
Human endothelial cells generate Th17 and regulatory T cells under inflammatory conditions

Frontline:
IL-23 and the Th17 pathway promote inflammationand impair antifungal immune resistance
Teresa Zelante1, Antonella De Luca1, Pierluigi Bonifazi1,Claudia Montagnoli1, Silvia Bozza1, Silvia Moretti1, Maria L. Belladonna1,Carmine Vacca1, Carmela Conte1, Paolo Mosci1, Francesco Bistoni1,Paolo Puccetti1, Robert A. Kastelein2, Manfred Kopf3 and Luigina Romani1
1 Luigina Romani, Department of Experimental Medicine and Biochemical Sciences,University of Perugia, Perugia, Italy
2 Schering-Plough Biopharma (formerly DNAX Research, Inc.), Palo Alto, USA3 Institute of Integrative Biology, Molecular Biomedicine, ETH Zurich, Switzerland
Although inflammation is an essential component of the protective response to fungi, itsdysregulation may significantly worsen fungal diseases. We found here that the IL-23/IL-17 developmental pathway acted as a negative regulator of the Th1-mediatedimmune resistance to fungi and played an inflammatory role previously attributed touncontrolled Th1 cell responses. Both inflammation and infection were exacerbated bya heightened Th17 response against Candida albicans and Aspergillus fumigatus, twomajor human fungal pathogens. IL-23 acted as a molecular connection betweenuncontrolled fungal growth and inflammation, being produced by dendritic cells inresponse to a high fungal burden and counter-regulating IL-12p70 production. BothIL-23 and IL-17 subverted the inflammatory program of neutrophils, which resulted insevere tissue inflammatory pathology associated with infection. Our data are the firstdemonstrating that the IL-23/IL-17 pathway promotes inflammation and susceptibilityin an infectious disease model. As IL-23-driven inflammation promotes infection andimpairs antifungal resistance, modulation of the inflammatory response represents apotential strategy to stimulate protective immune responses to fungi.
See accompanying commentary: http://dx.doi.org/10.1002/eji.200737804
Introduction
IL-12 and IL-23 are members of a family of pro-inflammatory heterodimeric cytokines that share acommon p40 subunit linked to the IL-12p35 chain orthe IL-23p19 chain. IL-23 functions through a receptorcomplex composed of the IL-12Rb1 subunit and aunique component, the IL-23R chain [1, 2]. While bothcytokines can induce IFN-c expression in CD4+ T cells,
Correspondence: Luigina Romani, Department of Experi-mental Medicine, and Biochemical Science, University ofPerugia, Via del Giochetto, Perugia 06122, ItalyFax: +39-755-857-411e-mail: [email protected]
Received 20/4/07Revised 28/6/07
Accepted 3/8/07
[DOI 10.1002/eji.200737409]
Key words:Dendritic cells
� Fungal infections� Inflammation� Neutrophils
� T helper cells
Abbreviations: FL-DC: FLT3L-induced dendritic cell �GM-DC: GM-CSF-induced dendritic cell � MMP9: matrixmetalloproteinase 9 � MPO: myeloperoxidase � TLN: thoraciclymph node
Eur. J. Immunol. 2007. 37: 2695–2706 Highlights 2695
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

IL-23, although not involved in Th17 differentiation,plays an important role in maintaining Th17 effectorfunction [3, 4]. Th17 cells are responsible for variousorgan-related autoimmune diseases [5, 6], and bothIL-23 and the Th17 pathway correlate with diseaseseverity and immunopathology in diverse infections[7–9]. These studies suggest that IL-12 and IL-23 havedistinct roles in promoting antimicrobial immuneresponses and diseases in vivo.
In this study, we have investigated the contribution ofIL-23 and the Th17 pathway to inflammation andimmunity to Candida albicans and Aspergillus fumigatus,two major human fungal pathogens. Although inflam-mation is required for a prompt control of fungalinfections, resolution of inflammation is essential formaintaining the balance between protection andimmunopathology in infections and associated diseases[10]. Prolonged inflammation is a hallmark of a widerange of chronic diseases and autoimmunity. ForCandida, the failure to resolve inflammation associatedwith defective fungal clearance characterizes chronicmucocutaneous candidiasis (CMC). Although associatedwith autoimmune polyendocrinopathy-candidiasis-ec-todermal dystrophy, a condition of dysfunctional T cellactivity [11], CMC encompasses a variety of clinicalentities, the immunopathogenesis of which is still largelyunknown [12]. For Aspergillus, the association ofpersistent inflammation with intractable infection iscommon in non-neutropenic patients after allogeneichematopoietic stem cell transplantation [13]. For thelast two decades, the immunopathogenesis of fungalinfections and associated inflammatory diseases hasbeen explained primarily in terms of Th1/Th2 balanceand as affected by a combination of different types ofTreg cells [14–16]. We report here that not only is theTh17 pathway occurring in response to fungi, as recentlydescribed for C. albicans [17, 18], but importantly thatIL-23 and the Th17 pathway acted as a negativeregulator of the Th1-mediated immune resistance toC. albicans and A. fumigatus and played an inflamma-tory role previously attributed to uncontrolled Th1 cellreactivity.
Results
IL-23 and the Th17 pathway mediatesusceptibility to candidiasis
To evaluate the contribution of the IL-23/IL-17 pathwayto C. albicans infection, we comparatively assessedp19–/–, p35–/–, p40–/– and C57BL/6 mice for survival,fungal growth, tissue pathology and parameters ofinflammatory and adaptive Th1/Th17 immunity in micewith gastrointestinal infection. Fig. 1 shows that
resistance to candidiasis was severely impaired inp35–/– mice, more than 50% of which succumbed tothe infection (A) with an elevated fungal growth in thestomach (B). In contrast, despite no differences insurvival (Fig. 1A), the ability to restrict the fungalgrowth was greatly increased in p19–/– mice ascompared to C57BL/6 or p40–/– mice. Notably, p40–/–
mice, deficient in both IL-12 and IL-23, were lesssusceptible than p35–/– mice and more susceptible thanp19–/– mice to candidiasis, emphasizing the importantrole of IL-12 for immunosurveillance against the fungus[10]. Similar results were observed after the intravenousinjection of 5 � 105 or 106 fungal cells, with a mediansurvival time of 6 � 2 vs. 20 � 3 days and 4 � 2 vs.15 � 3 days, comparing p35–/– vs. p19–/– mice, respec-tively. Histopathological examination of the stomachrevealed the presence of parakeratosis, acanthosis andlimited inflammatory reaction in C57BL/6, p19–/– orp40–/– mice, although p40–/–, and in particular p19–/–,mice showed infiltrates of mononuclear cells. Incontrast, numerous fungal hyphae were present in thekeratinized layer in association with a massive infiltrateof PMN, signs of epithelial necrosis and prominentacanthosis in the stomach of p35–/– mice (Fig.1C). Theseresults suggest that the IL-23- and IL-12-dependentpathways have divergent roles in candidiasis.
