
Activation of p53 transcriptional activity requires ATM's kinase domain and multiple N-terminal...
11
Activation of p53 transcriptional activity requires ATM’s kinase domain and multiple N-terminal serine residues of p53 Gaetan A Turenne 1 , Proma Paul 1 , Lareina Laflair 1 and Brendan D Price* ,1 Department of Radiation Oncology, D810A, Dana-Farber Cancer Institute, Harvard Medical School, 44 Binney Street, Boston, Massachusetts, MA 02115, USA The ATM protein kinase regulates the cell’s response to DNA damage by regulating cell cycle checkpoints and DNA repair. ATM phosphorylates several proteins involved in the DNA-damage response, including p53. We have examined the mechanism by which ATM regulates p53’s transcriptional activity. Here, we demon- strate that reintroduction of ATM into AT cells restores the activation of p53 by the radio-mimetic agent bleomycin. Further, p53 activation is lost when a kinase inactive ATM is used, or if the N-terminal of ATM is deleted. In addition, AT cells stably expressing ATM showed decreased sensitivity to Ionizing Radiation- induced cell killing, whereas cells expressing kinase inactive ATM or N-terminally deleted ATM were indistinguishable from AT cells. Finally, single point- mutations of serines 15, 20, 33 or 37 did not individually block the ATM-dependent activation of p53 transcrip- tional activity by bleomycin. However, double mutations of either serines 15 and 20 or serines 33 and 37 blocked the ability of ATM to activate p53. Our results indicate that the N-terminal of ATM and ATM’s kinase activity are required for activation of p53’s transcriptional activity and restoration of normal sensitivity to DNA damage. In addition, activation of p53 by ATM requires multiple serine residues in p53’s transactivation domain. Oncogene (2001) 20, 5100 – 5110. Keywords: ATM; p53; chk2; phosphorylation; bleomy- cin Introduction In response to DNA damage, mammalian cells activate cell cycle checkpoints (Canman and Lim, 1998a; Weinert, 1998). These checkpoints block DNA syn- thesis or chromosome segregation if the DNA is damaged (Jeggo et al., 1998; Meyn, 1999; Weinert, 1998), which may allow cells extra time to repair DNA damage. The ATM protein is a key regulator of the DNA-damage response (Savitsky et al., 1995). Muta- tions in the ATM protein give rise to the inherited disease Ataxia Telangiectasia (AT). AT is characterized by immunodeficiency, cerebellar ataxia, increased incidence of cancer and extreme sensitivity to Ionizing Radiation (Beamish and Lavin, 1994; Beamish et al., 1994; Luo et al., 1996; Meyn, 1999; Taylor et al., 1975). AT cells exhibit increased sensitivity to IR, radioresistant DNA synthesis and increased genetic instability (Cornforth and Bedford, 1985; Meyn, 1999; Taylor et al., 1975). The kinetics of DNA double- strand break repair is grossly normal in AT cells, although the cells may contain residual levels of double-strand breaks (Jeggo et al., 1998). AT cells also exhibit high rates of spontaneous DNA recombination, poor fidelity during repair (Luo et al., 1996), and do not undergo potentially lethal damage repair (Weich- selbaum et al., 1977). Further, AT cells have elevated levels of chromosomal abnormalities, including short- ened telomeres and chromosome end joining (Metcalfe et al., 1996; Pandita et al., 1995). The ATM protein is a 3056 amino-acid protein containing a C-terminal kinase domain (Savitsky et al., 1995). ATM can phosphorylate several proteins involved in DNA repair, including p95/nibrin, (which may regulate the S phase check point; Lim et al., 2000; Zhao et al., 2000) and Brca1, a protein involved in DNA repair (Cortez et al., 1999). ATM can also directly phosphorylate two key regulators of cell cycle checkpoints – the p53 tumor suppressor gene (Banin et al., 1998; Canman et al., 1998; Kastan et al., 1991; 1992) and the checkpoint kinase chk2 (Blasina et al., 1999a; Brown et al., 1999; Matsuoka et al., 1998). chk2 is involved in the activation of the G2- checkpoint (Blasina et al., 1999b; Brown et al., 1999; Matsuoka et al., 1999). Exit from G2 requires activation of cyclin B/cdc2 through the tyrosine dephosphorylation of cdc2 by cdc25 phosphatase (reviewed in Rhind and Russell, 1998). In cells exposed to Ionizing Radiation, ATM phosphorylates chk2, activating chk2’s kinase activity (Blasina et al., 1999a; Brown et al., 1999; Matsuoka et al., 1998). Activated chk2 then phosphorylates the cdc25 tyrosine phospha- tase, inhibiting its activity (Blasina et al., 1999b; Furnari et al., 1999). Cyclin B/cdc2 therefore remains inactive, and the cells are arrested in G2 (Blasina et al., 1999b; Furnari et al., 1999). If AT cells are irradiated in G2-M, they exhibit a much shorter G2 arrest than Oncogene (2001) 20, 5100 – 5110 ª 2001 Nature Publishing Group All rights reserved 0950 – 9232/01 $15.00 www.nature.com/onc *Correspondence: BD Price; E-mail: [email protected] Received 13 February 2001; revised 17 May 2001; accepted 24 May 2001
Transcript of Activation of p53 transcriptional activity requires ATM's kinase domain and multiple N-terminal...

Activation of p53 transcriptional activity requires ATM's kinase domainand multiple N-terminal serine residues of p53
Gaetan A Turenne1, Proma Paul1, Lareina La¯air1 and Brendan D Price*,1
Department of Radiation Oncology, D810A, Dana-Farber Cancer Institute, Harvard Medical School, 44 Binney Street, Boston,Massachusetts, MA 02115, USA
The ATM protein kinase regulates the cell's response toDNA damage by regulating cell cycle checkpoints andDNA repair. ATM phosphorylates several proteinsinvolved in the DNA-damage response, including p53.We have examined the mechanism by which ATMregulates p53's transcriptional activity. Here, we demon-strate that reintroduction of ATM into AT cells restoresthe activation of p53 by the radio-mimetic agentbleomycin. Further, p53 activation is lost when a kinaseinactive ATM is used, or if the N-terminal of ATM isdeleted. In addition, AT cells stably expressing ATMshowed decreased sensitivity to Ionizing Radiation-induced cell killing, whereas cells expressing kinaseinactive ATM or N-terminally deleted ATM wereindistinguishable from AT cells. Finally, single point-mutations of serines 15, 20, 33 or 37 did not individuallyblock the ATM-dependent activation of p53 transcrip-tional activity by bleomycin. However, double mutationsof either serines 15 and 20 or serines 33 and 37 blockedthe ability of ATM to activate p53. Our results indicatethat the N-terminal of ATM and ATM's kinase activityare required for activation of p53's transcriptionalactivity and restoration of normal sensitivity to DNAdamage. In addition, activation of p53 by ATM requiresmultiple serine residues in p53's transactivation domain.Oncogene (2001) 20, 5100 ± 5110.
Keywords: ATM; p53; chk2; phosphorylation; bleomy-cin
Introduction
In response to DNA damage, mammalian cells activatecell cycle checkpoints (Canman and Lim, 1998a;Weinert, 1998). These checkpoints block DNA syn-thesis or chromosome segregation if the DNA isdamaged (Jeggo et al., 1998; Meyn, 1999; Weinert,1998), which may allow cells extra time to repair DNAdamage. The ATM protein is a key regulator of theDNA-damage response (Savitsky et al., 1995). Muta-
tions in the ATM protein give rise to the inheriteddisease Ataxia Telangiectasia (AT). AT is characterizedby immunode®ciency, cerebellar ataxia, increasedincidence of cancer and extreme sensitivity to IonizingRadiation (Beamish and Lavin, 1994; Beamish et al.,1994; Luo et al., 1996; Meyn, 1999; Taylor et al.,1975). AT cells exhibit increased sensitivity to IR,radioresistant DNA synthesis and increased geneticinstability (Cornforth and Bedford, 1985; Meyn, 1999;Taylor et al., 1975). The kinetics of DNA double-strand break repair is grossly normal in AT cells,although the cells may contain residual levels ofdouble-strand breaks (Jeggo et al., 1998). AT cells alsoexhibit high rates of spontaneous DNA recombination,poor ®delity during repair (Luo et al., 1996), and donot undergo potentially lethal damage repair (Weich-selbaum et al., 1977). Further, AT cells have elevatedlevels of chromosomal abnormalities, including short-ened telomeres and chromosome end joining (Metcalfeet al., 1996; Pandita et al., 1995).
