC11-Int3 cells are stimulatedwith SCF, the number of colonies formed per wellincreased B1.5-fold over untreated HC11-Int3 cells(Figure 7, compare lane 4 with lanes 5 and 6). Moststriking was the rapid onset of HC11-Int3 colonyformation in the presence of SCF. By day 10 afterplating, scorable colonies were observed in the SCF-treated Int3 cells (data not shown). Knockdown of c-Kitreceptor activity via siRNA inhibits this rapid onset ofcolony formation. Because the HC11-Int3 cells havesuch a marked response to SCF, we determined whetherthe stimulation of these cells with SCF would besufficient to overcome the effects of PDGFR-a knock-down on Int3-induced soft agar growth. In thisexperiment, we used siRNA oligomers to knock downPDGFR-a in the presence of SCF. Interestingly, wefound that even with increasing doses of SCF, stimula-tion of the c-Kit receptor in the presence of PDGFR-aknockdown was not sufficient to induce soft agargrowth (Figure 7, compare lane 7 with lanes 8 and 9).Thus, in this setting, PDGFR-a activity is at leastequally as important as c-Kit receptor activity inregulating soft agar growth of HC11-Int3 cells.
Figure 7 Effect of SCF stimulation in the presence of c-Kit orPDGFR-a siRNA inhibition of soft agar growth. HC11 and HC11-Int3 cells were plated for soft agar growth assay with SCF (50–200ng/ml), in the presence of c-Kit and/or PDGFR-a siRNAoligomers or non-silencing siRNA control. HC11 cells with non-silencing siRNA (lane 1), HC11 cells with non-silencing siR-NAþSCF 50 ng/ml (lane 2), HC11 cells with non-silencingsiRNAþSCF 200 ng/ml, HC11-Int3 cells with non-silencingsiRNA (lane 4), HC11-Int3 cells with non-silencing siRNAþ SCF50ng/ml (lane 5), HC11-Int3 cells with non-silencing siRNAþ SCF200ng/ml (lane 6), HC11-Int3 cells with PDGFR-a siRNA (lane 7),HC11-Int3 cells with PDGFR-b siRNAþSCF 50ng/ml (lane 8)and HC11-Int3 cells with PDGFR-a siRNAþ SCF 200 ng/ml (lane9). SCF accelerates colony onset, numbers and size but is not ableto overcome PDGFR-a inhibition. PDGFR-a is essential forcolony formation. Columns, mean; Bars, 7s.e.m.
Figure 6 siRNA knockdown of c-Kit and PDGFR-a inhibits softagar growth. Western blot and soft agar assay of HC11 or HC11-Int3 cells transfected with the indicated siRNA oligomers. (a)Western blot analysis of c-Kit, PDGFR-a and c-Abl expression inHC11 and HC11-Int3 cells treated with non-silencing or silencingsiRNA. Only silencing siRNA reduced the expression of its targetprotein, confirming the specificity of the siRNA. Immunoblottingwas performed with the indicated antibodies. (b) Soft agar growthof HC11 cells with non-silencing siRNA (lane 1), HC11 cells withc-Kit siRNA (lane 2). HC11 cells with PDGFR-a siRNA (lane 3),HC11 cells with PDGFR-b siRNA (lane 4), HC11 cells with c-AblsiRNA (lane 5), HC11-Int3 cells with non-silencing siRNA (lane 6),HC11-Int3 cells with c-Kit siRNA (lane 7), HC11-Int3 cells withPDGFR-a siRNA (lane 8), HC11-Int3 cells with PDGFR-b siRNA(lane 9) and HC11-Int3 cells with c-Abl siRNA (Lane 10), (c) Theeffect of combinations of siRNA on soft agar growth: HC11 cellswith non-silencing siRNA (lane 1), HC11-Int3 cells with non-silencing siRNA (lane 2), HC11-Int3 cells with c-Kit siRNA (lane3), HC11-Int3 cells with PDGFR-a siRNA (lane 4), HC11-Int3cells with c-Kit 50 nM siRNA (lane 5), HC11-Int3 cells withPDGFR-a 50 nM siRNA (lane 6) and HC11-Int3 cells with 50 nMof c-Kit and 50 nM of PDGFR-a siRNA (lane 7). Unless indicated,the concentration of siRNA oligomer used was 100 nM. Only,groups 5–7 in (C) were transfected with 50 nM siRNA oligomer asindicated. *Po0.001, receptor siRNA transfected as compared tonon-silencing siRNA control. Columns, mean; Bars7s.e.m.
