
Evidence in oyster of a plasma extracellular superoxide dismutase which binds LPS

Identification of an Hexapeptide That Binds to a SurfacePocket in Cyclin A and Inhibits the Catalytic Activity of theComplex Cyclin-dependent Kinase 2-Cyclin A*
Received for publication, April 12, 2006, and in revised form, September 15, 2006 Published, JBC Papers in Press, September 25, 2006, DOI 10.1074/jbc.M603511200
Nuria Canela‡, Mar Orzaez§, Raquel Fucho‡, Francesca Mateo‡, Ricardo Gutierrez¶, Antonio Pineda-Lucena§,Oriol Bachs‡1, and Enrique Perez-Paya§�2
From the ‡Departament de Biologia Cellular i Anatomia Patologica, Facultat de Medicina, University of Barcelona, E-08036Barcelona, Spain, the §Departamento de Quımica Medica, Centro de Investigacion Prıncipe Felipe, E-46013 Valencia, the ¶Unitat deRecerca Farmacologica, Institut Municipal d’Investigacio Medica (IMIM), Departament de Ciencies Experimentals i de la Salut,Universitat Pompeu Fabra (UPF), E-08003 Barcelona, and the �Instituto de Biomedicina de Valencia, CSIC, E-46010 Valencia, Spain
The protein-protein complexes formed between differentcyclins and cyclin-dependent kinases (CDKs) are central to cellcycle regulation. These complexes represent interesting pointsof chemical intervention for the development of antineoplasticmolecules. Here we describe the identification of an all D-aminoacid hexapeptide, termed NBI1, that inhibits the kinase activityof the cyclin-dependent kinase 2 (cdk2)-cyclin A complexthrough selective binding to cyclin A. Themechanism of inhibi-tion is non-competitive for ATP and non-competitive for pro-tein substrates. In contrast to the existing CDKs peptide inhib-itors, the hexapeptide NBI1 interferes with the formation of thecdk2-cyclin A complex. Furthermore, a cell-permeable deriva-tive of NBI1 induces apoptosis and inhibits proliferation oftumor cell lines. Thus, the NBI1-binding site on cyclin A mayrepresent a new target site for the selective inhibition of activitycdk2-cyclin A complex.
The interactions between proteins represent points of chem-ical intervention for therapeutic gain in the biological processesassociated with disease. Because of the specificity of individualprotein-protein interactions the modulation of these com-plexes is viewed as a putative source of future new highly selec-tive drugs (1–3). The early experience in the design of peptidesand small non-peptidic compounds that bind to protein inter-faces (2, 4) encouraged the viability of protein-protein interac-tions as target for drug discovery. In particular, there has beenmuch progress in the discovery of small molecule inhibitors ofthe unregulated cell growth that characterize cancer cells (5).Proliferation of eukaryotic cells is under control of a series ofconcerted molecular mechanisms defined as the cell divisioncycle whose progression is tightly governed by members of the
cyclin-dependent kinase family (CDKs)3 (6, 7). Thus, CDKsmediated phosphorylation of the tumor suppressor proteinpRb is required for inactivation of the protein and for the sub-sequent release of E2F transcription factors, thus altering thestatus of E2F-regulated genes from fully repressed to inducedand leading progression through G1 to S phases (8, 9). ThepRb-E2F pathway is frequently deregulated in cancer cells. E2Ftranscription factors are the preferential pRb targets, however,their binding toDNA is also regulated by cdk2-cyclinA activity.E2F phosphorylation by cdk2-cyclin A inhibits its binding toDNA leading to the end of the transcription of E2F-regulatedgenes produced at the beginning of mitosis (10, 11). It has alsobeen shown that E2F overexpression increases cell proliferationbut paradoxically, it simultaneously also triggers apoptosis (12–14). All this information suggests that pharmacologic inhibitionof cdk2-cyclin A might be of potential interest as an antineo-plastic strategy because cancer cells show an increased activityof the E2F due to pRb inactivation. Furthermore, cdk2-cyclin Acomplex is also required for S phase progression regulating thepre-replicative complex formation and activation (15, 16).Moreover cyclin A is revealed as an essential gene becauseits deletion results in embryonic lethality shortly afterimplantation (17, 18).The protein-protein complexes formed between different
cyclins and cdks (we will refer to the complexes here as CDKs)are central to cell cycle regulation. These complexes and theirnatural inhibitors defined as CDK inhibitor proteins (CKIs)have been the object of extensive research. Considerable efforthas been focused on the development of ATP-competitivesmall molecule inhibitors of CDKs (19). However, in general,these compounds have several alternative protein targets thatcompromise their demanded selectivity. Then, new strategiesfor the inhibition of CDKs activity through blocking of sub-strate recruitment (20, 21) andmore recently, through interfer-ence with protein-protein conformational changes (22) havebeen proposed. The cyclin recruitment motif (CRM) is a bind-ing domain for a large number of cdk2-cyclin A substrates, asfor instance, the transcription factor E2F1 (10), the tumor sup-
* This work was supported by Merck Farma y Quımica, S.A. and Spanish Min-istry of Science and Education Grants (MEC) SAF2001-2811 and BIO2004-998, Fundacion Areces, and Centro de Investigacion Prıncipe Felipe. Thecosts of publication of this article were defrayed in part by the payment ofpage charges. This article must therefore be hereby marked “advertise-ment” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
1 To whom correspondence may be addressed: Institut d’InvestigacionsBiomediques August Pi i Sunyer (IDIBAPS), University of Barcelona,c/Casanova, 143 E-08036 Barcelona, Spain. Tel.: 34-934035286; E-mail:[email protected].
2 To whom correspondence may be addressed: Avda. Autopista del Saler, 16,E-46013 Valencia, Spain. Tel.: 34-963289680; E-mail: [email protected].
3 The abbreviations used are: CDK, cyclin-dependent kinase; CKI, CDK inhibi-tor proteins; CRM, cyclin recruitment motif; GST, glutathione S-transferase;PBS, phosphate-buffered saline; TBS, Tris-buffered saline; PARP, poly(ADP-ribose) polymerase; Ac, acetyl; MCM2, minichromosome maintenance 2.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 281, NO. 47, pp. 35942–35953, November 24, 2006© 2006 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
35942 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 47 • NOVEMBER 24, 2006
by on Septem
ber 11, 2008 w
ww
.jbc.orgD
ownloaded from

pressors pRb (23) and p53 (24), the CKIs p21Cip1 and p27Kip1(23, 25) andother substrates asMDM2 (26). All these substratescontain a highly homologous region of 12 amino acids, whichincludes the sequence RXLYY�, where X, Y, and Y� are any andhydrophobic amino acids, respectively, responsible for thebinding to the CRM.We are currently engaged in a discovery program that targets
the complex cdk2-cyclin A with the aim of finding new leadcompounds that could be developed in safety and selectiveantineoplasic drugs. Here we describe the identification of anhexapeptide that inhibits the kinase activity of the cdk2-cyclinA complex through selective binding to cyclin A. The charac-terization of the inhibitory mechanism revealed that thehexapeptide is non-competitive for eitherATPor histoneH1.Acell permeable derivative of the hexapeptide induces apoptosisand inhibits proliferation of tumor cell lines.
