
Role of p53 mutation in polyomavirus-induced tumorigenesis
11
Journal of General Virology (1997), 78, 893–903. Printed in Great Britain ........................................................................................................................................................................................................................................................................................... Role of p53 mutation in polyomavirus-induced tumorigenesis Wang Qian, Elena Kashuba, Kristinn P. Magnusson, Katja Pokrovskaja, Ismail Okan, George Klein and Klas G. Wiman Microbiology and Tumor Biology Center (MTC), Karolinska Institute, S-171 77 Stockholm, Sweden Small DNA tumour viruses, such as simian virus 40 (SV40), papilloma viruses and adenoviruses, encode proteins that form complexes with and in- activate the p53 and retinoblastoma (RB) proteins. This convergent evolution reflects the common need of these viruses to inactivate these two important regulators of cell cycle progression and cell survival. Polyomavirus, a close relative of SV40, is different. Its large T protein complexes only with RB, not with p53. We have examined whether this is com- pensated by the frequent appearance of p53 mutations in polyomavirus-induced tumours. We tested the p53 status of 15 polyomavirus-induced sarcomas. Two sarcomas were p53-negative while six carried mutant p53. Another six sarcomas expressed low levels of wild-type p53. One tumour expressed high levels of wild-type p53 protein as shown by DNA sequencing and immunofluorescence Introduction The p53 gene is often inactivated by point mutation or deletion in a wide variety of human and rodent tumours (Greenblatt et al., 1994). Moreover, p53 null mice develop spontaneous tumours with a dramatically increased incidence (Donehower et al., 1992), supporting the notion that p53 is a crucial tumour suppressor. p53 is expressed at low levels under normal conditions, but accumulates due to protein stabilization in response to DNA damage (Maltzman & Czyzyk, 1984 ; Kastan et al., 1991 ; Fritsche et al., 1993) or induction of cell proliferation by the E1A and c-myc oncogenes (Debbas & White, 1993 ; Hermeking & Eick, 1994 ; Lowe et al., 1994; Wagner et al., 1994). The increased p53 levels cause cell cycle arrest in the G " phase or cell death through apoptosis (for a review see Selivanova & Wiman, 1995). The p53 protein is a sequence-specific transcription factor that transactivates genes, such as those encoding WAF1}p21, Author for correspondence : Klas G. Wiman. Fax ›46 8 33 04 98. e-mail Klas.Wiman!mtc.ki.se staining. MDM2 amplification was not detected in any of the tumours, but Northern blotting showed that MDM2 was overexpressed in at least two tumours that expressed wild-type p53 and in one tumour that expressed both wild-type and mutant p53. Treatment with the DNA-damaging agent mitomycin C caused p53 protein accumulation fol- lowed by induction of MDM2 and WAF1/p21 mRNA in four of the tumours expressing wild-type p53, indicating that p53-mediated transcriptional acti- vation was unaltered in these tumours. However, p53-mediated transactivation of WAF1/p21 was impaired in the wild-type p53-expressing tumours that expressed elevated levels of MDM2. These results demonstrate that p53 mutation and inacti- vation are frequently but not invariably involved in polyomavirus-induced tumorigenesis. MDM2, GADD45, cyclin G, Bax and Fas (reviewed by Ko & Prives, 1996). Transactivation of WAF1}p21 is important for p53-induced cell cycle arrest but is dispensable for p53-induced apoptosis (Deng et al., 1995 ; Waldman et al., 1995). However, transactivation of bax and fas may facilitate apoptosis (see Ko & Prives, 1996). p53-mediated transactivation of MDM2 constitutes an autoregulatory feedback loop that modulates the activity of p53 (Wu et al., 1993). The MDM2 protein can inhibit p53-mediated transcriptional activation by binding an N-terminal region of p53 that coincides with the acidic transactivation domain (Oliner et al., 1993). Overexpression of MDM2 increases the tumorigenicity of NIH3T3 cells and overcomes wild-type p53-mediated suppression of cell growth (Fakharzadeh et al., 1991 ; Finlay, 1993). Amplification and overexpression of this gene are frequently observed in human sarcomas (Oliner et al., 1992). More recent data have indicated that the early embryonic lethality of MDM2-deficient mice can be rescued by lack of p53, suggesting that MDM2 is a vital negative regulator of p53 activity during embryonic development (de Oca Luna et al., 1995 ; Jones et al., 1995). 0001-4432 # 1997 SGM IJD
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Role of p53 mutation in polyomavirus-induced tumorigenesis

Journal of General Virology (1997), 78, 893–903. Printed in Great Britain. . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . . .
Role of p53 mutation in polyomavirus-induced tumorigenesis
Wang Qian, Elena Kashuba, Kristinn P. Magnusson, Katja Pokrovskaja, Ismail Okan, George Kleinand Klas G. Wiman
Microbiology and Tumor Biology Center (MTC), Karolinska Institute, S-171 77 Stockholm, Sweden
Small DNA tumour viruses, such as simian virus 40(SV40), papilloma viruses and adenoviruses,encode proteins that form complexes with and in-activate the p53 and retinoblastoma (RB) proteins.This convergent evolution reflects the common needof these viruses to inactivate these two importantregulators of cell cycle progression and cell survival.Polyomavirus, a close relative of SV40, is different.Its large T protein complexes only with RB, not withp53. We have examined whether this is com-pensated by the frequent appearance of p53mutations in polyomavirus-induced tumours. Wetested the p53 status of 15 polyomavirus-inducedsarcomas. Two sarcomas were p53-negative whilesix carried mutant p53. Another six sarcomasexpressed low levels of wild-type p53. One tumourexpressed high levels of wild-type p53 protein asshown by DNA sequencing and immunofluorescence
IntroductionThe p53 gene is often inactivated by point mutation or
deletion in a wide variety of human and rodent tumours(Greenblatt et al., 1994). Moreover, p53 null mice developspontaneous tumours with a dramatically increased incidence(Donehower et al., 1992), supporting the notion that p53 is acrucial tumour suppressor. p53 is expressed at low levels undernormal conditions, but accumulates due to protein stabilizationin response to DNA damage (Maltzman & Czyzyk, 1984 ;Kastan et al., 1991 ; Fritsche et al., 1993) or induction of cellproliferation by the E1A and c-myc oncogenes (Debbas &White, 1993 ; Hermeking & Eick, 1994 ; Lowe et al., 1994 ;Wagner et al., 1994). The increased p53 levels cause cell cyclearrest in the G
"phase or cell death through apoptosis (for a
review see Selivanova & Wiman, 1995).The p53 protein is a sequence-specific transcription factor
that transactivates genes, such as those encoding WAF1}p21,
Author for correspondence: Klas G. Wiman.
Fax 46 8 33 04 98. e-mail Klas.Wiman!mtc.ki.se
staining. MDM2 amplification was not detected inany of the tumours, but Northern blotting showedthat MDM2 was overexpressed in at least twotumours that expressed wild-type p53 and in onetumour that expressed both wild-type and mutantp53. Treatment with the DNA-damaging agentmitomycin C caused p53 protein accumulation fol-lowed by induction of MDM2 and WAF1/p21 mRNAin four of the tumours expressing wild-type p53,indicating that p53-mediated transcriptional acti-vation was unaltered in these tumours. However,p53-mediated transactivation of WAF1/p21 wasimpaired in the wild-type p53-expressing tumoursthat expressed elevated levels of MDM2. Theseresults demonstrate that p53 mutation and inacti-vation are frequently but not invariably involved inpolyomavirus-induced tumorigenesis.
