
Hemodynamic Changes in Splanchnic Blood Vessels in Portal Hypertension
15
Hemodynamic Changes in Splanchnic Blood Vessels in Portal Hypertension ISABELLE COLLE,* ANJA M. GEERTS, CHRISTOPHE VAN STEENKISTE, AND HANS VAN VLIERBERGHE Department of Hepatology and Gastroenterology, Ghent University Hospital, Ghent, Belgium ABSTRACT Portal hypertension (PHT) is associated with a hyperdynamic state characterized by a high cardiac output, increased total blood volume, and a decreased splanchnic vascular resistance. This splanchnic vasodilation is a result of an important increase in local and systemic vasodilators (ni- tric oxide, carbon monoxide, prostacyclin, endocannabinoids, and so on), the presence of a splanchnic vascular hyporesponsiveness toward vaso- constrictors, and the development of mesenteric angiogenesis. All these mechanisms will be discussed in this review. To decompress the portal circulation in PHT, portosystemic collaterals will develop. The presence of these portosystemic shunts are responsible for major complications of PHT, namely bleeding from gastrointestinal varices, encephalopathy, and sepsis. Until recently, it was accepted that the formation of colla- terals was due to opening of preexisting vascular channels, however, recent data suggest also the role of vascular remodeling and angiogenesis. These points are also discussed in detail. Anat Rec, 291:699–713, 2008. Ó 2008 Wiley-Liss, Inc. Key words: portal hypertension; splanchnic; angiogenesis; hemodynamic Portal hypertension (PHT) is the major hemodynamic complication of a variety of diseases that obstruct portal blood flow. Portal pressure gradient (DP) is the result of portal vascular resistance (R) and portal venous inflow (Q) in analogy with Ohm’s law: DP = R × Q. In normal circumstances the portal pressure gradient varies between 2 and 6 mmHg. Two major theories were put forward to explain portal hypertension. The ‘‘backward’’ theory assumes that the resistance to portal flow results in portal hypertension (Benoit et al., 1985). Portal vascular resistance consists of the sum of the serial resistance in portal vein and intrahepatic vascular bed and of the parallel resistance of the collaterals. The cause of enhanced vascular resist- ance can be localized pre-, intra-, and posthepatic. Pre- hepatic portal hypertension is mostly due to a portal vein thrombosis and is associated with a quite normal liver function. Posthepatically portal hypertension can be caused, for example, by thrombosis of the hepatic veins (Budd Chiari syndrome), occlusion of the caval vein (web), and constrictive pericarditis. The intrahe- patic form of portal hypertension can be subdivided into presinusoidal (hepatic schistosomiasis, granulomatosis, i.e., primary biliary cirrhosis and idiopathic portal hypertension), sinusoidal (cirrhosis and fibrosis) and postsinusoidal (veno-occlusive disease). The second theory, the ‘‘forward’’ theory, proposed an increase in portal inflow as the most important factor leading to portal hypertension (Vorobioff et al., 1984). Portal venous inflow includes the flow of the total portal venous system and the portosystemic collaterals. During the development of portal hypertension, as a result of a hyperdynamic circulation caused by enhanced plasma volume together with a decrease in the splanchnic arte- riolar vascular resistance and an increase in cardiac out- put, an increased blood flow in portal tributaries devel- ops maintaining portal hypertension. *Correspondence to: Isabelle Colle, Department of Hepatology and Gastroenterology, Ghent University Hospital, De Pintelaan 185, 9000 Gent, Belgium. Fax: 32-9-332-49-84. E-mail: [email protected] Received 14 May 2007; Accepted 19 December 2007 DOI 10.1002/ar.20667 Published online in Wiley InterScience (www.interscience.wiley. com). Ó 2008 WILEY-LISS, INC. THE ANATOMICAL RECORD 291:699–713 (2008)
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Hemodynamic Changes in Splanchnic Blood Vessels in Portal Hypertension

Hemodynamic Changes in SplanchnicBlood Vessels in Portal Hypertension
ISABELLE COLLE,* ANJA M. GEERTS, CHRISTOPHE VAN STEENKISTE, AND
HANS VAN VLIERBERGHEDepartment of Hepatology and Gastroenterology, Ghent University Hospital,
Ghent, Belgium
ABSTRACTPortal hypertension (PHT) is associated with a hyperdynamic state
characterized by a high cardiac output, increased total blood volume, anda decreased splanchnic vascular resistance. This splanchnic vasodilationis a result of an important increase in local and systemic vasodilators (ni-tric oxide, carbon monoxide, prostacyclin, endocannabinoids, and so on),the presence of a splanchnic vascular hyporesponsiveness toward vaso-constrictors, and the development of mesenteric angiogenesis. All thesemechanisms will be discussed in this review. To decompress the portalcirculation in PHT, portosystemic collaterals will develop. The presenceof these portosystemic shunts are responsible for major complicationsof PHT, namely bleeding from gastrointestinal varices, encephalopathy,and sepsis. Until recently, it was accepted that the formation of colla-terals was due to opening of preexisting vascular channels, however,recent data suggest also the role of vascular remodeling and angiogenesis.These points are also discussed in detail. Anat Rec, 291:699–713,2008. � 2008 Wiley-Liss, Inc.
Key words: portal hypertension; splanchnic; angiogenesis;hemodynamic
Portal hypertension (PHT) is the major hemodynamiccomplication of a variety of diseases that obstruct portalblood flow. Portal pressure gradient (DP) is the result ofportal vascular resistance (R) and portal venous inflow(Q) in analogy with Ohm’s law: DP = R × Q. In normalcircumstances the portal pressure gradient variesbetween 2 and 6 mmHg.Two major theories were put forward to explain portal
hypertension. The ‘‘backward’’ theory assumes that theresistance to portal flow results in portal hypertension(Benoit et al., 1985). Portal vascular resistance consistsof the sum of the serial resistance in portal vein andintrahepatic vascular bed and of the parallel resistanceof the collaterals. The cause of enhanced vascular resist-ance can be localized pre-, intra-, and posthepatic. Pre-hepatic portal hypertension is mostly due to a portalvein thrombosis and is associated with a quite normalliver function. Posthepatically portal hypertension canbe caused, for example, by thrombosis of the hepaticveins (Budd Chiari syndrome), occlusion of the cavalvein (web), and constrictive pericarditis. The intrahe-patic form of portal hypertension can be subdivided intopresinusoidal (hepatic schistosomiasis, granulomatosis,
i.e., primary biliary cirrhosis and idiopathic portalhypertension), sinusoidal (cirrhosis and fibrosis) andpostsinusoidal (veno-occlusive disease).The second theory, the ‘‘forward’’ theory, proposed an
increase in portal inflow as the most important factorleading to portal hypertension (Vorobioff et al., 1984).Portal venous inflow includes the flow of the total portalvenous system and the portosystemic collaterals. Duringthe development of portal hypertension, as a result of ahyperdynamic circulation caused by enhanced plasmavolume together with a decrease in the splanchnic arte-riolar vascular resistance and an increase in cardiac out-put, an increased blood flow in portal tributaries devel-ops maintaining portal hypertension.
*Correspondence to: Isabelle Colle, Department of Hepatologyand Gastroenterology, Ghent University Hospital, De Pintelaan185, 9000 Gent, Belgium. Fax: 32-9-332-49-84.E-mail: [email protected]
Received 14 May 2007; Accepted 19 December 2007
DOI 10.1002/ar.20667Published online in Wiley InterScience (www.interscience.wiley.com).
� 2008 WILEY-LISS, INC.
THE ANATOMICAL RECORD 291:699–713 (2008)

Presently, the consensus is that both the rise in resist-ance and the enhanced portal inflow play an importantrole in the development of portal hypertension. In thisreview, we will only discuss the hemodynamic changes inthe splanchnic vascular bed and the development of portalsystemic collaterals, associated with portal hypertension.
MECHANISM OF SPLANCHNICHYPERDYNAMIC CIRCULATION
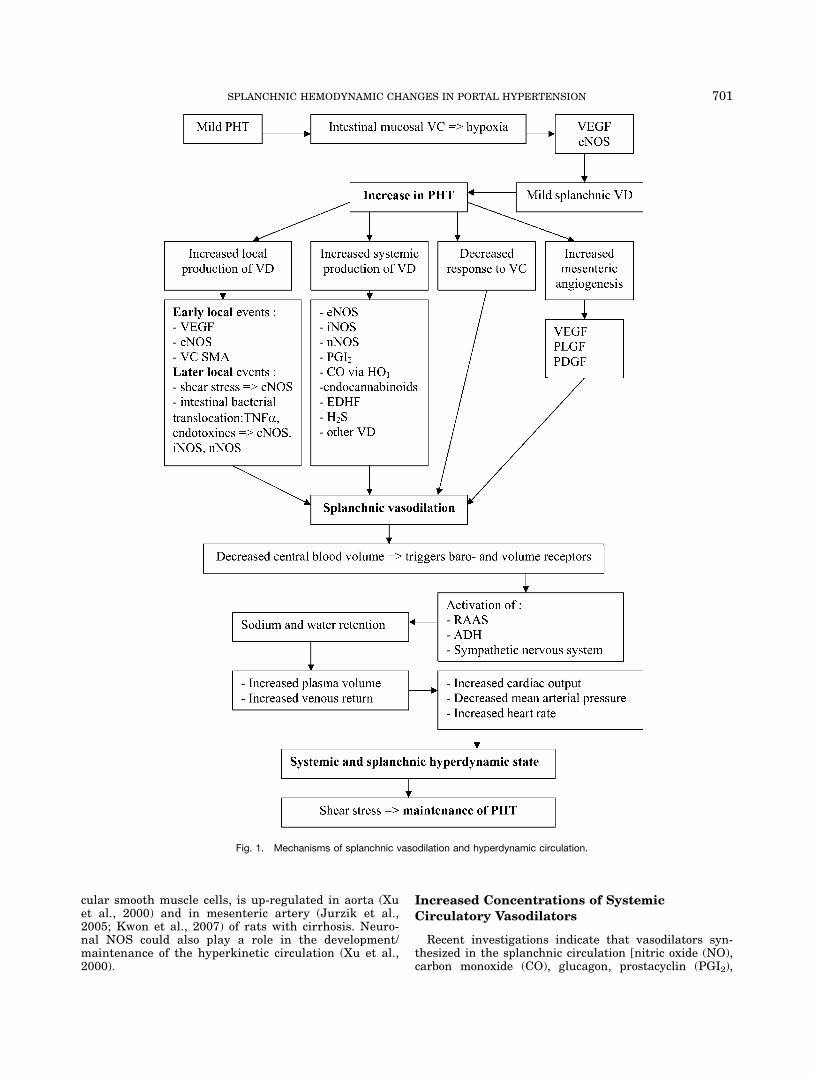
Portal hypertension is associated with a hyperdynamiccirculation characterized by increased cardiac outputand total blood volume in response to decreased systemicvascular resistance (Moreau et al., 1988; Groszmann,1993, 1994; Bosch and Garcia-Pagan, 2000; Fig. 1). Thisdiminished systemic vascular resistance is largely theresult of a decrease in splanchnic arterial resistanceowing to splanchnic vasodilation (Bosch and Garcia-Pagan, 2000). At least three mechanisms have been pro-posed to contribute to this splanchnic vasodilation inassociation with portal hypertension: (1) Increased localproduction of vasodilators; (2) Increased concentrationsof systemic circulatory vasodilators and; (3) Decreasedvascular response to vasoconstrictors (Groszmann andAbraldes, 2005). Recently, a fourth mechanism of adecrease in splanchnic arterial resistance has been pro-posed, namely mesenteric neoangiogenesis (Fernandezet al., 2004, 2005; Geerts et al., 2006a). Figure 1 showsthe different mechanisms playing a role in portal hyper-tension and hyperdynamic circulation.
