
Antiangiogenic and antitumor activity of a selective PDGFR tyrosine kinase inhibitor, CP-673,451
11
Antiangiogenic and Antitumor Activity of a Selective PDGFR Tyrosine Kinase Inhibitor, CP-673,451 W. Gregory Roberts, Pamela M. Whalen, Erik Soderstrom, Garrett Moraski, Joseph P. Lyssikatos, Huifen-F. Wang, Beth Cooper, Deborah A. Baker, Douglas Savage, Deepak Dalvie, James A. Atherton, Sherry Ralston, Ruby Szewc, John C. Kath, Jing Lin, Cathy Soderstrom, George Tkalcevic, Bruce D. Cohen, Vince Pollack, Wayne Barth, Will Hungerford, and Ethan Ung Pfizer Oncology, Pfizer Global Research and Development, Groton, Connecticut Abstract CP-673,451 is a potent inhibitor of platelet-derived growth factor B-receptor (PDGFR-B) kinase- and PDGF-BB-stimulated autophosphorylation of PDGFR-B in cells (IC 50 = 1 nmol/L) being more than 450-fold selective for PDGFR-B versus other angiogenic receptors (e.g., vascular endothelial growth factor receptor 2, TIE-2, and fibroblast growth factor receptor 2). Multiple models have been used to evaluate in vivo activity of CP-673,451 and to understand the pharmacology of PDGFR-B inhibition and the effect on tumor growth. These models include an ex vivo measure of PDGFR-B phosphorylation in glioblastoma tumors, a sponge model to measure inhibition of angiogenesis, and multiple models of tumor growth inhibition. Inhibition of PDGFR-B phosphorylation in tumors correlates with plasma and tumor levels of CP-673,451. A dose of 33 mg/kg was adequate to provide >50% inhibition of receptor for 4 hours corresponding to an EC 50 of 120 ng/mL in plasma at C max . In a sponge angiogenesis model, CP-673,451 inhibited 70% of PDGF-BB-stimulated angiogenesis at a dose of 3 mg/kg (q.d. 5, p.o., corresponding to 5.5 ng/mL at C max ). The compound did not inhibit vascular endothelial growth factor- or basic fibroblast growth factor-induced angiogenesis at concentrations which inhibited tumor growth. The antitumor efficacy of CP-673,451 was evaluated in a number of human tumor xenografts grown s.c. in athymic mice, including H460 human lung carcinoma, Colo205 and LS174T human colon carcinomas, and U87MG human glio- blastoma multiforme. Once-daily p.o. 10 days dosing routinely inhibited tumor growth (ED 50 V 33 mg/kg). These data show that CP-673,451 is a pharmacologically selective PDGFR inhibitor, inhibits tumor PDGFR-h phosphorylation, selectively inhibits PDGF-BB-stimulated angiogenesis in vivo , and causes significant tumor growth inhibition in multiple human xenograft models. (Cancer Res 2005; 65(3): 957-66) Introduction The ingrowth of host microvasculature into a solid tumor, referred to as angiogenesis, is now widely appreciated to be a necessary event for tumor growth beyond a few cubic millimeters in volume (1, 2). It is expected that drugs that block the molecular events responsible for tumor angiogenesis will be effective against a broad spectrum of tumor types. Angiogenesis is a highly regulated process and, although essential for embryogenesis, this process is restricted in adults to ovulation, cyclical endometrial proliferation, and wound repair (3). Therefore, inhibitors of angiogenesis are predicted to be better tolerated than conventional cytotoxic cancer therapies that affect all rapidly growing cells. Owing to the invading normal host vasculature being the target, another potential attribute of an antiangiogenesis approach may be the avoidance of drug resistance, commonly associated with conventional anticancer modalities that target genetically unstable tumor cells (4). Moreover, with oral or i.v. delivery, neovascular endothelial cells will be the first cell types encountered by extravasating drugs, providing the possibility that tumor penetration by these agents may not be necessary. Angiogenesis is a complex biological process that requires a variety of factors and signaling pathways to stimulate the migration and proliferation of the component cell types and to establish functional blood vessels (5). Platelet-derived growth factor (PDGF) was originally identified in platelets and serum as a potent mitogen for smooth muscle cells and fibroblasts in vitro (6, 7). The PDGF family consists of four polypeptides, A-D, forming dimeric proteins that signal through two tyrosine kinase receptors, PDGFR-a and PDGFR-h. The ligands and receptors can form homodimers or heterodimers depending on cell type, receptor expression, and ligand availability (8). PDGFR-h is a split kinase transmembrane tyrosine kinase receptor closely related to c-kit and CSF-1R (9). Signaling through PDGFR-h has been shown to initiate endothelial, pericyte, and smooth muscle cell migration and proliferation in vitro and in vivo (10). The PDGF-B and PDGFR-h system is critical for the migration and proliferation of pericytes and the development of a functional vasculature (11, 12). Disruption of PDGF-B or PDGFR-h genes in mice is lethal, resulting in microvascular hemorrhage and edema with kidney, cardiovascular, and hematologic abnormalities (13–16) PDGF-B / murine embryos are totally devoid of microvascular pericytes (17). Endothelial cells and pericytes are co-dependent and although pericytes are commonly associated with capillaries, they are most abundant on venules (18), the vessel subtype most commonly responsive to angiogenic stimuli. Through secretion of growth factors and modulation of the extracellular matrix, endothelial-pericyte interactions are critical for vascular matura- tion, remodeling, and maintenance (19). Pericytes have classically been believed to be absent from tumor vasculature, but recent evidence shows that they are common on tumor vasculature although their morphology and permeability function are per- turbed (20, 21). PDGF growth factors and receptors are involved in both autocrine and paracrine stimulation in many tumor types, in situ and in human xenografts. Evaluation of surgical specimens of Note: G. Moraski, J.P. Lyssikatos, V. Pollack, and W. Barth are former Pfizer employees. Requests for reprints: W. Gregory Roberts, Pfizer Oncology, Pfizer Global Research and Development, Eastern Point Road, Groton, CT 06340. Phone: 860-441- 8478; Fax: 860-686-2382; E-mail: [email protected]. I2005 American Association for Cancer Research. www.aacrjournals.org 957 Cancer Res 2005; 65: (3). February 1, 2005 Research Article Research. on February 16, 2016. © 2005 American Association for Cancer cancerres.aacrjournals.org Downloaded from
Transcript of Antiangiogenic and antitumor activity of a selective PDGFR tyrosine kinase inhibitor, CP-673,451

Antiangiogenic and Antitumor Activity of a Selective PDGFR
Tyrosine Kinase Inhibitor, CP-673,451
W. Gregory Roberts, Pamela M. Whalen, Erik Soderstrom, Garrett Moraski, Joseph P. Lyssikatos,Huifen-F. Wang, Beth Cooper, Deborah A. Baker, Douglas Savage, Deepak Dalvie,James A. Atherton, Sherry Ralston, Ruby Szewc, John C. Kath, Jing Lin, Cathy Soderstrom,George Tkalcevic, Bruce D. Cohen, Vince Pollack, Wayne Barth, Will Hungerford, and Ethan Ung
Pfizer Oncology, Pfizer Global Research and Development, Groton, Connecticut
Abstract
CP-673,451 is a potent inhibitor of platelet-derived growthfactor B-receptor (PDGFR-B) kinase- and PDGF-BB-stimulatedautophosphorylation of PDGFR-B in cells (IC50 = 1 nmol/L)being more than 450-fold selective for PDGFR-B versus otherangiogenic receptors (e.g., vascular endothelial growth factorreceptor 2, TIE-2, and fibroblast growth factor receptor 2).Multiple models have been used to evaluate in vivo activity ofCP-673,451 and to understand the pharmacology of PDGFR-Binhibition and the effect on tumor growth. These modelsinclude an ex vivo measure of PDGFR-B phosphorylation inglioblastoma tumors, a sponge model to measure inhibition ofangiogenesis, and multiple models of tumor growthinhibition. Inhibition of PDGFR-B phosphorylation in tumorscorrelates with plasma and tumor levels of CP-673,451. A doseof 33 mg/kg was adequate to provide >50% inhibition ofreceptor for 4 hours corresponding to an EC50 of 120 ng/mL inplasma at Cmax. In a sponge angiogenesis model, CP-673,451inhibited 70% of PDGF-BB-stimulated angiogenesis at a doseof 3 mg/kg (q.d. � 5, p.o., corresponding to 5.5 ng/mL atCmax). The compound did not inhibit vascular endothelialgrowth factor- or basic fibroblast growth factor-inducedangiogenesis at concentrations which inhibited tumor growth.The antitumor efficacy of CP-673,451 was evaluated in anumber of human tumor xenografts grown s.c. in athymicmice, including H460 human lung carcinoma, Colo205 andLS174T human colon carcinomas, and U87MG human glio-blastoma multiforme. Once-daily p.o. � 10 days dosingroutinely inhibited tumor growth (ED50 V 33 mg/kg). Thesedata show that CP-673,451 is a pharmacologically selectivePDGFR inhibitor, inhibits tumor PDGFR-h phosphorylation,selectively inhibits PDGF-BB-stimulated angiogenesis in vivo,and causes significant tumor growth inhibition in multiplehuman xenograft models. (Cancer Res 2005; 65(3): 957-66)
Introduction
The ingrowth of host microvasculature into a solid tumor,referred to as angiogenesis, is now widely appreciated to be anecessary event for tumor growth beyond a few cubic millimetersin volume (1, 2). It is expected that drugs that block the molecularevents responsible for tumor angiogenesis will be effective against a