MDM2, GADD45, cyclin G, Bax and Fas (reviewed by Ko &Prives, 1996). Transactivation of WAF1}p21 is important forp53-induced cell cycle arrest but is dispensable for p53-inducedapoptosis (Deng et al., 1995 ; Waldman et al., 1995). However,transactivation of bax and fas may facilitate apoptosis (see Ko& Prives, 1996). p53-mediated transactivation of MDM2constitutes an autoregulatory feedback loop that modulatesthe activity of p53 (Wu et al., 1993). The MDM2 protein caninhibit p53-mediated transcriptional activation by binding anN-terminal region of p53 that coincides with the acidictransactivation domain (Oliner et al., 1993). Overexpression ofMDM2 increases the tumorigenicity of NIH3T3 cells andovercomes wild-type p53-mediated suppression of cell growth(Fakharzadeh et al., 1991 ; Finlay, 1993). Amplification andoverexpression of this gene are frequently observed in humansarcomas (Oliner et al., 1992).
More recent data have indicated that the early embryoniclethality of MDM2-deficient mice can be rescued by lack ofp53, suggesting that MDM2 is a vital negative regulator ofp53 activity during embryonic development (de Oca Luna etal., 1995 ; Jones et al., 1995).
0001-4432 # 1997 SGM IJD

W. Qian and othersW. Qian and others
p53 can also be inactivated at the protein level bycomplexing with virus oncoproteins, including simian virus 40(SV40) large T (LT), adenovirus E1B 55 kDa protein (55Kprotein) and human papillomavirus (HPV)-16}18 E6 proteins(reviewed by Levine, 1990). The fact that the virus proteindomains that interact with p53 are essential for their trans-formation action indicates that inactivation of p53 is criticalfor the transformation capacity of these viruses. Virusoncoprotein-mediated inactivation of p53 may have evolvedto ensure S phase entry and virus DNA replication followinginfection, of which transformation is merely a pathologicalconsequence.
Polyoma is a DNA tumour virus that can induce a widevariety of tumours in newborn mice, hamsters, rats and rabbits(Grodzicker & Hopkins, 1981 ; Topp et al., 1981). The earlyregion of the polyomavirus genome encodes three T antigens,LT, middle T (MT) and small T (ST), with molecular masses of100, 55 and 22 kDa, respectively. The polyomavirus LTantigen shares 60% amino acid sequence identity and a widerange of biological and biochemical properties with the LTantigen encoded by the closely related SV40 (Soeda et al.,1980 ; Turler, 1980 ; Depamphilis & Bradley, 1986). However,polyoma LT differs from SV40 LT in that it cannot causemalignant transformation by itself, although it can immortalizeprimary rat embryo fibroblasts (Asselin & Bastin, 1985 ; Cowieet al., 1986 ; Jat & Sharp, 1986). Moreover, while both SV40and polyoma LT form complexes with the retinoblastoma (RB)protein, SV40 LT but not polyoma LT binds p53 (DeCaprio etal., 1988 ; Wang et al., 1989 ; Dyson et al., 1990 ; Lin &Simmons, 1991). The polyoma MT antigen can transformestablished cells in culture, but induction of tumours in rats orefficient transformation of primary rat embryo fibroblastsusually requires cooperation with either polyoma LT or ST(Treisman et al., 1981 ; Asselin et al., 1983, 1986). There is noevidence indicating that MT forms a physical complex withp53.
If polyomavirus does not encode any function that blocksp53, and inactivation of the p53 pathway is a requirement forthe development of polyomavirus-induced tumours, onewould expect to find p53 mutations and}or deletions, MDM2overexpression or other lesions that disrupt p53 function athigh frequency in polyomavirus-induced sarcomas. Here, wehave examined the status of the p53 and MDM2 genes as wellas the transcriptional transactivation function of p53 in 15polyomavirus-induced sarcomas and sarcoma cell lines. Ourdata demonstrate that p53 inactivation by point mutation,deletion or MDM2 overexpression occurs frequently inpolyomavirus-induced sarcomas.
Methods+ Tumours and cell culture. Three polyomavirus-induced sarcomacell lines from C3H}Bi Da mice, A1633, A4412 and A4455, and two suchlines from (BALB}cby J¬C3H}Bi Da)F
"mice, A4300 and A3377, were
provided by Thomas Benjamin, Harvard Medical School, USA. The other
polyomavirus-induced sarcomas (see Table 1) were established in ourlaboratory by inoculation of newborn mice (ABY, ACA, ASW, A}sn,CBA and C75BL strains) with polyomavirus. SEAC and SEWE are
primary tumours. The other tumours have been passaged in adult micebetween 5 and 47 times. Cells were grown at 37 °C in 5% CO
#in
Iscove’s medium supplemented with 10% heat-inactivated foetal calfserum, 1000 units}ml penicillin, 100 µg}ml streptomycin and 2 mM -glutamine. For mitomycin C (MMC) treatment, the cells were fed withthe same medium containing 5 µg}ml MMC (Sigma) and harvested 0, 5
or 18 h later.
+ Metabolic labelling and immunoprecipitation. Radiolabellingwith [$&S]methionine and immunoprecipitation were performed as
described by Wiman et al. (1991). The antibodies used were PAb246,specific for wild-type p53, PAb240, specific for mutant p53, and PAb421,recognizing both wild-type and mutant p53. All anti-p53 antibodies werefrom Oncogene Science.
+ Immunofluorescence staining. Monolayer cells were grown onsterile coverslips. Cells growing in suspension were spun on slides witha Cytospin cytocentrifuge at 900 r.p.m. for 10 min. Cells were fixed inmethanol-acetone (1 :1) at ®20 °C for 10 min, rehydrated with balancedsalt solution (BSS) for 60 min, and incubated in blocking solution (2%
bovine serum albumin, 5% glycerol, 0±2% Tween 20 and 0±02% azide inPBS) for 30 min. The anti-p53 antibody PAb421 or the anti-polyomavirusLT antibody 711 (provided by Stephen Dilworth, Royal PostgraduateMedical School, London, UK) was then incubated for 60 min at a 1 :50dilution in this blocking solution. After washing three times with BSS, thecells were incubated for 30 min with FITC-conjugated rabbit anti-mouse
IgG (Dako) or with FITC-conjugated rabbit anti-rat IgG (Dako) diluted1 :20 in the blocking solution. For double layer fluorescent staining, FITC-conjugated swine anti-rabbit IgG (Dako) was added at a dilution of 1 :200for an additional 30 min. All incubations were carried out at roomtemperature. The second antibody solution always contained 0±4 µg}mI
Hoechst 33258 (Farbwerke Hoechst) for DNA staining and to monitorthe absence of mycoplasma contamination. Finally, the cells were washedthree times with BSS and mounted with BSS-glycerol (1 :1) containing2±5% 1,4-diazabicyclo[2.2.2]octane (Sigma) anti-fading reagent.