Kit and PDGFR-a in Notch4/Int3-induced tumorigenesisA Raafat et al
667
Oncogene

Discussion
In the present work, we have shown that WAP-Int3mammary tumors contain high steady-state levels ofc-Kit and other members of the RTK III family. Thelinkage between Notch and Kit/PDGFR signaling is notunprecedented. For instance, endothelial-to-mesenchy-mal transformation is associated with Notch4 signaling,which leads to the upregulation of PDGFR expression(Noseda et al., 2004). Interestingly, human mammo-spheres that are enriched for early progenitor/stem cellsof the mammary gland coexpress Notch4 and PDGFR-a (Dontu et al., 2004). In other tissues such as ciliaryepithelium neural stem cells, c-Kit-mediated signalingupregulates Notch expression and signaling (Das et al.,2004). Enriched hematopoietic stem cells also coexpressNotch1 and c-Kit (Ramalho-Santos et al., 2002).
As members of the Kit/PDGFR RTK III family areinvolved in multiple tumor-associated processes, wequestioned whether their signaling contributes tomammary tumor development in the context of Int3signaling. Treatment of Int3 tumor-bearing mice withGleevec, an inhibitor specific for this family of RTK,caused the tumors to regress to a point that in manycases they were unpalpable. At a molecular level, aprimary consequence of Gleevec treatment on c-Kit,PDGFR-a and c-Abl was to decrease their phosphor-ylation. A secondary consequence of Gleevec treatmentwas the decrease in the number of proliferating cells andthe level of the microvascularization of the tumorscoupled with an increase in the level of apoptotic cells inthe tumors. The increase in apoptosis and decrease inproliferation in response to Gleevec might explain thereduction in tumor growth. Continuous release ofGleevec for a week resulted in 33% inhibition of tumorgrowth by day 2 and 66% by day 4. Gleevec treatmentproduced a complete inhibition of tumor growth,indicating that a continuous block of Gleevec targetsactivity is needed to produce complete tumor regression.Similar results were observed when Gleevec wasadministered every 8 h in mice bearing Leydig celltumors (Basciani et al., 2005) and in nude mice bearingBcr/Abl-positive human leukemia cell lines (le Coutreet al., 1999). Discontinuation of Gleevec treatmentresulted in rapid tumor re-growth, confirming the needfor a sustained blockage of Gleevec targets to inhibittumorigenesis. These results are in agreement withclinical data in CML patients (Cortes et al., 2004). Theantiangiogenic effect of Gleevec has been observed inseveral types of cancers (Hwang et al., 2003; Ueharaet al., 2003). This effect has been attributed to theinhibition of PDGFRs and c-Kit, which are importantsurvival factors for endothelial cells (Betsholtz,2003; Matsui et al., 2004). Gleevec inhibits vascularendothelial growth factor expression indirectly througha PDGF inhibition-mediated mechanism (Wang et al.,1999). In addition, both PDGFR-b and PDGF-b nullmutant mice die at late gestation from widespreadmicrovascular bleeding (Leveen et al., 1994; Soriano,1994). Taken together, we conclude from our datathat signaling by at least one RTK III member is
necessary for initiating or maintaining Int3 mammarytumorigenesis.
To determine which RTK III member or combinationof members is responsible for tumor promotion, we usedan in vitro model in which Int3 expression confers onHC11 mouse mammary epithelial cells the ability foranchorage independent-growth in soft agar. Treatmentof HC11-Int3 cells with Gleevec blocks, in a dose-dependent manner, their ability to grow in an ancho-rage-independent manner, whereas it is not cytotoxic toHC11 cells grown on monolayer. From this result, weconcluded that it was not RTK III signaling per se, but acollaboration between Int3 and RTK III signaling. Todefine more fully which RTK III family member(s) areinvolved in this collaboration, we used siRNA againsteach of the RTK III members in the HC11-Int3 softagar assay. As with Gleevec, siRNA against individualRTK III members is not toxic for HC11 cells. Inaddition, HC11 cells do not express PDGFR-b, andknockdown of c-Abl with siRNA had a minimal effecton the ability of HC11-Int3 cells to grow in soft agar.Therefore, we conclude that the primary contributors toHC11-Int3 soft agar growth are due to the activation ofc-Kit and PDGFR-a. Two observations suggest thatboth c-Kit and PDGFR-a are required to promote Int3-induced HC11 soft agar growth. First, suboptimal levelsof siRNA for c-Kit and PDGFR-a act cooperatively inthe inhibition of HC11-Int3 soft agar growth, andsecond, SCF induction of c-Kit activity was unable toovercome PDGFR-a siRNA inhibition of HC11-Int3soft agar growth. We conclude that there are compo-nents of the c-Kit and PDGFR-a signaling pathwaysthat are unique to each and that they collaborate withInt3 signaling in mammary tumor progression.
