Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens
18
LETTER doi:10.1038/nature13988 Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens Matthew M. Gubin 1 , Xiuli Zhang 2 , Heiko Schuster 3 , Etienne Caron 4 , Jeffrey P. Ward 1,5 , Takuro Noguchi 1 , Yulia Ivanova 1 , Jasreet Hundal 6 , Cora D. Arthur 1 , Willem-Jan Krebber 7 , Gwenn E. Mulder 7 , Mireille Toebes 8 , Matthew D. Vesely 1 , Samuel S. K. Lam 1 , Alan J. Korman 9 , James P. Allison 10 , Gordon J. Freeman 11 , Arlene H. Sharpe 12 , Erika L. Pearce 1 , Ton N. Schumacher 8 , Ruedi Aebersold 4,13 , Hans-Georg Rammensee 3 , Cornelis J. M. Melief 7,14 , Elaine R. Mardis 6,15 , William E. Gillanders 2 , Maxim N. Artyomov 1 & Robert D. Schreiber 1 The immune system influences the fate of developing cancers by not only functioning as a tumour promoter that facilitates cellular transformation, promotes tumour growth and sculpts tumour cell immunogenicity 1–6 , but also as an extrinsic tumour suppressor that either destroys developing tumours or restrains their expansion 1,2,7 . Yet, clinically apparent cancers still arise in immunocompetent indivi- duals in part as a consequence of cancer-induced immunosuppression. In many individuals, immunosuppression is mediated by cytotoxic T-lymphocyte associated antigen-4 (CTLA-4) and programmed death-1 (PD-1), two immunomodulatory receptors expressed on T cells 8,9 . Monoclonal-antibody-based therapies targeting CTLA-4 and/or PD-1 (checkpoint blockade) have yielded significant clinical benefits— including durable responses—to patients with different malignan- cies 10–13 . However, little is known about the identity of the tumour antigens that function as the targets of T cells activated by check- point blockade immunotherapy and whether these antigens can be used to generate vaccines that are highly tumour-specific. Here we use genomics and bioinformatics approaches to identify tumour- specific mutant proteins as a major class of T-cell rejection antigens following anti-PD-1 and/or anti-CTLA-4 therapy of mice bearing pro- gressively growing sarcomas, and we show that therapeutic synthetic long-peptide vaccines incorporating these mutant epitopes induce tumour rejection comparably to checkpoint blockade immunother- apy. Although mutant tumour-antigen-specific T cells are present in progressively growing tumours, they are reactivated following treat- ment with anti-PD-1 and/or anti-CTLA-4 and display some overlap- ping but mostly treatment-specific transcriptional profiles, rendering them capable of mediating tumour rejection. These results reveal that tumour-specific mutant antigens are not only important targets of checkpoint blockade therapy, but they can also be used to develop personalized cancer-specific vaccines and to probe the mechanistic underpinnings of different checkpoint blockade treatments. In this study, we used two distinct progressor 3-methylcholanthrene- induced (MCA) sarcoma cell lines (d42m1-T3 and F244) and asked whether they expressed sufficient immunogenicity to be controlled by checkpoint blockade immunotherapy. Both sarcoma lines were rejected in wild-type mice treated therapeutically with anti-PD-1 and/or anti- CTLA-4 (Fig. 1a). Rejection was immunologic because (1) it was ablated by administration of monoclonal antibodies (mAbs) that either deplete CD4 1 or CD8 1 cells or neutralize interferon-c (IFN-c); (2) it did not occur in Rag2 2/2 mice lacking T, B and natural killer T (NKT) cells or Batf3 2/2 mice lacking CD8a 1 CD103 1 dendritic cells required for tumour antigen cross-presentation to CD8 1 T cells (Extended Data Fig. 1a); and (3) it induced a memory response that protected mice against rechal- lenge with the same tumour cells that had been injected into naive mice (Extended Data Fig. 1b, c). On the basis of our previous success using genomics approaches to identify tumour-specific mutant antigens (TSMA) responsible for the spon- taneous rejection of highly immunogenic, unedited MCA sarcomas 14 , we asked whether a similar approach could identify antigens responsi- ble for anti-PD-1-mediated rejection of d42m1-T3 progressor tumours. To increase the robustness and accuracy of our epitope predictions, we modified our method as follows: (1) mutation calls from complementary DNA capture sequencing 14 were translated to corresponding protein sequences, pipelined through three major histocompatibility complex (MHC) class I epitope-binding algorithms and a median binding affin- ity was calculated for each predicted epitope; (2) epitopes were priori- tized on the basis of predicted median binding affinities; and (3) filters were applied to the prioritized epitope list to eliminate those predicted to be poorly processed by the immunoproteasome and to deprioritize those from hypothetical proteins or those that displayed lower binding affinity to class I than their corresponding wild-type sequences. Using this approach, many epitopes were predicted for H-2D b (49,677 9-mer and 10-mer epitopes) (Extended Data Fig. 2a) and H-2K b (44,215 8-mer and 9-mer epitopes) (Fig. 1b) based on the 2,796 non-synonymous mu- tations expressed in d42m1-T3 14 . Focusing on epitopes with the highest predicted binding affinity to H-2D b or H-2K b , we narrowed the list down to four H-2D b -binding epitopes (Extended Data Fig. 2b) and 62 H-2K b - binding epitopes (Fig. 1c). Applying the aforementioned filters elimi- nated two predicted strong-binding H-2D b epitopes (Extended Data Fig. 2c) and 20 predicted strong-binding H-2K b epitopes (Fig. 1d) (epi- tope binding affinity distributions to different class I alleles are distict 15 ). Based on the resulting in-silico-generated epitope landscape, two pre- dominant H-2K b restricted mutant epitopes were identified by their predicted binding affinities: an A506T mutation (ITY AWTRLRITY TW TRL) in asparagine-linked glycosylation 8 (alpha-1,3-glucosyltransferase) (Alg8) and a G1254V mutation ( GGFNFRTLR VGFNFRTL) in lami- nin alpha subunit 4 (Lama4). Based on H-2K b consensus binding, the mutations that produce these epitopes occur at positions p4 (Alg8) and p1 (Lama4). Neither functions as an anchor residue for H-2K b (these occur 1 Department of Pathology and Immunology, Washington University School of Medicine, 660 South Euclid Avenue, St Louis, Missouri 63110, USA. 2 Department of Surgery, Washington University School of Medicine, 660 South Euclid Avenue, St Louis, Missouri 63110, USA. 3 Department of Immunology, Institute of Cell Biology, and German Cancer Consortium (DKTK), German Cancer Research Center (DKFZ) Partner Site Tu ¨ bingen, Auf der Morgenstelle 15, 72076 Tu ¨ bingen, Germany. 4 Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, 8093 Zurich, Switzerland. 5 Department of Medicine, Division of Oncology, Washington University School of Medicine, 660 South Euclid Avenue, St Louis, Missouri 63110, USA. 6 The Genome Institute, Washington University School of Medicine, 4444 Forest Park Avenue, St Louis, Missouri 63108, USA. 7 ISA Therapeutics B.V., 2333 CH Leiden, The Netherlands. 8 Division of Immunology, The Netherlands Cancer Institute, 1066 CX Amsterdam, The Netherlands. 9 Bristol-Myers Squibb, 700 Bay Road, Redwood City, California 94063, USA. 10 Department of Immunology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA. 11 Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, Massachusetts 02115, USA. 12 Department of Microbiology and Immunobiology, Harvard Medical School, Boston, Massachusetts 02115, USA. 13 Faculty of Science, University of Zurich, Zurich, 8093 Zurich, Switzerland. 14 Department of Immunohematology and Blood Transfusion, Leiden University Medical Center, 2333ZA Leiden, The Netherlands. 15 Department of Genetics, Washington University School of Medicine, 660 South Euclid Avenue, St Louis, Missouri 63110, USA. 27 NOVEMBER 2014 | VOL 515 | NATURE | 577 Macmillan Publishers Limited. All rights reserved ©2014
-
Upload
independent -
Category
Documents
-
view
2 -
download
0
Transcript of Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens

LETTERdoi:10.1038/nature13988
Checkpoint blockade cancer immunotherapy targetstumour-specific mutant antigensMatthew M. Gubin1, Xiuli Zhang2, Heiko Schuster3, Etienne Caron4, Jeffrey P. Ward1,5, Takuro Noguchi1, Yulia Ivanova1,Jasreet Hundal6, Cora D. Arthur1, Willem-Jan Krebber7, Gwenn E. Mulder7, Mireille Toebes8, Matthew D. Vesely1,Samuel S. K. Lam1, Alan J. Korman9, James P. Allison10, Gordon J. Freeman11, Arlene H. Sharpe12, Erika L. Pearce1,Ton N. Schumacher8, Ruedi Aebersold4,13, Hans-Georg Rammensee3, Cornelis J. M. Melief7,14, Elaine R. Mardis6,15,William E. Gillanders2, Maxim N. Artyomov1 & Robert D. Schreiber1
The immune system influences the fate of developing cancers bynot only functioning as a tumour promoter that facilitates cellulartransformation, promotes tumour growth and sculpts tumour cellimmunogenicity1–6, but also as an extrinsic tumour suppressor thateither destroys developing tumours or restrains their expansion1,2,7.Yet, clinically apparent cancers still arise in immunocompetent indivi-duals in part as a consequence of cancer-induced immunosuppression.In many individuals, immunosuppression is mediated by cytotoxicT-lymphocyte associated antigen-4 (CTLA-4) and programmed death-1(PD-1), two immunomodulatory receptors expressed on T cells8,9.Monoclonal-antibody-based therapies targeting CTLA-4 and/or PD-1(checkpoint blockade) have yielded significant clinical benefits—including durable responses—to patients with different malignan-cies10–13. However, little is known about the identity of the tumourantigens that function as the targets of T cells activated by check-point blockade immunotherapy and whether these antigens can beused to generate vaccines that are highly tumour-specific. Here weuse genomics and bioinformatics approaches to identify tumour-specific mutant proteins as a major class of T-cell rejection antigensfollowing anti-PD-1 and/or anti-CTLA-4 therapy of mice bearing pro-gressively growing sarcomas, and we show that therapeutic syntheticlong-peptide vaccines incorporating these mutant epitopes inducetumour rejection comparably to checkpoint blockade immunother-apy. Although mutant tumour-antigen-specific T cells are present inprogressively growing tumours, they are reactivated following treat-ment with anti-PD-1 and/or anti-CTLA-4 and display some overlap-ping but mostly treatment-specific transcriptional profiles, renderingthem capable of mediating tumour rejection. These results reveal thattumour-specific mutant antigens are not only important targets ofcheckpoint blockade therapy, but they can also be used to developpersonalized cancer-specific vaccines and to probe the mechanisticunderpinnings of different checkpoint blockade treatments.
In this study, we used two distinct progressor 3-methylcholanthrene-induced (MCA) sarcoma cell lines (d42m1-T3 and F244) and askedwhether they expressed sufficient immunogenicity to be controlled bycheckpoint blockade immunotherapy. Both sarcoma lines were rejectedin wild-type mice treated therapeutically with anti-PD-1 and/or anti-CTLA-4 (Fig. 1a). Rejection was immunologic because (1) it was ablatedby administration of monoclonal antibodies (mAbs) that either depleteCD41 or CD81 cells or neutralize interferon-c (IFN-c); (2) it did not
occur in Rag22/2 mice lacking T, B and natural killer T (NKT) cells orBatf32/2 mice lacking CD8a1CD1031 dendritic cells required for tumourantigen cross-presentation to CD81 T cells (Extended Data Fig. 1a); and(3) it induced a memory response that protected mice against rechal-lenge with the same tumour cells that had been injected into naive mice(Extended Data Fig. 1b, c).
On the basis of our previous success using genomics approaches toidentify tumour-specific mutant antigens (TSMA) responsible for the spon-taneous rejection of highly immunogenic, unedited MCA sarcomas14,we asked whether a similar approach could identify antigens responsi-ble for anti-PD-1-mediated rejection of d42m1-T3 progressor tumours.To increase the robustness and accuracy of our epitope predictions, wemodified our method as follows: (1) mutation calls from complementaryDNA capture sequencing14 were translated to corresponding proteinsequences, pipelined through three major histocompatibility complex(MHC) class I epitope-binding algorithms and a median binding affin-ity was calculated for each predicted epitope; (2) epitopes were priori-tized on the basis of predicted median binding affinities; and (3) filterswere applied to the prioritized epitope list to eliminate those predictedto be poorly processed by the immunoproteasome and to deprioritizethose from hypothetical proteins or those that displayed lower bindingaffinity to class I than their corresponding wild-type sequences. Usingthis approach, many epitopes were predicted for H-2Db (49,677 9-merand 10-mer epitopes) (Extended Data Fig. 2a) and H-2Kb (44,215 8-merand 9-mer epitopes) (Fig. 1b) based on the 2,796 non-synonymous mu-tations expressed in d42m1-T314. Focusing on epitopes with the highestpredicted binding affinity to H-2Db or H-2Kb, we narrowed the list downto four H-2Db-binding epitopes (Extended Data Fig. 2b) and 62 H-2Kb-binding epitopes (Fig. 1c). Applying the aforementioned filters elimi-nated two predicted strong-binding H-2Db epitopes (Extended DataFig. 2c) and 20 predicted strong-binding H-2Kb epitopes (Fig. 1d) (epi-tope binding affinity distributions to different class I alleles are distict15).Based on the resulting in-silico-generated epitope landscape, two pre-dominant H-2Kb restricted mutant epitopes were identified by theirpredicted binding affinities: an A506T mutation (ITYAWTRLRITYTWTRL) in asparagine-linked glycosylation 8 (alpha-1,3-glucosyltransferase)(Alg8) and a G1254V mutation (GGFNFRTLRVGFNFRTL) in lami-nin alpha subunit 4 (Lama4). Based on H-2Kb consensus binding, themutations that produce these epitopes occur at positions p4 (Alg8) and p1(Lama4). Neither functions as an anchor residue for H-2Kb (these occur
1Department of Pathology and Immunology, Washington University School of Medicine, 660 South Euclid Avenue, St Louis, Missouri 63110, USA. 2Department of Surgery, Washington University School ofMedicine, 660 South Euclid Avenue, St Louis, Missouri 63110, USA. 3Department of Immunology, Institute of Cell Biology, and German Cancer Consortium (DKTK), German Cancer Research Center (DKFZ)Partner Site Tubingen, Auf der Morgenstelle 15, 72076 Tubingen, Germany. 4Department of Biology, Institute of Molecular Systems Biology, ETH Zurich, 8093 Zurich, Switzerland. 5Department ofMedicine, Divisionof Oncology, Washington UniversitySchool of Medicine, 660South EuclidAvenue, St Louis, Missouri 63110,USA. 6The Genome Institute, Washington UniversitySchool of Medicine, 4444Forest Park Avenue, St Louis, Missouri 63108, USA. 7ISA Therapeutics B.V., 2333 CH Leiden, The Netherlands. 8Division of Immunology, The Netherlands Cancer Institute, 1066 CX Amsterdam, TheNetherlands. 9Bristol-Myers Squibb, 700 Bay Road, Redwood City, California 94063, USA. 10Department of Immunology, The University of Texas MD Anderson Cancer Center, Houston, Texas 77030, USA.11Department of Medical Oncology, Dana-Farber Cancer Institute, Harvard Medical School, Boston, Massachusetts 02115, USA. 12Department of Microbiology and Immunobiology, Harvard MedicalSchool, Boston, Massachusetts 02115, USA. 13Faculty of Science, University of Zurich, Zurich, 8093 Zurich, Switzerland. 14Department of Immunohematology and Blood Transfusion, Leiden UniversityMedical Center, 2333ZA Leiden, The Netherlands. 15Department of Genetics, Washington University School of Medicine, 660 South Euclid Avenue, St Louis, Missouri 63110, USA.
2 7 N O V E M B E R 2 0 1 4 | V O L 5 1 5 | N A T U R E | 5 7 7
Macmillan Publishers Limited. All rights reserved©2014

at p5 and p8). The mutant Alg8 (mAlg8) and mutant Lama4 (mLama4)epitopes are predicted to bind 1.2- and 12.8-fold stronger to H-2Kb,respectively, compared to wild-type sequences.
To identify which of the predicted d42m1-T3 neoepitopes functionedas targets for CD81 T cells in anti-PD-1-treated, tumour-bearing mice,freshly explanted CD81 tumour-infiltrating lymphocytes (TIL) were iso-lated just before tumour rejection (day 11) and stained with fluorescentlylabelled H-2Kb or H-2Db tetramers loaded with their correspondingstrong-binding 66 predicted mutant epitopes. The only tetramer-positiveT cells consistently identified in the CD81 TIL population were thosereacting with mLama4-H-2Kb tetramers (13.1% of CD81 TIL in exper-iment shown; 15.6 6 2.7% as the mean of 6 experiments) or mAlg8-H-2Kb tetramers (4.2% of CD81 TIL in experiment shown; 2.8 6 1.1% asthe mean of 6 experiments) (Fig. 1e and Extended Data Fig. 2d). Similarresults were obtained when freshly explanted CD81 TIL from the samemice were co-cultured with naive irradiated splenocytes pulsed witheach of the 66 predicted H-2Kb and H-2Db epitopes. The mLama4 andmAlg8 epitopes were, again, the only significant hits, inducing IFN-cand tumour necrosis factor-a (TNF-a) production (Fig. 1f and Ex-tended Data Fig. 2e). These results demonstrate that d42m1-T3 expresses
two dominant TSMA epitopes for CD81 T cells following anti-PD-1immunotherapy.
To independently validate these observations, we established CD81
T cell lines from spleens of mice that had rejected d42m1-T3 tumoursafter anti-PD-1 treatment. These T cells produced IFN-c when co-cultured with d42m1-T3 but not when co-cultured with F244 or otherindependent sarcoma lines (Extended Data Fig. 3a). Stimulation wasrestricted by H-2Kb but not by H-2Db. The only predicted epitopes thatstimulated these T cell lines were mLama4 and mAlg8 (Extended DataFig. 3b), but not their wild-type forms (Extended Data Fig. 4a).
Four subsequent findings supported the conclusion that mLama4 andmAlg8 were the relevant antigens responsible for anti-PD-1-inducedrejection of d42m1-T3. First, mLama4 or mAlg8 epitopes stabilizedH-2Kb expression on RMA-S cells, which lack a functional antigen trans-porter and thus fail to stably express MHC class I proteins on the cellsurface (Extended Data Fig. 4b). Second, both epitopes were detectedby mass spectrometry in eluates of affinity-purified H-2Kb isolated fromd42m1-T3 tumours. Using a discovery mass spectrometry approach,we identified mLama4 in the H-2Kb eluate (Extended Data Fig. 5a) andverified its identity using an isotope-labelled synthetic mLama4 peptide
Days post-transplant
Ep
itop
e p
red
ictio
ns
40
20
40
20
Affi
nity v
alu
e
mSbf2(V511L)6430548M08Rik(E392K)
mAlg8(A506T)
mLama4(G1254V)
mAlg8(A506T)
0
0
d
b
Affi
nity v
alu
e
Mutant H-2Kb epitopes
To
p 6
2 e
pito
pesFilte
red
VGFNFRTL
mLama4(G1254V)
ITYTWTRL
Un
filte
red
emLama4(G1254V)
mAlg8(A506T)
20
10
0
IF
N-γ
+T
NF
-α+ (%
) 20
10
0
f mLama4(G1254V)
mAlg8(A506T)
Tet+
(% o
f C
D8
+)
Tetra
mer s
tain
ing
40
20
0
All p
red
icte
d
H-2
Kb e
pito
pesA
ffin
ity v
alu
e
c
CD
8+ T
ILC
D8
+ TIL
Functio
nal a
naly
sis
d42m1-T3
Mean
dia
mete
r (m
m)
Days post-transplant
03020100 40 302010 40
20
30
10
F244a
0
Mean
dia
mete
r (m
m)
0
20
30
10
Figure 1 | Mutations in Lama4 and Alg8 formtop predicted d42m1-T3 epitopes. a, Growthof d42m1-T3 or F244 tumours in five-mousecohorts treated with anti-PD-1 (closed circles),anti-CTLA-4 (open circles), anti-PD-11anti-CTLA-4 (open triangle) or control mAb (closedtriangle). b, Potential H-2Kb binding epitopespredicted by in silico analysis of all missensemutations in d42m1-T3. c, Median affinity valuesfor the top 62 predicted H-2Kb epitopes. d, Medianaffinity values of H-2Kb epitopes after filtering.e, Screening for specificities of CD81 TIL fromanti-PD-1-treated, d42m1-T3-tumour-bearingmice using H-2Kb tetramers loaded with top 62H-2Kb epitopes. f, IFN-c and TNF-a induction inCD81 TIL from anti-PD-1-treated, d42m1-T3-tumour-bearing mice following culture withirradiated splenocytes pulsed with the top62 H-2Kb peptides. Data are presented as per centCD81 TIL expressing IFN-c, TNF-a or forboth. Data are representative of twoindependent experiments.
RESEARCH LETTER
5 7 8 | N A T U R E | V O L 5 1 5 | 2 7 N O V E M B E R 2 0 1 4
Macmillan Publishers Limited. All rights reserved©2014

