Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis
Upload
khangminh22Category
view
0download
0
ORIGINAL ARTICLE
PKA signaling drives mammary tumorigenesis through SrcAG Beristain1, SD Molyneux1,2, PA Joshi1,2, NC Pomroy1, MA Di Grappa1, MC Chang3, LS Kirschner4, GG Privé2, MA Pujana5 andR Khokha1,2,3
Protein kinase A (PKA) hyperactivation causes hereditary endocrine neoplasias; however, its role in sporadic epithelial cancers isunknown. Here, we show that heightened PKA activity in the mammary epithelium generates tumors. Mammary-restricted biallelicablation of Prkar1a, which encodes for the critical type-I PKA regulatory subunit, induced spontaneous breast tumors characterizedby enhanced type-II PKA activity. Downstream of this, Src phosphorylation occurs at residues serine-17 and tyrosine-416 andmammary cell transformation is driven through a mechanism involving Src signaling. The phenotypic consequences of thesealterations consisted of increased cell proliferation and, accordingly, expansion of both luminal and basal epithelial cell populations.In human breast cancer, low PRKAR1A/high SRC expression defines basal-like and HER2 breast tumors associated with poor clinicaloutcome. Together, the results of this study define a novel molecular mechanism altered in breast carcinogenesis and highlight thepotential strategy of inhibiting SRC signaling in treating this cancer subtype in humans.
Oncogene (2015) 34, 1160–1173; doi:10.1038/onc.2014.41; published online 24 March 2014
INTRODUCTIONCyclic-AMP dependent protein kinase A (PKA) ubiquitouslyfunctions as a signaling hub downstream of G-protein coupledreceptors and cAMP to regulate a spectrum of biological processesacross tissues.1–6 PKA impacts multiple signaling networks in bothphysiological and pathological conditions by phosphorylatingtarget proteins on serine/threonine residues. The complexityassociated with PKA function stems from its presence as twodistinct heterotetramers, termed type-I and type-II PKA,4,7 witheach PKA isozyme varying with respect to protein subunitcomposition, cellular localization and turnover. Four regulatory(R) subunits (R1α, R1β, R2α and R2β) and four catalytic (C) subunits(Cα, Cβ, Cγ and Prkx) have been identified, where the presence ofR1 or R2 subunits defines the type of PKA isozyme as type-I ortype-II, respectively.8 The balance between type-I/-II PKA caninfluence cell cycle entry and terminal differentiation in multiplesystems.4,7 Dysregulated PKA activity leads to the development oftumors in cAMP-responsive endocrine tissues9,10 and this isthought to stem from imbalances in activities of either type-I ortype-II PKA;7,11 however, its role as a cancer driver in a widerspectrum of tissues is less well known.The discovery of autosomal dominant inactivating mutations of
the PRKAR1A gene as the cause of Carney complex syndrome firstlinked PKA dysregulation to carcinogenesis.12,13 PRKAR1A encodesthe PKA regulatory subunit R1α, and of the four PKA regulatorysubunits, only PRKAR1A is essential for tissue development andcAMP-dependent regulation.14 Mutations to this gene in humansand mice induce multiple endocrine tumors as well as myxomas,osteoblastic neoplasias and schwannomas.9,15 When combinedwith Tp53+/� or Rb1+/� backgrounds, Prkar1a+/� mice exhibit agenerally increased incidence of sarcomas, pituitary tumors,thyroid tumors and chemically induced skin papillomas.10,16,17 Inaddition, we have found that tissue-specific heterozygous deletion
of Prkar1a in mesenchymal lineage cells is sufficient forspontaneous osteosarcoma development.18 However, the role ofPKA signaling in other cancer types and, particularly, in mammarycarcinogenesis remains unknown.In this study, we show that altered PKA regulation leading to
increased PKA activity in mammary tissue promotes carcino-genesis. Prkar1a loss results in heightened PKA activity defined byan increase in type-II PKA isozyme in mammary epithelial cells andthis hyperactivation drives mammary cell transformation througha mechanism involving Src. We further find that low PRKAR1A/highSRC marks a tumor subset of poor-prognosis basal-like and HER2breast cancer.
RESULTSPrkar1a loss in the mammary gland is sufficient to causemammary tumorsTo explore the effects of PKA hyperactivation in epithelial cells,we selected the mammary gland, a tissue outside of classicalendocrine epithelium harboring an extensive ductal network withmarker-defined lineages. It can develop a diverse family ofmolecular cancer subtypes in humans.19,20 Several mouse modelsare available, including those that allow homozygous genedeletion in the majority of mammary ductal epithelium. Weadopted a genetic strategy (Figure 1a) in which mammary-specificdeletion of Prkar1a was created by crossing Prkar1alox/lox mice(Prkar1a exon 2 flanked by LoxP sites) with transgenic miceexpressing Cre recombinase under the mammary epithelial-specific MMTV promoter. Unexpectedly, MMTV-Cre deletion ofPrkar1a was sufficient to generate mammary tumors (Figure 1b);Cre-mediated excision of the Prkar1a gene in this cohort of mice isshown in Figure 1c. Prkar1aΔMam mice developed multiple tumorswith 100% penetrance and a latency of 9–15 months of age (18/18
1Ontario Cancer Institute, Princess Margaret Hospital, Toronto, Ontario, Canada; 2Department of Medical Biophysics, University of Toronto, Toronto, Ontario, Canada; 3Departmentof Laboratory Medicine and Pathobiology, University of Toronto, Toronto, Ontario, Canada; 4Division of Endocrinology, Diabetes and Metabolism, The Ohio State University,Columbus, OH, USA and 5Breast Cancer and Systems Biology Unit, Translational Research Laboratory, Catalan Institute of Oncology, IDIBELL, L’Hospitalet del Llobregat, Barcelona,Spain. Correspondence: Dr R Khokha, Department of Medical Biophysics and Department of Laboratory Medicine and Pathobiology, Ontario Cancer Institute, University ofToronto, Toronto, Ontario, Canada M5G 2M9.E-mail: [email protected] 1 August 2013; revised 20 December 2013; accepted 24 December 2013; published online 24 March 2014
Oncogene (2015) 34, 1160–1173© 2015 Macmillan Publishers Limited All rights reserved 0950-9232/15
www.nature.com/onc

mice aged o16 months; Figure 1b; Supplementary Figure 1A)and progression from ductal hyperplasia (4.2 months) to palpabletumors (13 months) was observed by whole-mount analysis(Figure 1d).Histologically, mammary glands of >10 month-old Prkar1aΔMam
mice had an abundance of lobular hyperplasia, back-to-backgrowth and areas of atypia (Figure 1e). The tumors hadcharacteristics of papillomas with gradual progression to ductalcarcinoma in situ (DCIS) and invasive ductal carcinoma (IDC)(Figure 1e). In the majority of mammary tumors, a profoundexpansion of keratin 18-positive luminal cells was observed, whichwas accompanied with scattered expression of the myoepithelialmarker keratin 14 (Figure 1f). Additionally, select Prkar1aΔMam
mammary tumors showed immunohistochemical positivity forestrogen receptor α (Erα) and progesterone receptor (Pgr),whereas less-differentiated tumors harboring an invasive pheno-type were immuno-negative for Erα (Supplementary Figure 1B).Next, to examine whether Prkar1a loss cooperates with
molecular pathways known to be activated in human breastcancer, Prkar1aΔMam mice were bred into the widely used MMTV-PyMT (polyoma virus middle T-antigen) model that inducesactivation of ErbB2, Src, c-Myc and Ras/PI3 kinase signalingnetworks (Figure 2a).21,22 Confirmation of Cre-mediated Prkar1agene excision in this cohort was confirmed by PCR (Figure 2b).Tumor development was faster, with increased tumor burden, inPrkar1aΔMam/PyMT mice compared with PyMT controls (Figures 2).
Figure 1. Conditional loss of Prkar1a is sufficient to generate mammary tumors. (a) Schematic describes mouse-breeding strategy for thegeneration of Prkar1aΔMam mice. Prkar1aΔMam (MMTV-Cre/Prkar1afl/fl) denotes homozygous deletion by Cre recombinase expressed under thecontrol of the MMTV promoter. (b) Survival plot of Prkar1aΔMam (n= 18; solid black line) and control MMTV-Cre mice (n= 6; dashed line).(c) Cre-mediated genomic excision of Prkar1a in primary mammary epithelial cells derived from Prkar1aΔMam mice assessed by PCR; MMTV-Cremammary epithelial cells do not exhibit Cre-directed Prkar1a excision shown by lack of 175-bp PCR product. Prkar1afl/+ mouse osteoblastcultures transduced with retroviral Cre-recombinase (p-Cre) or GFP (p-GFP) serve as positive or negative controls.19 ‘L’ indicates DNA ladder.(d) Mammary gland whole-mounts of Prkar1aΔMam from 1.4 to 13 months of age highlights tissue progression to tumors. LN denotes lymphnode; red arrows highlight progression to tumors, ‘mo’ indicates age in months. Representative (e) H&E staining of tumors from Prkar1aΔMam
mice showing papillary, mixed (mix) and invasive ductal carcinoma (IDC) mammary tumors. Scale bars, 100 μm. (f) Representativeimmunofluorescent images of Prkar1aΔMam mammary tumors dual-labeled with epithelial lineage markers keratin 14 (basal) and keratin 18(luminal). Merged images show the combination of keratin 14 (red), keratin 18 (green) and DAPI (blue) positivity. Scale bars, 50 μm. See alsoSupplementary Figure 1
PKA-induced Src drives mammary tumorigenesisAG Beristain et al
1161
© 2015 Macmillan Publishers Limited Oncogene (2015) 1160 – 1173

In these mice, tumors were palpable by 33 days of age, reachingend point earlier compared with PyMT controls (detection median,38 versus 70 days; end point median, 72 versus 95 days), as shownin Figure 2d and Supplementary Figure 2. Gross tumor images andmammary gland whole mounts displayed extensive hyperplasia asearly as 40 days of age in Prkar1aΔMam/PyMT mice (Figures 2). Thisdemonstrates that mammary tumorigenesis is accelerated uponconditional deletion of Prkar1a in an established breast cancermodel. Tumors in Prkar1aΔMam/PyMT cohort were classified asDCIS harboring similar characteristics to those known to developin the PyMT model (Figure 2g). Forty-day-old lesions inPrkar1aΔMam/PyMT had far greater Ki67 positivity than in controlcohorts at 40 or 55 day of age, indicating that mammary deletionof Prkar1a drives cellular proliferation (Figure 2h).Taken together, we demonstrate that mammary-specific
deletion of Prkar1a is sufficient to induce stochastic mammarytumorigenesis exhibiting step-wise stages of histological progres-sion. Further, in the aggressive MMTV-PyMT mouse model ofbreast cancer, Prkar1a loss profoundly accelerates mammarytumorigenesis. Our data show that Prkar1a have a tumorsuppressor-like function in the mammary epithelium.