+ PCR and DNA sequencing. Total RNA was extracted as describedby Chomczynski & Sacchi (1987). First-strand cDNA was synthesizedfrom 1–2 µg total RNA using avian myeloblastosis virus reversetranscriptase (Seikagaku America). PCR amplification of cDNA wascarried out using primers E4F (5« CACTGCATGGACGATCTGTT 3«)and E9RB (5« CGGATCTTGAGGGTGAAATA 3«), flanking exons 4–9in p53 and producing an 875 bp fragment that contains amino acidcodons 97–320. Primer E9RB was biotinylated at the 5« end. Thebiotinylated strand of the PCR product was isolated with streptavidin-coated iron beads (Dynal) and sequenced using the two primers E4F2 (5«GCCCCTGTCATCTTTTGTCCC 3«) and E6F (5« CTCCTCCCCAGC-
ATCTTATCC 3«), located in exons 4 and 6, respectively. DNAsequencing was performed using the Solid Phase sequencing method(Hultman et al., 1991) and using the Autoread kit together withfluorescein-labelled dATP on an ALF automated sequencer (Pharmacia)(Voss et al., 1992).
+ Southern blotting. High molecular mass DNA was prepared asdescribed by Sambrook et al. (1989). Five µg DNA from each sample wasdigested overnight with 25 units EcoRI. After electrophoresis on 0±8%agarose gels in TAE buffer, the DNA was transferred to Hybond-N nylon
filters (Amersham) and hybridized with a $#P-labelled mouse MDM2cDNA probe.
IJE

p53 status of polyomavirus-induced tumoursp53 status of polyomavirus-induced tumours
Table 1. p53 and MDM2 status of polyomavirus-induced tumours
ImmunoprecipitationTumour and IF* DNA sequencing Northern blot†
Group cell line p53 status PAb240 PAb421 PAb246 (PAb421) (exons 4–9) (MDM2 overexpression)
A SEBB Negative ® ® ® ® 1±2SECA Negative ® ® ® ® 0±4
B SESO Wild-type ® ® Wild-type 4±9SEBA Wild-type ® ® Wild-type 1±8SEAB Wild-type ® ® Wild-type 1±5SE7E Wild-type ® ® Wild-type 2±5A3377 Wild-type ® ® Wild-type 1±2A1633 Wild-type ® ® Wild-type 1±7
C SEYF Wild-type ® Wild-type 6±3D SEAC Mutant ® Mutant 0±6
A4412 Mutant ® Mutant 0±7SEWA-NA Mutant ® 0±5SEWA-ADH Mutant Mutant
E A4300 Mut and Wt ® Mutant and wild-type 6±5SEWE Mut and Wt 1±4A4455 Mut and Wt 0±7
* IF, immunofluorescence staining. ®, ! 5% p53-positive cells ; , " 80% p53-positive cells.† MDM2 mRNA levels were quantified by PhosphoImager analysis and are shown as fold overexpression relative to MDM2 mRNA levels inNIH3T3 cells., Not determined.
+ RNA preparation and Northern blot analysis. RNA was
prepared from approximately 5¬10' asynchronously growing cells as
described by Dooley et al. (1989), with the following modifications. To
release the nuclei, the cells were lysed in buffer I (1% NP40 ; 150 mM
NaCl ; 100 mM Tris-HCl, pH 7±5 ; 5 mM EDTA) containing 1 mM of the
RNase inhibitor aurintricarboxylic acid (ATA) (Sigma) (Williams &
Krawisz, 1989). The nuclei were removed by centrifugation and the
supernatant was mixed with an equal volume of buffer II (7 M urea ; 1%
SDS; 350 mM NaCl ; 10 mM EDTA; 10 mM Tris-HCl, pH 7±5), followed
by phenol-chloroform extraction and precipitation with 1±3 vol. 2-
propanol. ATA at a final concentration of 0±1 mM was added to 20¬SSC during transfer. Ten µg RNA from each sample was fractionated on
formaldehyde-agarose gels, transferred to nylon filters, and subsequently
hybridized overnight with a mouse MDM2 cDNA probe or a mouse
WAF1}p21 cDNA probe at 42 °C in 50% formamide, 6¬ SSC, 0±2%
SDS, 5¬ Denhardt’s solution, 200 µg herring sperm DNA and 0±1 mM
ATA. The integrity of the RNA and loading differences were assessed by
ethidium bromide staining and}or by hybridization with a human
GAPDH cDNA probe.
+ Western blot analysis. Total cell extracts were prepared by direct
lysis in Laemmli buffer without bromphenol blue. Protein concentration
was determined using the Bio-Rad protein assay kit. Samples containing
50 µg protein were separated on 10% SDS-PAGE gels and transferred to
Hybond-C extra nitrocellulose membranes (Amersham). Blocking and
incubation with antibodies were performed in 5% milk in PBS.
Immunodetection using the ECL system (Amersham) was performed
according to the manufacturer’s instructions. The antibodies used were
the rat monoclonal antibody 711 against the polyomavirus LT antigen
and the rat monoclonal antibody 815 against the polyomavirus MTantigen, provided by Thomas Benjamin, Harvard Medical School, USA.
Resultsp53 gene status and p53 protein levels
We examined p53 in 15 polyomavirus-induced sarcomasusing immunoprecipitation with the antibodies PAb421(recognizing both wild-type and mutant p53), PAb240 (specificfor mutant p53) and PAb246 (specific for wild-type p53),immunofluorescence staining with PAb421, and DNAsequencing of exons 4–9. The results are summarized in Table1. Two tumours, SEBB and SECA, were completely p53-negative. Six tumours expressed low levels of wild-type p53.One tumour, SEYF, expressed elevated levels of wild-type p53.The remaining six tumours carried mutant p53. The mutant-specific PAb240 failed to precipitate p53 from SEAC andA4300, and PAb246 reacted with p53 in both tumours ;however, both SEAC and A4300 expressed high levels of p53(Figs 1 and 2 ; data not shown). DNA sequencing revealed amutation in codon 272 in SEAC, resulting in a Pro!Hissubstitution. No wild-type sequence was detected, indicatingthat only the mutant allele was present. DNA sequencing alsorevealed that A4300 carried one wild-type p53 allele and onemutant allele with a mutation in codon 142 that resulted in aSer!Arg substitution and a mutation in codon 147 that
IJF
W. Qian and othersW. Qian and others
69
46
240 421 246PAb ...