(Extended Data Fig. 5b). We also found more than 200 wild-type pep-tides associated with H-2Kb (Supplementary Table 1), but we have noevidence that any of these function as d42m1-T3 antigens. Mutant Alg8,wild-type Lama4 and wild-type Alg8 peptides were not detected (Sup-plementary Table 1). In contrast, using the more sensitive targeted selectedreaction monitoring (SRM) mass spectrometry method, both mLama4and mAlg8 peptides were identified in the H-2Kb eluate (Fig. 2a, Ex-tended Data Fig. 6a and Supplementary Data 1). Notably, mLama4 andmAlg8 were the only predicted strong-binding mutant epitopes found.Peptides from wild-type Lama4 or wild-type Alg8 were not detected.Neither mLama4 nor mAlg8 were detected in H-2Kb eluates from F244cells (Extended Data Fig. 6b). Third, as detected by staining with H-2Kb
-mLama4 or -mAlg8 tetramers, CD81 T cells specific for either antigenaccumulated temporally in d42m1-T3 tumours of anti-PD-1-treatedmice, reaching maximal values just before tumour rejection (Fig. 2b andExtended Data Fig. 7a). No mLama4- or mAlg8-tetramer-positive TILwere observed in F244 sarcomas from anti-PD-1-treated mice. Fourth,the two H-2Kb-restricted epitopes induced antigen-specific CD81 T
cell responses in naive mice when injected together with polyinosinic-polycytidylic acid (poly(I:C)) as assessed by ELISPOT (Fig. 2c).
Since mLama4- and mAlg8-specific T cells were linked to anti-PD-1-induced d42m1-T3 rejection we asked whether a therapeutic vaccinecomprised of these antigens could protect against tumour outgrowth.When 10-member groups of mice bearing established d42m1-T3 tumourswere vaccinated with the combination of mLama4 (28-mer) and mAlg8(21-mer) synthetic long peptides (SLPs) with poly(I:C), 9 rejected theirtumours compared to control mice vaccinated with irrelevant humanpapilloma virus (HPV) (30-mer) SLP plus poly(I:C) (1/10 mice survived)or poly(I:C) alone (1/10 mice survived) (Fig. 2d and Extended DataFig. 8a). In multiple experiments, mice vaccinated with mLama4 1 mAlg8SLP 1 poly(I:C) displayed an 85% survival (17/20) whereas those treatedwith HPV SLP 1 poly(I:C) or poly(I:C) alone showed 10% (2/20) and15% (3/20) survival, respectively (Fig. 2e). Prophylactic administrationof the combined mLama4 and mAlg8 SLP vaccine induced 88% sur-vival (15/17) (Extended Data Fig. 8b, c). The combined mLama4 andmAlg8 prophylactic SLP vaccine induced superior protection comparedto either SLP alone (Extended Data Fig. 8c) or compared to vaccinescomprised of the minimal 8 amino acid epitopes (Extended Data Fig. 8c).The d42m1-T3-specific vaccines did not prevent outgrowth of unrelatedF244 sarcomas (Fig. 2e and Extended Data Fig. 8c). These results not onlydemonstrate that mLama4 and mAlg8 are major antigenic targets thatmediate checkpoint blockade-induced rejection of d42m1-T3 tumours,but also show that anti-PD-1 or therapeutic SLP vaccines consisting ofthe TSMA targeted by anti-PD-1 are similarly efficacious.
Anti-P
D-1
+
Anti-C
TLA-4
Anti-C
TLA-4
Con
trol
Anti-P
D-1
a 25
12.5
mL
am
a4
-po
sitiv
e
cells
(%
of
CD
8α+
) ****
25
12.5
0
mA
lg8
-po
sitiv
e
cells
(%
of
CD
8α+
)
**
b
0
15
7.5
0 0
mL
am
a4
+ c
ells
p
er
tum
ou
r (×
10
4)
*
**
mA
lg8
+ c
ells
per
tum
ou
r (×
10
4) 15
7.5
* *
Anti-PD-1+
Anti-CTLA-4
mLama4-Tet+ mLama4-Tet–
Anti-
PD-1
Anti-
CTLA-4Control
mAb Anti-PD-1+
Anti-CTLA-4Anti-
PD-1
Anti-
CTLA-4Control
mAb
Anti-PD-1+
Anti-CTLA-4:
Anti-PD-1:
Anti-CTLA-4:
387
25
31
227 160
217
21
c dMetabolism
Cell cycle andeffector memory T-cell effector
pathways
Anti-P
D-1
+
Anti-C
TLA-4
Anti-C
TLA-4
Con
trol
Anti-P
D-1
Figure 3 | Differential effects of checkpoint blockade therapy on tumour-antigen-specific CD81 T cells. a, b, Top, per cent of CD81 TIL specific formLama4 (a) or mAlg8 (b) following checkpoint blockade therapy. Bottom,mean number of mLama4- (a) or mAlg8-specific (b) CD81 TIL per tumourfollowing checkpoint blockade therapy. N 5 5 mice per group pooled. Data aremeans 6 s.e.m. of at least three independent experiments. Samples werecompared to control mAb treatment using unpaired, two-tailed Student’s t test(*P , 0.05, **P , 0.01). c, Venn diagram revealing relationships betweendifferentially expressed genes (P , 0.05) in mLama4-specific CD81 TILfrom mice treated with checkpoint-blocking mAbs versus control mAb. d, Heatmap showing differentially expressed genes (P , 0.05) in mLama4-specificCD81 TIL from mice treated with checkpoint-blocking versus control mAbs.Colour pattern is relative with respect to the row, with red indicating geneupregulation and blue indicating gene downregulation. n 5 15 mice per groupanalysed in triplicate.
25
a
b
wtLama4 (not observed) mAlg8 (observed) wtAlg8 (not observed)
Rt (min)36.4
ReferenceVGFNFRTL
0
Inte
nsity (
×10
3)
0.5
0
Inte
nsity (
×10
3)
80
Rt (min)34.5
0.5
0
Inte
nsity (
×10
3)
18
0.5
0
Inte
nsity (
×10
3)
100
Rt (min)34.9
Rt (min)34.9
Rt (min)34.1
ReferenceGGFNFRTL
ReferenceITYTWTRL
ReferenceITYAWTRL
0.5
c
HPV control
100
Days post-transplant
Per
cent
mo
use s
urv
ival
0
mLama4 + mAlg8
50
100
0 755025
Poly(I:C) alone
Mean
dia
mete
r (m
m)
Days post-transplant
Tet+
(%
of
CD
8α+
) 25
12.5
0
15
7.5
00 2010 30
**
**
**
**
*
mLama4-Kb
mAlg8-Kb
F244 mLama4-Kb
d42m1-T3
F244
d
Anti-PD-1
IFN
-γ s
po
t fo
rmin
g c
ells
per
10
6 c
ells
0
2,000
1,000
WT MU
+ +Poly(I:C) + + +
WT MU
Lama4 Alg8 PBS
–
**
**
e
Rt (min)36.3
Per
cent
tum
our
reje
ctio
n
0
50
100
mLama4+
mAlg8F244
mLama4+
mAlg8
HPVcontrol
Poly(I:C)alone
(0/7)
(17/20)
(2/20)(3/20)
mLama4 (observed)
Figure 2 | Mutant Lama4 and mAlg8 are therapeutically relevant d42m1-T3TSMA. a, Detection of mLama4 and mAlg8 bound to cellular H-2Kb bymass spectrometry. Rt, retention time. b, Top, time-dependent tumourinfiltration of mLama4- and mAlg8-specific CD81 T cells (n 5 5). Datarepresent means 6 s.e.m. of 5 independent experiments. Bottom, growthkinetics of d42m1-T3 and F244 during anti-PD-1 immunotherapy (n 5 5).Data represent average tumour diameter 6 s.e.m. and are representative of atleast three independent experiments. c, IFN-c ELISPOT analysis of peptide-stimulated splenocytes from mice immunized with mLama4 or mAlg8 SLP pluspoly(I:C) (n 5 3 mice per group). Data are means 6 s.e.m. Representative oftwo independent experiments. Samples were compared using unpaired,two-tailed Student’s t test (*P , 0.05, **P , 0.01). d, Kaplan–Meier survivalcurves of d42m1-T3-tumour-bearing mice (10 mice per group) therapeuticallyvaccinated with SLP vaccines plus poly(I:C). mLama4 plus mAlg8 comparedto HPV control: P 5 0.0002 (log-rank (Mantel–Cox) test). Representative oftwo independent experiments. e, Cumulative data from two independent SLPtherapeutic vaccine experiments using mice (7–10 per group) with d42m1-T3or F244 tumours.
LETTER RESEARCH
2 7 N O V E M B E R 2 0 1 4 | V O L 5 1 5 | N A T U R E | 5 7 9
Macmillan Publishers Limited. All rights reserved©2014