The ATM protein is a 3056 amino-acid proteincontaining a C-terminal kinase domain (Savitsky et al.,1995). ATM can phosphorylate several proteinsinvolved in DNA repair, including p95/nibrin, (whichmay regulate the S phase check point; Lim et al., 2000;Zhao et al., 2000) and Brca1, a protein involved inDNA repair (Cortez et al., 1999). ATM can alsodirectly phosphorylate two key regulators of cell cyclecheckpoints ± the p53 tumor suppressor gene (Baninet al., 1998; Canman et al., 1998; Kastan et al., 1991;1992) and the checkpoint kinase chk2 (Blasina et al.,1999a; Brown et al., 1999; Matsuoka et al., 1998).
chk2 is involved in the activation of the G2-checkpoint (Blasina et al., 1999b; Brown et al., 1999;Matsuoka et al., 1999). Exit from G2 requiresactivation of cyclin B/cdc2 through the tyrosinedephosphorylation of cdc2 by cdc25 phosphatase(reviewed in Rhind and Russell, 1998). In cells exposedto Ionizing Radiation, ATM phosphorylates chk2,activating chk2's kinase activity (Blasina et al., 1999a;Brown et al., 1999; Matsuoka et al., 1998). Activatedchk2 then phosphorylates the cdc25 tyrosine phospha-tase, inhibiting its activity (Blasina et al., 1999b;Furnari et al., 1999). Cyclin B/cdc2 therefore remainsinactive, and the cells are arrested in G2 (Blasina et al.,1999b; Furnari et al., 1999). If AT cells are irradiatedin G2-M, they exhibit a much shorter G2 arrest than
Oncogene (2001) 20, 5100 ± 5110ã 2001 Nature Publishing Group All rights reserved 0950 ± 9232/01 $15.00
www.nature.com/onc
*Correspondence: BD Price;E-mail: [email protected] 13 February 2001; revised 17 May 2001; accepted 24 May2001

normal cells and exit rapidly into G1 (Beamish et al.,1994; Nagasawa et al., 1994). Thus the defective G2checkpoint seen in AT cells may be seen, at least inpart, as the failure of ATM to activate chk2 followingDNA damage.
AT cells also fail to activate the p53-dependent G1checkpoint (Kastan et al., 1991; 1992). In normal cells,exposure to Ionizing Radiation increases p53 proteinlevels by decreasing the proteolytic degradation of p53(Haupt et al., 1997; Kastan et al., 1991; Kubbutata etal., 1998; Tishler et al., 1993). Degradation of p53 ismediated by the mdm2 oncogene (Haupt et al., 1997;Kubbutata et al., 1998), such that mdm2-p53 com-plexes are targeted for ubiquitin dependent degrada-tion. The full activation of p53 by Ionizing Radiationrequires the ATM protein (Kastan et al., 1992), andATM protein can phosphorylate serine 15 of the p53protein in vitro (Banin et al., 1998; Canman et al.,1998). Further, serines 15 and 20 of p53 arephosphorylated in vivo following exposure of normal,but not AT cells, to Ionizing Radiation (Chebab et al.,1999; Sakaguchi et al., 1998; Shieh et al., 1997, 1999;Siliciano et al., 1997; Unger et al., 1999a). Sincephosphorylation of serine 20 of p53 is carried out bychk2 kinase (Chebab et al., 2000; Hirao et al., 2000;Tominaga et al., 1999), ATM therefore mediates boththe direct phosphorylation of serine 15 and indirectlyregulates serine 20 phosphorylation by controllingactivation of chk2. There are additional sites forDNA damage induced phosphorylation at serines 33and 37 of p53 (Sakaguchi et al., 1998; Shieh et al.,1997; 1999; Siliciano et al., 1997), which may beregulated by Atr and other kinases (Hall-Jackson et al.,1999; Tibbets et al., 1999). The phosphorylation ofp53's N-terminal has been shown to block binding ofmdm2, and therefore increase the stability of the p53protein. More detailed work has shown that phosphor-ylation of serine-20 is the key-regulator of mdm2-p53interaction, with other residues, including serines 15, 33and 37 playing a lesser role (Chebab et al., 1999; Shiehet al., 1997; Unger et al., 1999a). mdm2 is alsophosphorylated by ATM, and this may furthercontribute to the stabilization of p53 followingexposure to Ionizing Radiation (Khosravi et al.,1999). Thus the control of p53 activation by ATM iscomplex, involving direct phosphorylation of serine 15by ATM, phosphorylation of serine 20 by chk2 (whichis activated by ATM) and further by direct phosphor-ylation of mdm2 by ATM. In addition, DNA damageincreases the phosphorylation of serines 33 and 37 ofp53 through an ATM-independent mechanism (Saka-guchi et al., 1998). These phosphorylations are thoughtto prevent mdm2 binding and leads to accumulation ofp53 protein in the cell. Although accumulation of p53protein is the initial step in the ATM-dependentactivation of p53, subsequent steps, including activa-tion of p53's DNA binding and changes in p53'stranscriptional regulatory activity are also involved.For example, p53's DNA binding activity is increasedby the Ionizing Radiation-induced acetylation of the C-terminal of p53, and this acetylation requires the prior
phosphorylation of the N-terminal of p53 (Sakaguchiet al., 1998). Serines 15, 20, 33 and 37 are all within theN-terminal transactivation domain of p53, and phos-phorylation of some or all of these serine residues maybe required to stimulate transcriptional activation ofp53 target genes. However, the role of serines 15, 20,33 and 37 in regulating the ATM-dependent activationof p53 transcriptional activity is not known.
In this study, we have determined how ATM'skinase activity regulates activation of p53's transcrip-tional activity, and identi®ed the serine residues in theN-terminal of p53 which mediate this e�ect. Using akinase inactive ATM construct, we demonstrate thatthe kinase activity of ATM is required for activation ofp53 and chk2 kinase, and for the restoration of normalradiosensitivity in AT cells. Further, we have identi®edan N-terminal region of ATM which is required forcorrect ATM functioning. In addition, we demonstratethat double-mutations of either serines 15 and 20 orserines 33 and 37 abolishes the ATM-dependentactivation of p53 by ATM.
Results
To examine how ATM activates p53, we generatedseveral deletion constructs of the ATM protein (Figure1a). ATM constructs were inserted into the pcDNA3.1/HisA expression vector, which adds the Omni tag tothe N-terminal of ATM. The construction of full-
Figure 1 Construction and expression of ATM constructs. (a)Map of ATM constructs showing location of key domains andsize of fragments. FL-AT encodes amino-acids 1-3056, FL-ATkdencodes the triple mutation D2879A/N2884K/D2898A, AT-DNencodes amino-acids 769-3056 and ATK encodes amino-acids2138-3056. =Omni Tag, =Leucine zipper, =kinasedomain. (b) Left panel. GM5849 cells were transiently transfectedwith vector (C) or FL-AT (AT), immunoprecipitated with goatIgG (lanes 1 and 2) or Omni-tag antibody (lanes 3 and 4) andATM detected by Western blotting with anti-Omni antibody.Center and right panels. GM5849 AT cells expressing vector(pcDNA3.1/HisA), FL-AT, FL-ATkd, AT-DN, ATK or ATKkdwere immunoprecipitated with anti-Omni antibody and analysedby Western blotting using Omni antibody
Oncogene
ATM's kinase activity is required to activate p53GA Turenne et al
5101

length ATM (FL-AT) and kinase inactive ATM (FL-ATkd) and the demonstration that FL-ATkd does notdisplay kinase activity have been previously reportedby us (Blasina et al., 1999a). Two further constructswere prepared, described in Figure 1. The N-terminalregion of ATM has been reported to contain a site forinteraction with both p53 (Khanna et al., 1998) andhistone deacetylase (Kim et al., 1999). In AT-DN, theN-terminal of ATM (amino-acids 1-768) was deleted toexamine the role of this region in p53 activation.Further reports have indicated that the kinase domainof ATM may be su�cient to restore normal radio-sensitivity to AT cells (Morgan et al., 1997). To explorethis possibility, ATK, which encodes amino-acids2138 ± 3056 of ATM, including the kinase domain,was constructed.