Mammary epithelial lineage expansion upon PKA hyperactivationWe and others have demonstrated that loss of the Prkar1a PKAregulatory subunit results in elevated PKA enzymatic activity inbone and endocrine tissues.18,23 We sought to test whether thisoccurs in mammary epithelium, following Prkar1a tissue-specificablation. Analyzing basal (unstimulated) and total (cAMP-induced)
PKA activity in protein lysates from age-matched non-tumor(MMTV-Cre) and tumor-bearing (Prkar1aΔMam) mammary tissuerevealed significantly higher baseline PKA activity in Prkar1aΔMam
relative to MMTV-Cre controls (P= 0.047). Treatment with the PKAinhibitor, PKI, abolished the effect of cAMP-induced PKA activitydemonstrating kinase specificity of the assay (Figure 3a). Inaddition, baseline and total PKA activity increased four- to fivefoldin 40-day Prkar1aΔMam/PyMT tumors compared with age-matchedPyMT mammary tissue lysates (Figure 3b). Notably, PKA activity innon-tumor bearing 40-day-old mammary glands of Prkar1aΔMam
mice or 55-day-old PyMT mammary tumors was not elevated.Accordingly, PyMT/Prkar1aΔMam tumors at 40 and 90 days of ageexhibited elevated phosphorylated CREB, a canonical downstreamtarget of PKA (Figures 3).We next explored the cellular makeup of Prkar1aΔMam
mammary tumors by flow cytometry to determine the effect ofPKA hyperactivation on distinct mammary epithelial lineages. Thisanalysis revealed a significant expansion of both the luminal CD24+/CD49flo and basal CD24+/CD49fhi epithelial subpopulations(Figure 3e). The basal fraction, known to contain mammary stemcells,24 was increased to >13% of total cells compared with 5% incontrol MMTV-Cre tissue. Luminal-type breast cancers thatdevelop in the PyMT model are reported to exhibit a profoundexpansion of luminal (CD24+/CD49flo) mammary epithelial cells,21
and we observed a more extreme expansion of this populationin the Prkar1aΔMam/PyMT compared with the PyMT cohort(Figure 3f). Biochemically, fluorescence-activated cell sorting(FACS)-purified CD24+/CD49flo luminal mammary epithelial cells
Figure 2. Prkar1a ablation in the PyMT mammary tumor model accelerates tumorigenesis. (a) Schematic describes mouse-breeding strategyfor the generation of Prkar1aΔMam/PyMTmice. (b) Cre-mediated genomic excision of Prkar1a in primary mammary epithelial cells derived fromPrkar1aΔMam/PyMTmice assessed by PCR; MMTV-PyMT mammary epithelial cells (PyMT) do not display Cre-directed Prkar1a excision shown bylack of 175-bp PCR product. Prkar1afl/+ mouse osteoblast cultures transduced with Cre-recombinase (p-Cre) or GFP (p-GFP) serve as positive ornegative controls.19 ‘L’ indicates DNA ladder. (c) Combined inguinal mammary gland weights (mg) of Prkar1aΔMam/PyMT (n= 8) and PyMT(n= 12) at 40 and 70 days of age. *Po0.05. (d) Survival plot for Prkar1aΔMam/PyMT (n= 15; red line) and PyMT (n= 14; gray line) mice.Representative gross (e), whole mount (f) and H&E (g) images of 40-day-old inguinal mammary glands of Prkar1aΔMam/PyMT and PyMT mice.Dotted line and red arrows highlight tumor periphery and location. (h) Ki67 immunohistochemistry in mammary tissue from age-matched40-day-old Prkar1aΔMam/PyMT and PyMT, as well as in 55-day-old PyMT mammary tumor. Bars, 100 μm. See also Supplementary Figure 2.
PKA-induced Src drives mammary tumorigenesisAG Beristain et al
1162
Oncogene (2015) 1160 – 1173 © 2015 Macmillan Publishers Limited

from Prkar1aΔMam/PyMT showed increased baseline and total PKAactivity (Figure 3g). These data demonstrate that mammary-specific loss of Prkar1a elevates PKA activity and this coincideswith substantial expansion of the luminal and basal epithelialsubpopulations.
Type-II PKA isozyme hyperactivation in Prkar1aΔMam tumorsWe next set out to determine the predominant PKA isozymesubtype in mammary tissues and mammary epithelial cells of
Prkar1aΔMam and Prkar1aΔMam/PyMT mice. Gene expressionanalysis of PKA regulatory (Prkar1a, Prkar1b, Prkar2a and Prkar2b)and catalytic (Prkaca) subunits was performed using FACS-purifiedluminal, basal and stromal mammary cells. The lineage markerskeratin 14 and keratin 18 were used to verify that sorted cells werepurely basal (keratin 14-positive) or luminal (keratin 18-positive)(Figure 4a). As expected, Prkar1a expression was reduced in boththe luminal and basal epithelial subpopulations from Prkar1aΔMam
mice, while remaining unchanged in the stromal cell subpopula-tion, confirming the specificity of the MMTV-directed conditional
Figure 3. Mammary-specific deletion of Prkar1a results in elevated PKA activity and mammary epithelial cell expansion. (a) Quantification oftotal (cAMP-treated) and baseline (unstimulated) PKA activity in adult age-matched (10–13 months of age) littermate Prkar1aΔMam mammarytumors (n= 5) and control MMTV-Cre mammary glands (n= 4). PKI inhibitor co-treatment demonstrates PKA activity specificity. (b) Baselineand total PKA activity of age-matched 40-day-old Prkar1aΔMam/PyMT tumors (n= 4), tumor-free mammary tissues from Prkar1aΔMam (n= 1),MMTV-Cre (Wt; n= 1) and PyMT (n= 4) mice and 55-day-old (55 d) PyMT mammary tumors (n= 3)* Po0.05. (c) Phosphorylated Creb (Ser 133)and total Creb protein levels in mammary protein lysates from 40- and 90-day-old Prkar1aΔMam/PyMT and MMTV-Cre mice. Wt= age-matchedMMTV-Cre control. Numbers indicate individual mice from each cohort. (d) Phosphorylated Creb immunohistochemistry in Prkar1aΔMam/PyMTmammary tumor and PyMT 40-day-old mammary gland. Bar, 100 μm. (e) Representative FACS plots segregated by cell surface markers CD24and CD49f identifying luminal (L), basal (B) and stromal (S) subpopulations in adult (4–6 months) age-matched Prkar1aΔMam and controlMMTV-Cre glands; percentage of cells representing each cell population is included. Bar graphs compare total numbers of luminal(lin�/CD24+/CD49flo) and basal (lin�/CD24+/CD49fhi) epithelial cells in each cohort. N= 5/cohort; *Po0.05. (f) FACS plots segregated by cellsurface markers CD24 and CD49f identifying luminal (L), basal (B) and stromal (S) subpopulations in 40-day-old Prkar1aΔMam/PyMT and PyMTmammary tissues; percentage of cells representing each cell population is included. Bar graphs compare total numbers of luminal(lin�/CD24+/CD49flo) and basal (lin�/CD24+/CD49fhi) epithelial cells in each cohort. N= 5/cohort. *Po0.05. (g) Baseline and total PKAactivities in FACS-purified luminal mammary cells from PyMT (n= 3) and Prkar1aΔMam/PyMT (n= 3) mice. *P⩽ 0.05.
PKA-induced Src drives mammary tumorigenesisAG Beristain et al
1163
© 2015 Macmillan Publishers Limited Oncogene (2015) 1160 – 1173

gene deletion in the epithelial compartment (Figure 4a). Next,while Prkar1b expression did not change in any cell type, theexpression of both Prkar2a and Prkar2b regulatory subunit genesincreased significantly in luminal and basal subpopulations, butwere unchanged in the stroma (Figure 4a). Expression of thecatalytic subunit, Prkaca, was higher in all cellular subpopulations(Figure 4a). Given the luminal cell expansion observed in thePyMT model (Figure 3f), the expression of PKA subunits inthis subpopulation was further analyzed in Prkar1aΔMam/PyMTmice. Consistent with the above observations, significant over-expression of both regulatory type-II and catalytic subunits wasobserved, while Prkar1a levels were reduced and Prkar1b levelsunchanged in the luminal fraction (Figure 4b). Altogether, thesechanges suggest a shift towards the type-II PKA holoenzyme inmammary epithelium following the loss of Prkar1a.
To biochemically profile PKA holoenzyme kinase activity,we fractionated total mammary tissue protein, and used acombination of kinase assays and immunoblotting to match PKAactivity with type-I and type-II regulatory subunit abundance(Figures 4). Control MMTV-Cre and Prkar1aΔMam (Figure 4c)or PyMT and Prkar1aΔMam/PyMT (Figure 4d) protein lysatesprepared from whole mammary tissues were subjected to anionexchange chromatography and activity assays were performedon each collected fraction. Immunoblotting against R1α and R2βproteins in fractionated samples confirmed the identity of eachPKA holoenzyme species (type-I or type-II PKA) eluting across thelinear salt gradient.15 In total profiles, MMTV-Cre mammaryglands exhibited comparable amounts of type-I and -II PKAactivity, with PyMT control glands showing more PKA type-Iactivity (Figures 4). Notably, Prkar1aΔMam and Prkar1aΔMam/PyMT
Figure 4. PKA-II isozyme defines Prkar1a mammary-specific loss. (a) qPCR expression analysis of keratin 14, keratin 18, Prkar1a, Prkar1b, Prkar2a,Prkar2b and Prkaca in luminal (L), basal (B) and stromal (S) FACS-purified cell subpopulations derived from adult age-matched Prkar1aΔMam
mammary tumors and control MMTV-Cre mammary glands (n= 5 mice per cohort); *Po0.05 among individual cell populations.(b) Expression of Prkar1a, Prkar1b, Prkar2a, Prkar2b and Prkaca PKA subunits in flow-purified luminal mammary epithelial cells derived from40-day-old age-matched Prkar1aΔMam/PyMT and PyMT mice (n= 5 mice/cohort); *Po0.05. β-actin was used for normalization in all cohorts.(c, d) Measurement of PKA activity in individual FPLC protein fractions and subsequent verification of PKA isozyme by western blotting.PKA activity in Prkar1aΔMam tumors (n= 3) and control MMTV-Cre mammary glands (n= 2) are shown in (c), while PKA activity in age-matched40-day-old Prkar1aΔMam/PyMT and PyMT mammary tumors (n= 4/cohort) are in shown in (d). Specifically, protein lysates from tissues werefractionated by DEAE column chromatography over a linear salt gradient. Activity peaks correspond to type-I PKA and type-II PKA isozymes,which elute at different salt concentrations. Fraction numbers (14–44) are specified on the x axis, while kinase activities measured byradioactive counts/min are shown on the y axis. Western blots verify the holoenzyme identity of eluted PKA heterotetramers based on theantibody specific for R1α (type-I PKA) or R2β (type-II PKA) PKA regulatory subunits in FPLC-fractionated protein lysates. Non-fractionatedprotein lysates served as positive (input) controls.