To correlate these findings with patterns of IL-12/Th1 and IL-23/Th17 immune responses, mice wereassessed for levels of cytokines in the stomach, p35, p19,frequencies of mesenteric lymph node (MLN) IFN-c-,IL-4- or IL-17-producing CD4+ cells, and IL-12b2R andIL-23RmRNA expression inMLN. At the site of infection,the levels of IL-12p70/IFN-c were higher and those ofIL-23/IL-17 lower in C57BL6 and particularly p19–/–
mice as compared to p35–/– mice (Fig. 1D). Interestingly,the levels of IL-17 were approximately eight- to tenfoldhigher in supernatants of in vitro stimulated CD3+ cellsfrom the stomachs of p35–/– mice, a finding suggestingthat actual cytokine production in the stomach could befurther increased by infection. In MLN, the levels of p35and IL-12Rb2mRNA, and numbers of IFN-c+ cells, wereincreased in p19–/– mice compared to C57BL/6 mice,demonstrating augmented IL-12/Th1 responses in theabsence of IL-23. In contrast, levels of p19 and IL-23RmRNA and numbers of IL-17+ cells were enhanced inp35–/– mice lacking IL-12. As expected, the number ofIL-4-producing cells was also considerably enhanced inp35–/– mice (Fig. 1E, F). These data demonstrate apredominant Th1 response promoted by IL-12 andlimited by IL-23 in C57BL/6mice. IL-12 suppresses IL-23and IL-17 production and, vice versa, IL-23 inhibits IL-12and IFN-c production, indicating cross-regulation be-tween the IL-23/Th17 and IL-12/Th1 pathways. Thesedata suggest that a heightened IL-23/Th17 responserenders mice highly susceptible to candidiasis.
Teresa Zelante et al. Eur. J. Immunol. 2007. 37: 2695–27062696
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
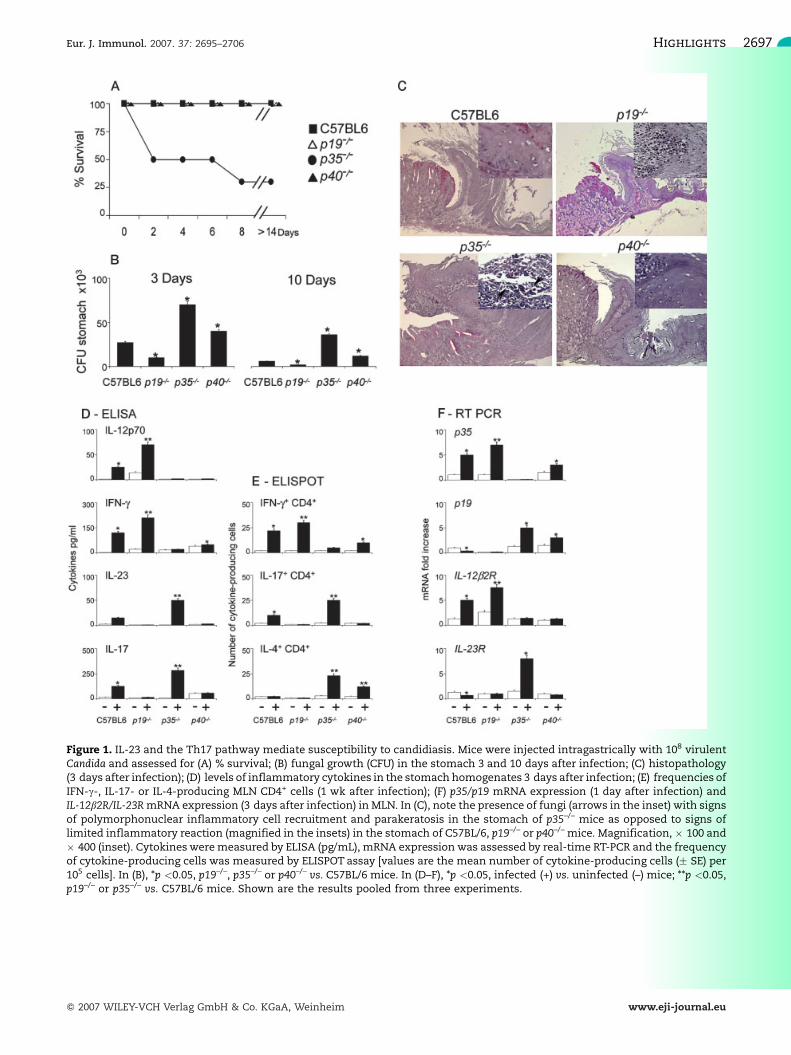
Figure 1. IL-23 and the Th17 pathway mediate susceptibility to candidiasis. Mice were injected intragastrically with 108 virulentCandida and assessed for (A) % survival; (B) fungal growth (CFU) in the stomach 3 and 10 days after infection; (C) histopathology(3 days after infection); (D) levels of inflammatory cytokines in the stomach homogenates 3 days after infection; (E) frequencies ofIFN-c-, IL-17- or IL-4-producing MLN CD4+ cells (1 wk after infection); (F) p35/p19 mRNA expression (1 day after infection) andIL-12b2R/IL-23RmRNA expression (3 days after infection) in MLN. In (C), note the presence of fungi (arrows in the inset) with signsof polymorphonuclear inflammatory cell recruitment and parakeratosis in the stomach of p35–/– mice as opposed to signs oflimited inflammatory reaction (magnified in the insets) in the stomach of C57BL/6, p19–/– or p40–/– mice. Magnification, � 100 and� 400 (inset). Cytokinesweremeasured by ELISA (pg/mL), mRNA expression was assessed by real-time RT-PCR and the frequencyof cytokine-producing cells was measured by ELISPOT assay [values are the mean number of cytokine-producing cells (� SE) per105 cells]. In (B), *p <0.05, p19–/–, p35–/– or p40–/– vs. C57BL/6 mice. In (D–F), *p <0.05, infected (+) vs. uninfected (–) mice; **p <0.05,p19–/– or p35–/– vs. C57BL/6 mice. Shown are the results pooled from three experiments.
Eur. J. Immunol. 2007. 37: 2695–2706 Highlights 2697
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

IL-23 and the Th17 pathway mediatesusceptibility to aspergillosis
To determine whether, similar to candidiasis, theactivation of IL-23 and the Th17 pathway correlatewith susceptibility to aspergillosis, p19–/–, p35–/–, p40–/–
or C57BL/6 mice were comparatively assessed forsusceptibility to pulmonary aspergillosis and parametersof inflammatory and adaptive Th1/Th17 immunity.Fig. 2A shows that the fungal burden was higher inC57BL6 than in the other types of mice, a findingsuggesting that neither IL-12 nor IL-23, alone ortogether, is essential in the early control of the infection.However, the higher fungal growth in p35–/– ascompared to p19–/– mice suggests a superior activityof IL-12 over IL-23 in opposing fungal infectivity.Histopathological examination of the lung revealedthe presence of a mild inflammatory pathology inC57BL/6, p40–/– or p19–/– mice, characterized by few
infiltrates of inflammatory mononuclear cells scatteredin an otherwise intact lung parenchyma. Although thenumber of infiltrating mononuclear cells was higher inp19–/– mice, no signs of parenchymal destruction wereobserved. In contrast, a massive infiltration of PMN(about 8–10-fold increase of Gr-1+CD11c– PMN) waspresent in the lungs of p35–/– mice, associated with signsof extensive interstitial pneumonia (Fig. 2B). Upon mildimmunosuppression, these mice eventually succumbedto the infection as opposed to WTcontrol mice (data notshown). Similar to infection with Candida, expression ofp35/IL-12b2R and p19/IL-23R were cross-regulated ininfection, with the p35 and IL-12Rb2 mRNA being up-regulated in thoracic lymph nodes (TLN) of p19–/– miceand the p19 and IL-23R mRNA being up-regulated inTLN of p35–/– mice compared to C57BL/6 mice. Incontrast, the absence of both IL-12 and IL-23 in p40–/–
mice did not significantly alter the expression of eitherp35 or p19 or their receptors IL-12b2 or IL-23R (Fig. 2C).
