
Mutant p53 gain of function: repression of CD95(Fas/APO1) gene expression by tumor-associated p53...
10
Mutant p53 gain of function: repression of CD95(Fas/APO-1) gene expression by tumor-associated p53 mutants Amir Zalcenstein 1 , Perry Stambolsky 1 , Lilach Weisz 1 , Martina Mu¨ ller 2 , David Wallach 3 , Tanya M Goncharov 3 , Peter H Krammer 4 , Varda Rotter 1 and Moshe Oren* ,1 1 Department of Molecular Cell Biology, Weizmann Institute of Science, PO Box 26, Rehovot, 76100, Israel; 2 Department of Internal Medicine IV, University Hospital, 69115 Heidelberg, Germany; 3 Department of Biological Chemistry, Weizmann Institute of Science, PO Box 26, Rehovot 76100, Israel; 4 Tumor Immunology Program, German Cancer Research Center, 69120 Heidelberg, Germany Tumor-associated mutant forms of p53 can exert an antiapoptotic gain of function activity, which probably confers a selective advantage upon tumor cells harboring such mutations. We report that mutant p53 suppresses the expression of the CD95 (Fas/APO-1) gene, encoding a death receptor implicated in a variety of apoptotic responses. Moderate (40–50%) downregulation of CD95 mRNA and surface protein expression by mutant p53 correlates with partial protection against CD95-dependent cell death. Excess mutant p53 represses the transcrip- tional activity of the CD95 promoter, with the extent of repression varying among different tumor-associated p53 mutants. Furthermore, mutant p53 protein binds the CD95 promoter in vitro, in a region distinct from the one implicated in tight interactions of the CD95 gene with wild-type p53. Hence, the CD95 promoter is likely to be a direct target for downregulation by mutant p53. This activity of mutant p53 may contribute to its gain of function effects in oncogenesis. Oncogene (2003) 22, 5667–5676. doi:10.1038/sj.onc.1206724 Keywords: p53; apoptosis; CD 95; Fas/APO-1; repression Introduction The p53 tumor suppressor gene plays a pivotal role in the prevention of cancer. Consequently, it is often a target of genetic alterations in tumors: about half of all human tumors carry direct alterations within the p53 locus, and in many of the other half, p53 function is impaired owing to mutations in proteins operating either upstream or downstream to p53 (Vogelstein et al., 2000; Ryan et al., 2001; Michael and Oren, 2002; Bargonetti and Manfredi, 2002). p53 exerts its tumor suppressor function through multiple mechanisms. However, the most prominent biochemical feature of p53, underlying the major part of its tumor-suppressor activities, is its ability to act as a sequence-specific transcription factor. This entails direct binding to specific target sequences within the DNA, denoted p53 response elements (p53REs), with conse- quent transcriptional activation of genes residing in the close vicinity of the p53REs. The importance of the transcriptional activity of p53 is underscored by the finding that the vast majority of p53 mutations encountered in cancer reside within the core sequence- specific DNA-binding domain of p53 (el-Deiry, 1998; Soussi, 2000). In addition, p53 can also repress the transcription of many other genes, although direct binding of p53 to specific sequences within such genes has not been implicated in most cases. Given p53’s pivotal role in tumor suppression, it is therefore logical that potentially tumorigenic cells should strive to inactivate p53, thereby eliminating an important stumbling block for the development of cancer. However, unlike other tumor suppressor genes, such as Rb (Harbour and Dean, 2000) or ARF (Serrano, 2000), which are typically deleted, truncated, silenced or otherwise downregulated in many cancers, selective pressures within tumors clearly favor missense muta- tions in p53 rather than its outright elimination, often leading to the constitutive expression of high amounts of mutant p53 in tumor cells (Sigal & Rotter, 2000). This evidence may suggest p53 mutations to be a double- edged sword; that is mutant p53 proteins may not only lose the tumor suppressor functions of wild-type p53 (wt p53), but may also acquire additional pro-oncogenic roles (Michalovitz et al., 1991). Indeed, different forms of mutant p53 vary with regard to their biological capabilities (Halevy et al., 1990), and there is now ample evidence that common tumor-associated p53 mutants often possess novel oncogenic ‘gain of function’ acti- vities (Sigal and Rotter, 2000; Cadwell and Zambetti, 2001). Of note, mutant p53 has been shown to have oncogenic properties in cells which are p53 null, ruling out the simple possibility of a dominant-negative effect over endogenous wt p53 (Dittmer et al., 1993; Wang et al., 1998; Liu et al., 2000). Several biochemical and biological functions of mutant p53 proteins, indepen- dent of the wt p53 protein, have been described. For instance, p53 mutants were shown to be potent activators of numerous genes, including c-Myc (Frazier et al., 1998), topoisomerase I (Albor et al., 1998), MDR- 1 (Chin et al., 1992; Zastawny et al., 1993; Sampath et al., 2001), the antiapoptotic BAG-1 (Yang et al., 1999), as well as of various viral promoters (Deb et al., Received 24 January 2003; revised 25 April 2003; accepted 25 April 2003 *Correspondence: M Oren, Department of Molecular Cell Biology, Weizmann Institute of Science, PO Box 26, Rehovot 76100, Israel; E-mail: [email protected] Oncogene (2003) 22, 5667–5676 & 2003 Nature Publishing Group All rights reserved 0950-9232/03 $25.00 www.nature.com/onc
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Mutant p53 gain of function: repression of CD95(Fas/APO1) gene expression by tumor-associated p53...

Mutant p53 gain of function: repression of CD95(Fas/APO-1) gene
expression by tumor-associated p53 mutants
Amir Zalcenstein1, Perry Stambolsky1, Lilach Weisz1, Martina Muller2, David Wallach3, Tanya MGoncharov3, Peter H Krammer4, Varda Rotter1 and Moshe Oren*,1
1Department of Molecular Cell Biology, Weizmann Institute of Science, PO Box 26, Rehovot, 76100, Israel; 2Department of InternalMedicine IV, University Hospital, 69115 Heidelberg, Germany; 3Department of Biological Chemistry, Weizmann Institute of Science,PO Box 26, Rehovot 76100, Israel; 4Tumor Immunology Program, German Cancer Research Center, 69120 Heidelberg, Germany
Tumor-associated mutant forms of p53 can exert anantiapoptotic gain of function activity, which probablyconfers a selective advantage upon tumor cells harboringsuch mutations. We report that mutant p53 suppresses theexpression of the CD95 (Fas/APO-1) gene, encoding adeath receptor implicated in a variety of apoptoticresponses. Moderate (40–50%) downregulation of CD95mRNA and surface protein expression by mutant p53correlates with partial protection against CD95-dependentcell death. Excess mutant p53 represses the transcrip-tional activity of the CD95 promoter, with the extent ofrepression varying among different tumor-associated p53mutants. Furthermore, mutant p53 protein binds theCD95 promoter in vitro, in a region distinct from theone implicated in tight interactions of the CD95 gene withwild-type p53. Hence, the CD95 promoter is likely to be adirect target for downregulation by mutant p53. Thisactivity of mutant p53 may contribute to its gain offunction effects in oncogenesis.Oncogene (2003) 22, 5667–5676. doi:10.1038/sj.onc.1206724
Keywords: p53; apoptosis; CD 95; Fas/APO-1; repression
Introduction
The p53 tumor suppressor gene plays a pivotal role inthe prevention of cancer. Consequently, it is often atarget of genetic alterations in tumors: about half of allhuman tumors carry direct alterations within the p53locus, and in many of the other half, p53 function isimpaired owing to mutations in proteins operatingeither upstream or downstream to p53 (Vogelsteinet al., 2000; Ryan et al., 2001; Michael and Oren,2002; Bargonetti and Manfredi, 2002).
p53 exerts its tumor suppressor function throughmultiple mechanisms. However, the most prominentbiochemical feature of p53, underlying the major part ofits tumor-suppressor activities, is its ability to act as asequence-specific transcription factor. This entails directbinding to specific target sequences within the DNA,
denoted p53 response elements (p53REs), with conse-quent transcriptional activation of genes residing in theclose vicinity of the p53REs. The importance of thetranscriptional activity of p53 is underscored by thefinding that the vast majority of p53 mutationsencountered in cancer reside within the core sequence-specific DNA-binding domain of p53 (el-Deiry, 1998;Soussi, 2000). In addition, p53 can also repress thetranscription of many other genes, although directbinding of p53 to specific sequences within such geneshas not been implicated in most cases.