broad spectrum of tumor types. Angiogenesis is a highly regulatedprocess and, although essential for embryogenesis, this process isrestricted in adults to ovulation, cyclical endometrial proliferation,and wound repair (3). Therefore, inhibitors of angiogenesis arepredicted to be better tolerated than conventional cytotoxic cancertherapies that affect all rapidly growing cells. Owing to the invadingnormal host vasculature being the target, another potentialattribute of an antiangiogenesis approach may be the avoidanceof drug resistance, commonly associated with conventionalanticancer modalities that target genetically unstable tumor cells(4). Moreover, with oral or i.v. delivery, neovascular endothelial cellswill be the first cell types encountered by extravasating drugs,providing the possibility that tumor penetration by these agentsmay not be necessary. Angiogenesis is a complex biological processthat requires a variety of factors and signaling pathways tostimulate the migration and proliferation of the component celltypes and to establish functional blood vessels (5).Platelet-derived growth factor (PDGF) was originally identified
in platelets and serum as a potent mitogen for smooth muscle cellsand fibroblasts in vitro (6, 7). The PDGF family consists of fourpolypeptides, A-D, forming dimeric proteins that signal throughtwo tyrosine kinase receptors, PDGFR-a and PDGFR-h. The ligandsand receptors can form homodimers or heterodimers dependingon cell type, receptor expression, and ligand availability (8).PDGFR-h is a split kinase transmembrane tyrosine kinase receptorclosely related to c-kit and CSF-1R (9). Signaling through PDGFR-hhas been shown to initiate endothelial, pericyte, and smoothmuscle cell migration and proliferation in vitro and in vivo (10).The PDGF-B and PDGFR-h system is critical for the migration andproliferation of pericytes and the development of a functionalvasculature (11, 12). Disruption of PDGF-B or PDGFR-h genes inmice is lethal, resulting in microvascular hemorrhage and edemawith kidney, cardiovascular, and hematologic abnormalities (13–16)PDGF-B�/� murine embryos are totally devoid of microvascularpericytes (17). Endothelial cells and pericytes are co-dependent andalthough pericytes are commonly associated with capillaries, theyare most abundant on venules (18), the vessel subtype mostcommonly responsive to angiogenic stimuli. Through secretion ofgrowth factors and modulation of the extracellular matrix,endothelial-pericyte interactions are critical for vascular matura-tion, remodeling, and maintenance (19). Pericytes have classicallybeen believed to be absent from tumor vasculature, but recentevidence shows that they are common on tumor vasculaturealthough their morphology and permeability function are per-turbed (20, 21).PDGF growth factors and receptors are involved in both
autocrine and paracrine stimulation in many tumor types, in situand in human xenografts. Evaluation of surgical specimens of
Note: G. Moraski, J.P. Lyssikatos, V. Pollack, and W. Barth are former Pfizeremployees.
Requests for reprints: W. Gregory Roberts, Pfizer Oncology, Pfizer GlobalResearch and Development, Eastern Point Road, Groton, CT 06340. Phone: 860-441-8478; Fax: 860-686-2382; E-mail: [email protected].
I2005 American Association for Cancer Research.
www.aacrjournals.org 957 Cancer Res 2005; 65: (3). February 1, 2005
Research Article
Research. on February 16, 2016. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

human breast carcinoma, colorectal adenocarcinoma, and lungcarcinoma revealed PDGFR-h staining in periepithelial stroma, butabsent in epithelial cells, whereas PDGF-BB was observed in tumorcells, suggesting paracrine stimulation (22–24). Autocrine stimula-tion has been observed in human prostate adenocarcinoma andastrocytic tumors, where PDGF-A or PDGF-B and their cognatereceptors were expressed in both stromal and tumor epithelial cells(6, 25). Although PDGF ligands A and B, as well as PDGFR-a, areexpressed by all stages of astrocytic tumors, PDGFR-h is expressedonly on the tumor vasculature (26, 27). PDGFR inhibition is anantiangiogenic approach potentially affecting all solid tumors dueto tumor vasculature having a pericyte coverage. However,depending on the tumor type, a PDGFR inhibitor would also beexpected to inhibit tumor growth by directly targeting those tumorcells driven by a PDGF autocrine loop as well as by affecting tumorvasculature.This manuscript describes the pharmacologic activity of
CP-673,451, a potent inhibitor of PDGFR kinase that is beinginvestigated for use as an anticancer agent. This compoundinhibits both PDGFR-h and PDGFR-a kinase (IC50 = 1 and 10nmol/L, respectively) but is >200-fold selective versus a variety ofother kinases [e.g., c-kit, vascular endothelial growth factorreceptor (VEGFR)-2, TIE-2, fibroblast growth factor receptor(FGFR)-2, epidermal growth factor receptor (EGFR), erbB2, andsrc]. CP-673,451 also inhibits PDGF-BB-stimulated autophosphor-ylation of dimeric PDGFR-h in transfected porcine aorticendothelial (PAE) cells with an IC50 value of 1 nmol/L. In vivo ,oral administration of CP-673,451 significantly inhibits PDGF-BB-induced angiogenesis as evaluated using a sponge implant model.CP-673,451 also potently inhibits the growth of multiple tumorxenografts despite a lack of PDGFR expression in tumor cells.Inhibition of angiogenesis or tumor growth is correlated withplasma and tumor concentration and inhibition of phospho-PDGFR in vivo .
Materials and Methods
Chemical Synthesis. CP-673,451 was prepared according to the
procedures described in PCT patent application WO 2001040217. The
structure and physical properties of this compound are shown in Fig. 1. The
tosylate salt form of this compound was used for the studies reported here.Kinase Inhibition Assay and Enzyme Kinetics. A glutathione
S-transferase–tagged kinase domain construct of the intracellular portion of
the PDGFR-h (amino acids 693-1,401, accession no. J03278) was expressed inSf-9 cells (baculovirus expression system, Invitrogen, Carlsbad, CA). Enzyme
kinetics were determined by incubating the enzyme with increasing
concentrations of ATP in phosphorylation buffer [50 mmol/L HEPES (pH
7.3), 125 mmol/L NaCl, 24 mmol/L MgCl>2 in Nunc Immuno MaxiSorp 96-well plates previously coated with 100 AL of 100 Ag/mL poly-Glu-Tyr (4:1
ratio) diluted in PBS. After 10 minutes, the plates were washed (PBS, 0.1%
Tween 20), incubated with anti-phosphotyrosine-horseradish peroxidase
antibody, and diluted in PBS, 0.05% Tween 20, 3% BSA for 30 minutes atroom temperature. The plates were washed as above and incubated with
3,3V,5,5V-tetramethylbenzidine. The reaction was stopped by adding an equal
volume of 0.09 N H2SO4. The phosphotyrosine-dependent signal was thenquantitated on a plate reader at 450 nm. For routine enzyme assays, the
enzyme was incubated with 10 Amol/L ( final) ATP in the presence of
compound diluted in DMSO (1.6% v/v DMSO assay final) for 30 minutes at
room temperature in plates, as above, previously coated with 100 AL of 6.25Ag/mL poly-Glu-Tyr. The remainder of the assay was carried out as above,
and IC50 values were calculated as percent inhibition of control. Selectivity
assays were done as described above using purified recombinant enzyme
(generated as described above) and ATP concentrations at or up to 3�
above the Km for each enzyme. CP-673,451 was also tested at aconcentration of 100 nmol/L in KinaseProfiler2 (Upstate Cell Signaling,
Waltham, MA). All enzymes in this panel are recombinant human and are
run at enzyme Km.
Cell-Based Phospho-PDGFR Inhibition Assay. PAE cells stably
expressing full-length PDGFR and VEGFR have been previously described(28). For cell-based selectivity assays, PAE cells were transfected with full-
length human PDGFR-a, PDGFR-h, or VEGFR-2. Cells were seeded at 4� 105
cells/mL in 50 AL growth medium (Ham’s F-12 media supplemented with
10% fetal bovine serum, 50,000 units each penicillin and streptomycin, and
500 Ag/mL gentamicin) per well in 96-well plates. After 6 to 8 hours, the
growth medium was replaced with 50 AL serum-depleted medium (as above,
but with 0.1% fetal bovine serum) and cells were incubated overnight.
Immediately before compound addition, the medium was replaced with
95 AL serum-depleted medium. Compounds were diluted in 100% DMSO,
added to the cells at a final DMSO concentration of 0.25% v/v, and
incubated at 37jC for 10 minutes. Cells were stimulated with theappropriate ligand (Becton Dickinson, Franklin Lakes, NJ, prepared in
serum-depleted supplemented medium) and incubated as above for an
additional 8 minutes. The medium was removed and the cells washed once
with PBS, then lysed with 50 AL HNTG buffer [20 mmol/L HEPES (pH 7.5),
150 mmol/L NaCl, 2% Triton X-100, 10% glycerol, 5 Amol/L EDTA, 2 mmol/
L NaVO4, and 1 EDTA-free complete protease inhibitor tablet per 25 mL]
for 5 minutes at room temperature. Lysates were then diluted with 50 ALHG buffer [20 mmol/L HEPES (pH 7.5), 10% glycerol]. The diluted cell
lysates were mixed thoroughly, 50 AL of supernatant were transferred to
the ELISA capture plate, and incubated at room temperature for 2 hourswith agitation. ELISA capture plates were prepared by coating 96-well
ReactiBind goat-antirabbit plates (Pierce, Rockville, IL) with 100 AL/well of5 Ag/mL rabbit anti-human PDGFR-h, anti-PDGFR-a, or anti-VEGFR-2
antibody (Santa Cruz, Santa Cruz, CA) for 60 to 90 minutes. At the end of
the 2-hour incubation the plates were washed (PBS, 0.1% Tween 20) before
incubation with anti-phosphotyrosine-horseradish peroxidase antibody
(diluted in PBS, 0.05% Tween 20) for 30 minutes at room temperature.
