
Construction of physical maps from oligonucleotide fingerprints data
Upload
independentCategory
view
2download
0
www.elsevier.com/locate/yexcr
Experimental Cell Researc
Enhanced oligonucleotide-directed gene targeting in mammalian cells
following treatment with DNA damaging agents
Luciana Ferraraa,b, Hetal Parekh-Olmedoa, Eric B. Kmieca,*
aDepartment of Biological Sciences, Delaware Biotechnology Institute, University of Delaware, Newark, DE 19711, United StatesbDepartment of Genetics, Biology, Biochemistry, University of Torino, Italy
Received 14 April 2004, revised version received 22 June 2004
Available online 8 August 2004
Abstract
Targeted gene repair, a form of oligonucleotide-directed mutagenesis, employs end-modified single-stranded DNA oligonucleotides to
mediate single-base changes in chromosomal DNA. In this work, we use a specific 72-mer to direct the repair of a mutated eGFP gene stably
integrated in the genome of DLD-1 cells. Corrected cells express eGFP that can be identified and quantitated by FACS. The repair of this
mutant gene is dependent on the presence of a specifically designed oligonucleotide and the frequency with which the mutation is reversed is
affected by the induction of DNA damage. We used hydroxyurea, VP16 (etoposide), and thymidine to modulate the rate of DNA replication
through the stalling of the replication forks or the introduction of lesions. Addition of hydroxyurea or VP16 before the electroporation of the
oligonucleotide, results in an accumulation of double-strand breaks (DSB) whose repair is facilitated by either nonhomologous end joining
(NHEJ) or homologous recombination (HR). The addition of thymidine results in DNA damage within replication forks, damage that is
repaired through the process of homologous recombination. Our data suggest that gene repair activity is elevated when DNA damage induces
or activates the homologous recombination pathway.
D 2004 Elsevier Inc. All rights reserved.
Keywords: DNA repair; DNA damage; Double-strand breaks; Single-stranded oligonucleotides; Nonhomologous end joining; Homologous recombination
Introduction
DNA oligonucleotides are being used to introduce single-
base changes into the genomes of prokaryotic and eukaryotic
cells [1]. The successful development of these kinds of
techniques will likely impact the fields of functional
genomics and gene therapy. While there are specific differ-
ences among the various approaches, including the strategies
used to deliver the oligonucleotide to the target cell or tissue,
the nucleotide alteration processes likely share common
features. It is currently believed that the reaction initializes
with a pairing phase in which the oligonucleotide is trans-
0014-4827/$ - see front matter D 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.yexcr.2004.06.021
* Corresponding author. Department of Biological Sciences, Delaware
Biotechnology Institute, University of Delaware, 15 Innovation Way,
Newark, DE 19711. Fax: +1 302 831 3427.
E-mail address: [email protected] (E.B. Kmiec).
ported to the nucleus and aligns in homologous register with a
complementary sequence in the genome. Once in alignment,
the complex attracts and engages endogenous DNA repair
systems that execute the base change. In prokaryotes, the
process of recombineering pioneered by Court et al. [2–4]
relies heavily on the activity of the E phage annealing protein
red h [5] to catalyze the pairing reaction. In eukaryotes,
triplex-forming oligonucleotides (TFOs) use nonenzymatic
activities to form the characteristic triple helix configuration
[6–8], while modified single-stranded oligonucleotides (non-
TFO) rely in all likelihood on recombinases such as Rad51 or
Rad52 [1,9–12].
While recombineering is achieving a remarkably high
level of nucleotide exchange (see Ref.[13] and references
therein), the frequency of gene repair in mammalian cells has
been unstable. There are numerous reasons for such
variability (see Ref. [1] and references therein), but one
h 300 (2004) 170–179

L. Ferrara et al. / Experimental Cell Research 300 (2004) 170–179 171
possibility is that the enzymes promoting the pairing or
repairing phases of the reaction are not present at sufficient
levels or in the required stoichiometry. Support for this notion
comes from recent data in which the overexpression of the
human RAD51 gene elevated the frequency of gene repair
only modestly (Hu et al., submitted for publication). In some
ways, this is not surprising because Rad51 participates in the
recombinational repair of DNA damage acting in conjunction
with a group of related proteins in a predetermined, optimal
stoichiometry [14]. In particular, this complex directs the
repair of double-stranded breaks that can occur during DNA
replication using the homologous recombination pathway in
some instances [15–19]. This response preserves and main-
tains genome stability.
Replicating DNA templates or at least cells in S phase
appears to be more amenable to alteration by single-stranded
oligonucleotides [20] for it is during this phase of the cell
cycle that the frequencies of gene repair are observed to be
higher [20] (Hu et al., submitted for publication). While
quiescent or nondividing cells contain a lower level of
proteins involved in HR and recombinational repair, cells in
S phase possess higher levels of these important proteins.
This relationship has recently been confirmed by studies
that detail correlations among cells in S phase, Rad51 foci
formation, and genetic recombination events [16,21,22].
Specifically, recombination between tandem repeats was
activated by replication arrest and the induction of double-
strand breaks (DSB) in a process known as recombination
induced by replication inhibition (RIRI) [22].