PKA-induced Src drives mammary tumorigenesisAG Beristain et al
1164
Oncogene (2015) 1160 – 1173 © 2015 Macmillan Publishers Limited
tumors exhibited dramatic increases in type-II PKA activity,and experienced a decrease in type-I PKA (Figures 4), aspreviously suggested by our mRNA expression analyses;modest type-I PKA activity observed in Prkar1aΔMam is likelyfrom non-epithelial (stromal) contributions from homogenizedmammary tissues. This series of gene/protein expressionand kinase activity studies demonstrate that mammary
epithelial deletion of Prkar1a results in type-II PKA isozymehyperactivation.
Heightened PKA activity drives mammary epithelial cellproliferationHaving showed that Prkar1a ablation in mammary epithelium issufficient to induce tumorigenesis, we next asked whether the
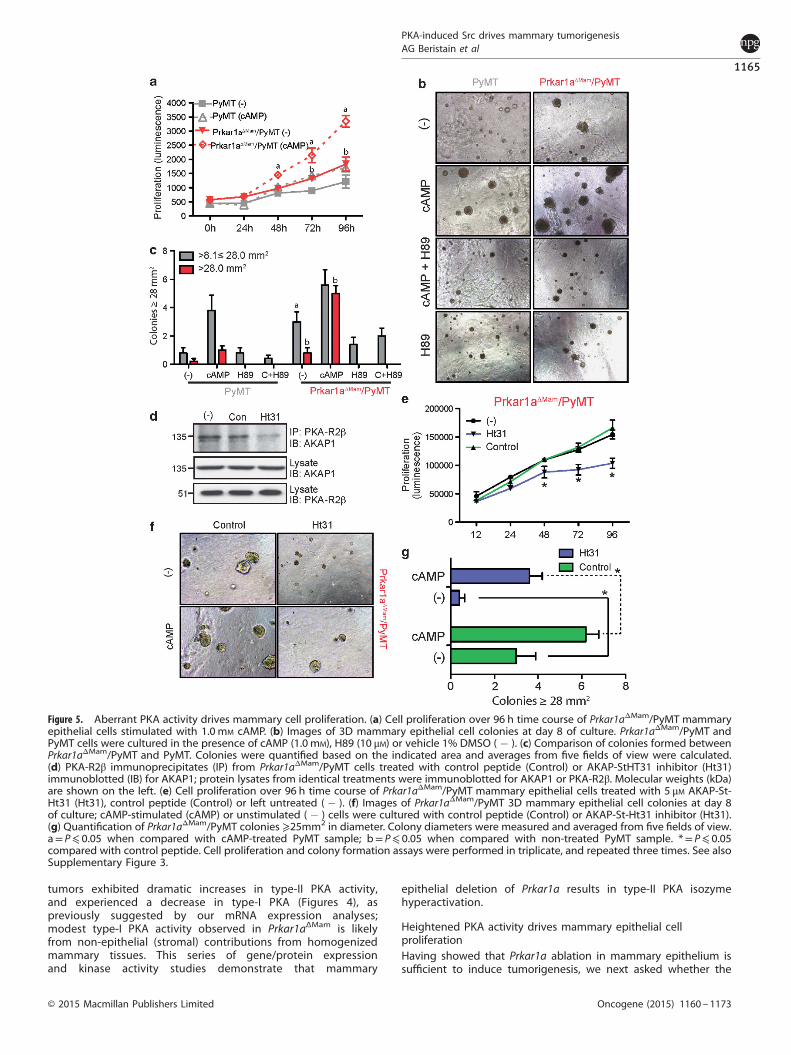
Figure 5. Aberrant PKA activity drives mammary cell proliferation. (a) Cell proliferation over 96 h time course of Prkar1aΔMam/PyMT mammaryepithelial cells stimulated with 1.0 mM cAMP. (b) Images of 3D mammary epithelial cell colonies at day 8 of culture. Prkar1aΔMam/PyMT andPyMT cells were cultured in the presence of cAMP (1.0 mM), H89 (10 μM) or vehicle 1% DMSO ( � ). (c) Comparison of colonies formed betweenPrkar1aΔMam/PyMT and PyMT. Colonies were quantified based on the indicated area and averages from five fields of view were calculated.(d) PKA-R2β immunoprecipitates (IP) from Prkar1aΔMam/PyMT cells treated with control peptide (Control) or AKAP-StHT31 inhibitor (Ht31)immunoblotted (IB) for AKAP1; protein lysates from identical treatments were immunoblotted for AKAP1 or PKA-R2β. Molecular weights (kDa)are shown on the left. (e) Cell proliferation over 96 h time course of Prkar1aΔMam/PyMT mammary epithelial cells treated with 5 μM AKAP-St-Ht31 (Ht31), control peptide (Control) or left untreated ( � ). (f) Images of Prkar1aΔMam/PyMT 3D mammary epithelial cell colonies at day 8of culture; cAMP-stimulated (cAMP) or unstimulated ( � ) cells were cultured with control peptide (Control) or AKAP-St-Ht31 inhibitor (Ht31).(g) Quantification of Prkar1aΔMam/PyMT colonies ⩾25mm2 in diameter. Colony diameters were measured and averaged from five fields of view.a= P⩽ 0.05 when compared with cAMP-treated PyMT sample; b= P⩽ 0.05 when compared with non-treated PyMT sample. *= P⩽ 0.05compared with control peptide. Cell proliferation and colony formation assays were performed in triplicate, and repeated three times. See alsoSupplementary Figure 3.
PKA-induced Src drives mammary tumorigenesisAG Beristain et al
1165
© 2015 Macmillan Publishers Limited Oncogene (2015) 1160 – 1173

increases in PKA activity downstream of Prkar1a deletion arerequired for mammary epithelial proliferation. Primary mammaryepithelial cells derived from Prkar1aΔMam/PyMT and control PyMTmice were subjected to proliferation assays stimulated with cAMPand treated with the PKA inhibitors H89 or PKI. To begin with,untreated Prkar1aΔMam/PyMT cells had higher baseline prolifera-tion rates than PyMT cells over 96 h of culture (Figure 5a;Supplementary Figure 3A). The addition of cAMP enhancedproliferation in both groups; however, Prkar1aΔMam/PyMT cellswere more sensitive to this stimulation, showing a two-foldinduction in proliferation over wild-type PyMT cells (Figure 5a).The PKA inhibitors H89 and PKI abolished the effect of cAMP onmammary epithelial cell proliferation across the time course inboth genotypes (Supplementary Figure 3).To assess the influence of PKA activity on cellular transforma-
tion, Prkar1aΔMam/PyMT and PyMT mammary epithelial cells weregrown in Matrigel to assess the potential of colony formation. Cellsfrom both models grew significantly more large colonies in thepresence of cAMP (Figures 5). Importantly, Prkar1aΔMam/PyMT cellsgenerated larger colonies than PyMT alone, even in the absence ofcAMP, and this was abrogated upon H89 co-treatment. To directlyexamine the importance of elevated type-II PKA in driving cellproliferation and transformation, we treated Prkar1aΔMam/PyMTcells with the peptide inhibitor, AKAP St-Ht31, which preventstype-II PKA regulatory subunit interaction with A-kinase anchoringproteins (AKAPs)25,26 important for optimal activity and cellularlocalization.27,28 AKAP St-Ht31 peptide mediated disruption ofPKA–R2AKAP interaction was verified by PKA–R2α immunopreci-pitation followed by AKAP1 immunoblotting, which showedreduced R2βAKAP1 interaction (Figure 5d). Importantly, AKAPSt-Ht31 peptide inhibitor treatment effectively decreased baselineproliferation and 3D colony formation, while treatment with a
control peptide (St-Ht31P) had no effect (Figures 5). AKAP St-Ht31modestly, but significantly inhibited cAMP-induced colony forma-tion (Figures 5). Together, these experiments demonstrate thattype-II PKA isozyme is fundamental in driving Prkar1aΔMam/PyMTcell growth. Thus, heightened type-II PKA activation is sufficientand required to promote mammary epithelial cell hyperprolifera-tion in the context of Prkar1a deletion.