The plates were washed again, then incubated with tetramethylbenzidine
and evaluated as described above. IC50 values were calculated as percent
inhibition of control. Cellular activity was also evaluated by Western
blotting. For PDGFR-h activity, PAE-h cells stimulated with PDGF-BB wereused. For c-kit selectivity, H526 small cell lung cancer cells stimulated with
stem cell factor were used (29). For both assays, cells were starved
overnight and treated the following day with increasing concentrations of
CP-673,451 for 30 minutes at 37jC. During the final 5 to 8 minutes of
incubation, either stem cell factor (50 ng/mL) or PDGF-BB (500 ng/mL)
was added. Unstimulated cells were used as phosphorylation controls.
Cells were washed, lysed, and equivalent protein levels were separated by
PAGE. Following transfer to nitrocellulose, membranes were blotted to
detect phospho-PDGFR (pY857 antibody, Santa Cruz) or phospho-c-kit
(pY719 antibody, Cell Signaling) antibody at 1: 200 overnight at 4jC.Densitometry of bands and IC50 calculation were measured using a
LumiImager (Roche, Indianapolis, IN).
Animals for In vivo Studies. Athymic female mice (CD-1 nu/nu, f20
grams) were used for all in vivo studies. Mice were obtained from Charles
River Laboratories (Wilmington, MA) and housed in specific pathogen-freeconditions, according to the guidelines of the Association for the
Assessment and Accreditation for Laboratory Animal Care, International.
All in vivo studies were carried out under approved institutionalexperimental animal care and use protocols.
Ex vivo ELISA. Female athymic mice were injected with 1 � 106 C6 rat
glioblastoma cells on day 1. On day 9, when tumors were approximately
300 mm3, the mice received compound or vehicle (5% Gelucire 44/14
in sterile water, Gattefosse, Paramus, NJ) orally. For pharmacokinetic/
pharmacodynamic analysis, blood and tumor samples were collected from
each animal (n z 4 mice/group/time point) into heparinized vacutainers
and liquid N2, respectively, at the indicated times post-dose. Blood and
tumors were harvested for evaluation of drug levels and tumor-associated
phospho-PDGFR-h. Plasma concentrations of CP-673,451 were determined
using reverse-phase high-performance liquid chromatography with mass
Cancer Research
Cancer Res 2005; 65: (3). February 1, 2005 958 www.aacrjournals.org
Research. on February 16, 2016. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

spectrometric (MS/MS) detection. Tumors were homogenized in 1 mLlysis buffer per 200 mg tumor [lysis buffer: 50 mmol/L HEPES (pH 7.5),150 mmol/L NaCl, 1.5 mmol/L MgCl2, 1 mmol/L EDTA, 1% glycerol, 1%Triton X-100, 1.6 mmol/L Na3VO4, 10 mmol/L NaF, 25 mg/L soy beantrypsin inhibitor, EDTA-free complete protease inhibitor tablets], spun for5 minutes at 14,000 rpm, and the supernatant aliquoted to 96-wellpolypropylene plates on dry ice. Total protein concentration wasdetermined using bicinchoninic acid protein assay (Pierce). Ninety-six-wellReactiBind goat-antirabbit plates (Pierce) were blocked with 100 AL/wellcold blocking buffer (TBS, 0.1% Tween 20, 3% BSA) for 60 minutes on a plateshaker at room temperature. The blocking buffer was replaced with 0.5 Aganti-PDGFR-h in 100 AL cold blocking buffer per well and incubated for60 minutes at room temperature with agitation. Plates were washed withTBS-T (TBS, 0.1% Tween 20) before addition of tumor lysate (100 ALdiluted to f5 mg/mL total protein in lysis buffer without proteaseinhibitors) and incubated for 2 hours at room temperature with agitation.The plates were washed as above, then incubated with 15 ng of anti-phosphotyrosine-horseradish peroxidase per well (in blocking buffer) for30 minutes at room temperature. The plates were washed as above andphosphotyrosine quantitated using tetramethylbenzidine as describedabove.
Tumor lysates were also evaluated by Western blotting for measurementof phospho-PDGFR-h. C6 tumor-bearing animals received doses of 10, 33, or100 mg/kg (p.o.) and tumors were harvested 1.5 hours post-dose (fTmax).Tumors were lysed and equivalent amounts of protein were separated byPAGE, transferred to nitrocellulose, and blotted for phospho-PDGFR-h.Band density was quantitated using a LumiImager. Multiple tumors wereevaluated at each dose.
Prediction of Efficacious Concentration. Blood and tumor sampleswere collected at each time point post-administration of CP-673,451 fordetermination of plasma drug concentration and PDGFR phosphotyr-osine reduction. The relationship between CP-673,451 concentration andPDGFR phosphotyrosine reduction has been explored in pharmacologicmodels (tumor-bearing athymic mice) with pooled experimental datafrom 12 individual studies. PDGFR phosphotyrosine reduction correlateswell with plasma concentrations of CP-673,451 in athymic mice andfollows a simple Emax pharmacodynamic model:
where E is the measured response (PDGFR phosphorylation reduction),Emax is the maximum response, EC50 is the concentration of CP-673,451required for half-maximal reduction of PDGFR phosphorylation, and C isthe plasma concentration of CP-673,451.
The predicted maximal inhibition of PDGFR tyrosine phosphorylation is
about 70% and the plasma concentration to achieve 35% inhibition of
PDGFR phosphotyrosine is about 28 ng/mL. The predicted efficacious
concentration of CP-673,451 to inhibit 50% PDGFR tyrosine phosphoryla-
tion using this model was calculated to be about 80 ng/mL (Fig. 4A).
In vivo Sponge Angiogenesis Assay. Lyophilized growth factors were
reconstituted in HBSS + 1% BSA and admixed with growth factor reduced
Matrigel 1:1 before adding to surgical gelatin sponges (4 � 4 � 2 mm). Mice
were anesthetized with isoflurane and a 5-mm incision was made in the
midline of the abdomen. For the dose response experiments, one PDGF-BB
spongewasused per animal. Animals received doses of 3, 10, or 33mg/kg q.d.
p.o.� 5 days. For the selectivity experiments, each mouse received basic FGF
(bFGF), VEGF, PDGF-BB, and control (no GF) sponges. Sponges were placed
s.c. in the axillary and inguinal areas. Incisions were stapled shut and 2 days
later mice began b.i.d. oral dosing with 5 mg/kg CP-673,451 (10 mg/kg total
daily dose) for a total of 5 days. Following the last oral dose of compound,
animals were sacrificed and sponges were removed. Sponges were homogen-
ized indouble-distilledwater (100AL/sponge)and lysateswere transferred toa96-well plate. Hemoglobin content was quantitated by measuring the
colorimetric change following addition of tetramethylbenzidine. The plates
were evaluated as described above. Four mice per dose group were used. A
Student’s t test was used to determine P value.
Tumor Growth Inhibition Studies. All tumor cell lines were obtainedfrom American Type Culture Collection (ATCC, Rockville, MD) and
propagated by standard tissue culture procedures in the medium as
suggested by the supplier. Exponentially growing cells were trypsinized
and resuspended in sterile PBS and inoculated s.c. (1 � 106 cells/mousein 200 AL) into the right flank of mice. Animals bearing tumors of
approximately 150 mm3 in size were divided into groups receiving either
vehicle (5% Gelucire) or CP-673,451 (diluted in vehicle), and dosed by oralgavage. Animal body weight and tumor measurements were obtained
every 2 days. Paclitaxel was diluted in saline and delivered i.p. at a dose
of 10 mg/kg/d for 5 consecutive days (30, 31). Equivalent volumes of
saline were injected for controls. Tumor volume (mm3) was measuredwith Vernier calipers and calculated using the formula: length (mm) �width (mm) � width (mm) � 0.5. Percent growth inhibition of an
individual tumor was calculated using the following formula: % growth
inhibition = (1�[(TL�T1)/(CL�C1)] � 100%), where TL and CL are thetreated and control tumor volumes on day last, and T1 and C1 are
treated and control tumor volumes on day 1. For all tumor growth
inhibition experiments, 8 to 10 mice per dose group were used. AStudent’s t test was used to determine P value.
Microscopy and Immunohistochemistry of TumorMicrovasculature.
Mice bearing Colo205 tumors were treated for 3 or 5 days with 5 mg/kg CP-
673,451 (b.i.d., p.o.). Following treatment, tumors were excised and quick
frozen in OCT media. Sections of 7 Am were cut and processed for
immunohistochemical detection of microvasculature using rat anti-murine
endothelial MECA32 antibody (anti-endoglin, PharMingen, San Diego, CA).
Tissue sections were counterstained with hematoxylin or methyl green and
examined using a Zeiss Axiophot microscope at 20� with a reticule grid. All
discreet, positively stained vascular profiles, with or without lumina, were
counted in 10 fields (200�) frommultiple sections of each tumor. Fields were
randomly chosen throughout the entire section. For each time point, four
mice were evaluated. A Student’s t test was used to determine P value.
