
Functional Analysis of Cyclin-Dependent Kinase Inhibitors of Arabidopsis
Upload
independentCategory
view
2download
0
J. Mol. Biol. (1996) 261, 646–657
A Mutation in the Human Cyclin-dependent KinaseInteracting Protein, CksHs2, Interferes WithCyclin-dependent Kinase Binding and BiologicalFunction, but Preserves Protein Structureand Assembly
Mark H. Watson, Yves Bourne, Andrew S. Arvai, Michael J. HickeyAlicia Santiago, Susan L. Bernstein, John A. Tainer andSteven I. Reed*
Department of Molecular A mutation directing an amino acid substitution in the conserved b-hingeregion of one of the human Cks isoforms, CksHs2, was constructed byBiology, MB-7, The Scrippssite-directed mutagenesis. Replacement of glutamine for glutamate 63Research Institute, 10550
North Torrey Pines Road (E63Q) was predicted to stabilize the b-interchanged dimeric and hex-americ assembly of CksHs2. However, such an effect was seen only at high,La Jolla, CA 92037, USAnon-physiological pH. Three-dimensional structures of the E63Qhexameric mutant protein were determined to 2.6 Å resolution in a P43212space group and 2.1 Å in the C2 space group isostructural with wild-type,and both were shown to be virtually identical to the refined 1.7 Å wild-typestructure. Thus, the E63Q mutation did not alter the wild-type structureand assembly of CksHs2 but, surprisingly, disrupted the essentialbiological function of the protein and significantly reduced its ability tobind to cyclin-dependent kinases. The Kd of wild-type CksHs2 for CDK2was 5.05 × 10−8 M, whereas the affinity of the mutant protein for CDK2 wastoo low to allow a determination. These data, coupled with the observationthat monomeric but not hexameric CksHs2 interacts with cyclin-dependentkinases, suggest that glutamine 63 is likely to be directly involved incyclin-dependent kinase binding in vitro and in vivo.
7 1996 Academic Press Limited
Keywords: cell cycle; CksHs2; cyclin-dependent kinase; site-directed*Corresponding author mutagenesis; crystal structure
Introduction
Cell duplication is regulated by controllingpassage through several key transitions within thecell cycle. The primary mode of regulation of thesetransitions is at the level of assembly and activationof cyclin-dependent kinases (Cdks; for reviewssee Pines & Hunter, 1991; Reed, 1992; Hunter,1993; Dunphy, 1994; Morgan, 1995). These proteinkinases consist of a catalytic subunit that, as
a monomer, possesses no intrinsic activity, and arequisite positive regulatory subunit known as acyclin. Since different cell cycle phase transitionsare likely to require the phosphorylation of distinctsets of target proteins, multiple Cdk activities arenecessary to allow any cell to transit through acomplete normal cell division cycle. This functionaldiversity is accomplished by the successive gen-eration of different Cdk activities as cells pro-gress through the various cell cycle phases (forreviews, see Hunter & Pines, 1994; King et al., 1994;Nasmyth, 1993; Reed, 1992; Sherr, 1994). In simpleeukaryotes, such as yeast, a single Cdk is activatedsuccessively by different groups of cyclins topromote the necessary phase transitions. Inmammalian cells, different Cdks as well as cyclinsare required to assemble the combinations necess-ary to achieve these ends.
Permanent address: Yves Bourne, CNRS, UPR 9039,Architecture et Fonction des MacromoleculesBiologiques, 31 Chemin Joseph Aigvier, 13402Marseille Cedex 20, France.
Abbreviations used: HA, haemaglutinin; PMSF,phenylmethylsulfonyl fluoride; Cdk, cyclin-dependentkinase.
0022–2836/96/350646–12 $18.00/0 7 1996 Academic Press Limited

Mutation in CksHs2 Disrupts Cdk Binding 647
Besides the activation of Cdks by cyclin binding,several other regulatory mechanisms modulateCdks in the context of cell cycle control. Theseinclude both positive (for review see Solomon,1994) and negative (Gould & Nurse, 1989) regu-latory phosphorylation of the Cdk subunit, theaccumulation of Cdk inhibitory proteins (for reviewsee Hunter & Pines, 1994; Morgan, 1995) and theregulated degradation of cyclins (for review seeDeshaies, 1995). These positive and negativecontrols are likely to represent the manifestation ofa complex web of interactions between Cdks andother cellular components, necessitated by theregulatory needs of the cell division process.
Some biological interactions between Cdks andother proteins, for example, the Cks proteins, havebeen difficult to explain. The Cks proteins havebeen shown both by genetic and biochemicalmethods to interact with Cdks, yet their functionremains mysterious. Initially identified in fissionyeast (as suc1+; Hayles et al., 1986; Hindley et al.,1987) and in budding yeast (as Cks1; Hadwigeret al., 1989) as the gene products of suppressors oftemperature-sensitive Cdk mutations, these highlyconserved proteins were later shown to bind tightlyto Cdks in vitro (Brizuela et al., 1987). Humanhomologs have subsequently been identified thatstructurally resemble the yeast proteins and canfunctionally substitute for them in vivo (Richardsonet al., 1990). Based on genetic analysis in the twoyeasts, it is clear that Cks proteins provide anessential function (Hayles et al., 1986; Hindley et al.,1987; Hadwiger et al., 1989; Tang & Reed, 1993).Beyond that, extensive genetic and biochemicalanalysis has yielded little insight into the biologicalrole that the Cks proteins play in the cell cycle.
In order to address this unresolved issue, wehave orchestrated a concerted structural andmutational analysis of the human and yeast Cksproteins. We have reported previously, based onX-ray diffraction structural determinations, thatCks proteins can exist in two distinct forms: a singledomain fold (Arvai et al., 1995) and a b-inter-changed dimer (Parge et al., 1993; Bourne et al.,1995). The latter form consists of two subunits heldtogether by an anti-parallel b strand exchangebetween their respective C termini. In the case ofone of the human isoforms, CksHs2, three of thesedimers can assemble to form a hexamer (Pargeet al., 1993). However, the determination of thesestructures has not revealed the nature of theCdk-binding interface or insight into the import-
ance of Cdk binding to Cks function. We thereforehave attempted to address these issues usingsite-directed mutagenesis. We report here a mutant,CksHs2E63Q, that does not interfere with Cksstructure or ability to multimerize, but whicheliminates Cdk binding and biological viability,allowing us, for the first time, to draw inferences onthe relationship between these various parameters.
Results
A mutation targeted at a conserved residue ofCksHs2 confers a loss of biological function
Based on the determination of the three-dimen-sional structure of CksHs2 (Parge et al., 1993), weused site-directed mutagenesis to target mutationsto various conserved domains of the protein. Oneof these mutations, Glu63 to Gln, falls within ahighly conserved region of b strand that forms ab-hinge motif (Bourne et al., 1995). The flexibleb-hinge facilitates an antiparallel exchange ofC-terminal b strands between subunits and thecreation of an interlocking dimer, three of whichform the hexamer observed in the crystal structure(Parge et al., 1993). Although hexamers have beenobserved for only one of several homologous Cksproteins analyzed, interlocking dimers have beendocumented for human and yeast Cks homologs(Bourne et al., 1995; Parge et al., 1993; Y. B., M. H.W, S. I. R. & J. A. T., unpublished results). Based onanalysis of these structures, the sequence conservedb-interchange region, particularly a motif known asthe b-hinge, was predicted to play a key role indimerization. Seeking mutations that might affectmultimerization, Glu63, which interacts symmetri-cally across the exchanged b-strands in the dimerand is centered in the b-hinge, was targeted formutagenesis.