Figure 2. IL-23 and the Th17 pathway mediate susceptibility to aspergillosis. Mice were infected intranasally with 2 � 107
Aspergillus resting conidia and evaluated for (A) fungal growth (chitin content, lg glucosamine/organ) in the lung 3 days afterinfection; (B) histopathology (3 days after infection) and (C) p35/p19 (1 day after infection) and IL-12b2R/IL-23R (3 days afterinfection)mRNA expression (RT-PCR) in TLN. In (B), note the presence of amild inflammatory pathology in C57BL/6, p40–/– or p19–/–
mice, characterized by few infiltrates of inflammatorymononuclear cells (magnified in the inset) scattered in an otherwise intactlung parenchyma as opposed to the massive infiltration of PMN in the lung of p35–/– mice associated with signs of interstitialpneumonia. Magnification, � 100 and � 400 (insets). In (A), *p <0.05, p19–/–, p35–/– or p40–/– vs. C57BL/6 mice. In (C), *p <0.05,infected (+) vs. uninfected (–) mice; **p <0.05, p19–/– or p35–/– vs. C57BL/6 mice. Shown are the results pooled from threeexperiments.
Teresa Zelante et al. Eur. J. Immunol. 2007. 37: 2695–27062698
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

Moreover, the numbers of IFN-c+ and IL-17+ CD4+
T cells were increased in p19–/– and p35–/– mice,respectively, at day 7 after infection (data not shown).In the lung, the levels of IL-12p70 were much higher inp19–/– (554 � 44 pg/mL) than in C57BL/6 mice(68 � 8 pg/mL), and IL-23 could be detected only inp35–/– mice (79 � 11 pg/mL). In contrast, IL-17 wasincreased in p35–/– (246 � 17 pg/mL) compared toC57BL/6 (37 � 7 pg/mL) mice. As in p35–/– mice withcandidiasis, the levels of IL-17 increased up to 1238 pg/mL on stimulation of lung cells in vitro with anti-CD3mAb. These data suggest that a heightened IL-23/IL-17-dependent inflammatory response is also associatedwith susceptibility to aspergillosis.
IL-23 and the Th17 pathway are causally linkedto susceptibility to fungal infections
To causally link the increased susceptibility to candi-diasis or aspergillosis with a heightened IL-23/IL-17pathway, we assessed fungal growth and the pattern ofTh1/Th17 cell development in C57BL6 mice withcandidiasis or aspergillosis upon IL-23 or IL-17 block-ade. Blockade of IL-23 and IL-17 greatly increasedresistance to both infections, as judged by a decreasedfungal growth in the relevant target organs (Fig. 3A).Either treatment greatly ameliorated signs of inflamma-tion, both clinically and at the tissue level, and decreasedPMN influx (data not shown). Resistance to bothinfections correlated with an increased frequency ofIFN-c+ Th1 cells, a decreased frequency of Th17 cells inthe MLN and a decreased actual production of IL-17(Fig. 3B, C). These results clearly demonstrate that theIL-23/IL-17 pathway confers susceptibility to fungalinfections by inhibition of protective Th1 immunity.
IL-23 and the Th17 pathway provide antifungalresistance in condition of IFN-c deficiency
The above data would suggest that one possiblemechanism through which the IL-23/IL-17 axis deter-mines susceptibility to fungal infections relies on therelative ability to restrain protective Th1 responses. Toformally prove it, blockade of IL-23, by means ofneutralizing antibodies, was done in intragastricallyinfected mice under conditions of either heightened(IL-4–/– mice) or deficient (IFN-c–/– mice) Th1 reactivity[10]. The fungal load was lower in IL-4–/– and higher inIFN-c–/– than BALB/c mice, a finding consistent with theheightened or impaired Th1 reactivity in IL-4–/– orIFN-c–/– mice, respectively. Similar to WT mice, block-ade of IL-23 greatly decreased the fungal burden in thestomach of IL-4–/– mice (Fig. 3D) and concomitantlyincreased the IL-12p70/IFN-c production in MLN (datanot shown), suggesting that both the Th2 and IL-23/
Figure 3. IL-23 and the Th17 pathway are causally linked tosusceptibility to fungal infections. Mice were infected as in thelegends to Fig. 1, 2 and treated with 200 lg of p19- or IL-17-neutralizing antibodies 5 h later. (A) Fungal growth in thestomach or lung of mice with candidiasis (CFU � 103) oraspergillosis, respectively, 3 days after infection. *p <0.05,treated vs. untreated mice. (B) Frequencies of IFN-c- or IL-17-producing CD4+ cells from MLN or TLN from mice withcandidiasis or aspergillosis, respectively, by ELISPOT assay[values are the mean number of cytokine-producing cells(� SE) per 105 cells] and (C) actual IL-17 production (1 wk afterinfection) in culture supernatants of antigen-stimulatedunfractionated MLN or TLN. *p <0.05, infected vs. uninfected(Ct) mice; **p <0.05, treated (+) vs. untreated (–) mice.(D) Candida growth (CFU � 103) in mice treated with p19-neutralizing antibodies as above, 3 days after infection.*p <0.05, treated (+) vs. untreated (–) mice; **p <0.05, IL-4–/–,IFN-c–/–, p35–/– or IFN-c–/–/p35–/– vs. BALB/c mice.
Eur. J. Immunol. 2007. 37: 2695–2706 Highlights 2699
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

Th17 pathway additively antagonize protective anti-fungal Th1 responses. Surprisingly, a beneficial effectwas observed in condition of IFN-c deficiency, as thefungal burden was actually further increased in IFN-c–/–
mice upon IL-23 neutralization (Fig. 3D), and this wasconcomitant with decreased IL-23/IL-17 production(from 229 pg/mL of IL-23 and 279 pg/mL of IL-17 to21 pg/mL of IL-23 and 95 pg/mL of IL-17 upon IL-23blockade). Therefore, in contrast with the observedability of IL-23 to confer antimicrobial protection byactivation of antigen-specific IFN-c-producing CD4+
T cells independently of IL-12p70 [19], our resultssuggest that IL-23 can have a protective role in fungalinfections in the absence of IFN-c. Interestingly, IL-23again turned out to be unfavorable in the absence ofIL-12 or IL-12 together with IFN-c, as demonstrated byreduced fungal burden upon its neutralization in p35–/–
or doubly deficient IFN-c–/–/p35–/– mice (Fig. 3D).These data suggest that the protective role of IL-23 in theabsence of IFN-c occurred through a p35-dependentmechanism.