Given p53’s pivotal role in tumor suppression, it istherefore logical that potentially tumorigenic cellsshould strive to inactivate p53, thereby eliminating animportant stumbling block for the development ofcancer. However, unlike other tumor suppressor genes,such as Rb (Harbour and Dean, 2000) or ARF (Serrano,2000), which are typically deleted, truncated, silenced orotherwise downregulated in many cancers, selectivepressures within tumors clearly favor missense muta-tions in p53 rather than its outright elimination, oftenleading to the constitutive expression of high amounts ofmutant p53 in tumor cells (Sigal & Rotter, 2000). Thisevidence may suggest p53 mutations to be a double-edged sword; that is mutant p53 proteins may not onlylose the tumor suppressor functions of wild-type p53 (wtp53), but may also acquire additional pro-oncogenicroles (Michalovitz et al., 1991). Indeed, different formsof mutant p53 vary with regard to their biologicalcapabilities (Halevy et al., 1990), and there is now ampleevidence that common tumor-associated p53 mutantsoften possess novel oncogenic ‘gain of function’ acti-vities (Sigal and Rotter, 2000; Cadwell and Zambetti,2001). Of note, mutant p53 has been shown to haveoncogenic properties in cells which are p53 null, rulingout the simple possibility of a dominant-negative effectover endogenous wt p53 (Dittmer et al., 1993; Wanget al., 1998; Liu et al., 2000). Several biochemical andbiological functions of mutant p53 proteins, indepen-dent of the wt p53 protein, have been described. Forinstance, p53 mutants were shown to be potentactivators of numerous genes, including c-Myc (Frazieret al., 1998), topoisomerase I (Albor et al., 1998), MDR-1 (Chin et al., 1992; Zastawny et al., 1993; Sampathet al., 2001), the antiapoptotic BAG-1 (Yang et al.,1999), as well as of various viral promoters (Deb et al.,Received 24 January 2003; revised 25 April 2003; accepted 25 April 2003
*Correspondence: M Oren, Department of Molecular Cell Biology,Weizmann Institute of Science, PO Box 26, Rehovot 76100, Israel;E-mail: [email protected]
Oncogene (2003) 22, 5667–5676& 2003 Nature Publishing Group All rights reserved 0950-9232/03 $25.00
www.nature.com/onc

1992). Mutant p53 was also shown to promote genomicinstability (Murphy et al., 2000) and to contribute to thedisruption of spindle checkpoint control (Gualbertoet al., 1998). Tumor-associated p53 mutants were shownto effectively block apoptosis induced by survival factordeprivation (Peled et al., 1996), and significantly were alsofound to be effective inhibitors of DNA damage-inducedapoptosis (Lotem & Sachs, 1995; Li et al., 1998; Blandinoet al., 1999; Matas et al., 2001). However, the exactmolecular mechanisms underlying the various gain of fun-ction activities of mutant p53 still remain to be resolved.
CD95(Fas/APO-1) is a type I transmembrane deathreceptor, shown to be crucial in a variety of physiologicaland pathological conditions leading to apoptosis (Walc-zak and Krammer, 2000). The apoptotic pathwayregulated by CD95 is triggered in response to engagementof this receptor by its cognate ligand, CD95L. A role forCD95 in p53-mediated apoptosis has been proposed onthe basis of the observation that the CD95 gene is atranscriptional target of wt p53 (Owen-Schaub et al.,1996), whose expression is induced through the bindingof wt p53 to a regulatory region residing within its firstintron, with a possible contribution also of sequenceswithin the CD95 upstream promoter (Muller et al., 1998;Munsch et al., 2000). Furthermore, there is evidence thatCD95 function is required for optimal cell killing byDNA-damaging agents (Friesen et al., 1996; Muller et al.,1997; Embree-Ku et al., 2002), although this may holdonly for some cell types and not for others (Newton andStrasser, 2000). Interestingly, while it is anticipated thatthe activation of wt p53 by DNA damage and other typesof chemotherapy can trigger CD95 expression in amanner that may contribute to cell killing by theseagents, CD95 has been shown to play a role in thecytotoxic effects of anticancer agents also in cells lackingwt p53 (O’Connor et al., 2000; Shao et al., 2001). Hence,downregulation of CD95 expression in such tumor cells islikely to render them more resistant to chemotherapy.
With this reasoning in mind, we asked whethermutant forms of p53 known to possess an antiapoptoticgain of function can modulate the expression of theCD95 gene. We report here that overexpression ofmutant p53 confers partial protection against CD95L-induced apoptosis, and that this is correlated with asignificant reduction in CD95 mRNA and surfaceprotein levels. Moreover, evidence is provided thatmutant p53 can downregulate the activity of the CD95promoter, in a manner probably entailing a physicalinteraction between mutant p53 and DNA sequenceswithin the promoter. These findings suggest a novelmechanism for the antiapoptotic gain of functionactivity of mutant p53, which may contribute to cancerprogression and therapy resistance.
Results
Mutant p53 attenuates CD95-induced apoptosis in PC3cells
Mutant p53 overexpression can attenuate apoptosis inp53 null cells following DNA damage or deprivation of
survival factors (Lotem and Sachs, 1995; Kremenets-kaya et al., 1997; Li et al., 1998; Blandino et al., 1999;Matas et al., 2001). This argues in favor of anantiapoptotic gain of function activity of mutant p53.In an attempt to elucidate the underlying mechanisms,we explored the possibility that mutant p53 mayinterfere with one or more endogenous apoptoticpathways. The CD95(Fas/APO-1) pathway has beenproposed to contribute to the killing of tumor cells byDNA-damaging agents, at least under some circum-stances (Friesen et al., 1996; Muller et al., 1997). Wetherefore asked whether overexpression of tumor-associated mutant p53 can affect cell killing by theCD95 pathway. The analysis was performed in p53-nullhuman prostate carcinoma PC3 cells, which are reason-ably sensitive to CD95-mediated apoptosis.
To assess the impact of mutant p53 on CD95-mediated apoptosis, PC3 cells were infected with arecombinant retrovirus encoding the p53R175H mu-tant, shown to exert a potent antiapoptotic gain offunction effect (Blandino et al., 1999). Staining of fixedcells with p53-specific antibodies revealed that over 90%of the cells in the infected culture expressed easilydetectable mutant p53 (data not shown). Following 8 hof exposure to a conditioned medium containing CD95ligand (CD95L) in the presence of the protein synthesisinhibitor cycloheximide (CHX), the rate of cell deathwas determined by propidium iodide (PI) uptake. Asseen in Figure 1a and b, the extent of PI uptake wasmarkedly reduced in PC3 cells infected with p53R175H,as compared to control p53-null counterparts. Hence,overexpression of tumor-associated mutant p53 canattenuate the induction of CD95-mediated cell death.