The speci®city of the Omni antibody was examinedby transiently expressing either vector or FL-AT in theAtaxia Telangiectasia cell line GM5849. ATM wasonly detected in cells transfected with FL-AT and wasspeci®cally immunoprecipitated by the Omni antibodybut not by IgG (Figure 1b, left panel). In Figure 1b,AT cells stably expressing the indicated construct wereimmunoprecipitated with Omni antibody and examinedby Western blotting. Vector transformed cells did notdisplay any immunoreactive protein (Figure 1b, centerpanel), whereas ATM was clearly detected in cellsexpressing FL-AT, FL-ATkd and AT-DN. Expressionof the kinase domain of ATM, ATK, and the kinaseinactive version, ATkd, was also seen. Note that ATkdhad a slightly increased mobility on SDS ±PAGE,presumably due to the triple mutation in the kinasedomain which results in the net loss of two negativelycharged amino-acids.
Prior to examining the ability of these ATMconstructs to activate p53, we examined the ability ofeach to complement the increased sensitivity of ATcells to IR. Cell lines stably expressing these ATMconstructs were exposed to increasing amounts ofIonizing Radiation and clonogenic cell survivalmeasured. GM5849 AT cells containing only theselection marker were very sensitive to radiation(Figure 2a,*). Cells expressing FL-AT showed astatistically signi®cant increase in cell survival follow-ing exposure to Ionizing Radiation (Figure 2a,*)whereas AT cells expressing the kinase inactive ATMconstruct were not statistically di�erent from controlcells (Figure 2a,D). AT cells typically display a 3 ± 8-fold increase in sensitivity to DNA damage (Beamishand Lavin, 1994; Beamish et al., 1994; Taylor et al.,1975). In our complementation studies, we were onlyable to achieve about a twofold increase in cell survivalfollowing exposure to Ionizing Radiation (Figure 2a).This is similar to other published results, where stabletransfection of ATM into AT cells increased cellsurvival by only 2 ± 3-fold (Zhang et al., 1997; Ziv etal., 1997). This low level of complementation mayderive from our use of pooled clonal populations,which may express heterogeneous levels of ATMprotein and therefore of radiosensitivity. In addition,AT cells are genetically unstable (Meyn, 1999) and will
have accumulated numerous genetic changes duringgrowth in culture. These changes may a�ect theinherent radiosensitivity of the AT cells, and reintro-duction of ATM may not completely correct this.
To examine if the kinase domain alone was su�cientto complement the increased sensitivity of AT cells,GM5849 AT cells expressing ATK or AT-DN wereexposed to increasing doses of Ionizing Radiation andclonogenic cell survival measured. Neither ATK,containing approximately 900 amino-acids from theC-terminal, or AT-DN increased the ability of AT cellsto survive radiation induced cell death (Figure 2B).Figure 2 therefore demonstrates that the kinase domainof ATM as well sequences in the N-terminal arerequired for ATM to mediate cell survival followingDNA damage.
The induction of transcriptionally active p53 byDNA damage involves at least two steps. First, p53'sstability is increased by inhibiting the interactionbetween mdm2 and p53 (Sakaguchi et al., 1998; Shiehet al., 1999; Siliciano et al., 1997; Unger et al., 1999a)and second, there is activation of p53's DNA bindingactivity and increased transcription of p53 target genes(Tishler et al., 1993). In AT cells, activation of p53 isdefective, and there is little or no increase intranscriptional activity (Kastan et al., 1991, 1992). Toanalyse the mechanism by which ATM activates p53'stranscriptional activity, we employed the SV40 trans-formed AT ®broblast cell line GM5849. AlthoughGM5849 AT cells express wtp53 (Jung et al., 1997),they also express the SV40Tag, which binds to theDNA binding domain of p53 and inactivates p53 (Hessand Brandner, 1997; Kohli and Jorgensen, 1999;O'Neil et al., 1996). However, several reports demon-strate that the p53 response to DNA damage is intactin SV40 transformed human ®broblast cells, includingthe activation of G1 arrest (Kohli and Jorgensen, 1999;O'Neil et al., 1996), phosphorylation of serines 15 and37 of p53 (Tibbets et al., 1999), activation of p53-DNAbinding activity and increased transcriptional activity(Hess and Brandner, 1997; Kohli and Jorgensen, 1999;O'Neil et al., 1996; Sheppard and Liu, 1999). Further,a signi®cant proportion of the p53 in these cells is notassociated with the SV40Tag (O'Neil et al., 1996),indicating that these cells contain a pool of free wtp53which can potentially be regulated in response to DNAdamage. In Figure 3a, we ®rst determined if GM5849AT cells contained p53 which was not bound toSV40Tag. AT cell extracts were immunodepleted ofSV40Tag by three sequential immunoprecipitationswith anti-SV40Tag antibody (Figure 3a, lanes 1 ± 3),followed by immunoprecipitation with anti-p53 anti-body (Figure 3a, lane 4). The immunoprecipitatedprotein was then examined by Western blot for bothSV40Tag and p53 protein (Figure 3a). The ®rstimmunoprecipitation with SV40Tag antibody pulleddown both Tag and p53, indicating that both proteinsexist as a complex, as previously described (O'Neil etal., 1996). Two further immunoprecipitations withSV40Tag antibody did not yield any additional p53or SV40Tag. When the SV40Tag depleted extracts were
ATM's kinase activity is required to activate p53GA Turenne et al
5102
Oncogene
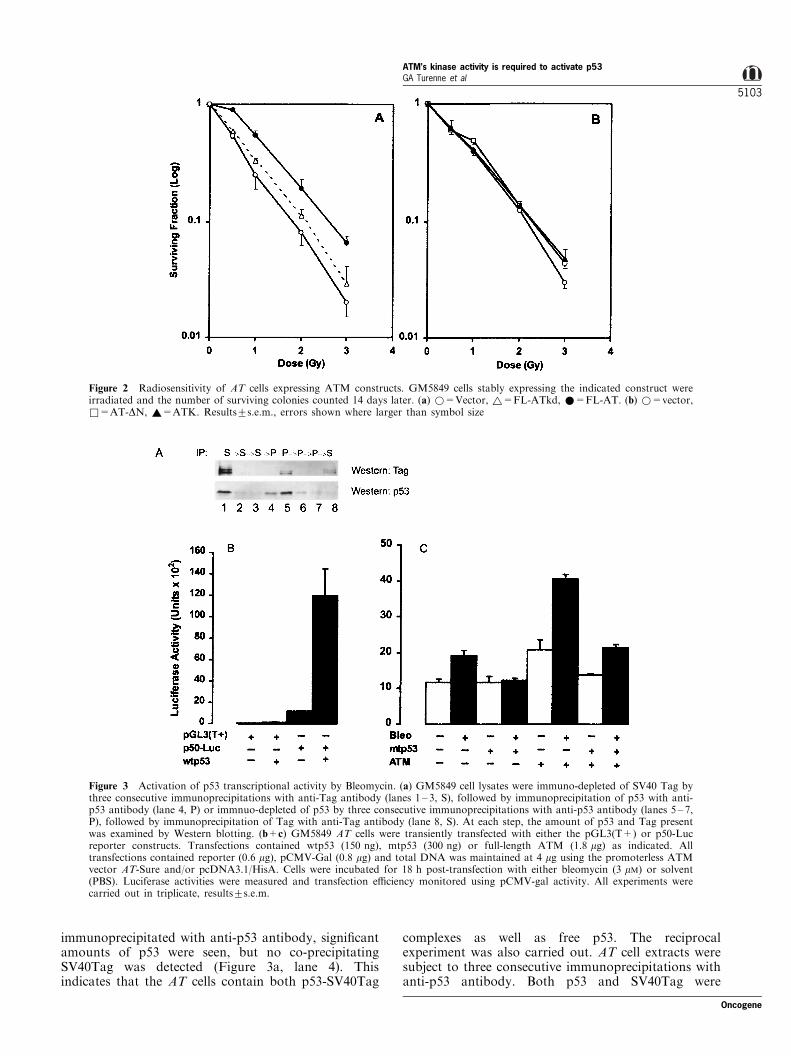
immunoprecipitated with anti-p53 antibody, signi®cantamounts of p53 were seen, but no co-precipitatingSV40Tag was detected (Figure 3a, lane 4). Thisindicates that the AT cells contain both p53-SV40Tag
complexes as well as free p53. The reciprocalexperiment was also carried out. AT cell extracts weresubject to three consecutive immunoprecipitations withanti-p53 antibody. Both p53 and SV40Tag were
Figure 3 Activation of p53 transcriptional activity by Bleomycin. (a) GM5849 cell lysates were immuno-depleted of SV40 Tag bythree consecutive immunoprecipitations with anti-Tag antibody (lanes 1 ± 3, S), followed by immunoprecipitation of p53 with anti-p53 antibody (lane 4, P) or immnuo-depleted of p53 by three consecutive immunoprecipitations with anti-p53 antibody (lanes 5 ± 7,P), followed by immunoprecipitation of Tag with anti-Tag antibody (lane 8, S). At each step, the amount of p53 and Tag presentwas examined by Western blotting. (b+c) GM5849 AT cells were transiently transfected with either the pGL3(T+) or p50-Lucreporter constructs. Transfections contained wtp53 (150 ng), mtp53 (300 ng) or full-length ATM (1.8 mg) as indicated. Alltransfections contained reporter (0.6 mg), pCMV-Gal (0.8 mg) and total DNA was maintained at 4 mg using the promoterless ATMvector AT-Sure and/or pcDNA3.1/HisA. Cells were incubated for 18 h post-transfection with either bleomycin (3 mM) or solvent(PBS). Luciferase activities were measured and transfection e�ciency monitored using pCMV-gal activity. All experiments werecarried out in triplicate, results+s.e.m.