PKA activates Src signaling during mammary tumorigenesisGiven our findings that aberrant PKA activity regulates cellproliferation, we sought the molecular mechanism through whichPKA drives breast carcinogenesis. To this end, we first investigatedthe Wnt and Erα signaling pathways, which are modulateddownstream of PKA in other cancer types.5,10,29 Expression ofseveral Wnt and Erα target genes (Axin2, Tcf4, Greb1 and Pra/b)were measured in 40-day-old Prkar1aΔMam/PyMT and controlPyMT FACS-purified luminal cells. This analysis failed to revealdifferences in the expression of these target genes between thesecohorts (Supplementary Figure 4), suggesting that type-II PKAhyperactivity accelerates PyMT breast cancers by mechanismsother than Erα or Wnt pathways.PKA can phosphorylate c-Src (herein referred to as Src) on
serine-17 to regulate its activity.30 Given that Src is involved inPyMT tumorigenesis,22,31 its activation was evaluated in 40-day-old PyMT cohort mammary tissues. PKA protein substrates wereimmunoprecipitated using an antibody specifically targetingphosphorylated serine/threonine epitopes (RRXS*/T*) of PKAsubstrates,32 followed by Src immunoblotting. This showed highlyelevated PKA-phosphorylated Src across Prkar1aΔMam/PyMTmammary tissues (Figure 6a). Since Ser17 phosphorylation of Srcby PKA directs its activation through auto-phosphorylation of
Figure 6. Mammary gland-specific loss of Prkar1a induces Src activation. (a) Phosphorylated PKA substrates from 40-day-old age-matchedPrkar1aΔMam/PyMT or PyMT mammary tissues were immunoprecipitated (IP) using an antibody-specific for PKA-phosphorylated serine/threonine (RRXS*/T*) epitopes, followed by immunoblotting (IB) with anti-Src antibody. Molecular weights (kDa) are shown on the left andnumbers represent individual mice. (b) Immunoblots of phosphorylated and total protein levels of Src (Ser17; Tyr416) and Akt (Ser473) inmammary protein lysates. β-actin indicates loading control. Numbers represent individual mice. (c) Immunoprecipitation (IP) ofphosphorylated PKA substrates from Prkar1aΔMam tumors and control MMTV-Cre mammary glands followed by Src immunoblotting (IB).Numbers indicate individual mice from respective cohorts. (d) Src Tyr416 phosphorylation indicates active Src in protein lysates fromPrkar1aΔMam and control mammary tissue. Protein lysate from Prkar1aΔMam/PyMT tumor are a positive control. See also SupplementaryFigure 4.
PKA-induced Src drives mammary tumorigenesisAG Beristain et al
1166
Oncogene (2015) 1160 – 1173 © 2015 Macmillan Publishers Limited

Tyr416,33 we examined whether this occurs in Prkar1aΔMam/PyMTmammary glands. Prkar1aΔMam/PyMT tissues exhibited consis-tently elevated Ser17 and Tyr416 Src phosphorylation (Figure 6b).Additionally, levels of total Src were also higher in Prkar1aΔMam/PyMT mice. Further, phospho-Akt (Ser473), a signaling effectorknown to be downstream of Src, was probed and found to behigher in Prkar1aΔMam/PyMT (Figure 6b).To determine whether Src is also phosphorylated by PKA in
Prkar1aΔMam tumors that develop spontaneously without thePyMT oncogene, we analyzed tissue bearing advanced tumorsfrom these mice. Immunoprecipitation of phosphorylated PKAsubstrates demonstrated highly elevated phosphorylated Src in 3of 4 tumors (Figure 6c). Furthermore, Tyr416 Src phosphorylationwas elevated in two of four Prkar1aΔMam mammary tumors,suggesting that Src is a frequent mediator in PKA-initiatedmammary tumors (Figure 6d). Together, these results highlightSrc as a critical pathway impacted in breast tumors linked withaberrant PKA activity.
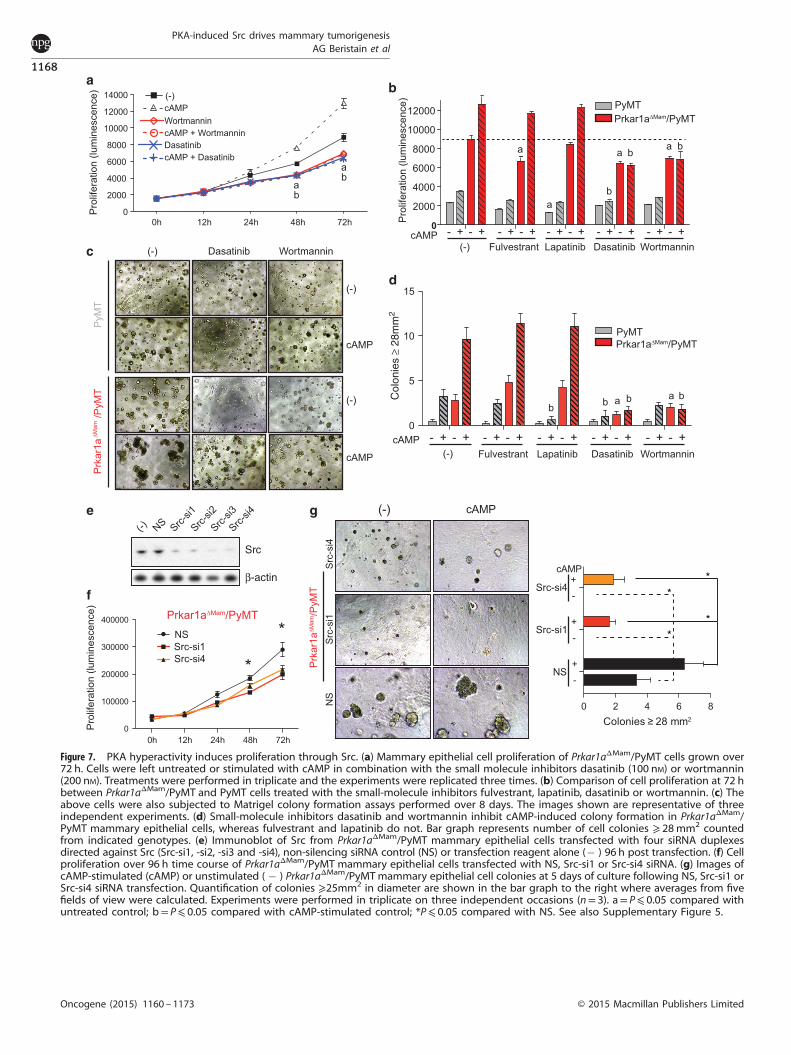
PKA-triggered Src activation is responsible for mammary cellgrowthNext, the importance of Src in PKA-driven mammary epithelial celltransformation was dissected by evaluating cell proliferation andcolony growth upon pathway modulation using inhibitors againstERα, ErbB2/EGFR, PI3K/Akt and/or Src. Prkar1aΔMam/PyMT mam-mary epithelial cells proliferated at a higher rate than PyMT, andcAMP promoted this phenotype (Figures 7a and b; SupplementaryFigure 5A), as previously observed. Addition of the ERα antagonistfulvestrant or the ErbB2/EGFR inhibitor lapatinib had incompleteinhibitory effects on baseline and cAMP-induced proliferation inboth PyMT and Prkar1aΔMam/PyMT genotypes (Figure 7b;Supplementary Figure 5A). In contrast, inhibition of Src withdasatinib reduced baseline, and profoundly prevented cAMP-induced proliferation of Prkar1aΔMam/PyMT mammary epithelialcells (Figures 7a and b). Wortmannin, which blocks PI3K/Akt,suppressed proliferation in a similar manner (Figures 7a and b).Mammary epithelial cell growth in Matrigel cultures provides a
readout of transformation. Here, ERα inhibition by fulvestrant hadno effect, whereas ErbB2/EGFR inhibition with lapatinib did notimpact baseline Prkar1aΔMam/PyMT or cAMP-induced colonyformation (Figures 7c and d; Supplementary Figure 5B). Incontrast, both dasatinib and wortmannin significantly reducedbaseline growth of colonies and additionally inhibited cAMP-induced colony formation in both genotypes (Figures 7c and d).To further dissect the importance of Src in Prkar1aΔMam/PyMT
cell transformation, small interfering RNA (siRNA)-directed deple-tion of endogenous Src was performed. Four siRNAs (Src-si1, -si2,-si3 and -si4) targeting Src were transfected into Prkar1aΔMam/PyMT mammary epithelial cells and Src knockdown was assayed96 h post transfection by immunoblotting; all four siRNAs resultedin reduced Src protein levels, whereas a control siRNA had noeffect (Figure 7e). Strikingly, Src depletion using two of theabove siRNA duplexes (Src-si1 or Src-si4) significantly inhibitedbaseline and cAMP-stimulated Prkar1aΔMam/PyMT cell proliferation(Figure 7f; Supplementary Figure 5C) and 3D colony growth(Figure 7g). Thus, aberrant PKA activity in the mammary glandpromotes epithelial cell proliferation and transformation through amechanism involving the activation of the tyrosine kinase Src.Further, Src activation may act via Akt signaling to provide agrowth advantage to these mammary epithelial cells.
Poor patient survival in low-PRKAR1A/high-SRC human breastcancersOur findings that PKA hyperactivation drives mammary tumor-igenesis in the mouse prompted us to examine this in humanbreast cancer. We first analyzed data from the TCGA data set34 andobserved a significant correlation between altered PRKAR1A
somatic copy number and expression (Pearson’s correlationcoefficient, PCC= 0.67; P= 2.2 × 10�16; Supplementary Figure 6A).High PRKAR1A copy number/expression was associated with highexpression of genes of the oxidative phosphorylation pathway,whereas low copy number/expression associated with highexpression of genes of the ribosome pathway (SupplementaryFigure 6B). Subsequent evaluation of PRKAR1A expressiondifferences across tumor subtypes as defined using the PAM50classifier indicated significant under-expression in the basal-likesubtype relative to all others (t-test P-values o0.001,Supplementary Figure 6C). Accordingly, PRKAR1A expression waspositively correlated with ESR1, which encodes for ERα, andto a lower degree with ERBB2, which encodes for the humanepidermal growth factor receptor 2 (HER2) (SupplementaryFigure 6D).Our above experiments in the mouse had established increased
Src signaling to be a main mechanism critical for PKA-directedmammary cell growth and transformation. To explore thismolecular link in human breast cancer, the TCGA series wasdivided in two tumor sets corresponding to low or high PRKAR1Aexpression values (based on tertile categorization). Analyzing theexpression differences between these sets identified an associa-tion with an oncogenic SRC signature previously derived in thenon-tumorigenic MCF10A mammary cell line:35 genes over-expressed with SRC activation in MCF10A cells were also foundto be over-expressed in breast tumors with low PRKAR1Aexpression (P= 0.023; Figure 8a, top left panel). This associationwas further confirmed by examining all genes over-expressed withSRC activation in MCF10A cells (at a false discovery rate o5%);P= 0.003; Figure 8a, top right panel). The genes under-expressedwith SRC oncogenic activation did not show significant results(Figure 8a, bottom panels). GSEA analysis in the TCGA data setrevealed that low-PRKAR1A/high-SRC tumors are enriched forpathways regulating GPI anchor biosynthesis and the peroxisomemetabolic pathways (Supplementary Figure 6E), while pathwaysregulating glycosphingolipid biosynthesis were under-expressed(Supplementary Figure 6F).Examining clinical characteristics in the TCGA tumors, those
with concurrent low PRKAR1A and high SRC expression (lowPRKAR1A/high SRC) were found to be more likely to recur (log-ranktest P= 0.007; Figure 8b). In addition, recurrence was also higher inthe low-PRKAR1A/high-SRC tumor subset compared with tumorsclassified as only low PRKAR1A, high SRC expression, or all others(log-rank test P-valueso0.05; Figure 8c). Subsequently, examiningthe concordance with PAM50-based subtypes, 33% of the basal-like and 17% of HER2 subtype had low-PRKAR1A/high-SRCexpression (Figure 8d). Analysis of recurrence in these subtypesshowed similar trends, although only HER2 reached significancedespite containing fewer cases (P= 0.021; Supplementary Figure6G). We repeated these analyses in the independent NKI-295 dataset36 both for the full data and the HER2 subtype (Figures 8e and f;Supplementary Figure 6H). This recapitulated our findings fromthe TCGA analysis, showing similar trends when examiningprognosis among tumors classified as low PRKAR1A/high SRC,low PRKAR1A, high SRC or others. Informed by our studies in mice,this work points to a distinct subset of human breast cancers withhigh rates of recurrence defined by low PRKAR1A/high SRCexpression, and these tumors are found particularly within thebasal-like and HER2 subtypes.