Because proteins involved in recombinational repair and
homologous recombination have been shown to increase the
frequency of gene repair in yeast [11,23], and DNA damage
stimulates homologous recombination pathways, we exam-
ined the effect of DNA damage on gene repair in
mammalian cells. Cells are grown under conditions where
the process of DNA replication is arrested or elongated and
double-strand breaks (DSB) are induced, the effects of
which supported a higher correction frequency. Our results
suggest that reducing the rate at which cells pass through S
phase, perhaps due to the accumulation of double-strand
breaks and the activation of the homologous recombination
pathway, leads to an improvement in the frequency of
targeted gene repair.
Materials and methods
Cell line and culture conditions
DLD-1 cells were obtained from American Type Cell
Culture (ATCC, Manassas, VA). DLD-1-integrated clone 1
(DLD-1-1) was obtained by integration of the vector
pEGFP-N3 containing a single point mutation (TAG) in
the eGFP gene. Cells were grown in RPMI 1640 medium
with 2 mM glutamine, 4.5 g/l glucose, 10 mM HEPES, 1
mM sodium pyruvate, and supplemented with 10% FBS.
Cells were maintained at 5% CO2, 378C and under selection
in 200 Ag/ml G418 (Gibco, Invitrogen Co., Carlsbad, CA).
eGFP gene targeting
Cells grown in complete medium supplemented with 10%
FBS were trypsinized and harvested by centrifugation. The
cell pellet was resuspended in serum-free medium at a density
of 1� 106 cells/100 Al and transferred to a 4 mm gap cuvette
(Fisher Scientific, Pittsburgh, PA). The oligonucleotide was
then added at a concentration of 4 AM and the cells were
electroporated (LV, 250 V, 13 ms, 2 pulses, 1 s interval) using
a BTX ECM830 apparatus (BTX, Holliston, MA). The cells
were then transferred to a 60-mm dish containing fresh
medium supplemented with 10% FBS and incubated for 48 h
at 378C before harvesting for FACS analysis.
Treatment of DLD-1 cell cultures with hydroxyurea, VP16,
or thymidine
Cells were seeded at a density of 0.8 � 106 cells 24 h
before addition of Hydroxyurea (HU) (1 mM), the eptop-
side, VP16 (3 AM), or thymidine (10 mM). The HU (Acros
Organics, Morris Plains, NJ) stock solution was prepared in
distilled water at a concentration of 500 mM and the VP16
(Sigma, St. Louis, MO) stock was prepared in DMSO
(100%) at a final concentration of 50 mM. Thymidine
(Sigma) stock was prepared in distilled water at a final
concentration of 200 mM. Hydroxyurea and VP16 were
added to the cells at the indicated concentration and time of
treatment was varied from 0–45 and 0–24 h, respectively.
Thymidine was added for a period of 24 h to synchronized
cells (see below).
Flow cytometry analysis
eGFP fluorescence of corrected cells was measured by a
Becton Dickinson FACS calibur flow cytometer (Becton
Dickinson, Rutherford, NJ). Cells were harvested 48 h after
electroporation and resuspended in FACS buffer (0.5%
BSA, 2 mM EDTA, 2 Ag/ml propidium iodide in PBS).
More specifically, the program was set for the appropriate
cell size (forward scatter versus side scatter) and the
population of single cells was gated for analysis. Using
the negative control (minus PI, minus GFP), the background
fluorescence was set by positioning the cells in the 101
decade of the dot plot by adjusting the voltage for FL1
(GFP) and FL2 (PI). The composition was then set for
multi-fluorochrome experiments using a GFP control
sample containing no PI and increasing the compensation
to bring the signal toward the FL1 parameter. Finally, the
last control, PI and no GFP, was used to increase the
compensation to bring the signal toward the FL2 parameter.
Samples of 50,000 cells each were analyzed and those cells
being GFP positive and PI negative were scored as corrected
cells. For cell cycle analysis, 1 � 106 cells were plated 24 h

L. Ferrara et al. / Experimental Cell Research 300 (2004) 170–179172
before the treatment with drugs and after 24 h of treatment,
cells were trypsinized, resuspended in 300 Al cold PBS, and
fixed by adding 700 Al cold ethanol. Cells were then
resuspended in 1 ml of PBS containing 50 Ag/ml RNaseA
and 2.5 Ag/ml propidium iodide, and analyzed for DNA
content. The number of cells possessing actively replicating
forks was determined by BrdU staining (In Situ Cell
Proliferation Kit, FLUOS, Roche Diagnostics, Indianapolis,
IN) following manufacturers suggestions.