Increased Production of Local Vasodilators
Early local events. In the early stages of PHT,Abraldes et al. demonstrated in rats with minimal portalhypertension (partial portal vein ligation PPVL over a16-gauge needle) an early increase (within 24 hr) in vas-cular endothelial growth factor (VEGF) and endothelialnitric oxide synthase (eNOS) expression in the jejunalmucosal microcirculation (Abraldes et al., 2006). Thisstudy confirms previous data in severe PHT, that up-reg-ulation of eNOS precedes the development of splanchnicarterial vasodilation and portal systemic shunts (Abra-ldes et al., 2006; Wiest et al., 1999a). The up-regulationof VEGF in the intestinal microcirculation accounts inlarge part for the initial eNOS activation (Abraldeset al., 2006). After venous pressure elevation, the redis-tribution of flow within the bowel from the mucosa tothe muscularis may cause a certain degree of hypoxia inthe mucosa that is sufficient to stimulate VEGF produc-tion (Granger et al., 1979, 1989; Davis and Gore, 1985).Because mucosal arterioles account for 25% of the totalmesenteric vascular resistance, NO activation at thislevel, can be the main site for the transduction of theincreased portal pressure into splanchnic vasodilationand being the initial step in the development of the hy-perdynamic circulation.A second event that occurs in rats with more severe
PHT after partial portal vein ligation (PPVL over a 20-gauge needle) is a myogenic response in the superiormesenteric artery (SMA) as early as 10 hr after induc-tion of PHT (Tsai et al., 2003). This SMA vasoconstric-tion triggers eNOS catalytic activity and thus NO hyper-
production (Tsai et al., 2003). In contrast, a milderincrease in portal pressure did not result in reflex SMAvasoconstriction and eNOS was not up-regulated at theSMA (Tsai et al., 2003).Iwakiri et al. showed that eNOS activity was
increased before eNOS expression, suggesting activationof eNOS at the posttranslational level (Iwakiri et al.,2002a). This increased eNOS activity is mediated by anAkt-dependent eNOS phosphorylation at the Serine1177
level (Iwakiri et al., 2002a). These phenomena may bethe early molecular signals that induce the cascade ofevents leading to the splanchnic hyperdynamic circula-tion.
Later local events. In PHT, once the hyperdy-namic circulation with high cardiac output is estab-lished, in vivo aortic levels of eNOS mRNA and eNOSprotein are increased, in response to shear stress(Pateron et al., 2000; Tazi et al., 2002). In both studies,treatment with propranolol significantly decreased invivo aortic eNOS mRNA and protein levels (Pateronet al., 2000; Tazi et al., 2002). Furthermore, in PHT ratsthere is an increased production of NO in response toshear stress in the superior mesenteric vascular bed(Hori et al., 1998; Wiest et al., 1999a). In cirrhotic rataortas, endothelial small calcium-dependent potassiumchannels (SKCa) are overexpressed and overactive inresponse to shear stress, stimulating the calcium/cal-modulin/eNOS pathway (Barriere et al., 2001). More-over, shear stress in portal hypertensive aortas resultsin activation of heat shock protein (Hsp)90, stimulatingeNOS catalytic activity. Hsp90 also contributes to thecontrol of the tone of the mesenteric vascular bed (Shahet al., 1999; Tazi et al., 2002).Intestinal bacterial translocation is common in cirrho-
sis and is followed by the production of tumor necrosisfactor alpha (TNFa) by mononuclear cells. Treatmentwith norfloxacin (an antibiotic that selectively decon-taminates the intestine) in cirrhotic patients and rats(Albillos et al., 2003; Tazi et al., 2005) decreases TNFaproduction and the hyperdynamic syndrome. Further-more, cirrhotic rats with bacterial translocation have amore pronounced systemic and splanchnic NO overpro-duction than those without (Wiest et al., 1999b). BothTNFa and endotoxins may activate inducible NOS(iNOS) and eNOS.On one hand, cirrhotic rats have increased levels of
aortic iNOS protein which decrease after treatment withnorfloxacin (Tazi et al., 2002, 2005). However, noenhanced levels of iNOS are found in the splanchnicvasculature of cirrhotic rats with bacterial translocation(Wiest et al., 1999b). Thus probably has iNOS a minorrole in the development of PHT.On the other hand, TNFa in the gastric mucosa of
portal hypertensive rats, activates Akt to phosphorylateeNOS at the Ser1177 level leading to eNOS activation(Kawanaka et al., 2002). Moreover, bacterial transloca-tion stimulates the endothelial gene expression of GTP-cyclohydrolase I, a key enzyme necessary for the produc-tion of tetrahydrobiopterin (BH4; Wiest et al., 2003).The BH4 production is a rate-limiting cofactor in theNO synthesis by eNOS in the mesenteric arterial bed(Wever et al., 1997; Wiest et al., 2003).Recently, it has been found that neuronal NOS
(nNOS) protein expression, present in neurons and vas-
700 COLLE ET AL.
cular smooth muscle cells, is up-regulated in aorta (Xuet al., 2000) and in mesenteric artery (Jurzik et al.,2005; Kwon et al., 2007) of rats with cirrhosis. Neuro-nal NOS could also play a role in the development/maintenance of the hyperkinetic circulation (Xu et al.,2000).
Increased Concentrations of Systemic
Circulatory Vasodilators
Recent investigations indicate that vasodilators syn-thesized in the splanchnic circulation [nitric oxide (NO),carbon monoxide (CO), glucagon, prostacyclin (PGI2),
Fig. 1. Mechanisms of splanchnic vasodilation and hyperdynamic circulation.
701SPLANCHNIC HEMODYNAMIC CHANGES IN PORTAL HYPERTENSION

vasoactive intestinal peptide (VIP), adenosine, bile salts,platelet activating factor, substance P, calcitonin gene-related peptide, adrenomedullin, atrial natriuretic pep-tide, endogenous cannabinoids, and others] might beresponsible for the hyperdynamic syndrome (Moreauand Lebrec, 1995). At the beginning of the 1990s, Val-lance and Moncada suggested the implication of NO, abiologically active gas, in the development of splanchnicvasodilation and the multiple organ malfunctions thatcharacterize the hyperkinetic syndrome (Vallance andMoncada, 1991). We discussed the local splanchnic pro-duction of NO in the previous sections, while we discusshere the systemic effects of NO and other vasodilatorspresent in the systemic circulation.
Nitric Oxide (NO). Most recent studies focusedtheir attention on the potential role of NO in the patho-genesis of splanchnic vasodilation (Vallance and Mon-cada, 1991; Hartleb et al., 1997; Martin et al., 1998; Liuand Lee, 1999). Nitric oxide is a very potent vasodilatorand is synthesized by NO synthase (NOS) from theamino acid L-arginine (Lowenstein et al., 1994). Threedifferent isoforms of NOS are known: neuronal (nNOS),endothelial (eNOS), and inducible NOS (iNOS; Moncadaet al., 1997). Tonic production of NO by eNOS is believedto play a major role in the maintenance of an activestate of vasodilation in the arterial circulation. A vastnumber of studies in human and experimental cirrhosis(Niederberger et al., 1995) suggest that NO synthesis isincreased and plays an important role in the pathogene-sis of arterial splanchnic vasodilation.In patients with ascites, the concentration of NO in
peripheral veins is higher than in controls and NO lev-els in the portal vein are higher than in the peripheralvein, suggesting that NO production is particularlyincreased in the splanchnic circulation (Battista et al.,1997). Serum nitrite and nitrate, metabolites of NO, andthe concentration of NO in exhaled air are alsoincreased in patients with cirrhosis and ascites (Guarneret al., 1993; Matsumoto et al., 1995; Sogni et al., 1995).Increased levels of NO may also play a role in the devel-opment of hepatopulmonary syndrome (Fallon et al.,1997). Finally, the infusion of a NOS inhibitor in a pe-ripheral artery of cirrhotic patients with ascites partiallyrestores the impaired reactivity to vasoconstrictors(Albillos et al., 1995; Campillo et al., 1995).Among the three isoforms, eNOS activation is the
major source of NO overproduction in the splanchnic cir-culation associated with PHT. Multiple factors such asshear stress, inflammatory cytokines (Iwakiri et al.,2002b; Tazi et al., 2003, 2005), and vascular endothelialgrowth factor (VEGF; Abraldes et al., 2006; Fernandezet al., 2005) stimulate eNOS-dependent NO productionin PHT leading to hyperdynamic circulation.Inducible NOS is produced in macrophages and vascu-
lar smooth muscle cells after stimulation by endotoxinsand inflammatory cytokines. However, iNOS is onlydetected in aorta of cirrhotic rats (Moreau et al., 2002;Tazi et al., 2005), but not in the splanchnic vasculatureof experimental animals with PHT and cirrhosis (Cahillet al., 1995; Martin et al., 1996; Morales-Ruiz et al.,1996; Wiest et al., 1999b).From recent data, there is also more evidence that
nNOS present in neurons and vascular smooth musclecells, can play an important role in PHT. Neuronal NOS
protein expression is up-regulated in cirrhotic rat aorta(Xu et al., 2000) and in the mesenteric artery (Jurziket al., 2005; Kwon et al., 2007). Treatment with a spe-cific nNOS inhibitor decreases the nNOS and cGMP lev-els in the aorta and normalizes the hyperdynamic circu-lation in cirrhotic rats, suggesting that nNOS also playsa role in the maintenance and/or development of thehyperkinetic mesenteric circulation (Xu et al., 2000).Inhibition of NO production only partially inhibits and
prevents the development of portosystemic shunts andthe hyperdynamic circulation (Lee et al., 1993; Garcia-Pagan et al., 1994; Pilette et al., 1996). This hypothesisis supported by the study of Iwakiri et al., showing thatthe hyperkinetic syndrome after portal vein ligation stilloccurs in mice lacking eNOS (eNOS2/2) and also devel-ops in double eNOS/iNOS knockout mice (Iwakiri et al.,2002b).
Prostacyclin (PGI2). Prostacyclin (PGI2) is aproduct of the metabolism of arachidonic acid by cycloox-ygenase (COX) and causes smooth muscle relaxation bystimulation of cyclic adenosine monophosphate (cAMP).COX-1 is the constitutive form and COX-2 is the induci-ble form of cyclooxygenase. Whole body production andportal venous levels of PGI2 are increased in portal hy-pertensive animals and cirrhotic patients and may playa role in the splanchnic hyperemia, collateral circula-tion, and portal hypertensive gastropathy (Sitzmannet al., 1994; Ohta et al., 1995). Studies using inhibitorsof COX and prostacyclin suggest a pathogenetic role forPGI2 in the hyperdynamic circulation associated withPHT (Munoz et al., 1999; Tsugawa et al., 1999).
Carbon monoxide (CO). Carbon monoxide (CO)is an endogenously produced gas molecule that in amanner similar to NO, activates soluble guanylate cy-clase leading to the generation of cGMP, which in turnmediates various physiological functions such as smoothmuscle cell relaxation (Wang et al., 1997). CO is pro-duced from the breakdown of heme to biliverdin andfree iron by means of the heme-oxygenase (HO) enzyme.Like NOS, HO has three isoforms: inducible HO (HO-1)and two constitutive forms (HO-2 and HO-3; Elbirt andBonkovsky, 1999).Fernandez et al. found that HO-1 protein levels and
activity is significantly increased in the mesentery,spleen, intestine, and liver of portal hypertensive rats(Fernandez and Bonkovsky, 1999; Fernandez et al.,2001). A progressively increased expression of HO-1 isfound in mesenteric arteries, aorta and cardiac ven-tricles from rats with secondary biliary cirrhosis (Liuet al., 2001; Chen et al., 2004). Administration of zincprotoporphyrin, a selective inhibitor of HO, normalizesaortic HO activity, partially restores vascular reactivityto vasoconstrictors and ameliorates the hyperdynamiccirculation associated with PHT (Fernandez et al., 2001;Chen et al., 2004).Carbon monoxide also plays a role in the pulmonary
vasodilation leading to hepatopulmonary syndrome, withincreased levels of carboxyhemoglobin in patientswith cirrhosis (Arguedas et al., 2003; Zhang et al., 2003;De Las et al., 2003). Heme oxygenase also mediateshyporeactivity to phenylephrine in the mesentericvessels of cirrhotic rats with ascites (Bolognesi et al.,2005).
702 COLLE ET AL.

Together with NO, carbon monoxide plays a role insplanchnic and pulmonary vasodilation, and both can beseen as portal hypertensive molecules. Whether HO-1plays a role as protective antioxidant enzyme remains tobe elucidated (Moreau, 2001). The precise mechanismwhereby HO-1 gene expression is induced is stillunknown. However several physical and chemical fac-tors that are present during portal hypertension, includ-ing cytokines, endotoxins, and shear stress, can activateHO-1 transcription (Elbirt and Bonkovsky, 1999; Boschand Garcia-Pagan, 2000).
Endocannabinoids. Anandamide is an endoge-nous lipid ligand that through binding with its cannabi-noid CB1 receptor leads to hypotension. Monocytes fromcirrhotic patients and rats contain increased levels ofanandamide (Batkai et al., 2001; Ros et al., 2002).Thereby, mesenteric vascular CB-1 receptors are maxi-mally activated in cirrhosis (Batkai et al., 2001; Roset al., 2002).Administration of a CB1 receptor antagonist SR
141716A increases mean arterial pressure (Batkai et al.,2001; Ros et al., 2002), peripheral resistance (Ros et al.,2002), and vascular tone in the mesenteric artery(Domenicali et al., 2005) and decreases portal hyperten-sion and mesenteric blood flow in cirrhotic rats (Batkaiet al., 2001). Activation of CB-1 receptors may lead onone hand to eNOS stimulation and production of NOand on the other hand to potassium channel activation,both leading to vasodilation (Batkai et al., 2001; Roset al., 2002; Howlett et al., 2002).