IL-23 and IL-12 are produced by distinct DCsubsets in response to fungi
It has already been shown that IL-23 is produced byhuman DC in response to Aspergillus in vitro [20]. Weevaluated here whether IL-23 is produced by DC inresponse to C. albicans and how it relates to theproduction of IL-12 and IL-10, two cytokines essentiallyrequired for the induction of protective tolerance to thefungus [15]. To this purpose, we generated bonemarrow DC in the presence of either GM-CSF + IL-4(GM-DC) or FLT3L (FL-DC), which share functionalcharacteristics of sorted conventional or plasmacytoidDC, respectively [21]. DC were stimulated in vitro withyeasts or hyphae of the fungus and assessed for cytokinemRNA expression and protein production. Zymosan andLPS were used as positive controls of GM-DC, and CpG-ODN as a positive control of FL-DC. The results showed adichotomy in the cytokine expression and production bythe two different DC subsets in response to the fungus.RT-PCR analysis revealed that p19 mRNA expressiononly increased in GM-DC in response to yeasts, morethan to hyphae; p35mRNA expression slightly increasedin GM-DC in response to yeasts but, similar to IL-10,greatly increased in FL-DC exposed to hyphae (Fig. 4A).The measurement of actual cytokine production inculture supernatants confirmed that IL-23 was producedby GM-DC in response to yeasts, particularly at highfungus/DC ratios, as well as to zymosan or LPS. Themaximum level of IL-23 production was observed at12 h of incubation (Fig. 4B) and declined thereafter(data not shown). Conversely, both IL-12p70 and IL-10were mainly produced by FL-DC stimulated with
Candida hyphae, LPS or CpG-ODN for 12 h (Fig. 4B),and continued to be elevated thereafter (data notshown). Together, these data suggest that IL-23 isproduced by conventional DC in response to the fungus,particularly in condition of high-level fungal growth andearlier than other directive cytokines. The ability ofdistinct DC subsets to produce directive cytokines inresponse to Candida may thus condition their anti-
Figure 4. IL-23 and IL-12 are produced by distinct DC subsets inresponse to fungi. BonemarrowDC obtained in the presence ofGM-CSF + IL-4 (GM-DC) or FLT3L (FL-DC) were stimulated withfungi and assessed for cytokinemRNA expression by real-timeRT-PCR (A) or for cytokine protein production by ELISA (pg/mL) (B). Zymosan or LPS (10 lg/mL) or CpG-ODN 2006 (0.06 lM)were used as positive controls. (1)DC exposed to yeasts at 10 : 1ratio. *p <0.05, exposed vs. unexposed (None) DC. (C) SplenicCD11c+ DC from p19–/– or p35–/– mice were stimulated withfungi as above before the assessment of cytokines in culturesupernatants. (D) Splenic CD11c+ DC from C57BL/6 mice wereexposed to fungi for 12 h in the presence (+) or not (–) ofcytokines (10 ng/mL) or neutralizing antibodies (10 lg/mL)before the assessment of cytokines in the culture super-natants.
Teresa Zelante et al. Eur. J. Immunol. 2007. 37: 2695–27062700
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

fungal-promoting immunity in vivo. As a matter of fact,as already shown for Aspergillus [21], Candida-pulsedFL-DC conferred protection and Candida-pulsed GM-DCexacerbated the infection upon adoptive transfer intorecipient mice with candidiasis (S. Bozza, unpublishedobservation).
To verify whether IL-12p70 and IL-23 productionsare cross-regulated in response to the fungus, wemeasured IL-12p70 and IL-23 secretion by splenic DCfrom p19–/– and C57BL/6 mice exposed to eitherIL-12p70 or IL-23 or the corresponding neutralizingantibodies. Fig. 4C shows that IL-12p70 and IL-23 areindeed cross-regulated, as the production of IL-12p70was higher in p19–/– DC and that of IL-23 higher inp35–/– DC as compared to C57BL/6 DC. Moreover, theexposure to either IL-12p70 or IL-23 significantlydecreased IL-23 or IL-12p70 secretion, respectively, byWT DC, and the reverse was true in condition of IL-12 orIL-23 neutralization (Fig. 4D). Because RT-PCR revealedthat unstimulated DC express both cytokine receptors(data not shown), these data suggest the existence of aparacrine loop by which IL-12p70 and IL-23 productionby DC is reciprocally regulated.
IL-23 production by DC is regulated by TLR andT cells and is associated with Th17 cell activation
To define the possible TLR dependency of IL-23production in response to fungi, we measured IL-23production in response to yeasts or conidia by GM-DCgenerated from TLR2–/– or TLR4–/– mice as well as fromMyD88–/– and TRIF–/– mice. Fig. 5A shows that both
TLR2 and TLR4 are essential for IL-23 production bysignaling through MyD88, but not TRIF. Notably, IL-23appeared to be even promoted in the absence of TRIF.Therefore, IL-23 is produced by conventional DC inresponse to fungi through the TLR/MyD88-dependentinflammatory pathway.
To define whether T cells may also regulate IL-23production, we assessed the levels of IL-23 produced insupernatants of DC cultured with CD4+ T cells. Theresults clearly showed that IL-23 production was up-regulated in cultures of T cells stimulated with Candida-pulsed DC from C57BL/6 and particularly p35–/– mice(groups 3 and 6; Fig. 5B), a finding suggesting thatactivated Tcells may provide a positive feedback loop forfurther induction of IL-23.
IL-23 and IL-17 impair antifungal effectorfunctions of PMN
PMN are essential in the initiation and execution of theacute inflammatory response to fungi [22]. The findingthat PMN were abundantly recruited to sites ofinfections in the face of a relevant fungal growth incondition of Th17 cell activation led us to hypothesizethat the IL-23/IL-17-dependent pathway could ad-versely affect the antifungal effector functions ofPMN. We evaluated therefore the fungicidal activity ofPMN from either p19–/– or p35–/– mice as well as fromC57BL/6 mice upon the exposure to IL-23 or IL-17,added either alone or together with IFN-c. The killingactivity was significantly increased in p19–/– PMN anddecreased in p35–/– mice as compared to C57BL6 PMN
Figure 5. IL-23 production by inflammatory DC in response to fungi is TLR and T cell dependent. (A) Splenic CD11c+ DC fromdifferent types of mice were exposed to fungi for 12 h before determination of IL-23 production (pg/mL). *p <0.05, exposed vs.unexposed (None) DC. Shown are the results pooled from four experiments. (B) Splenic CD4+ T cells from C57BL/6 or p35–/– micewere cultured in the presence of the corresponding splenic DC either unpulsed (groups 2 and 5) or pulsedwith Candida yeasts (Ag)(groups 3 and 6) for 5 days before cytokine quantification (pg/mL) in culture supernatants by ELISA. Groups 1 and 4, C57BL6 orp35–/– DC stimulated with fungi and no T cells; groups 7 and 8, p35–/– or C57BL/6 CD4+ T cells cultivated with C57BL/6 or p35–/– DC,respectively, in the presence of the fungus. *p <0.05, groups 2, 3 and 7 vs. 1; groups 5, 6 and 8 vs. 4.
Eur. J. Immunol. 2007. 37: 2695–2706 Highlights 2701
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

(Fig. 6A). Before exposing PMN to these cytokines, weverified whether, similar to IL-17R [23], IL-23R was alsoexpressed on murine PMN. Quantitative RT-PCRrevealed that unstimulated PMN express IL-23R, whoseexpression was further increased after stimulation withLPS (data not shown), a finding suggesting that PMN arealso responsive to IL-23. The exposure to either cytokineimpaired the killing activity of WT PMN in a dose-dependent manner and, importantly, even at dosesmimicking actual cytokine production in infected tissues(Fig. 6B). Remarkably, inhibition was still observed inthe presence of IFN-c (Fig. 6C). Therefore, IL-23 andIL-17 negatively regulated the antifungal effectorfunctions of PMN, which may account for the failureof p35–/– mice to efficiently restrict fungal growth asopposed to p19–/– mice.