DISCUSSIONHere we describe a pro-neoplastic role for PKA isozymehyperactivation in the mouse mammary gland. We show thatmammary-specific loss of Prkar1a leads to elevated type-II PKAisozyme activation and this is sufficient to drive breast carcino-genesis. Further, we show that heightened PKA activity associateswith Src pathway activation and this is in part responsible
PKA-induced Src drives mammary tumorigenesisAG Beristain et al
1167
© 2015 Macmillan Publishers Limited Oncogene (2015) 1160 – 1173
**
**
Figure 7. PKA hyperactivity induces proliferation through Src. (a) Mammary epithelial cell proliferation of Prkar1aΔMam/PyMT cells grown over72 h. Cells were left untreated or stimulated with cAMP in combination with the small molecule inhibitors dasatinib (100 nM) or wortmannin(200 nM). Treatments were performed in triplicate and the experiments were replicated three times. (b) Comparison of cell proliferation at 72 hbetween Prkar1aΔMam/PyMT and PyMT cells treated with the small-molecule inhibitors fulvestrant, lapatinib, dasatinib or wortmannin. (c) Theabove cells were also subjected to Matrigel colony formation assays performed over 8 days. The images shown are representative of threeindependent experiments. (d) Small-molecule inhibitors dasatinib and wortmannin inhibit cAMP-induced colony formation in Prkar1aΔMam/PyMT mammary epithelial cells, whereas fulvestrant and lapatinib do not. Bar graph represents number of cell colonies ⩾ 28mm2 countedfrom indicated genotypes. (e) Immunoblot of Src from Prkar1aΔMam/PyMT mammary epithelial cells transfected with four siRNA duplexesdirected against Src (Src-si1, -si2, -si3 and -si4), non-silencing siRNA control (NS) or transfection reagent alone (� ) 96 h post transfection. (f) Cellproliferation over 96 h time course of Prkar1aΔMam/PyMT mammary epithelial cells transfected with NS, Src-si1 or Src-si4 siRNA. (g) Images ofcAMP-stimulated (cAMP) or unstimulated ( � ) Prkar1aΔMam/PyMTmammary epithelial cell colonies at 5 days of culture following NS, Src-si1 orSrc-si4 siRNA transfection. Quantification of colonies ⩾25mm2 in diameter are shown in the bar graph to the right where averages from fivefields of view were calculated. Experiments were performed in triplicate on three independent occasions (n= 3). a= P⩽ 0.05 compared withuntreated control; b= P⩽ 0.05 compared with cAMP-stimulated control; *P⩽ 0.05 compared with NS. See also Supplementary Figure 5.
PKA-induced Src drives mammary tumorigenesisAG Beristain et al
1168
Oncogene (2015) 1160 – 1173 © 2015 Macmillan Publishers Limited

Figure 8. Association between PRKAR1A/SRC expression levels, SRC oncogenic activation and breast cancer recurrence. (a) Gene setenrichment analysis (GSEA) graphical outputs for the association analysis of expression differences between high and low PRKAR1A-expressingTCGA tumors and genes over-/under-expressed with SRC oncogenic activation in MCF10A cells. The top panels correspond to theover-expressed genes in the original SRC oncogenic signature (left panel) or to the over-expressed genes at false discovery rate o5% (that is,SRC-mediated perturbation, right panel). Similarly, the bottom panels correspond to the under-expressed genes with SRC activation ormediated perturbation. The GSEA enrichment score and the nominal P-values are shown. (b) Kaplan–Meier breast cancer recurrence curves forcategorization of TCGA tumors based on concurrent low PRKAR1A and high SRC expression versus the rest of cases with recurrenceinformation available. The log-rank test P value and number of cases (n) are indicated. (c) Kaplan–Meier breast cancer recurrence curves forcategorization of TCGA tumors based on low and/or high PRKAR1A/SRC expression values. (d) For the identification of low-PRKAR1A/high-SRCexpression TCGA tumors, the complete series was ordered according to PRKAR1A or SRC expression and, subsequently, the intersection wasidentified. The distribution of these tumors (that is, low PRKAR1A plus high SRC) across the PAM50-based subtypes is shown in the right panel,which reveals higher concordance with basal-like type. (e) Kaplan–Meier survival curves for categorization of tumors based on concurrent lowPRKAR1A and high SRC expression versus the rest of cases within each PAM50-based subtype in the full NKI-295 data set. (f) Kaplan–Meiersurvival curves for categorization of tumors based on low and/or high PRKAR1A/SRC expression values in the full NKI-295 data set. See alsoSupplementary Figure 6.
PKA-induced Src drives mammary tumorigenesisAG Beristain et al
1169
© 2015 Macmillan Publishers Limited Oncogene (2015) 1160 – 1173

for mammary epithelial cell proliferation and transformation.Our work additionally exposes a putative low-PRKAR1A/high-SRChuman tumor subset associated with poor patient outcome.PKA is the main mediator of the highly conserved cAMP
signaling pathway and aberrant activation of this kinase is knownto be causal in hereditary endocrine neoplasias of the Carneycomplex.12,13 However, its importance in sporadic tumorsoriginating from epithelial lineage is less understood. Mousemodeling of Prkar1a heterozygosity in the germ line settingrecapitulates the spectrum of endocrine tumors seen in Carneycomplex patients harboring autosomal dominant inactivatingPRKAR1A mutations.9 In mesenchymal cells, osteoblast-specificheterozygous Prkar1a deletion leads to spontaneous bonetumorigenesis.18,37 We now demonstrate that mammary epithelialcells lacking both copies of Prkar1a demonstrate aberrant type-IIPKA activation. This mirrors reports of type-II PKA activity increasesin Carney complex tumors.15,23 Importantly, we found thatheightened PKA activation is responsible for highly augmentedbaseline and cAMP-induced proliferation of primary mammarycells in multiple assays in vitro and mammary lineage expansionin vivo, enabling stochastic mammary cancer development by1 year of age. Although it is uncommon, breast ductal adenomashave been reported in women with Carney complex, indicatingthat this phenomenon is not limited to genetically engineeredmice.38 Our findings generalize the importance of PKA holoen-zyme homeostasis to epithelial tissue that impacts the mostcommon form of cancer in women.A major finding of this study highlights the importance of type-
II PKA as a critical mechanism in directing tumor cell proliferation.This observation is contrary to what has been previously reportedabout the roles of type-I and type-II PKA. Specifically, type-II PKApredominantly associates with well-differentiated non-prolifera-tive cell types, while a strong interaction exists between type-I PKAisozyme, cell proliferation and tumorigenesis.8 A possible explana-tion for our findings centers on the complexity of PKA holoenzymehomeostasis, where the Prkar1a regulatory subunit exertsdominance over other regulatory subunits in regulating PKAactivity. In our tumor model, where Prkar1a is absent in mammaryepithelia, the increase in PKA activity can be attributed by either acompensatory increase in type-II PKA isozyme or by an increasein the unbound (unregulated) PKA catalytic subunit. In the MMTV-PyMT background, hyperactive PKA activity (the result ofincreased type-II PKA or free PKA catalytic subunit) may cooperatewith underlying PyMT cancer drivers (that is, Src) leading toaccelerated tumorigenesis. The importance of Prkar1b in con-tributing to type-I PKA isozyme function cannot be overlooked inour mammary tumor model, even thought alterations in itsexpression were not significantly altered. It will be importantto determine whether human breast cancers defined by lowPRKAR1A exhibit an altered type-I/II PKA isozyme ratio.PKA has been studied as a mediator of ERα signaling in breast
cancer, including ligand-dependent/-independent induction andtamoxifen resistance.2,5,29,39–41 Using small-molecule inhibitors, weruled out the involvement of ERα and EGF receptor signaling inour PKA-driven mammary tumors. We also did not observeincreases in expression of ERα-responsive genes Pgr or Greb1, orWnt reporter genes Axin2 and Tcf4. Instead we found PKA-inducedSrc signaling to be a downstream mechanism in Prkar1aΔmam
cohorts. It has been shown that the development of PyMT tumorsdepends in part on Src activity, where mammary epithelialablation of Src significantly delays tumor onset.31 In our model,Prkar1aΔmam/PyMT tumors were associated with elevated phos-phorylated Src at both serine 17 and tyrosine 416 residues.PKA-directed regulation of Src has previously been observed infibroblasts and adrenal cells in vitro;33,42 our findings provide thefirst evidence of in vivo PKA-Src regulation. Although we did notdirectly examine Src activity in Prkar1aΔmam tumors, Src RNAiexperiments demonstrated the importance of Src as a
downstream mediator of PKA-directed cell proliferation andcolony growth. Thus, mammary-specific PKA hyperactivation inthis model cooperates with PyMT to augment Src-mediatedproliferation and cellular transformation. Importantly, we observedSrc activation even in the absence of the PyMT oncogene,highlighting the conserved signaling mechanism attributed toPrkar1a genetic ablation. However, our observation that not allPrkar1aΔmam tumors exhibited elevated phosphorylated Src (Y416)combined with our findings that Src inhibition partially blockedPKA-mediated cell proliferation suggests that aberrant PKA activitymay interact with other unidentified signaling pathways importantin tumorigenesis.Human breast cancers exhibit substantial complexity and
integrative genomic studies are now progressively stratifyingtumors into novel molecular subsets building on the PAM50designations.19,20,34,43 These studies have exposed considerableheterogeneity within the major types of breast cancer. Patientswith basal-like or HER2 breast cancers have adverse prognosiscompared to those categorized as luminal A and luminal B.Aberrant Src activity has been found to correlate with HER2positivity, higher breast tumor grade and inversely correlates withERα status.16,17 Informed by the observation that Prkar1a deletionactivates the Src pathway in the mouse, we probed for a putativelow-PRKAR1A/high-SRC breast cancer subset and uncovered asubstantial proportion of the patient population with tumorsharboring this signature. In both the TCGA and NKI-295 breastcancer data sets, low-PRKAR1A/high-SRC tumors exhibited distinctlypoor patient outcome, even when compared with low-PRKAR1A orhigh-SRC designations alone. Further, our analyses suggested thatlow-PRKAR1A/high-SRC tumors make up a notable proportion ofbasal-like as well as HER2 cancers. Given that SRC inhibitors such asdasatinib are in clinical trials for a variety of human cancersincluding advanced breast cancers, it will be important todetermine whether SRC inhibitors can provide benefits to thisputative novel low-PRKAR1A/high-SRC breast cancer subset.
