
Conditional expression of mutated K-ras accelerates intestinal tumorigenesis in Msh2-deficient mice
Upload
independentCategory
view
0download
0
[CANCER RESEARCH 59, 1301–1307, March 15, 1999]
Tumorigenesis inMlh1 and Mlh1/Apc1638NMutant Mice 1
Winfried Edelmann,2,3 Kan Yang,2 Mari Kuraguchi, Joerg Heyer, Marie Lia, Burkhard Kneitz, Kunhua Fan,Anthony M. C. Brown, Martin Lipkin, and Raju Kucherlapati 4
Department of Molecular Genetics, Albert Einstein College of Medicine, Bronx, New York 10461 [W. E., J. H., M. Lia, B. K., R. K.]; and Strang Cancer Prevention Center [K. Y.,M. K., K. F., A. M. C. B., M. Lip.] and Department of Cell Biology and Anatomy, Cornell University Medical College [A. M. C. B.], New York, New York 10021
ABSTRACT
MutL homologue 1 (MLH1) is a member of the family of proteinsrequired for DNA mismatch repair. Germ-line mutations in MLH1 lead tothe cancer susceptibility syndrome hereditary nonpolyposis colorectalcancer (HNPCC). We generated mice carrying a null mutation in theMlh1gene. We showed that mice heterozygous and homozygous for theMlh1gene are predisposed to developing tumors of the gastrointestinal (GI)tract, lymphomas, and a number of other tumor types. We also examinedthe role of adenomatous polyposis coli gene (Apc) gene mutations in the GItumors of Mlh1 mutant mice by different methods and showed that the GItumors in Mlh1 mice express little or no adenomatous polyposis coliprotein. When an Apcgene mutation was bred into theMlh1 mutant mice,the GI tumor incidence increased 40–100-fold. The wild-typeApc allele inthese tumors was found to contain mutations. Together, these results showthat we have developed two mouse models for human HNPCC and thatthe mechanisms of tumor development in the GI tract of these miceinvolve loss ofApc gene function in a manner very similar to that seen inthe GI tumors of HNPCC.
INTRODUCTION
CRC5 has emerged as a suitable model to identify genes involvedin the onset and progression of cancer. Two types of familial CRChave been studied extensively: (a) FAP is inherited as an autosomaldominant disorder, and individuals with this disorder are normal atbirth and progressively develop a large number of intestinal andduodenal tumors. It has been shown that germ-line mutations in thetumor suppressor geneAPC are responsible for FAP (1–3). Theseresults suggested thatAPCgene function is important to maintain thebalance of normal proliferative potential and loss of intestinal epithe-lial cells, andAPC is, therefore, termed a “gatekeeper” gene (4). Theobservation thatAPC gene mutations are detectable in virtually allcases of sporadic CRC suggested that the mechanism of initiation ofCRC involved mutation in theAPCgene. (b) HNPCC is also inheritedin an autosomal dominant fashion, and HNPCC individuals show apredisposition to colonic and other tumors (5–8). An important fea-ture of tumors in HNPCC patients is that they have a characteristicRER1 phenotype (5–9), which is manifested in the instability of DNAsequences containing short repeat sequences. Germ-line mutations ineach of several genes implicated in repair of mismatched nucleotides
in DNA have been shown to be responsible for HNPCC. Among themost prominent of these areMSH2andMLH1, which are homologuesof bacterial DNA MMR genesMutSandMutL, respectively. BecauseDNA MMR is an activity that is intrinsic to all replicating cells, it isimportant to understand how mutations in DNA MMR genes cancause a cancer predisposition. Knowledge of the mechanism of actionof the MMR complex suggests that mutations in the repair genescause cancer indirectly by increasing the frequency of oncogenicmutations in cells that lack MMR activity.
To understand the role of APC and the MMR genes in the onset andprogression of colon cancer and to understand the normal role of thesegenes in development, differentiation, and cell/tissue homeostasis, wedeveloped a series of mouse lines, each of which carries a mutation ina different gene. The first among these mutations was designatedApc1638N. In this model, we used gene targeting to introduce aneomycin phosphotransferase expression cassette at the position cor-responding to codon 1638 of the mouseApc gene. Mice that areheterozygous forApc1638Nare normal at birth but progressivelydevelop aberrant crypt foci, colonic polyps, and tumors of the smallintestine, including the duodenum. The mouse model is akin to humanpatients with attenuated FAP (10, 11).
A second line of mice was developed by targeting the mouseMlh1gene. Gene targeting allowed us to delete a portion of the 59 end of theMlh1 gene in mouse embryonic stem cells. In an initial report, weshowed that the mutation we introduced results in the failure toproduce any detectable MLH1 protein. Mice that are homozygous fortheMlh1 mutation are viable and their cells are defective in MMR andexhibit microsatellite instability; mice of both sexes are sterile (12).
Here, we examined the susceptibility ofMlh1 mutant mice to tumordevelopment. We present data showing thatMlh1 1/2 and2/2 micehave increased morbidity and develop lymphomas and tumors of theGI tract and a number of other organs. To assess the role of theApcgene in tumorigenesis inMlh1 mutant mice, we mated theMlh1mutant mice toApc1638N1/2 mice. Here, we show that the GItumors, indeed, result from inactivation of theApc gene.
MATERIALS AND METHODS
Mice. The animals used in this study were generated by crossingMlh1heterozygote males and females of the F1 and F2 generation to generatehomozygous, heterozygous, and wild-type mice. The animals were of a mixedgenetic background (C57BL/63 129/Ola). To generateMlh1/Apc1638Nmu-tant mice, we crossedMlh1 heterozygote F1 animals withApc1638Nheterozy-gote animals (in the C57BL/6 genetic background). TheMlh1/Apc1638Ndouble heterozygote offspring were interbred to generate mice homozygous forMlh1 and heterozygous forApc1638N.