Since the aforementioned analyses were conducted using anti-PD-1-treated mice bearing d42m1-T3 tumours, we asked whether the presenceof mLama4- and mAlg8-specific T cells in the TIL population was depen-dent on checkpoint blockade therapy. T cells specific for mLama4 ormAlg8 were detected in mice treated with control mAb or anti-PD-1 and/or anti-CTLA-4 (Fig. 3a, b and Extended Data Fig. 7b). The percentageand total number of mLama4- or mAlg8-specific CD81 TIL were similarin tumours from control mAb- and anti-PD-1-treated mice but wereelevated in mice treated with either anti-CTLA-4 or anti-CTLA-4 1
anti-PD-1.The observation that mLama4- and mAlg8-specific T cells were found
in tumours from mice treated with either control mAb or checkpointblockade mAbs prompted us to assess the resultant changes in the TILpopulation following anti-PD-1 and/or anti-CTLA-4 treatment. Weused RNA sequencing (RNA-Seq) to assess gene expression in freshlyisolated, mLama4-H-2Kb-tetramer1 TIL from groups of tumour-bearingmice treated with control mAb, anti-PD-1, anti-CTLA-4 or anti-PD-1 1 anti-CTLA-4. Since mLama4-specific T cells were seven times moreabundant in d42m1-T3 tumours than mAlg8-specific T cells in this seriesof experiments, we restricted our analysis to the former. Only a subsetof 25 genes was commonly regulated (either up or down) by treatmentwith anti-PD-1 and/or anti-CTLA-4 (Fig. 3c and Extended Data Table 1a).This group included a subset of genes whose enhanced expression issimilar to that observed in CD81 T cells from mice during acute second-ary viral infection and depressed in a manner similar to that of exhausted
CD81 T cells in chronic viral infection16 (Fig. 3c, Extended Data Table 1aand Supplementary Table 2). In contrast, antigen-specific CD81 TILisolated from anti-PD-1 and/or anti-CTLA-4 treated mice displayedmostly treatment-specific alterations of non-overlapping sets of genesinvolved in CD81 T cell effector functions (Supplementary Table 2).The effects of checkpoint blockade on gene expression were predomi-nantly observed on TSMA-specific T-cells and not in other CD81 TIL(Fig. 3d).
To determine which pathways were regulated by the different check-point blockade therapies, we performed gene set enrichment analysis(GSEA) using canonical-pathway- and immunological-signature-databases.When compared to mLama4-specific TIL from control mAb-treatedmice, tumour-antigen-specific TIL from mice treated with anti-PD-1and/or anti-CTLA-4 displayed a common set of alterations involvingeffector function, MAPK, chemokine and cytokine receptor signalling(Extended Data Table 1b and Supplementary Table 3). In contrast,mLama4-specific TIL from mice treated with anti-PD-1, anti-CTLA-4or both mAbs displayed profound treatment-specific pathway altera-tions (Extended Data Table 1b and Supplementary Table 3). Treatmentwith anti-PD-1 produced metabolic changes including those involvingoxidative phosphorylation, glycolysis, respiratory electron transport,tricarboxylic acid cycle and pentose phosphate pathways, as well as inpathways involved in IL-2 signalling. These cells also displayed a profileconsistent with response to type I IFN. Treatment with anti-CTLA-4increased NFAT and JAK-STAT signalling pathway activity, cellularproliferation/cell cycle, and activation of effector T cells. Treatment withboth anti-CTLA-4 and anti-PD-1 induced a synergistic pattern of meta-bolic and effector T-cell-specific functions, including those involvingT-cell-mediated anti-tumour activity. This was reflected in the mostsignificant enhancement of effector molecules such as IFN-c, GranzymeA, Granzyme B, and Fas ligand (Supplementary Table 2). Thus, whereasblockade of different inhibitory co-stimulators leads to a common bio-logical outcome—tumour eradication—the precise mechanisms by whichthis outcome is achieved differ.
We also assessed changes in expression of functionally relevant pro-teins on/in CD81 TIL in mice undergoing treatment with differentcheckpoint-blocking mAbs. TIL specific for mLama4 or mAlg8 frommice treated with anti-PD-1 and/or anti-CTLA-4 displayed lower cellsurface expression of lymphocyte-activation gene 3 (LAG-3) and T cellimmunoglobulin and mucin protein 3 (TIM-3) than those in progres-sively growing tumours in control mAb-treated mice (Fig. 4a and Ex-tended Data Fig. 9a, b). Elevated LAG-3 and TIM-3 expression is knownto mark antigen-experienced, dysfunctional (that is, exhausted) CD81
T cells16,17 in chronic viral infection. Conversely, TIL specific for mLama4or mAlg8 from anti-CTLA-4- or anti-CTLA-4 1 anti-PD-1-treated micedisplayed significantly higher levels of Granzyme B than antigen-specificTIL from mice treated with either anti-PD-1 alone or control mAb(Fig. 4b). Consistent with the RNA-Seq analysis, these changes wereobserved predominantly in antigen-specific TIL. In addition, whereasa low percentage of CD81 TIL from mice treated with control mAbproduced IFN-c and TNF-a (Fig. 4c and Extended Data Fig. 9c), thepercentage of IFN-c-producing TIL increased following treatment ofthe mice with anti-PD-1 and particularly with anti-CTLA-4 or the com-bination of anti-CTLA-4 1 anti-PD-1. TIL expressing both cytokines,which are likely to represent the most potent anti-tumour effectors,were most highly represented following treatment of tumour-bearingmice with the combination of anti-CTLA-4 1 anti-PD-1.
This report documents that TSMA are targets of checkpoint block-ade immunotherapy and can be used in vaccines that therapeuticallyinduce tumour rejection as effectively as checkpoint blockade therapy.The ability to rapidly and accurately identify TSMA using genomicsand bioinformatics approaches18–20 and use them to generate MHC tet-ramers to identify tumour-specific T cells provides a significant advan-tage to the fields of tumour immunology and cancer immunotherapy.This approach has not only facilitated the identification of the anti-genic targets of T cells affected by checkpoint blockade therapy21 but
**
b600
300
0
Gra
nzym
e B
MF
I ***
a
LA
G-3
MF
I
400
200
0
*
*
** **
TIM
-3 M
FI
2,000
1,000
0
****
mLama4-Tet+ mLama4-Tet–
mLama4-Tet+ mLama4-Tet–
*
Cyto
kin
e p
ositiv
e
(% o
f C
D8
α+) **
12.5
25
*
0
10
5
0
*
*
c
* *
IFN-γ-positive IFN-γ and TNF-α double-positive TNF-α-positive
Anti-P
D-1
+
Anti-C
TLA-4
Anti-C
TLA-4
Con
trol
Anti-P
D-1
Anti-P
D-1
+
Anti-C
TLA-4
Anti-C
TLA-4
Con
trol
Anti-P
D-1
Anti-P
D-1
+
Anti-C
TLA-4
Anti-C
TLA-4
Con
trol
Anti-P
D-1
Anti-P
D-1
+
Anti-C
TLA-4
Anti-C
TLA-4
Con
trol
Anti-P
D-1
Anti-P
D-1
+
Anti-C
TLA-4
Anti-C
TLA-4
Con
trol
Anti-P
D-1
Anti-P
D-1
+
Anti-C
TLA-4
Anti-C
TLA-4
Con
trol
Anti-P
D-1
Figure 4 | Checkpoint blockade therapy alters the functional phenotypes oftumour-antigen-specific CD81 T cells. a, TIM-3 or LAG-3 expression (MFI,mean fluorescent intensity) on CD81 TIL following checkpoint blockadetherapy. b, Granzyme B expression in CD81 TIL following checkpointblockade therapy. c, Per cent of CD81 TIL positive for IFN-c and/or TNF-afollowing checkpoint blockade therapy. n 5 5 mice per group pooled. Data aremeans 6 s.e.m. of at least three independent experiments. Samples werecompared to control mAb treated mice using an unpaired, two-tailed Student’st test (*P , 0.05, **P , 0.01).
RESEARCH LETTER
5 8 0 | N A T U R E | V O L 5 1 5 | 2 7 N O V E M B E R 2 0 1 4
Macmillan Publishers Limited. All rights reserved©2014