As both human CksHs1 and CksHs2 can rescuethe lethality of deleting the gene encoding theendogenous budding yeast homolog, Cks1(Richardson et al., 1990), we used this system to testwhether CksHs2E63Q could provide the essentialCks function in yeast. CksHs2 and CksHs2E63Qwere expressed in a yeast strain genomicallydisrupted for CKS1 and containing the wild-typegene on a plasmid. The CksHs2 alleles were underthe control of the inducible GAL1 promoter. Whenwild-type CksHs2 was expressed as a result of
Table 1. b-galactosidase activities of CDK/CksHs2 in the two-hybridassay and in vivo rescue of cks1 disruption by CksHs2 mutants
Rescue ofpACT2- pAS1-CDK1 pAS1-CDK2 cks1 disruption
CKSHS2: wild-type 100 100 +ckshs2: E63Q 3.58(21.10) 7.95(22.15) −
The b-galactosidase activities are mean values of four independent experimentsand are expressed as a percentage of the activity of the wild-type constructs.

Mutation in CksHs2 Disrupts Cdk Binding648
growth on medium containing galactose, theplasmid containing yeast CKS1 could be cured bygrowth on non-selective medium (Table 1). How-ever, when CksHs2E63Q was expressed in the samemanner, the plasmid containing yeast CKS1 couldnot be cured, even after prolonged growth underconditions non-selective for the plasmid marker(Table 1). Thus, the CksHs2E63Q protein isincapable of performing at least one essentialbiological function in yeast (Table 1). Because of thehighly conserved nature of Cks proteins, it isassumed that the biological functions in yeast aresimilar to those in mammalian cells. A similar resultwas obtained for the analogous mutation in theyeast homolog Cks1 (M. H. W., Y. B., S. I. R. &J. A. T., unpublished results).
CksHs2E63Q has a much lower affinity forhuman CDK1 and CDK2 in vivo and in vitro
The yeast Cks homologs, initially identifiedbased on genetic interactions with cyclin-depen-dent kinases, were subsequently shown to have ahigh affinity for Cdks in vitro (Brizuela et al., 1987).We wished to determine whether the inability ofCksHs2E63Q to rescue Cks1 function in yeast couldbe explained by a reduced ability to bind whatpresumably are natural targets, human CDK1 andCDK2. To assay binding in vivo, we used the yeasttwo-hybrid system (for review see Phizicky &Fields, 1995). CksHs2 and CksHs2E63Q were fusedto the Gal4 transcriptional activation domain whileCDK1 and CDK2 were fused to the yeast Gal4 DNAbinding domain. Interaction was assayed bymeasuring b-galactosidase activity produced froma reporter. Although CksHs2 and CksHs2E63Qproteins were produced at similar levels based onWestern blotting, the mutant protein interactedwith CDK1 and CDK2 at 4% and 8% of the level ofwild-type, respectively, indicating a drastic impair-ment of binding (Table 1). Similar results wereobtained for the equivalent yeast cks1 mutation(M. H. W., Y. B., J. A. T. & S. I. R, unpublishedresults). These results suggest that the inability ofthese mutant proteins to provide biologicalfunction is likely to be due to an impaired abilityto bind to cyclin-dependent kinases.
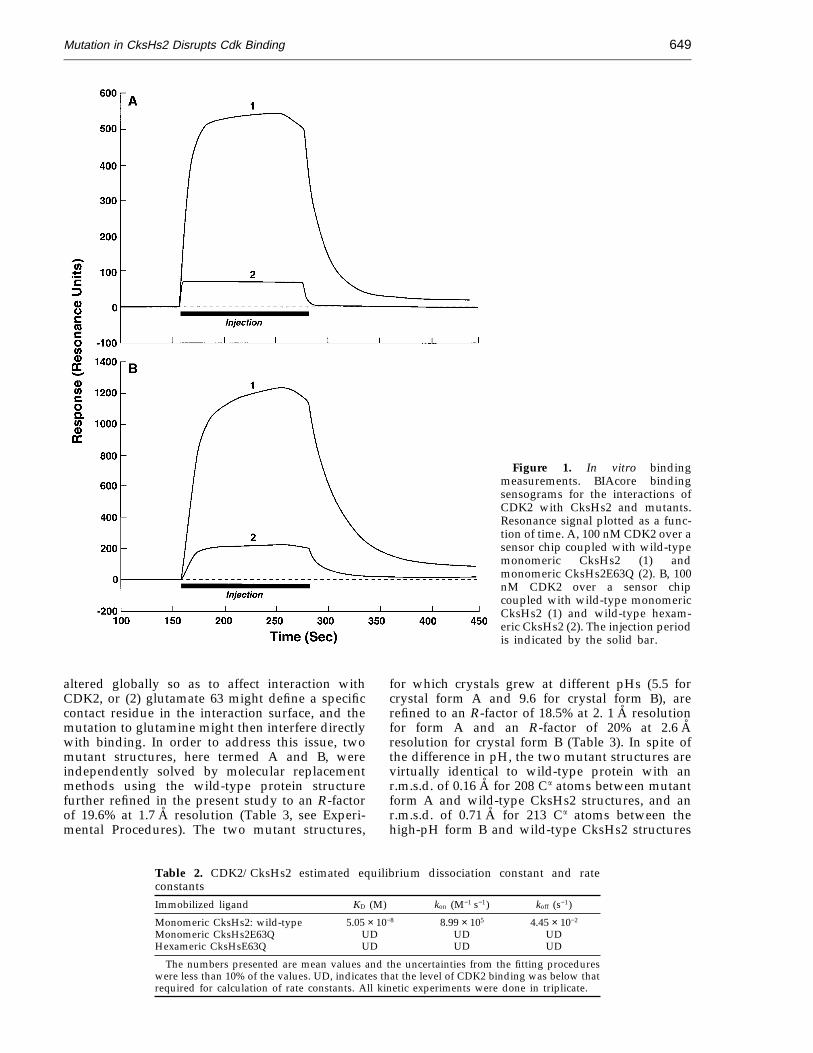
As a second test of CksHs2 mutant and wild-typeaffinities for CDK2, we measured the bindingconstants associated with these interactions in vitrousing a BIAcore surface plasmon resonancedetection system. Whereas a strong signal wasdetected when CDK2 and monomeric wild-typeCksHs2 were allowed to interact, giving anequilibrium dissociation constant of 5.05 × 10−8 M,only a slight interaction with CDK2 above back-ground was detected for the mutant protein (Fig-ure 1A; Table 2). It is, therefore, clear that the abilityof monomeric CksHs2E63Q to bind to CDK2 isseverely impaired compared to wild-type. Thisreduced ability to bind can, in principle, explain themutant protein’s biological defect.