Genotyping. The animals were genotyped by PCR of tail DNA. ForMlh1genotyping, the following primers were used: primer A, TGTCAATAGGCT-GCCCTAGG; primer B, TGGAAGGATTGGAGCTGCTG; and primer C,TTTTCAGTGCAGCCTATGCTC. The reaction was performed in a 50-mlreaction mixture containing 100 ng of DNA, 10 ng/ml primer A and 5 ng/mlprimers B and C, 1.3 mM MgCl2, 0.2 mM each dNTP, and 0.5 units of Taqpolymerase. Cycling conditions were 5 min at 94°C (1 cycle); 1 min at 94°C,1 min at 56°C, and 30 s at 72°C (35 cycles); and 5 min at 72°C (1 cycle). Thepresence of the wild-type allele is indicated by a 350-bp PCR fragment(primers A and C) and the mutant allele by a 450-bp PCR fragment (primers
Received 8/10/98; accepted 1/15/99.The costs of publication of this article were defrayed in part by the payment of page
charges. This article must therefore be hereby markedadvertisementin accordance with18 U.S.C. Section 1734 solely to indicate this fact.
1 This work was supported by NIH Grants CA76329 (to W. E.), CA67944, andNO1-CN-65031 (to M. Lip. and R. K.); American Cancer Society Grant RPG-95-022-03-CN (to R. K.); NIH Center Grant CA13330 to Albert Einstein College of Medicine; afellowship from the Cancer Research Foundation of America (to M. K.); funding from theIris Cantor Cancer Research Unit at Strang Cancer Prevention Center; and an Irma T.Hirschl Career Scientist Award (to A. M. C. B.).
2 The first two authors contributed equally to this work.3 Present address: Department of Cell Biology, Albert Einstein College of Medicine,
1300 Morris Park Avenue, Bronx, NY 10461.4 To whom requests for reprints should be addressed, at Department of Molecular
Genetics, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY10461. Phone: (718) 430-2069; Fax: (718) 430-8776.
5 The abbreviations used are: CRC, colorectal cancer; FAP, familial adenomatouspolyposis; APC, adenomatous polyposis coli; HNPCC, hereditary nonpolyposis colorectalcancer; RER, replication error; MMR, mismatch repair; MSH2, MutS homologue 2;MLH1, MutL homologue 1; GI, gastrointestinal; PTT, protein truncation test.
1301
on June 19, 2016. © 1999 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from

A and B). ForApc1638Ngenotyping, the following primers were used: primerA, TGCCAGCACAGAATAGGCTG; primer B, TGGAAGGATTGGAGCT-GCTG; and primer C, GTTGTCATCCAGGTCTGGTG. The reaction wasperformed in a 50-ml reaction mixture containing 100 ng of DNA, 4 ng/mlprimers A and C and 8 ng/ml primer B, 1.2 mM MgCl2, 0.2 mM each dNTP, and0.5 units of Taq polymerase. Cycling conditions were 5 min at 94°C (1 cycle);1 min at 94°C, 45 s at 58°C, and 30 s at 72°C (28 cycles); and 5 min at 72°C(1 cycle). The presence of the wild-type allele is indicated by a 300-bp PCRfragment (primers A and C) and the mutant allele by a 400-bp PCR fragment(primers A and B).
Analysis of Tumors. After sacrifice, the GI tract was opened and examinedunder a dissecting microscope for tumors. In our analysis, duodenum is fromthe pylorus, the junction of the stomach and the duodenum, through thesuspensory ligament of Treitz (;7 cm in length). The remaining part of thesmall intestine is divided in half: the upper part is the jejenum and the lowerpart is the ileum (each region is;14 cm in length). Tumors, if found, the GItract, and other organs, including the lungs, heart, liver, kidneys, and spleen,were removed and fixed in 10% neutral buffered formalin. Representativetissues from the tumors and organs were taken for processing and paraffinembedding, and others were frozen for subsequent DNA analysis. All tissuesections were prepared for H&E stain. The GI tumor tissues were studied forAPC protein expression by immunohistochemistry, and the nature of thelymphomas was ascertained by immunotyping. The antibodies used in thisstudy were: C-20, an antibody directed against the COOH terminus of the APCprotein (Santa Cruz Biotechnology, Santa Cruz, CA); B-lymphocyte CD45R/B220, an antibody against a B cell-specific CD45 antigen (PharMingen, SanDiego, CA); and an antibody against a T lymphocyte-specific antigen CD3(Vector Laboratories, Burlingame, CA). Avidin-biotin-peroxidase techniquewas used for immunohistochemical stain. Deparaffinized slides were incubatedwith primary antibody overnight at 4°C, followed by addition of biotinylatedgoat antirabbit IgG and avidin-biotin-peroxidase complex. Color was devel-oped in 3,39-diaminobenzidine.
Microsatellite Instability Analysis. DNA was extracted from tumor tissueand tails and subjected to PCR. End-labeled primer pairs were used to amplifysequences containing dinucleotide repeats D1Mit36, D7Mit91, D10Mit2,D14Mit15, and D18Mit15 (13), and two others, JH104 and U12235, were usedto amplify sequences containing mononucleotide repeats (primer sequenceswere as follows: JH104F, 59-AGGTGATTGTAACGGGCATC-39, andJH104R, 59-TATCCTCTCAGTGGTGAGTG-39; U12235F, 59-GCTCATCT-TCGTTCCCTGTC-39, and U12235R, 59-CATTCGGTGGAAAGCTCTGA-39). Amplified PCR products were separated on a denaturing polyacrylamidegel and autoradiographed for analysis.
Analysis of Apc Mutations. Protein truncation mutations within the seg-ment ofApc corresponding to codons 677–1233 were identified by a modifi-cation of the PTT described previously (14, 15). To amplify specifically thewild-type allele of Apc, PCR amplification was first performed using theforward primer 59-TACAGCACTTGAAATCTCACAG (nucleotides 1991–2012 of mouseApc; GenBank accession no. M88127) and a wild-type allele-specific reverse primer, 59-GTTGTCATCCAGGTCTGGTG (nucleotides5123–5142). Genomic DNA (150–200 ng) from frozen tumor samples wasamplified in 20-ml reactions containing 10 mM Tris-HCl (pH 8.6), 50 mM KCl,0.1% Triton X-100, 1 mM MgCl2, 0.2 mM dNTP, 0.2mM primers, and 0.05units/ml of Taq2000 (Stratagene). Cycling conditions were as described pre-viously (15), except that the annealing temperature was 60°C, and a total of 25cycles were used. This reaction gave a 3150-bp product spanningApccodons664–1714. One-ml aliquots of these reaction products were reamplified in50-ml reactions containing the forward primer 59-CGGGATCCTAATACGA-CTCACTATAGGGAGACCACCATGGATGCATGTGGAACTTTGTGG-39and the reverse primer 59-CGATCGAAGCTTGGACGCAGCTGATTCT-39,each at 0.3mM. Reaction conditions were as above, except that a total of 20cycles were used and the annealing temperature was 57°C. The purified finalPCR products were then used as templates in PTT assays performed with theTNT Quick coupled reticulocyte lysate system (Promega, Madison, WI) ac-cording to the manufacturer’s protocol, and [35S]methionine-labeled polypep-tides were analyzed by 12% SDS-PAGE and fluorography. For characteriza-tion of the tumor-specific mutations, the PCR products were ligated into anSP64 plasmid vector. Individual clones were screened by PTT to identify thosecarrying mutations, and their DNA sequence was determined.