Results
Selective Inhibition of PDGFR Kinase by CP-673,451. CP-673,451 is a highly selective benzimidazole inhibitor of PDGFR-aand PDGFR-h tyrosine kinases (Fig. 1). Increasing concentrations ofATP resulted in a Michaelis-Menten saturation with a calculated Km
of 3.3 Amol/L (not shown). The compound shows concentration-dependent inhibition of enzyme activity consistent with competitiveinhibition of ATP (Fig. 2). Although CP-673,451 is approximatelyequipotent between PDGFR-a and PDGFR-h kinase (IC50 = 10 and1 nmol/L, respectively), it is greater than 250� selective forPDGFR-h kinase relative to c-kit kinase (Table 1). Moreover, CP-673,451 shows 450-fold to more than 5,000-fold selectivity forPDGFR-h compared with other angiogenic tyrosine kinases,including VEGFR-1, VEGFR-2, TIE-2, and FGFR-2. Importantly,CP-673,451 is 1,000� to 10,000� selective relative to many otherreceptor tyrosine kinases (Tables 1 and 2). In cell-based assaysusing PAE cells stably transfected with recombinant humanreceptor and stimulated by the cognate ligand, CP-673,451 showsthe same 10� selectivity for PDGFR-h relative to PDGFR-a seen inthe kinase assays. Interestingly, this compound is >1,000� selectivefor PDGFR-h relative to VEGFR-2 when tested in PAE cell-basedassays. Cell activity of CP-673,451 was also visualized inimmunoblots (Fig. 3). PDGFR-h in PAE-h cells was inhibited withan IC50 of 6.4 nmol/L, comparable to results in the morequantitative cell-based ELISA (Fig. 3A). H526 cells expressingendogenous c-kit were also evaluated (Fig. 3B). CP-673,451incubation with these cells resulted in an IC50 of 1.1 Amol/Lagainst c-kit, showing f180� selectivity between these targets at
E ¼ Emax � C
EC50 þ C
Preclinical Pharmacology of CP-673,451
www.aacrjournals.org 959 Cancer Res 2005; 65: (3). February 1, 2005
Research. on February 16, 2016. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

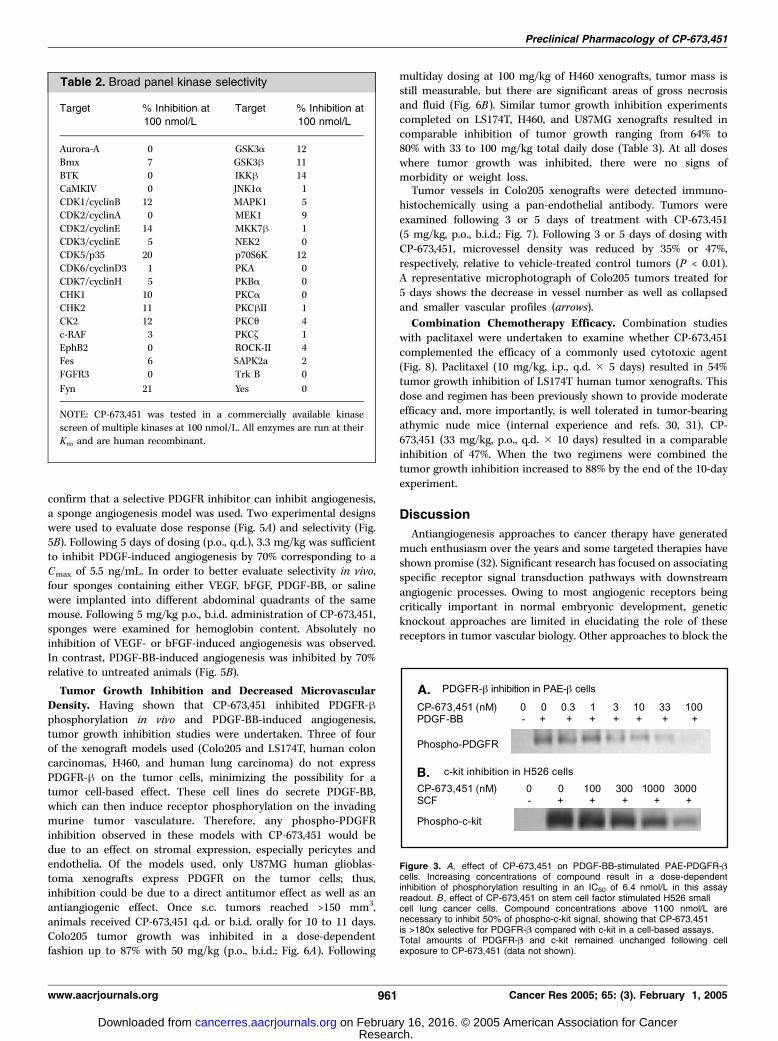
the cell level mimicking kinase selectivity. Levels of total PDGFR andc-kit remained unchanged with compound treatment (data notshown). CP-673,451 was also submitted to the KinaseProfiler2 at 100nmol/L (Table 2). These results show the extent of kinase selectivityof this compound against multiple kinases and kinase families.
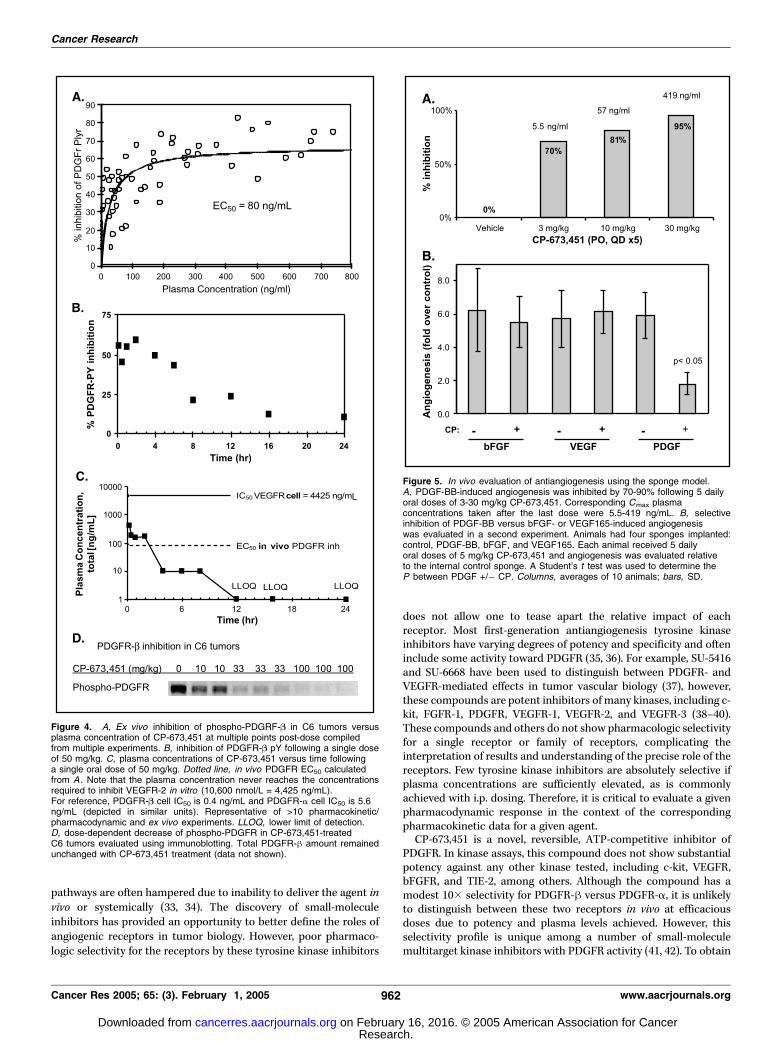
Inhibition of PDGFR Phosphorylation in Tumor Xenografts.In order to evaluate the pharmacologic modulation of phosphory-lated receptor in vivo , rat C6 glioblastoma xenograft models wereused. These tumors consistently express phosphorylated PDGFR. Allexperiments evaluate total PDGFR and phospho-PDGFR in theELISA to ensure consistent and reproducible endogenous expres-sion. Glioblastoma tumors are among the few solid tumor typeswhich express PDGFR on the tumor itself providing sufficient signal-to-noise for an ex vivo ELISA-based evaluation of phospho-PDGFRlevels in the tumor.Plasma levels generally increased with increasing dose from 3
to 100 mg/kg with a mean Tmax of 1 to 2 hours and a half-life of2 to 3 hours in mice. The highest dose tested was 100 mg/kg.A concomitant decrease of tumor-associated phosphorylatedPDGFR-h was measured with a calculated EC50 of 80 ng/mL
(Fig. 4A). Phosphorylated PDGFR-h was reduced >50% forf4 hours following a single 50 mg/kg oral dose (Fig. 4B).CP-673,451 Cmax plasma levels following a single oral dose of 50mg/kg never reached the cellular VEGFR-2 IC50 (10,600 nmol/L =4425 ng/mL; Fig. 4C). The IC50 for PDGFR-a and h kinaseactivity are 4 and 0.4 ng/mL, respectively. Tumor lysates(2-3 tumors/dose) were evaluated in immunoblots (Fig. 4D).The ED50 following a single dose of CP-673,451 at the fTmax was10 mg/kg. Levels of total PDGFR remained unchanged withcompound treatment (data not shown).