Mutant p53 downregulates CD95 mRNA and protein
One possible way in which mutant p53 might attenuateCD95-dependent apoptosis is by downregulating theexpression of the CD95 receptor itself. We thereforeinvestigated the effect of overexpressed p53R175H onthe levels of endogenous CD95 mRNA and protein.Real-time PCR analysis revealed a significant reductionin the amount of CD95 mRNA in PC3 cells stablyexpressing the mutant p53 (Figure 2). Although thereduction was relatively mild, it was highly reproduciblein multiple experiments (data not shown). Importantly,FACS analysis revealed that overexpression of mutantp53 also led to a measurable decrease in the amount ofCD95 death receptor displayed on the cell surface(Figure 3). In agreement with the RNA data, thedecrease was only partial and did not result in completeablation of CD95 surface protein expression. Thisobservation is also consistent with the fact that themutant p53 expressors exhibit only a delay in theinduction of CD95-mediated apoptosis, rather thanbeing completely refractory to it (Figure 1).
Mutant p53 represses the CD95 promoter
The downregulation of CD95 mRNA by mutant p53might be due to repression of the CD95 gene promoter.
Mutant p53 represses CD95(Fas/APO-1) expressionA Zalcenstein et al
5668
Oncogene

This possibility was explored in transient transfectionassays, employing a plasmid in which the expression of aluciferase reporter gene is driven by regulatory DNAelements derived from the human CD95 gene. Morespecifically, these regulatory sequences include theCD95 promoter, followed by a region from intron 1 ofthat gene encompassing the p53REs (Muller et al.,1998). This plasmid is denoted CD95(Pþ I)-luc accord-ing to the terminology of Muller et al. (1998). For thesake of simplicity, unless otherwise stated, it will bedesignated CD95-luc in the following sections.
As seen in Figure 4a, cotransfection of p53R175Hsignificantly reduced CD95-luc activity in PC3 cells,supporting the conjecture that mutant p53 can indeedrepress CD95 gene transcription, and that this repres-sion is responsible for the reduced levels of CD95
mRNA and protein and for the reduced susceptibility toCD95-mediated apoptosis.
Different tumor-associated p53 mutants vary in theextent of their antiapoptotic gain of function; forinstance, whereas p53R175H protects transfected p53-null cells very efficiently against etoposide-induceddeath, the p53R273H mutant does so less efficiently(Blandino et al., 1999). We therefore compared theabilities of these two hot-spot mutants to repress the
Figure 1 Mutant p53 reduces the rate of CD95-induced apoptosis.PC3 cells were stably transfected with a plasmid encoding themouse ecotropic receptor and selected for 2 weeks with 0.4mg/mlG418. The cells were subsequently infected with a retrovirusexpressing mutant p53 (p53R175H) and selected for 48 h in amedium containing 4mg/ml puromycin. Infected (175H) andnoninfected (ctrl) cells were seeded in six-well culture dishes, andexposed 24 h later to a conditioned medium containing secretedCD95L, in the presence of 2mg/ml CHX. Cells were collected 8 hlater, incubated with 1 mg/ml PI and subjected to FACS analysis.(a) depicts actual PI-exclusion staining data for a single experimentwith cells exposed to 25% CD95L-containing medium; R1 and R2represent the PI-negative and PI-positive subpopulations, respec-tively. (b) shows a compilation of PI exclusion data from severalruns under identical conditions
Figure 2 Mutant p53 downregulates CD95 mRNA. PC3 cellsinfected with a retrovirus expressing mutant p53 (p53 175H), aswell as control PC3 cells, were seeded in 6 cm culture dishes. After24 h, cells were harvested and total RNA was extracted. In total,2mg of each RNA was used as a template for reverse transcriptionby MMLV reverse transcriptase. Real-time PCR (Lightcycler,Roche) was performed with primers specific for either CD95 orGAPDH mRNA, the latter serving as a normalization reference.Relative CD95 mRNA levels were calculated by normalization ofthe CD95 value for the amount of GAPDH transcripts in the samesample
Figure 3 Mutant p53 represses CD95 surface receptor expression.PC3 cells infected with a retrovirus expressing mutant p53 (p53175H), as well as control PC3 cells (cont) were harvested andincubated with an anti-CD95 antibody (Bender) for 1 h at roomtemperature. Cells were then incubated with goat–anti-mouse Cy5-conjugated secondary antibody (Jackson) for 30min, and analysedfor surface CD95 staining by FACS
Mutant p53 represses CD95(Fas/APO-1) expressionA Zalcenstein et al
5669
Oncogene

Figure 4 Mutant p53 represses the transcriptional activity of the CD95 promoter. (a) PC3 cells were seeded in 24-well culture dishes.Each well was transfected with 0.2mg of CD95-luc, a reporter plasmid expressing the firefly luciferase gene under the transcriptionalcontrol of the CD95 gene regulatory region, which comprises the CD95 promoter as well as a segment of the first intron encompassingthe previously described p53RE (Muller et al., 1998), together with 25 ng of one of the following plasmids: pCMV-neo-Bam vectorcontrol (cont), mutant p53R175H expression plasmid (175H), and mutant p53R273H expression plasmid (273H). Luciferase activitywas assayed 48 h post-transfection. Each plasmid combination was transfected into six identical wells; the average and standard errorare shown. (b) H1299 lung adenocarcinoma cells were seeded as in (a). Each well was transfected with a CD95 luciferase reporter as in(a), together with 300 ng of one of the following plasmids: pCMV-neo-Bam vector control (cont), mutant p53R175H expressionplasmid (175H), and a plasmid encoding a derivative of p53R175H with two-point mutations in the transactivation domain (22,23).(c) Extracts from transfections in (b) were assayed for p53 protein expression by Western blot analysis with p53-specific antibodies. (d)H1299 cells were seeded and transfected as in (b) with a CD95 luciferase reporter together with either a control plasmid (cont), orplasmids encoding the 175H, 273H or 248W p53 mutants. (e) Extracts from transfections in (d) were assayed for p53 proteinexpression by Western blot analysis with p53-specific antibodies. (f) H1299 cells, seeded as in (a), were transfected with 0.2 mg of eitherCD95(Pþ I)-luciferase (referred to as CD95-luc in the previously described experiments) or CD95(Ps)-luciferase (f) upper panel,described in Muller et al., 1998), together with 0.3mg of either pCMV-neo-Bam (cont) or expression plasmid encoding p53R175H.Briefly, the CD95(Pþ I) construct contains the CD95 promoter as well as the p53RE (indicated by a box) located in the gene’s firstintron, while the CD95 (Ps) construct contains only the CD95 promoter
Mutant p53 represses CD95(Fas/APO-1) expressionA Zalcenstein et al
5670
Oncogene
CD95 promoter. Although p53R273H was still capableof repressing the CD95 promoter, it did so less efficientlythan p53R175H (Figure 4a and d), supporting theexistence of a correlation between the antiapoptoticactivity of a given p53 mutant and its ability todownregulate the CD95 promoter.