also has provided insights into the molecular changes that occur withinthe tumour-antigen-specific T-cell population that give rise to the anti-tumour effects of anti-PD-1 and/or anti-CTLA-4. Our findings pro-vide some of the first experimental support for the clinical observationsthat (1) checkpoint blockade therapy amplifies, in some cases, pre-existing anti-tumour T-cell responses8,9,21,22; (2) whereas anti-CTLA-4treatment eliminates regulatory T cells, promotes T-cell priming, andrenders the host more susceptible to autoimmunity13,22,23, anti-PD-1promotes T-cell activation acting as a rheostat of immune effectorfunction9,22,24,25; and (3) dual blockade of CTLA-4 and PD-1 is particularlyeffective in promoting enhanced anti-tumour effector functions10,22,26.
The mutational loads of the MCA sarcomas used in this study arehigh and similar to those of ultraviolet- and carcinogen-induced humancancers. For these types of tumours, it is likely that TSMA vaccinestargeting multiple antigens will be possible, thereby providing bettercoverage of the tumour cell population in part due to dealing more effec-tively with tumour heterogeneity27,28. Additionally, the combination ofa TSMA vaccine and checkpoint blockade may facilitate the immunesystem’s ability to recognize less immunogenic TSMA as well as shared,tumour-associated antigens (TAA) via mechanisms that mimic epitope-spreading29. However, recent studies have shown that human tumourscontaining far fewer mutations (for example, 26 mutations) can be sen-sitive to TSMA-based immunotherapy even for tumour-specific anti-gens that are targets for class II restricted CD41 T cells30. Our studythus provides a strong argument to actively pursue the use of TSMA astargets for cancer immunotherapy, as a means to identify patients whowould best benefit from such therapy, and as components of MHC tet-ramers that can be used to identify tumour-specific T cells as biomarkersof successful anti-tumour responses.
Online Content Methods, along with any additional Extended Data display itemsandSourceData, are available in the online version of the paper; references uniqueto these sections appear only in the online paper.
Received 14 May; accepted 22 October 2014.
1. Shankaran, V. et al. IFNc and lymphocytes prevent primary tumour developmentand shape tumour immunogenicity. Nature 410, 1107–1111 (2001).
2. Dunn, G. P., Bruce, A. T., Ikeda, H., Old, L. J. & Schreiber, R. D. Cancerimmunoediting: from immunosurveillance to tumor escape. Nature Immunol. 3,991–998 (2002).
3. Mantovani, A., Allavena, P., Sica, A. & Balkwill, F. Cancer-related inflammation.Nature 454, 436–444 (2008).
4. Grivennikov, S. I., Greten, F. R. & Karin, M. Immunity, inflammation, and cancer. Cell140, 883–899 (2010).
5. Trinchieri, G. Cancer and inflammation: an old intuition with rapidly evolving newconcepts. Annu. Rev. Immunol. 30, 677–706 (2012).
6. Coussens, L. M., Zitvogel, L. & Palucka, A. K. Neutralizing tumor-promoting chronicinflammation: a magic bullet? Science 339, 286–291 (2013).
7. Koebel, C. M. et al. Adaptive immunity maintains occult cancer in an equilibriumstate. Nature 450, 903–907 (2007).
8. Quezada, S. A., Peggs, K. S., Simpson, T. R. & Allison, J. P. Shifting the equilibrium incancer immunoediting: from tumor tolerance to eradication. Immunol. Rev. 241,104–118 (2011).
9. Pardoll, D. M. The blockade of immune checkpoints in cancer immunotherapy.Nature Rev. Cancer 12, 252–264 (2012).
10. Wolchok, J. D. et al. Nivolumab plus ipilimumab in advanced melanoma. N. Engl.J. Med. 369, 122–133 (2013).
11. Hamid, O. et al. Safety and tumor responses with lambrolizumab (anti-PD-1) inmelanoma. N. Engl. J. Med. 369, 134–144 (2013).
12. Topalian, S. L.et al. Safety, activity, and immune correlates of anti-PD-1antibody incancer. N. Engl. J. Med. 366, 2443–2454 (2012).
13. Hodi, F. S. et al. Improved survival with ipilimumab in patients with metastaticmelanoma. N. Engl. J. Med. 363, 711–723 (2010).
14. Matsushita,H.et al.Cancer exome analysis reveals a T-cell-dependent mechanismof cancer immunoediting. Nature 482, 400–404 (2012).
15. Paul, S. et al. HLA class I alleles are associated with peptide-binding repertoires ofdifferent size, affinity, and immunogenicity. J. Immunol. 191, 5831–5839 (2013).
16. West, E. E. et al. Tight regulation of memory CD81 T cells limits their effectivenessduring sustained high viral load. Immunity 35, 285–298 (2011).
17. Wherry, E. J. T cell exhaustion. Nature Immunol. 12, 492–499 (2011).18. Castle, J. C. et al. Exploiting the mutanome for tumor vaccination. Cancer Res. 72,
1081–1091 (2012).19. Robbins, P. F. et al. Mining exomic sequencing data to identify mutated antigens
recognized by adoptively transferred tumor-reactive T cells. Nature Med. 19,747–752 (2013).
20. Fritsch, E. F. et al. HLA-binding properties of tumor neoepitopes in humans. CancerImmunol. Res. 2, 522–529 (2014).
21. van Rooij, N. et al. Tumor exome analysis reveals neoantigen-specific T-cellreactivity in an ipilimumab-responsive melanoma. J. Clin. Oncol. 31, e439–e442(2013).
22. Curran, M. A., Montalvo, W., Yagita, H. & Allison, J. P. PD-1 and CTLA-4 combinationblockade expands infiltrating T cells and reduces regulatory T and myeloid cellswithin B16 melanoma tumors. Proc. Natl Acad. Sci. USA 107, 4275–4280 (2010).
23. Brunner, M. C. et al. CTLA-4-mediated inhibition of early events of T cellproliferation. J. Immunol. 162, 5813–5820 (1999).
24. Keir, M. E., Butte, M. J., Freeman, G. J. & Sharpe, A. H. PD-1 and its ligands intolerance and immunity. Annu. Rev. Immunol. 26, 677–704 (2008).
25. Okazaki, T., Chikuma, S., Iwai, Y., Fagarasan, S. & Honjo, T. A rheostat for immuneresponses: the unique properties of PD-1 and their advantages for clinicalapplication. Nature Immunol. 14, 1212–1218 (2013).
26. Duraiswamy, J., Kaluza, K. M., Freeman, G. J. & Coukos, G. Dual blockade of PD-1and CTLA-4 combined with tumor vaccine effectively restores T-cell rejectionfunction in tumors. Cancer Res. 73, 3591–3603 (2013).
27. Spiotto, M. T., Rowley, D. A. & Schreiber, H. Bystander elimination of antigen lossvariants in established tumors. Nature Med. 10, 294–298 (2004).
28. Wolkers, M. C., Brouwenstijn, N., Bakker, A. H., Toebes, M. & Schumacher, T. N.Antigen bias in T cell cross-priming. Science 304, 1314–1317 (2004).
29. Corbiere, V. et al. Antigen spreading contributes to MAGE vaccination-inducedregression of melanoma metastases. Cancer Res. 71, 1253–1262 (2011).
30. Tran, E. et al. Cancer immunotherapy based on mutation-specific CD41 T cells in apatient with epithelial cancer. Science 344, 641–645 (2014).
Supplementary Information is available in the online version of the paper.
Acknowledgements We are grateful to K. Murphy for the Batf32/2 mice, T. Hansen forproviding MHC class I antibodies and the H-2Kb construct, D. Fremont for the humanb2m construct, and the National Institutes of Health (NIH) Tetramer Core Facility forproducing MHC class I tetramers. We also thank R. Ahmed and M. Hashimoto for themultiplex staining strategy used to define functional and dysfunctional T cells. Wethank A. Bensimon, O. Schubert and P. Kouvonen for instrument maintenance and fortechnical support with the mass spectrometry measurements and R. Vanganipuram,M. Selby and J. Valle for generating and supplying anti-PD-1 and anti-CTLA-4 inendotoxin-free sterile form. We also thank K. Sheehan, P. Allen, G. Dunn and R. Chan forconstructive criticisms and comments, all members of the Schreiber laboratory fordiscussions, and the many members of TheGenome Institute atWashington UniversitySchool of Medicine. We would also like to thank W. Song for his assistance with thebioinformatics approaches, P. Kvistborg for assistance with tetramer combinatorialcoding, and Christopher Nelson for advice with peptide-MHC monomer purification.This work was supported by grants to R.D.S. from the National Cancer Institute (RO1CA043059, U01 CA141541), the Cancer Research Institute and the WWWWFoundation; to R.D.S. and W.E.G. from The Siteman Cancer Center/Barnes-JewishHospital (Cancer Frontier Fund); to W.E.G. from Susan G. Komen for the Cure (Promisegrant); to E.R.M. from the National Human Genome Research Institute; to G.J.F. fromthe National Institute of Health (P50 CA101942, P01 AI054456, P50 CA101942); toA.H.S. from the National Institute of Health (P50 CA101942); and to T.N.S. from theDutch Cancer Society (Queen Wilhelmina Research Award). E.C. is supported by aMarie Curie Intra-European Fellowship within the Seventh Framework Programme ofthe European Community for Research. M.M.G. was supported by a postdoctoraltraining grant (T32 CA00954729) from the National Cancer Institute and is currentlysupportedbyapostdoctoral training grant (IrvingtonPostdoctoral Fellowship) fromtheCancer Research Institute. Aspects of studiesatWashington University wereperformedwith assistance by the Immunomonitoring Laboratory of the Center for HumanImmunology and Immunotherapy Programs and the Siteman Comprehensive CancerCenter.
Author Contributions M.M.G. and R.D.S. were involved in all aspects of this studyincluding planning and performing experiments, analysing and interpreting data, andwriting the manuscript. X.Z. performed peptide binding experiments, helped designand perform the vaccine experiments. H.S., E.C., R.A. and H.-G.R. planned andperformed the mass spectrometry analyses, interpreted the data and were involved inwriting the manuscript. T.N., J.P.W., C.D.A., M.D.V., S.S.K.L. and E.L.P., participated inassessing the phenotypes of the tumour-specific T-cell lines, interpreting the data andin writing the manuscript. M.T. helped generate MHC class I multimers. A.J.K., J.P.A.,G.J.F. and A.H.S. provided mAbs, helped plan the checkpoint blockade therapyexperiments, and contributed to writing the manuscript. T.N.S. helped generate MHCclass I multimers, analysed data and was involved in writing the manuscript. W.-J.K.,G.E.M. and C.J.M.M. produced and purified the synthetic long peptides, participated inthe planningof the vaccine experiments, analysed data andwere involved inwriting themanuscript. J.H. and E.R.M. were responsible for genomic analyses and epitopeprediction and participated in writing the manuscript. W.E.G. contributed to the designand analysis of peptide binding and vaccine experiments and in writing themanuscript. Y.I. and M.N.A were responsible for optimizing the epitope predictionmethod, performing the RNA-sequencing analyses, analysing data and writing themanuscript. R.D.S. oversaw all the work performed.
Author Information RNA-sequencing data are available at Gene Expression Omnibus(GEO) repositoryathttp://www.ncbi.nlm.nih.gov/geo/ (accessionnumberGSE62771).Reprints and permissions information is available at www.nature.com/reprints. Theauthors declare no competing financial interests. Readers are welcome to comment onthe online version of the paper. Correspondence and requests for materials should beaddressed to R.D.S. ([email protected]).
LETTER RESEARCH
2 7 N O V E M B E R 2 0 1 4 | V O L 5 1 5 | N A T U R E | 5 8 1
Macmillan Publishers Limited. All rights reserved©2014