Pulsed-field gel electrophoresis
Twenty-four hours before treatment with HU orVP16, 1�106 cells were plated in tissue culture flasks, followed by
induction of DNA damage with HU or VP16 for 24 h. The
cells were released by trypsinization and melted in agarose
inserts. The agarose inserts were incubated in 0.5 M EDTA–
1% N-laurosylsarcosine-proteinase K (1 mg/ml) at 508C for
48 h and then washed four times in TE buffer before loading
on a 1% agarose gel (Pulse-Field Certified Agarose, Bio-Rad,
Hercules, CA) and DNA separation by pulsed-field gel
electrophoresis was carried out for 24 h (Bio-Rad, 1208 fieldangle, 60–240 s switch time, 4 V/cm). The gel was
subsequently stained with ethidium bromide and analyzed
Fig. 1. (A) DNA sequence of the target gene and the modified single-stranded oli
codon at position +67, is depicted with the (corrected) wild-type gene (lower pan
nonspecific (random) oligonucleotide (Hyg3S/74NT) are also shown. The asteri
represents modified phosphorothioate linkages. (B) FACS analysis of DLD-1 clon
(using propidium iodide staining). The control cells (left) were electroporated with
right represents mutant eGFP cells that were targeted with the specific oligonucleot
nonviable cells, while the lower right quadrant represents corrected eGFP (fluore
with AlphaImagerk 2200 (Alpha Innotech Corp., San
Leandro, CA).
Cell synchronization
Cells were synchronized in G1 or at the G1/S border by a
double thymidine block. Twenty-four hours before the
addition of any agent (HU, etc.), cells were plated at a
density of 0.5 � 106 cells per 100 mm dish, followed by
incubation in 2 mM thymidine (Sigma) for 16 h, washed and
released in fresh medium for 10 h, then incubated in 2 mM
thymidine for an additional 15 h. Cells were then washed
and either electroporated with oligonucleotide or treated
with HU (1 mM), VP16 (3 AM), or thymidine (10 mM) for
24 h before the electroporation step.
Results
Gene repair activity was assayed using a mutant eGFP
gene as a target. The wild-type gene was mutated at amino
acid 67 in the chromophore region so that no green
fluorescence is observed when it is expressed. The mutation
creates a stop codon (TAG) at a site that originally encoded a
gonucleotides used in this study. The mutant eGFP gene, containing a stop
el). The 72 base-specific oligonucleotide (EGFP3S/72NT) and the 74 base-
sk between the terminal of these bases in each oligonucleotide sequence
e-1-corrected cells. Cells were analyzed for eGFP expression and viability
a nonspecific oligonucleotide, Hyg3S/74NT, while the FACS graph on the
ide, EGFP3S/72NT (see text for details). The upper right quadrant represents
scent) cells.

L. Ferrara et al. / Experimental Cell Research 300 (2004) 170–179 173
tyrosine residue (TAC). We have previously used this mutant
gene as an episomal target for analyzing gene repair activity
[24], and Thorpe et al. [12] used a similar mutant eGFP target
to measure correction. For the present study, however, the
eGFP gene was integrated into DLD-1 cells using a pEGFP-
N3 vector generating a clonal cell line known as DLD-1-1
(clone-1) (Hu et al., submitted for publication). These cells
contain 2–4 copies of the mutant eGFP gene but do not
produce functional eGFP (see below). The DLD-1 cell line
was chosen because of its robust growth and its ability to
adapt to the transfer of oligonucleotides by square-wave
electroporation. This cell line has been reported to have a
deficiency in some aspects of homologous recombination
[25], but the cell line actually is heterogenous with several
subpopulations of cells [26], with varying responses to DNA
damaging agents [27]. To address this issue, we obtained
individual clonal isolates for our studies to avoid confusion
about its responsiveness to agents tested in the experiments
outlined in this paper.
The experimental strategy involved the introduction of
oligonucleotides into DLD-1-clone 1 cells by electroporation
followed by phenotypic readout of the corrected eGFP gene
48 h later. The targeting oligonucleotide is 72 bases in length
(72-mer), complementary to the nontranscribed strand of the
mutant eGFP gene but designed to create a single mismatch in
the third base of codon 67 (see Fig. 1). It directs conversion of
a TAGYTAC codon that enables phenotypic expression of
eGFP, which can be detected by FACS [see Ref. [24]. Fig. 1A
outlines the sequence of the target gene, the 72-mer, and a
nonspecific 74-mer used as a control. The time of addition of
certain agents such as hydroxyurea or VP16, relative to the
timing of electroporation of the oligonucleotide, is described
below (see Fig. 2).
Fig. 2. Both HU and VP16 stimulate gene repair in DLD-1 asynchronous cell cultu
added for 24 h to asynchronously growing cells. The percentage of green fluore
experimental point at various concentrations of either HU of VP16. (B) Cell vi
presented. Cell death was determined by propidium iodide staining and measured
each point are shown in the figure. CE, correction efficiency (*P value b 0.05; *
Fig. 1B demonstrates the usefulness and validity of the
eGFP system. Clone 1 cells were electroporated with either
EGFP3S/72NT or Hyg3S/74NT and the level of gene
correction measured 48 h later by FACS analysis. Approx-
imately 1.2% of the cells treated with EGFP3S/72NT score
positive for eGFP expression, but the frequency of correction
in any given experiment was observed to vary from 0.8% to
1.4%. No green fluorescence is observed in the control (lower
right quadrant) when the population of cells was treated with
the nonspecific oligonucleotide Hyg3S/74NT or with a
completely complementary oligonucleotide (data not
shown). As displayed in Fig. 1A, Hyg3S/74NT contains no
direct sequence complementarity to the mutant eGFP target
site.