EXPERIMENTAL PROCEDURES
Preparation of Synthetic Combinatorial Libraries and Indi-vidual Peptides—The dual defined position library, Ac-OOXXXX-NH2was synthesized in an iterative format using theprocess of divide, couple, and recombine in conjunction withsimultaneous peptide synthesis. All individual peptides wereprepared using simultaneous multiple peptide synthesis. Pep-tide mixtures and individual peptides were solubilized in H2Oor 5% Me2SO/H2O aliquoted and stored at �20 °C. Individualpeptides were characterized by laser desorption time-of-flightmass spectroscopy analyses and purified by preparative reversephase-high performance liquid chromatography.Protein Expression and Purification—Cdk2, cyclin A full-
length, and cyclin A fragment (A1 amino acids 1–88; A2 aminoacids 1–171; A3 amino acids 1–257; A4 amino acids 1–345; A5amino acids 171–432; and A6 amino acids 257–432) cDNAswere cloned into pGEX-6P plasmid; and a fragment of pRb,fpRb (amino acids 792–928), and p21Cip1 were cloned intopGEX-KG-2T plasmid. All were glutathione S-transferase(GST) fusion proteins and were expressed in Escherichia coliBL21(DE3). Cells were grown at 37 °C with shaking (200 rpm)to mid-log phase (A600 � 0.8). Expression was induced by theaddition of isopropyl 1-thio-�-D-galactopyranoside at a finalconcentration of 0.5 mM and the culture was incubated for afurther 4 h at room temperature. Bacteria were harvested bycentrifugation, and the cell pellet was resuspended in NENTbuffer (20 mM Tris, pH 8, 100 mM NaCl, 1 mM EDTA, 0.5%Nonidet P-40 (Igepal®, Sigma)) containing protease inhibitors(1 mM phenylmethylsulfonyl fluoride, 1 �g/ml aprotinin, and10 �g/ml leupeptin). Bacterial cells were lysed by sonication.The insoluble fraction was pelleted by centrifugation and thesupernatant was incubated on a glutathione-Sepharose columnaccording to the manufacturer’s instructions (GE Healthcare).In some cases GSTwas separated from theGST fusion proteinsby digestion with thrombin protease (Sigma) or PreScissionprotease (Amersham Biosciences).Kinase Assays for the Screening—Kinase assays were carried
out in enzyme-linked immunosorbent assay plates that wereblocked with 200 �l of blocking solution (phosphate-bufferedsaline (PBS) containing 1% bovine serum albumin, 0.02%Tween, and 0.02% sodium azide) overnight at 4 °C. Plates were
washed 3 timeswith 100�l of washing solution (PBS containing0.02% Tween and 0.02% sodium azide). Plates were then driedduring 2–4 h at room temperature, and stored at 4 °C. Underthese conditions, they are stable for 1 month. Assays were per-formed in a final volume of 60�l of kinase buffer (25mMHepes,pH 7.4 and 10mMMgCl2) containing 4�g of histoneH1 (RocheApplied Science), 30 �M ATP, 2 mM dithiothreitol, 0.2 �Ci of[�-32P]ATP (Amersham Biosciences, 3000 Ci/mmol, 10 mCi/ml), 800 nM GST-cdk2, and 800 nM GST-cyclin A. Assays werecarried out in the presence or absence of different concentra-tions of peptide mixtures to be checked. An inhibitory controlwas performed adding 800 nM GST-p21 to the reaction media.Mixtures were incubated for 30 min at 37 °C. After incubation,50�l of eachmixturewere filtered in nitrocellulosemembranesplaced in a dot blot apparatus. Samples were washed with 35 �lof 10% trichloroacetic acid, and finally with two washes of 100�l of 10% trichloroacetic acid followed by 100 �l of H2O. Afterthis process, membranes were dried at room temperature. Theradioactivity associated to the membranes was detected with aMolecular Imager FX; Bio-Rad Laboratories, Inc.Determination of cdk1, cdk2, and cdk6 IC50—Assays were
performed in duplicate in a 30-�l final volume containing 2 �gof histone H1 or histone H1-derived peptide, pepH1 (PKTP-KKAKKL) as a substrate and the CDK complex (25 ng of cdk2-cyclin A, 25 ng of cdk2-cyclin E, 20 ng of cdk1-cyclin B1, or 25ng of cdk6-cyclin D3), all purchased to Upstate Biotechnology,in kinase buffer (25 mM Hepes, pH 7.4, 10 mM MgCl2, 2 mMdithiothreitol, 30 �M ATP, and 0.2 �Ci of [�-32P]ATP). Reac-tions were incubated 30 min at 37 °C and then were stopped bytransferring 25 �l of each on Whatman P81 phosphocellulosepaper. The filters were washed three times with 1% phosphoricacid, air dried, and finally counted in liquid scintillation equip-ment (Wallac). In the substrate competitive assays thesequence of peptide inhibitor (referred to as pepH1-T3V) isPKVPKKAKKL.In some assays, the GST-pRb protein fragment, fpRb (amino
acids 792–928), was used as a substrate. In these cases, the reac-tion was stopped by adding 10 �l of Laemmli buffer. Sampleswere solved in a 10% SDS-PAGE gel and then the gel was dried.The radioactivity associated to the gel was detected with aPhosphorImager (GE Healthcare). In the assays describing anon-competitive action to the CRM of cyclin A, the pepp21(FYHSKRRLIFS), derived from p21Cip1 that targets the CRM,was used.Immunoprecipitation and CDK Activity Assays—HCT116
cells were lysed in IP buffer (50 mM Hepes, pH 7.4, 250 mMNaCl, 2.5mMEGTA, 1mMEDTA, 0.1%Tween 20, 0.1mMNaF,10% glycerol, 10 mM �-glycerolphosphate, 1 mM dithiothreitol,0.1 mM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 0.5�g/�l aprotinin, 10 �g/�l leupeptin) for 40 min on ice. Lysates(0.25–0.5mg of protein) were incubated with 1�g of anti-cdk1(Santa Cruz sc-54), anti-cdk2 (Santa Cruz sc-163), or anti-cdk4(Santa Cruz sc-749) overnight at 4 °C. Then, 40 �g of proteinA/G-agarose beads (Pierce) were added and sampleswere incu-bated for 1 h at 4 °C. After three washes in IP buffer and two inkinase buffer (50 mM Hepes, pH 7.4, 2.5 mM EGTA, 10 mMMgCl2) immunoprecipitates were resuspended in a final vol-ume of 30 �l of kinase buffer containing 15 �M ATP, 10 �Ci of
Cyclin A Surface Inhibitors
NOVEMBER 24, 2006 • VOLUME 281 • NUMBER 47 JOURNAL OF BIOLOGICAL CHEMISTRY 35943
by on Septem
ber 11, 2008 w
ww
.jbc.orgD
ownloaded from

[32P]ATP, 2mMdithiothreitol, and 3�g of histoneH1 (for cdk2and cdk1) or 3 �g of pRb (for cdk4 assays) for 30 min at 30 °C.Reactions were stopped by the addition of Laemmli buffer. Thesampleswere then electrophoresed on a 12%SDS-polyacrylam-ide gels and then stained with Coomassie Blue and dried. Theradioactivity associated to the gels was detected with aPhosphorImager.Determination of CK2 Activity—Protein kinase CK2 activity
was assayed as described previously (27) using 4 mg/ml �-ca-sein, and �-casein peptide only phosphorylated by CK2 as sub-strates. One unit of protein kinase activity is defined as theamount that catalyzes the transfer of 1 nmol of phosphate from[�-32P]GTP to �-casein per min at 30 °C.Determination of Mitogen-activated Protein Kinase II
Activity—The activity was measured by the incorporation ofradioactive phosphate into myelin basic protein, catalyzed bythe active mitogen-activated protein kinase 2/Erk2 (recombi-nant protein expressed in E. coli, catalog number 14-550Upstate Biotechnology) following the manufacturer’s protocol.Determination of Glycogen Synthase Kinase 3�, Glycogen
Synthase Kinase 3�Activity—The glycogen synthase kinase 3�,active (recombinant protein expressed in Sf21 cells) catalognumber 14-492, Upstate Biotechnology) and the Phospho-Gly-cogen Synthase Peptide-2 (glycogen synthase kinase 3 sub-strate), catalog number 12-241Upstate Biotechnology as a sub-strate, were used following the manufacturer’s protocol.Fluorescence PolarizationAssays—Fluorescence polarization