RESULTS
Mhl1 Mutant Mice Develop a Spectrum of Tumors. The initialMlh1 mutation was generated in E14-1 ES cells. The chimeric micewere mated with C57B1/6J (B6) mice. The F1 mice were continu-ously back-crossed to B6 mice. The analyses ofMlh1 mice wereconducted on mice in the second and third back-cross generations.The initial Apc1638Nmice were generated in E14-1 ES cells, whichwere derived from the 129/Ola strain of mice. We developed B6 micethat are congenic for theApc1638Nmutation by 10 or more genera-tions of repeated back-crossing to B6.
Mlh1 heterozygotes were inter-crossed to generateMlh1 1/1,Mlh1 1/2, andMlh1 2/2 mice. Each of the crosses yielded offspringof all three classes in the expected ratios. We have previously shownthat theMlh1 gene targeting resulted in a null mutant (12). Theseresults suggest that a reduction or complete absence of MLH1 proteinis not detrimental to the normal development of mice.
Mice of the differentMlh1 genotypes were monitored for survival.The results of this analysis are shown in Fig. 1. Both heterozygous andhomozygousMlh1 mutant mice had reduced longevity compared totheir wild-type littermates. Fifty % of the1/2 mice died by the ageof 18 months, whereas it took only 7 months for 50% of the2/2 miceto die.
Several of the mice withMlh1 mutation were sacrificed when theywere moribund to assess whether they had developed tumors and, if so,of what origin. The results of this analysis are presented in Table 1.
Seven of 22 (32%) ofMlh1 1/2 mice developed tumors, whereasa much larger proportion (13 of 18; 72%) ofMlh1 2/2 mice devel-oped tumors. Because some mice died prior to examination, it ispossible that we underestimated the tumor frequency. The fact that the
Table 1 Tumor incidence inMlh1 mutant mice
Genotype n Age (months) Sex (M;F) No. (%) of mice with tumors
No. (%) of mice with
GI tumors Extra-GI tumors
1/1 20 21.76 4.8 12;8 1 (5) 0 1 (5)1/2 22 9.86 4.4 10;12 7 (32) 3 (14) 5 (23)2/2 18 7.26 3.6 14;4 13 (72) 6 (33) 8 (44)
Fig. 1. Kaplan-Meier survival curves forMlh1 mutant mice.
1302
MOUSE MODELS FOR HNPCC
on June 19, 2016. © 1999 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from

1/2 mice were moribund at an average age of 9.8 months whereasthe 2/2 mice were moribund earlier (7.0 months) shows that theMlh1 2/2 mice develop tumors much faster than their heterozygouslittermates. In both cases, tumors outside the GI tract were as frequentas those within the GI tract. In contrast, 1 of 20 wild-type micedeveloped a single non-Hodgkin’s lymphoma at 15 months of age.
We examined the distribution and nature of the tumors in theMlh1mutant mice. We were able to detect tumors in the duodenum,jejunum, and colon in the1/2 mice, whereas the2/2 mice hadtumors in all parts of the GI tract tested. These included the stomach,
duodenum, jejunum, ileum, and colon. Histological examination ofthe seven GI tumors indicated that the1/2 mice had an equalproportion of adenocarcinomas and early invasive carcinomas,whereas 20 tumors in2/2 mice contained 13% carcinomas, 37%early invasive carcinomas, and 50% adenomas.
Tumors outside the GI tract inMlh1 mutant mice are of differenttypes (Fig. 2). In1/2 mice, we examined five tumors: one non-Hodgkin’s lymphoma, one T-cell lymphoblastic lymphoma, one skinsweat gland carcinoma, two cervical squamous cell carcinomas, andone lung bronchio-alveolar carcinoma. In2/2 mice, there was apreponderance of lymphomas (eight of nine) and one (one of nine)skin sweat gland carcinoma. Interestingly, the lymphomas were ofeither B- or T-cell origin. Of the eight lymphomas, seven werenon-Hodgkin’s lymphomas (five T-cell type and two B-cell type). Theremaining lymphoma was classified as Hodgkin’s disease.
The Combination of Mlh1 and Apc Gene Mutations DecreasesLongevity and Increases Tumorigenesis.We have shown previ-ously that mice with a mutation in theApcgene (Apc1638N) developtumors late in the first year of their life and that each mouse developsrelatively few (between one and three) tumors. All of these earlytumors are in the GI tract. To assess the role of Apc in the GI tumorsof Mlh1 mutant mice, we brought theApc1638Nmutation into micecarrying theMlh1 mutation. Apc1638Nheterozygotes were matedwith Mlh1 heterozygotes. The double heterozygotes were identifiedby genotyping and mated again withMlh1 heterozygotes. This matingprovided mice that were wild-type or heterozygous forApc1638Nandthat were either heterozygous or homozygous forMlh1. These twoclasses of mice were monitored for survival (Fig. 3), and several mice
Fig. 2. Extraintestinal tumors inMlh1 mice. A,non-Hodgkin’s lymphoma fromMlh1 2/2 mouse(scale bar, 40mm). B, non-Hodgkin’s lymphoma inA stained with B220 (scale bar, 40mm). C, non-Hodgkin’s lymphoma fromMlh1 2/2 mouse.D,non-Hodgkin’s lymphoma inC stained with CD3antibody (scale bar, 20mm). E, metastasis of skintumors in the lung fromApc1638N1/2, Mlh1 1/2mouse (scale bar, 1.25 mm).F, cross-section ofmetastatic lung (scale bar, 40mm).
Fig. 3. Kaplan-Meier survival curve forApc1638N/Mlh1mice.