SEBB
240 421 246
SE7E
240 421 246
A4412
240 421 246
A4455
p53
kDa
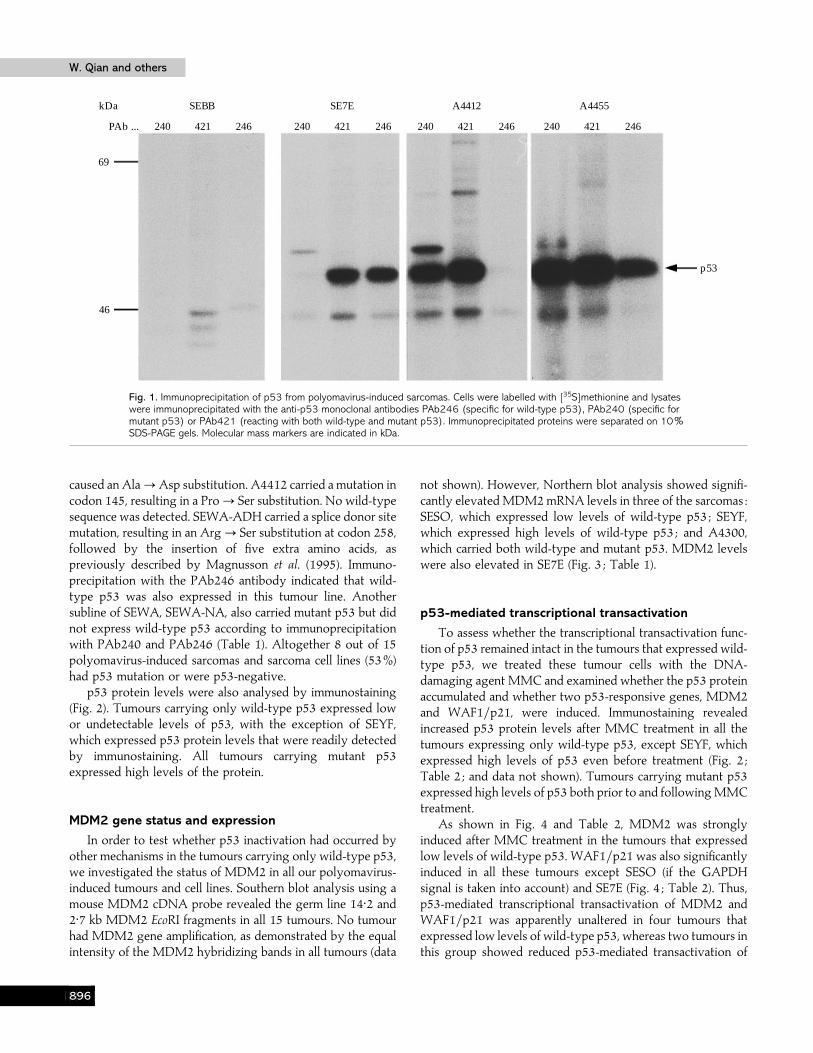
Fig. 1. Immunoprecipitation of p53 from polyomavirus-induced sarcomas. Cells were labelled with [35S]methionine and lysateswere immunoprecipitated with the anti-p53 monoclonal antibodies PAb246 (specific for wild-type p53), PAb240 (specific formutant p53) or PAb421 (reacting with both wild-type and mutant p53). Immunoprecipitated proteins were separated on 10%SDS-PAGE gels. Molecular mass markers are indicated in kDa.
caused an Ala!Asp substitution. A4412 carried a mutation incodon 145, resulting in a Pro! Ser substitution. No wild-typesequence was detected. SEWA-ADH carried a splice donor sitemutation, resulting in an Arg! Ser substitution at codon 258,followed by the insertion of five extra amino acids, aspreviously described by Magnusson et al. (1995). Immuno-precipitation with the PAb246 antibody indicated that wild-type p53 was also expressed in this tumour line. Anothersubline of SEWA, SEWA-NA, also carried mutant p53 but didnot express wild-type p53 according to immunoprecipitationwith PAb240 and PAb246 (Table 1). Altogether 8 out of 15polyomavirus-induced sarcomas and sarcoma cell lines (53%)had p53 mutation or were p53-negative.
p53 protein levels were also analysed by immunostaining(Fig. 2). Tumours carrying only wild-type p53 expressed lowor undetectable levels of p53, with the exception of SEYF,which expressed p53 protein levels that were readily detectedby immunostaining. All tumours carrying mutant p53expressed high levels of the protein.
MDM2 gene status and expression
In order to test whether p53 inactivation had occurred byother mechanisms in the tumours carrying only wild-type p53,we investigated the status of MDM2 in all our polyomavirus-induced tumours and cell lines. Southern blot analysis using amouse MDM2 cDNA probe revealed the germ line 14±2 and2±7 kb MDM2 EcoRI fragments in all 15 tumours. No tumourhad MDM2 gene amplification, as demonstrated by the equalintensity of the MDM2 hybridizing bands in all tumours (data
not shown). However, Northern blot analysis showed signifi-cantly elevated MDM2 mRNA levels in three of the sarcomas :SESO, which expressed low levels of wild-type p53 ; SEYF,which expressed high levels of wild-type p53 ; and A4300,which carried both wild-type and mutant p53. MDM2 levelswere also elevated in SE7E (Fig. 3 ; Table 1).
p53-mediated transcriptional transactivation
To assess whether the transcriptional transactivation func-tion of p53 remained intact in the tumours that expressed wild-type p53, we treated these tumour cells with the DNA-damaging agent MMC and examined whether the p53 proteinaccumulated and whether two p53-responsive genes, MDM2and WAF1}p21, were induced. Immunostaining revealedincreased p53 protein levels after MMC treatment in all thetumours expressing only wild-type p53, except SEYF, whichexpressed high levels of p53 even before treatment (Fig. 2 ;Table 2 ; and data not shown). Tumours carrying mutant p53expressed high levels of p53 both prior to and following MMCtreatment.
As shown in Fig. 4 and Table 2, MDM2 was stronglyinduced after MMC treatment in the tumours that expressedlow levels of wild-type p53. WAF1}p21 was also significantlyinduced in all these tumours except SESO (if the GAPDHsignal is taken into account) and SE7E (Fig. 4 ; Table 2). Thus,p53-mediated transcriptional transactivation of MDM2 andWAF1}p21 was apparently unaltered in four tumours thatexpressed low levels of wild-type p53, whereas two tumours inthis group showed reduced p53-mediated transactivation of
IJG

p53 status of polyomavirus-induced tumoursp53 status of polyomavirus-induced tumours
Tumourcells
–MMC +MMCp53status
SEBB –
A1633 Wt
SEYF Wt
SEAC Mutant
A4455 Mutantand Wt
Fig. 2. p53 protein accumulation inresponse to MMC treatment.Polyomavirus-induced tumour cells werestained with the anti-p53 monoclonalantibody PAb421 at 0 h (®MMC) or 5 h(MMC) after MMC treatment. Wt, wild-type. Magnification : 12±5¬63.
MDM2
GAPDH
SP2/
0SE
SOSE
BASE
AB
SE7E
A33
77A
1633
SEYF
A43
00N
IH3T
3
Fig. 3. MDM2 expression in polyomavirus-induced tumours. Ten µg RNAwas fractionated on a formaldehyde-agarose gel, transferred to nylon filter,and hybridized with a mouse MDM2 cDNA probe, or a human GAPDHcDNA probe as a control for equal loading.
WAF1}p21. SEYF, which expressed high levels of MDM2 andWAF1}p21 prior to MMC treatment, showed only a minorinduction of both genes. In contrast, all tumours carrying
mutant p53, except A4300, expressed low or undetectablelevels of WAF1}p21 and MDM2, and in most cases showedlittle or no induction of these genes after MMC treatment (Fig.4 ; Table 2). A4300 expressed relatively high levels ofWAF1}p21 and MDM2 both prior to and following treatment.WAF1}p21 was markedly induced in SEAC, but the absolutelevels were very low (Fig. 4). SEBB and SECA, the two p53-negative sarcomas, showed a minor induction of both MDM2and WAF1}p21 after MMC treatment.