The CksHs2E63Q mutation does not affectdimer/hexamer formation but thedimer/hexamer form does not bind CDK2
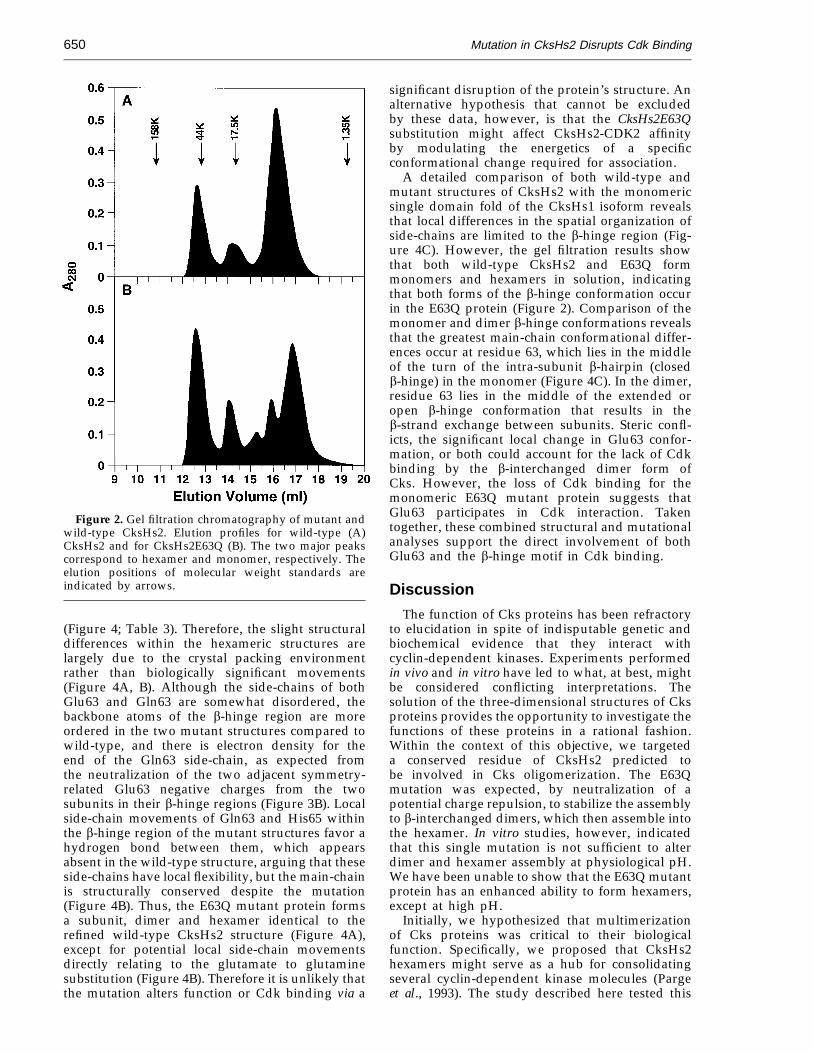
In the course of purifying CksHs2 for crystallo-graphic studies, it was observed that the proteinexisted in a mixture of assembly states includingmonomers and hexamers (Parge et al., 1993). Thefirst structure determined was that of the CksHs2hexamer, composed of three interlocked dimers(Parge et al., 1993). It was initially hypothesized thatthe hexamer (or dimer) was the active form of theprotein. We therefore sought to determine if theE63Q mutant protein was altered in multimerassembly properties. We discovered, however, thatCksHs2E63Q displayed an equivalent tendency toassemble into dimers and hexamers as compared tothe wild-type protein (Figure 2). Due to the removalof the charge repulsion that occurs between thesymmetry-related Glu63 side-chains in the wild-type protein dimer (Parge et al., 1993), theCksHs2E63Q protein appears to have equal orimproved stability over the wild-type protein interms of its purification and crystallization behav-ior, including maintaining its solubility andhexameric (trimer of dimers) assembly from pH 5.5to pH 9.6. Therefore, an altered ability to formmultimers was not likely to account for the loss ofbiological activity, consistent with the idea that thebiological defect of the mutant protein was due,instead, to a reduced ability to bind Cdks. Aparallel issue, however, pertains to the question ofwhether monomeric or hexameric or both forms ofCksHs2 can bind to CDK2. To address this, thebinding affinity between monomeric or hexamericCksHs2 and CDK2 was determined directly usingthe BIAcore instrument (Figure 1B; Table 2). Bothmonomeric and hexameric CksHs2 protein,purified by gel filtration chromatography, wereimmobilized and tested for interaction with CDK2.Monomeric CksHs2 protein binds strongly toCDK2, whereas the hexamer preparation showsonly a slight interaction. While we cannot discountthe possibility that the hexamer interacts veryweakly with CDK2, the signal detected likelyresults from dissociation of a small amount of thehexamer to monomer during the purification andcoupling procedures. These results suggest that thehexamer is an inactive form or has an alternativefunction to Cdk binding. Observations with thefission yeast homolog suc1 also suggest that thedimeric b-interchanged form is incapable ofbinding Cdks (data not shown). These data suggestthat the monomeric bent b-hinge form of Cks,rather than the extended b-hinge form character-istic of the b-interchanged dimer, binds Cdks.
The CksHs2E63Q mutation does not affectprotein structure
The reduced affinity of the monomeric CksHs2mutant protein for CDK2 could be explained in twoways: (1) the structure of the protein might be
Mutation in CksHs2 Disrupts Cdk Binding 649
Figure 1. In vitro bindingmeasurements. BIAcore bindingsensograms for the interactions ofCDK2 with CksHs2 and mutants.Resonance signal plotted as a func-tion of time. A, 100 nM CDK2 over asensor chip coupled with wild-typemonomeric CksHs2 (1) andmonomeric CksHs2E63Q (2). B, 100nM CDK2 over a sensor chipcoupled with wild-type monomericCksHs2 (1) and wild-type hexam-eric CksHs2 (2). The injection periodis indicated by the solid bar.
altered globally so as to affect interaction withCDK2, or (2) glutamate 63 might define a specificcontact residue in the interaction surface, and themutation to glutamine might then interfere directlywith binding. In order to address this issue, twomutant structures, here termed A and B, wereindependently solved by molecular replacementmethods using the wild-type protein structurefurther refined in the present study to an R-factorof 19.6% at 1.7 A resolution (Table 3, see Experi-mental Procedures). The two mutant structures,
for which crystals grew at different pHs (5.5 forcrystal form A and 9.6 for crystal form B), arerefined to an R-factor of 18.5% at 2. 1 A resolutionfor form A and an R-factor of 20% at 2.6 Aresolution for crystal form B (Table 3). In spite ofthe difference in pH, the two mutant structures arevirtually identical to wild-type protein with anr.m.s.d. of 0.16 A for 208 Ca atoms between mutantform A and wild-type CksHs2 structures, and anr.m.s.d. of 0.71 A for 213 Ca atoms between thehigh-pH form B and wild-type CksHs2 structures
Table 2. CDK2/CksHs2 estimated equilibrium dissociation constant and rateconstantsImmobilized ligand KD (M) kon (M−1 s−1) koff (s−1)
Monomeric CksHs2: wild-type 5.05 × 10−8 8.99 × 105 4.45 × 10−2
Monomeric CksHs2E63Q UD UD UDHexameric CksHsE63Q UD UD UD
The numbers presented are mean values and the uncertainties from the fitting procedureswere less than 10% of the values. UD, indicates that the level of CDK2 binding was below thatrequired for calculation of rate constants. All kinetic experiments were done in triplicate.
Mutation in CksHs2 Disrupts Cdk Binding650
Figure 2. Gel filtration chromatography of mutant andwild-type CksHs2. Elution profiles for wild-type (A)CksHs2 and for CksHs2E63Q (B). The two major peakscorrespond to hexamer and monomer, respectively. Theelution positions of molecular weight standards areindicated by arrows.
significant disruption of the protein’s structure. Analternative hypothesis that cannot be excludedby these data, however, is that the CksHs2E63Qsubstitution might affect CksHs2-CDK2 affinityby modulating the energetics of a specificconformational change required for association.