Endothelium-derived hyperpolarizing factor(EDHF). In normal arterial walls exposed to NOS/cy-clooxygenase (COX)-inhibitors, acetylcholine or shearstress induce the release of an endothelium-derivedrelaxing factor (EDRF; Cohen and Vanhoutte, 1995).This NOS/COX independent EDRF has been calledendothelium-derived hyperpolarizing factor or EDHF,because it induces hyperpolarization and arterial vascu-lar smooth muscle cell relaxation (Cohen and Vanhoutte,1995). The exact nature of EDHF is controversial; how-ever, the main molecules considered to explain EDHF-mediated vasodilation are monovalent cation potassium,arachidonic acid metabolites, components of gap junc-tions, and hydrogen peroxidase (Iwakiri and Groszmann,2006). EDHF is more prominent in smaller arteries andarterioles than in larger arteries, and its role becomesmore important in the absence of NO (Iwakiri andGroszmann, 2006). Thus, the presence of EDHF canexplain why hyperdynamic circulation still develops inportal hypertensive eNOS/iNOS knockout mice (Iwakiriet al., 2002b).In cirrhotic rats, EDHF is released by the superior
mesenteric artery but not in the aorta (Barriere et al.,2000). In this cirrhotic mesenteric artery, EDHF-inducedsmooth muscle cell relaxation is abolished by a combina-tion of apamin and charybdotoxin and decreased by bar-ium or ouabain (Barriere et al., 2000). Thus, in the supe-rior mesenteric artery from cirrhotic rats, EDHF may bea K1 ion released by endothelial apamin- and charybdo-toxin sensitive K1 channels, K1 then activating bariumsensitive K1 channels and Na1/K1 ATPase in thesmooth muscle cells leading to hyperpolarization andrelaxation of the vascular wall (Barriere et al., 2000).
Moreover, it has been shown that endothelial small-conductance Ca11-dependent K1 (SK1
Ca) channels areoverexpressed in cirrhotic aortas (Barriere et al., 2001a).In cirrhosis, selective SK1
Ca channel blockade by apa-min results in a decreased eNOS hyperactivity and NO-dependent smooth muscle relaxation (Barriere et al.,2001). Thus, in cirrhotic arterial walls, activation of K1
channels located in the plasma membrane of endothelialand smooth muscle cells, induces membrane hyperpolar-ization, which may contribute to systemic and splanch-nic arterial vasodilation.
Hydrogen sulfate (H2S). Recently it has beensuggested that H2S is a potent endogenous vasodilator(derived from L-cysteine) in mesenteric arteries andaorta (Hosoki et al., 1997; Zhao and Wang, 2002; Chenget al., 2004). This H2S-mediated vasodilation occurs bymeans of opening of K1
ATP channels and thus independ-ently of the cGMP pathway (Zhao et al., 2001). The roleof H2S is postulated in the vascular abnormalities seenin cirrhosis; however, more studies are needed to con-firm this hypothesis (Ebrahimkhani et al., 2005).
Other systemic vasodilators. In portal hyper-tension, despite NOS and COX inhibition, arterial vaso-dilation, and vascular hyporeactivity are not totally sup-pressed, suggesting the presence of NOS/COX independ-ent vasodilators (Moreau and Lebrec, 1995). Increasedplasma levels of natriuretic peptides, glucagon, adreno-medulin, calcitonin gene-related peptide, substance P,and vasoactive intestinal peptide have been described incirrhosis (Moreau and Lebrec, 1995, 2005). Probably,other vasodilators will be discovered in the future.
Vascular Hyporesponsiveness to
Vasoconstrictors
In cirrhosis and portal hypertension, the presence ofsplanchnic vasodilation in the face of highly elevatedlevels of circulating vasoconstrictors (angiotensin II, nor-epinephrin, endothelin, vasopressin, and so on), can beexplained by vascular hyporesponsiveness. This splanch-nic resistance to vasoconstrictor agents (Lee et al., 1992;Sieber et al., 1993) explains why the hyperdynamic cir-culation increases with progression of the disease de-spite the stimulation of renin-angiotensin, sympatheticnervous system, and vasopressin release. In contrast,these systems induce vasoconstriction in other organs,such as the brain and kidneys in patients with ascites(Fernandez-Seara et al., 1989; Guevara et al., 1998;Maroto et al., 1993; Maroto et al., 1994).A large number of studies have described hyporespon-
siveness in cirrhosis and portal hypertension to differentvasoconstrictors (methoxamine, potassiumchloride, phe-nylephrine, terlipressin, vasopressin, endothelin-1, an-giotensin-II, norepinephrine; Michielsen et al., 1995a;Atucha et al., 1996; Sogni et al., 1996, 1997; Heinemannet al., 1997; Schepke et al., 2001; Chu et al., 2000;Barriere et al., 2001; Colle et al., 2004b). Arterial hypo-reactivity to vasoconstrictors may also differ from onevascular bed to another: in cirrhotic rats vascular hypo-responsiveness occurs in the superior mesenteric arteryand in the aorta, but is normal in carotid artery(Pateron et al., 1999).
703SPLANCHNIC HEMODYNAMIC CHANGES IN PORTAL HYPERTENSION

The increased concentrations of local and systemicvasodilators (NO, HO, and adrenomedullin) as describedabove, are probably the cause of this hyporeactivity(Kojima et al., 2004; Bolognesi et al., 2005; Erario et al.,2005). However, the molecular mechanisms of this vas-cular hyporesponsiveness are not well understood andare multiple (Bomzon and Huang, 2001; Moreau, 2001).Vascular hyporeactivity to most relevant endogenous
vasoconstrictors in the hepatic artery of cirrhoticpatients is not caused by a down regulation of a1-adre-noreceptors a, b, and c; angiotensin II receptor I; vaso-pressin V1a receptor, and endothelin A and B receptors.These vasoconstrictor receptors are even up-regulated inthe hepatic artery (Neef et al., 2003).Under normal conditions, splanchnic arterioles are
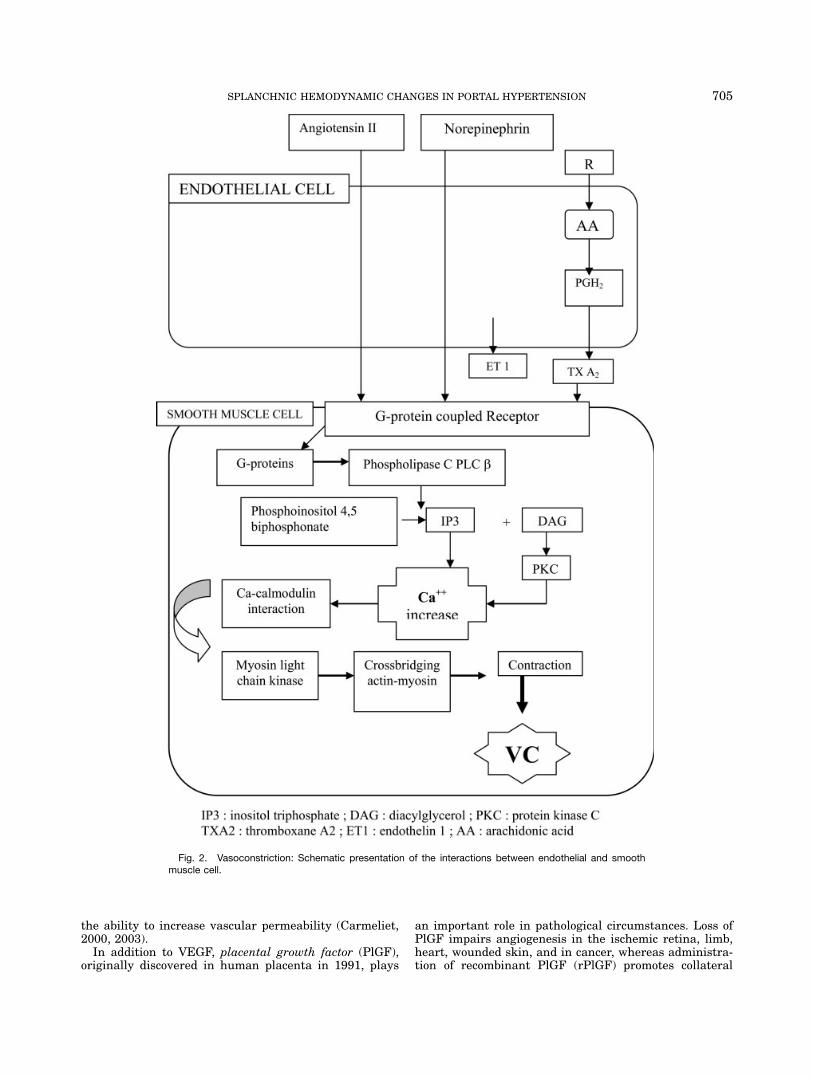
partially constricted and have the capacity to either fur-ther constrict or dilate. The basal contractile state (tone)of arteriolar smooth muscle cells reflects the balance ofmultiple influences that either cause relaxation or con-striction of the vascular smooth muscle cell. Importantvasoconstrictors influencing splanchnic arteriolesinclude some circulating agents (e.g., angiotensin II),myogenic factors, certain endothelium-derived substan-ces (e.g., endothelin), and some neurotransmitters (e.g.,norepinephrine). All vasoconstrictor receptors belong tothe superfamily of guanine nucleotide-binding protein(G-protein) -coupled receptors (GPCR). Stimulation ofGPCR on the vascular smooth muscle cell activates Gproteins and consequently phospholipase C (PLC) -b. PLChydrolyzes phosphatidylinositol 4,5-biphosphate into ino-sitol triphosphate (IP3) and diacylglycerol (DAG). IP3diffuses in the cytosol and DAG remains in the plasmamembrane activating protein kinase C (PKC). Bothproducts cause an increase in intracellular calcium inthe vascular smooth muscle cell. The released calciuminitiates a cascade of intracellular events, resulting incross bridging of actin and myosin, leading to contrac-tion (Bomzon and Huang, 2001; Cahill et al., 2001).Figure 2 shows schematically the mechanism of vasocon-striction.Different possible mechanisms contribute to this hypo-
reactivity to vasoconstrictors. In normal circumstancesNO stimulates the production of cGMP which activatescGMP dependent serine-threonine protein kinase called‘‘PKG’’ (Lincoln and Cornwell, 1993). PKG inhibits theGPCR signaling pathway used by vasoconstrictors at dif-ferent levels: (1) PKG increases GTPase activity termi-nating vasoconstrictor signalling (Tang et al., 2003); (2)PKG may phosphorylate GPCR and thus uncouple thereceptor and G-proteins (Lincoln and Cornwell, 1993;Moreau and Lebrec, 2005).In portal hypertension, there is a decreased ex vivo
production of IP3 and DAG in response to vasoconstric-tors using GPCR in portal hypertensive arteries (Moreauand Lebrec, 1995). Also ex vivo enzymatic activities ofPKC-a and PKC-d are decreased in cirrhotic vascularsmooth muscle cells (Tazi et al., 2000). Moreover, in vivoPKC-a protein levels are decreased in portal hyperten-sive aortas (Moreau and Lebrec, 1995; Bomzon andHuang, 2001). In endothelium-denuded hepatic arteriesfrom cirrhotic patients, ex vivo exposure to different vas-oconstrictors shows hyporeactivity to some but not toothers (Heller et al., 1999).These results suggest that both effects of vasodilators
on the vasoconstrictive system, but also that abnormal-
ities within the smooth muscle and endothelial cells maybe responsible for this vascular hyporesponsiveness tovasoconstrictors. This has been extensively reviewed intwo articles of Cahill et al. and Bomzon et al. (Bomzonand Huang, 2001; Cahill et al., 2001).Recently, the presence of neuropeptide Y1 (NPY) in
the superior mesenteric artery in PHT was observed.NPY becomes increasingly important and increasedrelease of NPY may represent a compensatory mecha-nism to counterbalance arterial vasodilation by restoringthe efficacy of endogenous cathecholamines, especially instates of high levels of alfa1-adrenergic activity (Wiestet al., 2006, 2007).