It has been shown that the cytoprotective effects ofmutant p53 rely on the integrity of its N-terminaltransactivation domain (Matas et al., 2001). We there-fore tested whether a mutant p53 protein impaired inthis transactivation domain can still repress the CD95promoter efficiently. These experiments were performedin human lung adenocarcinoma H1299 cells, which likePC3 are also p53-null. As shown in Figure 4b, aderivative of p53R175H carrying point mutations atresidues 22 and 23, which incapacitate the transactiva-tion domain (Lin et al., 1994), was also impaired inrepression of the CD95 promoter, although the levels ofmutant p53 protein achieved in the transfected cells werecomparable for both mutants (Figure 4c). Thus, theintegrity of the N-terminal transactivation domain isimportant for the ability of mutant p53 to repress CD95promoter activity as well as for its ability to protecttumor cells from drug-induced apoptosis.
Another common tumor-associated p53 mutant isp53R248W. Like p53R273H, the p53R248W hot-spotmutant was also capable of partial repression of theCD95 promoter, albeit not as efficiently as p53R175H(Figure 4d) when expressed at comparable levels(Figure 4e).
The reporter plasmid employed in all the previousexperiments contains a combination of the CD95promoter and an additional genomic DNA fragmentderived from intron 1 of the CD95 gene [CD95(Pþ I);Muller et al., 1998]. This intronic DNA fragmentcontains a potent p53-binding site, and is primarilyresponsible for the activation of the CD95 gene by wtp53 (Muller et al., 1998). Surprisingly, a reporterplasmid containing only the CD95 promoter, withoutaddition of the intronic p53RE [CD95(Ps), Figure 4f],was still repressed by mutant p53. Thus, the potentialfor mutant p53-mediated repression resides within theCD95 gene promoter, and does not require thepreviously defined intronic wt p53RE.
In recent years, it has become clear that many tumor-associated p53 mutants can bind and inactivate also theother members of the p53 family, namely p63 and p73(Di Como et al., 1999; Marin et al., 2000; Strano et al.,2000; Gaiddon et al., 2001; Strano et al., 2002). At leastfor p73, the efficacy with which mutant p53 exerts thisdominant negative effect has been shown to depend to agreat extent on the identity of the residue located atposition 72 of human p53. Owing to a commonpolymorphism, normal individuals can carry either aproline or an arginine at that position. However, whenp53 undergoes a mutation within its DNA binding coredomain, the presence of Arg at position 72 leads to amuch stronger inhibition of p73 function than a Pro atposition 72 (Marin et al., 2000). Of note, this differencehas been demonstrated convincingly for the twopolymorphic versions of p53R175H (Marin et al.,
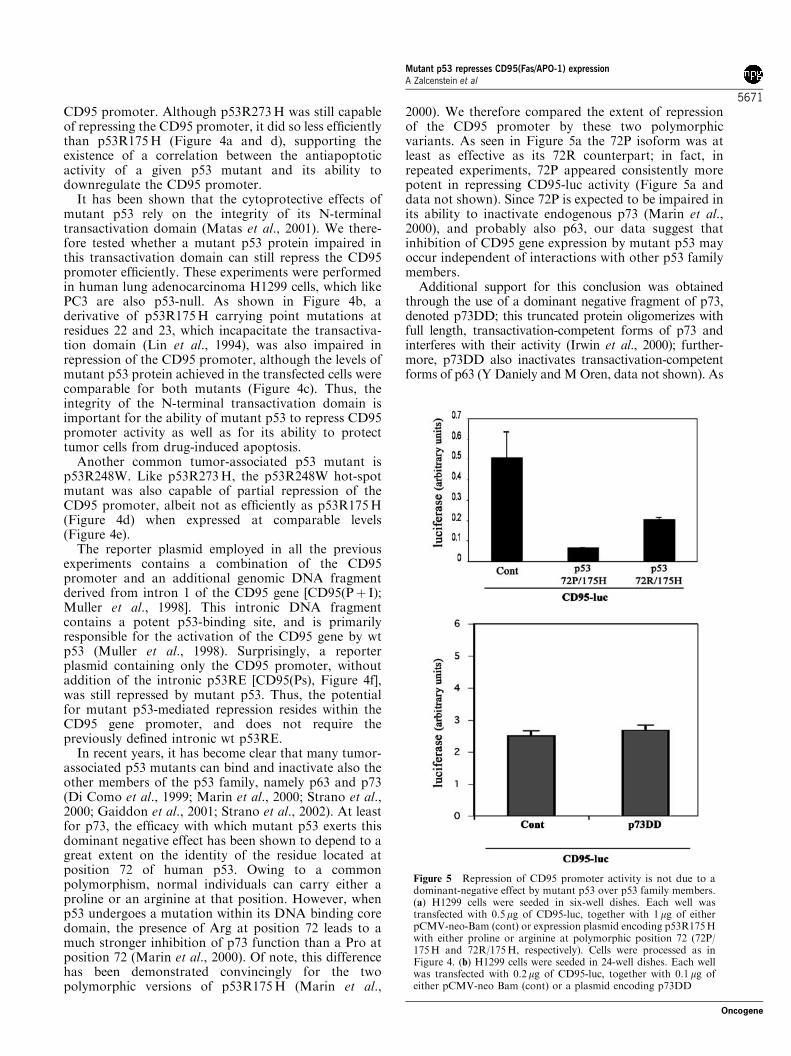
2000). We therefore compared the extent of repressionof the CD95 promoter by these two polymorphicvariants. As seen in Figure 5a the 72P isoform was atleast as effective as its 72R counterpart; in fact, inrepeated experiments, 72P appeared consistently morepotent in repressing CD95-luc activity (Figure 5a anddata not shown). Since 72P is expected to be impaired inits ability to inactivate endogenous p73 (Marin et al.,2000), and probably also p63, our data suggest thatinhibition of CD95 gene expression by mutant p53 mayoccur independent of interactions with other p53 familymembers.
Additional support for this conclusion was obtainedthrough the use of a dominant negative fragment of p73,denoted p73DD; this truncated protein oligomerizes withfull length, transactivation-competent forms of p73 andinterferes with their activity (Irwin et al., 2000); further-more, p73DD also inactivates transactivation-competentforms of p63 (Y Daniely andMOren, data not shown). As
Figure 5 Repression of CD95 promoter activity is not due to adominant-negative effect by mutant p53 over p53 family members.(a) H1299 cells were seeded in six-well dishes. Each well wastransfected with 0.5 mg of CD95-luc, together with 1mg of eitherpCMV-neo-Bam (cont) or expression plasmid encoding p53R175Hwith either proline or arginine at polymorphic position 72 (72P/175H and 72R/175H, respectively). Cells were processed as inFigure 4. (b) H1299 cells were seeded in 24-well dishes. Each wellwas transfected with 0.2mg of CD95-luc, together with 0.1 mg ofeither pCMV-neo Bam (cont) or a plasmid encoding p73DD
Mutant p53 represses CD95(Fas/APO-1) expressionA Zalcenstein et al
5671
Oncogene
seen in Figure 5b, p73DD failed to repress the CD95promoter, although it effectively blocked the induction ofseveral p53-responsive promoters, including the CD95promoter, under similar experimental conditions (data notshown). This further argues that the basal level of CD95promoter activity in this experimental system is notdependent on endogenous p63 or p73, and thus the effectof mutant p53 must be exerted through some mechanismother than dominant-negative repression of p63 and p73.