measurements were made in black 96-well plates using a Wal-lacVictor2 1420 Multilabel HTS counter with an excitationwavelength of 485 nm and observed emission wavelength of535 nm. The assay was carried out by adding 60 nM of the car-boxyfluorescein NBI1-labeled peptide (CF-NBI1) to differentconcentrations of cyclin A-(171–432) in PBS buffer and incu-bating the samples during 20 min at 30 °C before reading.Surface Plasmon Resonance Experiments—All the surface
plasmon resonance analysis were performed at room tempera-ture using a Biacore 3000 (Biacore International AB). The TAT(YGRKKRRQRRRG) or TAT-NBI1 peptides were immobilizedon a carboxymethylated dextran sensor chip (CM5) using theamine coupling method as described by the manufacturer. Tatpeptide was used to construct the reference surface. Purifiedcdk2, full-length cyclin A, cyclin A fragments, and the NBI1peptide were diluted to various concentrations with HBS-EPbuffer (Biacore International AB) and each sample was injectedseparately over the flow cells at a flow rate of 10 �l/min for180 s. Following a variable dissociation time, final regenerationof the surfaces was performed with a short pulse of 0.2% (w/v)SDS. The interaction between proteins and TAT-NBI1 wasdetected and presented as a sensorgram by plotting resonanceunits against time. The kinetic results were calculated usingBIAEvaluation software, version 3.0.2, and binding curveswerefitted to a simple 1:1 Langmuir reaction model.Cell Lines—HCT116 and HT29 are colon carcinoma cells
cultured in Dulbecco’s modified Eagle’s medium, F-12 supple-mentedwith 10% fetal calf serum. T98-G andA2780, a glioblas-toma and an ovarian carcinoma human cell line, respectively,were grown in RPMI 1640 medium supplemented with 10%fetal calf serum. All media contained 2 mM L-glutamine, 1%
nonessentials amino acids, 1 mM pyruvic acid, 50 units/ml pen-icillin, and 50 �g/ml streptomycin. Cells were maintained at37 °C in a humidified atmosphere containing 5% CO2.Viability Assays—Cells were seeded in 96-well dishes at 7 �
103 cells/well. After 24 h Tat or Tat-NBI1 were added at differ-ent concentrations and incubated for 24 h. Following incuba-tion, plates were rinsed with PBS and 100 �l of stock 3-(4,5-di-methylthiazol-2-yl)-2,5-diphenyltetrazolium bromide solution(0.5mg/ml in culturemedium)was added.Cellswere incubatedat 37 °C for 1 h; then 200 �l of Me2SO were added to each well.Plates were read on a MRX (Dynatech Laboratories) readerafter a 1-h incubation, at awavelength of 570nm.Results shownare from triplicates in at least three independent experiments.Flow Cytometry Analysis—Cells were plated in 60-mm2 cul-
ture dishes at 5 � 105 cells/plate 24 h prior treatments. Tat orTat-NBI1 were added for 24 or 48 h. Cells were fixed with 70%methanol for 2 h at room temperature, washed with PBS, andfinally incubated with 50 �g/ml of propidium iodide (Sigma)and 200�g/ml RNase for 10min at room temperature. Analysisof DNA content was carried out in a BD Biosciences FACSCalibur. Results shown are from triplicates in at least threeindependent experiments.Determination of DNA Synthesis, Thymidine Incorporation—
Cells were seeded at 5� 105 cells/plate and allowed to grow forat least 24 h. Tat or Tat-NBI1 was then added for 24 h and[methyl-3H]thymidine (4mCi/ml; Amersham Biosciences) wasincorporated 1 h before the cells were collected. DNA synthesiswas determined measuring the incorporation of [methyl-3H]thymidine into DNA as described (28).Cell Cycle Synchronization and Laser-scanning Confocal
Analysis—Cells were grown on coverslips for 24 h. G1 synchro-nization was achieved by maintaining at 80% of confluence thecultures with 0.5% fetal calf serummedium for 24 h. Cells weresynchronized at S phase with 1.5 mM hydroxyurea for 24 h. Forsynchronization in M phase, cells were treated with 100 ng/mlnocodazol during 18 h. After synchronization drugs wereremoved and cells were washed with fresh media and rinsedover 1 h. Treatments with TAT or TAT-NBI1 at 20 �M werethen added and time courses were performed.The DNA cellular content in synchronized cultures was ana-
lyzed by laser-scanning cytometry. Cells were fixed with 70%methanol for 2 h at room temperature and washed with phos-phate-buffered saline. Then, they were incubated with 50�g/ml propidium iodide (Sigma) and 200 �g/ml RNase for 10min. The coverslips were seeded onto a slide using mountingmedium containing 25% propidium iodide (100 �g/ml in PBS)and 75% glycerol. Slides were scanned using the �40 objectiveand 5milliwatts of Argon laser power. A red fluorescent thresh-old and a minimum cell size of 100 pixels identified cells formeasurement. At least 10,000 cells were analyzed.Protein Extraction and Western Blot Analysis—Treated cells
were washed twice in PBS, scraped, and lysed in lysis buffer (80mMTris-HCl, pH 6.8, 2% (v/v) SDS). Protein concentrationwasdetermined by the Lowry method. Lysates (25 �g) were loadedinto a SDS-PAGE gel and after that, proteins were transferredto Immobilon membranes. Subsequently, membranes wereblocked in 5% nonfat dry milk in TBS buffer for 1 h at roomtemperature and then incubated with different primary anti-
Cyclin A Surface Inhibitors
35944 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 47 • NOVEMBER 24, 2006
by on Septem
ber 11, 2008 w
ww
.jbc.orgD
ownloaded from
bodies overnight at 4 °C: anti-phospho(S32)-MCM2 1:20000(Abcam ab11897), anti-MCM2 1:50 (Abcam ab6153), anti-PARP1:1000 (BDPharmingen 556493), and anti-actin cloneC41:1000 (ICN Biomedicals). After three washes with TBST,membranes were incubated for 45minwith horseradish perox-
idase-coupled secondary antibodies diluted in TBS with 2.5%nonfat dry milk. The filter was then washed two times withTBST and once with TBS, and analyzed by enhanced chemi-luminescence with ECL detection reagents (AmershamBiosciences).Apoptosis Assays—For detection of cellular fragmentation
into apoptotic bodies, cells were seeded on coverslips, treated atdifferent times, fixed with 70% methanol, and subjected toDNA staining with propidium iodide.
RESULTS
The crystallographic structure of the complex cdk2-cyclin Ashows that cyclin A binds to and interacts with both the N- andC-lobes of cdk2 to form a continuous protein-protein interface(29, 30). The structure of cdk2-cyclin A complexed with theCKI protein p27Kip1 has also been resolved showing the inter-action between the CKI substrate and the complex at the CRM
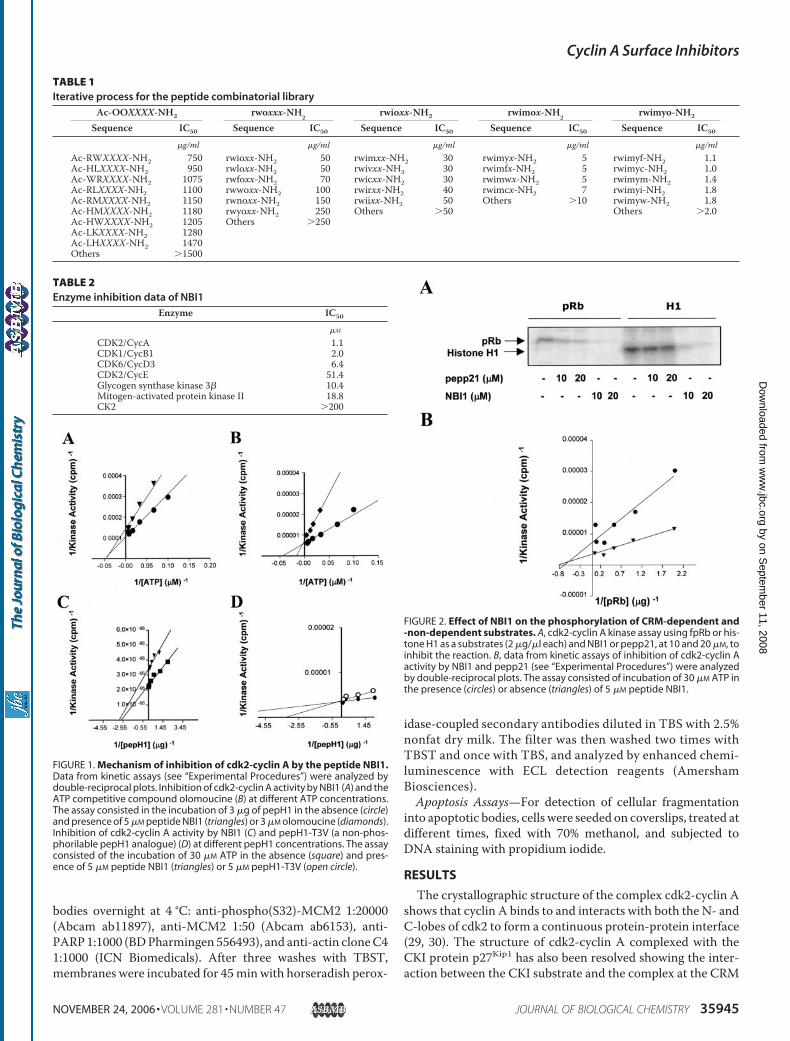
FIGURE 1. Mechanism of inhibition of cdk2-cyclin A by the peptide NBI1.Data from kinetic assays (see “Experimental Procedures”) were analyzed bydouble-reciprocal plots. Inhibition of cdk2-cyclin A activity by NBI1 (A) and theATP competitive compound olomoucine (B) at different ATP concentrations.The assay consisted in the incubation of 3 �g of pepH1 in the absence (circle)and presence of 5 �M peptide NBI1 (triangles) or 3 �M olomoucine (diamonds).Inhibition of cdk2-cyclin A activity by NBI1 (C) and pepH1-T3V (a non-phos-phorilable pepH1 analogue) (D) at different pepH1 concentrations. The assayconsisted of the incubation of 30 �M ATP in the absence (square) and pres-ence of 5 �M peptide NBI1 (triangles) or 5 �M pepH1-T3V (open circle).