1303
MOUSE MODELS FOR HNPCC
on June 19, 2016. © 1999 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from
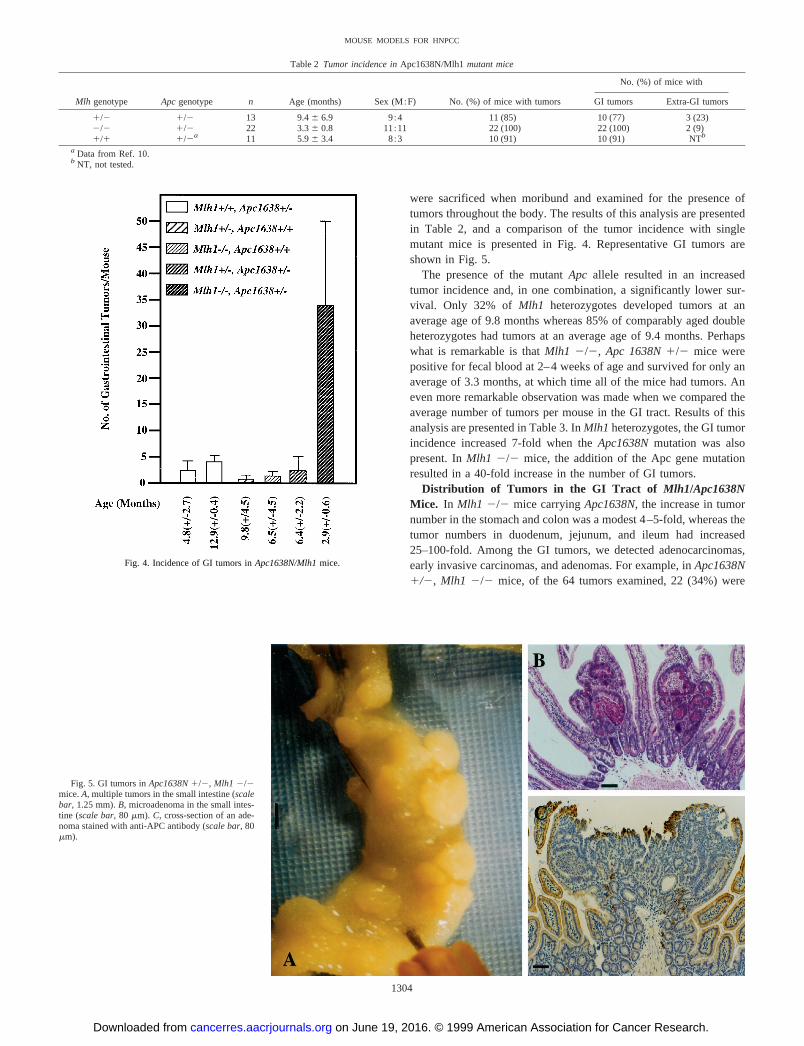
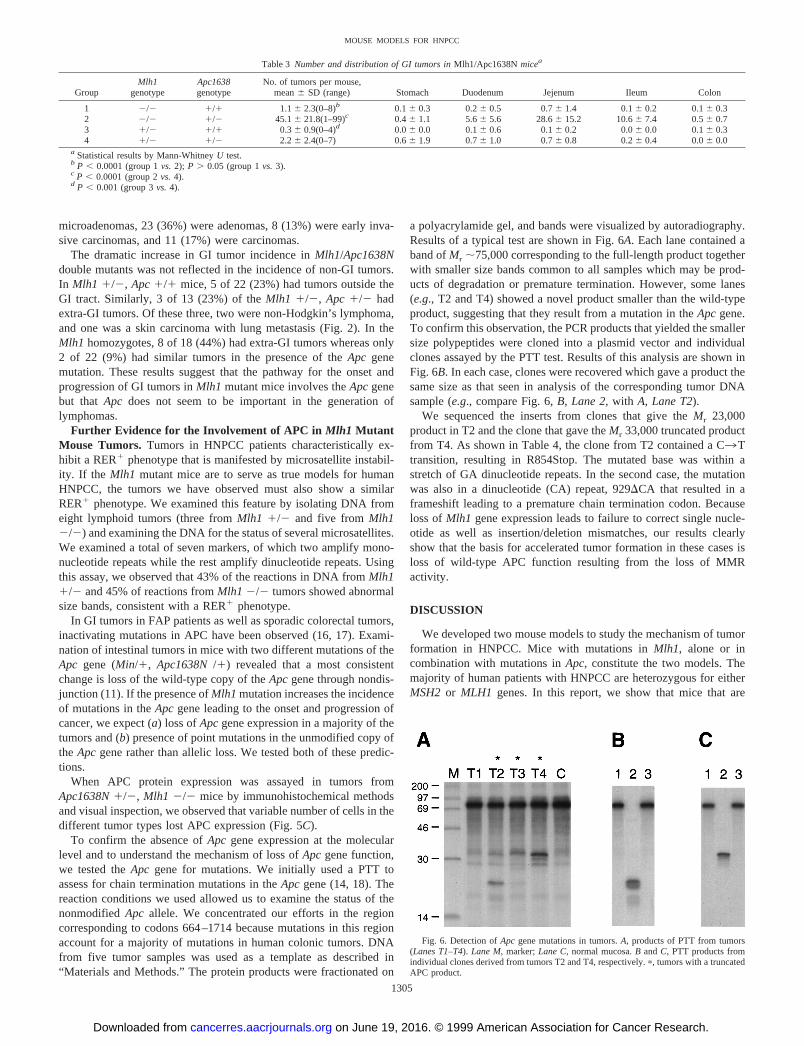
were sacrificed when moribund and examined for the presence oftumors throughout the body. The results of this analysis are presentedin Table 2, and a comparison of the tumor incidence with singlemutant mice is presented in Fig. 4. Representative GI tumors areshown in Fig. 5.