Notch1-ICD (ICD1) also confers on HC11 cells thecapability of growth in soft agar (Dievart et al., 1999).Like HC11-Int3 cells, Gleevec blocks HC11-ICD1 cellgrowth in soft agar, suggesting that c-Kit and PDGFR-aare the targets for the drug in these cells as well. It seemspertinent, therefore, that in human foreskin fibroblasts(BJ) and human embryonic kidney (HEK) epithelialcells, oncogenic Ras activates the expression of Notch1that in turn activates the expression of Notch4 (Weijzenet al., 2002). In this setting, oncogenic Ras is upstreamof Notch1, as suppression of Notch1 expression inhibitsthe growth of Ras-transformed cells.
At the present time, only a limited number of studieshave been undertaken to look at the expression ofNotch, c-Kit and PDGFR-a in normal human breastand in primary breast carcinomas. Reedijk et al.(2005)found that the expression of each member of the Notchgene family could be detected to varying degrees in asurvey of 184 breast carcinomas. In fact, high levels ofNotch1 and its ligand JAG1 were linked to poor overallsurvival. Imatani and Callahan (2000) identified atruncated Notch4 RNA species in certain human breast,colon and lung carcinoma cell lines. This Notch4 RNAspecies encodes a portion of the ICD of the protein.Expression of this Notch4 RNA species from atransgene in the mouse mammary gland is associatedwith mammary tumor development (Raafat et al., 2004).
Kit and PDGFR-a in Notch4/Int3-induced tumorigenesisA Raafat et al
668
Oncogene

High levels of c-Kit were observed in the normalmammary ductal epithelium but not in myoepithelialcells (Weijzen et al., 2002). Assessment of breastcarcinomas for c-Kit expression have shown that asthe tumors become more invasive, the levels of c-Kitexpression decrease; thus, 44–53% of ductal carcinomasin situ (DCIS) are c-Kit positive, whereas only 9–10% ofinvasive ductal carcinomas (IDC) are c-Kit positive(Ulivi et al., 2004; Tsuda et al., 2005a; b; Diallo et al.,2006). In contrast, 39% of IDC are positive forPDGFR-a expression (de Jong et al., 1998; Carvalhoet al., 2005). In these tumors, the stroma andendothelium also stain positive for PDGFR-a (de Jonget al., 1998). In conclusion, the reports of elevatedsteady-state levels of Notch, c-Kit and PDGFR-aexpression in human breast cancer tissue (Carvalhoet al., 2005; Diallo et al., 2006) suggest that Gleevec is apotential candidate drug for breast cancer treatment andprevention.
Materials and methods
Animals and experimental designPrimary mammary tumors in the WAP-Int3 females werepalpated weekly. The length, width and depth of palpabletumors were measured with Vernier calipers. TW wasdetermined using the equation TW (mg)¼ (S)2�L/2 whereS and L (le Coutre et al., 1999) were measured in millimeters.S and L are the shortest and longest diameters of the tumor,respectively. Primary tumors were allowed to grow until theyreached 500mg, at which time the WAP-Int3 tumor-bearingmice were killed and mammary tumors were collected as viabletissue. To reduce inter-tumor variations, virgin FVB/N femalemice from our colony were used at 10 weeks of age and theinguinal mammary glands of these FVB\N mice served as thetransplantation site of the primary WAP-Int3 viable tumortissue. Viable tissue from each WAP-Int3 mammary tumorwas placed in the inguinal mammary gland of two separateFVB/N mice. Once tumors reached the desired weight, onemouse with tissue from a single primary tumor receivedGleevec and its matching control received water (control).Alzet miniosmotic pumps (Model 2001, pumping rate 1ml/h,Durect Corp., Cupertino, CA, USA), implanted subcuta-neously on the dorsal surface of the mouse, were used todeliver a subcutaneous dose of Gleevec (10.5 or 21mg/mouse/week) or water (control). For studies longer than 7 days, freshpumps were used every 7 days. Gleevec was generouslyprovided by Novartis Pharma (Basel, Switzerland). Treatedmice were killed at various time points. Mice were kept understandard laboratory conditions according to the guidelines ofthe National Cancer Institute. This study was approved by theInstitutional Ethics Committee for Laboratory Animals usedin Experimental Research.