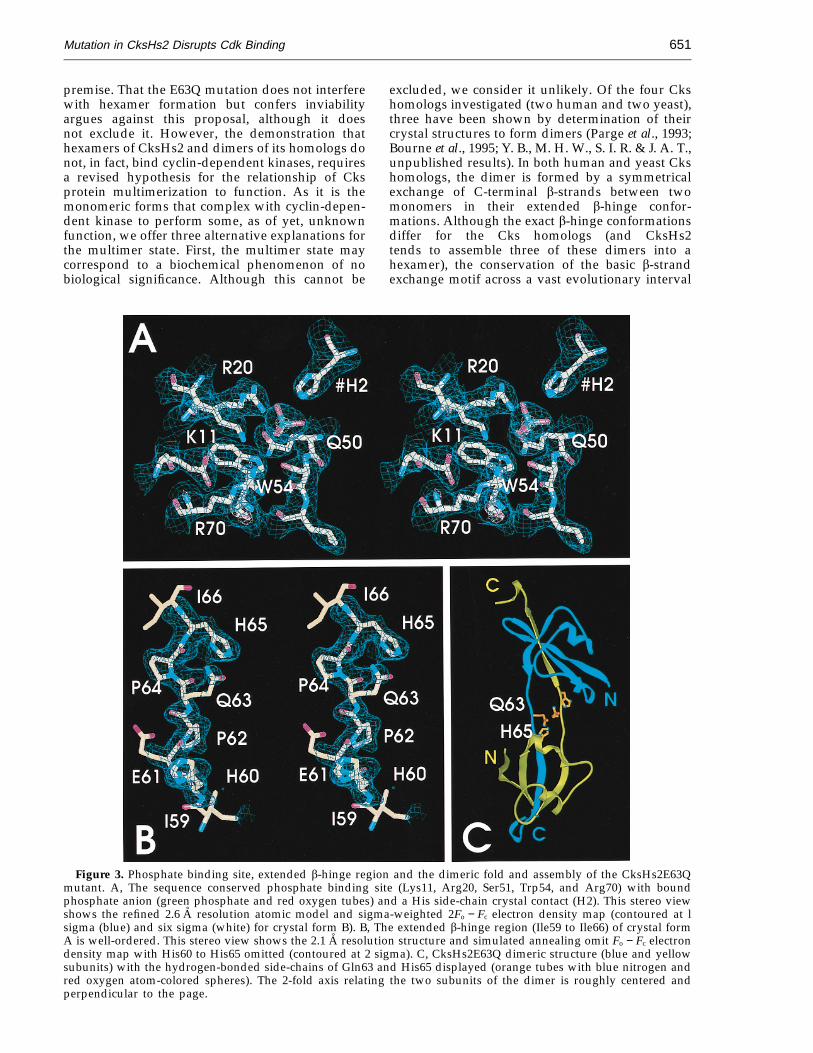
A detailed comparison of both wild-type andmutant structures of CksHs2 with the monomericsingle domain fold of the CksHs1 isoform revealsthat local differences in the spatial organization ofside-chains are limited to the b-hinge region (Fig-ure 4C). However, the gel filtration results showthat both wild-type CksHs2 and E63Q formmonomers and hexamers in solution, indicatingthat both forms of the b-hinge conformation occurin the E63Q protein (Figure 2). Comparison of themonomer and dimer b-hinge conformations revealsthat the greatest main-chain conformational differ-ences occur at residue 63, which lies in the middleof the turn of the intra-subunit b-hairpin (closedb-hinge) in the monomer (Figure 4C). In the dimer,residue 63 lies in the middle of the extended oropen b-hinge conformation that results in theb-strand exchange between subunits. Steric confl-icts, the significant local change in Glu63 confor-mation, or both could account for the lack of Cdkbinding by the b-interchanged dimer form ofCks. However, the loss of Cdk binding for themonomeric E63Q mutant protein suggests thatGlu63 participates in Cdk interaction. Takentogether, these combined structural and mutationalanalyses support the direct involvement of bothGlu63 and the b-hinge motif in Cdk binding.
Discussion
The function of Cks proteins has been refractoryto elucidation in spite of indisputable genetic andbiochemical evidence that they interact withcyclin-dependent kinases. Experiments performedin vivo and in vitro have led to what, at best, mightbe considered conflicting interpretations. Thesolution of the three-dimensional structures of Cksproteins provides the opportunity to investigate thefunctions of these proteins in a rational fashion.Within the context of this objective, we targeteda conserved residue of CksHs2 predicted tobe involved in Cks oligomerization. The E63Qmutation was expected, by neutralization of apotential charge repulsion, to stabilize the assemblyto b-interchanged dimers, which then assemble intothe hexamer. In vitro studies, however, indicatedthat this single mutation is not sufficient to alterdimer and hexamer assembly at physiological pH.We have been unable to show that the E63Q mutantprotein has an enhanced ability to form hexamers,except at high pH.
Initially, we hypothesized that multimerizationof Cks proteins was critical to their biologicalfunction. Specifically, we proposed that CksHs2hexamers might serve as a hub for consolidatingseveral cyclin-dependent kinase molecules (Pargeet al., 1993). The study described here tested this
(Figure 4; Table 3). Therefore, the slight structuraldifferences within the hexameric structures arelargely due to the crystal packing environmentrather than biologically significant movements(Figure 4A, B). Although the side-chains of bothGlu63 and Gln63 are somewhat disordered, thebackbone atoms of the b-hinge region are moreordered in the two mutant structures compared towild-type, and there is electron density for theend of the Gln63 side-chain, as expected fromthe neutralization of the two adjacent symmetry-related Glu63 negative charges from the twosubunits in their b-hinge regions (Figure 3B). Localside-chain movements of Gln63 and His65 withinthe b-hinge region of the mutant structures favor ahydrogen bond between them, which appearsabsent in the wild-type structure, arguing that theseside-chains have local flexibility, but the main-chainis structurally conserved despite the mutation(Figure 4B). Thus, the E63Q mutant protein formsa subunit, dimer and hexamer identical to therefined wild-type CksHs2 structure (Figure 4A),except for potential local side-chain movementsdirectly relating to the glutamate to glutaminesubstitution (Figure 4B). Therefore it is unlikely thatthe mutation alters function or Cdk binding via a
Mutation in CksHs2 Disrupts Cdk Binding 651
premise. That the E63Q mutation does not interferewith hexamer formation but confers inviabilityargues against this proposal, although it doesnot exclude it. However, the demonstration thathexamers of CksHs2 and dimers of its homologs donot, in fact, bind cyclin-dependent kinases, requiresa revised hypothesis for the relationship of Cksprotein multimerization to function. As it is themonomeric forms that complex with cyclin-depen-dent kinase to perform some, as of yet, unknownfunction, we offer three alternative explanations forthe multimer state. First, the multimer state maycorrespond to a biochemical phenomenon of nobiological significance. Although this cannot be
excluded, we consider it unlikely. Of the four Ckshomologs investigated (two human and two yeast),three have been shown by determination of theircrystal structures to form dimers (Parge et al., 1993;Bourne et al., 1995; Y. B., M. H. W., S. I. R. & J. A. T.,unpublished results). In both human and yeast Ckshomologs, the dimer is formed by a symmetricalexchange of C-terminal b-strands between twomonomers in their extended b-hinge confor-mations. Although the exact b-hinge conformationsdiffer for the Cks homologs (and CksHs2tends to assemble three of these dimers into ahexamer), the conservation of the basic b-strandexchange motif across a vast evolutionary interval
Figure 3. Phosphate binding site, extended b-hinge region and the dimeric fold and assembly of the CksHs2E63Qmutant. A, The sequence conserved phosphate binding site (Lys11, Arg20, Ser51, Trp54, and Arg70) with boundphosphate anion (green phosphate and red oxygen tubes) and a His side-chain crystal contact (H2). This stereo viewshows the refined 2.6 A resolution atomic model and sigma-weighted 2Fo − Fc electron density map (contoured at lsigma (blue) and six sigma (white) for crystal form B). B, The extended b-hinge region (Ile59 to Ile66) of crystal formA is well-ordered. This stereo view shows the 2.1 A resolution structure and simulated annealing omit Fo − Fc electrondensity map with His60 to His65 omitted (contoured at 2 sigma). C, CksHs2E63Q dimeric structure (blue and yellowsubunits) with the hydrogen-bonded side-chains of Gln63 and His65 displayed (orange tubes with blue nitrogen andred oxygen atom-colored spheres). The 2-fold axis relating the two subunits of the dimer is roughly centered andperpendicular to the page.