MATERIALS AND METHODSReagentsThe mouse mammary epithelial cell line (NMuMG) was obtained fromATCC (CRL-1636). Ongoing cultures were maintained in DMEM containing25mM glucose, L-glutamine, antibiotics (100 U/ml penicillin, 100 μg/mlstreptomycin) and supplemented with 10% FBS. 8-bromoadenosine 3′,5′-cyclic monophosphate (cAMP), cholera toxin, hydrocortisone, insulinand fulvestrant were purchased from Sigma Aldrich (St Louis, MO, USA).Phenol red-free, growth factor-reduced Matrigel was purchased from BDBiosciences (San Jose, CA, USA). EGF was purchased from Peprotech (RockHill, NJ, USA). Dasatinib and wortmannin were generously provided by DrsShereen Ezzat and Vuk Stambolic, Ontario Cancer Institute, Toronto.Lapatinib was purchased from Selleck Chemicals. H89 was purchased fromCell Signaling Technology (Boston, MA, USA). PKI, InCELLect AKAP-St-Ht31inhibitor and InCELLect St-Ht31P control peptide were purchasedfrom Promega (Sunnyvale, CA, USA). For cell signaling experiments, cellswere washed in PBS and starved in DMEM media for 24 h; cells weretreated with recombinant mouse EGF (10 nM) or insulin (50 nM) for 15min.The following small molecules were used at the given concentrations: H89(10 μM), PKI (20 μM), cAMP (1.0 mM), InCELLect AKAP-St-Ht31inhibitor andcontrol peptide (5μM), fulvestrant (100 nM), lapatinib (100 nM), wortmannin(200 nM) and dasatinib (100 nM).
MiceMMTV-PyMT mice and MMTV-Cre mice in the FVB background wereobtained from Dr W Muller (McGill University, Canada). The generation ofthe Prkar1alox/lox mice and genotyping of the WT, loxP and deletion allelehave been described.16 To generate the experimental mice, the followingbreeding pairs were set: Prkar1alox/lox/MMTV-PYMT+/MMTV-Cre+ maleswere bred with Prkar1alox/lox/MMTV-PYMT–/MMTV-Cre� females. Theproper Mendelian ratios were obtained. Mammary tumor onset wasdetermined by manual palpation of mammary glands. Tumor endpoints,determined by Animal Care Committee guidelines, were reached when
PKA-induced Src drives mammary tumorigenesisAG Beristain et al
1170
Oncogene (2015) 1160 – 1173 © 2015 Macmillan Publishers Limited

mammary tumor diameter measured 1.5 cm. All animal experiments wereapproved by the Animal Care Committee, Ontario Cancer Institute.
Whole-mount analysis, immunohistochemistry andimmunofluorescenceThoracic mammary glands from 40- or 90-day-old mice were analyzedfor mammary morphology by carmine-alum whole-mount staining asdescribed previously.44 For immunohistochemistry, 4% paraformaldehyde-fixed paraffin-embedded tissue sections were de-paraffinized in xylene,gradually rehydrated in descending concentrations of ethanol andsubsequently treated in Borg Decloaker antigen retrieval solution (pH 9)for 30min at 121 °C and 10 seconds at 90 °C using a Decloaking chamber(Biocare Medical, Concord, CA, USA). Tissue sections were stained usingHRP-AEC tissue staining kit according to manufacturer’s instructions (R&DSystems, Minneapolis, MN, USA). For dual immunofluorescence micro-scopy, paraffin-imbedded sections were hydrated and subjected toantigen retrieval as described above. Tissues were permeabilized with0.2% Tritin X-100 and 0.1% sodium borohydride, and blocked in 5% normalgoat serum containing 0.1 mg/ml saponin. Primary antibodies (anti-keratin14 and anti-kertain 18) diluted in PBS were incubated overnight at 4 °C in ahumidified chamber. Primary antibodies were labeled with Alexa Fluor 488goat anti-mouse and Alexa Fluor 594 goat anti-rabbit secondaryantibodies. Slides were mounted in DAPI mounting medium (Invitrogen,Carlsbad, CA, USA) and imaged using an Axio Observer invertedmicroscope (Carl Zeiss, Jena, Germany). Antibodies used were anti-mouse antibodies against keratin 14 (Covance, Princeton, NJ, USA), keratin18 (Fitzgerald, Burlington, ON, Canada), Ki67 (Novus Biologicals, Oakville,ON, Canada), PR (hPRa7; generated in-house), ERα (clone M20, Santa CruzBiotechnology; Dallas, TX, USA) and phosphorylated CREB (p-Creb Ser133)from Cell Signaling Technology (Beverly, MA, USA).
Mammary epithelial cell preparation, FACS analysis and cellsortingSingle mammary cell suspensions were generated from freshly isolatedpairs of fourth inguinal mammary glands of individual mice by enzymaticdigestion and analyzed by flow cytometry as reported previously44 orcultured on collagen-coated tissue culture plates. Briefly, mammary glandswere digested for 2.5 h at 37 °C in mouse Epicult-B with 5% FBS, 750 U/mlcollagenase and 250 U/ml hyaluronidase. Organoids obtained aftervortexing were subjected to red blood cell lysis in 0.8% NH4Cl, furtherdissociation in 0.25% trypsin for 2 min, 5 mg/ml dispase with 0.1 mg/mlDNase I for 2 min, and filtered through a 40-μmmesh to obtain single cells.All reagents were from StemCell Technologies Inc. (Vancouver, BC, Canada)and antibodies were from BD Pharmingen (San Diego, CA, USA) unlessotherwise stated. Cells were blocked with Fc receptor antibody, incubatedwith biotinylated StemSep mouse/human chimera cocktail and anti-CD31in order to label hematopoietic CD45+/Ter119+ cells and endothelial cellsrespectively, which were excluded by secondary conjugation withstreptavidin-PE-Cy7 using flow cytometry. Dead cells were excluded fromanalysis by staining with propidium iodide (Sigma). Anti-CD49f-FITC (cloneGoH3) and anti-CD24-R-PE (clone M1/69) were used to identify themammary epithelial cell populations. FACS analysis was performed usingFACSCalibur (BD) and FlowJo software (Tree Star Inc., Ashland, OR, USA).Cell sorting was performed on a FACSAria (BD). The purity of sortedpopulations was routinely greater than 96%.
RNA isolation and real-time PCR analysisTotal RNA was prepared from FACS-sorted primary mammary cellsubpopulations using the PicoPure RNA Isolation Kit (Arcturus, Carlsbad,CA, USA) as described in Joshi et al.44 The quality and concentration of RNAwas determined by visualizing purified RNA samples on SyBr Green II(Invitrogen)-stained formaldehyde agarose gels and by analysis with aNanoDrop 2000 Spectrometer (260/280 ratio; Thermo Scientific, Waltham,MA, USA). Isolated and purified total RNA was reverse transcribed into firststrand cDNA and amplified using the SMARTer PCR cDNA Synthesis Kit andAdvantage2 PCR Kit (Clontech). Amplified cDNA aliquots (5 μl) wereanalyzed on ethidium bromide-stained agarose gels (1.2%) to determinethe optimal number of LD-PCR cycles for cDNA amplification that rangedfrom 18–21 cycles. Relative quantification real-time PCR (ΔΔCt) wasperformed on 4 ng of cDNA generated from FACS-purified primarymammary cells using an ABI PRISM 7900HT Sequence Detection System(Applied Biosystems, Foster City, CA, USA). TaqMan gene expression assay
mix containing unlabeled PCR primers and FAM labeled TaqMan MGBprobes were used to detect expression of specific genes as listed by thecatalog numbers in Supplementary Table 1. All raw data were analyzedusing Sequence Detection System software Version 2.1 (Applied Biosys-tems). The threshold cycle (CT) values were used to calculate relative RNAexpression levels. Expression levels of target genes were normalized toendogenous β-actin transcripts and compared with control MMTV-Cre orPyMT luminal population (relative expression= 1).