The presence of the mutantApc allele resulted in an increasedtumor incidence and, in one combination, a significantly lower sur-vival. Only 32% of Mlh1 heterozygotes developed tumors at anaverage age of 9.8 months whereas 85% of comparably aged doubleheterozygotes had tumors at an average age of 9.4 months. Perhapswhat is remarkable is thatMlh1 2/2, Apc 1638N1/2 mice werepositive for fecal blood at 2–4 weeks of age and survived for only anaverage of 3.3 months, at which time all of the mice had tumors. Aneven more remarkable observation was made when we compared theaverage number of tumors per mouse in the GI tract. Results of thisanalysis are presented in Table 3. InMlh1 heterozygotes, the GI tumorincidence increased 7-fold when theApc1638Nmutation was alsopresent. InMlh1 2/2 mice, the addition of the Apc gene mutationresulted in a 40-fold increase in the number of GI tumors.
Distribution of Tumors in the GI Tract of Mlh1/Apc1638NMice. In Mlh1 2/2 mice carryingApc1638N, the increase in tumornumber in the stomach and colon was a modest 4–5-fold, whereas thetumor numbers in duodenum, jejunum, and ileum had increased25–100-fold. Among the GI tumors, we detected adenocarcinomas,early invasive carcinomas, and adenomas. For example, inApc1638N1/2, Mlh1 2/2 mice, of the 64 tumors examined, 22 (34%) were
Table 2 Tumor incidence inApc1638N/Mlh1mutant mice
Mlh genotype Apc genotype n Age (months) Sex (M;F) No. (%) of mice with tumors
No. (%) of mice with
GI tumors Extra-GI tumors
1/2 1/2 13 9.46 6.9 9;4 11 (85) 10 (77) 3 (23)2/2 1/2 22 3.36 0.8 11;11 22 (100) 22 (100) 2 (9)1/1 1/2a 11 5.96 3.4 8;3 10 (91) 10 (91) NTb
a Data from Ref. 10.b NT, not tested.
Fig. 4. Incidence of GI tumors inApc1638N/Mlh1mice.
Fig. 5. GI tumors inApc1638N1/2, Mlh1 2/2mice.A, multiple tumors in the small intestine (scalebar, 1.25 mm).B, microadenoma in the small intes-tine (scale bar, 80mm). C, cross-section of an ade-noma stained with anti-APC antibody (scale bar, 80mm).
1304
MOUSE MODELS FOR HNPCC
on June 19, 2016. © 1999 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from
microadenomas, 23 (36%) were adenomas, 8 (13%) were early inva-sive carcinomas, and 11 (17%) were carcinomas.
The dramatic increase in GI tumor incidence inMlh1/Apc1638Ndouble mutants was not reflected in the incidence of non-GI tumors.In Mlh1 1/2, Apc 1/1 mice, 5 of 22 (23%) had tumors outside theGI tract. Similarly, 3 of 13 (23%) of theMlh1 1/2, Apc 1/2 hadextra-GI tumors. Of these three, two were non-Hodgkin’s lymphoma,and one was a skin carcinoma with lung metastasis (Fig. 2). In theMlh1 homozygotes, 8 of 18 (44%) had extra-GI tumors whereas only2 of 22 (9%) had similar tumors in the presence of theApc genemutation. These results suggest that the pathway for the onset andprogression of GI tumors inMlh1 mutant mice involves theApcgenebut that Apc does not seem to be important in the generation oflymphomas.
Further Evidence for the Involvement of APC in Mlh1 MutantMouse Tumors. Tumors in HNPCC patients characteristically ex-hibit a RER1 phenotype that is manifested by microsatellite instabil-ity. If the Mlh1 mutant mice are to serve as true models for humanHNPCC, the tumors we have observed must also show a similarRER1 phenotype. We examined this feature by isolating DNA fromeight lymphoid tumors (three fromMlh1 1/2 and five fromMlh12/2) and examining the DNA for the status of several microsatellites.We examined a total of seven markers, of which two amplify mono-nucleotide repeats while the rest amplify dinucleotide repeats. Usingthis assay, we observed that 43% of the reactions in DNA fromMlh11/2 and 45% of reactions fromMlh1 2/2 tumors showed abnormalsize bands, consistent with a RER1 phenotype.
In GI tumors in FAP patients as well as sporadic colorectal tumors,inactivating mutations in APC have been observed (16, 17). Exami-nation of intestinal tumors in mice with two different mutations of theApc gene (Min/1,Apc1638N/1) revealed that a most consistentchange is loss of the wild-type copy of theApcgene through nondis-junction (11). If the presence ofMlh1 mutation increases the incidenceof mutations in theApc gene leading to the onset and progression ofcancer, we expect (a) loss ofApcgene expression in a majority of thetumors and (b) presence of point mutations in the unmodified copy ofthe Apc gene rather than allelic loss. We tested both of these predic-tions.
When APC protein expression was assayed in tumors fromApc1638N1/2, Mlh1 2/2 mice by immunohistochemical methodsand visual inspection, we observed that variable number of cells in thedifferent tumor types lost APC expression (Fig. 5C).
To confirm the absence ofApc gene expression at the molecularlevel and to understand the mechanism of loss ofApc gene function,we tested theApc gene for mutations. We initially used a PTT toassess for chain termination mutations in theApc gene (14, 18). Thereaction conditions we used allowed us to examine the status of thenonmodifiedApc allele. We concentrated our efforts in the regioncorresponding to codons 664–1714 because mutations in this regionaccount for a majority of mutations in human colonic tumors. DNAfrom five tumor samples was used as a template as described in“Materials and Methods.” The protein products were fractionated on
a polyacrylamide gel, and bands were visualized by autoradiography.Results of a typical test are shown in Fig. 6A. Each lane contained aband ofMr ;75,000 corresponding to the full-length product togetherwith smaller size bands common to all samples which may be prod-ucts of degradation or premature termination. However, some lanes(e.g., T2 and T4) showed a novel product smaller than the wild-typeproduct, suggesting that they result from a mutation in theApcgene.To confirm this observation, the PCR products that yielded the smallersize polypeptides were cloned into a plasmid vector and individualclones assayed by the PTT test. Results of this analysis are shown inFig. 6B. In each case, clones were recovered which gave a product thesame size as that seen in analysis of the corresponding tumor DNAsample (e.g., compare Fig. 6,B, Lane 2, withA, Lane T2).
We sequenced the inserts from clones that give theMr 23,000product in T2 and the clone that gave theMr 33,000 truncated productfrom T4. As shown in Table 4, the clone from T2 contained a C3Ttransition, resulting in R854Stop. The mutated base was within astretch of GA dinucleotide repeats. In the second case, the mutationwas also in a dinucleotide (CA) repeat, 929DCA that resulted in aframeshift leading to a premature chain termination codon. Becauseloss ofMlh1 gene expression leads to failure to correct single nucle-otide as well as insertion/deletion mismatches, our results clearlyshow that the basis for accelerated tumor formation in these cases isloss of wild-type APC function resulting from the loss of MMRactivity.