Mutation in CksHs2 Disrupts Cdk Binding652
Table 3. Crystallographic data and refinement statisticsCofactor Wild-type sulfate E63Q form A sulfate E63Q form B phosphate
Data collectionSpace group C2 C2 P43212Cell dimensions (A) a = 100.1, b = 57.9, a = 100.1, b = 57.5, a = b = 53.9,
c = 51.3, b = 117.6 c = 51.1, b = 117.3 c = 177.3N. monomer per a.u. 3 3 3Resolution limit (A) 17–1.7 19–2.1 26–2.6Observations/Reflections 169,395/26,743 86,437/14,530 67,040/8414Completeness (%)/Rsym (%) 93/6.9 96/5.4 96/8.3
Final refinement parametersN. protein atoms 1984 1975 1931N. cofactor atomsa 35 30 10N. solvent atoms 195 167 78Resolution range (A) 5–1.7 6–2.1 6–2.6N. reflections (all data) 25,411 13,864 7634R-factor/R-free (%) 19.6/24.6 18.5/26.0 20.0/29.0
Geometry of the final modelRMS dev. bond distances (A) 0.007 0.007 0.007RMS dev. bond angles (°) 1.6 1.9 1.9Residues in most allowed regions (%) 93 92 82G score + 0.25 + 0.24 + 0.15
B-factor value (A2)main-chain atoms 32 27 31side-chain atoms 38 32 34r.m.s. main-chain atoms 1.3 1.8 2.6r.m.s. side-chain atoms 2.2 3.1 3.3cofactor atoms 60 56 56solvent atoms 50 43 46
RMS deviations from superpositionb
Subunit 1 to 2 0.43 0.58 0.60Subunit 2 to 3 0.60 0.64 0.48a Cofactor atoms within the crystallographic asymmetric unit differ for the three structures. For wild-type protein, there are seven
sulfate ions: one bound to each subunit anion-binding site, one near Thr35 side-chain and backbone nitrogen atoms of Lys34 andThr35 in each subunit, plus one additional sulfate ion near Glu61 for only two of the subunits. For E63Q crystal form A, there aresix sulfate ions: one bound to each subunit anion-binding site, plus bound near Thr35 side-chain and backbone nitrogen atoms ofLys34 and Thr35 in each subunit, but none near Glu61. For the E63Q high pH crystal form B, there are only two bound phosphateions: one bound to each of two anion-binding sites.
b Superpositions were done with the program SUPERPOSE (Collaborative Computational Project, number 4 (1996)) using the Ca
atoms of residues 5 to 74 in each structure.
suggests an underlying function. Secondly, multi-mers may constitute a negative regulatory state.Based on the observation that dimers and hexamersdo not bind cyclin-dependent kinase, the regulatedoligomerization of Cks proteins is a plausiblemeans of negatively regulating their interactionswith cyclin-dependent kinases without requiringchanges in Cks protein levels. This model suggeststhat intracellular signals, as yet unknown, canefficiently mediate that transition from monomer tomultimer, as well as the reverse transition. Finally,the Cks oligomers may have a cellular functionunrelated to cyclin-dependent kinase binding.Further mutational analysis will be required toclarify this issue.
Lethality of E63Q
The results presented here, from biological,biochemical and crystallographic characterization,suggest that CksHs2E63Q has only one biochemicalproperty that is likely to be relevant to thebiological phenotype conferred. Specifically, itsmonomer has a reduced affinity for cyclin-dependent kinase. Since the mutation does not
appear to affect the fold of the molecule, asdetermined crystallographically, and by all avail-able criteria there is no difference in stabilitybetween the mutant and wild-type protein, thesimplest hypothesis consistent with all the data isthat glutamate 63 defines an important contactresidue for interactions with Cdks. If this interpret-ation is correct, then inability of Cks dimers (andhexamers) to bind Cdks can be easily rationalized.The structures of the CksHs2 hexamer (Parge et al.,1993) and the suc1 (Bourne et al., 1995) and Cks1(Y. B., M. H. W., S. I. R. & J. A. T., unpublishedresults) dimers indicate that glutamate 63 (and itsequivalent in the other structures) is directlyinvolved in the interlocked exchange of b strandsand therefore likely to be unavailable or in anincorrect b-hinge conformation for Cdk inter-actions. After completing the work reported here,we solved the structure of the closely relatedCksHs1 isoform in complex with CDK2 (Bourneet al., 1996). Although similar to CksHs2 in primarystructure, CksHs1 has never been shown to forman interlocked dimer, suggesting some inherentdifferences in subunit interactions. Nevertheless,the conclusions drawn from the structure of theCksHs1/CDK2 heterodimer are consistent with the

Mutation in CksHs2 Disrupts Cdk Binding 653
inferences drawn from the genetic, biochemical andstructural studies described in this report.
The CksHs2 wild-type vs . E63Q structure
The virtual identity of the wild-type and mutantproteins is apparent from the combination of sizingcolumn chromatography and X-ray crystallo-graphic results. Both wild-type and E63Q mutantproteins form monomers and hexamers in solution(Figure 2), although we have been able to crystallizeonly the hexamer form assembled from threeb-interchanged dimers. To test that the mutant didnot alter Cks structure, we refined the wild-typestructure to 1.7 A resolution and solved mutant
structures in the same space group and conditionsas wild-type and also in a different space group atpH 9.6, where the Glu to Gln charge neutralizationmight be expected to show maximal differences. Allthree structures are identical within experimentalerror, except for small side-chain rotations of Gln63and His65 to form hydrogen bonds. Comparisonof these b-interchanged dimer structures to theCksHs1 monomer structure reveals that the onlysignificant conformational differences occur locallywithin the b-hinge motif and include Glu63. Thusthe structural and biochemical results reported hereimplicate the Glu63 side-chain and the closed,monomer conformation of the b-hinge in Cdkbinding.