Immunoprecipitation and immunoblot analysisProtein (500 μg) from whole mammary gland extracted by RIPA extractionbuffer (Tris–HCl pH 7.6, 1% Triton X-100, 0.1% SDS, 1% NP-40, 1% sodiumdeoxycholate, 5 mM EDTA, 50mM NaCl) supplemented with 200 μMNa3VO4, 200 μM NaF, 2 mM PMSF and an appropriate dilution of CompleteMini, EDTA-free protease inhibition cocktail tablets (Roche, Indianapolis, IN,USA) was subjected to immunoprecipitation overnight at 4 °C using (1) anantibody that specifically recognizes RRXS*/T*-phosphorylated PKA sub-strate epitopes (Cell Signaling Technology) or (2) a polyclonal antibodyrecognizing the R2β PKA regulatory subunit (ab38949, Abcam, Cambridge,UK). Immunoprecipitated protein was purified following incubation withprotein G plus protein A/G agarose beads (Invitrogen) for 1 h. Theprecipitated protein complexes were washed at 4 °C in RIPA buffer lackingsodium deoxycholate and SDS and was then subjected to SDS–PAGEfollowed by immunoblotting. For immunoblotting, cells were washed inPBS and incubated in RIPA cell extraction buffer, for 30 min. Proteinconcentrations were determined using a BCA kit (Pierce Chemicals;Rockford, IL, USA). Cell protein lysate (30 μg) was resolved by SDS–PAGEand transferred to nitrocellulose membranes. The membranes wereprobed using the following mouse antibodies: anti-AKAP1, anti-phosphorylated CREB (pCreb, Ser133) anti-Creb, Akt/PKB (p-Akt, Ser473),anti-Akt/PKB, anti-phosphorylated Src (Ser17), anti-phosphorylated Src(Tyr416), anti-Src (all from Cell Signaling Technology). The blots werestripped and re-probed with an HRP-conjugated monoclonal antibodydirected against mouse β-actin (Santa Cruz Biotechnology).
siRNA transfectionFour ON-TARGETplus siRNAs (Thermo Scientific; LQ-040877-00-0002; 25 nM)targeting the mouse c-Src mRNA transcript (Src-si1 50-CCAAGGGCCUCAACGUGAA-30 ; Src-si2 50-CCUCAGGCAUGGCGUACGU-30 ; Src-si350-CGUCCAAGCCGCAGACUCA-30 ; Src-si4 50-GAGAACCUGGUGUGCAAAG-30)were transfected into primary mammary epithelial cells using Lipofecta-mine RNAi Max transfection reagent (Life Technologies, Carlsbad, CA, USA)according to the manufacturer’s protocol. Cells transfected with ON-TARGETplus Non-silencing siRNA#1 (NS; Cat# D-001810-01-20) or culturedin the presence of transfection reagent alone, served as negative controls.
Mammary cell proliferation assaysCells were seeded at 2 × 103 cells/well into opaque collagen-coated 96-wellmicroplates in 100 μl of DMEM/F12 1:1 medium containing 2% charcoal-stripped FBS, EGF (5 ng/ml), insulin (10 ng/ml), LA complex (5 μg/ml),penicillin/streptomycin and anti-microbiotic (Wisent, St Bruno, QC,Canada). Cells were cultured for 0, 24, 48, 72 or 96 h in the presence ofcAMP (1.0 mM) or the small molecules H89, PKI, fulvestrant, lapatinib,wortmannin or dasatinib, after which cell proliferation was measured usingthe CellTiter-Glo Luminescent Cell Viability Assay (Promega) following themanufacturer’s instructions on a POLARstar Omega plate reader (BMG;Offenburg, Germany). In vitro Matrigel colony forming assays wereperformed as described45 with modifications. Single mammary cellsuspensions were prepared from individual mice, and 5000 total mammarycells from the inguinal glands of PyMT or Prkar1aΔMam /PyMT were seededonto 80 μL of pre-coated Matrigel in eight-well chamber slides andcultured in DMEM/F12 1:1 medium containing 2% charcoal-stripped FBS,2.5% Matrigel, EGF, insulin and cholera toxin (200 ng/ml). After 24 h, culturemedia was replaced with media containing the small-molecule inhibitorsdescribed above. All untreated cells were supplemented with 1.0% DMSO.Cell colonies were fixed at day 8 of culture in 4% paraformaldehyde for 30min at 4 °C. Colonies were imaged using an inverted light microscopeusing a × 4 objective; colonies were counted in five random fields of view.Colony size (μm2) was determined by measuring the height and width ofeach colony using Image J Software.
PKA-induced Src drives mammary tumorigenesisAG Beristain et al
1171
© 2015 Macmillan Publishers Limited Oncogene (2015) 1160 – 1173

FPLC chromatographyAll procedures were performed at 4 °C. Samples (1.5 mg protein in 500 μlPKA protein buffer) were separated using a 5ml Bio-Scale Mini Macro-PrepDEAE cartridge (Bio-Rad, Mississauga, ON, Canada) on an Akta FPLC System(GE Healthcare, Buckinghamshire, UK) at a flow rate of 1.0 ml/min. Sampleswere loaded onto the column in low-salt buffer (10mM Tris–HCl, 1 mM
EDTA, 1 mM dithiothreitol, pH 7.1). The column was washed with 25ml oflow-salt buffer (five column volumes), collecting 5-ml fractions. The columnwas then eluted with a linear NaCl gradient (0.0–0.8 M) from low-salt bufferto high-salt buffer (10mM Tris-HCl, 1 mM EDTA, 1 mM dithiothreitol, 1.0 MNaCl, pH 7.1) over 25ml (five column volumes), collecting 0.5-ml fractions.The column was cleaned with 25ml (five column volumes) of high-saltbuffer before re-equilibrating to low-salt buffer for the following run.A total of 100 μl of each fraction was precipitated with 200 μl
trichloroacetic acid (50%) at 4 °C for 20min. The precipitated fractionswere centrifuged at 14 000× g, 15 min, 4 °C. Supernatants were discarded,the pellets were washed with 500 μl cold acetone and centrifuged again.The acetone wash was removed, the pellets were air-dried andresuspended in Laemmli buffer (Tris–HCl 375mM, pH 6.8; SDS 9%; glycerol50%; betamercaptoethanol 9%; bromophenol blue 0.03%) where sampleswere resolved by PAGE followed by immunoblotting using antibodiesdirected against R1α (BD Transduction Labs, Lexington, KY, USA) and R2β(BD Transduction Labs). The remaining 400 μl of each fraction wasconcentrated down to 40 μl using 10 -kDa-cuttoff Microcon Centrifugalfilters (Millipore, Billerica, MA, USA). These concentrated protein sampleswhere then used to determine PKA activities (10 μg/reaction).
PKA activity assayThe SignaTech protein kinase (PKA) assay system (Promega) was used tomeasure basal (untreated) and total (cAMP-treated) PKA activities fromtotal mammary gland protein lysates, FACS-purified mammary epithelialcells and concentrated FPLC protein fractions. Briefly, 10 μg of proteinlysate containing 32P-labeled phosphate was incubated with biotinylatedKemptide, a PKA-specific substrate, in the absence or presence of 0.01mM
cAMP. PKA activity specificity was determined using the PKA inhibitor PKI(20 μM). The 32P-labeled biotinylated substrate is recovered from thereaction mix using perforated SAM2 Biotin Capture Membranes that bindto and immobilize the substrate. PKA activity is then measured usingstandard scintillation to determine radioactive counts/min, reflecting theamount of phosphorylated substrate. All determinations of PKA activitywere performed in duplicate, corrected for protein concentration, and anaverage value was calculated for each experiment.
BioinformaticsThe publicly available TCGA data were downloaded from the correspond-ing repository (tcga-data.nci.nih.gov/tcga/tcgaDownload) on July 2012.This set contained expression data for 509 primary tumors, of which 241have also recurrence information available. The TCGA-normalized and pre-processed data were used in subsequent analyses. The PAM50 predictorwas applied as previously described.46 Of the 241 tumors set, 65 wereclassified as basal-like, 30 as HER2, 73 as luminal A, 57 as luminal B and 16as normal-like. The raw MCF10A expression data were downloaded fromthe Gene Expression Omnibus reference GSE3151. Data were normalizedand the significance analysis of microarrays (SAM) algorithm47 was appliedto identify differentially expressed probes at a ⩽ 5% false discovery rate(over-expressed, n=1600; under-expressed, n= 261). Gene expression andclinical data for the NKI data set were downloaded from the StanfordMicrorray Database. The PRKAR1A and SRC genes were represented by asingle probe each in the NKI data set (NM_002734 and NM_005417,respectively). Correlation, clustering and survival analyses were carried outusing R/Bioconductor software (Seattle, WA, USA). The GSEA tool48 was runusing default values for all parameters.
StatisticsData are reported as mean± standard error of the mean. All calculationswere carried out using GraphPad Prism software (La Jolla, CA, USA).Comparisons were made by two-tailed Student’s t-test and ANOVA. Thedifferences were accepted as significant at Po0.05.
CONFLICT OF INTERESTThe authors declare no conflict of interest.
ACKNOWLEDGEMENTSWe thank Megan K Barker and Paul Waterhouse for critical reading of the manuscript.We would also like to acknowledge Shareen Ezzat for sharing the small moleculeDasatinib, Vuk Stambolic for offering insight into biochemistry experiments and theIDIBELL’s Biostatistics Unit for help in analyzing breast cancer data sets. This work wassupported by a Canadian Breast Cancer Foundation (CBCF) grant to RK and theSpanish Ministry of Health grant FIS-PI12/01528 and RD12/0036/0008 to MAP. AGBholds a CBCF fellowship.
REFERENCES1 Nadella KS, Jones GN, Trimboli A, Stratakis CA, Leone G, Kirschner LS. Targeted
deletion of Prkar1a reveals a role for protein kinase A in mesenchymal-to-epithelial transition. Cancer Res 2008; 68: 2671–2677.
2 Tharakan R, Lepont P, Singleton D, Kumar R, Khan S. Phosphorylation of estrogenreceptor alpha, serine residue 305 enhances activity. Mol Cell Endocrinol 2008;295: 70–78.
3 Yin Z, Jones GN, Towns WH, Zhang X, Abel ED, Binkley PF et al. Heart-specificablation of Prkar1a causes failure of heart development and myxomagenesis.Circulation 2008; 117: 1414–1422.
4 Almeida MQ, Stratakis CA. How does cAMP/protein kinase A signaling lead totumors in the adrenal cortex and other tissues? Mol Cell Endocrinol 2011; 336:162–168.
5 Kok M, Zwart W, Holm C, Fles R, Hauptmann M, Van’t Veer LJ et al. PKA-inducedphosphorylation of ERα at serine 305 and high PAK1 levels is associated withsensitivity to tamoxifen in ER-positive breast cancer. Breast Cancer Res Treat 2011;125: 1–12.
6 Moujalled D, Weston R, Anderton H, Ninnis R, Goel P, Coley A et al. Cyclic-AMP-dependent protein kinase A regulates apoptosis by stabilizing the BH3-onlyprotein Bim. EMBO Rep 2011; 12: 77–83.
7 Neary CL, Nesterova M, Cho YS, Cheadle C, Becker KG, Cho-Chung YS. Proteinkinase A isozyme switching: eliciting differential cAMP signaling and tumorreversion. Oncogene 2004; 23: 8847–8856.