DISCUSSION
We developed two mouse models to study the mechanism of tumorformation in HNPCC. Mice with mutations inMlh1, alone or incombination with mutations inApc, constitute the two models. Themajority of human patients with HNPCC are heterozygous for eitherMSH2 or MLH1 genes. In this report, we show that mice that are
Fig. 6. Detection ofApc gene mutations in tumors.A, products of PTT from tumors(Lanes T1–T4).Lane M, marker;Lane C, normal mucosa.B andC, PTT products fromindividual clones derived from tumors T2 and T4, respectively.p, tumors with a truncatedAPC product.
Table 3 Number and distribution of GI tumors inMlh1/Apc1638Nmicea
GroupMlh1
genotypeApc1638genotype
No. of tumors per mouse,mean6 SD (range) Stomach Duodenum Jejenum Ileum Colon
1 2/2 1/1 1.16 2.3(0–8)b 0.16 0.3 0.26 0.5 0.76 1.4 0.16 0.2 0.16 0.32 2/2 1/2 45.16 21.8(1–99)c 0.46 1.1 5.66 5.6 28.66 15.2 10.66 7.4 0.56 0.73 1/2 1/1 0.36 0.9(0–4)d 0.06 0.0 0.16 0.6 0.16 0.2 0.06 0.0 0.16 0.34 1/2 1/2 2.26 2.4(0–7) 0.66 1.9 0.76 1.0 0.76 0.8 0.26 0.4 0.06 0.0
a Statistical results by Mann-WhitneyU test.b P , 0.0001 (group 1vs.2); P . 0.05 (group 1vs.3).c P , 0.0001 (group 2vs.4).d P , 0.001 (group 3vs.4).
1305
MOUSE MODELS FOR HNPCC
on June 19, 2016. © 1999 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from

heterozygous or homozygous for a null mutation in theMlh1 genehave a predisposition to cancer. There are differences between the1/2 and2/2 mice in terms of tumor incidence. Only 32% of1/2mice developed easily identifiable tumors at a mean age of 9.8months, whereas 72% of2/2 mice developed tumors at a mean ageof 7.0 months. We have shown previously that cells fromMlh1heterozygotes are capable of repairing mismatched DNA, whereascells from2/2 mice are not. These results indicate that the loss ofMMR activity is probably a prerequisite for initiation of cancer inMlh1 1/2 mice. This notion was confirmed by our observations thattumors fromMlh1 1/2 or Mlh12/2 mice show similarly high levelsof microsatellite instability. It is of interest to note that olderMsh21/2 mice (19), in a different genetic background than ourMlh1mutant mice, developed 40% higher level of tumors compared towild-type with no effect on survival.
The spectrum of tumors observed in theMlh1 mutant mice issomewhat different from that seen in HNPCC. Although a largenumber of tissue types are involved in the tumors of HNPCC patients,lymphoid tumors are not very frequent. However, in theMlh1 mutantmice, lymphoid tumors predominate the non-GI tumor category. It isalso of interest that we have detected both T- and B-cell lymphomas.The presence of a large number of lymphoid tumors is not limited toMlh1 mutant mice.Msh2 and Msh6 mutant mice also develop lym-phomas, although the lymphomas inMsh2 mutant mice are exclu-sively of T-cell origin (19–21), whereas theMsh6 mutant micedevelop B- as well as T-cell lymphomas (22). T-cell lymphomas werealso detected in mice withp53 mutations (23). These results mightreflect some intrinsic differences between humans and mice in termsof lymphoma susceptibility. Prollaet al. (24) also observed a tumorsusceptibility phenotype in an independently derivedMlh1 mutanthomozygote.
Because APC plays an important gatekeeper role in colorectaltumorigenesis, we examined whether the cause of GI tumors in theMlh1 mutant mice is through loss of APC function. A number ofdifferent experimental results support this view. WhenMlh1 andApcdouble mutants were generated by breeding, we observed that the timerequired for mortality, the number of tumors per mouse, and the stageof tumors were all affected. IfApc and Mlh1 acted in differentpathways, the time of tumor onset would not be expected to change,and the tumor number would be additive. If inactivation of theApcgene is an important prerequisite for tumor development, the time ofonset and the number of tumors would be expected to change. Thiswas, indeed, what we observed. InMlh1 2/2 mice, a mutatedApcgene resulted in a very significant (40-fold) increase in tumor num-bers. These results clearly show thatApc gene mutation is an impor-tant early step in the onset of GI cancer inMlh1 mutant mice.
Our results also showed that the mechanism of GI tumor develop-ment and lymphoid tumor development are distinct. When theApcmutation was crossed into theMlh1 mutant background, the lymphoidtumor incidence was unaffected. This observation, together with thefact thatApcmutant mice do not develop lymphoid tumors, suggeststhat theApc gene does not play a role in lymphomagenesis in thesemice. Alternatively, the double mutant mice could succumb to the GItumors before the lymphoid tumors have an opportunity to develop.
Loss of Apc gene function is an important and early event in thedevelopment of GI tumors in mice and humans (25). Tumors in theMlh1/Apc1638Nmice also follow the same pattern. We have shownthe loss ofApcgene function by histochemical and molecular biolog-ical methods. However, the mechanism of loss ofApc gene functionis different inApc1638NandMlh1/Apc1638Nmice. We have previ-ously shown (11) that, inApc1638Nmice, loss ofApc gene functionis mediated through loss of the wild-type copy ofApcgene, apparentlythrough chromosomal nondisjunction. In contrast, the tumors in theMlh1/Apc1638Nmice show mutations in the wild-type copy of theApc gene. This mechanism is consistent with the known function ofMlh1 in DNA MMR.