Figure 4. Highly similar CksHs2E63Q and wild-type dimeric structures share an extended b-hinge conformationdistinct from the closed b-hinge turn conformation of the monomeric Cks fold. A, Stereo view of the wild-typehexameric CksHs2 structure (yellow Ca trace) superimposed with that of CksHs2E63Q crystal form B (blue Catrace).The side-chains of Gln63 are displayed for each subunit, together with the CksHs2E63Q phosphate anion (magentaspheres). In this view down the non-crystallographic 3-fold axis of the hexamer, the crystallographic 2-fold, which runsdiagonally from the lower left to the upper right, relates the top three subunits to the bottom three subunits with eachdimer containing one top and one bottom subunit. Note the near identity of the wild-type pH 5.5 and mutant pH 9.6structures despite the large pH difference. B, Superimposed structures of the 1.7 A resolution refined wild-type CksHs2dimer structure (yellow subunits) and the 2.1 A refined CksHs2E63Q form A dimer (magenta subunits). The view isroughly the crystallographic 2-fold, which relates the two subunits forming the b-interchanged dimer. The backbonefold for the b-strand swapped dimers is shown as ribbons with Glu63, Gln63 and His65 side-chains (white tubes withblue nitrogen and red oxygen atom-colored spheres for wild-type CksHs2 and orange tubes with atom-colored spheresfor CksHs2E63Q crystal form A). The close superposition reveals that these structures are identical within experimentalerror. C, The b-interchanged dimeric CksHs2E63Q of crystal form A (yellow and blue subunits with orange carbontubes plus blue nitrogen and red oxygen atom-colored spheres for Gln63 and His65) superimposed upon themonomeric fold of CksHs1 (magenta subunit with white tubes for side-chain carbons of Glu63 and His65), and foldsshown by ribbons. Superposition was done using Ca atoms of the yellow subunit of the dimer. White circle, centeredon the Glu63 side-chain of the closed b-hinge within the CksHs1 monomeric fold, emphasizes the difference in residue63 main-chain and side-chain conformation between the Cks monomer and dimer folds. The sizing column resultssuggest that both mutant and wild-type CksHs2 can similarly switch between monomer and b-interchanged dimericforms.

Mutation in CksHs2 Disrupts Cdk Binding654
Experimental Procedures
Site-directed mutagenesis
The E63Q mutation was introduced into the humanCKSHS2 gene, contained in the bacterial expressionplasmid pRK171 (Rosenberg et al., 1987), using theTransformer Mutagenesis Kit (Clontech Laboratories Inc.,Palo Alto, CA) as described by the manufacturer. Themutagenic primer was: 5'-GAA TAT GTG GTT GTG GCTCAT G-3'. The mutant sequences were amplified by PCRusing flanking oligonucleotides containing BamH1restriction sites, and the products were cloned into theyeast galactose-inducible plasmid YCpG2 (Richardsonet al., 1989) and the two-hybrid GAL4 transcriptionalactivation domain plasmid pACT2. The resultingconstructs were confirmed by DNA sequencing, and themutant CksHs2 protein was purified as described for thewild-type protein (Parge et al., 1993).
Two-hybrid assays
CKSHS2 wild-type and mutants were cloned intothe BamH1 restriction site in the polylinker of theplasmid pACT2 to yield GAL4 transcriptional activ-ation domain fusion constructs tagged with thehaemaglutinin (HA) epitope and carrying the LEU2gene. The human CDK1 and CDK2 open reading framewas cloned into the BamH1 restriction site in thepolylinker of the plasmid pAS1 (Durfee et al., 1993)to produce GAL4 DNA-binding domain fusion con-structs tagged with the HA epitope and carrying theTRP1 gene. The pACT2 constructs were transformed intothe haploid Saccharomyces cerevisiae strain Y190 (MATagal4 gal80 his3 trp1-901 ade2-101 ura3-52 leu2-3,112 +URA3::GAL : lacZ, LYS2::GAL(UAS) : HIS3cyhr) andselected on plates lacking leucine. The pAS1 constructswere transformed into the haploid strain Y187 (MATagal4 gal80 his3 trp1-901 ade2-101 ura3-52 leu2-3,112 met−
URA3::GAL : lacZ) and selected on plates lackingtryptophan. Yeast transformations were performed asdescribed by Elble (1992). The expression of the fusionproteins was confirmed by Western blotting using amonoclonal antibody (12CA5) to the HA epitope.
Yeast cells containing both pAS1-CDK1 or pAS1-CDK2and pACT2-CKSHS2 are not viable making it impossibleto maintain strains containing both plasmids (M. H. W.& S. I. R., unpublished results). Therefore the method of‘‘quantitative mating’’ was used to measure theinteraction between the fusion proteins in newly formedzygotes produced from mating haploid cells containingthe individual plasmids. Y187 cells (5 × 107) containingthe pAS1-CDK1 or pAS1-CDK2 construct were vacuumfiltered onto 0.45 mm Metricel membranes (GelmanSciences). Y190 cells (5 × 107) carrying the pACT2-CK-SHS2 constructs were filtered over the layer of Y187 cells,and the filter was then placed onto a YEPD platecontaining 1 M sorbitol with the cell side up andincubated at 30°C. After five hours, the filter was placedinto a 50 ml conical tube with 10 ml of YEPD mediumand vortexed to resuspend cells. The filter was discarded,and a sample of the cells was fixed in formaldehyde formicroscopic scoring of the percentage of zygotes. Themating efficiency typically ranged between 10 and 20%.The cells were pelleted by a ten minute spin at 1000 rpmin a clinical centrifuge. Extracts were prepared byresuspending the pellets in 150 ml of 100 mM Tris-HCl(pH 8), 20% glycerol (v/v), 1 mMb-mercaptoethanol,1 mg/ml leupeptin, 1 mM phenylmethylsulfonyl chloride
(PMSF), 1 mg/ml pepstain A and 2 mg/ml aprotinin.Glass beads (0.5 mm diameter) were added to the levelof the meniscus and vortexed 5 × 30 seconds with 30second intermissions on ice. Extracts were clarified by a15 minute spin at 14,000 g and then frozen in liquidnitrogen and stored at −70°C. Protein concentrationswere measured using the Bradford assay (Bradford,1976). b-Galactosidase activity was measured using achemiluminescence assay with the Galacto-Light Plussystem (Tropix, Inc.) following the manufacturer’sprocedure. Briefly, 5 to 20 ml of extract was added to100 ml of diluted Galacton and incubated at roomtemperature for 30 minutes. 100 ml of accelerator solutionwas added and chemiluminescence was measured induplicate for five seconds using a Lumat LB9501luminometer, The chemiluminescent signal was normal-ized for the amount of protein and the percentage ofzygotes and expressed as a percentage of b-galactosidaseactivity relative to wild-type CksHs2. Three to fiveindependent matings were done for each experiment.
In vivo rescue of cks1 disruption
The ability of the CksHs2E63Q allele to function in vivowas tested in a yeast strain that has the chromosomal cks1gene disrupted and is kept alive with a plasmidcontaining a wild-type copy of CKS1, cks1::LEU2(CEN1::TRP1::CKS1) (Richardson et al., 1990). Haploidcells were transformed with the inducible vector YCpG2containing either wild-type or mutant CksHs2 under thecontrol of the GAL1 promoter and carrying the URA3gene. Transformants were selected based on tryptophanand uracil prototrophy and were switched to growth forseveral generations on medium containing galactose (toallow expression of the mutant or wild-type CKSHS2allele from the GAL1 promoter) and tryptophan (to allowloss of the wild-type CKS1 gene and the TRP1 marker).Loss of the plasmid containing the wild-type CKS1 genewas monitored by plating and testing colonies fortryptophan auxotrophy.