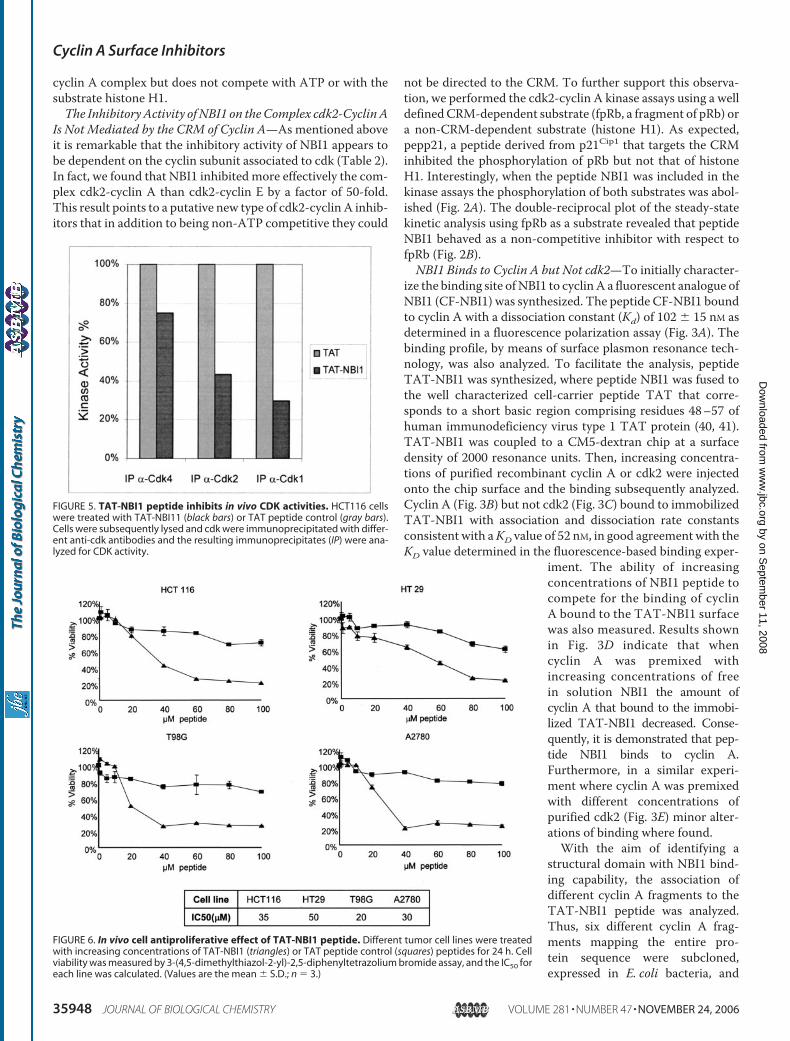
FIGURE 2. Effect of NBI1 on the phosphorylation of CRM-dependent and-non-dependent substrates. A, cdk2-cyclin A kinase assay using fpRb or his-tone H1 as a substrates (2 �g/�l each) and NBI1 or pepp21, at 10 and 20 �M, toinhibit the reaction. B, data from kinetic assays of inhibition of cdk2-cyclin Aactivity by NBI1 and pepp21 (see “Experimental Procedures”) were analyzedby double-reciprocal plots. The assay consisted of incubation of 30 �M ATP inthe presence (circles) or absence (triangles) of 5 �M peptide NBI1.
TABLE 1Iterative process for the peptide combinatorial library
Ac-OOXXXX-NH2 rwoxxx-NH2 rwioxx-NH2 rwimox-NH2 rwimyo-NH2
Sequence IC50 Sequence IC50 Sequence IC50 Sequence IC50 Sequence IC50
�g/ml �g/ml �g/ml �g/ml �g/mlAc-RWXXXX-NH2 750 rwioxx-NH2 50 rwimxx-NH2 30 rwimyx-NH2 5 rwimyf-NH2 1.1Ac-HLXXXX-NH2 950 rwloxx-NH2 50 rwivxx-NH2 30 rwimfx-NH2 5 rwimyc-NH2 1.0Ac-WRXXXX-NH2 1075 rwfoxx-NH2 70 rwicxx-NH2 30 rwimwx-NH2 5 rwimym-NH2 1.4Ac-RLXXXX-NH2 1100 rwwoxx-NH2 100 rwirxx-NH2 40 rwimcx-NH2 7 rwimyi-NH2 1.8Ac-RMXXXX-NH2 1150 rwnoxx-NH2 150 rwiixx-NH2 50 Others �10 rwimyw-NH2 1.8Ac-HMXXXX-NH2 1180 rwyoxx-NH2 250 Others �50 Others �2.0Ac-HWXXXX-NH2 1205 Others �250Ac-LKXXXX-NH2 1280Ac-LHXXXX-NH2 1470Others �1500
TABLE 2Enzyme inhibition data of NBI1
Enzyme IC50
�M
CDK2/CycA 1.1CDK1/CycB1 2.0CDK6/CycD3 6.4CDK2/CycE 51.4Glycogen synthase kinase 3� 10.4Mitogen-activated protein kinase II 18.8CK2 �200
Cyclin A Surface Inhibitors
NOVEMBER 24, 2006 • VOLUME 281 • NUMBER 47 JOURNAL OF BIOLOGICAL CHEMISTRY 35945
by on Septem
ber 11, 2008 w
ww
.jbc.orgD
ownloaded from

(31). A number of groups have reported the identification ofinhibitors of cdk2-cyclin A through the design of moleculesthat bind to the CRM (20, 32, 33). As stated above the goal ofthis study was to identify inhibitors of cdk2-cyclin A complexesbut not targeting the ATP binding site nor the CRM. To thisaim we screened combinatorial libraries in the appropriateexperimental conditions, thus we adjusted the ATP concentra-tion to be high enough to difficult ATP competition while min-imizing the use of [�-32P]ATP. As substrate we used histoneH1that is phosphorylated directly and without the required bind-ing to the cyclin A CRM that have other substrates as pRb andE2F protein families (32).
Screening of a Dual DefinedPosition Peptide CombinatorialLibrary—We initially screened adiversity oriented positional scan-ning library of trimers ofN-alkylgly-cines that have previously allowedthe identification of lead com-pounds in different biological assaysin our laboratories (34–37). How-ever, we did not find any suitablelead compound that satisfied theinitial goals. Thus we decided tofocus on a putatively more reliablelibrary that encompasses a morestep-by-step definition of the func-tionalities required by the experi-mental conditions. Thus, we syn-thesized a dual defined positionpeptide combinatorial library as ini-tially proposed by Houghten et al.(38). The peptide combinatoriallibrary consisted of 400 separatehexapeptide mixtures having aCOOH-terminal amide and anacetylatedNH2 terminus. Eachmix-ture (represented by the formulaAc-OOXXXX-NH2) had the firsttwo positions defined with one ofthe 20 naturally occurring aminoacids (referred to as “O”) and theremaining four positions are closeto equimolar mixtures of 19 of thenaturally occurring amino acids(referred to as “X,” cysteine wasomitted). The peptide mixtureswere screened for the ability toinhibit cdk2-cyclin A activity. Asshown in Table 1 a number of pep-tide mixtures assayed were found tohave significant inhibitory activity.From these assays the peptidemixture Ac-RWXXXX-NH2 wasselected for further deconvolution.Furthermore, from this initialscreening we learned that the pres-ence of positive charges at the NH2
terminus of the peptide mixtures was recognized as one of thecharacteristic of all of the most active peptide mixtures, thuswe decided to proceed with the deconvolution process using anon-acetylated version of the initial library. Moreover, due to thepotential interest in performing in vivo studies with the putativeinhibitory peptides defined after the deconvolution process, wedecided to synthesize the peptide mixtures using the more pro-tease-stable D-stereoisomers of the amino acids (that will bereferred here using lowercase letters). Nevertheless, prior to a fur-ther deconvolution step we synthesized the peptide mixturerwxxxx-NH2 that was shown to posses an inhibitory activity sim-ilar to that of the mixture Ac-RWXXXX-NH2.
FIGURE 3. Analysis of the binding of NBI1 to cyclin A and cdk2. A, a 60 nM solution of CF-NBI1 in PBS bufferwas titrated with increasing concentrations of cyclin A-(171– 432) and fluorescence polarization was measuredat 30 °C. The data (mean � S.D.; n � 3) were recorded in millipolarization units and adjusted to a two-statebinding model. B–D, binding of NBI1 peptide to cyclin A and cdk2 by surface plasmon resonance. Sensorgramsof immobilized TAT-NBI1 and TAT CM-5 sensor chips exposed to increasing concentrations (0.2, 0.5, 1, and 2�M) of cyclin A (B) or cdk2 (C). Binding of cyclin A to TAT-NBI1 is concentration dependent. To analyze the effectof NBI1-free peptide or cdk2 on the interaction between cyclin A and TAT-NBI1; free NBI1 (D) or purified cdk2 (E)at different concentrations (0, 0.1, 0.2, 0.5, 1, and 2 �M matches with lines 1– 6, respectively) were preincubatedwith 0.5 �M cyclin A in HBS-EP buffer. Each reaction mixture was then loaded in running buffer over the chip,after the dissociation period we regenerated the surface chip with 0.2% (w/v) SDS and the amount of cyclin Athat remained bound to the immobilized TAT-NBI1 was measured. RU, resonance unit.