Northern Blot Analysis and RT–PCRTo detect c-Kit, c-Abl, PDGFR-a and PDGFR-b expressionin WAP-Int3 tumors, total RNA extracts were preparedfrom WAP-Int3 mammary tumors using TRIzol (InvitrogenLife Technologies, Carlsbad, CA, USA), according to themanufacturer’s instructions.Twenty-five micrograms of total RNA from WAP-Int3 and
MMTV LTR-Int3 mammary tumors as well as mammarytissue from normal FVB/N and WAP-Int3 females was used in
Northern blot analysis as previously described (Robbins et al.,1992; Gallahan and Callahan, 1997). Full-length cDNAs forc-KIT (ATCC 10699933) and GAPDH (ATCC 10539048)were used for hybridization as described previously (Robbinset al., 1992; Gallahan and Callahan, 1997).The nucleotide sequences of synthetic oligonucleotides for
RT–PCR are as follows: c-Kit: 50-CCAGTGCTTCCGTGACATTC-30 and 50-CGTCCACTGGTGAGACAGGA-30; c-Abl: 50-CGCATGTTCCGGGACAAAAGC-30 and 50-CCATTTTCTCATCTCCAA GCC-30; PDGFR-a: 50-CGACTCCAGATGGGAGTTCCC-30 and 50-TGCCATCCACTTCACAGGCA-30; PDGFR-b: 50-AGCTACATGGCCCCTTATGA-30
and 50-GGATCCCAAAAGACCAGACA-30. For each experi-ment, a control reaction without reverse transcriptase wasperformed. A thermal cycle of 581C annealing temperature wasrepeated 32 times using Superscript One-Step RT–PCR withPlatinum Taq (Invitrogen Life Technologies, Carlsbad, CA,USA) according to the manufacturer’s protocol. RT–PCRproducts were separated on a 1.5% agarose gel and visualizedwith ethidium bromide.
Preparation of tissue for histologyMammary glands were routinely fixed in 4% freshly preparedparaformaldehyde in phosphate-buffered saline (PBS) (pH 7.4)for 2 h, rinsed through several changes of buffer, dehydrated inethanol and embedded in paraffin. Paraffin sections (5 mm)were placed on slides, deparaffanized and stained with H&E.
ImmunohistochemistryImmunohistochemistry was carried out with the ABC methodaccording to the manufacturer’s protocol (Vector LaboratoriesInc., Burlingame, CA, USA). Primary antibody incubationwas carried out overnight at 41C: PCNA (sc-9857; Santa Cruz,Santa Cruz, CA, USA), c-Kit (sc-168), PDGFR-a (sc-338),PDGFR-b (sc-1627), c-Abl antibody (2862, Cell Signaling Inc.,Technologies Inc., Beverly, MA, USA). For phosphorylated(P) proteins, P-c-Kit (sc-180676R), P-PDGFR-a (sc-12911R),P-PDGFR-b (3161, Cell Signaling Technologies, Inc., Beverly,MA, USA) and P-c-Abl antibody (2861, Cell SignalingTechnologies Inc., Beverly, MA, USA) were used. Primaryantibodies were diluted 100� in PBS–1% BSA and appro-priate biotinylated secondary anti-goat (PK-6105, VectorLaboratories Inc., Burlingame, CA, USA) or anti-rabbit(PK-6101, Vector Laboratories, Inc., Burlingame, CA, USA)antibodies were diluted according to the manufacturer’srecommendations. For apoptosis and angiogenesis assay, theRoche in situ cell detection POD kit (1684817, Roche,Indianapolis, IN, USA) and the Chemicon, blood vesselstaining kit (ECM590, Temecula, CA, USA) were usedaccording to the manufacturer’s recommendations, respec-tively. Labeling index was determined in at least a total of 3000cells in each experimental condition.