Vascular Responsiveness to Vasodilators
While for most vasoconstrictors there is an impairedvascular response, conflicting results concerning theresponse (hypo-, hyper-, and normal response) of thesplanchnic vascular bed to different vasodilators havebeen reported (Tables 1 and 2). By using endothelium-dependent (acetylcholine) and -independent agents[pinacidil, potassium ATP (KATP) channel opener; deta-NONOate and sodiumnitroprusside as NO donors], theintegrity of the endothelium and the vascular smoothmuscle cell can be evaluated. Figure 3 shows a simpli-fied scheme of different action mechanisms of vasoactiveagents. Our group found also an in vivo hyporeactivityof the mesenteric vascular bed for acetylcholine, pinaci-dil, detaNONOate and sildenafil, which persisted afterNOS and COX inhibition (Colle et al., 2004a,b). Hyper-response to vasodilators can contribute to furthersplanchnic vasodilation, while hyporesponse to vasodila-tors can be seen as a defence against further aggrava-tion of the hyperdynamic circulation.
Increased Splanchnic Angiogenesis
Introduction to angiogenesis. During embryo-genesis and organogenesis blood vessel formation occursby aggregation of de novo forming angioblasts or endo-thelial progenitor cells into a primitive vascular plexus(vasculogenesis), which then undergoes a complexremodeling process, in which growth, migration andsprouting lead to the development of a functional circu-latory system (angiogenesis; Carmeliet, 2000). Duringadulthood in normal situations, angiogenesis occurs onlyin the cycling ovary and in the placenta during preg-nancy. In pathological situations in response to hypoxiaand/or inflammation, angiogenesis is reactivated duringwound healing and repair. However, this stimulus canbecome excessive, and the balance between stimulatorsand inhibitors turns to a pathological angiogenic switch(best known condition is malignant tumor formation)(Carmeliet, 2000, 2003).The vascular endothelial growth factor (VEGF) family
and their VEGF receptors (VEGFRs) are considered asthe most important factors involved in angiogenesis andreceive thereby most of the attention in the current lit-erature (Carmeliet, 2003; Ferrara et al., 2003). VEGF-Ahas been recognized as the major relatively specificgrowth factor for endothelial cells and exhibits twomajor biological activities: first is the capacity to stimu-late vascular endothelial cell proliferation, and second is
704 COLLE ET AL.
the ability to increase vascular permeability (Carmeliet,2000, 2003).In addition to VEGF, placental growth factor (PlGF),
originally discovered in human placenta in 1991, plays
an important role in pathological circumstances. Loss ofPlGF impairs angiogenesis in the ischemic retina, limb,heart, wounded skin, and in cancer, whereas administra-tion of recombinant PlGF (rPlGF) promotes collateral
Fig. 2. Vasoconstriction: Schematic presentation of the interactions between endothelial and smoothmuscle cell.
705SPLANCHNIC HEMODYNAMIC CHANGES IN PORTAL HYPERTENSION
vessel growth in models of myocardial and limb ischemia(Carmeliet et al., 2001; Carmeliet, 2003; Autiero et al.,2003).Three receptor tyrosine kinases, binding the VEGF
family members have thus far been identified. VEGFreceptor-1 (or Flt-1, Fms-like tyrosine kinase-1) bindsVEGF-A and PlGF; VEGF receptor-2 (or Flk-1, fetalliver kinase-1) binds VEGF-A and VEGF-C and VEGFreceptor-3 (or Flt-4) binds VEGF-C. The major mediator
of the mitogenic, angiogenic, and permeability-enhanc-ing effects of VEGF-A is VEGFR-2 (Carmeliet et al.,2001, 2003; Autiero et al., 2003).
Role of VEGF in splanchnic hyperdynamic cir-culation. Fukumura et al. demonstrated that VEGFinduces NO production by means of activation of eNOSprotein expression and activity (Fukumura et al., 2001).Recently, Abraldes et al. showed that VEGF up-regula-
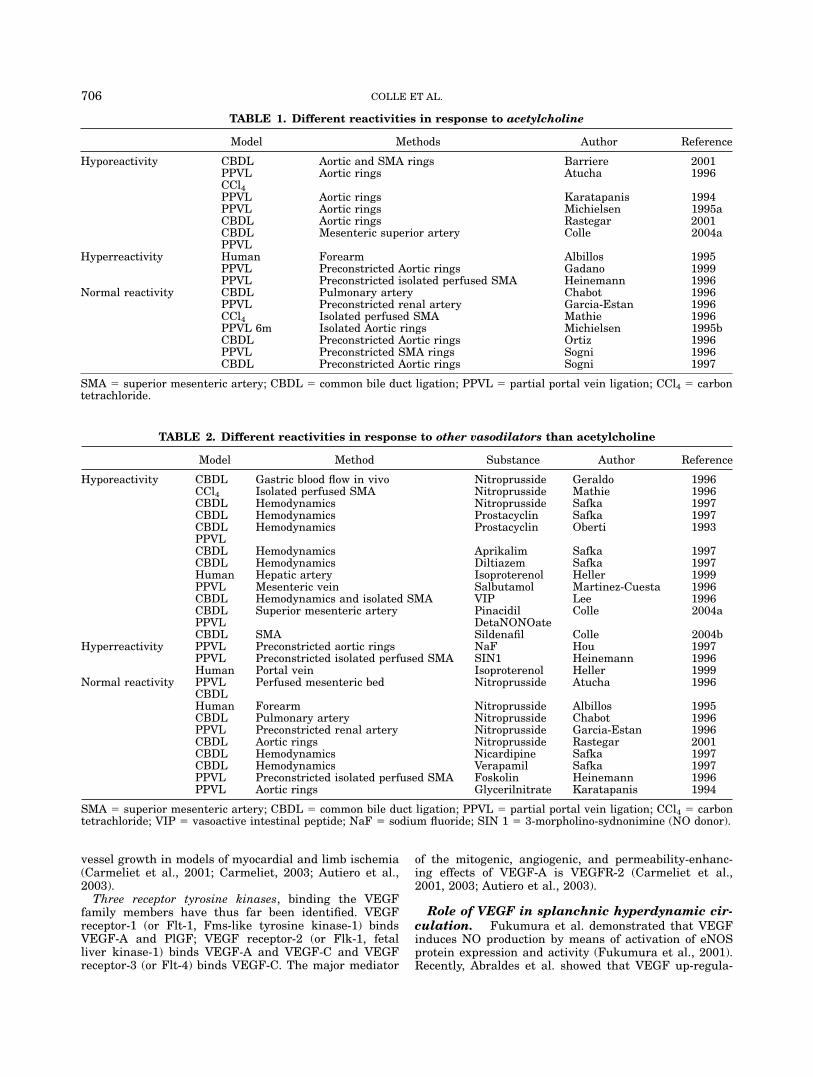
TABLE 1. Different reactivities in response to acetylcholine
Model Methods Author Reference
Hyporeactivity CBDL Aortic and SMA rings Barriere 2001PPVL Aortic rings Atucha 1996CCl4PPVL Aortic rings Karatapanis 1994PPVL Aortic rings Michielsen 1995aCBDL Aortic rings Rastegar 2001CBDL Mesenteric superior artery Colle 2004aPPVL
Hyperreactivity Human Forearm Albillos 1995PPVL Preconstricted Aortic rings Gadano 1999PPVL Preconstricted isolated perfused SMA Heinemann 1996
Normal reactivity CBDL Pulmonary artery Chabot 1996PPVL Preconstricted renal artery Garcia-Estan 1996CCl4 Isolated perfused SMA Mathie 1996PPVL 6m Isolated Aortic rings Michielsen 1995bCBDL Preconstricted Aortic rings Ortiz 1996PPVL Preconstricted SMA rings Sogni 1996CBDL Preconstricted Aortic rings Sogni 1997
SMA 5 superior mesenteric artery; CBDL 5 common bile duct ligation; PPVL 5 partial portal vein ligation; CCl4 5 carbontetrachloride.
TABLE 2. Different reactivities in response to other vasodilators than acetylcholine
Model Method Substance Author Reference
Hyporeactivity CBDL Gastric blood flow in vivo Nitroprusside Geraldo 1996CCl4 Isolated perfused SMA Nitroprusside Mathie 1996CBDL Hemodynamics Nitroprusside Safka 1997CBDL Hemodynamics Prostacyclin Safka 1997CBDL Hemodynamics Prostacyclin Oberti 1993PPVLCBDL Hemodynamics Aprikalim Safka 1997CBDL Hemodynamics Diltiazem Safka 1997Human Hepatic artery Isoproterenol Heller 1999PPVL Mesenteric vein Salbutamol Martinez-Cuesta 1996CBDL Hemodynamics and isolated SMA VIP Lee 1996CBDL Superior mesenteric artery Pinacidil Colle 2004aPPVL DetaNONOateCBDL SMA Sildenafil Colle 2004b
Hyperreactivity PPVL Preconstricted aortic rings NaF Hou 1997PPVL Preconstricted isolated perfused SMA SIN1 Heinemann 1996Human Portal vein Isoproterenol Heller 1999
Normal reactivity PPVL Perfused mesenteric bed Nitroprusside Atucha 1996CBDLHuman Forearm Nitroprusside Albillos 1995CBDL Pulmonary artery Nitroprusside Chabot 1996PPVL Preconstricted renal artery Nitroprusside Garcia-Estan 1996CBDL Aortic rings Nitroprusside Rastegar 2001CBDL Hemodynamics Nicardipine Safka 1997CBDL Hemodynamics Verapamil Safka 1997PPVL Preconstricted isolated perfused SMA Foskolin Heinemann 1996PPVL Aortic rings Glycerilnitrate Karatapanis 1994
SMA 5 superior mesenteric artery; CBDL 5 common bile duct ligation; PPVL 5 partial portal vein ligation; CCl4 5 carbontetrachloride; VIP 5 vasoactive intestinal peptide; NaF 5 sodium fluoride; SIN 1 5 3-morpholino-sydnonimine (NO donor).
706 COLLE ET AL.

tion in the intestinal mucosal microcirculation accountslargely for the initial eNOS up-regulation in mild PHT,which precedes the development of vasodilation and thedevelopment of portosystemic shunting in mild PHT(Abraldes et al., 2006). Moreover, our group also foundincreased levels of VEGF in mesenteric tissue of ratswith PHT and cirrhosis (Geerts et al., 2006a). This wasassociated with an increased vascular permeability, onlydetected in cirrhotic rats but not in pure portal hyper-tensive rats (Geerts et al., 2006a).