Figure 2 Radiosensitivity of AT cells expressing ATM constructs. GM5849 cells stably expressing the indicated construct wereirradiated and the number of surviving colonies counted 14 days later. (a) *=Vector, ~=FL-ATkd, *=FL-AT. (b) *=vector,&=AT-DN, ~=ATK. Results+s.e.m., errors shown where larger than symbol size
Oncogene
ATM's kinase activity is required to activate p53GA Turenne et al
5103

detected after the ®rst immunoprecipitation with p53antibody, and subsequent immunoprecipitations re-vealed only small amounts of p53 (Figure 3a, lanes5 ± 7). When the extracts which were immunodepletedof p53 were further immunoprecipitated with SV40Tagantibody, free SV40tAG was detected but no copreci-pitating p53 was seen (Figure 3a, lane 8). Thisinteraction between p53 and SV40Tag was una�ectedby either reintroduction of the ATM gene or exposureof the cells to the DNA-damaging agent bleomycin,indicating that ATM and DNA damage do notmeasurably alter the association between p53 andSV40 Tag (B Price, unpublished results). ThereforeGM5849 AT cells contain a pool of free wtp53 with thepotential to be activated by DNA-damaging agents.
p53's transcriptional activity was monitored in atransient transfection experiment using the p53 lucifer-ase reporter construct p50-Luc (Youmell et al., 1998).p50-Luc contains two copies of a p53-consensusbinding site linked to the TATA box and minimalpromoter of the pGL3(T+) luciferase reporter con-struct. AT cells transfected with pGL3(T+) alonedisplayed negligible luciferase activity, and cotransfec-tion with a wtp53 expression vector did not increasethis activity (Figure 3b). GM5849 AT cells transfectedwith p50-Luc had increased basal activity, anddisplayed strong activation of the reporter followingcotransfection with a wtp53 expression vector (Figure3b). Since the only di�erence between pGL3(T+) andp50-Luc is the presence of the p53-consensus bindingsite in p50-Luc, Figure 3b demonstrates that p50-Luccan be activated by wtp53. In Figure 3c, AT cells weretransfected with p50-Luc and exposed to the radio-mimetic agent bleomycin. Bleomycin induces strandbreaks in DNA that are similar to those caused byIonizing Radiation, and AT cells show increasedsensitivity to bleomycin compared to normal cells(Beamish and Lavin, 1994; Meyn, 1999). Bleomycincaused a small increase in transcription from the p50-Luc reporter which was abolished by mtp53, whichfunctions as a dominant negative inhibitor of wtp53function. When AT cells were cotransfected withFL-AT, there was an increase in basal activity fromthe promoter which was increased further by bleo-mycin treatment. When cells were cotransfected withATM plus the dominant negative mtp53, both basaland bleomycin induced transcription were decreased(Figure 3c). The results in Figure 3 demonstrate thatp50-Luc is speci®cally responsive to wild type, but notmutant p53. Further, bleomycin activates transcriptionfrom p50-Luc, and this activation is increased in thepresence of ATM, but inhibited by mtp53. Theseresults are consistent with bleomycin increasing wtp53transcriptional activity through an ATM-dependentmechanism.
In Figure 4, GM5849 AT cells were transientlytransfected with either FL-AT, FL-ATkd or wtp53 inthe indicated combinations and the ability of bleomy-cin to increase the transcriptional activity of p50-Lucmeasured. Control cells (Figure 4, *) showed anincrease in activity from the reporter as the bleomycin
concentration was increased. When FL-AT wastransfected into the GM5849 AT cells, bleomycincaused a signi®cant increase in activity from thereporter, with maximal activation between 1 ± 5 mMbleomycin (Figure 4, ~). In contrast, the full-lengthkinase inactive ATM construct was indistinguishablefrom control cells (Figure 4, &). Because activation ofthe reporter was only seen in the presence of both FL-AT and a DNA-damage signal from bleomycin, thisindicates that there is speci®c activation of the p53reporter by the ATM protein, presumably throughactivation of endogenous p53. To determine if thisactivation was indeed the result of p53 activationrather than through some alternative mechanism,GM5849 cells were transiently transfected with anexpression vector for wtp53. In the absence ofbleomycin, the exogenous p53 induced a fourfoldincrease in basal transcriptional activity, and, whencombined with the DNA-damage signal from bleomy-cin, gave a further twofold increase in activity (Figure4, *). Since GM5849 cells do not express ATMprotein, this increase in p53 activity by bleomycintreatment may re¯ect activation of other regulators ofp53, such as Atr or DNA-PK. If cells werecotransfected with FL-ATkd and p53, no furtherincrease in transcriptional activity was seen comparedto cells transfected with p53 alone (Figure 4, &).However, when cells were transfected with both wtp53and FL-AT, strong activation of the p53 reporterconstruct was seen. We interpret this to mean thatATM can activate the transcriptional activity of p53 inthese cells when exposed to an appropriate DNA-
Figure 4 ATM's kinase activity is required to activate p53transcription. GM5849 AT cells were transiently transfected withp50-Luc reporter (0.6 mg), pCMV-Gal (0.8 mg) and either thepromoterless AT-Sure vector (Con, *), pFL-AT (FL-AT, ~),pFL-ATkd (ATkd, &), in the absence (open symbols) or presence(closed symbols) of pcDwtp53 (p53; 150 ng). Cells were exposedto bleomycin for 18 h, and extracts assayed for luciferase activity.b-galactosidase activity from the constitutive pCMV-Gal vectorwas used to control for transfection e�ciencies. All data pointswere carried out in triplicate. Error bars indicated where greaterthan symbol size+s.e.m.
ATM's kinase activity is required to activate p53GA Turenne et al
5104
Oncogene

damage signal. Further, Figure 4 indicates thatactivation of p53's transcriptional activity by ATMrequires an intact kinase domain.
In Figure 5a, we further examined the role of ATM'skinase domain in activating p53's transcriptionalactivity. GM5849 AT cells were transfected withwtp53 in the presence of the indicated construct. Theper cent increase in reporter activity followingbleomycin exposure was then calculated. WhenGM5849 AT cells transfected with p53 were exposedto bleomycin, the activity of the p50-Luc reporter wasincreased (Figure 5a: Vector), similar to that seen inFigure 4. When GM5849 cells were transfected withboth p53 and FL-AT (Figure 5a: FL-AT), strongactivation of the p53 reporter construct by bleomycinwas observed. However, cells transfected with p53 andeither AT-DN or the kinase domain of ATM, ATK,failed to stimulate the transcriptional activity of wtp53when exposed to bleomycin. The results in Figures 4and 5a indicate that both the kinase activity andsequences in the N-terminal of the ATM protein arerequired for the e�cient activation of p53 by ATMfollowing exposure to a DNA-damaging signal.