Cell cultureHC11 (Ball et al., 1988) and HC11-Int3 mouse mammaryepithelial cells were grown in RPMI medium (Gibco BRL,Grand Island, NY, USA) supplemented with 10% FBS(Mediatech, Herndon, VA, USA), 5 mg/ml insulin (GibcoBRL, Grand Island, NY, USA), 10 ng/ml EGF (Gibco BRL,Grand Island, NY, USA) and 1% penicillin–streptomycin(Gibco BRL, Grand Island, NY, USA). The HC11-Int3 cellline was generated as described previously (Raafat et al., 2004).Human Notch1 intracellular domain (hNotch1-ICD) was agenerous gift from Dr Sean Jeffries (Jeffries and Capobianco,2000).
Kit and PDGFR-a in Notch4/Int3-induced tumorigenesisA Raafat et al
669
Oncogene

Colony formation in soft agarFive thousand cells in 2� growth medium with or withoutSCF (50 mM; Peprotech, Rocky Hill, NJ, USA) or c-Kitactivity-blocking antibody, Clone K44.2 (200 mg/ml; Sigma StLouis, MO, USA) were mixed 1 : 1 with 0.4% agar and thenanalysed for colony formation as described previously (Ghataket al., 2002). After 20 days of growth in agar, 1ml of nitrobluetetrazolium vital dye (Sigma, St. Louis, MO, USA) was addedto each well to visualize viable colonies. The following day,colonies were counted at a magnification of � 10 using amanufactured ocular scale (Electron Microscopy Sciences,Fort Washington, PA, USA). Colonies measuring larger than150 mm in diameter were counted. Each experiment was carriedout in duplicate and performed at least three times.
RNAi gene silencingc-Kit, c-Abl, PDGFR-a and PDGFR-b gene silencing wasperformed using Qiagen-designed siRNA duplexes for c-Kit,c-Abl, PDGFR-a and PDGFR-b (Qiagen Inc., Valencia, CA,USA). Two double-stranded siRNAs were generated for eachtarget, except for c-Abl, which was generated and pre-validated by the manufacturer. All oligomers were tested. Anasterisk indicates the siRNA that showed the greatestinhibition and was subsequently used in the experiment.DNA target sequences or regions are as follows: c-Kit (1)TTCCGTGACATTCAACGTTTA, c-Kit (2)* CCCACTGTGATTCCGCCTTTA, PDGFR-a (1) CGGCGACTACATGGACATGAA, PDGFR-a (2)* ACGGATGAGAGTGAGATC GAA, PGDFR-b (1)* CCGGTACGTGTCAGAACTGAT, PGDFR-b (2) GCGGGTGGTGTTCGAGGCTTAand c-Abl* 1818–1863. In all experiments, a non-silencingduplex siRNA was used as a control. HC11 cells were platedthe day before transfection to reach 60–80% confluency thenext day. All transfections were carried out in six-well platesaccording to the Qiagen protocol, using a 1:3 ratio ofsiRNA:RNAi Fect Reagent. Gene silencing was monitoredat the protein level by Western blotting of cell lysates collected48 h following transfection. Cells were harvested for soft agarassay 48 h following transfection.
ImmunoblottingCells were harvested with trypsin-ethylene diaminetetraaceticacid (1� ) (Gibco BRL) and collected as a pellet bycentrifugation at 41C. Cells were lysed in buffer containing1% Nonidet-40, 0.5mM ethylene glycol tetraacetate, 5mM
sodium orthovanadate, 10% glycerol, 1� concentration ofCalbiochem Protease Inhibitor Cocktail I (La Jolla, CA, USA)
and 50mM HEPES, pH 7.5. SDS–polyacrylamide gel electro-phoresis (SDS—PAGE), followed by transfer to nitrocellulose.After blocking for 1 h and washing with Tris-buffered saline–0.1% Tween 20, the membranes were probed overnight withprimary antibodies to c-Kit, c-Abl, PDGFR-a, PDGFR-b (all1:100; Santa Cruz, Santa Cruz, CA, USA), or tubulin (1:5000.Sigma, St Louis, MO, USA). Membranes were washed asdescribed above, followed by incubation with horseradishperoxidase (HRP)-conjugated secondary antibody (1:5000;Amersham Biosciences, Piscataway, NJ, USA). ECL reagent(Amersham Biosciences, Piscataway, NJ, USA) was used fordetection.