Cyclin A Surface Inhibitors
35946 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 47 • NOVEMBER 24, 2006
by on Septem
ber 11, 2008 w
ww
.jbc.orgD
ownloaded from

To define the third amino acid from the hexapeptide, thelibrary rwoxxx-NH2 (containing 20 different mixtures) wassynthesized and screened. The most active mixtures wererwixxx-NH2, rwlxxx-NH2, and rwfxxx-NH2 (Table 1). Due tothe similar chemical character of the third amino acid, onlyrwixxx-NH2 was tracked in the iterative process. The Leu-Phepair is one of the most abundant residue pairs found at thecyclin A CRM (39) (i.e. RXLFY�). Taking into account our goalof finding non-CRM ligands together with the slightly higheractivity observed for themixture rwixxx-NH2, we selected D-Ileat the third position. A new library was synthesized with thegeneral formula rwioxx-NH2 and the next position was identi-fied. Themost activemixtures were rwimxx-NH2, rwivxx-NH2,rwicxx-NH2, rwirxx-NH2, and rwiixx-NH2 (Table 1). All fiveshowed similar inhibitory activity then, at this point to makethe selection we incorporated a secondary assay directedtoward kinase selectivity. All five mixtures were screenedagainst the CDK non-related kinases calmodulin-dependentkinase II and p38 SAPK. The mixtures rwicxx-NH2, rwiixx-NH2, rwirxx-NH2, and rwivxx-NH2 but not rwimxx-NH2showed some inhibitory activity against these two kinases, thenD-Met was selected at the fourth position of the hexapeptide. Anew set of mixtures, rwimox-NH2, was synthesized to definethe fifth amino acid. The library was screened and four mix-tures, rwimyx-NH2, rwimfx-NH2, rwimwx-NH2, and rwimcx-NH2, were shown to have similar IC50 values (Table 1). Thus, inthis case the criterion used to define the amino acid was based
in the chemical nature of the mostactive amino acid that defines eachsub-library. D-Tyrosinewas selectedas representative of the aromaticamino acids that appeared to be themost active at this particular posi-tion. Finally, a collection of 20 indi-vidual hexapeptides was synthe-sized. The sequences rwimyc-NH2and rwimyf-NH2with IC50 values of1.0 � 0.1 �g/ml were shown to bethe most active compounds. Thepeptides were extensively purifiedand analyzed by high performanceliquid chromatography and massspectrometry. We noticed that thepeptide rwimyc-NH2 showed atendency to form dimers in thekinase assay experimental condi-tions, we then decided to continueour study with the purified rwimyf-NH2 (peptide NBI1). In addition,a scrambled version of NBI1(sequence yirwfm-NH2, namedNBIrd) and a peptide derived from adifferent study that shares four ofsix of the amino acids of NBI1(sequence ykrwqm-NH2, namedNBInr) did not inhibit the kinaseactivity of the cdk2-cyclin A com-plex in our experimental conditions
(results not shown), suggesting that the inhibitory activity ofNBI1 is sequence dependent and specific. Furthermore, NBI1was found to inhibit both cdk1-cyclin B1 and cdk6-cyclin D3,although more specifically the former (Table 2). In contrast,NBI1 was a weak inhibitor of cdk2-cyclin E and showedremarkable selectivity with respect to a representative set ofother serine/threonine kinases, thus CK2 was not inhibited,whereas mitogen-activated protein kinase II and glycogensynthase kinase 3� were only slighted inhibited (Table 2).The Inhibition of cdk2-Cyclin A Activity by NBI1 Does Not
Compete with ATP or Substrates—We next investigated thesteady-state kinetics of cdk2-cyclin A inhibition by NBI1. Inthese experiments phosphorylation of a histone H1-derivedpeptide (pepH1, see “Experimental Procedures”) by cdk2-cy-clin A was measured in the presence of increasing amounts ofATP and in the absence or presence of 5 �MNBI1. The double-reciprocal plot revealed that NBI1 behaves as a non-competi-tive inhibitor with respect to ATP (Fig. 1A). The ATP-compet-itive inhibitor olomoucine was used as a control (Fig. 1B). Wenext examined inhibition by NBI1 with respect to pepH1 sub-strate in the presence of saturating concentrations of ATP. Thedouble-reciprocal plot is consistent with a non-competitivemode of inhibition (Fig. 1C). As a control for a competitiveinhibition we used a peptide derived from pepH1 (pepH1-T3V,see “Experimental Procedures”) where the susceptible Thr res-idue of phosphorylation has been replaced by Val (Fig. 1D).Thus peptide NBI1 inhibits the catalytic activity of the cdk2-
FIGURE 4. Analysis of the binding of NBI1 to cyclin A fragments. A, design of the six cyclin A fragments. B,Biacore sensorgrams representing binding experiments using a CM-5 chip containing immobilized TAT-NBI1and TAT exposed to 0.5 �M of each cyclin A fragment. Binding was demonstrated by fragments A4, A5, and A6.aa, amino acid.
Cyclin A Surface Inhibitors
NOVEMBER 24, 2006 • VOLUME 281 • NUMBER 47 JOURNAL OF BIOLOGICAL CHEMISTRY 35947
by on Septem
ber 11, 2008 w
ww
.jbc.orgD
ownloaded from
cyclin A complex but does not compete with ATP or with thesubstrate histone H1.The Inhibitory Activity of NBI1 on theComplex cdk2-CyclinA
Is Not Mediated by the CRM of Cyclin A—Asmentioned aboveit is remarkable that the inhibitory activity of NBI1 appears tobe dependent on the cyclin subunit associated to cdk (Table 2).In fact, we found that NBI1 inhibited more effectively the com-plex cdk2-cyclin A than cdk2-cyclin E by a factor of 50-fold.This result points to a putative new type of cdk2-cyclin A inhib-itors that in addition to being non-ATP competitive they could
not be directed to the CRM. To further support this observa-tion, we performed the cdk2-cyclin A kinase assays using a welldefinedCRM-dependent substrate (fpRb, a fragment of pRb) ora non-CRM-dependent substrate (histone H1). As expected,pepp21, a peptide derived from p21Cip1 that targets the CRMinhibited the phosphorylation of pRb but not that of histoneH1. Interestingly, when the peptide NBI1 was included in thekinase assays the phosphorylation of both substrates was abol-ished (Fig. 2A). The double-reciprocal plot of the steady-statekinetic analysis using fpRb as a substrate revealed that peptideNBI1 behaved as a non-competitive inhibitor with respect tofpRb (Fig. 2B).NBI1 Binds to Cyclin A but Not cdk2—To initially character-
ize the binding site ofNBI1 to cyclinA a fluorescent analogue ofNBI1 (CF-NBI1) was synthesized. The peptide CF-NBI1 boundto cyclin A with a dissociation constant (Kd) of 102 � 15 nM asdetermined in a fluorescence polarization assay (Fig. 3A). Thebinding profile, by means of surface plasmon resonance tech-nology, was also analyzed. To facilitate the analysis, peptideTAT-NBI1 was synthesized, where peptide NBI1 was fused tothe well characterized cell-carrier peptide TAT that corre-sponds to a short basic region comprising residues 48–57 ofhuman immunodeficiency virus type 1 TAT protein (40, 41).TAT-NBI1 was coupled to a CM5-dextran chip at a surfacedensity of 2000 resonance units. Then, increasing concentra-tions of purified recombinant cyclin A or cdk2 were injectedonto the chip surface and the binding subsequently analyzed.Cyclin A (Fig. 3B) but not cdk2 (Fig. 3C) bound to immobilizedTAT-NBI1 with association and dissociation rate constantsconsistent with aKD value of 52 nM, in good agreement with theKD value determined in the fluorescence-based binding exper-
iment. The ability of increasingconcentrations of NBI1 peptide tocompete for the binding of cyclinA bound to the TAT-NBI1 surfacewas also measured. Results shownin Fig. 3D indicate that whencyclin A was premixed withincreasing concentrations of freein solution NBI1 the amount ofcyclin A that bound to the immobi-lized TAT-NBI1 decreased. Conse-quently, it is demonstrated that pep-tide NBI1 binds to cyclin A.Furthermore, in a similar experi-ment where cyclin A was premixedwith different concentrations ofpurified cdk2 (Fig. 3E) minor alter-ations of binding where found.With the aim of identifying a
structural domain with NBI1 bind-ing capability, the association ofdifferent cyclin A fragments to theTAT-NBI1 peptide was analyzed.Thus, six different cyclin A frag-ments mapping the entire pro-tein sequence were subcloned,expressed in E. coli bacteria, and
FIGURE 5. TAT-NBI1 peptide inhibits in vivo CDK activities. HCT116 cellswere treated with TAT-NBI11 (black bars) or TAT peptide control (gray bars).Cells were subsequently lysed and cdk were immunoprecipitated with differ-ent anti-cdk antibodies and the resulting immunoprecipitates (IP) were ana-lyzed for CDK activity.