Hydroxyurea and VP16 stimulate gene repair activity
Hydroxyurea can be used to synchronize growing cells in
S phase by blocking or retarding the movement of the
replication fork [28,29]. The effect on replication leads to
double-strand breaks at or near the fork [15], both of which
are repaired primarily by homologous recombination (HR)
[16,18]. VP16 (etoposide) is an anti-cancer drug that induces
DNA double-strand breaks through specific inhibition of the
resealing activity of topoisomerase II [30]. It is still not clear
if VP16-induced breaks occur preferentially at replication
forks or at random sites, but both treatments have been shown
to induce HR pathways and elevate the frequency of HR as a
result of DNA damage [15,16,18,22,31–33]. Because HR in
mammalian cells is induced by DNA damage [18] and our
previous data implicate HR in the process of gene repair
[11,34], we tested the hypothesis that the addition of DNA
damaging agents to mammalian cell cultures influences the
res. (A) Hydroxyurea (0, 0.3, 1, 2, 5 mM) or VP16 (0, 0.5, 1, 3, 10 AM) was
scent cells is presented as a function of the 50,000 cells assayed for each
ability posttreatment with HU or VP16 at the indicated concentrations is
by FACS analysis. Means and standard deviations for four experiments for
*P value b 0.01).

Fig. 3. Treatment with HU and VP16 increases the correction efficiency as a
function of exposure time to the drug. Cells were plated 24 h before adding
the addition of (A) HU (1 mM) or (B) VP16 (3 AM) for the time indicated.
Correction efficiency was determined as a percentage of eGFP-positive
cells after analyzing 50,000 cells. The means and standard deviations of two
to four experiments are shown (*P value b 0.05; **P value b 0.01).
L. Ferrara et al. / Experimental Cell Research 300 (2004) 170–179174
process of gene repair. DLD-1 cells were incubated with
varying concentrations of hydroxyurea or VP16 and the level
of eGFP expression, a phenotypic measure of gene repair,
was analyzed. Cells were plated for 24 h before the addition
of HU or VP16 followed by electroporation 24 h later, and the
percentage of corrected cells was determined by FACS. As
shown in Fig. 2A, HU stimulated gene repair activity at each
concentration with a statistically significant difference
observed at 1, 2, and 5 mM, respectively. Similar results
were found in the VP16 experiments; stimulation is observed
at each concentration with statistically significant differences
observed at 1 and 3AM, respectively. VP16 elevates the gene
repair frequency up to 5% while the highest frequency
observed with HU treatment is 2.2%. The concentrations of
HU and VP16 that stimulate gene repair activity fall within
the range of concentration known to induce double-strand
breaks in mammalian cells [15,16,22].
The normal FACS assay used to detect gene repair is also
designed to measure cell death through the uptake of
propidium iodide and Fig. 2B displays cell survival after
treatment with increasing concentrations of HU or VP16.
While a modest amount of cell death is observed in cultures
treated with either agent, viability is slightly reduced at
higher concentrations, as observed previously [35–41].
We next evaluated enhancement of the frequency of gene
repair as a function of the time of exposure to either HU or
VP16. We incubated cells with 1 mM HU or 3 AM VP16 for
various times before the introduction of the oligonucleotide
under conditions where the overall time for each treatment
was the same but the time of exposure was varied. For
example, in the case of the 6 versus 24 h (VP16 treatment),
two cultures were grown for equal time (24 h), but in this
case the cells were treated with VP16 for the entire 24 h
while in the other case the cells were treated with VP16 for
the final 6 h. As shown in Figs. 3A and B, a gradual rise in
correction efficiency is observed up through 30–35 h with
HU and 12 h of exposure with VP16, respectively. The
asterisks in the figure indicate points that exhibit a statisti-
cally significant difference from the (zero) control: one
asterisk (*) = P value b 0.05; whereas two asterisks (**) = P
value b 0.01. These results indicate that longer contact time
with either agent leads to elevated frequencies of gene
repair, and using the terminology of Saintigny et al. [18], the
increase in gene repair activity can be classified as a blateresponseQ. Such a late response to double-strand DNA
breaks is catalyzed in large part by homologous recombi-
nation more so than nonhomologous end joining (NHEJ).
The concentration range of HU and VP16 used in our
experiments has been reported previously to induce DNA
damage, most often double-stranded DNA breaks [16,22].
But these conclusions were drawn from experiments con-
ducted in other cell lines, not the DLD-1 line. Thus, we
monitored the formation or accumulation of double-strand
breaks in DLD-1 cells by pulse-field gel electrophoresis
(PFGE) to assess the degree of DNA damage resulting from
the addition of HU or VP16. Cultures of asynchronously
growing DLD-1 cells were incubated with varying concen-
trations of HU or VP16 for 24 h and DNA breakage was then
assessed by PFGE. As shown in Fig. 4, a progressive increase
in the amount of damaged DNA, as a function of HU or VP16
concentration, is observed. Importantly, double-strand breaks
are found at the concentration of HU andVP16 that have been
shown coincidentally to stimulate the frequency of gene
repair. Our results are consistent with the observations of
Lundin et al. [16] and Saintigny et al. [18], and taken together
suggest a correlation between the two phenomena.