SinceWAF1}p21 is probably themain downstream effectorof p53-induced cell cycle arrest, it is conceivable that this genemight be inactivated by some mechanism in the polyomavirus-induced tumours that express wild-type p53. However, bothSouthern blot analysis (not shown) and the Northern blotanalysis shown in Fig. 4 demonstrated that WAF1}p21 wasnot deleted in any of the tumours. Furthermore, the Northernblot analysis revealed normal size WAF1}p21 transcripts in alltumours studied.
Expression of polyomavirus LT and MT antigens
Although neither polyomavirus LT nor MT can bind p53(Villareal & Fan, 1989 ; Wang et al., 1989), it remains possible
IJH

W. Qian and othersW. Qian and others
Table 2. Accumulation of p53 and induction of WAF1/p21 and MDM2 after MMC treatment
Northern blot†Tumour and IF*
Group cell line p53 status (p53 accumulation) MDM2 induction WAF1 induction
A SEBB Negative ® 1±5 3±0SECA Negative ® 2±8 4±4
B SESO Wild-type 7±0 1±9SEBA Wild-type 17±9 6±2SEAB Wild-type 22±9 23±5SE7E Wild-type 11±1 4±0A3377 Wild-type 43±4 10±7A1633 Wild-type 17±8 11±5
C SEYF Wild-type ‡ 2±0 1±7D SEAC Mutant ‡ 4±5 11±3
A4412 Mutant ‡ 1±5 4±0SEWA-NA Mutant ‡ 1±9 3±7SEWA-ADH Mutant
E A4300 Mutant and wild-type ‡ 3±1 1±8SEWE Mutant and wild-type ‡ 1±2 1±6A4455 Mutant and wild-type ‡ 1±5 1±0
* Immunofluorescence staining. ®, No p53-positive cells ; , strong p53 staining in the majority of cells.† MDM2 and WAF1}p21 mRNA levels after MMC treatment were quantified by PhosphoImager analysis and are shown as fold inductionrelative to the levels prior to treatment.‡ p53 protein levels were very high, even prior to treatment., Not determined.
SEA
B
SE7E
A33
77
A16
33
SEYF
A43
00WAF1/p21
MDM2
GAPDH
0 18 0 18 0 18 0 18 0 18 0 18 0 18 0 18 0 18 0 18 0 18 0 18
SEBA
SESO
SEBB
SECA
SEA
C
A44
55
MMC ...(h)
Fig. 4. WAF1/p21 and MDM2 induction in response to MMC treatment. Ten µg RNA was fractionated on a formaldehyde-agarose gel, transferred to nylon filter, and hybridized with mouse WAF1/p21 or MDM2 cDNA probes, or a human GAPDHcDNA probe as a control for equal loading.
IJI
p53 status of polyomavirus-induced tumoursp53 status of polyomavirus-induced tumours
LT
220
97
66
46
kDa
(a)
NIH3T
3
SEBB
SESO
SEBA
A3377
SEW
A-NA
A4412
A4300
SECA
SEAB
SE7E
A1633
SEYF
SEAC
SEW
EA44
55
SE7E A1633 SEAC A4455
SEAB SEYF SECA SEWE
66
46
kDa
(b)
NIH3T
3
SEBB
SESO
SEBA
A3377
SEW
E
A4412
A4300
SECA
SEAB
SE7E
A1633
SEYF
SEAC
SEW
A-NA
A4455
MT
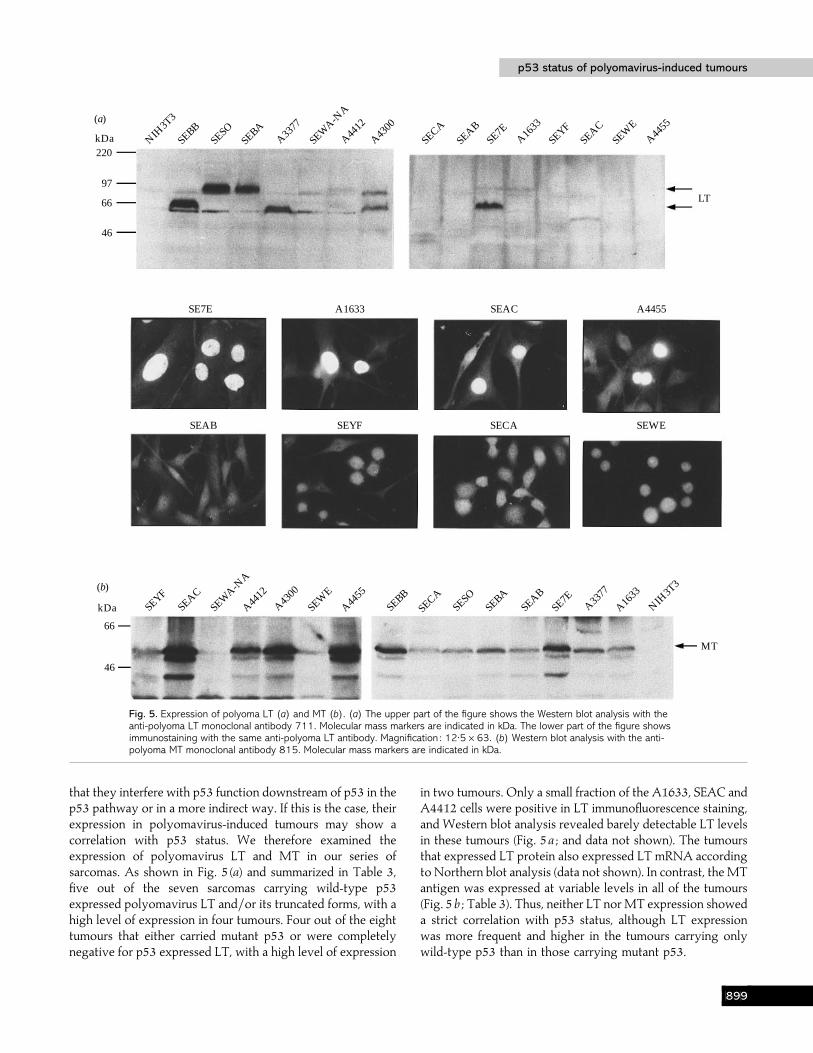
Fig. 5. Expression of polyoma LT (a) and MT (b). (a) The upper part of the figure shows the Western blot analysis with theanti-polyoma LT monoclonal antibody 711. Molecular mass markers are indicated in kDa. The lower part of the figure showsimmunostaining with the same anti-polyoma LT antibody. Magnification : 12±5¬63. (b) Western blot analysis with the anti-polyoma MT monoclonal antibody 815. Molecular mass markers are indicated in kDa.
that they interfere with p53 function downstream of p53 in thep53 pathway or in a more indirect way. If this is the case, theirexpression in polyomavirus-induced tumours may show acorrelation with p53 status. We therefore examined theexpression of polyomavirus LT and MT in our series ofsarcomas. As shown in Fig. 5 (a) and summarized in Table 3,five out of the seven sarcomas carrying wild-type p53expressed polyomavirus LT and}or its truncated forms, with ahigh level of expression in four tumours. Four out of the eighttumours that either carried mutant p53 or were completelynegative for p53 expressed LT, with a high level of expression
in two tumours. Only a small fraction of the A1633, SEAC andA4412 cells were positive in LT immunofluorescence staining,and Western blot analysis revealed barely detectable LT levelsin these tumours (Fig. 5a ; and data not shown). The tumoursthat expressed LT protein also expressed LT mRNA accordingto Northern blot analysis (data not shown). In contrast, the MTantigen was expressed at variable levels in all of the tumours(Fig. 5b ; Table 3). Thus, neither LT nor MT expression showeda strict correlation with p53 status, although LT expressionwas more frequent and higher in the tumours carrying onlywild-type p53 than in those carrying mutant p53.