ATM activation of p53 is thought to occur throughtwo distinct phosphorylation steps. First, ATM canphosphorylate serine 15 of p53 (Banin et al., 1998;Canman et al., 1998). Second, ATM activates chk2kinase which can phosphorylate serine 20 of p53
(Chebab et al., 2000; Hirao et al., 2000; Tominaga etal., 1999). Thus ATM activation leads to phosphoryla-tion of p53 at serines 15 and 20. The inability of FL-ATkd, AT-DN or ATK to activate p53 might thereforestem from their inability to phoshorylate serine 15 orto activate chk2. To examine the relative importance ofthese two events in activating p53, we examined theability of ATM constructs to activate chk2 kinasefollowing exposure to bleomycin.
chk2 activation was examined by transfecting cellswith expression vectors for chk2 or a kinase inactivechk2 termed chk2kd. Both contain a HA-epitope tag atthe N-terminal, which increases their apparent mass byapproximately 2 kd. These constructs were thentransiently expressed in GM637 cells, an SV40transformed ®broblast cell line which expresses normalATM protein. In Figure 5b (top panel), bothendogenous (lower band) and transfected chk2 con-structs (upper band) were readily detected by Westernblotting. GM637 cells were then transfected with chk2or chk2kd and immunokinase assays carried out tomonitor chk2 autophosphorylation after exposurebleomycin. chk2 anti-sera immunoprecipitated a64 kd protein whose autophosphorylation was stimu-lated by bleomycin (Figure 5b, lanes 3 and 4). Thisactivity was absent from immunoprecipitates using IgG(Figure 5b, lanes 1 and 2), and was not detected inGM5849 AT cells (lanes 9 and 10). Cells transfected
Figure 5 Activation of chk2 requires the ATM protein. (a) p53 activation requires the N-terminal of ATM. GM5849 cells weretransiently transfected with p50-Luc reporter (0.6 mg), pCMVGal (0.8 mg), wtp53 (100 ng) and either vector, FL-AT, AT-DN orATK. Cells were exposed to bleomycin (2 mM) for 16 h, and extracts assayed for luciferase activity. b-galactosidase activity from theconstitutive pCMV-Gal vector was used to control for transfection e�ciencies. Results are expressed as the % increase in reporteractivity by bleomycin in the presence of the indicated ATM construct. Results+s.e.m. (b) Chk2 expression. Upper panel. NormalGM637 ®broblasts were transiently transfected with HA tagged chk2 (chk2), HA tagged kinase inactive chk2 (chk2kd) or vector andexamined by Western blotting using an anti-chk2 antibody. Middle panel. GM637 cells (lanes 1 ± 8) were transiently transfected withchk2 (C), chk2kd (kd) or vector (V). Non-transfected GM5849 AT cells were used as controls (lanes 9 and 10). Cells were exposedto bleomycin (2 mM,+) or solvent (PBS, 7) for 10 min and extracts immunoprecipitated with anti-chk2 anti-sera (lanes 3 ± 10) orIgG (lanes 1 and 2) and chk2 immunokinase assays carried out as described in Materials and methods. Bottom panel. GM5849 ATcells were transfected with chk2 (lanes 11 ± 18) and either vector (V, lanes 11 and 12), pFL-AT (A, lanes 13 and 14), pFL-ATkd (Ak,lanes 15 and 16) or AT-DN (DN, lanes 17 and 18). Cells were treated with bleomycin (2 mM,+) or solvent (PBS, 7) for 10 min andextracts immunoprecipitated with anti-chk2 anti-sera (lanes 11 ± 18). Chk2 was immunoprecipitated and immunokinase assays werecarried out as described in Materials and methods
Oncogene
ATM's kinase activity is required to activate p53GA Turenne et al
5105

with exogenous chk2 (which has a 2 kd HA tag)displayed activation of both endogenous and trans-fected chk2 after bleomycin (lanes 7 and 8), whereascells transfected with chk2kd exhibited only endogen-ous kinase activity (lanes 5 and 6). Figure 5demonstrates that chk2 is activated in an ATMdependent manner, as previously shown (Blasina etal., 1999a; Brown et al., 1999; Matsuoka et al., 1998).GM5489 AT cells were then transfected with chk2 inthe presence of the indicated ATM construct. Transfec-tion of GM5849 AT cells with chk2 alone did notrestore bleomycin induced activation of chk2 (Figure5b, lanes 11 and 12). However, cotransfection of FL-AT with chk2 restored bleomycin dependent activation(lanes 13 and 14), whereas both FL-ATkd and AT-DNwere inactive (lanes 15 ± 18). Interestingly, the unsti-mulated chk2 kinase activity in FL-AT transfected cellswas slightly increased (Figure 5, lane 13). The data inFigure 5b is consistent with Figures 4 and 5a,indicating that both the kinase activity and the N-terminal of the ATM protein are required for thecorrect functioning of the ATM protein. Further, theydemonstrate that chk2 is activated by FL-AT, but notby kinase inactive versions or by the AT-DN construct.
Figures 4 and 5 demonstrate that the kinase activityof ATM is required for the activation of p53 and chk2by bleomycin. In vitro studies demonstrate that ATMphosphorylates both serine 15 of p53 and threonine 68of chk2 kinase (Banin et al., 1998; Canman et al.,1998b; Melchionna et al., 2000). Phosphorylation ofchk2 by ATM activates its kinase activity, and chk2then phosphorylates serine 20 of p53. In vivo, theDNA-damage induced phosphorylation of serines 15and 20 of p53 requires functional ATM protein(Chebab et al., 1999; Sakaguchi et al., 1998; Shieh etal., 1997, 1999; Siliciano et al., 1997; Unger et al.,1999a). These observations imply that ATM regulatesp53 transcriptional activity through the phosphoryla-tion of serines 15 and 20 of p53. However, serines 33and 37 of p53 are also phosphorylated, in vivo,following exposure to DNA damage, although thesephosphorylations are not known to be ATM-depen-dent (Sakaguchi et al., 1998; Shieh et al., 1997, 1999;Siliciano et al., 1997). Thus activation of p53'stranscriptional activity is likely to involve both theATM-dependent phosphorylation of serines 15 and 20as well as the phosphorylation of serines 33 and 37 byother kinases. In the following experiments, we set outto determine which of the N-terminal phosphorylationsites in p53 are required for the ATM-dependentactivation of p53 by bleomycin.
Single serine to alanine mutations in serines 15, 20,33 and 37, as well as double mutations in serines 15and 20 (directly regulated by ATM) and serines 33 and37 (not directly regulated by ATM). Each of theconstructs had similar basal transcriptional activity,indicating that the amino-acid substitutions introduceddid not signi®cantly alter the transcriptional activity ofthese mutant p53 proteins (B Price, unpublishedresults). This is similar to data reported by others(Ashcroft et al., 1999; Blattner et al., 1999; Unger et
al., 1999b), who showed that single or doublemutations in N-terminal serine residues did notsigni®cantly a�ect the basal transcriptional activity ofthese p53 constructs. In Figure 6, each of the p53constructs was examined for the ability of bleomycin toactivate p53's transcriptional activity in the absence orpresence of ATM. In the absence of ATM, wtp53 andp53 with single mutations in serines 15, 20, 33 or 37 allactivated the p53 reporter to similar levels whenexposed to bleomycin, with p53S33A showing thehighest activation (Figure 6, ± ). When each of thesep53 mutants were cotransfected with FLAT andexposed to bleomycin, bleomycin increased the de-tected p53 transcriptional activity to similar levels.Mutation of residues 15, 20, 33 and 37 therefore didnot a�ect the ATM dependent activation of p53 bybleomycin. Next, we examined double mutations in theATM regulated phosphorylation sites (serines 15 and30), or in the ATM-independent sites (serines 33 and37). Exposure of cells transfected with only p53S15/20A to bleomycin increased p53's transcriptionalactivity to a higher level than that seen with wtp53.However, when p53S15/20A was co-transfected withATM, bleomycin did not induce any further increase intranscriptional activity (Figure 6). Further, when cellswere transfected with p53S33/37A and exposed tobleomycin, no signi®cant activation of the p53-reporterconstruct was seen. This is in contrast to wtp53, whichshowed a 20% increase in p53 activity followingexposure to bleomycin. When p53S3/37A was cotrans-fected with ATM, bleomycin was unable to furtheractivate p53's transcriptional activity seen. Figure 6demonstrates that mutation of either serines 15 and 20or 33 and 37 of p53 inhibits the ATM-dependentactivation of p53 by bleomycin.
Figure 6 Activation of p53 by ATM requires N-terminal serineresidues. GM5849 cells were transiently transfected with thepromoterless ATM vector AT-Sure (7) or FL-AT (+) in thepresence of either wtp53 expression vector or the indicated serineto alanine mutation (150 ng each). p50-Luc reporter (0.6 mg) andpCMV-Gal (0.8 mg) were also added. Cells were to exposed tobleomycin (3 mM:+) for 18 h, and extracts assayed for luciferaseactivity. b-galactosidase activity from the constitutive pCMVGalvector was used to control for transfection e�ciencies. Results areexpressed as the % increase in reporter activity caused bybleomycin relative to solvent exposed cells. Results+s.e.m.