CP-673,451 Selectively Blocks PDGF-BB Induced Angio-genesis. Unlike FGFR-2 and VEGFR-2, the critical nature ofPDGFR signaling in angiogenesis is not as well established. To
Figure 1. Chemical features and physical properties. Chemical name: 1-{2-[5-(2-methoxy-ethoxy)-benzoimidazol-1-yl]-quinolin-8-yl}-piperidin-4-ylamine.Molecular formula: C24H27N5O2. Molecular weight: 417.52 (free base);589.71 (tosylate salt). Physical properties: white crystalline solid (tosylate salt).Melting point: 216jC (tosylate salt). pKa: 9.16, 4.88 (free base). Log P : 3.66(free base). Log D (pH 7.4): 2.92 (free base). Solubility: >30 mg/mL inPBS (tosylate salt).
Figure 2. Kinetic analysis of PDGFR-h enzymatic activityin the presence of varied concentrations of ATP andPDGFR inhibitor CP-673,451. Increasing concentrationsof the kinase inhibitor resulted in a concentration-dependentinhibition of enzyme activity. The Vmax could be restoredby increasing the concentration of the ATP substrate.This enzymatic profile describes the competitiveinhibition of ATP in the presence of the PDGFR inhibitorCP-673,451 [0.37 Amol/L (*); 0.1 Amol/L (�); 0.04 Amol/L (E);0.01 Amol/L (n); 0.0 Amol/L (x)].
Table 1. Kinase and cell selectivity
Target Kinase
(IC50, nmol/L)
Cell*(IC50, nmol/L)
Cellc
(IC50, nmol/L)
PDGFR-h 1 1 6
PDGFR-a 10 14
c-kit 252 ND >1100
VEGFR-2 450 10,600VEGFR-1 450 ND
Lck >1,000 ND
Src >2,000 ND
v-abl >5,000 NDTIE-2 >5,000 ND
bFGFR >5,000 ND
EGFR >10,000 NDerbB2 >10,000 ND
c-met >10,000 ND
IGF-1R >10,000 ND
NOTE: All IC50 data are a mean of three assays run with triplicate wells
on five- or six-point curves, except for PDGFR-a and PDGFR-h kinaseand cell, which were run more than 20 times with triplicate wells and
five- or six-point IC50 curves.*ELISA-based cell assay.cImmunoblotting-based cell assay.
Cancer Research
Cancer Res 2005; 65: (3). February 1, 2005 960 www.aacrjournals.org
Research. on February 16, 2016. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from
confirm that a selective PDGFR inhibitor can inhibit angiogenesis,a sponge angiogenesis model was used. Two experimental designswere used to evaluate dose response (Fig. 5A) and selectivity (Fig.5B). Following 5 days of dosing (p.o., q.d.), 3.3 mg/kg was sufficientto inhibit PDGF-induced angiogenesis by 70% corresponding to aCmax of 5.5 ng/mL. In order to better evaluate selectivity in vivo,four sponges containing either VEGF, bFGF, PDGF-BB, or salinewere implanted into different abdominal quadrants of the samemouse. Following 5 mg/kg p.o., b.i.d. administration of CP-673,451,sponges were examined for hemoglobin content. Absolutely noinhibition of VEGF- or bFGF-induced angiogenesis was observed.In contrast, PDGF-BB-induced angiogenesis was inhibited by 70%relative to untreated animals (Fig. 5B).
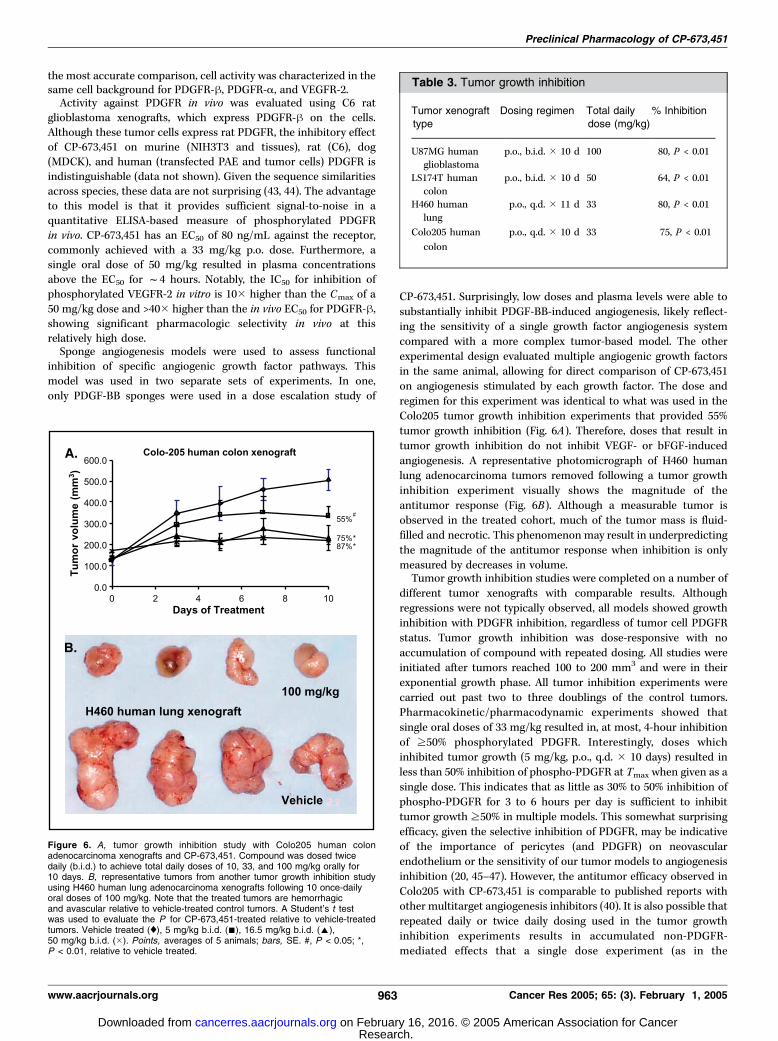
Tumor Growth Inhibition and Decreased MicrovascularDensity. Having shown that CP-673,451 inhibited PDGFR-hphosphorylation in vivo and PDGF-BB-induced angiogenesis,tumor growth inhibition studies were undertaken. Three of fourof the xenograft models used (Colo205 and LS174T, human coloncarcinomas, H460, and human lung carcinoma) do not expressPDGFR-h on the tumor cells, minimizing the possibility for atumor cell-based effect. These cell lines do secrete PDGF-BB,which can then induce receptor phosphorylation on the invadingmurine tumor vasculature. Therefore, any phospho-PDGFRinhibition observed in these models with CP-673,451 would bedue to an effect on stromal expression, especially pericytes andendothelia. Of the models used, only U87MG human glioblas-toma xenografts express PDGFR on the tumor cells; thus,inhibition could be due to a direct antitumor effect as well as anantiangiogenic effect. Once s.c. tumors reached >150 mm3,animals received CP-673,451 q.d. or b.i.d. orally for 10 to 11 days.Colo205 tumor growth was inhibited in a dose-dependentfashion up to 87% with 50 mg/kg (p.o., b.i.d.; Fig. 6A). Following
multiday dosing at 100 mg/kg of H460 xenografts, tumor mass isstill measurable, but there are significant areas of gross necrosisand fluid (Fig. 6B). Similar tumor growth inhibition experimentscompleted on LS174T, H460, and U87MG xenografts resulted incomparable inhibition of tumor growth ranging from 64% to80% with 33 to 100 mg/kg total daily dose (Table 3). At all doseswhere tumor growth was inhibited, there were no signs ofmorbidity or weight loss.Tumor vessels in Colo205 xenografts were detected immuno-
histochemically using a pan-endothelial antibody. Tumors wereexamined following 3 or 5 days of treatment with CP-673,451(5 mg/kg, p.o., b.i.d.; Fig. 7). Following 3 or 5 days of dosing withCP-673,451, microvessel density was reduced by 35% or 47%,respectively, relative to vehicle-treated control tumors (P < 0.01).A representative microphotograph of Colo205 tumors treated for5 days shows the decrease in vessel number as well as collapsedand smaller vascular profiles (arrows).
Combination Chemotherapy Efficacy. Combination studieswith paclitaxel were undertaken to examine whether CP-673,451complemented the efficacy of a commonly used cytotoxic agent(Fig. 8). Paclitaxel (10 mg/kg, i.p., q.d. � 5 days) resulted in 54%tumor growth inhibition of LS174T human tumor xenografts. Thisdose and regimen has been previously shown to provide moderateefficacy and, more importantly, is well tolerated in tumor-bearingathymic nude mice (internal experience and refs. 30, 31). CP-673,451 (33 mg/kg, p.o., q.d. � 10 days) resulted in a comparableinhibition of 47%. When the two regimens were combined thetumor growth inhibition increased to 88% by the end of the 10-dayexperiment.
Discussion
Antiangiogenesis approaches to cancer therapy have generatedmuch enthusiasm over the years and some targeted therapies haveshown promise (32). Significant research has focused on associatingspecific receptor signal transduction pathways with downstreamangiogenic processes. Owing to most angiogenic receptors beingcritically important in normal embryonic development, geneticknockout approaches are limited in elucidating the role of thesereceptors in tumor vascular biology. Other approaches to block the
Table 2. Broad panel kinase selectivity
Target % Inhibition at
100 nmol/L
Target % Inhibition at
100 nmol/L
Aurora-A 0 GSK3a 12
Bmx 7 GSK3h 11
BTK 0 IKKh 14CaMKIV 0 JNK1a 1
CDK1/cyclinB 12 MAPK1 5
CDK2/cyclinA 0 MEK1 9
CDK2/cyclinE 14 MKK7h 1CDK3/cyclinE 5 NEK2 0
CDK5/p35 20 p70S6K 12
CDK6/cyclinD3 1 PKA 0CDK7/cyclinH 5 PKBa 0
CHK1 10 PKCa 0
CHK2 11 PKChII 1
CK2 12 PKCu 4c-RAF 3 PKC~ 1
EphB2 0 ROCK-II 4
Fes 6 SAPK2a 2
FGFR3 0 Trk B 0
Fyn 21 Yes 0
NOTE: CP-673,451 was tested in a commercially available kinase
screen of multiple kinases at 100 nmol/L. All enzymes are run at theirKm and are human recombinant.