S phase has been observed to be the optimal period of the
cell cycle in which the highest levels of gene targeting are
obtained [20]. Besides inducing DNA damage, the addition
of HU or VP16 tomammalian cells in culture causes a stalling
of replication forks, as the cells respond to the DNA damage
and the metabolic stress. Because an enhancement of gene
repair activity is seen as a function of the addition of HU or
VP16 to growing cultures, it was of interest to determine what
percentage of the cells was actually in S phase at the time of
electroporation. The population of cells in this phase might
enable gene repair to take place more efficiently due to the
presence of actively replicating DNA forks and an overall
open chromatin structure. A modified nucleosomal config-
uration would likely be more accessible to the oligonucleo-
tide, which, in turn, would lead to higher levels of gene repair.
In this experiment, the cell cycle position of the treated and

Fig. 4. DSBs are induced in DLD-1 cells by exposure to HU and VP16, respectively. Double-strand breaks were visualized by PFGE after treatment with the
indicated concentrations of HU or VP16. Cells were treated for 24 h with HU (0.3, 1, 5 mM) or VP16 (0.5, 1, 3 AM) before collection and processing for gel
electrophoresis. Lane M indicates chromosomes of S. cerevisiae used as a marker, and lane C is a control of cells undergoing the same experimental
manipulation but without addition of HU or VP16.
L. Ferrara et al. / Experimental Cell Research 300 (2004) 170–179 175
untreated cells was determined by FACS after exposing cells
to VP16 or HU for 24 h. The results are shown in Fig. 5 (left
vertical column). While the profile of the untreated sample
depicts cells in all three phases, there is a substantial reduction
of cells in G2 as shown in the profiles of the HU or VP16 cell-
treated population. A coincident elevation in the percentage
of cells in S phase is also observed. These data are similar to
that obtained by Kumari et al. [42] in a similar series of
experiments measuring replication arrest associated with
double-strand breaks. Cells treated with VP16 also exhibit a
small peak of nonviable cells that represents a heightened
level of sensitivity of cells in S phase to this particular drug.
The percentage of cells in S phase was also determined by
BrdU staining in which BrdU becomes incorporated into
growing or replicating DNA strands. For each treatment
illustrated in FACS profile, the percentage of BrdU incorpo-
rating cells is presented in Fig. 5 (right column). The addition
of HU or VP16 to DLD-1 cells induces a higher percentage to
enter, remain, or stall in S phase. Our results correlate with the
cell cycle profiles obtained by Saintigny and Lopez [22] in
which a variety of replication inhibitors were tested for the
induction of HR activities. Thus, agents that induce DNA
damage simultaneously result in an accumulation of cells in S
phase; these two cellular responses may act synergistically to
elevate the frequency of gene repair.
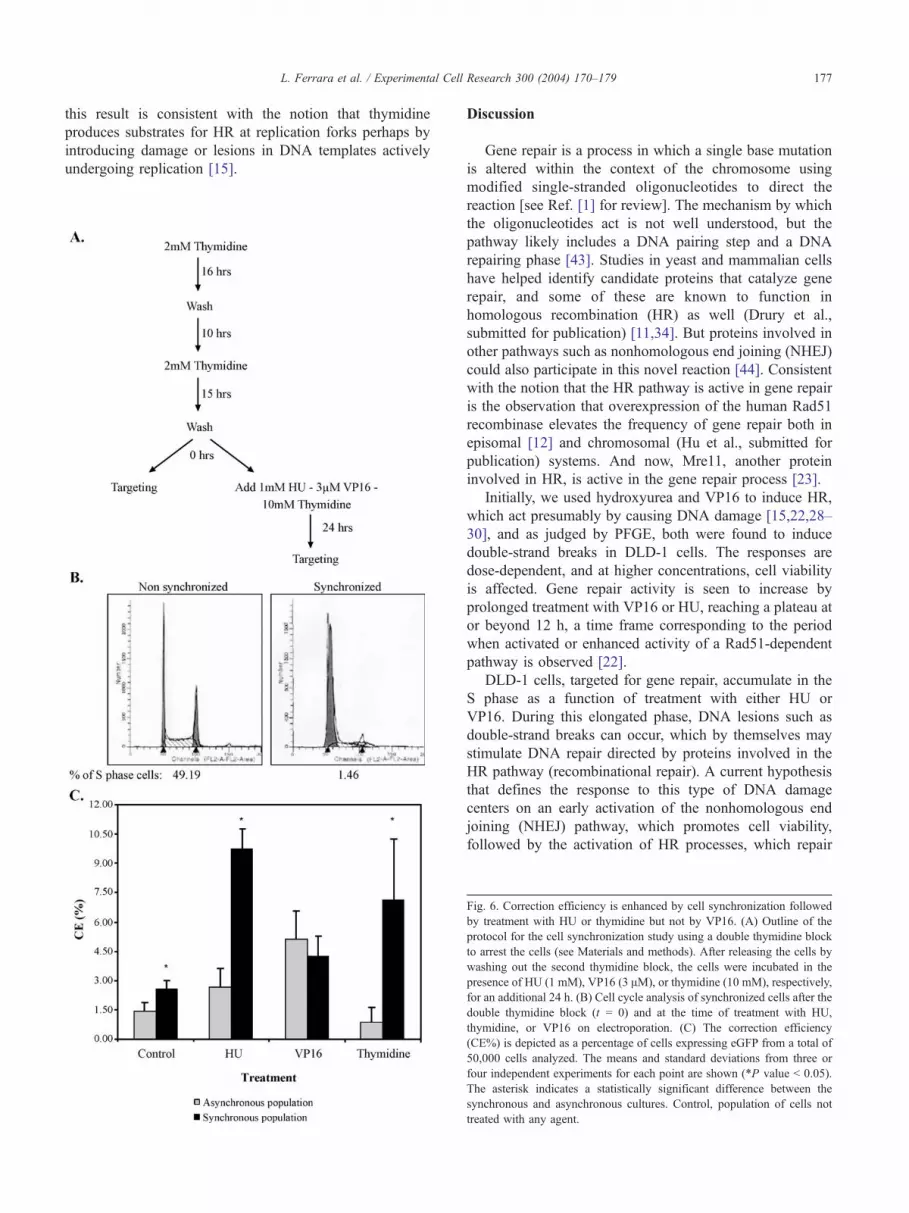
Gene repair activity within a population of cells enriched
in S phase can be assessed directly by synchronizing the
cells using a double thymidine block followed by release
with subsequent electroporation of the targeting oligonu-
cleotide. If gene repair is more active on cells in S phase or
is elevated by a block in DNA replication incurred by
treatment with various agents, then a higher level of activity
would be predicted as more cells enter S phase together.