IL-23 and IL-17 subvert the indoleamine2,3-dioxygenase-dependent anti-inflammatoryprogram of PMN
We have already shown that indoleamine 2,3-dioxygen-ase (IDO) negatively regulates the inflammatoryprogram of PMN against Candida, such that IDOblockade resulted in the promotion of an inflammatorystate of PMN [24]. We evaluated here the effects of bothIL-23 and IL-17 on IDO expression and on theinflammatory response of WT PMN. As IFN-c is onepotent inducer of IDO on PMN [24], we measured theeffect of IL-23 or IL-17 on the IFN-c-mediated IDOactivation on PMN as well as on matrixmetalloproteinase 9 (MMP9)/myeloperoxidase (MPO)production, both linked to IL-17 activation [25], in
Figure 6. IL-23 and IL-17 impair antifungal effector functions and subvert the anti-inflammatory program of PMN. (A) PMN fromC57BL/6, p19–/– or p35–/– mice were assessed for fungicidal activity after incubation with unopsonized yeasts or conidia at 37�C(PMN/fungus ratio of 10 : 1). For fungicidal activity, the percentage of CFU inhibition (mean� SE) is shown. *p <0.05, p19–/– or p35–/–
PMN vs. C57BL/6 PMN. Shown are the results pooled from three experiments. (B) Fungicidal activity of PMN fromC57BL/6 exposedto different concentrations of IL-23 or IL-17. *p <0.05, cytokine-exposed vs. unexposed PMN. (C, E) PMN from C57BL/6 wereexposed to 50 ng/mL IFN-c and/or 100 ng/mL IL-23/IL-17 for 60 min before the assessment of fungicidal activity against Candidayeasts or Aspergillus conidia or MMP9 and MPO production. Production of gelatinase and MPO were assessed by gelatinzymography, andWestern blot analysiswas performed on culture supernatants. The bands show the active 92-kDaMMP9 and the60-kDa MPO. *p <0.05, cytokine-exposed vs. unexposed PMN; **p <0.05, IFN-c + IL-23- or IFN-c + IL-17-exposed PMN vs.IFN-c-exposed PMN. (D) IDO protein expression by Western blotting on PMN exposed to cytokines as described above. IDO-expressing MC24 transfectants and mock-transfected MC22 cells served as positive and negative controls, respectively.
Teresa Zelante et al. Eur. J. Immunol. 2007. 37: 2695–27062702
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

response to fungi. Both cytokines completely antag-onized the induction of IDO by IFN-c (Fig. 6D).Moreover, IL-23 and in particular IL-17 considerablyincreased MMP-9 and MPO (Fig. 6E). Interestingly, theexposure to IL-23 or IL-17 also modified PMN viability,as the number of apoptotic PMN was significantlydecreased upon the exposure to both cytokines (data notshown). This could be a further mechanism by whichinflammation is perpetrated by the Th17 pathway.Therefore, the ability to subvert the inflammatoryprogram of PMN along with the increased netproteolytic load in inflamed tissues may account forthe inflammatory pathology associated with Th17 cellactivation in fungal infections.
Discussion
Prolonged inflammation is a hallmark of a wide range ofchronic diseases and autoimmunity. Before the discoveryof IL-23 and its recently documented role in autoimmuneinflammation [26, 27], IL-12 was thought to beresponsible for overreacting Th1-mediated immuneand autoimmune disorders. This was also true of fungalinfections and diseases where immunoregulation provedto be essential in fine-tuning inflammation anduncontrolled Th1/Th2 antifungal reactivity [10, 11,15]. The results of the present study show that the Th17pathway, and not an uncontrolled Th1 response, isassociated with defective pathogen clearance, failure toresolve inflammation and to initiate protective immuneresponses to Candida and Aspergillus. Thus, the newfindings may serve to accommodate the paradoxicalassociation of chronic inflammatory responses withintractable forms of fungal infections where fungalpersistence occurs in the face of an ongoing inflamma-tion. Both IL-23 and IL-17 impaired the antifungaleffector activities of PMNeven in the presence of IFN-c, afinding suggesting that the Th17 effector pathwayprevails over the Th1 pathway. This may translate intoconcomitant IL-4+ Th2 cell activation, known to bestrictly dependent on high levels of fungal growth [10]and further preventingTh1 functioning. Inaddition,bothcytokines activated the inflammatoryprogramofPMNbyinducing the release of MMP9 and MPO, which likelyaccounts for the high inflammatory pathology and tissuedestruction associated with Th17 cell activation, and bycounteracting the IFN-c-dependent activation of IDO, anenzyme that is highly responsive to signals from themammalian host immune system [28] and is known tolimit the inflammatory status of PMN against fungi [24].
Although signaling through IL-17R is required forPMN recruitment and host defense in bacterial pneu-monia [29], the involvement of IL-17A in inflammatorylung disorders has also been reported [30]. In support of
this observation, our results point to a detrimental effectfor IL-17 on PMN function. In candidiasis, this wouldpredict that the relative contribution of IL-17 to hostdefense might vary depending on the infectious setting.This appeared to be the case, as the decreased influx ofPMN to infected organs accounted for the highsusceptibility of IL-17AR-deficient mice to systemic[31] but not of p19–/– or IL-17-depleted WT mice tomucosal (this study) candidiasis. This is consistent withthe general view that a defect in PMN counts is onemajor predisposing factor to disseminated but notmucosal candidiasis [10]. Even though a comparativeanalysis of susceptibility to systemic or mucosalcandidiasis in IL-17-deficient mice is needed to clarifythis issue, it is quite conceivable that IL-17, by controllingboth the quantity and the quality of PMN effector cells,may impact differently on various infection settings.
As already described for other infections [32, 33], theTh1 or Th17 developmental pathways were reciprocallyregulated in both fungal infections. This finding suggeststhat the occurrence of either pathway in response tofungi is under strict environmental control. Regulationmay occur at different stages. One obvious level ofregulation is represented by IFN-c which is known toregulate the induction of Th17 cells [32, 33]. The IL-23and IL-17 production were indeed heightened incondition of IFN-c deficiency in both infections, andthe number of IFN-c-producing cells increased uponIL-17 neutralization. These data are in line with thenotion that IFN-c is required for IL-12 responsiveness inmice with candidiasis [10]. More important, theproduction of IL-12 was higher in p19–/– DC and thatof IL-23 higher in p35–/– DC, and both cytokines werecross-regulated in WT DC. These findings suggest thatthese cytokines are reciprocally regulated at the level ofDC production [34]. However, because inflammatoryDCmore than tolerogenic DC appear to produce IL-23 inresponse to fungi, this implies that the Th1/Th17balance also depends on the reciprocal regulationbetween DC subsets at different body sites.
The finding that IL-23 is produced in response tofungi in condition of high-threat inflammation, i.e. byinflammatory DC in response to high yeast numbersthrough the TLR/MyD88 pathway, has importantimplications. Not only does it point to IL-23 as animportant molecular link between the inflammatoryprocesses and fungal virulence, but it also establishes ascenario whereby a vicious circle may be at work. Byperpetrating the activation of pathogenic Th17 cells,IL-23 may contribute to the uncontrolled fungal growthand the resulting concomitant activation of non-protective Th2 cells. In addition, as IL-23 secretion byDC was dramatically increased in the presence of Tcells,this finding suggests that activated T cells may provide apositive feedback loop for further induction of IL-23.