Role of angiogenesis in splanchnic hyperdy-namic circulation. Nevertheless, these functionalalterations (especially vasodilation) described above cannot fully explain the observed sustained splanchnic vaso-dilation. In addition to these functional changes, prob-ably also structural vascular changes are implicated inportal hypertension.Previous studies provided evidence for increased
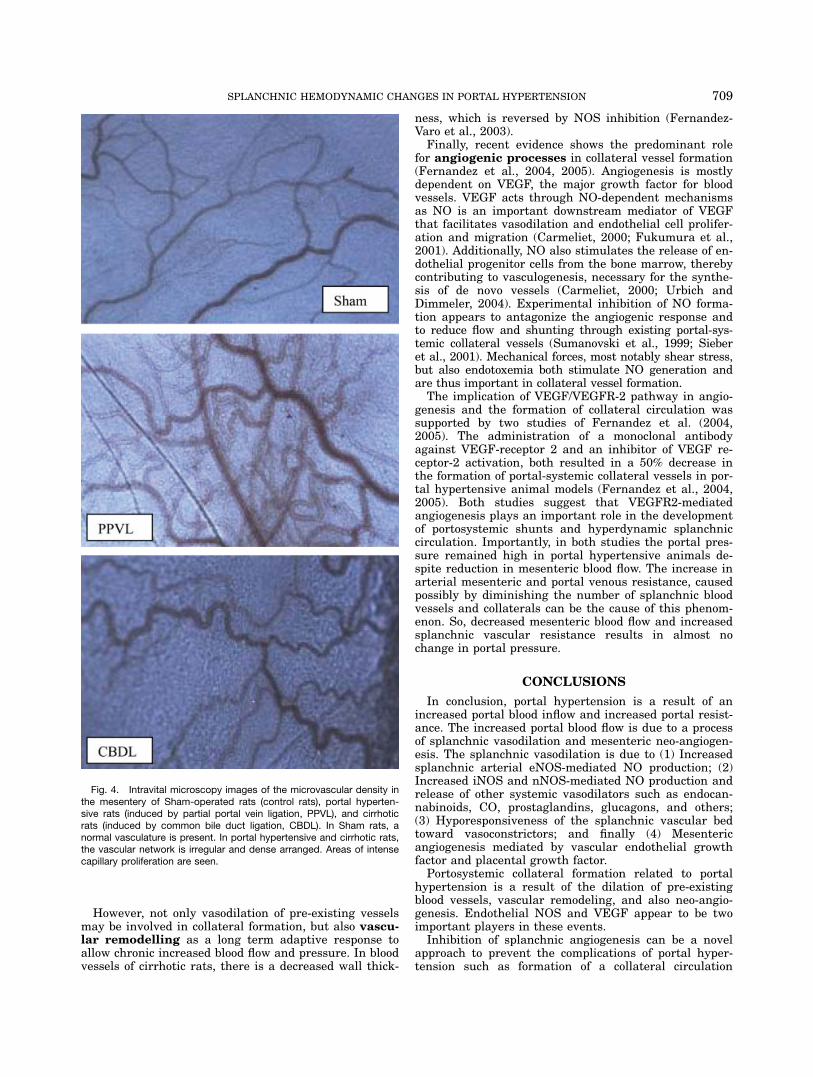
angiogenesis and VEGF production in the splanchnicterritory of portal hypertensive rats and cirrhoticpatients (Perez-Ruiz et al., 1999; Sumanovski et al.,1999; Cejudo-Martin et al., 2001; Sieber et al., 2001).Recently, our group was able to show an in vivoincreased angiogenesis in the mesenteric microcircula-tion of rats with PHT with and without cirrhosis (Fig.4)(Geerts et al., 2006a). This increased mesenteric angio-genesis was associated with an increased VEGF andeNOS protein expression (Geerts et al., 2006a). We couldalso demonstrate that neo-angiogenesis is present in themesentery of portal hypertensive mice, and is associatedwith an up-regulation of VEGF and PlGF protein levelsin the mesentery (Geerts et al., 2006a,b)(both oral pre-sentations). PlGF knockout (PlGF2/2) portal hyperten-sive mice do not develop neo-angiogenesis in the mesen-tery and have lower portal venous pressures comparedwith the control portal hypertensive mice (Geerts et al.,2006a,b).These findings confirm the assumption that chronic
portal hypertension induces structural, as well as thewell-described functional vascular changes. Sieber dem-onstrated that this increased mesenteric angiogenesiscould be reversed by chronically inhibiting NO formation(Sieber et al., 2001).Recently, Fernandez et al. showed that blocking of the
VEGF-receptor 2 signaling pathway in portal vein-ste-nosed mice and rats (using anti-VEGFR-2 monoclonalantibodies or using VEGFR-2–autophosphorylation inhi-bition, both for 5–7 days after surgery) resulted in a sig-nificant decrease in the number of mesenteric blood ves-sels (shown by splanchnic protein levels of CD31) andVEGFR2 protein expression (Fernandez et al., 2004,2005). These results were accompanied by an increase insplanchnic arteriolar and portal venous resistanceresulting in a decreased portal venous inflow, however,portal pressure remained high (Fernandez et al., 2004,2005). These studies further contribute to the hypothesisthat a decrease in VEGF- and PlGF-dependent angio-genesis could reduce vascular density, splanchnic bloodflow and thus portal venous inflow in partial portal vein-ligated animals (Fernandez et al., 2004, 2005; Geertset al., 2006b).The mechanisms by which VEGF protein expression
in portal hypertension is increased are probably multi-factorial. Indeed, several factors relevant to the pathoge-
nesis of portal hypertension, such as hypoxia, cytokines,and mechanical stress, have been shown to promoteVEGF expression in various cell types and tissues (Car-meliet, 2000, 2003). We can assume that increased por-tal pressure (even if it is minimal) is the initial factorthat triggers VEGF and eNOS expression in the portalsystem (Abraldes et al., 2006), which is followed byincreased blood flow further exaggerating VEGF overex-pression, resulting in enhanced angiogenesis. Recently, arole for NAD(P)H oxidase (a major source of reactive ox-ygen species) and for the enzyme heme-oxygenase-1(HO) was suggested to contribute to the angiogenic stim-ulus in PHT (Abraldes et al., 2006; Angermayr et al.,2006, 2007). Chronic HO inhibition significantlydecreased VEGF protein expression in the mesentery ofportal hypertensive rats, suggesting that HO enzymaticactivity is an important stimulus for VEGF productionin portal hypertension (Angermayr et al., 2006). Also,hypoxia inducible factor (HIF) plays probably an initiat-ing role in the activation of VEGF.
Role of angiogenesis in other organs associ-ated with PHT. Besides mesenteric angiogenesisthere is evidence for up-regulation of angiogenic factorsin other splanchnic organs. In patients and in animalmodels there is an increased expression of VEGF in por-tal hypertensive gastric mucosa and can be involved inthe development of portal hypertensive gastropathy(Tsugawa et al., 2000, 2001). Moreover, Yin et al. demon-strated higher levels of VEGF in the esophagus of portalhypertensive rats (Yin et al., 2005). At this moment, thisdomain needs further investigation.
COLLATERAL CIRCULATION
The development of portal hypertension is associatedwith changes in both the venous and arterial splanchniccirculation. In the venous circulation, portosystemic col-laterals are formed which cause shunting of blood fromthe portal to the systemic circulation. The developmentof portosystemic shunts, as a compensatory mechanismto decompress the portal circulation and pressure, is re-sponsible for major complications such as encephalop-athy, sepsis and bleeding from gastrointestinal varices.At the arterial side, there is an important vasodilationincreasing portal venous inflow. By this mechanism, por-tal pressure remains high, despite the formation of anextensive network of collaterals.Until recently, it was thought that the development of
collateral circulation was due to the opening of pre-existing vascular channels in response to increasedportal pressure, a physiological process which includesNO-mediated vasodilation activated by shear stress andVEGF. Accordingly, all therapeutic strategies are aimedto decrease portal blood inflow and thus pressure. Porto-systemic shunting was inhibited by NOS inhibitors inportal vein stenosed rats (Mosca et al., 1992; Lee et al.,1993; Chan et al., 1999). Nonselective b-blockers notonly reduce cardiac output but also constrict the collat-eral circulation (azygos blood flow; Cales et al., 1985a,b)leading to a decreased portal pressure. The administra-tion of propranolol also decreases shear stress and con-sequently attenuates eNOS production and systemicarterial vasodilation (Tazi et al., 2002).
707SPLANCHNIC HEMODYNAMIC CHANGES IN PORTAL HYPERTENSION
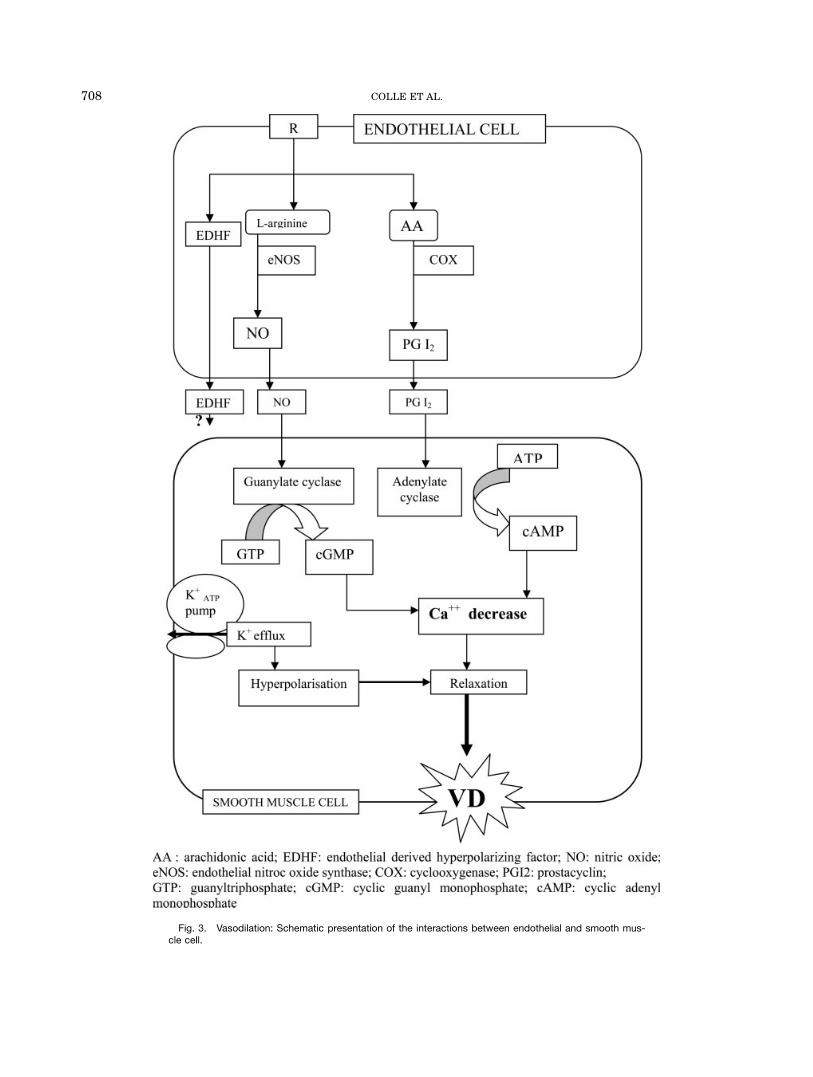
Fig. 3. Vasodilation: Schematic presentation of the interactions between endothelial and smooth mus-cle cell.
708 COLLE ET AL.
However, not only vasodilation of pre-existing vesselsmay be involved in collateral formation, but also vascu-lar remodelling as a long term adaptive response toallow chronic increased blood flow and pressure. In bloodvessels of cirrhotic rats, there is a decreased wall thick-
ness, which is reversed by NOS inhibition (Fernandez-Varo et al., 2003).Finally, recent evidence shows the predominant role
for angiogenic processes in collateral vessel formation(Fernandez et al., 2004, 2005). Angiogenesis is mostlydependent on VEGF, the major growth factor for bloodvessels. VEGF acts through NO-dependent mechanismsas NO is an important downstream mediator of VEGFthat facilitates vasodilation and endothelial cell prolifer-ation and migration (Carmeliet, 2000; Fukumura et al.,2001). Additionally, NO also stimulates the release of en-dothelial progenitor cells from the bone marrow, therebycontributing to vasculogenesis, necessary for the synthe-sis of de novo vessels (Carmeliet, 2000; Urbich andDimmeler, 2004). Experimental inhibition of NO forma-tion appears to antagonize the angiogenic response andto reduce flow and shunting through existing portal-sys-temic collateral vessels (Sumanovski et al., 1999; Sieberet al., 2001). Mechanical forces, most notably shear stress,but also endotoxemia both stimulate NO generation andare thus important in collateral vessel formation.The implication of VEGF/VEGFR-2 pathway in angio-
genesis and the formation of collateral circulation wassupported by two studies of Fernandez et al. (2004,2005). The administration of a monoclonal antibodyagainst VEGF-receptor 2 and an inhibitor of VEGF re-ceptor-2 activation, both resulted in a 50% decrease inthe formation of portal-systemic collateral vessels in por-tal hypertensive animal models (Fernandez et al., 2004,2005). Both studies suggest that VEGFR2-mediatedangiogenesis plays an important role in the developmentof portosystemic shunts and hyperdynamic splanchniccirculation. Importantly, in both studies the portal pres-sure remained high in portal hypertensive animals de-spite reduction in mesenteric blood flow. The increase inarterial mesenteric and portal venous resistance, causedpossibly by diminishing the number of splanchnic bloodvessels and collaterals can be the cause of this phenom-enon. So, decreased mesenteric blood flow and increasedsplanchnic vascular resistance results in almost nochange in portal pressure.
CONCLUSIONS
In conclusion, portal hypertension is a result of anincreased portal blood inflow and increased portal resist-ance. The increased portal blood flow is due to a processof splanchnic vasodilation and mesenteric neo-angiogen-esis. The splanchnic vasodilation is due to (1) Increasedsplanchnic arterial eNOS-mediated NO production; (2)Increased iNOS and nNOS-mediated NO production andrelease of other systemic vasodilators such as endocan-nabinoids, CO, prostaglandins, glucagons, and others;(3) Hyporesponsiveness of the splanchnic vascular bedtoward vasoconstrictors; and finally (4) Mesentericangiogenesis mediated by vascular endothelial growthfactor and placental growth factor.Portosystemic collateral formation related to portal
hypertension is a result of the dilation of pre-existingblood vessels, vascular remodeling, and also neo-angio-genesis. Endothelial NOS and VEGF appear to be twoimportant players in these events.Inhibition of splanchnic angiogenesis can be a novel
approach to prevent the complications of portal hyper-tension such as formation of a collateral circulation
Fig. 4. Intravital microscopy images of the microvascular density inthe mesentery of Sham-operated rats (control rats), portal hyperten-sive rats (induced by partial portal vein ligation, PPVL), and cirrhoticrats (induced by common bile duct ligation, CBDL). In Sham rats, anormal vasculature is present. In portal hypertensive and cirrhotic rats,the vascular network is irregular and dense arranged. Areas of intensecapillary proliferation are seen.