Mutant p53 binds preferentially to the CD95 promoter
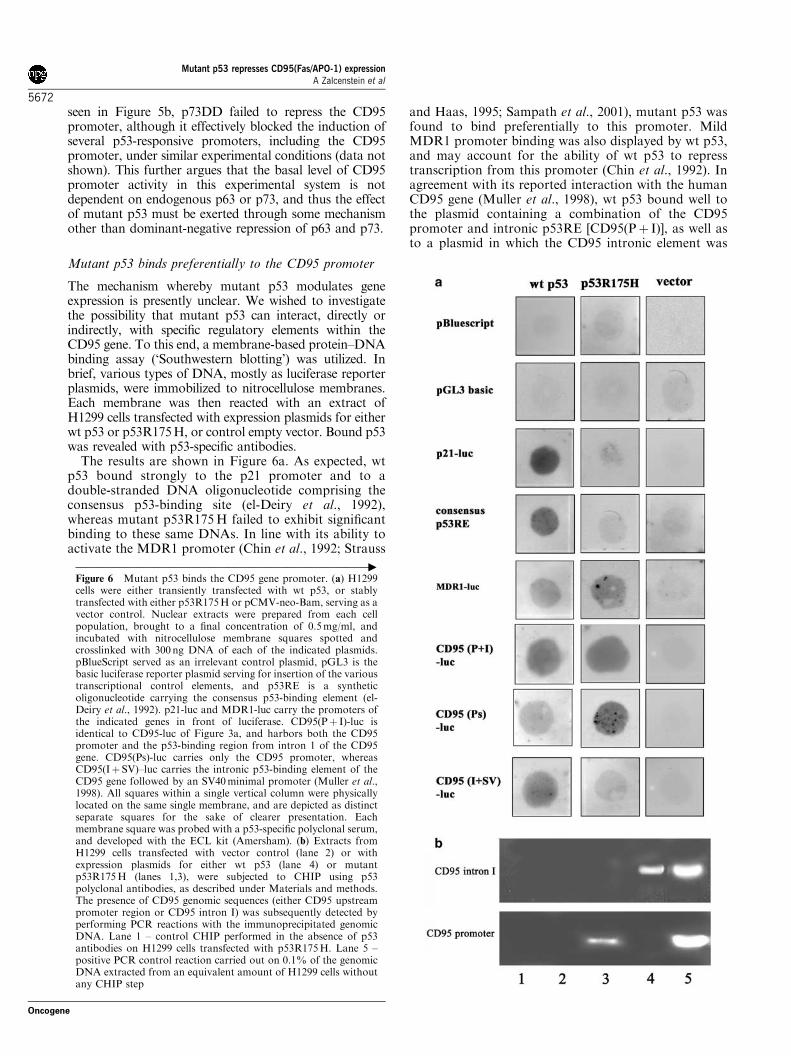
The mechanism whereby mutant p53 modulates geneexpression is presently unclear. We wished to investigatethe possibility that mutant p53 can interact, directly orindirectly, with specific regulatory elements within theCD95 gene. To this end, a membrane-based protein–DNAbinding assay (‘Southwestern blotting’) was utilized. Inbrief, various types of DNA, mostly as luciferase reporterplasmids, were immobilized to nitrocellulose membranes.Each membrane was then reacted with an extract ofH1299 cells transfected with expression plasmids for eitherwt p53 or p53R175H, or control empty vector. Bound p53was revealed with p53-specific antibodies.
The results are shown in Figure 6a. As expected, wtp53 bound strongly to the p21 promoter and to adouble-stranded DNA oligonucleotide comprising theconsensus p53-binding site (el-Deiry et al., 1992),whereas mutant p53R175H failed to exhibit significantbinding to these same DNAs. In line with its ability toactivate the MDR1 promoter (Chin et al., 1992; Strauss
and Haas, 1995; Sampath et al., 2001), mutant p53 wasfound to bind preferentially to this promoter. MildMDR1 promoter binding was also displayed by wt p53,and may account for the ability of wt p53 to represstranscription from this promoter (Chin et al., 1992). Inagreement with its reported interaction with the humanCD95 gene (Muller et al., 1998), wt p53 bound well tothe plasmid containing a combination of the CD95promoter and intronic p53RE [CD95(Pþ I)], as well asto a plasmid in which the CD95 intronic element was
––––––––––––––––––––––––––––––––––––––––––––––––––––––––"
Figure 6 Mutant p53 binds the CD95 gene promoter. (a) H1299cells were either transiently transfected with wt p53, or stablytransfected with either p53R175H or pCMV-neo-Bam, serving as avector control. Nuclear extracts were prepared from each cellpopulation, brought to a final concentration of 0.5mg/ml, andincubated with nitrocellulose membrane squares spotted andcrosslinked with 300 ng DNA of each of the indicated plasmids.pBlueScript served as an irrelevant control plasmid, pGL3 is thebasic luciferase reporter plasmid serving for insertion of the varioustranscriptional control elements, and p53RE is a syntheticoligonucleotide carrying the consensus p53-binding element (el-Deiry et al., 1992). p21-luc and MDR1-luc carry the promoters ofthe indicated genes in front of luciferase. CD95(Pþ I)-luc isidentical to CD95-luc of Figure 3a, and harbors both the CD95promoter and the p53-binding region from intron 1 of the CD95gene. CD95(Ps)-luc carries only the CD95 promoter, whereasCD95(Iþ SV)–luc carries the intronic p53-binding element of theCD95 gene followed by an SV40minimal promoter (Muller et al.,1998). All squares within a single vertical column were physicallylocated on the same single membrane, and are depicted as distinctseparate squares for the sake of clearer presentation. Eachmembrane square was probed with a p53-specific polyclonal serum,and developed with the ECL kit (Amersham). (b) Extracts fromH1299 cells transfected with vector control (lane 2) or withexpression plasmids for either wt p53 (lane 4) or mutantp53R175H (lanes 1,3), were subjected to CHIP using p53polyclonal antibodies, as described under Materials and methods.The presence of CD95 genomic sequences (either CD95 upstreampromoter region or CD95 intron I) was subsequently detected byperforming PCR reactions with the immunoprecipitated genomicDNA. Lane 1 – control CHIP performed in the absence of p53antibodies on H1299 cells transfected with p53R175H. Lane 5 –positive PCR control reaction carried out on 0.1% of the genomicDNA extracted from an equivalent amount of H1299 cells withoutany CHIP step
Mutant p53 represses CD95(Fas/APO-1) expressionA Zalcenstein et al
5672
Oncogene

linked to a minimal SV40 promoter instead of the CD95promoter [CD95(Iþ SV)]. It can be noted that wt p53also exhibited some binding to the CD95 promoter[CD95(Ps)], supporting the possible existence of one ormore weak p53REs within the CD95 promoter (Mulleret al., 1998).
Importantly, p53R175H bound efficiently to theCD95 regulatory sequences [CD95(Pþ I)]; the signalintensity was comparable to that obtained with wt p53.Further mapping revealed that this interaction targetsthe upstream promoter [CD95(Ps)], rather than theintronic region containing the wt p53RE. This isconsistent with the fact that the CD95 promoter isrepressed by mutant p53 in reporter assays (Figure 4f).
To determine whether mutant p53 associates with theCD95 promoter also within living cells, chromatinimmunoprecipitation (CHIP) analysis was performed.H1299 cells expressing either no p53 at all, orp53R175H or wt p53, were crosslinked, harvested, andsubjected to CHIP with an anti-p53 polyclonal anti-body, followed by RT–PCR analysis with primer pairsderived either from the CD95 promoter (about 1 kbupstream of the transcription initiation site), or from theintronic region spanning the wt p53RE of the CD95gene. The results are shown in Figure 6b. As expected,wt p53 was found to be associated specifically with theregion containing the p53RE (lane 4). Mutant p53, onthe other hand, showed no in vivo binding to the intronicp53RE; instead, it exhibited selective binding to theCD95 promoter region (lane 3).