FIGURE 6. In vivo cell antiproliferative effect of TAT-NBI1 peptide. Different tumor cell lines were treatedwith increasing concentrations of TAT-NBI1 (triangles) or TAT peptide control (squares) peptides for 24 h. Cellviability was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay, and the IC50 foreach line was calculated. (Values are the mean � S.D.; n � 3.)
Cyclin A Surface Inhibitors
35948 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 47 • NOVEMBER 24, 2006
by on Septem
ber 11, 2008 w
ww
.jbc.orgD
ownloaded from

purified (Fig. 4A). The binding profile to TAT-NBI1 was ana-lyzed by means of surface plasmon resonance (Fig. 4B). Theresults suggest that theminimal structural domain that binds toNBI1 is defined at a highly conserved region between aminoacids 257 and 345 that comprises helices �3, �4, and �5. Thesehelices formpart of the structural domain named the cyclin box(residues 209–310), a region conserved amongmembers of thecyclin family and implicated in the interaction with cdks (30).NBI1 Inhibits Cell Cycle Progression and Induces Apoptosis—
We further aimed to study the effect of the hexapeptide on cellviability. To ensure an efficient uptake ofNBI1 across cellmem-braneswe employed theTAT-NBI1 peptide in cell based assays.Preliminary studies showed that TAT-NBI1 had the sameinhibitory activity as NBI1 in the in vitro cdk2-cyclin A assay(data not shown). Furthermore, immunoprecipitation experi-
ments on cells treated with TAT-NBI1were performed to analyze thein vivo effect of the peptide on cdkactivity. Thus, immunoprecipita-tion using anti-cdk1, anti-cdk2, oranti-cdk4 antibodies were carriedout on cells treated with TAT-NBI1or TAT control peptide. Then, thedifferent cdk activities were ana-lyzed in the immunoprecipitates. Inagreement with the in vitro results(Table 2), we observed that cdk2and cdk1 activities were inhibited incells treated with TAT-NBI1,whereas control cdk4 activity wasonly slightly inhibited (Fig. 5).Thus, after ensuring that TAT-
NBI1 is able to enter the cell andexert is inhibitory activity on cdk2activity, the effect of different dosesof TAT-NBI1 or TAT were studiedin a number of tumor cell lines(HCT116,HT29, T98G, andA2780)using a standard 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazo-lium bromide cell viability assay.Results revealed that the hexapep-tidedecreasedcell viability inadose-dependent fashion (Fig. 6).Flow cytometric analysis of asyn-
chronously growing tumor cellstreated with TAT-NBI1 resulted ina cell cycle arrest mainly at the Sphase, clearly accompanied by anincrease in the sub-G1 population(Fig. 7). The blockage in S phase wasfurther confirmed in two cell lines(HCT116 and A2780) by measuringthe rate of DNA synthesis in thepresence of the TAT-NBI1 peptide(Fig. 8A). These effects were corre-lated with a NBI1-induced in vivodecrease of cdk2-cyclin A activity,
as measured by the low rate of phosphorylation of the CDKprotein substrate minichromosome maintenance 2(MCM2), a nuclear protein involved in the onset of DNAreplication (Fig. 8B) (42). We subsequently analyzed theeffect of TAT-NBI1 and TAT peptide control on cells syn-chronized at different phases of the cell cycle. Interestingly,TAT-NBI1 did not affect cell cycle progression in G1 cells,whereas in contrast a strong increase in the sub-G1 popula-tion was observed when S or G2/M cells were treated withTAT-NBI1 (Fig. 9A). These results indicate that cells in S orG2/M are highly sensitive to the peptide and are in agree-ment with the fact that cdk2-cyclin A and cdk1-cyclin A/Bare the complexes that govern S and G2/M progression andthat are strongly inhibited by TAT-NBI1. The treatment ofcells (HCT116 and A2780) with the hexapeptide caused apo-
FIGURE 7. Cell cycle arrest. Cell cycle distribution of HCT116 (A), HT29 (B), T98G (C), and A2780 (D) exposed todifferent doses of TAT-NBI1 (black bars) or TAT control peptide (gray bars) for 24 h, as assessed by propidiumiodide staining and analysis by flow cytometry. Results are represented as the mean values of the percentageof cells in the different phases of the cell cycle of three independent experiments.
Cyclin A Surface Inhibitors
NOVEMBER 24, 2006 • VOLUME 281 • NUMBER 47 JOURNAL OF BIOLOGICAL CHEMISTRY 35949
by on Septem
ber 11, 2008 w
ww
.jbc.orgD
ownloaded from

ptosis as observed by caspase 3-dependent cleavage of PARP(Fig. 9B) and by the induction of apoptotic bodies (Fig. 9C).
DISCUSSION
Natural inhibitors of CDKs are either mutated or deletedin primary tumor cells. In an attempt to regulate aberrantcell cycle pathways, new synthetic agents that can restore thefunctions of altered tumor suppressor proteins, as CKIs, arecentral targets in current cancer research. Active sites ofenzymes such as kinases have challenged drug discoveryapproaches. However, ATP competitive drug-like com-pounds have proven difficult to define kinase specificitybecause most of such enzymes share a high degree ofsequence similarity within the ATP active site. Alternativeinhibitory pathways were found derived from studiesdirected to elucidate the structure and activation mecha-nism of CDKs (43, 44). Thus, the CRM, a consensus shortrecognition motif present in many cell cycle regulatory pro-teins that interact with a hydrophobic patch on the surface ofcyclins (23), inspired the design of new inhibitors. In thissense, peptides derived from the COOH terminus of theCDK inhibitor p21Cip1 (45) or from the transcription factorE2F1 (23, 46) were shown to have antiproliferative effects inmultiple cell lines. More recently, Gondeau et al. (22) havedescribed another strategy to inhibit the activity of CDKs bytargeting the interface between the cdk and cyclin. Thus, a22-residue long peptide derived from the amino acidsequence of the �5 helix of cyclin A was shown to bind to thecdk2-cyclin A complex and in turn, it was demonstrated toblock the proliferation of tumor cell lines (22). These pep-tides, besides their own activity, are valuable tools that could
drive the discovery of new families of small molecule inhib-itors of CDKs (1, 5, 47, 48).To explore alternate binding sites that may inhibit the
activity of CDKs we screened chemical libraries underrestrictive assay conditions. The imposition of such screen-ing conditions would limit the putative hits to molecules thatcould not compete with ATP and could bind to cyclin A andinhibit the productive protein-protein interaction that couldrender the active cdk2-cyclin A complex. The peptide NBI1(rwimyf-NH2) was selected as lead compound for the devel-opment of this new class of inhibitors (Table 1). The peptideshowed an IC50 value of 1.1 �M for the inhibition of theactivity of the cdk2-cyclin A complex, whereas was lessactive against a panel of other kinases (Table 2). The kineticanalysis showed that the NBI1 peptide neither competeswith ATP nor with CRM substrates as pRb nor with sub-strates as histone H1 (Figs. 1 and 2). Furthermore, we haveshown that the NBI1 peptide selectively binds to cyclin A(Fig. 3). Thus, it is tempting to hypothesize that the NBI1-binding site on cyclin A may represent a new target site forthe selective inhibition of activity for the cdk2-cyclin A com-plex. In contrast to the existing CDKs peptide inhibitors, thehexapeptide NBI1 could disrupt the cdk2-cyclin A complex.The cdk2-cyclin A complex x-ray crystal structure (30)showed that at the complex interface several structural ele-ments from both the cdk2 and the cyclin A subunits adapteach other resulting in a buried surface area larger than 3000Å2. Thus, the NBI1 peptide should recognize and bind to acritical structural element of cyclin A that in turn shouldinduce a putative conformational change that diminishes itsaffinity to bind to the cdk2 subunit. It would be of interest toidentify such a structural element. Human cyclin A aggre-gates at the concentrations required for structural studies.Nevertheless, the reported x-ray crystal structures of thecdk2-cyclin A complexes were determined using a func-tional cyclin A domain comprised by residues 173–432 (30,47). However, this domain is still very unstable for futurestructural determinations in the absence of the cdk2 subunit.In an attempt to get a deeper insight into the structural
basis of the interaction, we docked the NBI1 peptide onto thecyclin A molecule. Fig. 10 shows the results obtained usingthe deposited coordinates for the complex cdk2-cyclin A,and a peptide (structure on red in Fig. 10) bound to the CRM(Protein Data Bank code 2C5V), and the results obtainedusing AutoDock (49) for the peptide NBI1 (structure on bluein Fig. 10). Although it is still speculative, the NBI1 peptidecould bind to a cleft on the surface of cyclin A that is impor-tant for the binding of cyclin A to cdk2. From the output ofthe docking procedure, it looks like residues Gln228, Asn229(helix �3), Asn312, Gln313 (helix �6); Met334 (helix �7); andLys417 (at the COOH-terminal end) could contribute to thedefinition of this new binding site. These results are in agree-ment with those obtained from the analysis by surface plas-mon resonance of the minimal structural domain that bindsto NBI1 (Fig. 4). Interestingly, the docking experiments alsoexplain the selectivity found for NBI1 against different cyc-lins. As it was shown in Table 2, NBI1 shows a remarkable
FIGURE 8. TAT-NBI1 peptide inhibits DNA synthesis. A, effect of TAT-NBI1on DNA synthesis in HCT116 (left panel) and A2780 (right panel) cells. Thepercentages on the left represent [3H]thymidine incorporation percentage.Cultured cells were treated with 20 �M TAT-NBI1 or TAT peptide control by24 h, and DNA synthesis was measured by [3H]thymidine incorporation asdescribed under “Experimental Procedures.” In both cases, the values are themean � S.D. from three independent experiments. B, Western blot analysis ofMCM2 phosphorylation. HCT116 (upper blots) and A2780 (lower blots) weretreated at 24 h with 20 �M TAT peptide control, or three different doses ofTAT-NBI1 (10, 20, and 40 �M). Representative Western blots using specificantibodies, anti-phospho-MCM2, anti-MCM2 total protein, and loading con-trol with anti-actin are shown.