METHODSMice. Wild type and Rag22/2 mice were purchased from Taconic Farms. Batf32/2
mice31 on a 129S6 background were provided by K. M. Murphy and were bred inour specific-pathogen free animal facility. All in vivo experiments used 8–12-week-old male, 129S6 background mice (to match the sex and strain of the tumours)housed in our specific-pathogen free animal facility. All studies were performed inaccordance with procedures approved by the AAALAC accredited Animal StudiesCommittee of Washington University in St Louis.Tumour transplantation. MCA-induced sarcomas used in this study were gen-erated in male 129S6 strain wild-type or Rag22/2 mice and were banked as low-passage tumour cells as previously described1. All cell lines used in this study weretested for mycoplasma. Tumour cells derived from frozen stocks and propagatedin vitro in RPMI media (Hyclone) supplemented with 10% FCS (Hyclone) werewashed extensively, resuspended at a density of 6.67 3 106 cells ml21 in endotoxin-free PBS and then 150ml were injected subcutaneously into the flanks of recipientmice. Tumour cells were .90% viable at the time of injection as assessed by trypanblue exclusion. Tumour growth was quantified by calliper measurements and expressedas the average of two perpendicular diameters. Tumour growth measurementswere performed blinded. Sample size was chosen based on extensive previous workwith this animal model. No randomization was used. For antibody depletion studies,250mg of control mAb (PIP), anti-CD4 (GK1.5) or anti-CD8a (YTS169.4) wasinjected intraperitoneally into mice at day 21 and every 7 days thereafter.MHC class I epitope prediction. All missense mutations for d42m1-T3 were ana-lysed for the potential to form MHC class I epitopes that bind to either H-2Db orH-2Kb molecules. The Stabilized Matrix Method (SMM) algorithm32, the ArtificialNeural Network (ANN) algorithm33, and the NetMHCpan algorithm34 provided bythe Immune Epitope Database and Analysis Resource (http://www.immuneepitope.org) was used to predict epitope processing and binding affinities and the resultswere ultimately expressed as affinity values (1/IC50 3 100; where IC50 is the half-maximum inhibitory concentration). The median affinity value was calculated bytaking the median of the predicted affinity values from SMM, ANN, and NetMHCpanalgorithms. For the prediction of epitope processing, the NetChop algorithm35 avail-able from http://www.immuneepitope.org was used and potential epitopes werefiltered with those with a NetChop score of 0.6 or higher being prioritized. Filterswere also applied to eliminate mutations occurring in hypothetical Riken proteinsand to de-prioritize mutations resulting in affinity values that were less than thatpredicted for the wild-type sequence.Antibodies. Anti-H-2Kb (B8-24-3) and anti-H-2Db (B22/249) antibodies wereprovided by T. H. Hansen (Washington University School of Medicine). Anti-PD-1(rat chimaeric murine IgG1 clone 4H2) and anti-CTLA-4 (murine IgG2b clone 9D9)antibodies were provided by A. J. Korman (Bristol-Myers Squibb). Isotype controlantibodies (mouse IgG2a OKT3 and mouse IgG1 GIR-208) were purchased fromBio X Cell and Leinco Technologies, respectively. Anti-CD4 (GK1.5), anti-CD8a(YTS169.4), anti-IFN-c (H22) monoclonal antibodies and control immunoglobulin(PIP, a monoclonal antibody specific for bacterial glutathione S-transferase) wereproduced from hybridoma supernatants and purified in endotoxin-free form byProtein G affinity chromatography (Leinco Technologies). Fluorescently conju-gated antibodies against CD3e, CD4, CD8, CD45, LAG-3, TIM-3, PD-1, IFN-c,and TNF-awere purchased from BioLegend. For tetramer staining, an antibody toCD8 was purchased from Accurate Chemical (clone CT-CD8a) as recommendedfor use with tetramer staining by the NIH Tetramer Core Facility at Emory. Fc block(anti-CD16/32) was purchased from BD Bioscience.Peptides. All 8-, 9-, and 10-mer peptides were purchased from Peptide 2.0. Allpeptides were HPLC-purified to .95% purity. Peptides for vaccine experimentswere synthesized at ISA Therapeutics B.V. The peptides were synthesized usingsolid phase Fmoc/tBu chemistry on a PTI Prelude peptide synthesizer and purifiedon a Gilson preparative HPLC system to .95% purity. The identity and purity ofthe peptides for vaccine experiments were confirmed with UPLC-mass spectrometryon a Waters Acquity UPLC/TQD system.Checkpoint blockade immunotherapy. Tumour cells were subcutaneously trans-planted into mice at 1 3 106 cells in 150ml into the flank. Mice were treated intra-peritoneally with 200mg anti-PD-1 or anti-CTLA-4, used alone or in combinationon days 3, 6, and 9 post-tumour transplant. For controls, mice were injected with200mg each of IgG2a and IgG1 isotype control antibodies.Generation of CTL lines. To generate the d42m1-T3-specific CTL lines, wild-typemice were injected with 1 3 106 d42m1-T3 tumour cells and treated with anti-PD-1that induced tumour rejection. Alternatively, wild-type mice were injected with1 3 106 d42m1-T3 tumour cells and were not treated with anti-PD-1, leading toformation of progressively growing tumours that were then surgically resected.Fifty days later, these mice were subsequently rechallenged with 1 3 106 d42m1-T3tumour cells, which were then rejected. In the case of either protocol, spleens fromindependent mice that rejected the tumour were harvested two weeks after rejec-tion and CTL lines were established by stimulating 40 3 106 splenocytes with
2 3 106 d42m1-T3 tumour cells pre-treated for 48 h with 100 U ml21 of recom-binant murine IFN-c and then irradiated (100 Gy). Cultures were stimulated twomore times with irradiated, IFN-c stimulated tumour cells plus irradiated spleno-cytes and then CD81 T cells were purified using magnetic beads (Miltenyi Biotec).Peptide binding assay. The peptide-MHC class I binding assay with RMA-S cells36
was performed by incubating RMA-S cells with serial dilutions of peptides for 24 h.Cells were stained with mAbs against H-2Kb and H-2Db followed by secondaryPE-conjugated goat anti-mouse Ig (BD Biosciences) and analysed by flow cytometry.Tetramers. For determining kinetics of mLama4- or mAlg8-specific T-cell infiltra-tion into tumours, H-2Kb tetramers conjugated to phycoerythrin (PE) were pre-pared with mutant Lama4 or Alg8 peptides and produced by the NIH TetramerCore Facility (Emory University). For screening H-2Kb or H-2Db predicted epi-topes, we generated peptide-MHC Class I complexes in house. The peptide–MHCclass I complexes refolded with an ultraviolet-cleavable conditional ligand were pre-pared as described with modifications37. Briefly, recombinant H-2Kb and H-2Db
heavy chains and humanb2 microglobulin light chain were produced in Escherichiacoli, isolated as inclusion bodies, and dissolved in 4 M urea, 20 mM Tris pH 8.0.MHC Class I refolding reactions were performed by dialyzing a molar ratio of heavychain:light chain:peptide of 1:1:8 against 10 mM potassium phosphate, pH 7.4 for48 h. The ultraviolet-cleavable peptide SIINFEJL used to refold H-2Kb was pur-chased from Peptide 2.0. Refolded peptide-MHC class I complexes were capturedby ion exchange (HiTrap Q HP, GE), biotinylated, and purified by gel filtrationFPLC. Ultraviolet-induced ligand exchange and combinatorial encoding of MHCClass I multimers was performed as described38, except that the peptide-MHC mul-timers used for flow cytometry staining were prepared by the addition of titratedamounts of streptavidin-fluorochrome in a 10ml format39.Measurement of IFN-c production. To generate target cells, tumour cells weretreated with 100 U ml21 IFN-c for 48 h and irradiated with 100 Gy before use. CTLcells (20,000 cells) were co-cultured with tumour target cells (20,000 cells) in 96-wellround-bottomed plates for 48 h in a total volume of 200ml. For peptide stimulationof CTL lines, 100,000 naive irradiated (30 Gy) splenocytes were pulsed with various8- or 9-mer peptides at 37u C for 1 h. Splenocytes were washed and 20,000 CTLswere added and the culture was incubated for 48 h. IFN-c in supernatants wasquantified using an IFN-cELISA kit (eBioscience). For blocking assays, 25mg ml21
of anti-H-2Kb (B8-24-3) or anti-H-2Db (B22/249) were added to the target cells(tumours) for 1 h before addition of CTL cells. For IFN-cELISPOT, pre-coated 96-well PVDF filtration plates (Millipore) were washed with PBS and conditioned withmedium containing 10% FCS. Erythrocyte-free single-cell suspensions from thespleen were added in triplicate and incubated for 20 h with 1mM mutant or wild-type peptide. After extensive washes, 1mg ml21 biotinylated detection antibody wasadded. Streptavidin-alkaline phosphatase and 5-bromo-4-chloro-3-indolyl phos-phate/nitro blue tetrazolium (BCIP/NBT) (Moss Substrates) were subsequently usedfor colour development. Plates were scanned and analysed on an ImmunoSpot reader(C.T.L.). Reagents for ELISPOT were purchased from Mabtech.Tumour and spleen harvest. Established tumours were excised from mice, mincedand treated with 1 mg ml21 type IA collagenase (Sigma) in HBSS (Hyclone) for 1 hat 37 uC. Spleens were also harvested, crushed and vigorously resuspended to makesingle-cell suspensions. To remove aggregates and clumps, cells were filtered througha 40-mm strainer.CD81 TIL peptide restimulation. Cells from tumours were enriched for CD81
cells using Miltenyi mouse CD81 enrichment kit by following manufacturer’s pro-tocol. Splenocytes harvested from naive mice were labelled with CFSE and irradiatedat 30 Gy. 100,000 labelled irradiated splenocytes were then pulsed with 1 mM ofpeptide and 100,000 CD81 TIL were subsequently added and incubated at 37 uC.After 1 h, BD GolgiPlug (BD Bioscience) was added and cells were incubated foran additional 4 h. Surface staining was performed and cells were then fixed andpermeabilized with BD Cytofix/Cytoperm kit. Cells were then stained for IFN-cand TNF-a.Isolation of H-2Kb presented peptides. For H-2Kb isolation, sarcoma cell linesd42m1-T3 and F244 were each expanded to 5 3 108 cells. Prior to harvesting, cellswere stimulated with 300 U ml21 IFN-c for 48 h to increase MHC expression. Detach-ment of cells was facilitated by incubation with 100 U ml21 Collagenase IV (Gibco)in PBS for 10 min at 37 uC in a 5% CO2 incubator. Cells were washed twice withPBS and cell pellets were subsequently snap frozen. MHC class I molecules wereisolated as previously described40. In brief, cell pellets were taken up in 1 ml of lysisbuffer (1.2% CHAPS (Applichem, Darmstadt, Germany), 13 Protease InhibitorCocktail (Roche) in PBS) and homogenized by sonication. Lysates were clearedfrom remaining cell debris by centrifugation (2,500g, 30 min) and passing througha 0.2-mm filter (Sartorius, Goettingen, Germany). MHC class I molecules were iso-lated by immunoaffinity purification using H-2Kb-specific antibody Y3 covalentlycoupled to cyanogen bromide-activated Sepharose 4B (GE Healthcare). MHCmolecules were eluted with 0.2% trifluoroacetic acid (TFA) and released peptideswere further isolated by ultrafiltration through centricons with a 10-kDa cut-off
RESEARCH LETTER
Macmillan Publishers Limited. All rights reserved©2014