Taken together, these findings imply that the mechan-ism mediating the inhibitory effect of mutant p53 onCD95 gene transcription is distinct from that underlyingthe stimulatory effect of wt p53 on the same gene.Moreover, our observations lend strong support to theconjecture that the selective physical association ofmutant p53 with the CD95 promoter underlies its abilityto repress CD95 gene expression.
Discussion
The data presented in this study demonstrate that, in thepresence of mutant p53, the CD95L-CD95 apoptoticpathway is markedly attenuated. This is likely to be due,at least in part, to repression of CD95 gene transcriptionby mutant p53. The data further suggest that suchrepression may rely on the binding of mutant p53 to theCD95 promoter, in a region distinct from the intronicsite serving as the primary binding target of wt p53 inthe CD95 gene.
Our findings may help explain some recent observa-tions relating to the expression of CD95 in humancancer. Thus, a survey of nonsmall cell lung carcinomatumors revealed a strong correlation between thepresence of p53 mutations and reduced CD95 mRNAlevels (Boldrini et al., 2001). Similarly, p53 mutations inhepatocellular carcinoma were shown to correlate withloss of CD95 expression, measured by immunohisto-chemistry (Volkmann et al., 2001). Interestingly, the
latter study suggested that reduced CD95 expression ischaracteristic of tumors harboring only some, but notall, forms of mutant p53. Our findings predict that thisobservation may be explained by a differential capacityof distinct p53 mutants to downregulate CD95 promoteractivity. It is noteworthy that common tumor-associatedp53 mutants indeed differ markedly with regard to theirantiapoptotic gain of function (Blandino et al., 1999). Atleast for three hot-spot mutants, p53R175H,p53R273H and p53R248W, differential inhibition ofdrug-induced apoptosis correlates well with differentialrepression of the CD95 promoter: p53R175H is aneffective inhibitor of apoptosis and CD95 promoteractivity, whereas both p53R273H and p53R248Winhibit apoptosis (Blandino et al., 1999) and CD95promoter activity (Figure 4d) to a lesser degree thanp53R175H. This suggests that a common mechanismmay underlie both these activities of mutant p53. Thisconclusion is also supported by the apparent require-ment of the N-terminal transactivation domain for bothCD95 repression (Figure 4b) and cytoprotection (Mataset al., 2001) by mutant p53.
Our findings also provide several clues on themolecular mechanism whereby mutant p53 repressesCD95 transcription. One likely explanation might havebeen that mutant p53 exerts its impact on the CD95promoter owing to its ability to bind and inactivatetranscription-competent (TA) forms of the p53 familymembers p63 and p73 (Di Como et al., 1999; Marinet al., 2000; Strano et al., 2000; Gaiddon et al., 2001;Strano et al., 2002). Indeed, TAp73 can activatetranscription from the CD95 promoter/intronic p53REin transient transfection assays (M Muller, G Melinoand PHK, in preparation). Thus, if p73 or p63 plays arole in maintaining basal CD95 mRNA levels, theirinactivation by excess mutant p53 might explain ourobservations. Although we cannot presently rule outcompletely a contribution of this mechanism to ourresults, the data in Figure 5 argue strongly at leastagainst its being the only explanation. Moreover, we didnot observe significant differences between the R72 andP72 isoforms of p53R175H with regard to their abilityto augment the resistance of H1299 cells to killing byetoposide (data not shown), further suggesting that bothrepression of CD95 transcription and inhibition ofDNA-damage induced apoptosis by mutant p53 donot rely on inactivation of endogenous p73, and perhapsalso p63. Thirdly, activation of CD95 gene expressionby TA forms of p63 and p73 is expected to rely on theinteraction of these proteins with the intronic p53RE;yet, mutant p53-mediated repression is independent ofthe p53RE (Figure 4f). All these argue in favor of arepression mechanism that relies on some other mole-cular property of mutant p53, distinct from itsdominant-negative effect on TA isoforms of p53 familymembers.
Indeed, we found a physical interaction betweenmutant p53 and regulatory elements within the CD95promoter. Such a specific interaction could be confirmedto occur not only in vitro (Figure 6a), but also withinliving cells (Figure 6b). As the experiment in Figure 6a
Mutant p53 represses CD95(Fas/APO-1) expressionA Zalcenstein et al
5673
Oncogene

was performed with crude nuclear extracts rather thanwith purified mutant p53, the data do not prove directbinding of mutant p53 to CD95 promoter DNA. It thusremains to be established whether mutant p53 recog-nizes directly specific sequence elements within theCD95 DNA, or rather is tethered to them throughprotein–protein interactions with one or more DNA-binding proteins. Of note, activation of the MDR1promoter by mutant p53 has been shown to require itsassociation with transcription factors of the ETS family(Sampath et al., 2001). Either way, it is reasonable toassume that the ability of mutant p53 to associate withthe CD95 promoter underlies the repression of thispromoter.
Our findings may have important clinical implica-tions. In view of the observations that CD95 activity isrequired for the efficient killing of many types of tumorcells by chemotherapy (Friesen et al., 1996; Muller et al.,1997; O’Connor et al., 2000; Shao et al., 2001),downregulation of endogenous CD95 expression bymutant p53 proteins, often accumulated in cancer cells,might contribute to increased chemotherapy resistance.Furthermore, it is highly conceivable that this inhibitoryeffect of mutant p53 is not restricted to the CD95 gene.There may thus exist additional proapoptotic geneswhose expression is downregulated by mutant p53,further contributing to a worse patient outcome. Whilesuch additional mutant p53 target genes remain to beidentified, one might predict that interfering with theexpression or function of mutant p53 in tumor cells mayrender such tumors more responsive to therapy andperhaps reduce their aggressiveness and metastaticcapacity.
Materials and methods
Cells, plasmids, infections and transfections
H1299 cells were obtained from ATCC. H1299 cells weremaintained in an RPMI medium (Sigma) supplemented with10% fetal calf serum (FCS, Sigma). PC3 cells were maintainedin Dulbecco’s modified Eagle medium (DMEM, Gibco)supplemented with 10% FCS.Plasmid pCMV-neo-Bam-p53R273H and expression plas-
mids for p53R175H, carrying either Arg or Pro at position 72,were obtained from B Vogelstein and W Kaelin, respectively.A pcDNA3-based expression plasmid for p53R175H(22,23)was constructed by G Blandino. Luciferase reporter plasmidscarrying various portions of the human CD95 gene have beendescribed before (Muller et al., 1998). Plasmid pM5neo-ecotropic-R, encoding the mouse ecotropic receptor wasprovided by S Benchimol.Infections for FACS, real-time PCR analysis, and surface
CD95 protein quantification were carried out using a standardprotocol. Briefly, PC3 cells were stably transfected withpM5neo-ecotropic-R, encoding the mouse ecotropic receptor,and selected for two weeks in 0.4mg/ml G418. Concurrently,293T cells were transfected with the retroviral constructpBabe-175H, using JetPEI (Polyplus transfection) accordingto the manufacturer’s instructions. Virus-containing culturesupernatants were harvested 24–48 h post-transfection at 6 hintervals and pooled together. The stably transfected PC3 cells
were then infected with the filtered supernatants, and selectedfor 48 h in 40 mg/ml of puromycin, resulting in cell populationsstably expressing mutant p53R175H. For luciferase assays, allcells were seeded and transfected using Fugene (Roche) at aratio of 2 mg DNA:3 ml Fugene, according to the manufac-turer’s instructions.