Figure 3. A, effect of CP-673,451 on PDGF-BB-stimulated PAE-PDGFR-Bcells. Increasing concentrations of compound result in a dose-dependentinhibition of phosphorylation resulting in an IC50 of 6.4 nmol/L in this assayreadout. B , effect of CP-673,451 on stem cell factor stimulated H526 smallcell lung cancer cells. Compound concentrations above 1100 nmol/L arenecessary to inhibit 50% of phospho-c-kit signal, showing that CP-673,451is >180x selective for PDGFR-B compared with c-kit in a cell-based assays.Total amounts of PDGFR-B and c-kit remained unchanged following cellexposure to CP-673,451 (data not shown).
Preclinical Pharmacology of CP-673,451
www.aacrjournals.org 961 Cancer Res 2005; 65: (3). February 1, 2005
Research. on February 16, 2016. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from
pathways are often hampered due to inability to deliver the agent invivo or systemically (33, 34). The discovery of small-moleculeinhibitors has provided an opportunity to better define the roles ofangiogenic receptors in tumor biology. However, poor pharmaco-logic selectivity for the receptors by these tyrosine kinase inhibitors
does not allow one to tease apart the relative impact of eachreceptor. Most first-generation antiangiogenesis tyrosine kinaseinhibitors have varying degrees of potency and specificity and ofteninclude some activity toward PDGFR (35, 36). For example, SU-5416and SU-6668 have been used to distinguish between PDGFR- andVEGFR-mediated effects in tumor vascular biology (37), however,these compounds are potent inhibitors of many kinases, including c-kit, FGFR-1, PDGFR, VEGFR-1, VEGFR-2, and VEGFR-3 (38–40).These compounds and others do not show pharmacologic selectivityfor a single receptor or family of receptors, complicating theinterpretation of results and understanding of the precise role of thereceptors. Few tyrosine kinase inhibitors are absolutely selective ifplasma concentrations are sufficiently elevated, as is commonlyachieved with i.p. dosing. Therefore, it is critical to evaluate a givenpharmacodynamic response in the context of the correspondingpharmacokinetic data for a given agent.CP-673,451 is a novel, reversible, ATP-competitive inhibitor of
PDGFR. In kinase assays, this compound does not show substantialpotency against any other kinase tested, including c-kit, VEGFR,bFGFR, and TIE-2, among others. Although the compound has amodest 10� selectivity for PDGFR-h versus PDGFR-a, it is unlikelyto distinguish between these two receptors in vivo at efficaciousdoses due to potency and plasma levels achieved. However, thisselectivity profile is unique among a number of small-moleculemultitarget kinase inhibitors with PDGFR activity (41, 42). To obtain
Figure 4. A, Ex vivo inhibition of phospho-PDGRF-B in C6 tumors versusplasma concentration of CP-673,451 at multiple points post-dose compiledfrom multiple experiments. B, inhibition of PDGFR-B pY following a single doseof 50 mg/kg. C, plasma concentrations of CP-673,451 versus time followinga single oral dose of 50 mg/kg. Dotted line, in vivo PDGFR EC50 calculatedfrom A . Note that the plasma concentration never reaches the concentrationsrequired to inhibit VEGFR-2 in vitro (10,600 nmol/L = 4,425 ng/mL).For reference, PDGFR-B cell IC50 is 0.4 ng/mL and PDGFR-a cell IC50 is 5.6ng/mL (depicted in similar units). Representative of >10 pharmacokinetic/pharmacodynamic and ex vivo experiments. LLOQ, lower limit of detection.D, dose-dependent decrease of phospho-PDGFR in CP-673,451-treatedC6 tumors evaluated using immunoblotting. Total PDGFR-h amount remainedunchanged with CP-673,451 treatment (data not shown).
Figure 5. In vivo evaluation of antiangiogenesis using the sponge model.A, PDGF-BB-induced angiogenesis was inhibited by 70-90% following 5 dailyoral doses of 3-30 mg/kg CP-673,451. Corresponding Cmax plasmaconcentrations taken after the last dose were 5.5-419 ng/mL. B, selectiveinhibition of PDGF-BB versus bFGF- or VEGF165-induced angiogenesiswas evaluated in a second experiment. Animals had four sponges implanted:control, PDGF-BB, bFGF, and VEGF165. Each animal received 5 dailyoral doses of 5 mg/kg CP-673,451 and angiogenesis was evaluated relativeto the internal control sponge. A Student’s t test was used to determine theP between PDGF +/� CP. Columns, averages of 10 animals; bars, SD.
Cancer Research
Cancer Res 2005; 65: (3). February 1, 2005 962 www.aacrjournals.org
Research. on February 16, 2016. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from
the most accurate comparison, cell activity was characterized in thesame cell background for PDGFR-h, PDGFR-a, and VEGFR-2.Activity against PDGFR in vivo was evaluated using C6 rat
glioblastoma xenografts, which express PDGFR-h on the cells.Although these tumor cells express rat PDGFR, the inhibitory effectof CP-673,451 on murine (NIH3T3 and tissues), rat (C6), dog(MDCK), and human (transfected PAE and tumor cells) PDGFR isindistinguishable (data not shown). Given the sequence similaritiesacross species, these data are not surprising (43, 44). The advantageto this model is that it provides sufficient signal-to-noise in aquantitative ELISA-based measure of phosphorylated PDGFRin vivo. CP-673,451 has an EC50 of 80 ng/mL against the receptor,commonly achieved with a 33 mg/kg p.o. dose. Furthermore, asingle oral dose of 50 mg/kg resulted in plasma concentrationsabove the EC50 for f4 hours. Notably, the IC50 for inhibition ofphosphorylated VEGFR-2 in vitro is 10� higher than the Cmax of a50 mg/kg dose and >40� higher than the in vivo EC50 for PDGFR-h,showing significant pharmacologic selectivity in vivo at thisrelatively high dose.Sponge angiogenesis models were used to assess functional
inhibition of specific angiogenic growth factor pathways. Thismodel was used in two separate sets of experiments. In one,only PDGF-BB sponges were used in a dose escalation study of
CP-673,451. Surprisingly, low doses and plasma levels were able to
substantially inhibit PDGF-BB-induced angiogenesis, likely reflect-ing the sensitivity of a single growth factor angiogenesis system
compared with a more complex tumor-based model. The other
experimental design evaluated multiple angiogenic growth factorsin the same animal, allowing for direct comparison of CP-673,451
on angiogenesis stimulated by each growth factor. The dose and
regimen for this experiment was identical to what was used in theColo205 tumor growth inhibition experiments that provided 55%
tumor growth inhibition (Fig. 6A). Therefore, doses that result in
tumor growth inhibition do not inhibit VEGF- or bFGF-induced
angiogenesis. A representative photomicrograph of H460 humanlung adenocarcinoma tumors removed following a tumor growth
inhibition experiment visually shows the magnitude of the
antitumor response (Fig. 6B). Although a measurable tumor isobserved in the treated cohort, much of the tumor mass is fluid-
filled and necrotic. This phenomenon may result in underpredicting
the magnitude of the antitumor response when inhibition is onlymeasured by decreases in volume.Tumor growth inhibition studies were completed on a number of
different tumor xenografts with comparable results. Although
regressions were not typically observed, all models showed growthinhibition with PDGFR inhibition, regardless of tumor cell PDGFR
status. Tumor growth inhibition was dose-responsive with no
accumulation of compound with repeated dosing. All studies wereinitiated after tumors reached 100 to 200 mm3 and were in their
exponential growth phase. All tumor inhibition experiments were
carried out past two to three doublings of the control tumors.Pharmacokinetic/pharmacodynamic experiments showed that
single oral doses of 33 mg/kg resulted in, at most, 4-hour inhibition
of z50% phosphorylated PDGFR. Interestingly, doses whichinhibited tumor growth (5 mg/kg, p.o., q.d. � 10 days) resulted in
less than 50% inhibition of phospho-PDGFR at Tmax when given as a
single dose. This indicates that as little as 30% to 50% inhibition of
phospho-PDGFR for 3 to 6 hours per day is sufficient to inhibittumor growth z50% in multiple models. This somewhat surprising
efficacy, given the selective inhibition of PDGFR, may be indicative
of the importance of pericytes (and PDGFR) on neovascularendothelium or the sensitivity of our tumor models to angiogenesis
inhibition (20, 45–47). However, the antitumor efficacy observed in
Colo205 with CP-673,451 is comparable to published reports withother multitarget angiogenesis inhibitors (40). It is also possible that
repeated daily or twice daily dosing used in the tumor growth
inhibition experiments results in accumulated non-PDGFR-mediated effects that a single dose experiment (as in the
Figure 6. A, tumor growth inhibition study with Colo205 human colonadenocarcinoma xenografts and CP-673,451. Compound was dosed twicedaily (b.i.d.) to achieve total daily doses of 10, 33, and 100 mg/kg orally for10 days. B, representative tumors from another tumor growth inhibition studyusing H460 human lung adenocarcinoma xenografts following 10 once-dailyoral doses of 100 mg/kg. Note that the treated tumors are hemorrhagicand avascular relative to vehicle-treated control tumors. A Student’s t testwas used to evaluate the P for CP-673,451-treated relative to vehicle-treatedtumors. Vehicle treated (x), 5 mg/kg b.i.d. (n), 16.5 mg/kg b.i.d. (E),50 mg/kg b.i.d. (�). Points, averages of 5 animals; bars, SE. #, P < 0.05; *,P < 0.01, relative to vehicle treated.