Eur. J. Immunol. 2007. 37: 2695–2706 Highlights 2703
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

The above considerations may help to accommodatefungi, either commensal or ubiquitous, within theimmune homeostasis and its dysregulation. If the abilityto subvert the inflammatory program through theactivation of the IL-23/IL-17 axis may eventually leadto immune dysregulation, their ability to activate Tregcells, integral and essential components of protectiveimmunity to either Candida [14] or Aspergillus [15], mayrepresent a mechanism whereby deregulated immunityis prevented. In this regard, a functional antagonismbetween Th17 and Treg cells has been described [2],including the inhibitory role of IL-10 in the developmentof IL-17-producing cells in vivo [35]. It is thereforepossible that a reciprocal developmental pathway for thegeneration of Th17 and Treg cells may also take place infungal infections.
Another important observation of the present studyis that the IL-23/IL-17-dependent pathway may providesome antifungal resistance in condition of IFN-cdeficiency, through a p35-dependent pathway. ThatIL-23 may serve a protective role in condition of IL-12deficiency has already been reported in chroniccryptococcosis [36], mycobacterial infection [20] andacute pulmonary Klebsiella pneumoniae infection [7],where the protection correlated with the ability of IL-23to activate antigen-specific IFN-c-producing CD4+
T cells, independently of IL-12p70, and to recruitPMN mediating pathogen clearance [7]. As a matterof fact, in experimental Helicobacter hepaticus-inducedcolitis, IL-23 has clearly been shown to drive both IFN-c-and IL-17-producing cells [35]. Our results seem tosuggest a further level of cross-regulation between theTh1 and the Th17 pathways in infections that implicatesa p35-dependent pathway in the action of IL-23.Ultimately, the ability of IL-23 to process initialinflammatory danger signals before the onset of theappropriate immune effector functions dominated bythe IL-12-dependent axis [37] is consistent withantagonist as well as collaborative activities betweenthis pair of cytokines.
Collectively, the data presented in this studydemonstrate a previously undefined role for IL-23 andthe Th17 lineage in fungal infections that has importantimplications for mechanisms of host defense, immunehomeostasis and immunity to fungi. Moreover, theyshow a molecular connection between the failure toresolve inflammation and lack of antifungal immuneresistance. The current results might aid in thedevelopment of strategies for immune therapy of fungalinfections that attempt to limit inflammation tostimulate an effective immune response.
Materials and methods
Mice
Female C57BL/6 and BALB/c mice, 8–10 wk old, werepurchased from Charles River (Calco, Italy). HomozygousIL-12p35-, IL-23p19- or IL-12p40-deficient (hereafter referredto as p35–/–, p19–/– or p40–/–), TLR2-, TLR4-, MyD88- or TRIF-deficient (hereafter referred to as TLR2–/–, TLR4–/–, MyD88–/–
or TRIF–/–) mice on the C57BL/6 background were bred underspecific pathogen-free conditions at the Animal Facility ofPerugia University, Perugia, Italy. Breeding pairs of IFN-c–/–/p35–/– mice, on the BALB/c background, were provided by Dr.M. Colombo (IstitutoTumori, Milan, Italy). IFN-c–/– and IL-4–/–
mice, on the BALB/c background, were also bred at the AnimalFacility of Perugia University. Experiments were performedaccording to the Italian Approved Animal Welfare AssuranceA-3143-01.
Fungi, infections and treatments
The origin and characteristics of the C. albicans strain used inthis study have already been described [38]. For gastro-intestinal infection, 108 Candida cells were injected intragas-trically, and quantification of fungal growth was expressed asCFU per organ (mean � SE) as described [38]. For the i.v.infection, mice received different numbers of the fungus/0.5 mL, intravenously. The strain of A. fumigatus and theculture conditions were as described [39]. Mice received2 � 107 Aspergillus resting conidia intranasally twice. Fungiwere suspended in endotoxin-free (Detoxi-gel; Pierce, Rock-ford, IL) solutions (<1.0 EU/mL, as determined by the LALmethod). Quantification of fungal growth was done by thechitin assay and results are expressed as micrograms ofglucosamine/organ. For histology, sections (3–4 lm) ofparaffin-embedded tissues were stained with periodic acid-Schiff reagent. Treatments with 200 lg (as by initial experi-ments) of p19-neutralizing antibody [40] or IL-17-neutralizingmAb (clone 50104, mAb 421; R&D Systems, Space Import-Export srl, Milan, Italy) were done i.p. 5 h after infection.Control mice were injected with PBS as no differences wereobserved between PBS-treated and isotype control-treated(each treatment) animals (n >6 for each group).
Purification of cells
Gr-1+ CD11b+ PMN (>98% pure on FACS analysis) wereisolated from the peritoneal cavity of mice by magnetic bead-activated sorting using Ly-6G MicroBeads and MidiMacs(Miltenyi Biotec, Bergisch Gladbach, Germany). CD4+
T cells were purified from the MLN, TLN and spleens bymagnetic bead-activated sorting using CD4 MicroBeads andMidiMacs (Miltenyi Biotech). DC were obtained from bonemarrow cells cultured in Iscove's modified medium in thepresence of 150 U/mL mouse rGM-CSF (Sigma) and 75 U/mLrIL-4 (R&D Systems) for 7 days to obtain CD11b+ DC or200 ng/mL FLT3L (R&D Systems) for 9 days to obtain FL-DC[21]. Splenic DC (>99% CD11c+ and<0.1% CD3+) consistingof 90–95% CD8–, 5–10% CD8+, and 1–5% B220+ cells) werepurified by magnetic bead-activated sorting using CD11c
Teresa Zelante et al. Eur. J. Immunol. 2007. 37: 2695–27062704
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

MicroBeads and MidiMacs (Miltenyi Biotec). Zymosan fromSaccharomyces cerevisiae (10 lg/mL; Sigma, St. Louis, MO),ultra-pure LPS from Salmonella minnesota Re 595 (10 lg/mL;Labogen, Rho, Milan, Italy) and CpG-ODN 2006 (0.06 lM)were used as described [41].
Pulsing of DC and culture of cells
DC were exposed to live unopsonized fungi, with and without10 ng/mL cytokines or neutralizing antibodies (10 lg/mL), ata 1 : 1 cell/fungus ratio, as described [38, 39]. At 12 h ofculture, cells were harvested for RT-PCR and supernatantswere assessed for cytokine contents by ELISA. Splenic CD4+
T cells (106/mL) were cultured in flat-bottom 96-well plates inthe presence of 5 � 105 Candida-pulsed splenic DC for 5 days,before cytokine quantification in culture supernatants. Un-fractionated MLN or TLN cells were cultured with inactivatedfungi as described [38, 39], before cytokine determination inculture supernatants 5 days later.
Phagocytosis, antifungal effector activity,zymography and MPO assays
Assays of PMN phagocytosis of unopsonized Candida yeasts orAspergillus conidia and fungicidal activity were conducted asdescribed [22]. Results are expressed as the percentage of CFUinhibition (mean � SE). PMN were exposed to differentconcentrations of rIL-17 or rIL-23 (R&D Systems) or to 50 ng/mL IFN-c � IL-23/IL-17 (100 ng/mL) for 12 h before Westernblotting for IDO, or for 60 min before the addition of fungi foran additional 60 min for studies of fungicidal activity andMMP9/MPO determination. Gelatin zymography was per-formed as described [22]. The gelatinolytic activity of MMP9was determined by scanning the lysis band in the 72-kD area.For MPO determination, samples were probed with rabbitpolyclonal anti-human MPO antibody (Calbiochem, SanDiego, CA) and visualized using ECL (Amersham PharmaciaBiotech).