IJJ

W. Qian and othersW. Qian and others
Table 3. Polyomavirus LT antigen (PyLT) and MT antigen (PyMT) expression
PyLT expressionTumour and PyMT expression
Group cell line p53 status IF* Northern Western (Western)
A SEBB Negative SECA Negative ® ® ®
B SESO Wild-type SEBA Wild-type SEAB Wild-type ® ® ® SE7E Wild-type A3377 Wild-type A1633 Wild-type few
C SEYF Wild-type ® ® ® D SEAC Mutant few
A4412 Mutant few SEWA-NA Mutant ® ® ® SEWA-ADH Mutant
E A4300 Mutant and wild-type SEWE Mutant and wild-type ® ® ® A4455 Mutant and wild-type few ®
* IF, Immunofluorescence staining. ®, No PyLT-positive cells ; , majority of the cells are PyLT-positive, except where indicated.
DiscussionThe p53 and RB proteins play critical roles in the regulation
of cell proliferation and survival. RB is probably an importantcomponent of the restriction point, R, in the G
"phase of the
cell cycle (reviewed by Weinberg, 1995). p53 can induceexpression of the cyclin-cdk inhibitor WAF1}p21, whichinhibits cyclin D-CDK4}6-mediated phosphorylation of RB,and thus prevents inactivation of the gate-keeping function ofRB in G
". In addition, p53 can induce cell death by apoptosis.
The adenovirus E1A protein that binds and inactivates RBinduces p53 protein accumulation followed by apoptosis(Debbas & White, 1993 ; Lowe et al., 1994). The p53-triggeredapoptosis in response to E1A is inhibited by the adenovirusE1B 19 kDa (19K) and 55K proteins (Debbas & White, 1993).This reflects the general need of DNA tumour viruses to bothstimulate progression of the infected cell into S phase byblocking RB, and to prevent apoptosis triggered by theaccumulation of p53 in response to induction of cell cycleprogression.
Like the LT antigen encoded by the closely related SV40virus, and the E1A and E7 proteins encoded by oncogenicsubtypes of adenovirus and HPV, respectively, the polyoma-virus LT antigen targets and inactivates the RB protein. This isan essential requirement for the ability of the virus to inducehost DNA synthesis and timely synthesis of virus DNA, andto immortalize primary rat embryo fibroblasts (Dyson et al.,1990 ; Larose et al., 1991 ; Freund et al., 1992, 1994). However,unlike the other three viruses, polyomavirus does not encode
any protein that complexes with p53 and directly blocks itsfunction. Polyomavirus LT does not bind p53 (Wang et al.,1989), and there are no published data indicating thatpolyomavirus MT and ST might bind p53. This raises thepossibility that polyomavirus has evolved other mechanismsto escapewild-type p53-induced cell cycle arrest and apoptosis.Despite their similar genomic structure, polyomavirus, but notSV40, encodes the MT antigen, a transforming protein knownto bind several cellular proteins, including pp60-c-Src, pp62-c-Yes, pp59-c-Fyn, PP2A and PI-3 kinase, and capable ofactivating the mitogen-activated protein kinase pathway(Dilworth, 1995). Conceivably, MT could act as a downstreaminhibitor of the p53 pathway.
The question of whether polyomavirus interferes with thep53 pathway is relevant for the assessment of the p53 status ofpolyomavirus-induced tumours. If polyomavirus does notblock p53 function and if inactivation of p53 function isimportant or even essential for the development ofpolyomavirus-induced tumours, p53 mutations and}ordeletions would be expected to occur at high frequency inpolyomavirus-induced tumours. We found that 6 out of 15polyomavirus-induced sarcomas expressed low levels of wild-type p53 and retained p53-mediated transcriptional trans-activation of at least the p53-responsive MDM2 genefollowing MMC treatment (Tables 1 and 2). Four tumours inthis group retained p53-mediated transactivation of WAF1}p21 as well. The low induction of WAF1}p21 in SESO andSE7E indicates that the transcriptional transactivation functionof p53 is partially deficient in these two tumours. It is
JAA

p53 status of polyomavirus-induced tumoursp53 status of polyomavirus-induced tumours
noteworthy that SESO overexpressed MDM2, which mayinterfere with p53 function by binding the N-terminaltransactivation domain of p53 (Oliner et al., 1993). MDM2levels were lower in SE7E, but still higher than the MDM2levels in the other tumours that expressed low levels of wild-type p53 (Table 1). SEYF, which expressed elevated levels ofwild-type p53, also overexpressed the p53 antagonist MDM2.WAF1}p21 and MDM2 were only weakly induced followingDNA damage in this tumour.
Eight out of the 15 tumours (53%) carried mutant p53 orwere p53-negative (Table 1), and in general showed nosignificant induction of WAF1}p21 and MDM2 followingMMC treatment. Although WAF1}p21 mRNA levelsincreased following MMC treatment in SEAC, the levels weremuch lower than those observed in the tumours that expressedwild-type p53. The moderate induction of MDM2 andWAF1}p21 in several of the tumours lacking wild-type p53 isconsistent with work demonstrating that both genes are alsoregulated by p53-independent mechanisms (Barak et al., 1994 ;Michieli et al., 1994 ; Parker et al., 1995).
Nevertheless, the fact that all tumours that carried mutantp53 were deficient in p53-mediated transcriptional trans-activation supports the notion that the p53 point mutationsidentified indeed inactivate p53 function. Altogether 8 out ofthe 15 tumours examined (53%) were p53-negative or carriedp53 mutation, and another 3 tumours were deficient intranscriptional transactivation of one or two p53-responsivegenes in response to p53 accumulation. This frequency of p53mutation and}or inactivation is similar to that found in manynon-virus-induced human and rodent tumours (Greenblatt etal., 1994), indicating that inactivation of p53 is important forthe development of polyomavirus-induced sarcomas, and thatpolyomavirus is unlikely to block p53 function in a mannersufficient to reduce the selective pressure for p53 pointmutation or deletion during polyomavirus-induced tumori-genesis. It cannot be excluded, however, that polyomavirusinterferes with the apoptosis-inducing effect of p53 in thecontext of lytic infection, a possibility not addressed here.
The majority of sarcomas in our study have been passagedserially in vivo. p53 mutations may have occurred during thatpassage. However, the two primary tumours examined, SEACand SEWE, both carried mutant p53, indicating a selection forp53 mutation prior to in vivo passage.