709SPLANCHNIC HEMODYNAMIC CHANGES IN PORTAL HYPERTENSION

(varices, encephalopathy) and splanchnic vasodilation,thereby reducing morbidity and mortality in patientswith chronic liver diseases. However, further studies areneeded to explore if other proangiogenic factors (e.g.,PLGF, and so on) and receptors (VEGFR1, and so on)are involved in portal hypertension.
LITERATURE CITED
Abraldes JG, Iwakiri Y, Loureiro-Silva M, Haq O, Sessa WC, Grosz-mann RJ. 2006. Mild increases in portal pressure upregulatevascular endothelial growth factor and endothelial nitric oxidesynthase in the intestinal microcirculatory bed, leading to a hy-perdynamic state. Am J Physiol Gastrointest Liver Physiol290:G980–G987.
Albillos A, Rossi I, Cacho G, Martinez MV, Millan I, Abreu L, Bar-rios C, Escartin P. 1995. Enhanced endothelium-dependent vaso-dilation in patients with cirrhosis. Am J Physiol 268:G459–G464.
Albillos A, de la Hera A, Gonzalez M, Moya JL, Calleja JL, Monser-rat J, Ruiz-del-Arbol L, Alvarez-Mon M. 2003. Increased lipopoly-saccharide binding protein in cirrhotic patients with markedimmune and hemodynamic derangement. Hepatology 37:208–217.
Angermayr B, Mejias M, Gracia-Sancho J, Garcia-Pagan JC, BoschJ, Fernandez M. 2006. Heme oxygenase attenuates oxidativestress and inflammation, and increases VEGF expression in por-tal hypertensive rats. J Hepatol 44:1033–1039.
Angermayr B, Fernandez M, Mejias M, Gracia-Sancho J, Garcia-Pagan JC, Bosch J. 2007. NAD(P)H oxidase modulates angiogene-sis and the development of portosystemic collaterals and splanch-nic hyperaemia in portal hypertensive rats. Gut 56:560–564.
Arguedas MR, Abrams GA, Krowka MJ, Fallon MB. 2003. Prospec-tive evaluation of outcomes and predictors of mortality in patientswith hepatopulmonary syndrome undergoing liver transplanta-tion. Hepatology 37:192–197.
Atucha NM, Shah V, Garcia-Cardena G, Sessa WE, Groszmann RJ.1996. Role of endothelium in the abnormal response of mesentericvessels in rats with portal hypertension and liver cirrhosis. Gas-troenterology 111:1627–1632.
Autiero M, Waltenberger J, Communi D, Kranz A, Moons L, Lam-brechts D, Kroll J, Plaisance S, De Mol M, Bono F, Kliche S, Fell-brich G, Ballmer-Hofer K, Maglione D, Mayr-Beyrle U, Dewer-chin M, Dombrowski S, Stanimirovic D, Van Hummelen P, DehioC, Hicklin DJ, Persico G, Herbert JM, Communi D, Shibuya M,Collen D, Conway EM, Carmeliet P. 2003. Role of PlGF in theintra- and intermolecular cross talk between the VEGF receptorsFlt1 and Flk1. Nat Med 9:936–943.
Barriere E, Tazi KA, Pessione F, Heller J, Poirel O, Lebrec D, Mor-eau R. 2001. Role of small-conductance Ca21-dependent K1 chan-nels in in vitro nitric oxide-mediated aortic hyporeactivity toalpha-adrenergic vasoconstriction in rats with cirrhosis. J Hepa-tol 35:350–357.
Barriere E, Tazi KA, Rona JP, Pessione F, Heller J, Lebrec D, Mor-eau R. 2000. Evidence for an endothelium-derived hyperpolariz-ing factor in the superior mesenteric artery from rats with cirrho-sis. Hepatology 32:935–941.
Batkai S, Jarai Z, Wagner JA, Goparaju SK, Varga K, Liu J, WangL, Mirshahi F, Khanolkar AD, Makriyannis A, Urbaschek R, Gar-cia N Jr, Sanyal AJ, Kunos G. 2001. Endocannabinoids acting atvascular CB1 receptors mediate the vasodilated state in advancedliver cirrhosis. Nat Med 7:827–832.
Battista S, Bar F, Mengozzi G, Zanon E, Grosso M, Molino G. 1997.Hyperdynamic circulation in patients with cirrhosis: direct mea-surement of nitric oxide levels in hepatic and portal veins. J Hep-atol 26:75–80.
Benoit JN, Womack WA, Hernandez L, Granger DN. 1985.‘‘Forward’’ and ‘‘backward’’ flow mechanisms of portal hyperten-sion. Relative contributions in the rat model of portal vein steno-sis. Gastroenterology 89:1092–1096.
Bolognesi M, Sacerdoti D, Di PM, Angeli P, Quarta S, Sticca A, Pon-tisso P, Merkel C, Gatta A. 2005. Haeme oxygenase mediates
hyporeactivity to phenylephrine in the mesenteric vessels of cir-rhotic rats with ascites. Gut 54:1630–1636.
Bomzon A, Huang YT. 2001. Vascular smooth muscle cell signalingin cirrhosis and portal hypertension. Pharmacol Ther 89:255–272.
Bosch J, Garcia-Pagan JC. 2000. Complications of cirrhosis. I. Por-tal hypertension. J Hepatol 32:141–156.
Cahill PA, Foster C, Redmond EM, Gingalewski C, Wu Y, SitzmannJV. 1995. Enhanced nitric oxide synthase activity in portal hyper-tensive rabbits. Hepatology 22:598–606.
Cahill PA, Redmond EM, Sitzmann JV. 2001. Endothelial dysfunc-tion in cirrhosis and portal hypertension. Pharmacol Ther89:273–293.
Cales P, Braillon A, Girod C, Lebrec D. 1985a. Acute effect of pro-pranolol on splanchnic circulation in normal and portal hyperten-sive rats. J Hepatol 1:349–357.
Cales P, Braillon A, Jiron MI, Lebrec D. 1985b. Superior portosyste-mic collateral circulation estimated by azygos blood flow inpatients with cirrhosis. Lack of correlation with oesophageal vari-ces and gastrointestinal bleeding. Effect of propranolol. J Hepatol1:37–46.
Campillo B, Chabrier PE, Pelle G, Sediame S, Atlan G, Fouet P,Adnot S. 1995. Inhibition of nitric oxide synthesis in the forearmarterial bed of patients with advanced cirrhosis. Hepatology22:1423–1429.
Carmeliet P. 2000. Mechanisms of angiogenesis and arteriogenesis.Nat Med 6:389–395.
Carmeliet P. 2003. Angiogenesis in health and disease. Nat Med9:653–660.
Carmeliet P, Moons L, Luttun A, Vincenti V, Compernolle V, De MolM, Wu Y, Bono F, Devy L, Beck H, Scholz D, Acker T, DiPalma T,Dewerchin M, Noel A, Stalmans I, Barra A, Blacher S, Vanden-driessche T, Ponten A, Eriksson U, Plate KH, Foidart JM,Schaper W, Charnock-Jones DS, Hicklin DJ, Herbert JM, CollenD, Persico MG. 2001. Synergism between vascular endothelialgrowth factor and placental growth factor contributes to angio-genesis and plasma extravasation in pathological conditions. NatMed 7:575–583.
Cejudo-Martin P, Ros J, Navasa M, Fernandez J, Fernandez-VaroG, Ruiz-del-Arbol L, Rivera F, Arroyo V, Rodes J, Jimenez W.2001. Increased production of vascular endothelial growth factorin peritoneal macrophages of cirrhotic patients with spontaneousbacterial peritonitis. Hepatology 34:487–493.
Chabot F, Mestiri H, Sabry S, Dall’Ava-Santucci J, Lockhart A,Dinh-Xuan AT. 1996. Role of NO in the pulmonary artery hypo-reactivity to phenylephrine in experimental biliary cirrhosis. EurRespir J 9:560–564.
Chan CC, Lee FY, Wang SS, Chang FY, Lin HC, Chu CJ, Tai CC,Lai IN, Lee SD. 1999. Effects of vasopressin on portal-systemiccollaterals in portal hypertensive rats: role of nitric oxide andprostaglandin. Hepatology 30:630–635.
Chen YC, Gines P, Yang J, Summer SN, Falk S, Russell NS, SchrierRW. 2004. Increased vascular heme oxygenase-1 expression con-tributes to arterial vasodilation in experimental cirrhosis in rats.Hepatology 39:1075–1087.
Cheng Y, Ndisang JF, Tang G, Cao K, Wang R. 2004. Hydrogen sul-fide-induced relaxation of resistance mesenteric artery beds ofrats. Am J Physiol Heart Circ Physiol 287:H2316–H2323.
Chu CJ, Wu SL, Lee FY, Wang SS, Chang FY, Lin HC, Chan CC,Lee SD. 2000. Splanchnic hyposensitivity to glypressin in a haem-orrhage/transfused rat model of portal hypertension: role of nitricoxide and bradykinin. Clin Sci (Lond) 99:475–482.
Cohen RA, Vanhoutte PM. 1995. Endothelium-dependent hyperpo-larization. Beyond nitric oxide and cyclic GMP. Circulation 92:3337–3349.
Colle I, De Vriese AS, Van Vlierberghe H, Lameire NH, Devos M.2004a. Systemic and splanchnic haemodynamic effects of sildena-fil in an in vivo animal model of cirrhosis support for a risk in cir-rhotic patients. Liver Int 24:63–68.
Colle IO, De Vriese AS, Van Vlierberghe HR, Lameire NH, De VosMM. 2004b. Vascular hyporesponsiveness in the mesenteric ar-tery of anaesthetized rats with cirrhosis and portal hypertension:an in-vivo study. Eur J Gastroenterol Hepatol 16:139–145.
710 COLLE ET AL.

Davis MJ, Gore RW. 1985. Capillary pressures in rat intestinalmuscle and mucosal villi during venous pressure elevation. Am JPhysiol 249:H174–H187.
De Las HD, Fernandez J, Gines P, Cardenas A, Ortega R, NavasaM, Barbera JA, Calahorra B, Guevara M, Bataller R, Jimenez W,Arroyo V, Rodes J. 2003. Increased carbon monoxide productionin patients with cirrhosis with and without spontaneous bacterialperitonitis. Hepatology 38:452–459.
Domenicali M, Ros J, Fernandez-Varo G, Cejudo-Martin P, CrespoM, Morales-Ruiz M, Briones AM, Campistol JM, Arroyo V, Vila E,Rodes J, Jimenez W. 2005. Increased anandamide induced relaxa-tion in mesenteric arteries of cirrhotic rats: role of cannabinoidand vanilloid receptors. Gut 54:522–527.
Ebrahimkhani MR, Mani AR, Moore K. 2005. Hydrogen sulphideand the hyperdynamic circulation in cirrhosis: a hypothesis. Gut54:1668–1671.
Elbirt KK, Bonkovsky HL. 1999. Heme oxygenase: recent advancesin understanding its regulation and role. Proc Assoc Am Physi-cians 111:438–447.
Erario MA, Gonzales S, Romay S, Eizayaga FX, Castro JL, LembergA, Tomaro ML. 2005. Role of heme oxygenase/carbon monoxidepathway on the vascular response to noradrenaline in portal hy-pertensive rats. Clin Exp Pharmacol Physiol 32:196–201.
Fallon MB, Abrams GA, Luo B, Hou Z, Dai J, Ku DD. 1997. Therole of endothelial nitric oxide synthase in the pathogenesis of arat model of hepatopulmonary syndrome. Gastroenterology 113:606–614.
Fernandez M, Bonkovsky HL. 1999. Increased heme oxygenase-1gene expression in liver cells and splanchnic organs from portalhypertensive rats. Hepatology 29:1672–1679.
Fernandez M, Lambrecht RW, Bonkovsky HL. 2001. Increasedheme oxygenase activity in splanchnic organs from portal hyper-tensive rats: role in modulating mesenteric vascular reactivity. JHepatol 34:812–817.
Fernandez M, Vizzutti F, Garcia-Pagan JC, Rodes J, Bosch J. 2004.Anti-VEGF receptor-2 monoclonal antibody prevents portal-sys-temic collateral vessel formation in portal hypertensive mice.Gastroenterology 126:886–894.
Fernandez M, Mejias M, Angermayr B, Garcia-Pagan JC, Rodes J,Bosch J. 2005. Inhibition of VEGF receptor-2 decreases the devel-opment of hyperdynamic splanchnic circulation and portal-sys-temic collateral vessels in portal hypertensive rats. J Hepatol43:98–103.
Fernandez-Seara J, Prieto J, Quiroga J, Zozaya JM, Cobos MA,Rodriguez-Eire JL, Garcia-Plaza A, Leal J. 1989. Systemic and re-gional hemodynamics in patients with liver cirrhosis and asciteswith and without functional renal failure. Gastroenterology 97:1304–1312.