Western blotting for IDO
Immunoblotting was performed with a rabbit polyclonal IDO-specific antibody, as described [24]. The positive controlconsisted of IDO-expressing MC24 transfectants and thenegative control of mock-transfected MC22 cells.
Quantification of cytokines by real-time RT-PCR,ELISA and ELISPOT assay
Real-time RT-PCR was performed using the iCycler iQdetection system (Bio-Rad) and SYBR Green chemistry(Finnzymes Oy, Espoo, Finland). Cells were lysed and totalRNAwas extracted using the RNeasy Mini Kit (QIAGEN, Milan,Italy) and was reverse transcribed with Sensiscript ReverseTranscriptase (QIAGEN) according to the manufacturer'sdirections. PCR primers were obtained from Invitrogen(Carlsbad, CA). The PCR primers used were: forward primer50-CACCCTTGCCCTCCTAAACC and reverse primer 50-CAAGG--CAAGGCACAGGGTCATCATC for mouse IL-12p35; forwardprimer 50-CCAGCAGCTCTCTCGGAATC and reverse primer
50-TCATATGTCCCGCTGGTGC for mouse IL-23p19; forwardprimer 50-CTTCTTAACAGCACGTCCTGG and reverse primer50-GGTCTCAGATCTCGCAGGTCA for IL-12Rb2; forward pri-mer 50-TGAAAGAGACCCTACATCCCTTGA and reverse primer50-CAGAAAATTGGAAGTTGGGATATGTT for IL-23R; forwardprimer 50-CGCAAAGACCTGTATGCCAAT and reverse primer50-GGGCTGTGATCTCCTTCTGC for mouse c-actin. PCR am-plification of the housekeeping c-actin gene was performed foreach sample (triplicates) to control for sample loading and toallow normalization between samples as per the manufac-turer's instructions (Applied Biosystems, Foster City, CA).Water controls were included to ensure specificity. The thermalprofile for SYBR Green real-time PCR was at 95�C for 3 min,followed by 40 cycles of denaturation for 15 s at 95�C and anannealing/extension step of 1 min at 60�C. Each data pointwas examined for integrity by analysis of the amplificationplot. The mRNA normalized data were expressed as relativecytokine mRNA in treated cells compared to that of mock-infected cells. Cytokine content was assessed by ELISA (R&DSystems; for IL-23, eBioscience, Societ� Italiana Chimici,Rome, Italy) on tissue homogenates or supernatants ofcultured cells. The detection limits (pg/mL) of the assayswere <16 for IL-12p70, <30 for IL-23, <10 for IFN-c, <3 forIL-10, <10 for IL-17, and <4 for TGF-b1. AID EliSpot assay kits(Amplimedical, Buttigliera Alta, Turin, Italy) were used onpurified MLN CD4+ T cells co-cultured with Candida-pulsedDC for 3 days to enumerate cytokine-producing cells.
Statistical analysis
The log-rank test was used for paired data analysis of theKaplan–Meier survival curves. Student's t-test or analysis ofvariance (ANOVA) and Bonferroni's test were used todetermine the statistical significance of differences in organclearance and in vitro assays. Significance was defined asp <0.05. The data reported are either from one representativeexperiment out of three independent experiments or pooledfrom three to five experiments. The in vivo groups consisted ofsix to eight mice per group.
Acknowledgements: This study was supported by theNational Research Project on AIDS, contract 30G.28“Opportunistic Infections and Tuberculosis”, Italy, andby the Specific Targeted Research Project “EURAPS”(LSHM-CT-2005), contract number 005223 and “MAN-ASP” (LSHE-CT-2006), contract number 037899 (FP6).
References
1 Trinchieri, G., Pflanz, S. and Kastelein, R. A., The IL-12 family ofheterodimeric cytokines: New players in the regulation of T cell responses.Immunity 2003. 19: 641–644.
2 Bettelli, E. and Kuchroo, V. K., IL-12- and IL-23-induced T helper cellsubsets: Birds of the same feather flock together. J. Exp. Med. 2005. 201:169–171.
3 Veldhoen, M., Hocking, R. J., Atkins, C. J., Locksley, R. M. andStockinger, B., TGF-b in the context of an inflammatory cytokine milieusupports de novo differentiation of IL-17-producing T cells. Immunity 2006.24: 179–189.
Eur. J. Immunol. 2007. 37: 2695–2706 Highlights 2705
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu

4 Stockinger, B. and Veldhoen, M., Differentiation and function of Th17T cells. Curr. Opin. Immunol. 2007. 19: 281–286.
5 Dong, C., Diversification of T-helper-cell lineages: Finding the family root ofIL-17-producing cells. Nat. Rev. Immunol. 2006. 6: 329–333.
6 Harrington, L. E., Mangan, P. R. andWeaver, C. T., Expanding the effectorCD4 T-cell repertoire: The Th17 lineage. Curr. Opin. Immunol. 2006. 18:349–356.
7 Happel, K. I., Dubin, P. J., Zheng, M., Ghilardi, N., Lockhart, C., Quinton,L. J., Odden, A. R. et al., Divergent roles of IL-23 and IL-12 in host defenseagainst Klebsiella pneumoniae. J. Exp. Med. 2005. 202: 761–769.
8 Hunter, C. A., New IL-12-family members: IL-23 and IL-27, cytokines withdivergent functions. Nat. Rev. Immunol. 2005. 5: 521–531.
9 Mangan, P. R., Harrington, L. E., O'Quinn, D. B., Helms,W. S., Bullard, D.C., Elson, C. O., Hatton, R. D. et al., Transforming growth factor-betainduces development of the T(H)17 lineage. Nature 2006. 441: 231–234.
10 Romani, L., Immunity to fungal infections. Nat. Rev. Immunol. 2004. 4:1–23.
11 Ryan, K. R., Lawson, C. A., Lorenzi, A. R., Arkwright, P. D., Isaacs, J. D.and Lilic, D., CD4+CD25+ T-regulatory cells are decreased in patients withautoimmune polyendocrinopathy candidiasis ectodermal dystrophy. J.Allergy Clin. Immunol. 2005. 116: 1158–1159.
12 Lilic, D., New perspectives on the immunology of chronic mucocutaneouscandidiasis. Curr. Opin. Infect. Dis. 2002. 15: 143–147.
13 Ortega, M., Rovira, M., Filella, X., Martinez, J. A., Almela, M., Puig, J.,Carreras, E. andMensa, J., Prospective evaluation of procalcitonin in adultswith non-neutropenic fever after allogeneic hematopoietic stem celltransplantation. Bone Marrow Transplant. 2006. 37: 499–502.
14 Netea, M. G., Sutmuller, R., Hermann, C., Van der Graaf, C. A., Van derMeer, J. W., van Krieken, J. H., Hartung, T. et al., Toll-like receptor 2suppresses immunity against Candida albicans through induction of IL-10and regulatory T cells. J. Immunol. 2004. 172: 3712–3718.
15 Romani, L. and Puccetti, P., Protective tolerance to fungi: The role of IL-10and tryptophan catabolism. Trends Microbiol. 2006. 14: 183–189.
16 Hohl, T. M., Rivera, A. and Pamer, E. G., Immunity to fungi. Curr. Opin.Immunol. 2006. 18: 465–472.
17 Leibundgut-Landmann, S., Gross, O., Robinson, M. J., Osorio, F., Slack,E. C., Tsoni, S. V., Schweighoffer, E. et al., Syk- and CARD9-dependentcoupling of innate immunity to the induction of T helper cells that produceinterleukin 17. Nat. Immunol. 2007. 8: 630–638.