Alternatively, more cells could simply reside in S phase at
the time of the addition of the agent. The experiments
outlined in Fig. 6A were carried out by first synchronizing
the cells using a double thymidine block then releasing the
cells before electroporation. In the control reaction,
EGFP3S/72NT was electroporated at the time of release
and repair of the mutant eGFP gene was quantified 48 h
later by FACS. In some reactions, HU, VP16, or thymidine
was added to the culture at the time of release and EGFP3S/
72NT was electroporated after 24 h of treatment. HU and
VP16 had already been shown to enhance gene repair
activity when added for this amount of time to an
asynchronous population of DLD-1 cells (see Figs. 2 and
3). The effect of the double thymidine block on cell
synchrony is shown in Fig. 6B as the synchronized culture
exhibits a high percentage of cells in G1 or at the G1/S
border with concurrent reduction in the percentage of cells
in S phase (as judged by BrdU staining). The results of these
experiments indicate that the cell synchronization process
alone leads to a modest increase in the correction efficiency
as compared to asynchronously growing cultures (Fig. 6C).
The addition of HU stimulates gene repair activity
significantly in the synchronized culture raising the fre-
quency to greater than 9%. But, in contrast, the addition of
VP16 did not lead to a statistically significant difference in
the correction frequency of the cells in the synchronized
culture as compared to the asynchronously growing culture.
The single asterisk represents a statistically significant
difference between the synchronized and nonsynchronized
paired samples. We suggest that HU acts to stimulate gene
repair preferentially on a population of cells enriched in S
phase while VP16 exerts an equally stimulatory effect on
asynchronously or synchronously growing cultures.
Based on several criteria, including the temporal
response of gene repair observed in these studies, we
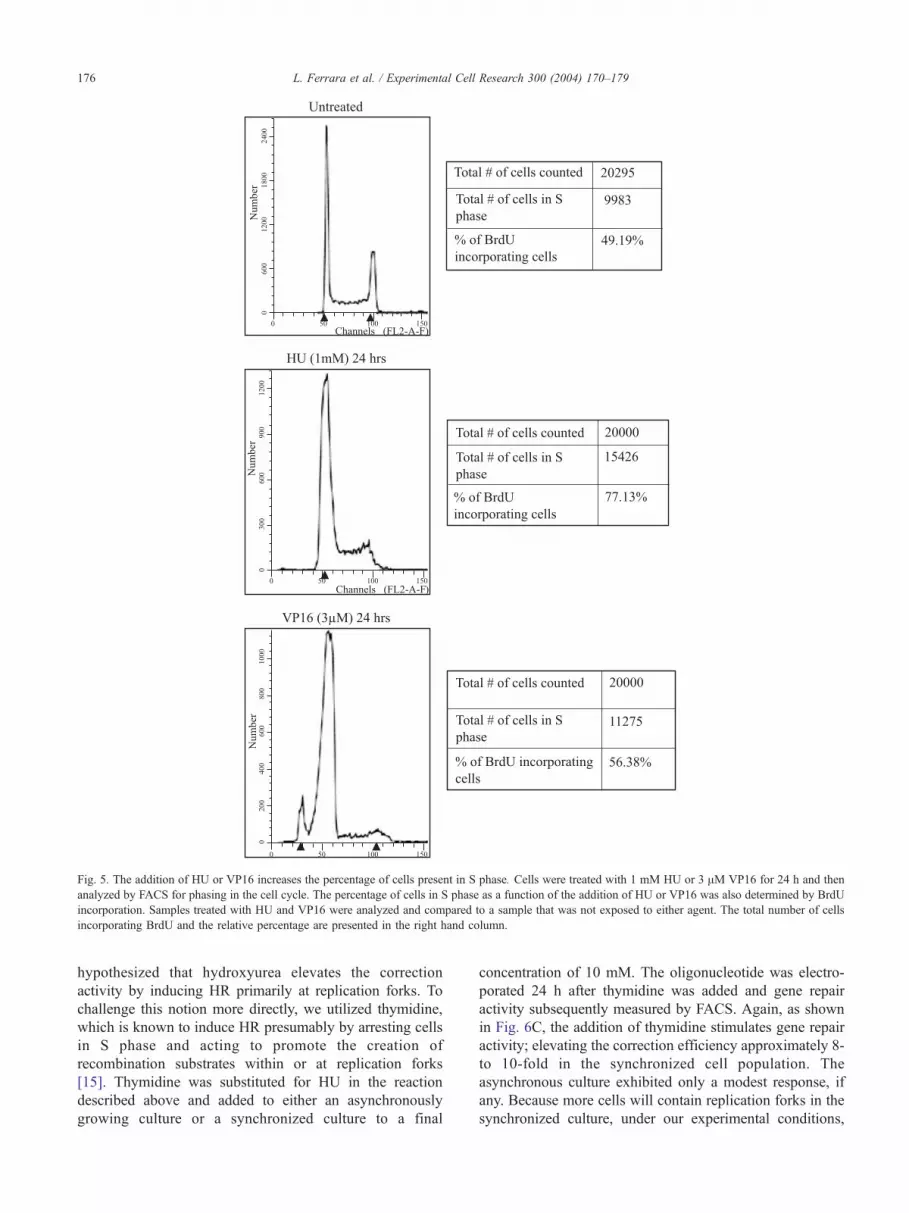
Fig. 5. The addition of HU or VP16 increases the percentage of cells present in S phase. Cells were treated with 1 mM HU or 3 AM VP16 for 24 h and then
analyzed by FACS for phasing in the cell cycle. The percentage of cells in S phase as a function of the addition of HU or VP16 was also determined by BrdU
incorporation. Samples treated with HU and VP16 were analyzed and compared to a sample that was not exposed to either agent. The total number of cells
incorporating BrdU and the relative percentage are presented in the right hand column.
L. Ferrara et al. / Experimental Cell Research 300 (2004) 170–179176
hypothesized that hydroxyurea elevates the correction
activity by inducing HR primarily at replication forks. To