ImmunoprecipitationCells were harvested as described above and lysates werenormalized to 1mg/ml total protein. Anti-phosphotyrosine(clone 4G10) immunoprecipitating antibody (4 mg; UpstateLake Placid, NY, USA) was added to the cell lysate andincubated on a rotator at 41C overnight. Protein G agarosebeads (Roche Diagnostics, Indianapolis, IN, USA) were addedto capture the immunocomplex and incubated on a rotator at41C for 2 h. The agarose beads were collected, washed oncewith ice-cold PBS, twice with 1� Tris-buffered saline–0.25Msodium chloride and resuspended in 5� sample buffer. Beadswere heated at 1001C for 5min, centrifuged and the super-natant was subjected to SDS–PAGE and transfer. Membraneswere incubated with antibodies against c-Kit, c-Abl, PDGFR-aor PDGFR-b (1:100; all Santa Cruz), followed by HRP-conjugated secondary antibody as described above.
StatisticsQuantitative values are represented as the mean of at leastthree experiments. All in vivo experiments were repeated atleast three times, and at least five mice were used in eachexperiment. The statistical significance of the differencebetween groups was determined by the Wilcoxon rank sumtest. Comparisons resulting in P-values less than 0.05 wereconsidered statistically significant and identified in the figureswith an asterisk (*).
Acknowledgements
We thank Dr Barbara Vonderhaar and Dr Gilbert H Smith fortheir critical review of the manuscript. We specially thankNovartis Pharmaceuticals for providing Gleevec.
References
Allenspach EJ, Maillard I, Aster JC, Pear WS. (2002). Notchsignaling in cancer. Cancer Biol Ther 1: 466–476.
Ball RK, Friis RR, Schoenenberger CA, Doppler W, GronerB. (1988). Prolactin regulation of beta-casein gene expressionand of a cytosolic 120-kDa protein in a cloned mousemammary epithelial cell line. EMBO J 7: 2089–2095.
Basciani S, Brama M, Mariani S, De Luca G, Arizzi M, VesciL et al. (2005). Imatinib mesylate inhibits Leydig cell tumorgrowth: evidence for in vitro and in vivo activity. Cancer Res65: 1897–1903.
Besmer P, Murphy JE, George PC, Qiu FH, Bergold PJ,Lederman L et al. (1986). A new acute transforming felineretrovirus and relationship of its oncogene v-kit with theprotein kinase gene family. Nature 320: 415–421.
Betsholtz C. (2003). Biology of platelet-derived growth factors indevelopment. Birth Defects Res C Embryo Today 69: 272–285.
Callahan R, Raafat A. (2001). Notch signaling in mammary glandtumorigenesis. J Mammary Gland Biol Neoplasia 6: 23–36.
Carvalho I, Milanezi F, Martins A, Reis RM, Schmitt F.(2005). Overexpression of platelet-derived growth factorreceptor alpha in breast cancer is associated with tumourprogression. Breast Cancer Res 7: R788–R795.
Chiaramonte R, Basile A, Tassi E, Calzavara E, Cecchinato V,Rossi V et al. (2005). A wide role for NOTCH1 signaling inacute leukemia. Cancer Lett 219: 113–120.
Cortes J, O’Brien S, Kantarjian H. (2004). Discontinuation ofimatinib therapy after achieving a molecular response. Blood104: 2204–2205.
Dang TP, Eichenberger S, Gonzalez A, Olson S, Carbone DP.(2003). Constitutive activation of Notch3 inhibits terminalepithelial differentiation in lungs of transgenic mice.Oncogene 22: 1988–1997.
Kit and PDGFR-a in Notch4/Int3-induced tumorigenesisA Raafat et al
670
Oncogene

Dang TP, Gazdar AF, Virmani AK, Sepetavec T, Hande KR,Minna JD et al. (2000). Chromosome 19 translocation,overexpression of Notch3, and human lung cancer. J NatlCancer Inst 92: 1355–1357.