Table 3. Tumor growth inhibition
Tumor xenografttype
Dosing regimen Total dailydose (mg/kg)
% Inhibition
U87MG humanglioblastoma
p.o., b.i.d. � 10 d 100 80, P < 0.01
LS174T human
colon
p.o., b.i.d. � 10 d 50 64, P < 0.01
H460 human
lung
p.o., q.d. � 11 d 33 80, P < 0.01
Colo205 human
colon
p.o., q.d. � 10 d 33 75, P < 0.01
Preclinical Pharmacology of CP-673,451
www.aacrjournals.org 963 Cancer Res 2005; 65: (3). February 1, 2005
Research. on February 16, 2016. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

pharmacokinetic/pharmacodynamic experiments) underrepre-sents. For example, in glioblastoma multiforme where EGFR andPDGFR are both overexpressed and can be amplified (48), acoordination of multiple growth factor receptor pathways mayresult in PDGFR transactivation of EGFR (49). In this circumstance,it is possible that repeated inhibition of PDGFR-h results in greatertumor growth inhibition due to additional downstream inhibitoryaffects on another receptor due to PDGFR cross-talk. Similarcomplementary effects can occur with other growth factors andreceptors in tumor stroma, such as PDGFR inhibition of VEGFproduction (50). There were no signs of morbidity or weight loss atany efficacious dose in the tumor growth inhibition studies,suggesting that selective PDGFR inhibition is well tolerated atefficacious doses.
To further show a measurable antiangiogenesis effect with CP-673,451, tumors were evaluated for microvessel density following amultiday dosing regimen. Tumors at the end of a typical 10-daytumor growth inhibition experiment were necrotic and unsatis-factory for immunohistochemical analysis of microvessel density.Therefore, tumors were removed and evaluated after 3 or 5 days ofdosing. Interestingly, doses that resulted in 47% inhibition ofmicrovessel density on day 5 yielded a 55% tumor growthinhibition with continued dosing by day 10.It is likely that targeted agents such as CP-673,451 will be
combined with conventional chemotherapeutic agents. It isanticipated that the mechanism of action of each agent will becomplementary, toxicities will not be additive, and combination of
the agents will result in greater efficacy (51). In testing thishypothesis, CP-673,451 was combined with paclitaxel in a tumorgrowth inhibition study on LS174T human xenografts. The plasmaconcentration of CP-673,451 was unchanged regardless of coad-ministration with paclitaxel, showing the increased tumor growthinhibition in combination is not due to increased CP-673,451plasma levels. Although paclitaxel concentrations were notmeasured, there is a low probability of drug-drug interaction withCP-673,451 because of the compound having greater than 40 Amol/L IC50 against all the major cytochrome P450 enzymes (data notshown). Single daily doses of CP resulted in comparable tumorgrowth inhibition (49%) to paclitaxel (54%). Paclitaxel was not usedat the maximally tolerated dose in order to better evaluate anyadditive efficacy and toxicities. When CP-673,451 was combinedwith paclitaxel, 88% tumor growth inhibition was observed withno increase in morbidity or weight loss versus vehicle- or singleagent-treated animals. When given together, these compoundswere roughly additive.In this report, data were presented on CP-673,451 showing it is a
highly selective PDGFR tyrosine kinase inhibitor in vitro and in vivo.
At doses and plasma concentrations where tumor growth
inhibition was observed in multiple models, there was no
inhibition of VEGF- or bFGF-stimulated angiogenesis. At all
efficacious doses, the compound was well tolerated in tumor-
bearing athymic mice. The significant selectivity of this compound
versus VEGFR, in particular, should allow dissection of angio-
genesis signal transduction pathways in vitro and in vivo as well as
Figure 7. Immunohistochemical evaluationof microvascular density using apan-endothelial antibody (MECA32)on Colo205-bearing mice dosed withCP-673,451 for 3 or 5 days (twice daily dosesof 5 mg/kg). Representative micrographsfrom 5-day cohorts are shown with stainedvessels (arrows ). Microvascular densityin treated tumors was inhibited 35% by day 3,which increased to 47% by day 5 comparedwith vehicle-treated control tumors.A Student’s t test was used to determinedthe P of treated relative to control tumors.Inhibition of microvessel density: 3-day dosing,35%, P < 0.01; 5-day dosing, 47%, P < 0.01.
Figure 8. Combination therapy with paclitaxel and CP-673,451in mice bearing LS174T human colon adenocarcinoma xenografts.Paclitaxel was dosed once daily on days 1 to 5 whereas CP-673,451 was dosed once daily for 10 days. The combination of thetwo agents together resulted in substantially more tumor growthinhibition than either agent alone. Vehicle treated (x), 33 mg/kgCP-673,451 (E), 10 mg/kg paclitaxel (�), 33 mg/kg CP-673451,and 10 mg/kg paclitaxel (.). Points, averages of 8 animals; bars,SE. *, P < 0.05 versus vehicle-treated animals.
Cancer Research
Cancer Res 2005; 65: (3). February 1, 2005 964 www.aacrjournals.org
Research. on February 16, 2016. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

increase our understanding of the role of pericytes in tumor
vascular biology. The oral availability of this compound allows for a
meaningful evaluation of the role of PDGFR in tumor biology as
well as other pathologies where PDGFR figures prominently, such
as restenosis, lung fibrosis, atherosclerosis, or glomerulonephritis
(10, 52, 53). The oral dosing regimen has the added benefit of
avoiding an extremely high Cmax often observed with i.p. dosing,
resulting in cross-inhibition of multiple kinases. Finally, the true
benefit of having a pharmacologically selective PDGFR inhibitor
must be appreciated in the clinic to determine whether selectivity
can avoid unwanted side effects, such as hypertension, associated
with other targets unable to be avoided with nonselective
multitarget tyrosine kinase inhibitors (54).
Acknowledgments
Received 5/28/2004; revised 11/3/2004; accepted 11/16/2004.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.
We thank Jim Moyer, Jean Beebe, and Rick Connell for comments and for review ofthe manuscript.
References1. Folkman J. Antiangiogenesis: new concept for theoryof solid tumors. Ann Surg 1972;175:409–16.
2. Folkman J. Tumor angiogenesis. In: Klein G,Weinhouse S, editors. Adv Cancer Res. 11008th ed.San Diego: Academic Press; 1985. p. 175–203.
3. Folkman J, Shing Y. Angiogenesis. J Biol Chem1992;267:10931–4.
4. Boehm T, Folkman J, Browder T, O’Reilly MS.Antiangiogenic therapy of experimental cancer doesnot induce acquired drug resistance. Nature1997;390:404–7.
5. Carmeliet P, Jain RK. Angiogenesis and cancer in otherdiseases. Nature 2000;407:249–57.
6. Hermanson M, Funa K, Hartman M, et al. Platelet-derived growth factor and its receptors in humanglioma tissue: expression of messenger RNA andprotein suggests the presence of autocrine and para-crine loops. Cancer Res 1992;52:3213–9.
7. Heldin C-H, Westermark B. Mechanism of action andin vivo role of platelet-derived growth factor. PhysiolRev 1999;79:1283–316.
8. Betsholtz C, Karlsson L, Lindahl P. Developmentalroles of platelet-derived growth factors. BioEssays2001;23:494–507.
9. Yarden Y, Escobedo JA, Kuang W-J, et al. Structure ofthe receptor for platelet-derived growth factor helpsdefine a family of closely related growth factorreceptors. Nature 1986;323:226–32.
10. Ostman A, Heldin C-H. Involvement of platelet-derived growth factor in disease: development ofspecific antagonists. Adv Cancer Res 2001;80:1–38.
11. Holmgren L, Glaser A, Pfeifer-Ohlsson S, Ohlsson R.Angiogenesis during human extraembryonic develop-ment involves the spatiotemporal control of PDGFligand and receptor gene expression. Development1991;113:749–54.
12. Hellstrom M, Kalen M, Lindahl P, Abramsson A,Betsholtz C. Role of PDGF-B and PDGFR-h in recruit-ment of vascular smooth muscle cells and pericytesduring embryonic blood vessels formation in themouse. Development 1999;126:3047–55.
13. Shinbrot E, Peters KG, Williams LT. Expression of theplatelet-derived growth factor h-receptor during orga-nogenesis and tissue differentiation in the mouseembryo. Dev Dyn 1994;199:169–75.
14. Leveen P, Pekny M, Gebre-Medhin S, Swolin B,Larsson E, Betsholtz C. Mice deficient in PDGF B showrenal, cardiovascular, and hematological abnormalities.Genes Dev 1994;8:1875–87.
15. Soriano P. Abnormal kidney development andhematological disorders on PDGF h-receptor mutantmice. Genes Dev 1994;8:1888–96.