challenge this notion more directly, we utilized thymidine,
which is known to induce HR presumably by arresting cells
in S phase and acting to promote the creation of
recombination substrates within or at replication forks
[15]. Thymidine was substituted for HU in the reaction
described above and added to either an asynchronously
growing culture or a synchronized culture to a final
concentration of 10 mM. The oligonucleotide was electro-
porated 24 h after thymidine was added and gene repair
activity subsequently measured by FACS. Again, as shown
in Fig. 6C, the addition of thymidine stimulates gene repair
activity; elevating the correction efficiency approximately 8-
to 10-fold in the synchronized cell population. The
asynchronous culture exhibited only a modest response, if
any. Because more cells will contain replication forks in the
synchronized culture, under our experimental conditions,
L. Ferrara et al. / Experimental Cell Research 300 (2004) 170–179 177
this result is consistent with the notion that thymidine
produces substrates for HR at replication forks perhaps by
introducing damage or lesions in DNA templates actively
undergoing replication [15].
Discussion
Gene repair is a process in which a single base mutation
is altered within the context of the chromosome using
modified single-stranded oligonucleotides to direct the
reaction [see Ref. [1] for review]. The mechanism by which
the oligonucleotides act is not well understood, but the
pathway likely includes a DNA pairing step and a DNA
repairing phase [43]. Studies in yeast and mammalian cells
have helped identify candidate proteins that catalyze gene
repair, and some of these are known to function in
homologous recombination (HR) as well (Drury et al.,
submitted for publication) [11,34]. But proteins involved in
other pathways such as nonhomologous end joining (NHEJ)
could also participate in this novel reaction [44]. Consistent
with the notion that the HR pathway is active in gene repair
is the observation that overexpression of the human Rad51
recombinase elevates the frequency of gene repair both in
episomal [12] and chromosomal (Hu et al., submitted for
publication) systems. And now, Mre11, another protein
involved in HR, is active in the gene repair process [23].
Initially, we used hydroxyurea and VP16 to induce HR,
which act presumably by causing DNA damage [15,22,28–
30], and as judged by PFGE, both were found to induce
double-strand breaks in DLD-1 cells. The responses are
dose-dependent, and at higher concentrations, cell viability
is affected. Gene repair activity is seen to increase by
prolonged treatment with VP16 or HU, reaching a plateau at
or beyond 12 h, a time frame corresponding to the period
when activated or enhanced activity of a Rad51-dependent
pathway is observed [22].