Das AV, James J, Zhao X, Rahnenfuhrer J, Ahmad I. (2004).Identification of c-Kit receptor as a regulator of adult neuralstem cells in the mammalian eye: interactions with Notchsignaling. Dev Biol 273: 87–105.
de Jong JS, van Diest PJ, van der Valk P, Baak JP. (1998).Expression of growth factors, growth inhibiting factors, andtheir receptors in invasive breast cancer. I: an inventory insearch of autocrine and paracrine loops. J Pathol 184: 44–52.
Diallo R, Rody A, Jackisch C, Ting E, Schaefer KL, Kissler Set al. (2006). C-KIT expression in ductal carcinoma in situ ofthe breast: co-expression with HER-2/neu. Hum Pathol 37:205–211.
Dievart A, Beaulieu N, Jolicoeur P. (1999). Involvement ofNotch1 in the development of mouse mammary tumors.Oncogene 18: 5973–5981.
Dontu G, Jackson KW, McNicholas E, Kawamura MJ,Abdallah WM, Wicha MS. (2004). Role of Notch signalingin cell-fate determination of human mammary stem/pro-genitor cells. Breast Cancer Res 6: R605–R615.
Gallahan D, Callahan R. (1997). The mouse mammary tumorassociated gene INT3 is a unique member of the NOTCHgene family (NOTCH4). Oncogene 14: 1883–1890.
Gallahan D, Jhappan C, Robinson G, Hennighausen L, SharpR, Kordon E et al. (1996). Expression of a truncated Int3gene in developing secretory mammary epithelium specifi-cally retards lobular differentiation resulting in tumorigen-esis. Cancer Res 56: 1775–1785.
Gallahan D, Kozak C, Callahan R. (1987). A new commonintegration region (int-3) for mouse mammary tumor viruson mouse chromosome 17. J Virol 61: 218–220.
Ghatak S, Misra S, Toole BP. (2002). Hyaluronan oligosac-charides inhibit anchorage-independent growth of tumorcells by suppressing the phosphoinositide 3-kinase/Akt cellsurvival pathway. J Biol Chem 277: 38013–38020.
Heinrich MC, Corless CL, Duensing A, McGreevey L, ChenCJ, Joseph N et al. (2003). PDGFRA activating mutationsin gastrointestinal stromal tumors. Science 299: 708–710.
Hwang RF, Yokoi K, Bucana CD, Tsan R, Killion JJ, EvansDB et al. (2003). Inhibition of platelet-derived growth factorreceptor phosphorylation by STI571 (Gleevec) reducesgrowth and metastasis of human pancreatic carcinoma inan orthotopic nude mouse model. Clin Cancer Res 9:6534–6544.
Imatani A, Callahan R. (2000). Identification of a novelNOTCH-4/INT-3 RNA species encoding an activated geneproduct in certain human tumor cell lines. Oncogene 19:223–231.
Jeffries S, Capobianco AJ. (2000). Neoplastic transformationby Notch requires nuclear localization. Mol Cell Biol 20:3928–3941.
Jhappan C, Gallahan D, Stahle C, Chu E, Smith GH, MerlinoG et al. (1992). Expression of an activated Notch-related int-3 transgene interferes with cell differentiation and inducesneoplastic transformation in mammary and salivary glands.Genes Dev 6: 345–355.
Kim MK, Higgins J, Cho EY, Ko YH, Oh YL. (2000).Expression of CD34, bcl-2, and kit in inflammatory fibroidpolyps of the gastrointestinal tract. Appl ImmunohistochemMol Morphol 8: 147–153.
Lasota J, Jasinski M, Sarlomo-Rikala M, Miettinen M. (1999).Mutations in exon 11 of c-Kit occur preferentially inmalignant versus benign gastrointestinal stromal tumors
and do not occur in leiomyomas or leiomyosarcomas. Am JPathol 154: 53–60.
le Coutre P, Mologni L, Cleris L, Marchesi E, Buchdunger E,Giardini R et al. (1999). In vivo eradication of human BCR/ABL-positive leukemia cells with an ABL kinase inhibitor.J Natl Cancer Inst 91: 163–168.
Leveen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E,Betsholtz C. (1994). Mice deficient for PDGF B show renal,cardiovascular, and hematological abnormalities. Genes Dev8: 1875–1887.