The Apc1638Nmice we developed are a good model for theattenuated form of FAP. Compared toMin/1 mice, Apc1638Nmicedevelop fewer tumors and at a later time; these tumors progress intoadenocarcinomas. As a result, the mice live longer, providing anopportunity to examine the role of environmental factors, such as diet,on the onset and progression of CRC. The combination ofMlh1 andApc1638Nmutations results in a large number of tumors at an earlyage. Although, in this respect, these mice are similar toMin/1 (26),they have important differences. These mice are a model of HNPCCbecause of the MMR phenotype associated with them, whereas theMin/1 are not. Another significant difference is that theMin/1 micedo not develop carcinomas. It has been suggested that the number oftumors and their time of onset would preclude them from becomingcarcinomas. However, theMlh1/Apc1638Nmice have a very highincidence of carcinomas. Therefore, these mice are uniquely suited forexamining the development of carcinomas. Mice that are doublemutant forMin andPms2, a close relative ofMlh1, have been reportedrecently (27). Although these mice show increased numbers of ade-nomas, they do not seem to progress to carcinomas. This differencealso makes theMlh1/Apc1638Nmice excellent models for the studyof the full spectrum of progression of the GI tumors.
REFERENCES
1. Kinzler, K. W., Nilbert, M. C., Su, L. K., Vogelstein, B., Bryan, T. M., Levy, D. B.,Smith, K. J., Preisinger, A. C., Hedge, P., McKechnie, D., Finniaer, R., Markham, A.,Groffen, J., Boguski, M. S., Altschul, S. F., Horii, A., Ando, H., Miyoshi, Y., Miki,Y., Nishisho, I., and Naklamura, Y. Identification of FAP locus genes from chromo-some 5q21. Science (Washington DC),253: 661–665, 1991.
2. Groden, J., Thliveris, A., Samowitz, W., Carlson, M., Gelbert, L., Albertsen, H.,Joslyn, G., Stevens, J., Spirio, L., Robertson, M., Sargeant, L., Krapcho, K., Wolff, E.,Burt, R., Hughes, J. P., Warrington, J., McPherson, P., Wasmuth, J., LePaslier, D.,Abderrahim, H., Cohen, D., Leppert, M., and White, R. Identification and character-ization of the familial adenomatous polyposis coli gene. Cell,66: 589–600, 1991.
3. Joslyn, G., Carlson, M., Thliveris, A., Albertsen, H., Gelbert, L., Samowitz, W., Groden,J., Stevens, J., Spirio, L., Robertson, M., Sargeant, L., Krapcho, K., Wolff, E., Burt, R.,Hughes, J. P., Warrington, J., McPherson, P., Wasmuth, J., LePaslier, D., Abderrahim, H.,Cohen, D., Leppert, M., and White, R. Identification of deletion mutations and three newgenes at the familial polyposis locus. Cell,66: 601–613, 1991.
4. Kinzler, K. W., and Vogelstein, B. Landscaping the cancer terrain. Science (Wash-ington DC),280: 1036–1037, 1998.
5. Aaltonen, L. A., Peltomaki, P., Leach, F., Sistonen, P., Pylkkanen, S. M., Mecklin,J-P., Jarvinen, H., Powell, S., Jen, J., Hamilton, S. R., Petersen, G. M., Kinzler, K. W.,Vogelstein, B., and de la Chapelle, A. Clues to the pathogenesis of familial colorectalcancer. Science (Washington DC),260: 812–816, 1993.
6. Parsons, R., Li, G-M., Longley, M. J., Fang, W-H., Papadopoulos, N., Jen, J., de laChapelle, A., Kinzler, K. W., Vogelstein, B., and Modrich, P. Hypermutability andmismatch repair deficiency in RER1 tumor cells. Cell,75: 1227–1236, 1993.
Table 4 Examples of somaticApc truncation mutations in intestinal tumors fromMlh1 2/2, Apc1638Nmice
Apparent size oftruncated product (Mr) 59 position Wild-type sequencea 39 position Mutation
;23,000 850 TTG GAG AGA GAGCGA GGT 855 R854StopL E R E R G (C3T)
;33,000 925 GCC TCCCAC ACA CAC TCA 930 929DCAA S H T H S (frameshift)
a The site of mutation is shown in boldface type.
1306
MOUSE MODELS FOR HNPCC
on June 19, 2016. © 1999 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from

7. Lothe, R. A., Peltomaki, P., Meling, G. I., Aaltonen, L. A., Nystrom-Lahti, M.,Pylkkanen, L., Heimdal, K., Andersen, T. I., Moller, P., Rognum, T. O., Fossa, S. D.,Haldorsen, T., Langmark, F., Brogger, A., de la Chapelle, A., and Borresen, A-L.Genomic instability in colorectal cancer: relationship to clinicopathological variablesand family history. Cancer Res.,53: 5849–5852, 1993.
8. Lynch, H. T., Smyrk, T. C., Watson, P., Lanspa, S. J., Lynch, J. F., Lynch, P. M.,Cavalieri, R. J., and Boland, C. R. Genetics, natural history, tumor spectrum, andpathology of hereditary nonpolyposis colorectal cancer: an updated review. Gastro-enterology,104: 1535–1549, 1993.
9. Aaltonen, L. A., Peltomaki, P., Mecklin, J. P., Jarvinen, H., Jass, J. R., Green, J. S.,Lynch, H. T., Watson, P., Tallqvist, G., Juhola, M., Sistonen, P., Hamilton, S. R.,Kinzler, K. W., Vogelstein, B., and de la Chapelle, A. Replication errors in benignand malignant tumors from hereditary nonpolyposis colorectal cancer patients. Can-cer Res.,54: 1645–1648, 1994.
10. Fodde, R., Edelmann, W., Yang, K., van Leeuwen, C., Carlson, C., Renault, B.,Breukel, C., Alt, E., Lipkin, M., Khan, P. M., and Kucherlapati, R. A targetedchain-termination mutation in the mouseApc gene results in multiple intestinaltumors. Proc. Natl. Acad. Sci. USA,91: 8969–8973, 1994.
11. Smits, R., Kartheuser, A., Jagmohan-Changur, S., Leblanc, V., Breukel, C., deVries, A.,van Kranen, H., van Krieken, J. H., Williamson, S., Edelmann, W., Kucherlapati, R.,Meera Khan, P., and Fodde, R. Loss ofApcand the entire chromosome 18 but absenceof mutations at the Ras and Tp53 genes in intestinal tumors fromApc 1638, a mousemodel forApc-driven carcinogenesis. Carcinogenesis (Lond.),18: 321–327, 1997.