Inactivation of p53 function may occur at different stagesduring multistep tumorigenesis or tumour progression indifferent tumours. Among the in vivo passaged tumours, two,SEWA and SEYF, are ascites sarcomas. SEWA carries mutantp53 whereas SEYF expresses high levels of MDM2 which canblock p53 function. Previous work indicated that anaerobicadaptation is a characteristic of ascites tumours but not oftumours in general (Warburg & Hiepler, 1952). Graeber et al.(1996) have recently reported that hypoxia induces apoptosisin tumour cells and that further genetic alterations, such as lossof p53 function or overexpression of Bcl-2, substantially
reduce hypoxia-induced cell death. Our data are thus consistentwith the possibility that p53 inactivation is an importantrequirement for the growth of originally polyomavirus-induced ascites sarcomas.
Polyomavirus LT, both full-length and truncated forms,was expressed in a subset of the sarcomas studied here,including sarcomas carrying only wild-type p53, sarcomascarrying mutant p53 and p53-negative sarcomas. In contrast,MT was expressed in all tumours examined, albeit at variablelevels. Thus, neither LT nor MT expression correlated withp53 status. This further supports the idea that neither LT norMT inactivate p53 in the course of polyomavirus-inducedtumour development, although at least MT is required forexpression and maintenance of the neoplastic phenotype.
We thank Donna L. George, University of Pennsylvania, for themouse MDM2 cDNA, Thomas L. Benjamin, Harvard Medical School, forthe polyomavirus-induced sarcoma cell lines and the anti-polyoma MTmonoclonal antibody 815, Stephen M. Dilworth, Royal PostgraduateMedical School, London, for the anti-polyoma LT monoclonal antibody711 and the polyomavirus LT cDNA, Bert Vogelstein, Johns HopkinsOncology Center, for the mouse WAF1}p21 cDNA, and Yisong Wang,Laszlo Szekely and Galina Selivanova, Microbiology and TumourBiology Center, Karolinska Institute, for fruitful discussions and technicalhelp. This work was supported by grants to K.G.W. from the SwedishCancer Society, Magn. Bergvalls Stiftelse and AI ke Wibergs Stiftelse.W.Q., E.K. and K.P. are recipients of fellowships from the ConcernFoundation, Los Angeles, and the Cancer Research Institute, New York.
ReferencesAsselin, C. & Bastin, M. (1985). Sequences from polyomavirus andsimian virus 40 large T genes capable of immortalizing primary ratembryo fibroblasts. Journal of Virology 56, 958–968.
Asselin, C., Gelinas, C. & Bastin, M. (1983). Role of the threepolyomavirus early proteins in tumorigenesis. Molecular and CellularBiology 3, 1451–1459.
Asselin, C., Vass-Marego, J. & Bastin, M. (1986). Mutation in thepolyomavirus genome that activates the properties of large T associatedwith neoplastic transformation. Journal of Virology 57, 165–172.
Barak, Y., Gottlieb, E., Juven-Gershon, T. & Oren, M. (1994).Regulation of mdm2 expression by p53 : alternative promoters producetranscripts with nonidentical translational potential. Genes & Development8, 1739–1749.
Chomczynski, P. & Sacchi, N. (1987). Single-step method of RNAisolation by acid guanidinium thiocyanate-phenol-chloroform extraction.Analytical Biochemistry 162, 156–159.
Cowie, A., de Villiers, J. & Kamen, R. (1986). Immortalization of ratembryo fibroblasts by mutant polyomavirus large T antigen deficient inDNA binding. Molecular and Cellular Biology 6, 4344–4352.
Debbas, M. & White, E. (1993). Wild type p53 mediates apoptosis byE1A, which is inhibited by E1B. Genes & Development 7, 546–554.
DeCaprio, J. A., Ludlow, J. W., Figge, J., Shew, J. Y., Huang, C. M., Lee,W. H., Marsilio, E., Paucha, E. & Livingston, D. M. (1988). SV40 largeT tumor antigen forms a specific complex with the product of theretinoblastoma susceptibility gene. Cell 54, 275–283.
Deng, C., Zhang, P., Harper, J. W., Elledge, S. J. & Leder, P. (1995).Mice lacking p21-CIP1}WAF1 undergo normal development, but aredefective in G1 checkpoint control. Cell 82, 675–684.
JAB

W. Qian and othersW. Qian and others
de Oca Luna, R. M., Wagner, D. S. & Lozano, G. (1995). Rescue of earlyembryonic lethality in mdm-2 deficient mice by deletion of p53. Nature378, 203–206.
Depamphilis, M. L. & Bradley, M. K. (1986). Replication of SV40 andpolyoma virus chromosomes. In The Papovaviridae, vol. 1, pp. 99–244.Edited by N. P. Salzman. New York : Plenum.
Dilworth, S. M. (1995). Polyoma virus middle T antigen : meddler ormimic? Trends in Microbiology 3, 31–35.
Donehower, L. A., Harvey, M., Slagle, B. L., McArthur, M. J.,Montgomery, C. A., Jr, Butel, J. S. & Bradley, A. (1992). Mice deficientfor p53 are developmentally normal but susceptible to spontaneoustumors. Nature 356, 215–221.
Dooley, S., Ehrhart, E., Radtke, J., Unteregger, G. & Blin, N. (1989). Aprocedure for simultaneous isolation of undegraded RNA, DNA andnuclear proteins suitable for interaction analysis in homologous systems.Methods in Molecular and Cellular Biology 1, 95–105.
Dyson, N., Bernards, R., Friend, S. H., Gooding, L. R., Hassell, J. A.,Major, E. O., Pipas, J. M., Van Dyke, T. & Harlow, E. (1990). Large Tantigens of many polyomaviruses are able to form complexes with theretinoblastoma protein. Journal of Virology 64, 1353–1356.
Fakharzadeh, S. S., Trusko, S. P. & George, D. L. (1991). Tumorigenicpotential associated with enhanced expression of a gene that is amplifiedin a mouse tumor cell line. EMBO Journal 10, 1565–1569.
Finlay, C. A. (1993). The mdm-2 oncogene can overcome wild-type p53suppression of transformed cell growth. Molecular and Cellular Biology 13,301–306.
Freund, R., Bronson, R. T. & Benjamin, T. L. (1992). Separation ofimmortalization from tumor induction with polyoma large T mutants thatfail to bind the retinoblastoma gene product. Oncogene 7, 1979–1987.
Freund, R., Bauer, P. H., Crissman, H. A., Bradbury, E. M. & Benjamin,T. L. (1994). Host range and cell cycle activation of properties ofpolyomavirus large T-antigen mutants defective in pRB binding. Journalof Virology 68, 7227–7234.
Fritsche, M., Haessler, C. & Brandner, G. (1993). Induction of nuclearaccumulation of the tumor-suppressor protein p53 by DNA-damagingagents. Oncogene 8, 307–318.
Graeber, T. G., Osmanian, C., Jacks, T., Housman, D. E., Koch, C. J.,Lowe, S. W. & Giaccia, A. J. (1996). Hypoxia-mediated selection of cellswith diminished apoptotic potential in solid tumors. Nature 379, 88–91.
Greenblatt, M. S., Bennett, W. P., Hollstein, M. & Harris, C. C. (1994).Mutations in the p53 tumor suppressor gene : clues to cancer etiologyand molecular pathogenesis. Cancer Research 54, 4855–4878.
Grodzicker, T. & Hopkins, N. (1981). Origins of contemporary DNAtumor virus research. In DNA Tumor Viruses : Molecular Biology of TumorViruses, pp. 7–8. Edited by J. Tooze. Cold Spring Harbor, NY: ColdSpring Harbor Laboratory.