18 Acosta-Rodriguez, E. V., Rivino, L., Geginat, J., Jarrossay, D., Gattorno,M., Lanzavecchia, A., Sallusto, F. and Napolitani, G., Surface phenotypeand antigenic specificity of human interleukin 17-producing T helpermemory cells. Nat. Immunol. 2007. 8: 639–646.
19 Khader, S. A., Pearl, J. E., Sakamoto, K., Gilmartin, L., Bell, G. K., Jelley-Gibbs, D. M., Ghilardi, N. et al., IL-23 compensates for the absence ofIL-12p70 and is essential for the IL-17 response during tuberculosis but isdispensable for protection and antigen-specific IFN-c responses if IL-12p70is available. J. Immunol. 2005. 175: 788–795.
20 Gafa, V., Lande, R., Gagliardi, M. C., Severa, M., Giacomini, E., Remoli,M. E., Nisini, R. et al.,Human dendritic cells following Aspergillus fumigatusinfection express the CCR7 receptor and a differential pattern ofinterleukin-12 (IL-12), IL-23, and IL-27 cytokines, which lead to a Th1response. Infect. Immun. 2006. 74: 1480–1489.
21 Romani, L., Bistoni, F., Perruccio, K., Montagnoli, C., Gaziano, R., Bozza,S., Bonifazi, P. et al., Thymosin a1 activates dendritic cell tryptophancatabolism and establishes a regulatory environment for balance ofinflammation and tolerance. Blood 2006. 108: 2265–2274.
22 Bellocchio, S., Moretti, S., Perruccio, K., Fallarino, F., Bozza, S.,Montagnoli, C., Mosci, P. et al., TLRs govern neutrophil activity inaspergillosis. J. Immunol. 2004. 173: 7406–7415.
23 Yao, Z., Fanslow, W. C., Seldin, M. F., Rousseau, A. M., Painter, S. L.,Comeau, M. R., Cohen, J. I. and Spriggs, M. K., Herpesvirus Saimiriencodes a new cytokine, IL-17, which binds to a novel cytokine receptor.Immunity 1995. 3: 811–821.
24 Bozza, S., Fallarino, F., Pitzurra, L., Zelante, T., Montagnoli, C.,Bellocchio, S., Mosci, P. et al., A crucial role for tryptophan catabolismat the host/Candida albicans interface. J. Immunol. 2005. 174: 2910–2918.
25 Kolls, J. K. and Linden, A., Interleukin-17 family members andinflammation. Immunity 2004. 21: 467–476.
26 Cua, D. J., Sherlock, J., Chen, Y., Murphy, C. A., Joyce, B., Seymour, B.,Lucian, L. et al., Interleukin-23 rather than interleukin-12 is the criticalcytokine for autoimmune inflammation of the brain. Nature 2003. 421:744–748.
27 Langrish, C. L., Chen, Y., Blumenschein, W. M., Mattson, J., Basham, B.,Sedgwick, J. D., McClanahan, T. et al., IL-23 drives a pathogenic T cellpopulation that induces autoimmune inflammation. J. Exp. Med. 2005. 201:233–240.
28 Mellor, A. L. and Munn, D. H., IDO expression by dendritic cells: Toleranceand tryptophan catabolism. Nat. Rev. Immunol. 2004. 4: 762–774.
29 Ye, P., Rodriguez, F. H., Kanaly, S., Stocking, K. L., Schurr, J.,Schwarzenberger, P., Oliver, P. et al., Requirement of interleukin 17receptor signaling for lung CXC chemokine and granulocyte colony-stimulating factor expression, neutrophil recruitment, and host defense.J. Exp. Med. 2001. 194: 519–527.
30 Linden, A., Laan, M. and Anderson, G. P.,Neutrophils, interleukin-17A andlung disease. Eur. Respir. J. 2005. 25: 159–172.
31 Huang, W., Na, L., Fidel, P. L. and Schwarzenberger, P., Requirement ofinterleukin-17A for systemic anti-Candida albicans host defense in mice. J.Infect. Dis. 2004. 190: 624–631.
32 Cruz, A., Khader, S. A., Torrado, E., Fraga, A., Pearl, J. E., Pedrosa, J.,Cooper, A. M. and Castro, A. G., Cutting Edge: IFN-c regulates theinduction and expansion of IL-17-producing CD4 Tcells during mycobacter-ial infection. J. Immunol. 2006. 177: 1416–1420.
33 Park, H., Li, Z., Yang, X. O., Chang, S. H., Nurieva, R., Wang, Y. H., Wang,Y. et al., A distinct lineage of CD4 T cells regulates tissue inflammation byproducing interleukin 17. Nat. Immunol. 2005. 6: 1133–1141.
34 Becker, C., Dornhoff, H., Neufert, C., Fantini, M. C., Wirtz, S., Huebner,S., Nikolaev, A. et al., Cutting Edge: IL-23 cross-regulates IL-12 productionin Tcell-dependent experimental colitis. J. Immunol. 2006. 177: 2760–2764.
35 Kullberg, M. C., Jankovic, D., Feng, C. G., Hue, S., Gorelick, P. L.,McKenzie, B. S., Cua, D. J. et al., IL-23 plays a key role in Helicobacterhepaticus-induced T cell-dependent colitis. J. Exp. Med. 2006. 203:2485–2494.
36 Kleinschek, M. A., Muller, U., Brodie, S. J., Stenzel, W., Kohler, G.,Blumenschein, W. M., Straubinger, R. K. et al., IL-23 enhances theinflammatory cell response in Cryptococcus neoformans infection andinduces a cytokine pattern distinct from IL-12. J. Immunol. 2006. 176:1098–1106.
37 McKenzie, B. S., Kastelein, R. A. and Cua, D. J., Understanding the IL-23-IL-17 immune pathway. Trends Immunol. 2006. 27: 17–23.
38 Montagnoli, C., Bacci, A., Bozza, S., Gaziano, R., Mosci, P., Sharpe, A. H.and Romani, L., B7/CD28-dependent CD4+CD25+ regulatory T cells areessential components of the memory-protective immunity to Candidaalbicans. J. Immunol. 2002. 169: 6298–6308.
39 Montagnoli, C., Fallarino, F., Gaziano, R., Bozza, S., Bellocchio, S.,Zelante, T., Kurup, W. P. et al., Immunity and tolerance to Aspergillusinvolve functionally distinct regulatory T cells and tryptophan catabolism. J.Immunol. 2006. 176: 1712–1723.
40 Belladonna, M. L., Vacca, C., Volpi, C., Giampietri, A., Fioretti, M. C.,Puccetti, P., Grohmann, U. and Campanile, F., IL-23 neutralizationprotects mice from gram-negative endotoxic shock. Cytokine 2006. 34:161–169.
41 Bellocchio, S., Montagnoli, C., Bozza, S., Gaziano, R., Rossi, G.,Mambula, S. S., Vecchi, A. et al., The contribution of the Toll-like/IL-1receptor superfamily to innate and adaptive immunity to fungal pathogensin vivo. J. Immunol. 2004. 172: 3059–3069.
Teresa Zelante et al. Eur. J. Immunol. 2007. 37: 2695–27062706
f 2007 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim www.eji-journal.eu
Copyright © 2022 FDOKUMEN




