12. Edelmann, W., Cohen, P. E., Kane, M., Lau, K., Morrow, B., Bennett, S., Umar, A.,Kunkel, T., Cattoretti, G., Chaganti, R., Pollard, J. W., Kolodner, R. D., andKucherlapati, R. Meiotic pachytene arrest inMLH1-deficient mice. Cell,85:1125–1134, 1996.
13. Dietrich, W. F., Miller, J. C., Steen, R. G., Merchant, M., Damron, D., Nahf, R.,Gross, A., Joyce, D. C., Wessel, M., Dredge, R. D., Marquis, A., Stein, L. D.,Goodman, N., Page, D. C., and Lander, E. C. A genetic map of the mouse with 4,006simple sequence length polymorphisms. Nat. Genet.,7: 220–245, 1994.
14. Powell, S. M., Petersen, G. M., Krush, A. J., Booker, S., Jen, J., Giardiello, F. M.,Hamilton, S. R., Vogelstein, B., and Kinzler, K. W. Molecular diagnosis of familialadenomatous polyposis. N. Engl. J. Med.,329: 1982–1987, 1993.
15. Shoemaker, A. R., Luongo, C., Moser, A. R., Marton, L. J., and Dove, W. F. Somaticmutational mechanisms involved in intestinal tumor formation in Min mice. CancerRes.,57: 1999–2006, 1997.
16. Lazar, V., Grandjouan, S., Bognel, C., Couturier, D., Rougier, P., Bellet, D., andBressac-de Paillerets, B. Accumulation of multiple mutations in tumour suppressorgenes during colorectal tumorigenesis in HNPCC patients. Hum. Mol. Genet.,3:2257–2260, 1994.
17. Huang, J., Papadopoulos, N., McKinley, A. J., Farrington, S. M., Curtis, L. J., Wyllie,A. H., Zheng, S., Willson, J. K., Markowitz, S. D., Morin, P., Kinzler, K. W.,Vogelstein, B., and Dunlop, M. G. APC mutations in colorectal tumors with mismatchrepair deficiency. Proc. Natl. Acad. Sci. USA,93: 9049–9054, 1996.
18. van der Luijt, R., Khan, P. M., Vasen, H., van Leeuwen, C., Tops, C., Roest, P., denDunnen, J., and Fodde, R. Rapid detection of translation-terminating mutations at theadenomatous polyposis coli (APC) gene by direct protein truncation test. Genomics,20: 1–4, 1994.
19. de Wind, N., Dekker, M., van Rossum, A., van der Valk, M., and te Riele, H. Mousemodels for hereditary nonpolyposis colorectal cancer. Cancer Res.,58: 248–255,1998.
20. Reitmair, A. H., Schmits, R., Ewel, A., Bapat, B., Redston, M., Mitri, A., Waterhouse,P., Mittrucker, H. W., Wakeham, A., Liu, B., Thomason, A., Griesser, S., Gallinger,S., Ballhausen, W. G., Fishel, R., and Mak, T. W. MSH2 deficient mice are viable andsusceptible to lymphoid tumours. Nat. Genet.,11: 64–70, 1995.
21. Lowsky, R., DeCoteau, J. F., Reitmair, A. H., Ichinohasama, R., Dong, W. F., Xu, Y.,Mak, T. W., Kadin, M. E., and Minden, M. D. Defects of the mismatch repair geneMSH2 are implicated in the development of murine and human lymphoblasticlymphomas and are associated with the aberrant expression of rhombotin-2 (Lmo-2)and Tal-1 (SCL). Blood,89: 2276–2282, 1997.
22. Edelmann, W., Yang, K., Umar, A., Heyer, J., Lau, K., Fan, K., Liedtke, W., Cohen,P. E., Kane, M. F., Lipford, J. R., Yu, N., Crouse, G. F., Pollard, J. W., Kunkel, T.,Lipkin, M., Kolodner, R., and Kucherlapati, R. Mutation in the mismatch repair geneMsh6causes cancer susceptibility. Cell,91: 467–477, 1997.
23. Donehower, L. A., Harvey, M., Slagle, B. L., McArthur, M. J., Montgomery, C. A.,Jr., Butel, J. S., and Bradley, A. Mice deficient for p53 are developmentally normalbut susceptible to spontaneous tumours. Nature (Lond.),356: 215–221, 1992.
24. Prolla, T. A., Baker, S. M., Harris, A. C., Tsao, J. L., Yao, X., Bronner, C. E., Zheng,B., Gordon, M., Reneker, J., Arnheim, N., Shibata, D., Bradley, A., and Liskay, R. M.Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 andPms2 DNA mismatch repair. Nat. Genet.,18: 276–279, 1998.
25. Kinzler, K. W., and Vogelstein, B. Lessons from hereditary colorectal cancer. Cell,87: 159–170, 1996.
26. Su, L. K., Kinzler, K. W., Vogelstein, B., Preisinger, A. C., Moser, A. R., Luongo, C.,Gould, K. A., and Dove, W. F. Multiple intestinal neoplasia caused by a mutation inthe murine homolog of the APC gene. Science (Washington DC),256: 668–670,1992.
27. Baker, S. M., Harris, A. C., Tsao, J. L., Flath, T. J., Bronner, C. E., Gordon, M.,Shibata, D., and Liskay, R. M. Enhanced intestinal adenomatous polyp formation inPms22/2;Min mice. Cancer Res.,58: 1087–1089, 1998.
1307
MOUSE MODELS FOR HNPCC
on June 19, 2016. © 1999 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from

1999;59:1301-1307. Cancer Res Winfried Edelmann, Kan Yang, Mari Kuraguchi, et al.
Mutant Mice Apc1638N/Mlh1 and Mlh1Tumorigenesis in
Updated version
http://cancerres.aacrjournals.org/content/59/6/1301
Access the most recent version of this article at:
Cited articles
http://cancerres.aacrjournals.org/content/59/6/1301.full.html#ref-list-1
This article cites 27 articles, 14 of which you can access for free at:
Citing articles
/content/59/6/1301.full.html#related-urls
This article has been cited by 28 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
To request permission to re-use all or part of this article, contact the AACR Publications
on June 19, 2016. © 1999 American Association for Cancer Research.cancerres.aacrjournals.org Downloaded from
Copyright © 2022 FDOKUMEN




