FACS analysis of CD95 surface protein
Noninfected PC3 cells and cells infected with mutantp53R175H were plated in six-well dishes. At 24 h after plating,cells were trypsinized and washed once in PBS. Cells were thenincubated with an anti-CD95 monoclonal antibody (BMS140,Bender) for 1 h, washed, and incubated for 30min with a Cy-5conjugated goat–anti-mouse secondary antibody (Jackson).Cells were then placed on ice and analysed by FACS.
Nuclear extracts
Nuclear extracts were prepared as described (Offer et al.,1999). Cells (106–107) were washed twice with cold PBS andharvested. Cell pellets were resuspended by gentle pipetting in400 ml of buffer A (10mm HEPES–KOH pH 7.9; 1.5mm
MgCl2; 10mm KCl; 0.5mm DTT; 0.2mm PMSF; 10 mg/mlleupeptin; 1mg/ml pepstatin and 10mg/ml aprotinin). After15min incubation on ice, 25ml of 10% NP-40 was added andvortexing was performed vigorously for 10min. After centri-fugation, cell pellets were resuspended in 150–300ml of bufferC (20mm HEPES–KOH pH 7.9; 25% glycerol; 420mm NaCl;1.5mm MgCl2; 0.2mm EDTA; 0.5mm DTT; 0.2mm PMSF;10 mg/ml leupeptin; 1mg/ml pepstatin and 10mg/ml aprotinin).Tubes were transferred to a rotating platform for 15min at41C, and then centrifuged and the protein content of thesupernatant determined by the Bradford procedure. Aliquotswere stored at �701C.
Western blot analysis
Cells were lysed in passive lysis buffer (Promega). Lysatealiquots were resolved by SDS–PAGE on a 10% polyacryla-mide gel, transferred onto a nitrocellulose membrane, andprobed sequentially with a mixture of the p53-specificmonoclonal antibodies PAb1801 and DO-1, followed byantivinculin antibody (Sigma). Membranes were then reactedwith secondary goat–anti-mouse HRP-conjugated antibodies(1:10 000, Jackson), and developed using the ECL kit(Amersham).
‘Southwestern’ analysis
In total, 300 ng of each DNA sample was mounted onto anitrocellulose membrane and crosslinked with 120 J/m2 ofUVC light. Membranes were washed twice in PBS and blockedwith 5% skim milk in PBS for 3 h. Nuclear extracts wereprepared as described, and diluted to a protein concentrationof 0.5mg/ml in gel-shift buffer (12.5mm Tris-HCl pH 7.9;3.1mm MgCl2; 25mm KCl; 0.5mm DTT; 10% glycerol;0.25mm EDTA; 0.2mm PMSF; 10 mg/ml leupeptin; 1 mg/mlpepstatin, 10 mg/ml aprotinin and 1mg/ml of pGL3-basicplasmid DNA for blocking). Nuclear extracts were incubatedovernight at 41C with the membrane segments, followed by3� 5min washes in PBS-T. The membrane was next incubatedfor 1 h with a p53-specific polyclonal serum under standardWestern blot analysis conditions, followed by three consecu-tive 5min washes in PBS-T and incubation for 30min withhorseradish peroxidase-conjugated goat–anti-rabbit secondaryantibody. The membranes were then developed with an ECLkit (Amersham).
Mutant p53 represses CD95(Fas/APO-1) expressionA Zalcenstein et al
5674
Oncogene

CHIP analysis
CHIP was performed essentially as described previously(Weinmann and Farnham, 2002), except that lysates from3� 107 H1299 cells stably expressing p53R175H or transientlytransfected with wt p53 were diluted fivefold in a CHIPdilution buffer (0.01%. SDS, 1.1% Triton X-100, 1.2mm
EDTA, 16.7mm Tris pH 8.1, 167mm NaCl), and boundcomplexes were collected with protein A-agarose beads cross-linked to anti-p53 rabbit polyclonal antibody by DMP (Bailonet al., 2000). PCR of the CD95 intronic p53RE (wt p53-binding site) was performed on immunoprecipitated chromatinusing the following pair of oligonucleotide primers: 50-GGATAATTAGACGTACGTGGGC-30 (forward) and 50-GGACAATTGACAAAATCAGTATC-30 (reverse).PCR of the CD95 promoter (mutant p53-binding site) was
performed using the oligonucleotides 50-CCGCTGGGCA-GGCGGGGCAGCTCC-30 (forward) and 50-TGCAGGCT-CTCTCCCCGCCCCCGC-30 (reverse).
Luciferase assays
Luciferase assays were performed using (d)-luciferin (Roche).Luminescence was determined with the aid of a Rosys-AnthosLucy 3 luminometer.
Apoptosis assays
An expression vector for soluble CD95L (lz-FASL) wasdesigned similar to the plasmid used for preparing solubleCD40L (Fanslow et al., 1994). Briefly, CD95L was fused to amutant leucine zipper (LZ) domain of the yeast transcriptionfactor GCN4. In total, 293 cells were transfected with theCD95L expression vector, and the medium collected 3 dayslater as a source of CD95L.
For analysis of apoptosis by PI exclusion, cells were washedtwice with PBS, treated for 1min with 0.05% trypsin–EDTAsolution (Bet-Haemek, Israel), and rapidly washed again withPBS. Cells were dislodged from the dish by pipetting,centrifuged and resuspended in PBS containing 1 mg/ml PI(Sigma) for 30min before being analysed with the aid of aFACSORT double laser flow cytometer.
Real-time RT–PCR analysis
Total RNA was extracted using the Nucleo-Spin RNA II kit(Macherey Nagel). In total, 2mg of each RNA sample wasreverse transcribed using MMLV reverse transcriptase (Pro-mega). Real-time PCR was performed on a Roche Lightcyclersystem using CD95-specific primers (sense: CAAGGGG-GAATTGAGGA, antisense: CACTTGGTGTTGCTGGT-GAG) to yield a 150 bp PCR product. A touch-down PCRmethod was employed, where the annealing temperature isgradually reduced by 0.51C each cycle during the first 16 cycles(from 65 to 571C), in order to increase CD95 primer specificity.cDNA levels were normalized to GAPDH, amplified withappropriate primers (sense: ACCACAGTCGCCATCAC,antisense: TCCACCACCCTGTTGCTGTA), yielding a400 bp product.
AcknowledgementsWe thank B Vogelstein and W Kaelin for p53 expressionplasmids and S Benchimol for the ecotropic receptor expres-sion plasmid. This work was supported in part by GrantsQLG1-1999-00273 and QLK6-2000-00159 from the EuropeanCommission, and by grants from the Cooperation Program inCancer Research of the DKFZ and Israel’s Ministry of Science(MOS), the German-Israel Project Cooperation (DIP), theKadoorie Charitable Foundations, and the Yad AbrahamCenter for Cancer Diagnosis and Therapy.
References
Albor A, Kaku S and Kulesz-Martin M. (1998). Cancer Res.,58, 2091–2094.
Bailon P, Ehrlich G, Fung W and Berthold W. (2000).Methods in Molecular Biology, Vol. 147. Humana Press:Totowa, pp. 41–49.
Bargonetti J and Manfredi JJ. (2002). Curr. Opin. Oncol., 14,86–91.
Blandino G, Levine AJ and Oren M. (1999). Oncogene, 18,477–485.