ATM's kinase activity is required to activate p53GA Turenne et al
5106
Oncogene

Discussion
We have shown that the kinase activity of the ATMprotein is required for activation of p53 and chk2 byDNA damage, and is also required for the restorationof normal cellular radiosensitivity by the ATM protein.This implies that the ability of ATM to activateappropriate downstream signal transduction pathwaysin response to DNA damage is dependent on thekinase activity of ATM. We also expressed an ATMfragment containing only the kinase domain plussequences N-terminal to this (amino-acids 2138 ±3056) in AT cells; this fragment was inactive and didnot activate p53, chk2 or alter the radiosensitivity ofAT cells. In contrast, others have reported that asmaller, approximately 400 amino-acid fragment con-taining the kinase domain could complement theradiosensitivity of AT cells (Morgan et al., 1997),suggesting that their fragment retained some intrinsickinase activity. The reason for the discrepancy betweenthese observations is unclear, but may stem from thesize di�erences in the fragments employed. However,our studies demonstrate that an intact kinase domain isrequired for restoration of normal sensitivity to DNAdamage and for activation of p53 and chk2. Previousstudies have shown that ATM's kinase activity isincreased in cells exposed to DNA damage (Banin etal., 1998; Blasina et al, 1999a; Canman et al., 1998),and that ATM can phosphorylate multiple proteinsinvolved in the DNA damage response, including p53(Banin et al., 1998; Canman et al., 1998b), chk2 kinase(Chebab et al, 2000; Hirao et al., 2000; Tominaga etal., 1999), p95/nibrin (Lim et al., 2000; Zhao et al.,2000) and Brca1 (Cortez et al., 1999). The diversephenotype expressed by AT cells, including aberrantcheckpoint activation, genomic instability and defectiverepair may therefore derive from the inability of ATcells to correctly phosphorylate and upregulate keyproteins involved in all aspects of DNA repair.
We also examined the role of the N-terminal ofATM. Using an ATM construct lacking the N-terminal, we did not observe any activation of eitherthe p53 or chk2 proteins, or any increase in cellsurvival following exposure to lonizing Radiation,indicating that an intact N-terminal is essential forcorrect ATM functioning. The N-terminal of ATM canassociate with both p53 (Khanna et al., 1998) andhistone deacetylase in vitro (Kim et al., 1999), and bothof these proteins co-precipitate with ATM in vivo(Khanna et al., 1998; Kim et al., 1999). ATM has alsobeen shown to bind to dsDNA (Smith et al., 1999a;Suzuki et al., 1999), and this may involve sequences inthe N-terminal of ATM (Smith et al., 1999b). WhetherATM binds directly to DNA or if this interaction ismediated through a speci®c DNA-binding sub-unit isnot known. However, we speculate that the N-terminalof ATM may contain sites for interaction with DNA aswell as for association with p53 and other ATMe�ectors. Deletion of this region would be predicted toinactivate the ATM protein. An alternative explanationis that deletion of the N-terminal may disrupt the
overall structure of the ATM protein and inactivate it.A more detailed deletion and mutagenesis approachwill be needed to address this issue.
We have also determined which of several serinephosphorylation sites in the N-terminal of p53 wererequired for activation of p53's transcriptional activityby ATM. We used a minimal p53 reporter constructcontaining only a p53 consensus binding site linked toa TATA box and basal promoter (Youmell et al.,1998). This minimal reporter was speci®cally responsiveto p53. Although the promotor regions of p53regulated genes contain binding sites for multipletranscription factors, this minimal promoter was usedsince it is more likely to be regulated by p53 alone,rather than in concert with other DNA-damageactivated transcription factors. The role of N-terminalserine residues in the activation of p53 has been thesubject of intense investigation, but these experimentshave generally examined only the basal (unstimulated)transcriptional activity of the p53 protein. Thisapproach has lead to con¯icting results, with mostexperiments showing no alteration in p53 transcrip-tional activity (Ashcroft et al., 1999; Blattner et al.,1999). Here, we have addressed this issue by examiningthe interaction between ATM, a DNA damage signaland p53 transcriptional activity. Using this system, wehave shown that bleomycin can increase p53'stranscriptional activity by a process which dependson both ATM's kinase activity and on multiple serineresidues in the N-terminal of p53. A potentialconfounding factor in these experiments is the presenceof the SV40 Tag protein in the AT cells used. It ispossible that ATM regulates the interaction betweenp53 and SV40Tag, such that activation of ATM causesrelease of p53 bound to SV40Tag, which in turnincreases transcription from the p53 reporter construct.This is unlikely for several reasons. First, neitherbleomycin nor ATM status alters the amount of p53bound to SV40Tag in GM5849 AT cell (B Price,unpublished results). Second, a recent report demon-strates that the interaction between p53 and SV40Tagis una�ected by phosphorylation of serines 15 and 37of p53 (Sheppard and Liu, 1999). Further, previouslypublished data has shown that the p53-DNA damageresponse is intact in many SV40 transformed ®broblastcells, including the phosphorylation of p53, increasedp53-DNA binding and transcription of the p21 gene(Hess and Brandner, 1997; Kohli and Jorgensen, 1999;O'Neil et al., 1996; Tibbets et al., 1999). We haveshown that approximately 50% of the p53 in GM5849cells is not bound to SV40Tag (Figure 3) and thetransient expression of p53 into these cells will furtherincrease the levels of free p53. Thus we feel it isunlikely that ATM is regulating p53-SV40Tag interac-tions, but is more likely to be a�ecting the transcrip-tional activity of the p53 protein directly. We haveattempted to replicate the results in non-transformedAT ®broblasts or in transformed AT lymphoblasts, butfor technical reasons we are unable to obtain highenough transfection e�ciencies to perform theseexperiments. We interpret our data to mean that
Oncogene
ATM's kinase activity is required to activate p53GA Turenne et al
5107

ATM can directly a�ect the transcriptional activity ofthe p53 protein.
In our experimental system, single mutations ofeither serines 15, 20, 33 or 37 had minimal e�ect on theability of ATM to increase p53 transcriptional activity.This indicates that there is some redundancy in the roleof these serine residues, such that loss of a single serinedoes not impair p53 function. However, doublemutation of either serines 15 and 20 or serines 33and 37 blocked the ability of ATM to activate p53.This is consistent with our observation that ATM'skinase activity is required for p53 activation, andimplies that phosphorylation of p53 is one of the keysteps in activation of p53. Serines 15 and 20 have beenimplicated in regulating the p53-mdm2 interaction(Chebab et al., 1999; Kubbutata et al, 1998; Unger etal., 1999a), with serine 20 being the main regulator ofp53-mdm2 interaction (Unger et al., 1999a). Phosphor-ylation of serines 15 and 20 is proposed to block theinteraction between mdm2 and p53, leading toaccumulation of the p53 protein. Since phosphoryla-tion of serines 15 and 20 is ATM-dependent (Chebabet al., 1999; Sakaguchi et al., 1998; Shieh et al., 1997;1999; Siliciano et al., 1997), our results are consistentwith the hypothesis that serines 15 and 20 of p53 arerequired for the ATM-dependent activation of the p53protein.
Surprisingly, mutation of serines 33 and 37 alsoblocked the ability of ATM to activate p53. Phosphor-ylation of serines 33 and 37 is not known to bedependent on the ATM protein, indicating thatadditional DNA-damage activated kinases are requiredfor the activation of p53, speci®cally kinases whichphosphorylate serines 33 and 37. Potential proteinkinases which might phosphorylate serines 33 and 37include the Jun and Atr kinases respectively (Tibbets etal., 1999). There are also other phosphorylation siteswithin p53 N-terminal which can be regulated by DNAdamage, including serines 6 and 9 (Higashimoto et al.,2000) and serine 46 (Oda et al., 2000), which maycontribute to the overall activation of p53. Theseobservations demonstrate that a complex pattern ofphosphorylations within the N-terminal of p53 arerequired for the full activation of p53's transcriptionalactivity by DNA damage. In addition, whilst ATM andserines 15 and 20 of p53 are essential, they are notsu�cient to elicit full activation of p53's transcriptionalactivity. The requirement for serines 33 and 37 of p53strongly suggests a role for additional, DNA-damageactivated protein kinases in the activation of p53.
The activation of p53's transcriptional activity byDNA damage involves several steps. These includedecreased degradation of the p53 protein (Sakaguchi etal., 1998; Shieh et al., 1997; 1999; Siliciano et al., 1997),increases in p53 DNA-binding activity (Tishler et al.,1993) and changes in the actual transcriptional activityof the p53 protein. The activation of p53 by ATM inour study re¯ects the overall relative contributions ofeach of these steps to the formation of transcriptionallyactive p53 complexes. However, in our system, thepresence of stable SV40 Tag-p53 complexes as well as
endogenous wtp53 prevents us from monitoring thehalf-life and DNA-binding activity of the p53 mutantswe have examined. Thus we are unable to determine ifthe increased p53 transcriptional activity is due tochanges in p53 stability, increased DNA binding ortranscriptional activity. The most likely explanation isthat all three contribute to this e�ect.