Cyclin A Surface Inhibitors
35950 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 47 • NOVEMBER 24, 2006
by on Septem
ber 11, 2008 w
ww
.jbc.orgD
ownloaded from

selectivity for the cdk2-cyclin Acomplex, but it is a weak inhibitorof the cdk2-cyclin E complex.Because both cyclins bind to thesame Cdk, the only explanation forthe different selectivity has to befound in structural differencesbetween both cyclins at the bind-ing region suggested by the dock-ing experiments. Fig. 11 shows asurface representation of cyclin A(Fig. 11A) and cyclin E (Fig. 11B)structures, together with the posesuggested by Autodock for NBI1in cyclin A (Fig. 11C). The figureclearly shows that the nature of thecavity is different in both proteins,and that NBI1 cannot make thesame contacts with the two pro-teins. In fact, from the enzymeinhibition data, it seems clear that
FIGURE 9. Effect of TAT-NBI1 peptide on cell cycle and apoptosis. A, TAT-NBI1 affects cell cycle progression. HCT116 cells synchronized at different phases of the cellcycle were treated with TAT-NBI1 or TAT peptide control at 20 �M for 6 h and then, the cell cycle distribution was analyzed by laser-scanning cytometry (black square,cells in G1 phase; empty square, cells in S phase; gray square, cells in M phase and crossed square, cells in sub-G1 phase). Cells were synchronized in G1 (left panel), S phase(middle panel), or mitosis (right panel). B, detection of cleaved PARP in cell extracts. HCT116 or A2780 cells were treated during 24 h with 20 �M TAT control peptide, orthree different doses of TAT-NBI1 (5, 10, and 20 �M). Representative Western blots using anti-PARP revealed the presence of PARP cleavage products on the blot(85-kDa PARP fragment). C, TAT-NBI1 peptide induces formation of apoptotic bodies in HCT116 and A2780 cells. Images show HCT116 and A2780 cells subjected to48 h of TAT-NBI1 or TAT control peptide (40 �M) treatment. Cells were stained with propidium iodide and loss of integrity of nuclear envelope and visualization ofapoptotic bodies were detected (white arrows).
FIGURE 10. Surface representation of the cdk2-cyclin A structure and location of the NBI1 bindingsite on cyclin A. A, surface representation of the structure of cyclin A (soft gray) with a CRM bindingpeptide in red (coordinates were extracted from Protein data bank code 2C5V). The L-amino acid versionof the peptide NBI1 (in blue) was docked on the structure of cyclin A. In this docking approach, the peptidebackbone of NBI1 was kept rigid, whereas all the side chains were defined as flexible using the deftorsmodule. Cyclin A was treated as the macromolecule part of the docking calculation as was kept rigid.B, surface representation of the structure of the cdk2 (soft blue)-cyclin A (soft gray) complex. The surfacerepresentation of a CRM-binding and NBI1 peptides are shown in red and blue, respectively.
Cyclin A Surface Inhibitors
NOVEMBER 24, 2006 • VOLUME 281 • NUMBER 47 JOURNAL OF BIOLOGICAL CHEMISTRY 35951
by on Septem
ber 11, 2008 w
ww
.jbc.orgD
ownloaded from

the interaction between NBI1 and cyclin A is more efficientthan with cyclin E. On the other hand, cdk1 possesses a veryhigh similarity with cdk2 (66% sequence identity), and it isknown that cyclin A can bind, in addition to cdk2, to cdk1. Inthis context, it is not surprising that NBI1 could also disruptthe cdk1-cyclin A interaction. To the best of our knowledge,the structure of cyclin B1 is not yet known, so it is difficult toexplain in detail why NBI1 can inhibit cdk1-cyclin B1 activ-ity. However, results obtained by comparative homologymodeling using different software packages (data not shown)have not been able to reveal big differences between cyclinB1 and cyclin A structures, thereby explaining why NBI1 isalso able to disrupt the cdk1-cyclin B1 complex. Finally, thelow degree of sequence similarity between cdk2 and cdk6
(49% sequence identity) andamong cyclin D3 and cyclin A(30% sequence identity) makes itvery difficult to structurally inter-pret the results obtained for thecdk6-cyclin D3 (Table 2).Supporting the data obtained
from the in vitro model systems,NBI1 in its TAT-NBI1 form wasshown to have activity in livingcells. In the cell cycle S phase,cdk2-cyclin A complexes areimportant for phosphorylationand inactivation of the E2F/DP1transcription factor. Inhibition ofcdk2-cyclin A results in an ele-vated E2F concentration leadingto S phase arrest and apoptosis.TAT-NBI1 was able to inhibit cellproliferation (Fig. 6) in a concen-tration- and time-dependent man-ner, and induce both, specificblockade of cell cycle at S phase(Fig. 7) and apoptosis (Fig. 9) inthe glioblastoma (T98G) colon(HCT116) and ovarian (A2780)cancer cells evaluated.The peptide NBI1 could define a
new class of cdk2-cyclin A inhibi-tors binding at a different site in thecyclin A molecule than other avail-able pharmacological inhibitors. Infact, in contrast to most reportedCDKs inhibitors that are competi-tive to known substrates of CDKcomplexes, the mechanism ofaction of NBI1 appeared to be non-competitive to ATP and non-com-petitive to CRM substrates. Theability ofNBI1 to specifically bind toa new binding site on cyclin A andinhibit the formation of the cdk2-cyclin A complex provide new alter-natives for drug discovery because
the chemical properties for binding are different from activesite-directed compounds and would also allow selectivity.
Acknowledgment—We thank Ana Gimenez (Centro de InvestigacionPrıncipe Felipe) for technical assistance.
REFERENCES1. Berg, T. (2003) Angew. Chem. Int. Ed. Engl. 42, 2462–24812. Cochran, A. G. (2001) Curr. Opin. Chem. Biol. 5, 654–6593. Cochran, A. G. (2000) Chem. Biol. 7, R85–R944. Clackson, T., and Wells, J. A. (1995) Science 267, 383–3865. Arkin, M. (2005) Curr. Opin. Chem. Biol. 9, 317–3246. Loog, M., and Morgan, D. O. (2005) Nature 434, 104–1087. Morgan, D. O. (1995) Nature 374, 131–134
FIGURE 11. Comparison of cyclin A and cyclin E structures. A, surface representation of the cyclin A structure(soft gray) displaying the protein interface (yellow) delimited by the residues suggested by Autodock for theinteraction with NBI1. B, surface representation of the cyclin E structure (cyan) displaying the correspondingprotein interface (yellow) for this protein. C, surface representation of the cyclin A structure (soft gray) with theputative protein interface (yellow) for the interaction with NBI1, and the NBI1 pose (blue) suggested byAutodock for this peptide.