Binding affinity measurements
Experiments were performed using surface plasmonresonance technology on a BioSensor BIAcore instrument(Pharmacia). Purified CksHs2 wild-type and E63Qmutant proteins were coupled through primary aminegroups to the activated carboxyl groups of CM5 sensorchips by the manufacturer’s procedure. The matrixsurface was activated with a mixture of 50 mMN-hydroxysuccinimide (NHS) and 200 mM N-ethyl-N '-(3-diethylaminopropyl)-carbodiimide (EDC) for sevenminutes at a flow rate of 5 ml/minute. Wild-type or E63Qmutant CksHs2 at a concentration of 0.1 mg/ml in10 mM N-2-hydroxyethylpiperazine-N '-2-ethanesulfonicacid (Hepes) (pH 7.4) and 3.4 mM ethylenediaminetetra-acetic acid (EDTA) was injected over the matrix forseven minutes at 5 ml/minute. De-activation of the matrixwas achieved with 1 M ethanolamine hydrochloride(pH 8.5) for seven minutes at 5 ml/minute. Theimmobilization yield of the protein was monitored fromthe increase in resonance units during the couplingprocedure. After each binding experiment, the surface ofthe sensor chip was regenerated by washing with 50%ethylene glycol (w/v), 0.5 M NaCl (pH 7.4) (Kusubataet al., 1992). Binding sensorgrams for the interaction ofCksHs2 wild-type and E63Q mutant protein were createdusing concentrations of CDK2 (a gift of W. Holmes andW. Rocque, Glaxo-Wellcome Research Institute) ranging

Mutation in CksHs2 Disrupts Cdk Binding 655
from 5 nM to 5 mM in 10 mM Hepes (pH 7.4), 3.4 mMEDTA, 150 mM NaCl and 0.001% surfactant P-20 (w/v).The sensorgrams were analyzed using the BIAevaluationsoftware (Pharmacia). Association (kon) and dissociation(koff) rate constants were calculated using the associationand dissociation phases of the sensorgrams, respectively.The Kd values were calculated using the equationKd = koff/kon. All kinetic experiments were done intriplicate.
Gel filtration chromatography of mutant andwild-type CksHs2
Gel filtration chromatography of CksHs2 andCksHs2E63Q were carried out as in Parge et al. (1993).150 mg of protein in 50 mM K2HPO4, 150 mM NaCl(pH 7.8), was loaded onto a Superose-75 column andeluted with the same buffer at a flow rate of 1 ml/minute.The identity of the eluted peaks was confirmed by 15%SDS-PAGE (w/v).
Crystallization
Crystals of native CksHs2 (0.5 × 0.5 × 0.8 mm) wereobtained by the vapor diffusion technique at roomtemperature from a protein solution (8 to 9 mg/ml)equilibrated with 60% saturated ammonium sulphate(w/v) and 0.1 M sodium citrate (pH 5.5). During theinitial crystallization trials, it appeared that CksHs2crystallized much more efficiently if the protein solutionwas left on ice for 1 to 4 days before setting up in drops.Further gel filtration experiments on CksHs2 revealedthat this procedure favored a hexameric assembly. Thecrystals belong to the space group C2 with celldimensions: a = 100.1 A, b = 57.7 A, c = 51.1 A andb = 117.4° giving a Vm value of 2.3 A3/Da (47% solvent)for a CksHs2 trimer in the asymmetric unit. Acomplete data set extending to 1.7 A resolution wascollected at beamline 7-1 of the Stanford SynchrotronRadiation Laboratory (SSRL), using a Mar-researchImage Plate. This new high resolution data providedincreased accuracy of the key Glu63 b-strand exchangeregion, which showed more disorder in the initialstructure.
Isostructural crystals of the mutant CksHs2E63Q, heretermed crystal form A, were obtained using the sameconditions as described above. A 2.1 A complete data setwas collected from a single crystal using a Mar-researchImage Plate mounted on a Siemens Rotating Anodeoperating a 50 kV × 100 mA. In addition, a second crystalform, form B, was obtained from 25% polyethylene glycol4 K and 5% ammonium phosphate (v/v from a saturatedsolution) as precipitant and 0.1 M Tris-HCl (pH 9.6). Thecrystals belong to the space group P43212 with celldimensions: a = b = 53.9 A and c = 177.3 A giving a Vm
value of 2.3 A3/Da (47% solvent) for a CksHs2 trimer inthe asymmetric unit. We were unable to grow wild-typecrystals at high pH, suggesting that the E63Q mutation,which does not affect monomer to hexamer equilibria atneutral pH, does stabilize the hexameric form atnon-physiologically high pH. Data were collected ona single crystal using a Mar-research image mounted on ahigh brilliance Rikagu rotating anode source operating a50 kVa × 100 mA. In all cases, oscillation images wereintegrated, scaled, and merged using DENZO andSCALEPACK (Otwinowski, 1993).
Molecular replacement
In the case of the E63Q mutant form B crystal, initialphases were obtained with the molecular replacementmethod using a CksHs2 trimer as a search model (Pargeet al., 1993). Solvent and sulfate molecules were removedfrom the initial model. Self-rotation functions calculatedusing data between 10 and 4 A and an integration radiusof 25 A showed a small peak (1 sigma level) in the 120°kappa section, confirming the presence of a CksHs2trimer in the asymmetric unit. The orientation of aCksHs2 trimer molecule was found with AMoReprogram packages (Navaza, 1994). The peak heights ofthe translation function corresponding to the CksHs2trimer were used to solve the space group ambiguity.After rigid-body refinement on the three subunits, thecorrelation and the R-factor were 57% and 40.5%,respectively, in the 15 A to 4 A resolution range.
Structure refinement
Native CksHs2
Crystallographic refinement was carried with X-PLOR(Brunger et al., 1987), starting from our refined 2.1 Aresolution model (Parge et al., 1993), with all water andsulfate molecules. The initial R-factor was 33% in the5 A to 1.7 A resolution range and multiple roundsof positional and individual B-factor refinement wereapplied. At the end of the first round, an anisotropicB-factor correction was applied to the data and the modelwas examined with both 2Fo − Fc and Fo − Fc electrondensity maps using TURBO-FRODO (Roussel & Cambi-lau, 1989). Manual rebuilding of side-chains coupled withpositional and B-factor refinement was continued untilthe R-factor converged. The final refinement model hasa crystallographic R-factor of 19.6% and consists of aminoacids 5 to 77, 4 to 79 and 4 to 79 in the three subunits,respectively, seven sulfate anions and 195 solventmolecules. Non-crystallographic symmetry was notrestrained during refinement, and no attempt was madeto model alternative conformations for side-chains,which were, however, clearly visible for residues Met23,Val55 and Leu67.
CksHs2E63Q: crystal form A
The isostructural CksHs2 model refined to 2.1 Aresolution was used as a starting model (Parge et al.,1993) with all water and sulfate molecules and Glu63truncated to Cb. Prior to positional refinement steps, arigid-body procedure was performed to compensate forthe small changes observed in the cell dimensionsbetween the two crystals. After this step, the R-factordropped from 44% to 33% in the 6 A to 3.5 A resolutionrange. During the course of the refinement, watermolecules and side-chains were fitted into both the2Fo − Fc and difference electron density maps.
CksHs2E63Q: crystal form B
Rigid-body refinement in the 6 A to 2.6 A resolutionrange of the molecular replacement solution with eachsubunit as a separate group was performed prior topositional refinement. Subsequent refitting and refine-ment cycles with addition of the bound phosphategroups and a conservative number of solvent moleculeswere performed until the R-factor value converged.

Mutation in CksHs2 Disrupts Cdk Binding656
The final models were examined with PROCHECK(Laskowski et al., 1993). The statistics of the lastrefinement cycle for the two mutant CksHs2 structuresare given in Table 3. The atomic coordinates of thesestructures will be deposited with the Protein Data Bank,Chemistry Department, Brookhaven National Labora-tory, Upton, NY 11973, USA and are available directlyfrom the authors on request until they have beenprocessed and released.
AcknowledgementsWe thank N.-H. Xuong for access to the University of
California, San Diego, Research Resource for ProteinCrystallography, for X-ray data collection on the E63Qform B crystals. We thank Carlos F. Barbas and RichardLerner for generously allowing us to use their BIAcoreInstrument and William Holmes and Warren Rocquefor purified recombinant human CDK2. M. H. W. wassupported by a postdoctoral fellowship from the MedicalResearch Council of Canada. This work was supported inpart by the Wellcome–Glaxo Research Institute.