Marley SB, Gordon MY. (2005). Chronic myeloid leukaemia:stem cell derived but progenitor cell driven. Clin Sci(London) 109: 13–25.
Matsui J, Wakabayashi T, Asada M, Yoshimatsu K, OkadaM. (2004). Stem cell factor/c-kit signaling promotes thesurvival, migration, and capillary tube formation ofhuman umbilical vein endothelial cells. J Biol Chem 279:18600–18607.
Noseda M, McLean G, Niessen K, Chang L, Pollet I,Montpetit R et al. (2004). Notch activation results inphenotypic and functional changes consistent with endothe-lial-to-mesenchymal transformation. Circ Res 94: 910–917.
Pear WS, Aster JC. (2004). T cell acute lymphoblasticleukemia/lymphoma: a human cancer commonly associatedwith aberrant NOTCH1 signaling. Curr Opin Hematol 11:426–433.
Raafat A, Bargo S, Anver MR, Callahan R. (2004). Mammarydevelopment and tumorigenesis in mice expressing atruncated human Notch4/Int3 intracellular domain (h-Int3sh). Oncogene 23: 9401–9407.
Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC,Melton DA. (2002). ‘Stemness’: transcriptional profiling ofembryonic and adult stem cells. Science 298: 597–600.
Reedijk M, Odorcic S, Chang L, Zhang H, Miller N,McCready DR et al. (2005). High-level coexpression ofJAG1 and NOTCH1 is observed in human breast cancer andis associated with poor overall survival. Cancer Res 65:8530–8537.
Robbins J, Blondel BJ, Gallahan D, Callahan R. (1992).Mouse mammary tumor gene int-3: a member of the notchgene family transforms mammary epithelial cells. J Virol 66:2594–2599.
Smith GH, Gallahan D, Diella F, Jhappan C, Merlino G,Callahan R. (1995). Constitutive expression of a truncatedINT3 gene in mouse mammary epithelium impairs differ-entiation and functional development. Cell Growth Differ 6:563–577.
Soriano P. (1994). Abnormal kidney development andhematological disorders in PDGF beta-receptor mutantmice. Genes Dev 8: 1888–1896.
Taniguchi M, Nishida T, Hirota S, Isozaki K, Ito T, NomuraT et al. (1999). Effect of c-kit mutation on prognosis ofgastrointestinal stromal tumors. Cancer Res 59: 4297–4300.
Tsuda H, Morita D, Kimura M, Shinto E, Ohtsuka Y,Matsubara O et al. (2005a). Correlation of KIT and EGFRoverexpression with invasive ductal breast carcinoma of thesolid-tubular subtype, nuclear grade 3, and mesenchymal ormyoepithelial differentiation. Cancer Sci 96: 48–53.
Tsuda H, Tani Y, Weisenberger J, Kitada S, Hasegawa T,Murata T et al. (2005b). Frequent KIT and epidermalgrowth factor receptor overexpressions in undifferentiated-type breast carcinomas with ‘stem-cell-like’ features. CancerSci 96: 333–339.
Uehara H, Kim SJ, Karashima T, Shepherd DL, Fan D, TsanR et al. (2003). Effects of blocking platelet-derived growthfactor-receptor signaling in a mouse model of experimental
Kit and PDGFR-a in Notch4/Int3-induced tumorigenesisA Raafat et al
671
Oncogene

prostate cancer bone metastases. J Natl Cancer Inst 95:458–470.
Ulivi P, Zoli W, Medri L, Amadori D, Saragoni L, Barbanti Fet al. (2004). c-kit and SCF expression in normal and tumorbreast tissue. Breast Cancer Res Treat 83: 33–42.
Wang D, Huang HJ, Kazlauskas A, Cavenee WK. (1999).Induction of vascular endothelial growth factor expression
in endothelial cells by platelet-derived growth factor throughthe activation of phosphatidylinositol 3-kinase. Cancer Res59: 1464–1472.
Weijzen S, Rizzo P, Braid M, Vaishnav R, Jonkheer SM,Zlobin A et al. (2002). Activation of Notch-1 signalingmaintains the neoplastic phenotype in human Ras-trans-formed cells. Nat Med 8: 979–986.
Kit and PDGFR-a in Notch4/Int3-induced tumorigenesisA Raafat et al
672
Oncogene




