membrane (Millipore, Schwalbach, Germany). Prior to LC-mass spectrometryanalysis peptides were desalted using C18 Zip Tips (Millipore) according to manu-facturer’s instructions and volumes were adjusted by vacuum centrifugation.Identification of H-2Kb peptides by discovery mass spectrometry. For the dis-covery mass spectrometry41, mass spectrometry analysis was performed on an LTQOrbitrapXL hybrid mass spectrometer (Thermo Fisher Scientific, Waltham, MA,USA) equipped with a nanoelectron spray ion source coupled to a reverse-phaseliquid chromatography UHPLC system (Ultimate 3000 RSLCnano, Dionex, Sunny-vale, CA, USA). Samples were injected onto a 75 mm 3 2 cm trapping column(Acclaim PapMap RSLC, Dionex) at a flow rate of 4ml min21 for 5.75 min. Peptideswere then separated on a 50mm 3 50 cm separation column (Acclaim PapMapRSLC, Dionex) at 50 uC in a column oven with a flow rate of 175 nl min21 and a gra-dient ranging from 2.5 to 32% acetonitrile over 140 min. Data dependent acquisi-tion was enabled using a top five method (the five most abundant ions with chargesof 21 or 31 were selected for fragmentation during each scan cycle). Survey scanswere performed in the Orbitrap at a resolution of 60,000 and a mass range of 350–600 m/z. Peptides were fragmented and analysed in the ion trap using collision-induced dissociation (CID, normalized collision energy 35%, activation time 30 ms,isolation width, 2 m/z). Doubly charged masses for mLama4 and mAlg8 wereprioritized for fragmentation using an inclusion list.
Data processing was performed with Proteome Discoverer 1.3 (Thermo Fisher)and Mascot search engine (Mascot 2.2.04, Matrix Science). Peak lists were searchedagainst Swiss-Prot Mus musculus database (exported 10 September 2013, 16,633reviewed protein sequences) extended with the 62 mutated H-2Kb candidate sequences.Search restrictions were 5 p.p.m. precursor mass tolerance and 0.5 Da fragmentmass tolerance with no enzymatic cleavage specificity selected. Percolator algori-thm was used to evaluate false discovery rate with a target value of 0.05 (5% FDR).Additional post processing filters were an ion score $ 20, search engine rank 1 andpeptide length between 8 and 12 amino acids.Identification of H-2Kb peptides by targeted SRM. To generate the SRM42 assaylibrary, the synthetic peptides were first analysed on an AB Sciex QTRAP 5500mass spectrometer in SRM-triggered MS2 mode. Peptides (PEPotec SRM Grade 2)were synthesized by Thermo Fischer Scientific GmbH (Ulm, Germany). 51 of the62 strong binding H-2Kb peptides were selected for analysis because they hadphysiochemical properties that would allow their detection by mass spectrometryif present. Chromatographic separations of peptides were performed by a nanoLCultra 2Dplus system (Eksigent) coupled to a 15 cm fused silica emitter, 75mm innerdiameter, packed with a Magic C18 AQ 5 mm resin (Michrom BioResources). Pep-tides were loaded on the column from a cooled (4 uC) nanoLC-AS2 autosampler(Eksigent) and separated in 60 min by a linear gradient of acetonitrile (5–35%) andwater, containing 0.1% formic acid at a flow rate of 300 nl min21. The mass spectro-meter was operated in SRM mode, triggering acquisition of a full fragment ionspectrum upon detection of an SRM trace (threshold 1,000 ion counts). SRM acqui-sition was performed with Q1 and Q3 operated at unit resolution (0.7 m/z halfmaximum peak width) with a dwell time of 20 ms for each transition. For eachpeptide the first fragment ion of the y-series with an m/z above the m/z precursor 1 20Thomson (Th), for the doubly charged peptide precursors, were used as triggeringtransitions. Tandem mass spectrometry spectra were acquired in enhanced pro-duct ion mode, using quadrupole (q2) fragmentation, low Q1 resolution, scan speed10,000 Da s21, and an m/z range of 250–1,000. Collision energies (CEs) were cal-culated according to the formulas: CE 5 0.044 3 m/z precursor 1 5.5 for doublycharged precursor ions. Raw data files (wiff) were converted into mzXML formatusing msConvert from ProteoWizard (version 1.6.1455). Qtrap MS2 spectra wereassigned to peptide sequences using the SEQUEST algorithm and PeptideProphet.The software Skyline43 was used to generate a spectral library from the Qtrap mzXMLfiles. Optimal SRM transition conditions (collision energy and fragment ion selec-tion) were determined for individual peptides as previously described by using syn-thetic peptides44. Retention times were extracted from the spectra and convertedinto a system-independent retention time (iRT) using spiked-in calibration pep-tides (Biognosys)45. For each target mutant peptides, a heavy isotope-labelled ref-erence peptides was spiked in the H-2Kb peptide mixture for identification. For eachpeptide, three to seven transitions were monitored for the heavy and light versionand the resulting SRM data were visualized and analysed manually by using thesoftware Skyline. The unequivocal detection of mutant MHC Class I H-2Kb-associatedpeptides was achieved by comparing three properties between the reference heavyisotope-labelled peptide and its endogenous version: (1) their retention time mustbe consistent, (2) they must trigger all SRM transitions concurrently and (3) SRMtransitions must be in the correct abundance hierarchy.Fluorescence-activated cell sorting analysis. For flow cytometry, cells were stainedfor 5 min at room temperature with 500 ng of Fc block (anti-CD16/32) and thenstained with 200 ng of antibodies to CD45, CD4 or CD8a in 100ml of staining buffer(PBS with 2% FCS and 0.05% NaN3 (Sigma)) for 20 min at 4 uC. Propidium iodide(PI) (Sigma) was added at 1mg ml21 immediately before FACS analysis. For tetramer
staining, cells were stained for 5 min at room temperature with 500 ng of Fc block(anti-CD16/32). Cells were then stained with anti-CD8 antibody (clone CT-CD8afrom Accurate Chemical) for 20 min at 4 uC at a 1:100 dilution. Antibodies to CD45and CD3e along with H-2Kb tetramers conjugated to PE were then added at a con-centration of 1:100 for 30 min at 4 uC. For intracellular cytokine staining of tumour-infiltrating lymphocytes, cells were isolated incubated for 5 h with GolgiPlug (BDbioscience). Cells were then harvested, washed, stained with anti-CD3e and CD8afor 15 min, and then fixed and permeabilized with BD fixation and permeabiliza-tion kit (BD Biosciences). Cells with then stained with anti-IFN-c or anti-TNF-a.For sorting of mLama4-specific cells, tumour-infiltrating cells were enriched forCD451 cells using CD45 cell purification magnetic beads (Miltenyi Biotec). CD45-enriched cells were then sorted gating for PI2 CD3e1 CD8a1 mLama4-tetramer-PE1 or PI2 CD3e1 CD8a1 mLama4-tetramer-PE2 cells. Sorting was performedon a BD FACSAria II (BD Biosciences). Sorted cells were pelleted and processed forRNA analysis. The purity of the sorted cells for input RNA was greater than 90% asassessed during post-sort cellular analysis. All flow cytometry was performed onthe FACSCalibur (BD Biosciences) or LSR Fortessa (BD Biosciences) and analysedusing FlowJo software (TreeStar).Vaccination. Mice were vaccinated subcutaneously with 50mg peptides corres-ponding to mLama4 or mAlg8 in combination with 100mg poly(I:C) (Sigma-Aldrich).For SLP, the mLama4 peptide sequence used was QKISFFDGFEVGFNFRTLQPNGLLFYYT (epitope underlined) and the mAlg8 peptide sequence used was AVGITYTWTRLYASVLTGSLV (epitope underlined). Two weeks later spleens were har-vested and splenocytes were restimulated with wild-type or mutant Lama4 or Alg88-mer peptides. Cells were then collected for ELISPOT analysis. For prophylacticvaccination, mice were subcutaneously injected with 50mg of peptides correspond-ing to mLama4 or mAlg8, used alone or in combination, along with 100mg of poly(I:C) in a total volume of 200ml diluted in HBSS on day -10, -3, and 14 post-tumourtransplant. d42m1-T3 tumour cells were transplanted at 1 3 106 tumour cells onday 0. As controls, mice were immunized with either HBSS alone, poly(I:C) aloneor a long-peptide vaccine corresponding to human papilloma virus (HPV) withpoly(I:C). Therapeutic vaccination experiments were performed in identical man-ner except vaccines were administered on day 13, 19, and 115 post-tumour trans-plant. Lack of survival was defined as mouse death or tumour size of 20 mm.RNA sequencing. The purity of the sorted cell preparations were greater than 90%as assessed by post-sort cellular analysis. mRNA was extracted from cell lysatesusing oligo-dT beads (Invitrogen). For cDNA synthesis, we used custom oligo-dTprimers with a barcode and adaptor-linker sequence (CCTACACGACGCTCTTCCGATCT—XXXXXXXX-T15). After first strand synthesis, samples were pooledtogether based on Actb qPCR values and RNA-DNA hybrids were degraded usingconsecutive acid-alkali treatment. Then, a second sequencing linker (AGATCGGAAGAGCACACGTCTG) was ligated using T4 ligase (NEB) followed by SPRI clean-up. The mixture then was enriched by 16 cycles of PCR and SPRI purified to yieldfinal strand specific RNA-seq libraries. The libraries were sequenced on a HiSeq2500 instrument using 50 bp 3 25 bp pair-end sequencing. Second read-mate wasused for sample demultiplexing, at which point individual single-end fastqs files werealigned to mm9 genome using STAR aligner with the following options–runThreadN8–outFilterMultimapNmax 15–outFilterMismatchNmax 6–outReadsUnmappedFastx–outSAMstrandField intronMotif–outSAMtype BAM SortedByCoordinate.Gene expression was quantitated using ht-seq and differentially expressed geneswere defined using DESeq2 R package at P value ,0.01. Gene Set Enrichment Anal-ysis (GSEA) using canonical pathway- and immunological signature-databases wasperformed as previously described46.Statistical analysis. Samples were compared using an unpaired, two-tailed Student’st-test, unless specified.
31. Hildner, K. et al. Batf3 deficiency reveals a critical role for CD8a1 dendritic cells incytotoxic T cell immunity. Science 322, 1097–1100 (2008).
32. Peters, B. & Sette, A. Generating quantitative models describing the sequencespecificity of biological processes with the stabilized matrix method. BMCBioinformatics 6, 132 (2005).
33. Lundegaard, C. et al. NetMHC-3.0: accurate web accessible predictions of human,mouse and monkeyMHC class I affinities for peptides of length8-11. NucleicAcidsRes. 36, W509–W512 (2008).
34. Hoof, I. et al. NetMHCpan, a method for MHC class I binding prediction beyondhumans. Immunogenetics 61, 1–13 (2009).
35. Nielsen, M., Lundegaard, C., Lund, O. & Kesmir, C. The role of the proteasome ingenerating cytotoxic T-cell epitopes: insights obtained from improved predictionsof proteasomal cleavage. Immunogenetics 57, 33–41 (2005).
36. Esquivel, F., Yewdell, J. & Bennink, J. RMA/S cells present endogenouslysynthesized cytosolic proteins to class I-restricted cytotoxic T lymphocytes. J. Exp.Med. 175, 163–168 (1992).
37. Toebes, M. et al. Design anduse of conditionalMHC class I ligands. NatureMed. 12,246–251 (2006).
38. Andersen, R. S. et al. Parallel detection of antigen-specific T cell responses bycombinatorial encoding of MHC multimers. Nature Protocols 7, 891–902 (2012).
LETTER RESEARCH
Macmillan Publishers Limited. All rights reserved©2014

39. Kvistborg, P. et al. TIL therapy broadens the tumor-reactive CD81 T cellcompartment in melanoma patients. OncoImmunology 1, 409–418 (2012).
40. Kowalewski, D. J. & Stevanovic, S. Biochemical large-scale identification of MHCclass I ligands. Methods Mol. Biol. 960, 145–157 (2013).
41. Thommen, D. S. et al. Two preferentially expressed proteins protect vascularendothelial cells from an attack by peptide-specific CTL. J. Immunol. 188,5283–5292 (2012).
42. Domon, B. & Aebersold, R. Options and considerations when selecting aquantitative proteomics strategy. Nature Biotechnol. 28, 710–721 (2010).
43. MacLean, B. et al. Skyline: an open source document editor for creating andanalyzing targeted proteomics experiments. Bioinformatics 26, 966–968 (2010).
44. Lange, V., Picotti, P., Domon, B. & Aebersold, R. Selected reaction monitoring forquantitative proteomics: a tutorial. Mol. Syst. Biol. 4, 222 (2008).
45. Escher, C. et al. Using iRT, a normalized retention time for more targetedmeasurement of peptides. Proteomics 12, 1111–1121 (2012).
46. Subramanian, A. et al. Gene set enrichment analysis: a knowledge-basedapproach for interpreting genome-wide expression profiles. Proc. Natl Acad. Sci.USA 102, 15545–15550 (2005).
RESEARCH LETTER
Macmillan Publishers Limited. All rights reserved©2014

Extended Data Figure 1 | Innate and adaptive immune components arerequired for rejection of d42m1-T3 after checkpoint blockade therapy.a, Cohorts of Rag22/2, Batf32/2 or wild-type mice were treated with controlmAb, anti-CD4, anti-CD8a or anti-IFN-c mAbs and then were injectedwith 1 3 106 d42m1-T3 tumour cells subcutaneously and subsequently treatedwith anti-CTLA-4 on days 3, 6 and 9 post-transplant. b, c, d42m1-T3 (b) or
F244 (c) tumour cells were injected subcutaneously into wild-type mice (n 5 5)that were subsequently treated with anti-PD-1 on days 3, 6, and 9. Fiftydays after tumours were rejected, mice were rechallenged with d42m1-T3 orF244 tumour cells. Data are presented as average tumour diameter 6 s.e.m. of5 mice per group and are representative of at least two independentexperiments.
LETTER RESEARCH
Macmillan Publishers Limited. All rights reserved©2014