Fernandez-Varo G, Ros J, Morales-Ruiz M, Cejudo-Martin P, ArroyoV, Sole M, Rivera F, Rodes J, Jimenez W. 2003. Nitric oxide syn-thase 3-dependent vascular remodeling and circulatory dysfunc-tion in cirrhosis. Am J Pathol 162:1985–1993.
Ferrara N, Gerber HP, LeCouter J. 2003. The biology of VEGF andits receptors. Nat Med 9:669–676.
Fukumura D, Gohongi T, Kadambi A, Izumi Y, Ang J, Yun CO,Buerk DG, Huang PL, Jain RK. 2001. Predominant role of endo-thelial nitric oxide synthase in vascular endothelial growth fac-tor-induced angiogenesis and vascular permeability. Proc NatlAcad Sci U S A 98:2604–2609.
Gadano AC, Sogni P, Heller J, Moreau R, Bories PN, Lebrec D.1999. Vascular nitric oxide production during the development oftwo experimental models of portal hypertension. J Hepatol30:896–903.
Garcia-Estan J, Atucha NM, Groszmann RJ. 1996. Renal responseto methoxamine in portal hypertensive rats: role of prostaglan-dins and nitric oxide. J Hepatol 25:206–211.
Garcia-Pagan JC, Fernandez M, Bernadich C, Pizcueta P, PiqueJM, Bosch J, Rodes J. 1994. Effects of continued NO inhibition onportal hypertensive syndrome after portal vein stenosis in rat.Am J Physiol 267:G984–G990.
Geerts AM, De Vriese AS, Vanheule E, Van Vlierberghe H, MortierS, Cheung KJ, Demetter P, Lameire N, De Vos M, Colle I. 2006a.
Increased angiogenesis and permeability in the mesenteric micro-vasculature of rats with cirrhosis and portal hypertension: an invivo study. Liver Int 26:889–898.
Geerts AM, Vanheule E, Carmeliet P, Van Vlierberghe H, LeybaertL, De Vos M, Colle I. 2006b. Placental growth factor plays a rolein the mesenteric neo-angiogenesis of portal hypertensive mice.Hepatology 44:216A.
Geraldo J, Ferraz P, Wallace JL. 1996. Prostaglandins modulate theresponsiveness of the gastric microcirculation of sodium nitro-prusside in cirrhotic rats. Hepatology 23:123–129.
Granger DN, Richardson PD, Taylor AE. 1979. Volumetric assess-ment of the capillary filtration coefficient in the cat small intes-tine. Pflugers Arch 381:25–33.
Granger DN, Kvietys P, Korthuis RJ, Premen A. 1989. Microcircula-tion of the intestinal mucosa. In: Wood J, editor. Handbook of gas-trointestinal physiology. Bethesda, MD: American Physiology So-ciety. p 1405–1474.
Groszmann RJ. 1993. Hyperdynamic state in chronic liver diseases.J Hepatol 17(suppl 2):S38–S40.
Groszmann RJ. 1994. Hyperdynamic circulation of liver disease 40years later: pathophysiology and clinical consequences. Hepato-logy 20:1359–1363.
Groszmann RJ, Abraldes JG. 2005. Portal hypertension: from bed-side to bench. J Clin Gastroenterol 39:S125–S130.
Guarner C, Soriano G, Tomas A, Bulbena O, Novella MT, BalanzoJ, Vilardell F, Mourelle M, Moncada S. 1993. Increased serum ni-trite and nitrate levels in patients with cirrhosis: relationship toendotoxemia. Hepatology 18:1139–1143.
Guevara M, Bru C, Gines P, Fernandez-Esparrach G, Sort P, Batal-ler R, Jimenez W, Arroyo V, Rodes. 1998. Increased cerebrovascu-lar resistance in cirrhotic patients with ascites. Hepatology28:39–44.
Hartleb M, Michielsen PP, Dziurkowska-Marek A. 1997. The role ofnitric oxide in portal hypertensive systemic and portal vascularpathology. Acta Gastroenterol Belg 60:222–232.
Heinemann A, Stauber RE. 1996. Vasodilator responses to nitric ox-ide are enhanced in mesenteric arteries of portal hypertensiverats. Eur J Clin Invest 26:824–826.
Heinemann A, Wachter CH, Holzer P, Fickert P, Stauber RE. 1997.Nitric oxide-dependent and -independent vascular hyporeactivityin mesenteric arteries of portal hypertensive rats. Br J Pharmacol121:1031–1037.
Heller J, Schepke M, Gehnen N, Molderings GJ, Muller A,Erhard J, Spengler U, Sauerbruch T. 1999. Altered adrenergicresponsiveness of endothelium-denuded hepatic arteries andportal veins in patients with cirrhosis. Gastroenterology 116:387–393.
Hori N, Wiest R, Groszmann RJ. 1998. Enhanced release of nitricoxide in response to changes in flow and shear stress in the supe-rior mesenteric arteries of portal hypertensive rats. Hepatology28:1467–1473.
Hosoki R, Matsuki N, Kimura H. 1997. The possible role of hydro-gen sulfide as an endogenous smooth muscle relaxant in synergywith nitric oxide. Biochem Biophys Res Commun 237:527–531.
Hou MC, Cahill PA, Zhang S, Redmond EM, Sitzmann JV. 1997.Enhanced G-protein-induced relaxation in portal hypertensiverats: role of nitric oxide. Hepatology 26:27–33.
Howlett AC, Barth F, Bonner TI, Cabral G, Casellas P, Devane WA,Felder CC, Herkenham M, Mackie K, Martin BR, Mechoulam R,Pertwee RG. 2002. International Union of Pharmacology. XXVII.Classification of cannabinoid receptors. Pharmacol Rev 54:161–202.
Iwakiri Y, Groszmann RJ. 2006. The hyperdynamic circulation ofchronic liver diseases: from the patient to the molecule. Hepato-logy 43:S121–S131.
Iwakiri Y, Tsai MH, McCabe TJ, Gratton JP, Fulton D, GroszmannRJ, Sessa WC. 2002a. Phosphorylation of eNOS initiates exces-sive NO production in early phases of portal hypertension. Am JPhysiol Heart Circ Physiol 282:H2084–H2090.
Iwakiri Y, Cadelina G, Sessa WC, Groszmann RJ. 2002b. Mice withtargeted deletion of eNOS develop hyperdynamic circulation asso-ciated with portal hypertension. Am J Physiol Gastrointest LiverPhysiol 283:G1074–G1081.
711SPLANCHNIC HEMODYNAMIC CHANGES IN PORTAL HYPERTENSION

Jurzik L, Froh M, Straub RH, Scholmerich J, Wiest R. 2005. Up-regulation of nNOS and associated increase in nitrergic vasodila-tion in superior mesenteric arteries in pre-hepatic portal hyper-tension. J Hepatol 43:258–265.
Karatapanis S, McCormick PA, Kakad S, Chin JK, Islam M, JeremyJ, Harry D, McIntyre N, Burroughs AK, Jacobs M. 1994. Altera-tion in vascular reactivity in isolated aortic rings from portalvein-constricted rats. Hepatology 20:1516–1521.
Kawanaka H, Jones MK, Szabo IL, Baatar D, Pai R, Tsugawa K,Sugimachi K, Sarfeh IJ, Tarnawski AS. 2002. Activation of eNOSin rat portal hypertensive gastric mucosa is mediated by TNF-alpha via the PI 3-kinase-Akt signaling pathway. Hepatology35:393–402.
Kojima H, Sakurai S, Uemura M, Satoh H, Nakashima T, Mina-mino N, Kangawa K, Matsuo H, Fukui H. 2004. Adrenomedullincontributes to vascular hyporeactivity in cirrhotic rats with asci-tes via a release of nitric oxide. Scand J Gastroenterol 39:686–693.
Kwon SY, Groszmann RJ, Iwakiri Y. 2007. Increased neuronal nitricoxide synthase interaction with soluble guanylate cyclase contrib-utes to the splanchnic arterial vasodilation in portal hypertensiverats. Hepatol Res 37:58–67.
Lee FY, Albillos A, Colombato LA, Groszmann RJ. 1992. The role ofnitric oxide in the vascular hyporesponsiveness to methoxaminein portal hypertensive rats. Hepatology 16:1043–1048.
Lee FY, Colombato LA, Albillos A, Groszmann RJ. 1993. Adminis-tration of N omega-nitro-L-arginine ameliorates portal-systemicshunting in portal-hypertensive rats. Gastroenterology 105:1464–1470.
Lee SS, Huang M, Ma Z, Rorstad O. 1996. Vasoactive intestinalpeptide in cirrhotic rats: hemodynamic effects and mesenteric ar-terial receptor characteristics. Hepatology 23:1174–1180.
Lincoln TM, Cornwell TL. 1993. Intracellular cyclic GMP receptorproteins. FASEB J 7:328–338.
Liu H, Lee SS. 1999. Cardiopulmonary dysfunction in cirrhosis. JGastroenterol Hepatol 14:600–608.
Liu H, Song D, Lee SS. 2001. Role of heme oxygenase-carbon mon-oxide pathway in pathogenesis of cirrhotic cardiomyopathy in therat. Am J Physiol Gastrointest Liver Physiol 280:G68–G74.
Lowenstein CJ, Dinerman JL, Snyder SH. 1994. Nitric oxide: aphysiologic messenger. Ann Intern Med 120:227–237.
Maroto A, Gines A, Salo J, Claria J, Gines P, Anibarro L, JimenezW, Arroyo V, Rodes J. 1994. Diagnosis of functional kidney failureof cirrhosis with Doppler sonography: prognostic value of resistiveindex. Hepatology 20:839–844.
Maroto A, Gines P, Arroyo V, Gines A, Salo J, Claria J, Jimenez W,Bru C, Rivera F, Rodes J. 1993b. Brachial and femoral arteryblood flow in cirrhosis: relationship to kidney dysfunction. Hepa-tology 17:788–793.
Martin PY, Xu DL, Niederberger M, Weigert A, Tsai P, St John J,Gines P, Schrier RW. 1996. Upregulation of endothelial constitu-tive NOS: a major role in the increased NO production in cir-rhotic rats. Am J Physiol 270:F494–F499.
Martin PY, Gines P, Schrier RW. 1998. Nitric oxide as a mediator ofhemodynamic abnormalities and sodium and water retention incirrhosis. N Engl J Med 339:533–541.
Martinez-Cuesta MA, Moreno L, Pique JM, Bosch J, Rodrigo J,Esplugues JV. 1996. Nitric oxide-mediated beta 2-adrenoceptorrelaxation is impaired in mesenteric veins from portal-hyperten-sive rats. Gastroenterology 111:727–735.
Mathie RT, Ralevic V, Moore KP, Burnstock G. 1996. Mesenteric va-sodilator responses in cirrhotic rats: a role for nitric oxide? Hepa-tology 23:130–136.
Matsumoto A, Ogura K, Hirata Y, Kakoki M, Watanabe F, Taken-aka K, Shiratori Y, Momomura S, Omata M. 1995. Increased ni-tric oxide in the exhaled air of patients with decompensated livercirrhosis. Ann Intern Med 123:110–113.
Michielsen PP, Boeckxstaens GE, Sys SU, Herman AG, PelckmansPA. 1995a. Role of nitric oxide in hyporeactivity to noradrenalineof isolated aortic rings in portal hypertensive rats. Eur J Pharma-col 273:167–174.
Michielsen PP, Boeckxstaens GE, Sys SU, Herman AG, PelckmansPA. 1995b. The role of increased nitric oxide in the vascular hypo-reactivity to noradrenaline in long-term portal vein ligated rats. JHepatol 23:341–347.
Moncada S, Higgs A, Furchgott R. 1997. International Union ofPharmacology Nomenclature in Nitric Oxide Research. PharmacolRev 49:137–142.
Morales-Ruiz M, Jimenez W, Perez-Sala D, Ros J, Leivas A, LamasS, Rivera F, Arroyo V. 1996. Increased nitric oxide synthaseexpression in arterial vessels of cirrhotic rats with ascites. Hepa-tology 24:1481–1486.
Moreau R. 2001. Heme oxygenase: protective enzyme or portal hy-pertensive molecule? J Hepatol 34:936–939.
Moreau R, Lebrec D. 1995. Endogenous factors involved in the con-trol of arterial tone in cirrhosis. J Hepatol 22:370–376.
Moreau R, Lebrec D. 2005. Molecular mechanisms of systemic vaso-dilation and hyperdynamic circulatory state of cirrhosis. In: SanyalAJ, Shah V, editors. Clinical gastroenterology: portal hypertension.Totowa, NJ: Humana Press Inc. p 51–64.