Cyclin A Surface Inhibitors
35952 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 47 • NOVEMBER 24, 2006
by on Septem
ber 11, 2008 w
ww
.jbc.orgD
ownloaded from

8. Dynlacht, B. D., Flores, O., Lees, J. A., and Harlow, E. (1994)Genes Dev. 8,1772–1786
9. Dyson, N. (1998) Genes Dev. 12, 2245–226210. Krek, W., Ewen, M. E., Shirodkar, S., Arany, Z., Kaelin, W. G., Jr., and
Livingston, D. M. (1994) Cell 78, 161–17211. Xu, M., Sheppard, K. A., Peng, C. Y., Yee, A. S., and Piwnica-Worms, H.
(1994)Mol. Cell. Biol. 14, 8420–843112. Qin, X. Q., Livingston, D. M., Kaelin, W. G., Jr., and Adams, P. D. (1994)
Proc. Natl. Acad. Sci. U. S. A. 91, 10918–1092213. Shan, B., and Lee, W. H. (1994)Mol. Cell. Biol. 14, 8166–817314. Kowalik, T. F., DeGregori, J., Schwarz, J. K., andNevins, J. R. (1995) J. Virol.
69, 2491–250015. Fotedar, A., Cannella, D., Fitzgerald, P., Rousselle, T., Gupta, S., Doree,M.,
and Fotedar, R. (1996) J. Biol. Chem. 271, 31627–3163716. Frouin, I., Montecucco, A., Biamonti, G., Hubscher, U., Spadari, S., and
Maga, G. (2002) EMBO J. 21, 2485–249517. Murphy, M., Stinnakre, M. G., Senamaud-Beaufort, C., Winston, N. J.,
Sweeney, C., Kubelka, M., Carrington, M., Brechot, C., and Sobczak-The-pot, J. (1997) Nat. Genet. 15, 83–86
18. Winston, N., Bourgain-Guglielmetti, F., Ciemerych, M. A., Kubiak, J. Z.,Senamaud-Beaufort, C., Carrington, M., Brechot, C., and Sobczak-The-pot, J. (2000) Dev. Biol. 223, 139–153
19. Huwe, A., Mazitschek, R., and Giannis, A. (2003) Angew. Chem. Int. Ed.Engl. 42, 2122–2138
20. McInnes, C., and Fischer, P. M. (2005) Curr. Pharm. Des. 11, 1845–186321. Fischer, P. M. (2001) Curr. Opin. Drug Discov. Dev. 4, 623–63422. Gondeau, C., Gerbal-Chaloin, S., Bello, P., Aldrian-Herrada, G., Morris,
M. C., and Divita, G. (2005) J. Biol. Chem. 280, 13793–1380023. Adams, P. D., Sellers, W. R., Sharma, S. K., Wu, A. D., Nalin, C. M., and
Kaelin, W. G., Jr. (1996)Mol. Cell. Biol. 16, 6623–663324. Luciani, M. G., Hutchins, J. R., Zheleva, D., and Hupp, T. R. (2000) J. Mol.
Biol. 300, 503–51825. Chen, J., Saha, P., Kornbluth, S., Dynlacht, B. D., and Dutta, A. (1996)Mol.
Cell Biol. 16, 4673–468226. Zhang, T., and Prives, C. (2001) J. Biol. Chem. 276, 29702–2971027. Riera, M., Roher, N., Miro, F., Gil, C., Trujillo, R., Aguilera, J., Plana, M.,
and Itarte, E. (1999)Mol. Cell. Biochem. 191, 97–10428. Cripps-Wolfman, J., Henshaw, E. C., and Bambara, R. A. (1989) J. Biol.
Chem. 264, 19478–1948629. Russo, A. A., Jeffrey, P. D., and Pavletich, N. P. (1996) Nat. Struct. Biol. 3,
696–70030. Jeffrey, P. D., Russo, A. A., Polyak, K., Gibbs, E., Hurwitz, J., Massague, J.,
and Pavletich, N. P. (1995) Nature 376, 313–320
31. Russo, A. A., Jeffrey, P. D., Patten, A. K., Massague, J., and Pavletich, N. P.(1996) Nature 382, 325–331
32. Andrews, M. J., McInnes, C., Kontopidis, G., Innes, L., Cowan, A., Plater,A., and Fischer, P. M. (2004) Org. Biomol. Chem. 2, 2735–2741
33. Fischer, P. M. (2004) Curr. Med. Chem. 11, 1563–158334. Garcia-Martinez, C., Humet, M., Planells-Cases, R., Gomis, A., Caprini,
M., Viana, F., De La Pena, E., Sanchez-Baeza, F., Carbonell, T., De Felipe,C., Perez-Paya, E., Belmonte, C., Messeguer, A., and Ferrer-Montiel, A.(2002) Proc. Natl. Acad. Sci. U. S. A. 99, 2374–2379
35. Humet, M., Carbonell, T., Masip, I., Sanchez-Baeza, F., Mora, P., Canton,E., Gobernado, M., Abad, C., Perez-Paya, E., and Messeguer, A. (2003)J. Comb. Chem. 5, 597–605
36. Montoliu, C., Humet, M., Canales, J. J., Burda, J., Planells-Cases, R.,Sanchez-Baeza, F., Carbonell, T., Perez-Paya, E., Messeguer, A., Ferrer-Montiel, A., and Felipo, V. (2002) J. Pharmacol. Exp. Ther. 301, 29–36
37. Planells-Cases, R., Montoliu, C., Humet, M., Fernandez, A. M., Garcia-Martinez, C., Valera, E., Merino, J. M., Perez-Paya, E., Messeguer, A.,Felipo, V., and Ferrer-Montiel, A. (2002) J. Pharmacol. Exp. Ther. 302,163–173
38. Houghten, R. A., Pinilla, C., Blondelle, S. E., Appel, J. R., Dooley, C. T., andCuervo, J. H. (1991) Nature 354, 84–86
39. Lowe, E. D., Tews, I., Cheng, K. Y., Brown, N. R., Gul, S., Noble, M. E.,Gamblin, S. J., and Johnson, L. N. (2002) Biochemistry 41, 15625–15634
40. Dietz, G. P., and Bahr, M. (2004)Mol. Cell. Neurosci. 27, 85–13141. Wadia, J. S., Stan, R. V., and Dowdy, S. F. (2004) Nat. Med. 10, 310–31542. Forsburg, S. L. (2004)Microbiol. Mol. Biol. Rev. 68, 109–13143. Brown, E. J., Beal, P. A., Keith, C. T., Chen, J., Shin, T. B., and Schreiber,
S. L. (1995) Nature 377, 441–44644. Morgan, D. O. (1997) Annu. Rev. Cell Dev. Biol. 13, 261–29145. Zheleva, D. I., McInnes, C., Gavine, A. L., Zhelev, N. Z., Fischer, P.M., and
Lane, D. P. (2002) J. Pept. Res. 60, 257–27046. Mendoza, N., Fong, S., Marsters, J., Koeppen, H., Schwall, R., and Wick-
ramasinghe, D. (2003) Cancer Res. 63, 1020–102447. Kontopidis, G., Andrews,M. J.,McInnes, C., Cowan, A., Powers, H., Innes,
L., Plater, A., Griffiths, G., Paterson, D., Zheleva, D. I., Lane, D. P., Green,S., Walkinshaw, M. D., and Fischer, P. M. (2003) Structure (Camb.) 11,1537–1546
48. Wu, S. Y., McNae, I., Kontopidis, G., McClue, S. J., McInnes, C., Stewart,K. J., Wang, S., Zheleva, D. I., Marriage, H., Lane, D. P., Taylor, P., Fischer,P. M., and Walkinshaw, M. D. (2003) Structure (Camb.) 11, 399–410
49. Morris, G.M., Goodsell, D. S., Halliday, R. S., Huey, R., Hart,W. E., Belew,R. K., and Olson, A. J. (1998) J. Comp. Chem. 19, 1639–1662
Cyclin A Surface Inhibitors
NOVEMBER 24, 2006 • VOLUME 281 • NUMBER 47 JOURNAL OF BIOLOGICAL CHEMISTRY 35953
by on Septem
ber 11, 2008 w
ww
.jbc.orgD
ownloaded from
Copyright © 2022 FDOKUMEN




