Extended Data Figure 2 | H-2Db mutant epitopes of d42m1-T3 tumours.a, Missense mutations in d42m1-T3 were subjected to in silico analysis for thepotential to form H-2Db-binding epitopes using three epitope predictionalgorithms. The median predicted epitope-binding affinity for each peptidewas calculated and expressed as ‘median affinity value’ where affinityvalue 5 1/IC50 3 100. Predicted epitopes are arrayed along the x-axis inalphabetical order based on their protein of origin. b, Unfiltered median affinityvalues for the four predicted H-2Db epitopes. c, Median affinity values ofremaining two H-2Db epitopes after filtering. d, Tetramer staining of CD81
TIL from tumour-bearing mice treated with anti-PD-1 using H-2Db tetramersloaded with top 4 H-2Db synthetic peptides. e, IFN-c and TNF-a intracellularcytokine staining of CD81 TIL from tumour-bearing mice treated withanti-PD-1 immunotherapy following co-culture with naive irradiatedsplenocytes pulsed with the top four H-2Db synthetic peptides added at 1mMfinal concentration. Data are presented as per cent of CD81 TIL positive forIFN-c, TNF-a or both cytokines. Data are representative of two independentexperiments.
RESEARCH LETTER
Macmillan Publishers Limited. All rights reserved©2014

Extended Data Figure 3 | mLama4 and mAlg8 stimulate CD81 T cell linesgenerated against d42m1-T3 following anti-PD-1 immunotherapy.a, CD81 T cell lines generated from splenocytes of individual d42m1-T3-tumour-bearing mice that rejected their tumours after anti-PD-1 therapy wereincubated with irradiated d42m1-T3 tumour cells (or F244 tumour cells)treated with blocking mAb specific for H-2Kb, and/or H-2Db and IFN-c
production was quantitated. Data are presented as means 6 s.e.m. and arerepresentative of two independent experiments. Samples were compared usingan unpaired, two-tailed Student’s t test (***P , 0.001). b, IFN-c release bythe CTL 74 T cell line following co-culture with naive irradiated splenocytespulsed with the top 62 H-2Kb synthetic peptides added at 1 mM finalconcentration.
LETTER RESEARCH
Macmillan Publishers Limited. All rights reserved©2014

Extended Data Figure 4 | mLama4 and mAlg8 bind H-2Kb and stimulateCD81 T cell lines generated against d42m1-T3 following anti-PD-1immunotherapy. a, IFN-c release by CTL 62, CTL 73 or CTL 74 T cell linesfollowing stimulation with naive irradiated splenocytes pulsed with wild-typeor mutant forms of Lama4 or Alg8 peptides. b, RMA-S cells were incubated
with 8 amino acid peptides of mLama4 or mAlg8 and surface expression ofH-2Kb or H-2Db was assessed by flow cytometry. Mean fluorescent intensity ofH-2Kb and H-2Db was expressed as peptide binding score. Data presented arerepresentative of at least two independent experiments.
RESEARCH LETTER
Macmillan Publishers Limited. All rights reserved©2014
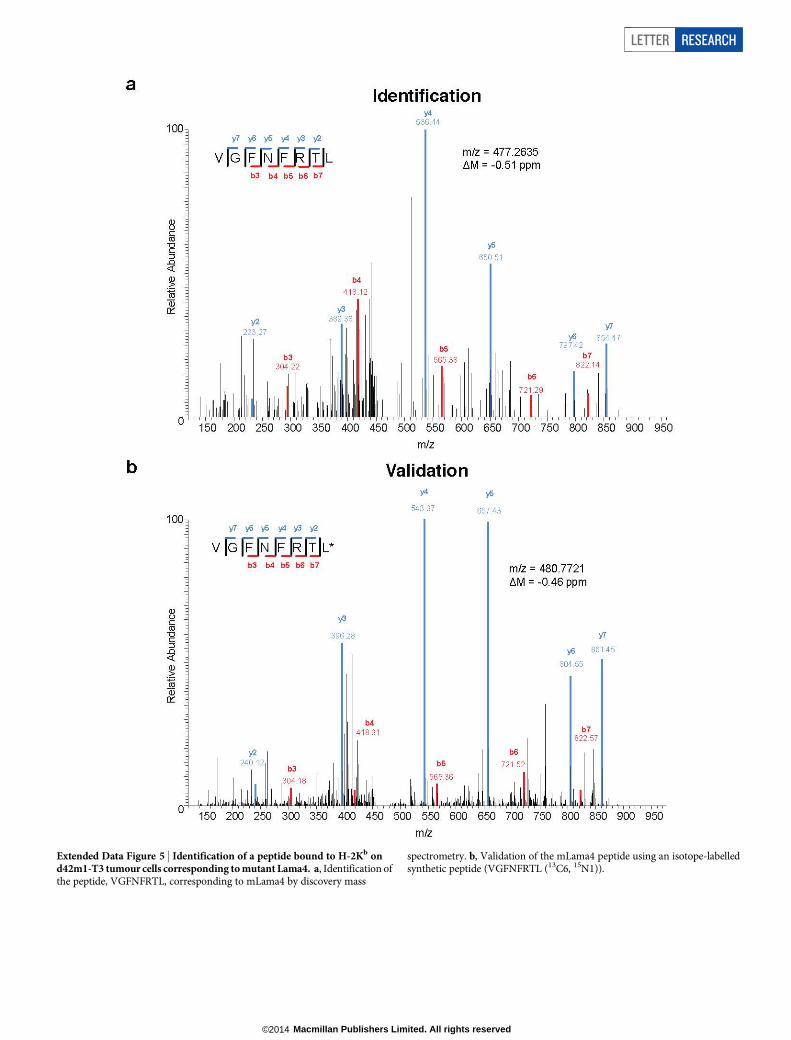
Extended Data Figure 5 | Identification of a peptide bound to H-2Kb ond42m1-T3 tumour cells corresponding to mutant Lama4. a, Identification ofthe peptide, VGFNFRTL, corresponding to mLama4 by discovery mass
spectrometry. b, Validation of the mLama4 peptide using an isotope-labelledsynthetic peptide (VGFNFRTL (13C6, 15N1)).
LETTER RESEARCH
Macmillan Publishers Limited. All rights reserved©2014

Extended Data Figure 6 | Generation of SRM assay library for the detectionof mutant H-2Kb peptides on d42m1-T3. a, SRM transitions were optimizedfor 51 of the 62 top predicted H-2Kb peptides. The 51 peptides chosen wereselected based on having physiochemical properties that would allow theirdetection by mass spectrometry if present. Only Lama4 and Alg8 are shownhere for simplicity. The 51 peptides were synthesized and LC-tandem massspectrometry acquisition was performed on each peptide to determine the bestcollision energy and to obtain the full fragment ion spectrum (left panel);
three to seven of the highest intensity peaks were selected to be built intoSRM transitions. Optimal SRM transitions displayed as extracted ionchromatograms are shown (right panel). Q1–Q3 transitions are indicated inparenthesis. The mutated amino acid in the peptide sequence is marked inred. b, F244 tumour cells, which lack the mLama4 and mAlg8 d42m1-T3epitope, lack detectable mLama4 or mAlg8 in complex with H-2Kb as assessedby SRM.
RESEARCH LETTER
Macmillan Publishers Limited. All rights reserved©2014

Extended Data Figure 7 | CD81 T cells specific for mutant forms of Lama4and Alg8 infiltrate d42m1-T3, but not F244, tumours. a, Detection oftumour-infiltrating mLama4- or mAlg8-specific T cells infiltrating d42m1-T3or F244 tumours of mice treated with anti-PD-1. Tumours were harvestedon day 12 post-transplant. Cells were gated on live CD451 and CD8a1
tumour-infiltrating lymphocytes. Detection of mLama4- or mAlg8-specificT cells was achieved by staining with peptide-MHC H-2Kb PE-labelledtetramers. Data are representative of at least five independent experiments.
b, Detection of mLama4-specific tumour-infiltrating T cells from tumour-bearing mice treated with anti-PD-1, anti-CTLA-4, both anti-PD-1 plusanti-CTLA-4 or control mAb. Detection of mLama4-specific T cells wasachieved by staining with mLama4-MHC H-2Kb PE-labelled tetramers. Datapresented are plotted as the mean mLama4 tetramer-positive as a percent ofCD8a1 tumour-infiltrating cells and are representative of at least threeindependent experiments.
LETTER RESEARCH
Macmillan Publishers Limited. All rights reserved©2014

Extended Data Figure 8 | mAlg8 and mLama4 SLP vaccine controld42m1-T3 tumour outgrowth when administered therapeutically orprophylactically. a, Tumour growth of d42m1-T3 tumours from micetherapeutically vaccinated with mLama4 and mAlg8 SLP plus poly(I:C),HPV control SLP plus poly(I:C) or poly(I:C) alone. Data shown aremean 6 s.e.m. Mutant Lama4 and mAlg8 SLP vaccine group was compared toHPV control SLP vaccine group using an unpaired, two-tailed Student’s t test
(*P , 0.05 and **P , 0.01). b, Kaplan–Meier survival curves of d42m1-T3-tumour-bearing mice (7 per group) prophylactically vaccinated with SLPvaccines plus poly(I:C). mLama4 plus mAlg8 compared to HPV control:P 5 0.0003 (log-rank (Mantel–Cox) test). Representative of two independentexperiments. c, Cumulative number of mice (7–10 per group) from at least twoindependent experiments rejecting d42m1-T3 or F244 tumours as aconsequence of SLP or minimal epitope peptide prophylactic vaccination.
RESEARCH LETTER
Macmillan Publishers Limited. All rights reserved©2014

Extended Data Figure 9 | Detection of TIM-3, LAG-3, IFN-c and TNF-aexpression in tumour-infiltrating CD81 T cells. a, Representative histogramof TIM-3 or LAG-3 expression on mLama4-specific CD81 tumour-infiltratingT cells from tumour-bearing mice treated with anti-PD-1, anti-CTLA-4,both anti-PD-1 and anti-CTLA-4 or control mAbs. b, TIM-3 and LAG-3 arereduced in mAlg8-specific CD81 TIL from tumour-bearing mice treated withanti-PD-1, anti-CTLA-4, or both anti-PD-1 and anti-CTLA-4 compared to
mice treated with control mAb. n 5 5 mice per group pooled. Data arepresented as mean 6 s.e.m. of at least three independent experiments. Sampleswere compared using an unpaired, two-tailed Student’s t test (*P , 0.05,**P , 0.01). c, Representative dot plots of IFN-c and TNF-a stained CD81
tumour-infiltrating T cells from tumour-bearing mice following treatment withanti-PD-1, anti-CTLA-4, both anti-PD-1 and anti-CTLA-4 or control mAbs.Data presented are representative of at least three independent experiments.
LETTER RESEARCH
Macmillan Publishers Limited. All rights reserved©2014

Extended Data Table 1 | Commonly differentially regulated genes and GSEA pathways in mLama41 TIL during checkpoint blockade therapy
a, 25 differentially expressed genes (P , 0.05) that are commonly upregulated (red) or downregulated (blue) in mLama4-specific CD81 TIL from mice treated with all checkpoint blockade mAbs versus TIL fromcontrol-mAb-treated mice. Genes underlined and in bold are those involved in CD81 T effector cells in acute versus chronic infection. b, GSEA pathway analysis showing selected pathways (and their P values)altered in mLama4-specific CD81 TIL during different checkpoint blockade treatments.
RESEARCH LETTER
Macmillan Publishers Limited. All rights reserved©2014