In conclusion, we suggest that phosphorylation ofp53 by ATM is the initiating event in a series of post-translational modi®cations which leads to the forma-tion of a transcriptionally active p53 complex. ATMdirectly regulates the phosphorylation of serines 15 and20, inhibiting mdm2 binding. This leads to decreasedp53 degradation and accumulation of p53. Phosphor-ylation of serines 33 and 37 (by unknown kinases) hasbeen shown to increase the binding of HistoneAcetytransferase to the N-terminal of p53 (Sakaguchiet al., 1998). Histone Acetyltransferase then acetylateslysines in the C-terminal of p53, which in turn activatesthe DNA-binding activity of the p53 protein (Saka-guchi et al., 1998). Loss of serines 15 and 20 or 33 and37 would then impair the activation of the p53 proteinby inhibiting either of the two key steps in p53activation ± protein stabilization and increased DNAbinding. This is the ®rst demonstration that the ATMprotein can directly in¯uence the transcriptionalactivity of the p53 protein through a mechanisminvolving N-terminal phosphorylation site.
Materials and methods
Cell lines, plasmids and antibodies
GM637 (normal) or GM5849 (AT) human ®broblast celllines were obtained from the Coriel Institute, Camden, NJ,USA. Cells were maintained in Minimal Eagle's Media withEarles Balanced Salt Solution supplemented with 7% FetalCalf Serum. Irradiation was carried out using a 250 kVp X-ray machine under temperature controlled conditions. Forclonogenic cell survival assays, cells were plated in triplicate24 h prior to irradiation. Cells were allowed to recover for 14days post-irradiation, and stained with crystal violet.Surviving colonies containing 450 cells were counted. Platinge�ciency was 30 ± 50%. ATM cDNA was provided by YShiloh, Tel Aviv, Israel. chk2 constructs and antibody wereprovided by C McGowan, Scripps, CA, USA. ATMantibodies and anti-Omni Tag were purchased from SantaCruz Biotech, CA, USA and SV40 and p53 antibodies fromOncogene Science, NY, USA. pAT-DN was constructed byinserting an EcoRI fragment spanning bases 2309 ± 4310 ofATM, in frame, into pcDNA3.1/HisA to generate pAT-CD.pAT-CD was then digested with KpnI/XhoI (to remove bases3909 ± 4310), and a KpnI/XhoI fragment containing bases3901 ± 9197 of ATM inserted. pAT-DN contains amino-acids769 ± 3056 of ATM. pATK was constructed by inserting a2781bp EcoRI fragment of the ATM protein (amino-acids2138 ± 3056), in frame, into the pcDNA3.1/HisA expressionvector. The construction of pcDwtp53, p50-Luc, FL-AT, FL-ATkd have been previously described by us (Blasina et al.,1999a; Youmell et al., 1998). Single and double pointmutations were generated using the p-Alter I mutagenesissystem or ExSite PCR site-directed mutagenesis kit (Strata-gene, CA, USA). All mutations were veri®ed by sequencing.
ATM's kinase activity is required to activate p53GA Turenne et al
5108
Oncogene

Transfection and Iuciferase reporter assays
Stably transfected cell lines. 36106 GM5849 cells werecultured on 100 mm dishes for 18 h prior to the addition ofa DNA-Lipofectin (Gibco ±BRL) mix (10 mg DNA/20 mglipofectin in 1.62 ml Dulbecco's Modi®ed Eagle's Media;DMEM). Six hours later, cells were washed in DMEM andrefed 48 h after transfection, cells were trypsinized, replatedat 1 : 5 to 1 : 20 dilution, allowed to reattach for 8 h, and thenexposed to G418 (400 mg/ml) for 12 ± 15 days, with mediumchanges every 3 days. Surviving colonies (approximately100 ± 150) were trypsinized, pooled, grown up and tested forexpression of the appropriate gene by Western blotting.Transient transfections. For chk2 kinase assays, GM5849 orGM637 cells were transfected as described above with chk2(2 mg) or AT constructs (8 mg) and adjusted to 10 mg of DNAwith a promoterless ATM vector. Cells were allowed torecover for 44 h before chk2 kinase assays were carried out.For luciferase reporter assays, 36105 GM5849 cells weretransfected with pCMV-b-galatosidase (0.8 mg), p50-Luc(0.5 mg), AT construct (1.8 mg) or p53 (150 ng) and adjustedto 4 mg of DNA with pcDNA/HisA in a ®nal volume of 2 mlcontaining 6.5 mg of lipofectin. After 6 h exposure to theDNA-lipofectin mix, cells were washed, and then exposed toeither solvent (PBS) or bleomycin for 16 h. Preparation ofcell extracts and assays for luciferase and b-galactosidaseactivity are as described in (Youmell et al., 1998). Somevariation between di�erent bleomycin batches was noted,with stimulation of p53 transcriptional activity varyingbetween 100 ± 300%. For this reason, each experiment wascarried out with all appropriate controls and with each pointperformed in triplicate.
Immunoprecipitation, Western blotting and kinase assays
ATM Cells were lyzed directly in 1 ml of RIPA [50 mM TrispH 7.4; 150 mM NaCl; 1% Triton X-100; 0.1% SDS; 0.5%
Deoxycholate; 1 mM PMSF; 1 mg/ml leupeptin; 1 mg/mlaprotinin; 500 mM Na-vanadate; 50 mM NaF] and centrifugedat (35 kg/15 min). Cell lysates are pre-cleared for 1 h withprotein-A/G agarose beads, then immunoprecipitated withprotein-A/G agarose beads (50% w/v) pre-coated with anti-Omni Tag antibody (2 mg: Santa Cruz, CA, USA) for 4 h.Beads were washed four times in RIPA, and protein removedfrom the beads in 30 ml of sample bu�er (2% sodium dodecylsulfate; 10% glycerol; 1% mercaptoethanol; 0.125 M TrispH 6.8) at 928C.chk2 Cells were lyzed on the dish in 1 ml of RIPA bu�er.Extracts were centrifuged (15 kg/10 min), and the super-natant immunoprecipitated with anti-cds1 antibodies pre-coated onto protein-A/G agarose beads. Immunoprecipitateswere washed in 461 ml of bu�er RIPA bu�er, twice inkinase bu�er (50 mM Tris, pH 7.4, 10 mM MgCl2) and kinasereactions carried out in 20 ml of kinase bu�er containing10 mCi 32P-ATP. Reactions were terminated by the additionof 46SDS sample bu�er. Phosphorylated products weredetected by SDS±PAGE and autoradiography.For Western blotting, proteins were transferred to
Immobilon PVDF membranes, blocked in 10% milk, andincubated in primary antibody for 2 ± 18 h. For analysis ofATM, proteins are separated by 6% SDS ±PAGE andtransferred for 30 min at 12 V in methanol free bu�er toPVDF membranes. Membranes were incubated with Omniantibody, and antigens then detected with by a coupledbiotin-avidin immuno-enzyme method (Vector Laboratories,Burlingame CA, USA) or by ECL (Amersham PLC, UK).
AcknowledgmentsWe thank Dr Y Shiloh for providing ATM cDNA and DrC McGowan for providing chk2 plasmids and antibodies.This work was supported by grant number CA64585 fromthe NCl and by funds from the AJCRT foundation.
References
Ashcroft M, Kubbutata HG and Vousden KH. (1999). Mol.Cell Biol., 19, 1751 ± 1758.
Banin S, Moyal L, Shieh S-Y, Taya Y, Anderson CW,Chessa L, Smorodinsky N-I, Prives C, Reiss Y, Shiloh Yand Ziv Y. (1998). Science, 281, 1674 ± 1678.
Beamish H and Lavin MF. (1994). Int. J. Radiat. Biol., 65,175 ± 184.
Beamish H, Khanna KK and Lavin MF. (1994). Rad. Res.,138, S130 ± S133.
Blasina A, Price BD, Turenne GA and McGowan CH.(1999a). Current Biology, 9, 1135 ± 1138.
Blasina A, Van de Weyer I, Laus MC, Luyten WH, ParkerAE and McGowan CH. (1999b). Curr. Biol., 9, 1 ± 10.
Blattner C, Tobiasch E, Litfen M, Rahmsdorf HJ andHerrlich P. (1999). Oncogene, 18, 1723 ± 1732.