Hermeking, H. & Eick, D. (1994). Mediation of c-Myc-induced apoptosisby p53. Science 265, 2091–2093.
Hultman, T., Bergh, S., Moks, T. & Uhle! n, M. (1991). Bidirectional solidphase sequencing of in vitro amplified plasmid DNA. BioTechniques 10,84–93.
Jat, P. S. & Sharp, P. A. (1986). Large T antigens of simian virus 40 andpolyomavirus efficiently establish primary fibroblasts. Journal of Virology59, 746–750.
Jones, S. N., Roe, A. E., Donehower, L. A. & Bradley, A. (1995). Rescueof embryonic lethality in Mdm2-deficient mice by absence of p53. Nature378, 206–208.
Kastan, M. B., Onyekwere, O., Sidransky, D., Vogelstein, B. & Craig,
R. W. (1991). Participation of p53 protein in the cellular response toDNA damage. Cancer Research 51, 6304–6311.
Ko, L. J. & Prives, C. (1996). p53 : puzzle and paradigm. Genes &Development 10, 1054–1072.
Larose, A., Dyson, N., Sullivan, M., Harlow, E. & Bastin, M. M. (1991).Polyomavirus large T mutants affected in retinoblastoma protein bindingare defective in immortalization. Journal of Virology 65, 2308–2313.
Levine, A. J. (1990). The p53 protein and its interactions with theoncogene products of the small DNA tumor viruses. Virology 177,419–426.
Lin, J. Y. & Simmons, D. T. (1991). The ability of large T antigen tocomplex with p53 is necessary for the increased life span and partialtransformation of human cells by simian virus 40. Journal of Virology 65,6447–6453.
Lowe, S. W., Jacks, T., Housman, D. E. & Ruley, H. E. (1994).Abrogation of oncogene-associated apoptosis allows transformation ofp53-deficient cells. Proceedings of the National Academy of Sciences, USA 91,2026–2030.
Magnusson, K. P., Minarovits, J., Klein, G. & Wiman, K. G. (1995). Asplice donor site mutation results in the insertion of five extra amino acidsinto p53 from SEWA mouse sarcoma cells. Gene 162, 231–234.
Maltzman, W. & Czyzyk, L. (1984). UV irradiation stimulates levels ofp53 cellular tumor antigen in nontransformed mouse cells. Molecular andCellular Biology 4, 1689–1694.
Michieli, P., Chedid, M., Lin, D., Pierce, J. H., Mercer, W. E. & Givol, D.(1994). Induction of WAF1}CIP1 by a p53-independent pathway.Cancer Research 54, 3391–3395.
Oliner, J. D., Kinzler, K. W., Meltzer, D. L., George, D. L. & Vogelstein,B. (1992). Amplification of a gene encoding a p53-associated protein inhuman sarcomas. Nature 358, 80–83.
Oliner, J. D., Pietenpol, J. A., Thiagalingams, S., Gyuris, J., Kinzler,K. W. & Vogelstein, B. (1993). Oncoprotein MDM2 conceals theactivation domain of tumor suppressor p53. Nature 362, 857–860.
Parker, S. B., Eichele, G., Zhang, P., Rawls, A., Sands, A. T., Bradley,A., Olson, E. N., Harper, J. W. & Elledge, S. J. (1995). p53-independentexpression of p21-Cip1 in muscle and other terminally differentiatingcells. Science 267, 1024–1027.
Sambrook, J., Fritsch, E. F. & Maniatis, T. (1989). Analysis and cloningof eukaryotic genomic DNA. In Molecular Cloning : A Laboratory Manual,2nd edn, pp. 9.16–9.19. Cold Spring Harbor, NY: Cold Spring HarborLaboratory.
Selivanova, G. & Wiman, K. G. (1995). p53 : a cell cycle regulatoractivated by DNA damage. Advances in Cancer Research 66, 143–180.
Soeda, E., Arrand, J. R., Smolar, N., Walsh, J. E. & Griffin, B. E. (1980).Coding potential and regulatory signals of the polyoma virus genome.Nature 283, 445–453.
Topp, W. C., Lane, D. P. & Poolack, R. (1981). Transformation bysimian virus 40 & polyoma virus. In DNA Tumor Viruses : MolecularBiology of Tumor Viruses, pp. 206–207. Edited by J. Tooze. Cold SpringHarbor, NY: Cold Spring Harbor Laboratory.
Treisman, R., Noval, U., Favaloro, J. & Kamen, R. (1981). Trans-formation of rat cells by an altered polyoma virus genome expressingonly the middle-T protein. Nature 292, 595–600.
Turler, H. (1980). The tumor antigens and the early functions ofpolyoma virus. Molecular and Cellular Biology 32, 63–93.
Villareal, L. P. & Fan, H. (1989). History and biological strategy ofpolyomavirus, adenovirus and papillomavirus. In Common Mechanisms ofTransformation by Small DNA Tumor Viruses, pp. 1–17. Edited by L. P.Villareal. Washington, DC: American Society for Microbiology.
JAC

p53 status of polyomavirus-induced tumoursp53 status of polyomavirus-induced tumours
Voss, H., Wiemann, S., Wirkner, S., Schwager, C., Zimmermann, J.,Stegemann, J., Erfle, H., Hewitt, N. A., Rupp, T. & Ansorge, W. (1992).Automated DNA sequencing system resolving 1000 bases withfluorescein-15*dATP as internal label. Methods in Molecular and CellularBiology 3, 153–155.
Wagner, A. J., Kokontis, J. M. & Hay, N. (1994). Myc-mediatedapoptosis requires wild type p53 in a manner independent of cell cyclearrest and the ability of p53 to induce p21-Waf1}Cip1. Genes &Development 8, 2817–2830.
Waldman, T., Kinzler, K. W. & Vogelstein, B. (1995). p21 is necessaryfor the p53-mediated G1 arrest in human cancer cells. Cancer Research 55,5187–5190.
Wang, E. H., Friedman, P. N. & Prives, C. (1989). The murine p53protein blocks replication of SV40 DNA in vitro by inhibiting theinitiation functions of SV40 large T antigen. Cell 57, 379–392.
Warburg, O. & Hiepler, E. (1952). Versuche mit Ascites-Tumorzellen.Zeitschrift fuX r Naturforschung Section B Chemical Sciences 7, 193–194.
Weinberg, R. A. (1995). The retinoblastoma protein and cell cyclecontrol. Cell 81, 323–330.
Williams, C. A. & Krawisz, B. R. (1989). Aurintricarboxylic acid as aribonuclease inhibitor. Methods in Molecular and Cellular Biology 1, 35–40.
Wiman, K. G., Magnusson, K. P., Ramqvist, T. & Klein, G. (1991).Mutant p53 detected in a majority of Burkitt lymphoma cell lines bymonoclonal antibody PAb240. Oncogene 6, 1633–1639.
Wu, X., Bayle, J. H., Olson, D. & Levine, A. J. (1993). The p53-mdm-2auto-regulatory feedback loop. Genes & Development 7, 1126–1132.
Received 26 September 1996; Accepted 15 November 1996
JAD




