ReferencesArvai, A. S., Bourne, Y., Hickey, M. J. & Tainer, J. A.
(1995). Crystal structure of the human cell cycleprotein CksHs1: single domain fold with similarityto kinase N-lobe domain. J. Mol. Biol. 249, 835–842.
Bourne, Y., Arvai, A. S., Bernstein, S. L., Watson, M. H.,Reed, S. I., Endicott, J. E., Noble, M. E., Johnson,L. N. & Tainer, J. A. (1995). Crystal structure of thecell cycle-regulatory protein sucl reveals a b-hingeconformational switch. Proc. Natl Acad. Sci. USA, 92,10232–10236.
Bourne, Y., Watson, M. H., Hickey, M. J., Holmes, W.,Rocque, W., Reed, S. I. & Tainer, J. A. (1996). Crystalstructure and mutational analysis of the humanCDK2 kinase complex with cell cycle-regulatoryprotein CksHs1. Cell, 84, 863–874.
Bradford, M. M. (1976). A rapid and sensitive method forthe quantification of microgram quantities of proteinutilizing the principle of protein-dye binding. Anal.Biochem. 72, 248–254.
Brizuela, L., Draetta, G. & Beach, D. (1987). p13suc1acts in the fission yeast cell division cycle as acomponent of the p34cdc2 protein kinase. EMBO J.6, 3507–3514.
Brunger, A. T., Kuriyan, J. & Karplus. M. (1987).Crystallographic R-factor refinement by moleculardynamics. Science, 235, 458–460.
Collaborative Computational Project, number 4 (1994).The CCP4 suite: programs for protein crystallogra-phy. Acta Crystallog. sect. D, 50, 760–763.
Deshaies, R. J. (1995). The self-destructive personality ofa cell cycle in transition. Curr. Opin. Cell Biol. 7,781–789.
Dunphy, W. G. (1994). The decision to enter mitosis.Trends Cell Biol. 4, 202–207.
Durfee, T., Becherer, K., Chen, P.-L., Yeh, S.-H., Yang, Y.,Kilburn, A. E., Lee, W.-H. & Elledge, S. J. (1993). Theretinoblastoma protein associates with the proteinphosphatase type I catalytic subunit. Genes Dev. 7,555–569.
Elble, R. (1992). A simple and efficient procedure fortransformation of yeasts. BioTechniques, 13, 18–20.
Gould, K. L. & Nurse, P. (1989). Tyrosine phosphoryl-ation of the fission yeast cdc2+ protein kinaseregulates entry into mitosis. Nature, 342, 39–45.
Hadwiger, J. A., Wittenberg, C., Mendenhall, M. D. &Reed, S. I. (1989). The Saccharomyces cerevisiae CKS1gene, a homolog of the Schizosaccharomyces pombesuc1+ gene, encodes a subunit of the Cdc28 proteinkinase complex. Mol. Cell. Biol. 9, 2034–2041.
Hayles, J., Aves, S. & Nurse, P. (1986). suc1 is an essentialgene involved in both the cell cycle and growth infission yeast. EMBO J. 5, 3373–3379.
Hindley, J., Phear, G., Stein, M. & Beach, D. (1987). suc1+
encodes a predicted 13-kilodalton protein that isessential for cell viability and is directly involved inthe division cycle of Schizosaccharomyces pombe. Mol.Cell. Biol. 7, 504–511.
Hunter, T. (1993). Braking the cycle. Cell, 75, 839–841.Hunter, T. & Pines, J. (1994). Cyclins and cancer. Cyclin
D and CDK inhibitors come of age. Cell, 79, 573–582.King, R. W., Jackson, P. K. & Kirschner, M. W. (1994).
Mitosis in transition. Cell, 79, 563–571.Kusubata, M., Tokui, T., Matsuoka, Y., Okumura, E.,
Tachibana, K., Hisanaga, S., Kishimoto, T., Yasuda,H., Kamijo, M., Ohba, Y., Tsujimura, K., Yatani, R. &Ingaki, M. (1992). p13 suc1 suppresses the catalyticfunction of p34cdc2 kinase for intermediate filamentproteins, in vivo. J. Biol. Chem. 276, 20937–20942.
Laskowski, R. A., MacArthur, M, W., Moss, D. S. &Thornton, J. M. (1993). PROCHECK: a program tocheck the stereochemical quality of protein struc-tures. J. Appl. Crystallog. 26, 283–291.
Morgan, D. O. (1995). Principles of CDK regulation.Nature, 374, 131–133.
Nasmyth, K. (1993). Control of the yeast cell cycle by theCdc28 protein kinase. Curr. Opin. Cell Biol. 5,166–179.
Navazza, J. (1994). AMoRe: an automated package formolecular replacement. Acta Crystallog. sect. A, 50,157–163.
Otwinowski, Z. (1993). Oscillation data reductionprogram. In Proc. CCP4 Study Weekend (Sawyer, L.,Isaacs, N. & Burley, S., eds), pp. 56–62, SERCDaresbury Laboratory, UK.
Parge, H. E., Arvai, A. S., Murtari, D. J., Reed, S. I. &Tainer, J. A. (1993). Human CksHs2 atomic structure:a role for its hexameric assembly in cell cycle control.Science, 262, 387–395.
Phizicky, E. M. & Fields, S. (1995). Protein–proteininteractions: methods for detection and analysis.Microbiol. Rev. 59, 94–123.
Pines, J. & Hunter, T. (1991). Cyclin-dependent kinases:a new cell cycle motif? Trends Cell Biol. 1, 117–121.
Reed, S. I. (1992). The role of p34 kinases in the G1to S-phase transition. Annu. Rev. Cell Biol. 8, 529–561.
Richardson, H. E., Wittenberg, C., Cross, F. & Reed, S. I.(1989). An essential G1 function for cyclin-likeproteins in yeast. Cell, 59, 1127–1133.
Richardson, H. E., Stueland, C. S., Thomas, J., Russell,P. & Reed, S. I. (1990). Human cDNAs encodinghomologs of the small p34Cdc28/cdc2-associated proteinof Saccharomyces cerevisiae and Schizosaccharomycespombe. Genes Dev. 4, 1332–1344.
Rosenberg, A. H., Lade, B. N., Chui, D. S., Lin, S. W.,Dunn, J. J. & Studier, F. W. (1987). Vectors forselective expression of cloned DNAs by T7 RNApolymerase. Gene, 56, 125–135.
Roussel, A. & Cambillau, C. (1989). TURBO-FRODO. InSilicon Graphics Geometry Partners Directory (Silicon

Mutation in CksHs2 Disrupts Cdk Binding 657
Graphics, ed.), pp. 77–78, Silicon Graphics, MountainView, CA.
Sherr, C. J. (1994). G1 Phase progression: cycling on cue.Cell, 79, 551–555.
Solomon, M. J. (1994). The functions(s) of CAK, the
p34cdc2-activating kinase. Trends Biochem. Sci. 19,496–500.
Tang, Y. & Reed, S. I. (1993). The Cdk-associated proteinCks1 functions both in G1 and G2 in Saccharomycescerevisiae. Genes Dev. 7, 822–832.
Edited by F. E. Cohen
(Received 27 February 1996; received in revised form 16 June 1996; accepted 24 June 1996)
Copyright © 2022 FDOKUMEN




