Moreau R, Lee SS, Soupison T, Roche-Sicot J, Sicot C. 1988. Abnor-mal tissue oxygenation in patients with cirrhosis and liver fail-ure. J Hepatol 7:98–105.
Moreau R, Barriere E, Tazi KA, Lardeux B, Dargere D, UrbanowiczW, Poirel O, Chauvelot-Moachon L, Guimont MC, Bernuau D,Lebrec D. 2002. Terlipressin inhibits in vivo aortic iNOS expres-sion induced by lipopolysaccharide in rats with biliary cirrhosis.Hepatology 36:1070–1078.
Mosca P, Lee FY, Kaumann AJ, Groszmann RJ. 1992. Pharmacologyof portal-systemic collaterals in portal hypertensive rats: role ofendothelium. Am J Physiol 263:G544–G550.
Munoz J, Albillos A, Perez-Paramo M, Rossi I, Alvarez-Mon M.1999. Factors mediating the hemodynamic effects of tumor necro-sis factor- alpha in portal hypertensive rats. Am J Physiol276:G687–G693.
Neef M, Biecker E, Heller J, Schepke M, Nischalke HD, Wolff M,Spengler U, Reichen J, Sauerbruch T. 2003. Portal hypertensionis associated with increased mRNA levels of vasopressor G-pro-tein-coupled receptors in human hepatic arteries. Eur J ClinInvest 33:249–255.
Niederberger M, Gines P, Tsai P, Martin PY, Morris K, Weigert A,McMurtry I, Schrier RW. 1995. Increased aortic cyclic guanosinemonophosphate concentration in experimental cirrhosis in rats:evidence for a role of nitric oxide in the pathogenesis of arterialvasodilation in cirrhosis. Hepatology 21:1625–1631.
Oberti F, Sogni P, Cailmail S, Moreau R, Pipy B, Lebrec D. 1993.Role of prostacyclin in hemodynamic alterations in conscious ratswith extrahepatic or intrahepatic portal hypertension. Hepatology18:621–627.
Ohta M, Kishihara F, Hashizume M, Kawanaka H, Tomikawa M,Higashi H, Tanoue K, Sugimachi K. 1995. Increased prostacyclincontent in gastric mucosa of cirrhotic patients with portal hyper-tensive gastropathy. Prostaglandins Leukot Essent Fatty Acids53:41–45.
Ortiz MC, Fortepiani LA, Martinez C, Atucha NM, Garcia-Estan J.1996. Vascular hyporesponsiveness in aortic rings from cirrhoticrats: role of nitric oxide and endothelium. Clin Sci (Lond) 91:733–738.
Pateron D, Oberti F, Lefilliatre P, Veal N, Tazi KA, Sogni P, PoirelO, Heller J, Moreau R, Cales P, Lebrec D. 1999. Relationshipbetween vascular reactivity in vitro and blood flows in rats withcirrhosis. Clin Sci (Lond) 97:313–318.
Pateron D, Tazi KA, Sogni P, Heller J, Chagneau C, Poirel O, Phil-ippe M, Moreau R, Lebrec D. 2000. Role of aortic nitric oxide syn-thase 3 (eNOS) in the systemic vasodilation of portal hyperten-sion. Gastroenterology 119:196–200.
Perez-Ruiz M, Ros J, Morales-Ruiz M, Navasa M, Colmenero J,Ruiz-del-Arbol L, Cejudo P, Claria J, Rivera F, Arroyo V, Rodes J,Jimenez W. 1999. Vascular endothelial growth factor productionin peritoneal macrophages of cirrhotic patients: regulation bycytokines and bacterial lipopolysaccharide. Hepatology 29:1057–1063.
712 COLLE ET AL.

Pilette C, Kirstetter P, Sogni P, Cailmail S, Moreau R, Lebrec D.1996. Dose-dependent effects of a nitric oxide biosynthesis inhibi-tor on hyperdynamic circulation in two models of portal hyperten-sion in conscious rats. J Gastroenterol Hepatol 11:1–6.
Rastegar H, Jorjani M, Roushanzamir F, Ahmadiani A, NamiranianK, Dehpour AR. 2001. Time-dependent reduction of acetylcholine-induced relaxation in aortic rings of cholestatic rats. PharmacolRes 44:519–525.
Ros J, Claria J, To-Figueras J, Planaguma A, Cejudo-Martin P, Fer-nandez-Varo G, Martin-Ruiz R, Arroyo V, Rivera F, Rodes J, Jime-nez W. 2002. Endogenous cannabinoids: a new system involved inthe homeostasis of arterial pressure in experimental cirrhosis inthe rat. Gastroenterology 122:85–93.
Safka V, Moreau R, Gadano A, Lebrec D. 1997. Vascular hypores-ponsiveness to vasodilators in rats with cirrhosis. J Hepatol26:382–386.
Schepke M, Heller J, Paschke S, Thomas J, Wolff M, Neef M,Malago M, Molderings GJ, Spengler U, Sauerbruch T. 2001. Con-tractile hyporesponsiveness of hepatic arteries in humans withcirrhosis: evidence for a receptor-specific mechanism. Hepatology34:884–888.
Shah V, Wiest R, Garcia-Cardena G, Cadelina G, Groszmann RJ,Sessa WC. 1999. Hsp90 regulation of endothelial nitric oxide syn-thase contributes to vascular control in portal hypertension. Am JPhysiol 277:G463–G468.
Sieber CC, Lopez-Talavera JC, Groszmann RJ. 1993. Role of nitricoxide in the in vitro splanchnic vascular hyporeactivity in asciticcirrhotic rats. Gastroenterology 104:1750–1754.
Sieber CC, Sumanovski LT, Stumm M, van der KM, Battegay E.2001. In vivo angiogenesis in normal and portal hypertensiverats: role of basic fibroblast growth factor and nitric oxide. J Hep-atol 34:644–650.
Sitzmann JV, Campbell K, Wu Y, St Clair C. 1994. Prostacyclin pro-duction in acute, chronic, and long-term experimental portalhypertension. Surgery 115:290–294.
Sogni P, Garnier P, Gadano A, Moreau R, Dall’Ava-Santucci J,Dinh-Xuan AT, Lebrec D. 1995. Endogenous pulmonary nitric ox-ide production measured from exhaled air is increased in patientswith severe cirrhosis. J Hepatol 23:471–473.
Sogni P, Sabry S, Moreau R, Gadano A, Lebrec D, Dinh-Xuan AT.1996. Hyporeactivity of mesenteric resistance arteries in portalhypertensive rats. J Hepatol 24:487–490.
Sogni P, Smith AP, Gadano A, Lebrec D, Higenbottam TW. 1997.Induction of nitric oxide synthase II does not account for excessvascular nitric oxide production in experimental cirrhosis. J Hep-atol 26:1120–1127.
Sumanovski LT, Battegay E, Stumm M, van der Kooji M, SieberCC. 1999. Increased angiogenesis in portal hypertensive rats: roleof nitric oxide. Hepatology 29:1044–1049.
Tang KM, Wang GR, Lu P, Karas RH, Aronovitz M, Heximer SP, Kal-tenbronn KM, Blumer KJ, Siderovski DP, Zhu Y, Mendelsohn ME.2003. Regulator of G-protein signaling-2 mediates vascular smoothmuscle relaxation and blood pressure. Nat Med 9:1506–1512.
Tazi KA, Moreau R, Heller J, Poirel O, Lebrec D. 2000. Changes inprotein kinase C isoforms in association with vascular hyporeac-tivity in cirrhotic rat aortas. Gastroenterology 119:201–210.
Tazi KA, Barriere E, Moreau R, Heller J, Sogni P, Pateron D, PoirelO, Lebrec D. 2002. Role of shear stress in aortic eNOS up-regula-tion in rats with biliary cirrhosis. Gastroenterology 122:1869–1877.
Tazi KA, Moreau R, Herve P, Dauvergne A, Cazals-Hatem D, BertF, Poirel O, Rabiller A, Lebrec D. 2005. Norfloxacin reduces aorticNO synthases and proinflammatory cytokine up-regulation in cir-rhotic rats: role of Akt signaling. Gastroenterology 129:303–314.
Tsai MH, Iwakiri Y, Cadelina G, Sessa WC, Groszmann RJ. 2003.Mesenteric vasoconstriction triggers nitric oxide overproduction
in the superior mesenteric artery of portal hypertensive rats. Gas-troenterology 125:1452–1461.
Tsugawa K, Hashizume M, Migou S, Kishihara F, Kawanaka H,Tomikawa M, Sugimachi K. 1999. A selective cyclo-oxygenase-2inhibitor, NS-398, may improve portal hypertension withoutinducing gastric mucosal injury. J Gastroenterol Hepatol 14:642–651.
Tsugawa K, Hashizume M, Migou S, Kishihara F, Kawanaka H,Tomikawa M, Sugimachi K. 2000. Role of vascular endothelialgrowth factor in portal hypertensive gastropathy. Digestion61:98–106.
Tsugawa K, Hashizume M, Tomikawa M, Migou S, Kawanaka H,Shiraishi S, Sueishi K, Sugimachi K. 2001. Immunohistochemi-cal localization of vascular endothelial growth factor in the ratportal hypertensive gastropathy. J Gastroenterol Hepatol 16:429–437.
Urbich C, Dimmeler S. 2004. Endothelial progenitor cells: charac-terization and role in vascular biology. Circ Res 95:343–353.
Vallance P, Moncada S. 1991. Hyperdynamic circulation in cirrhosis:a role for nitric oxide? Lancet 337:776–778.
Vorobioff J, Bredfeldt JE, Groszmann RJ. 1984. Increased bloodflow through the portal system in cirrhotic rats. Gastroenterology87:1120–1126.
Wang R, Wang Z, Wu L. 1997. Carbon monoxide-induced vasorelax-ation and the underlying mechanisms. Br J Pharmacol 121:927–934.
Wever RM, van Dam T, van Rijn HJ, de Groot F, Rabelink TJ.1997. Tetrahydrobiopterin regulates superoxide and nitric oxidegeneration by recombinant endothelial nitric oxide synthase. Bio-chem Biophys Res Commun 237:340–344.
Wiest R, Shah V, Sessa WC, Groszmann RJ. 1999a. NO overproduc-tion by eNOS precedes hyperdynamic splanchnic circulation inportal hypertensive rats. Am J Physiol 276:G1043–G1051.
Wiest R, Das S, Cadelina G, Garcia-Tsao G, Milstien S, GroszmannRJ. 1999b. Bacterial translocation in cirrhotic rats stimulateseNOS-derived NO production and impairs mesenteric vascularcontractility. J Clin Invest 104:1223–1233.
Wiest R, Cadelina G, Milstien S, McCuskey RS, Garcia-Tsao G,Groszmann RJ. 2003. Bacterial translocation up-regulates GTP-cyclohydrolase I in mesenteric vasculature of cirrhotic rats. Hepa-tology 38:1508–1515.
Wiest R, Jurzik L, Moleda L, Froh M, Schnabl B, von HS,Scholmerich J, Straub RH. 2006. Enhanced Y1-receptor-medi-ated vasoconstrictive action of neuropeptide Y (NPY) in supe-rior mesenteric arteries in portal hypertension. J Hepatol 44:512–519.
Wiest R, Jurzik L, Herold T, Straub RH, Scholmerich J. 2007. Roleof NPY for vasoregulation in the splanchnic circulation duringportal hypertension. Peptides 28:396–404.
Xu L, Carter EP, Ohara M, Martin PY, Rogachev B, Morris K, Cad-napaphornchai M, Knotek M, Schrier RW. 2000. Neuronal nitricoxide synthase and systemic vasodilation in rats with cirrhosis.Am J Physiol Renal Physiol 279:F1110–F1115.
Yin ZH, Liu XY, Huang RL, Ren SP. 2005. Expression of TNF-alphaand VEGF in the esophagus of portal hypertensive rats. World JGastroenterol 11:1232–1236.
Zhang J, Ling Y, Luo B, Tang L, Ryter SW, Stockard CR, GrizzleWE, Fallon MB. 2003. Analysis of pulmonary heme oxygenase-1and nitric oxide synthase alterations in experimental hepatopul-monary syndrome. Gastroenterology 125:1441–1451.
Zhao W, Wang R. 2002. H(2)S-induced vasorelaxation and underly-ing cellular and molecular mechanisms. Am J Physiol Heart CircPhysiol 283:H474–H480.
Zhao W, Zhang J, Lu Y, Wang R. 2001. The vasorelaxant effect ofH(2)S as a novel endogenous gaseous K(ATP) channel opener.EMBO J 20:6008–6016.
713SPLANCHNIC HEMODYNAMIC CHANGES IN PORTAL HYPERTENSION




