Boldrini L, Faviana P, Pistolesi F, Gisfredi S, Di Quirico D,Lucchi M, Mussi A, Angeletti CA, Baldinotti F, Fogli A,Simi P, Basolo F and Fontanini G. (2001). Oncogene, 20,6632–6637.
Cadwell C and Zambetti GP. (2001). Gene, 277, 15–30.Chin KV, Ueda K, Pastan I and Gottesman MM. (1992).
Science, 255, 459–462.Deb S, Jackson CT, Subler MA and Martin DW. (1992). J.
Virol., 66, 6164–6170.Di Como CJ, Gaiddon C and Prives C. (1999). Mol. Cell. Biol.,19, 1438–1449.
Dittmer D, Pati S, Zambetti G, Chu S, Teresky AK,Moore M, Finlay C and Levine AJ. (1993). Nat. Genet., 4,42–46.
el-Deiry WS. (1998). Semin. Cancer. Biol., 8, 345–357.el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW andVogelstein B. (1992). Nat. Genet., 1, 45–49.
Embree-Ku M, Venturini D and Boekelheide K. (2002). BiolReprod., 66, 1456–1461.
Fanslow WC, Srinivasan S, Paxton R, Gibson MG, SpriggsMK and Armitage RJ. (1994). Semin. Immunol., 6, 267–278.
Frazier MW, He X, Wang J, Gu Z, Cleveland JL and ZambettiGP. (1998). Mol. Cell Biol., 18, 3735–3743.
Friesen C, Herr I, Krammer PH and Debatin KM. (1996).Nat. Med., 2, 574–577.
Gaiddon C, Lokshin M, Ahn J, Zhang T and Prives C. (2001).Mol. Cell Biol., 21, 1874–1887.
Gualberto A, Aldape K, Kozakiewicz K and Tlsty TD. (1998).Proc. Natl. Acad. Sci. USA, 95, 5166–5171.
Halevy O, Michalovitz D and Oren M. (1990). Science, 250,113–116.
Harbour JW and Dean DC. (2000). Genes Dev., 14, 2393–2409.Irwin M, Marin MC, Phillips AC, Seelan RS, Smith DI, LiuW, Flores ER, Tsai KY, Jacks T, Vousden KH and KaelinJr WG. (2000). Nature, 407, 645–648.
Kremenetskaya OS, Logacheva NP, Baryshnikov AY, Chu-makov PM and Kopnin BP. (1997). Oncol. Res., 9, 155–166.
Li R, Sutphin PD, Schwartz D, Matas D, Almog N,Wolkowicz R, Goldfinger N, Pei H, Prokocimer M andRotter V. (1998). Oncogene, 16, 3269–3277.
Lin JY, Chen JD, Elenbaas B and Levine AJ. (1994). GenesDev., 8, 1235–1246.
Liu G, McDonnell TJ, Montes de Oca Luna R, Kapoor M,Mims B, El-Naggar AK and Lozano G. (2000). Proc. Natl.Acad. Sci. USA, 97, 4174–4179.
Lotem J and Sachs L. (1995). Proc. Natl. Acad. Sci. USA, 92,9672–9676.
Mutant p53 represses CD95(Fas/APO-1) expressionA Zalcenstein et al
5675
Oncogene

Marin MC, Jost CA, Brooks LA, Irwin MS, O’Nions J, TidyJA, James N, McGregor JM, Harwood CA, Yulug IG,Vousden KH, Allday MJ, Gusterson B, Ikawa S, Hinds PW,Crook T and Kaelin Jr WG. (2000). Nat. Genet., 25, 47–54.
Matas D, Sigal A, Stambolsky P, Milyavsky M, Weisz L,Schwartz D, Goldfinger N and Rotter V. (2001). EMBO J.,20, 4163–4172.
Michael D and Oren M. (2002). Curr. Opin. Genet. Dev., 12,53–59.
Michalovitz D, Halevy O and Oren M. (1991). J. CellBiochem., 45, 22–29.
Muller M, Strand S, Hug H, Heinemann EM, Walczak H,Hofmann WJ, Stremmel W, Krammer PH and Galle PR.(1997). J. Clin. Invest., 99, 403–413.
Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K,Li-Weber M, Friedman SL, Galle PR, Stremmel W,Oren M and Krammer PH. (1998). J. Exp. Med., 188,
2033–2045.Munsch D, Watanabe-Fukunaga R, Bourdon JC, Nagata S,May E, Yonish-Rouach E and Reisdorf P. (2000). J. Biol.Chem., 275, 3867–3872.
Murphy KL, Dennis AP and Rosen JM. (2000). FASEB J., 14,2291–2302.
Newton K and Strasser A. (2000). J. Exp. Med., 191, 195–200.O’Connor L, Harris AW and Strasser A. (2000). Cancer Res.,60, 1217–1220.
Offer H, Wolkowicz R, Matas D, Blumenstein S, Livneh Z andRotter V. (1999). FEBS Lett., 450, 197–204.
Owen-Schaub LB, Zhang W, Cusack JC, Angelo LS, SanteeSM, Fujiwara T, Roth JA, Deisseroth AB, Zhang WW,Kruzel E and Radinsky R. (1995). Mol. Cell Biol., 15,3032–3040.
Peled A, Zipori D and Rotter V. (1996). Cancer Res., 56, 2148–2156.
Ryan KM, Phillips AC and Vousden KH. (2001). Curr. Opin.Cell Biol., 13, 332–337.
Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH,Wang Q, Zambetti GP and Schuetz JD. (2001). J. Biol.Chem., 276, 39359–39367.
Serrano M. (2000). Carcinogenesis, 21, 865–869.Shao RG, Cao CX, Nieves-Neira W, Dimanche-Boitrel MT,Solary E and Pommier Y. (2001). Oncogene, 20, 1852–1859.
Sigal A and Rotter V. (2000). Cancer Res., 60, 6788–6793.Soussi T. (2000). Ann NY Acad Sci., 910, 121–137; discussion137–139.
Strano S, Fontemaggi G, Costanzo A, Rizzo MG, Monti O,Baccarini A, Del Sal G, Levrero M, Sacchi A, Oren M andBlandino G. (2002). J. Biol. Chem., 277, 18817–18826.
Strano S, Munarriz E, Rossi M, Cristofanelli B, Shaul Y,Castagnoli L, Levine AJ, Sacchi A, Cesareni G, Oren M andBlandino G. (2000). J. Biol. Chem., 275, 29503–29512.
Strauss BE and Haas M. (1995). Biochem. Biophys. Res.Commun., 217, 333–340.
Vogelstein B, Lane D and Levine AJ. (2000). Nature, 408, 307–310.
Volkmann M, Schiff JH, Hajjar Y, Otto G, Stilgenbauer F,Fiehn W, Galle PR and Hofmann WJ. (2001). J. Mol. Med.,79, 594–600.
Walczak H and Krammer PH. (2000). Exp. Cell Res., 256, 58–66.
Wang XJ, Greenhalgh DA, Jiang A, He D, Zhong L, BrinkleyBR and Roop DR. (1998). Mol. Carcinog., 23, 185–192.
Weinmann AS and Farnham PJ. (2002). Methods, 26,
37–47.Yang X, Pater A and Tang SC. (1999). Oncogene, 18, 4546–4553.
Zastawny RL, Salvino R, Chen JM, Benchimol S and Ling V.(1993). Oncogene, 8, 1529–1535.
Mutant p53 represses CD95(Fas/APO-1) expressionA Zalcenstein et al
5676
Oncogene




