8 Miller WR. Regulatory subunits of PKA and breast cancer. Ann NY Acad Sci 2002;968: 37–48.
9 Kirschner LS, Kusewitt DF, Matyakhina L, Towns WH, Carney JA, Westphal H et al.A mouse model for the Carney complex tumor syndrome develops neoplasia incyclic AMP-responsive tissues. Cancer Res 2005; 65: 4506–4514.
10 Almeida MQ, Muchow M, Boikos S, Bauer AJ, Griffin KJ, Tsang KM et al. MousePrkar1a haploinsufficiency leads to an increase in tumors in the Trp53+/� orRb1+/� backgrounds and chemically induced skin papillomas by dysregulation ofthe cell cycle and Wnt signaling. Hum Mol Genet 2010; 19: 1387–1398.
11 Bossis I, Stratakis CA. Minireview: PRKAR1A: normal and abnormal functions.Endocrinology 2004; 145: 5452–5458.
12 Kirschner LS, Carney JA, Pack SD, Taymans SE, Giatzakis C, Cho YS et al. Mutationsof the gene encoding the protein kinase A type I-alpha regulatory subunit inpatients with the Carney complex. Nat Genet 2000; 26: 89–92.
13 Kirschner LS, Sandrini F, Monbo J, Lin JP, Carney JA, Stratakis CA. Genetic het-erogeneity and spectrum of mutations of the PRKAR1A gene in patients with thecarney complex. Hum Mol Genet 2000; 9: 3037–3046.
14 Amieux PS, Mcknight GS. The essential role of RI alpha in the maintenance ofregulated PKA activity. Ann NY Acad Sci 2002; 968: 75–95.
15 Griffin KJ, Kirschner LS, Matyakhina L, Stergiopoulos SG, Robinson-White A,Lenherr SM et al. A transgenic mouse bearing an antisense construct of regulatorysubunit type 1A of protein kinase A develops endocrine and other tumours:comparison with Carney complex and other PRKAR1A induced lesions. J MedGenet 2004; 41: 923–931.
16 Wilson GR, Cramer A, Welman A, Knox F, Swindell R, Kawakatsu H et al. Activatedc-SRC in ductal carcinoma in situ correlates with high tumour grade, high pro-liferation and HER2 positivity. Br J Cancer 2006; 95: 1410–1414.
17 Elsberger B, Tan BA, Mitchell TJ, Brown SBF, Mallon EA, Tovey SM et al. Isexpression or activation of Src kinase associated with cancer-specific survival inER-, PR- and HER2-negative breast cancer patients? Am J Pathol 2009; 175:1389–1397.
18 Molyneux SD, Di Grappa MA, Beristain AG, Mckee TD, Wai DH, Paderova J et al.Prkar1a is an osteosarcoma tumor suppressor that defines a molecular subclassin mice. J Clin Invest 2010; 120: 3310–3325.
19 Gatza ML, Lucas JE, Barry WT, Kim JW, Wang Q, Crawford MD et al. A pathway-based classification of human breast cancer. Proc Natl Acad Sci USA 2010; 107:6994–6999.
20 Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ et al. Thegenomic and transcriptomic architecture of 2,000 breast tumours reveals novelsubgroups. Nature 2012; 486: 346–352.
PKA-induced Src drives mammary tumorigenesisAG Beristain et al
1172
Oncogene (2015) 1160 – 1173 © 2015 Macmillan Publishers Limited

21 Lin EY, Jones JG, Li P, Zhu L, Whitney KD, Muller WJ et al. Progression to malig-nancy in the polyoma middle T oncoprotein mouse breast cancer model providesa reliable model for human diseases. Am J Pathol 2003; 163: 2113–2126.
22 Guy CT, Webster MA, Schaller M, Parsons TJ, Cardiff RD, Muller WJ. Expression ofthe neu protooncogene in the mammary epithelium of transgenic mice inducesmetastatic disease. Proc Natl Acad Sci USA 1992; 89: 10578–10582.
23 Griffin KJ, Kirschner LS, Matyakhina L, Stergiopoulos S, Robinson-White A,Lenherr S et al. Down-regulation of regulatory subunit type 1A of protein kinase Aleads to endocrine and other tumors. Cancer Res 2004; 64: 8811–8815.
24 Smalley MJ, Kendrick H, Sheridan JM, Regan JL, Prater MD, Lindeman GJ et al.Isolation of mouse mammary epithelial subpopulations: a comparison of leadingmethods. J Mammary Gland Biol Neoplasia 2012; 17: 91–97.
25 Fink MA, Zakhary DR, Mackey JA, Desnoyer RW, Apperson-Hansen C, Damron DSet al. AKAP-mediated targeting of protein kinase a regulates contractility incardiac myocytes. Circulation Res 2001; 88: 291–297.
26 Burton KA, Johnson BD, Hausken ZE, Westenbroek RE, Idzerda RL, Scheuer T et al.Type II regulatory subunits are not required for the anchoring-dependent mod-ulation of Ca2+ channel activity by cAMP-dependent protein kinase. Proc NatlAcad Sci USA 1997; 94: 11067–11072.
27 Lim CJ, Han J, Yousefi N, Ma Y, Amieux PS, Mcknight GS et al. Alpha4 integrins aretype I cAMP-dependent protein kinase-anchoring proteins. Nat Cell Biol 2007; 9:415–421.
28 Gold MG, Fowler DM, Means CK, Pawson CT, Stephany JJ, Langeberg LK et al.Engineering A-kinase anchoring protein (AKAP)-selective regulatory subunits ofprotein kinase A (PKA) through structure-based phage selection. J Biol Chem 2013;288: 17111–17121.
29 Subbaramaiah K, Hudis C, Chang S-H, Hla T, Dannenberg AJ. EP2 and EP4receptors regulate aromatase expression in human adipocytes and breast cancercells. Evidence of a BRCA1 and p300 exchange. J Biol Chem 2008; 283: 3433–3444.
30 Baker MA, Hetherington L, Aitken RJ. Identification of SRC as a key PKA-stimulatedtyrosine kinase involved in the capacitation-associated hyperactivation of murinespermatozoa. J Cell Sci 2006; 119: 3182–3192.
31 Marcotte R, Smith HW, Sanguin-Gendreau V, McDonough RV, Muller WJ.Mammary epithelial-specific disruption of c-Src impairs cell cycle progression andtumorigenesis. Proc Natl Acad Sci USA 2012; 109: 2808–2813.
32 Soulard A, Cremonesi A, Moes S, Schutz F, Jenö P, Hall MN. The rapamycin-sensitive phosphoproteome reveals that TOR controls protein kinase A towardsome but not all substrates. Mol Biol Cell 2010; 21: 3475–3486.
33 Obara Y, Labudda K, Dillon TJ, Stork PJS. PKA phosphorylation of Src mediatesRap1 activation in NGF and cAMP signaling in PC12 cells. J Cell Sci 2004; 117:6085–6094.
34 Cancer Genome Atlas Network, Comprehensive molecular portraits of humanbreast tumours. Nature 2012; 490: 61–70.
35 Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D et al. Oncogenic pathwaysignatures in human cancers as a guide to targeted therapies. Nature 2006; 439:353–357.
36 van de Vijver MJ, He YD, Van’t Veer LJ, Dai H, Hart AAM, Voskuil DW et al. A gene-expression signature as a predictor of survival in breast cancer. New Engl J Med2002; 347: 1999–2009.
37 Pavel E, Nadella K, Towns WH, Kirschner LS. Mutation of Prkar1a causes osteoblastneoplasia driven by dysregulation of protein kinase A. Mol Endocrinol 2008; 22:430–440.
38 Carney JA, Toorkey BC. Ductal adenoma of the breast with tubular features.A probable component of the complex of myxomas, spotty pigmentation,endocrine overactivity, and schwannomas. Am J Surg Pathol 1991; 15:722–731.
39 Michalides R, Griekspoor A, Balkenende A, Verwoerd D, Janssen L, Jalink K et al.Tamoxifen resistance by a conformational arrest of the estrogen receptor alphaafter PKA activation in breast cancer. Cancer Cell 2004; 5: 597–605.
40 Subbaramaiah K, Morris PG, Zhou XK, Morrow M, Du B, Giri D et al. Increasedlevels of COX-2 and prostaglandin E2 contribute to elevated aromataseexpression in inflamed breast tissue of obese women. Cancer Discov 2012; 2:356–365.
41 Subbaramaiah K, Howe LR, Bhardwaj P, Du B, Gravaghi C, Yantiss RK et al. Obesityis associated with inflammation and elevated aromatase expression in the mousemammary gland. Cancer Prev Res 2011; 4: 329–346.
42 Schmitt JM, Stork PJS. PKA phosphorylation of Src mediates cAMP's inhibition ofcell growth via Rap1. Mol Cell 2002; 9: 85–94.
43 Prat A, Parker JS, Fan C, Perou CM. PAM50 assay and the three-gene model foridentifying the major and clinically relevant molecular subtypes of breast cancer.Breast Cancer Res Treat 2012; 135: 301–306.
44 Joshi PA, Jackson HW, Beristain AG, Di Grappa MA, Mote PA, Clarke CL et al.Progesterone induces adult mammary stem cell expansion. Nature 2010; 465:803–807.
45 Muthuswamy SK, Li D, Lelievre S, Bissell MJ, Brugge JS. ErbB2 but not ErbB1,reinitiates proliferation and induces luminal repopulation in epithelial acini. NatCell Biol 2001; 3: 785–792.
46 Parker JS, Mullins M, Cheang MCU, Leung S, Voduc D, Vickery T et al. Supervisedrisk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 2009; 27:1160–1167.
47 Tusher VG, Tibshirani R, Chu G. Significance analysis of microarraysapplied to the ionizing radiation response. Proc Natl Acad Sci USA 2001; 98:5116–5121.
48 Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA et al.Gene set enrichment analysis: a knowledge-based approach for interpretinggenome-wide expression profiles. Proc Natl Acad Sci USA 2005; 102: 15545–15550.
Supplementary Information accompanies this paper on the Oncogene website (http://www.nature.com/onc)
PKA-induced Src drives mammary tumorigenesisAG Beristain et al
1173
© 2015 Macmillan Publishers Limited Oncogene (2015) 1160 – 1173
Copyright © 2022 FDOKUMEN