16. Lindahl P, Hellstrom M, Kalen M, et al. ParacrinePDGF-B/PDGF-R h signaling controls mesangial celldevelopment in kidney glomeruli. Development1998;125:3301–12.
17. Lindahl P, Johansson BR, Leveen P, Betsholtz C.Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 1997;277:242–5.
18. Hirschi KK, D’Amore PA. Pericytes in the micro-vasculature. Cardiovasc Res 1996;32:687–98.
19. Allt G, Lawrenson JG. Pericytes: cell biology andpathology. Cells Tissues Organs 2001;169:1–11.
20. Morikawa S, Baluk P, Kaidoh T, Haskell A, Jain RK,McDonald DM. Abnormalities in pericytes on bloodvessels and endothelial sprouts in tumors. Am J Pathol2002;160:985–1000.
21. Gerhardt H, Betsholtz C. Endothelial-pericyteinteractions in angiogenesis. Cell Tissue Res 2003;314:15–23.
22. Bhardwaj B, Klassen J, Cossette N, et al. Localizationof platelet-derived growth factor h receptor expressionin the periepithelial stroma of human breast carcinoma.Clin Cancer Res 1996;2:773–82.
23. Sundberg C, Branting M, Bengt G, Rubin K.Tumor cell and connective tissue cell interactionsin human colorectal adenocarcinoma. Am J Pathol1997;151:479–92.
24. Kawai T, Hiroi S, Torikata C. Expression in lungcarcinomas of platelet-derived growth factor and itsreceptors. Lab Invest 1997;77:431–6.
25. Fudge K, Bostwick DG, Stearns ME. Platelet-derivedgrowth factor A and B chains and the a and h receptorsin prostatic intraepithelial neoplasia. Prostate1996;29:282–6.
26. Hermanson M, Funa K, Koopman J, et al. Associ-ation of loss of heterozygosity on chromosome 17pwith high platelet-derived growth factor a receptorexpression in human malignant gliomas. Cancer Res1996;56:164–71.
27. Plate KH, Breier G, Farrell CL, Risau W. Platelet-derived growth factor receptor-h is induced duringtumor development and up-regulated during tumorprogression in endothelial cells in human gliomas. LabInvest 1992;67:529–34.
28. Waltenberger J, Claesson-Welsh L, Siegbahn A,Shibuya M, Heldin C-H. Different signal transductionproperties of KDR and Flt1, two receptors for vascularendothelial growth factor. J Biol Chem 1994;269:26988–95.
29. Wolff NC, Randle DE, Wegorin MJ, Minna JD,Ilaria RL. Imatinib mesylate efficiently achievestherapeutic intratumor concentrations in vivo buthas limited activity in a xenograft model of smallcell lung cancer. Clin Cancer Res 2004;10:3528–34.
30. Knick VC, Eberwein DJ, Miller CG. Vinorelbinetartrate and paclitaxel combinations: enhanced activityagainst in vivo P388 murine leukemia cells. J NatlCancer Inst 1995;87:1072–7.
31. Yamori T, Sata S, Chikazawa H, Kadota T. Antitumorefficacy of paclitaxel against human lung cancerxenografts. Jpn J Cancer Res 1997;88:1205–10.
32. Hurwitz H, Fehrenbacher L, Novotny W, et al.Bevacizumab plus irinotecan, fluorouracil, and leuco-vorin for metastatic colorectal cancer. N Engl J Med2004;350:2335–42.
33. Deveraux QL, Aza-Blanc P, Wagner KW,Bauerschlag D, Cooke MP, Hampton GM. Exposingoncogenic dependencies for cancer drug targetdiscovery and validation using RNAi. Semin CancerBiol 2003;13:293–300.
34. Im SA, Kim JS, Gomez-Manzano C, et al. Inhibition ofbreast cancer growth in vivo by antiangiogenesis gene
therapy with adenovirus-mediated antisense-VEGF. Br JCancer 2001;84:1252–7.
35. Yu JC, Lokker NA, Hollenbach S, et al. Efficacyof the novel selective platelet-derived growth factorreceptor antagonist CT52923 on cellular prolifer-ation, migration, and suppression of neointimafollowing vascular injury. J Pharmacol Exp Ther2001;298:1172–8.
36. Zaman GJ, Vink PM, van den Doelen AA, VeenemanGH, Theunissen HJ. Tyrosine kinase activity of purifiedrecombinant cytoplasmic domain of platelet-derivedgrowth factor h-receptor (h-PDGFR) and discovery ofa novel inhibitor of receptor tyrosine kinases. BiochemPharmacol 1999;57:57–64.
37. Bergers G, Song S, Meyer-Morse N, Bergsland E,Hanahan D. Benefits of targeting both pericytes andendothelial cells in the tumor vasculature with kinaseinhibitors. J Clin Invest 2003;111:1287–95.
38. Fong TA, Shawver LK, Sun L, et al. SU5416 is apotent and selective inhibitor of the vascular endothe-lial growth factor receptor (Flk-1/KDR) that inhibitstyrosine kinase catalysis, tumor vascularization, andgrowth of multiple tumor types. Cancer Res 1999;59:99–106.
39. Laird AD, Christensen JG, Li G, et al. SU6668 inhibitsFlk-1/KDR and PDGFR-h in vivo , resulting in rapidapoptosis of tumor vasculature and tumor regression inmice. FASEB J 2002;16:681–90.
40. Mendel DB, Laird AD, Xin X, et al. In vivoantitumor activity of SU11248, a novel tyrosinekinase inhibitor targeting vascular endothelialgrowth factor and platelet-derived growth factorreceptors: Determination of a pharmacokinetic/phar-macodynamic relationship. Clin Cancer Res 2003;9:327–37.
41. Manley PW, Cowan-Jacob SW, Buchdunger E, et al.Imatinib: a selective tyrosine kinase inhibitor. Eur JCancer 2002;38:S19–27.
42. Shawver LK, Slamon D, Ullrich A. Smart drugs:tyrosine kinase inhibitors in cancer therapy. Cancer Cell2002;1:117–23.
43. Claesson-Welsh L, Eriksson A, Moren A, et al. cDNAcloning and expression of a human platelet-derivedgrowth factor (PDGF) receptor specific for B-chain-containing PDGF molecules. Mol Cell Biol 1988;8:3476–86.
44. Herren B, Weyer KA, Rouge M, Lotscher P, Pech M.Conservation in sequence and affinity of human androdent PDGF ligands and receptors. Biochim BiophysActa 1993;1173:294–302.
45. Beebe JS, Jani JP, Knauth E, et al. Pharmaco-logical characterization of CP-547,632, a novelvasular endothelial growth factor receptor-2 tyrosinekinase inhibitor for cancer therapy. Cancer Res2003;63:7301–9.
46. Bilodeau MT, Fraley ME, Hartman GD. Kinase insertdomain-containing receptor tyrosine kinase inhibitorsas antiangiogenic agents. Expert Opin Investig Drugs2002;11:737–45.
47. Kilic T, Alberta JA, Zdunek PR, et al. Intracranialinhibition of platelet derived growth factor-mediatedglioblastoma cell growth by an orally active kinase
Preclinical Pharmacology of CP-673,451
www.aacrjournals.org 965 Cancer Res 2005; 65: (3). February 1, 2005
Research. on February 16, 2016. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

inhibitor of the 2-phenylaminopyrimidine class. CancerRes 2000;60:5143–50.
48. Fleming TP, Saxena A, Clark WC, et al. Amplifi-cation and/or overexpression of platelet-derivedgrowth receptors and epidermal growth factorreceptor in human glial tumors. Cancer Res 1992;52:4550–3.
49. Saito Y, Haendeler J, Hojo Y, U Yamamoto K, Berk BC.Receptor heterodimerization: essential mechanism forplatelet-derived growth factor-induced epidermal
growth factor receptor transactivation. Mol Cell Biol2001;21:6387–94.
50. Wang D, Huang H-JS, Kazlauskas A, CaveneeWK. Induction of vascular endothelial growthfactor expression in endothelial cells by platelet-derived growth factor through activation ofphosphatidylinositol 3-kinase. Cancer Res 1999;59:1464–72.
51. Pietras K, Rubin K, Sjoblom T, et al. Inhibition ofPDGF receptor signaling in tumor stroma enhances
antitumor effect of chemotherapy. Cancer Res 2002;62:5476–84.
52. Laskey JA, Brody AR. Interstitial fibrosis and growthfactors. Environ Health Perspect 2000;108:751–62.
53. Bult H. Restenosis: a challenge for pharmacology.Trends Pharmacol Sci 2000;21:274–9.
54. Manley PW, Martiny-Baron G, Schlaeppi JM,Wood JM. Therapies directed at vascular endothelialgrowth factor. Expert Opin Investig Drugs 2002;11:1715–36.
Cancer Research
Cancer Res 2005; 65: (3). February 1, 2005 966 www.aacrjournals.org
Research. on February 16, 2016. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from

2005;65:957-966. Cancer Res W. Gregory Roberts, Pamela M. Whalen, Erik Soderstrom, et al. Tyrosine Kinase Inhibitor, CP-673,451Antiangiogenic and Antitumor Activity of a Selective PDGFR
Updated version
http://cancerres.aacrjournals.org/content/65/3/957
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/65/3/957.full.html#ref-list-1
This article cites 52 articles, 25 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/65/3/957.full.html#related-urls
This article has been cited by 12 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
Research. on February 16, 2016. © 2005 American Association for Cancercancerres.aacrjournals.org Downloaded from




