Brown AL, Lee C-H, Schwarz JK, Mitiku N, Piwnica-Worms H and Chung JH. (1999). J. Biol. Chem., 96,3745 ± 3756.
Canman CE and Lim D-S. (1998). Oncogene, 17, 3301 ± 3308.Canman CE, Lim D-S, Cimprich KA, Taya Y, Tamai K,
Sakaguchi K, Appella E, Kastan MB and Siliciano JD.(1998). Science, 281, 1677 ± 1682.
Chebab NH, Malikzay A, Stavridi ES and Halozonetis TD.(1999). Proc. Natl. Acad. Sci., 96, 13777 ± 13782.
Chebab NH, Malikzay A, Appel M and Halozonetis TD.(2000). Genes Dev., 14, 278 ± 288.
Cornforth MN and Bedford JS. (1985). Science, 227, 1589 ±1591.
Cortez D, Wang Y, Qin J and Elledge SJ. (1999). Science,286, 1162 ± 1166.
Furnari B, Blasina A, Boddy MN, McGowan CH andRussell P. (1999). Mol. Biol. Cell, 10, 833 ± 845.
Hall-Jackson CA, Cross DAE, Morrice N and Smythe C.(1999). Oncogene, 18, 6707 ± 6713.
Haupt Y, Maya R, Kazaz A and Oren M. (1997). Nature,387, 296 ± 299.
Hess RD and Brandner G. (1997). Oncogene, 15, 2501 ± 2504.Higashimoto Y, Saito S, Tong X, Homg A, Sakaguchi K,
Apella E and Anderson CW. (2000). J. Biol. Chem., 275,23199 ± 23203.
Hirao A, Kong Y-Y, Matsuoka S, Wakeham A, Ruland J,Yoshida H, Liu D, Elledge SJ and Mak TW. (2000).Science, 287, 1824 ± 1827.
Jeggo PA, Carr AM and Lehmann AR. (1998). Trends Gen.,14, 312 ± 316.
Jung M, Lee AS, Zhang Y and Dritschilo A. (1997). Int. J.Radiat. Oncol. Biol. Phys., 37, 417 ± 422.
Kastan MB, Onyekwere D, Sidransky D, Vogelstein B andCraig RW. (1991). Cancer Res, 51, 6304 ± 6311.
Kastan MB, Zhan Q, El-Deiry WS, Carrier F, Jacks T,Walsh W, Plunkett BS, Vogelstein B and Fornace AJ.(1992). Cell, 71, 587 ± 597.
Oncogene
ATM's kinase activity is required to activate p53GA Turenne et al
5109

Khanna KK, Keating KE, Kozlov S, Scott S, Gatei M,Hobson K, Taya Y, Gabrielli B, Lees-Miller SP and LavinMF. (1998). Nature Genetics, 20, 398 ± 400.
Khosravi R, Maya R, Gottleib T, Oren M, Shiloh Y andShkedy D. (1999). Proc. Natl. Acad. Sci., 96, 14973 ±14977.
Kim GD, Choi YH, Dimtchev A, Jeong SJ, Dritschilo A andJung M. (1999). J. Biol. Chem., 274, 31127 ± 31130.
Kohli M and Jorgensen TJ. (1999). Biochem. Biophys. Res.Comm., 257, 168 ± 176.
Kubbutata MHG, Ludwig RL, Ashcroft M and VousdenKH. (1998). Mol. Cell Biol., 18, 5690 ± 5698.
Lim D-S, Kim S-T, Maser RS, Lin J, Petrini JHJ and KastanMB. (2000). Nature, 404, 613 ± 617.
Luo C-M, Tang W, Mekeel KL, DeFrank JS, Anne PR andPowell SN. (1996). J. Biol. Chem., 271, 4497 ± 4503.
Matsuoka S, Huang M and Elledge SJ. (1998). Science, 282,1893 ± 1897.
Melchionna R, Chen XB, Blasina A and McGowan CH.(2000). Nature Cell Biology, 2, 762 ± 765. Meyn SM.(1993). Science, 260, 1327 ± 1330.
Metcalfe JA, Parkhill J, Campbell L, Stacey M, Biggs P,Byrd PJ and Taylor MR. (1996). Nature Genetics, 13,350 ± 353.
Meyn MS. (1999). Clin. Genet., 55, 289 ± 304.Morgan SE, Lovly C, Pandita TK, Shiloh Y and Kastan MB.
(1997). Mol. Cell Biol., 17, 2020 ± 2029.Nagasawa H, Keng P, Dahlberg W and Little JB. (1994). Int.
J. Rad. Biol., 66, 373 ± 379.Pandita TK, Pathak S and Geard CR. (1995). Cytogenet.
Cell Genet., 71, 86 ± 93.Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C,
Mori T, Nishimori H, Tamai K, Tokino T, Nakamura Yand Taya Y. (2000). Cell, 102, 849 ± 862.
O'Neil FJ, Hu Y, Chen T and Carney H. (1996). Oncogene,14, 955 ± 965.
Rhind N and Russell P. (1998). Curr. Opin. Cell Biol., 10,749 ± 758.
Savitsky K, Sfez S, Tagle DA, Ziv Y, Sartiel A, Collins FS,Shiloh Y and Rotman G. (1995). Hum. Mol. Genet., 4,2025 ± 2032.
Sakaguchi K, Herrera JE, Saito S, Miki T, Bustin M,Vassilev A, Anderson CW and Appella E. (1998). GenesDev., 12, 2831 ± 2841.
Sheppard HM and Liu X. (1999). Anticancer Res., 19, 2079 ±2083.
Shieh S-Y, Ikeda M, Taya Y and Prives C. (1997). Cell, 91,325 ± 334.
Shieh S-Y, Taya Y and Prives C. (1999). EMBO J., 18,1815 ± 1823.
Siliciano JD, Canman CE, Taya Y, Sakaguchi K, Appella Eand Kastan MB. (1997). Genes Dev., 11, 3471 ± 3481.
Smith GCM, Cary RB, Lakin ND, Hann BC, Teo S-H, ChenDJ and Jackson SP. (1999a). J. Biol. Chem., 96, 11134 ±11139.
Smith GCM, d'Adda F, Lakin ND and Jackson SP. (1999b).Mol. Cell Biol., 19, 6076 ± 6084.
Suzuki K, Kodama S and Watanabe M. (1999). J. Biol.Chem., 274, 25571 ± 25575.
Taylor AMR, Harnden DG, Arlett CF, Harcourt SA,Lehmann AR, Stevens S and Bridges BA. (1975). Nature,258, 427 ± 429.
Tibbets RS, Brumbaugh KM, Williams JM, Sarkaria JN,Cliby WA, Shieh S-Y, Taya Y, Prives C and Abraham RT.(1999). Genes Dev., 13, 152 ± 157.
Tishler RB, Calderwood SK, Coleman CN and Price BD.(1993). Cancer Research, 53, 2212 ± 2216.
Tominaga K, Morisaki H, Kaneko Y, Fujimoto A, TanakeT, Ohtsubo M, Hirai M, Okayama H, Ikeda K andNakanishi M. (1999). J. Biol. Chem., 274, 31463 ± 31467.
Unger T, Juvan-Goshen T, Moallem E, Berger M, SionovRV, Lozano G, Oren M and Haupt Y. (1999a). EMBO J.,18, 1805 ± 1814.
Unger T, Sionov RV, Moallen E, Yee CL, Howley PM, OrenM and Haupt Y. (1999b). Oncogene, 18, 3205 ± 3212.
Weichselbaum RR, Nove J and Little JB. (1977). Nature,271, 261 ± 262.
Weinert T. (1998). Cell, 94, 555 ± 558.Youmell MY, Park SJ, Basu S and Price BD. (1998).
Biochem. Biophys. Res. Comm., 245, 514 ± 518.Zhang N, Chen P, Khanna KK, Scott S, Gatei M, Kozlov S,
Watters D, Spring K, Yen T and Lavin MF. (1997). Proc.Natl. Acad. Sci., 94, 8021 ± 8026.
Zhao S, Weng Y-C, Yuan S-S, Lin Y-T, Hsu H-C, Lin S-C,Gerbino E, Song M-H, Zdienicka MZ, Gatti RA, ShayJW, Ziv Y, Shiloh S and Lee EY. (2000). Nature, 405,473 ± 477.
Ziv Y, Bar-Shira A, Pecker I, Russell P, Jorgensen TJ,Tsarfati L and Shiloh Y. (1997). Oncogene, 15, 159 ± 167.
ATM's kinase activity is required to activate p53GA Turenne et al
5110
Oncogene




