DLD-1 cells, targeted for gene repair, accumulate in the
S phase as a function of treatment with either HU or
VP16. During this elongated phase, DNA lesions such as
double-strand breaks can occur, which by themselves may
stimulate DNA repair directed by proteins involved in the
HR pathway (recombinational repair). A current hypothesis
that defines the response to this type of DNA damage
centers on an early activation of the nonhomologous end
joining (NHEJ) pathway, which promotes cell viability,
followed by the activation of HR processes, which repair
Fig. 6. Correction efficiency is enhanced by cell synchronization followed
by treatment with HU or thymidine but not by VP16. (A) Outline of the
protocol for the cell synchronization study using a double thymidine block
to arrest the cells (see Materials and methods). After releasing the cells by
washing out the second thymidine block, the cells were incubated in the
presence of HU (1 mM), VP16 (3 AM), or thymidine (10 mM), respectively
for an additional 24 h. (B) Cell cycle analysis of synchronized cells after the
double thymidine block (t = 0) and at the time of treatment with HU
thymidine, or VP16 on electroporation. (C) The correction efficiency
(CE%) is depicted as a percentage of cells expressing eGFP from a total o
50,000 cells analyzed. The means and standard deviations from three o
four independent experiments for each point are shown (*P value b 0.05)
The asterisk indicates a statistically significant difference between the
synchronous and asynchronous cultures. Control, population of cells no
treated with any agent.
,
,
f
r
.
t

L. Ferrara et al. / Experimental Cell Research 300 (2004) 170–179178
the subsequent (remaining) DNA damage [45–47]. Our
results do not directly address or exclude a role for NHEJ
in damage repair, but rather support a participatory role for
HR in gene repair. A previous report, however, did
indicate that HU activated both the nonhomologous and
the homologous recombination pathways, albeit to very
different levels; HR predominated in the overall response
[15]. But in other studies, double-strand breaks induced by
the action of the I-SceI enzyme in human glioma cells
were repaired predominantly by NHEJ between 24 and 48
h [48]. Thus, the dividing line, if one exists, between the
two pathways of chromosomal repair has not been made
concerning gene repair. The fact that the level of gene
repair continues to rise for a prolonged period after HU
addition, however, lends support to the involvement of
HR. Furthermore, the addition of thymidine to a popula-
tion of synchronized cells led to a stimulation of gene
repair activity and under these conditions, thymidine
induces homologous recombination exclusively [15].
Because we observe similar increases in correction
efficiency by the addition of either HU or thymidine, a
plausible conclusion is that gene repair activity is enhanced
when the HR pathway is induced.
Hydroxyurea appears to induce double-strand breaks
specifically at replication forks [15] while VP16 induces
breaks in a more random fashion [30]. Synchronized cells
treated with HU exhibit a higher level of gene repair while
synchronized cells treated with VP16 do not show increased
frequency beyond the levels obtained when VP16 is simply
added to asynchronously growing culture. The effect of HU
is best explained by the fact that more cells contain
replication forks when the culture is synchronized and
subsequently released into S, providing an increased number
of targets and accessibility for oligonucleotide-directed gene
repair activity. Because VP16 does not display a significant,
biased preference for replication forks, the level of
stimulation is predictably lower in synchronized cells
compared to that observed when HU is added.
DNA aberrations, lesions, or replication fork obstructions
result in the stalling of the replication process and cells
respond by extending their time in S phase where they
engage NHEJ or HR to help repair the damage. Coincident
with the repair of the structural damage in the DNA is an
increase in activity of gene repair, thus the results of our
study are consistent with an involvement of HR in the gene
repair reaction, at the very least as a bstimulatory agent.QThese conclusions are bolstered by new observations in
which the enhancement of gene repair activity induced by
the addition of VP16 was completely blocked when caffeine
was added to the reaction (Ferrara et al., in preparation).
Caffeine, a radiosensitizing agent, specifically inhibits
homologous recombination by blocking the activity of
ATM and ATR kinases, but it does not influence the process
of NHEJ [49,50]. In contrast, vanillin, an inhibitor of DNA-
PK activity in NHEJ, has no detectable effect on gene repair
reaction. Perhaps, the same complex of proteins, induced by
lesions accumulated during the process of DNA replication,
increases the activity of gene repair by its juxtaposition to
the site of DNA damage. Alternatively, the increase in HR
function may exhibit a more global effect on gene repair by
producing higher cellular concentrations of these critical
proteins. It is critical to note that our assay system does not
require a complete response of the HR pathway to generate
a positive signal. As mentioned above, certain clonal
isolates of DLD-1 cells may have differential HR activities
[25,27], but at present we have not yet determined if the
same proteins are required for both HR repair and gene
repair. What we can suggest is that stimulating the HR
response, as opposed to activating the NHEJ response, leads
to enhanced levels of gene repair. Elucidating the protein
participants will provide clues to the mechanism of this
interesting reaction that might eventually provide a basis for
utilizing gene repair as therapeutic.
Acknowledgments
This work was supported by grants from the National
Institutes of Health (R01 CA89325) and Tapestry
Pharmaceuticals.
References
[1] L. Liu, H. Parekh-Olmedo, E.B. Kmiec, The development and
regulation of gene repair, Nat. Rev., Genet. 4 (2003) 679–689.
[2] N.G. Copeland, N.A. Jenkins, D.L. Court, Recombineering: a
powerful new tool for mouse functional genomics, Nat. Rev., Genet.
2 (2001) 769–779.
[3] D.L. Court, J.A. Sawitzke, L.C. Thomason, Genetic engineering using
homologous recombination, Annu. Rev. Genet. 36 (2002) 361–388.
[4] D. Yu, H.M. Ellis, E.C. Lee, N.A. Jenkins, N.G. Copeland, D.L.
Court, An efficient recombination system for chromosome engineer-
ing in Escherichia coli, Proc. Natl. Acad. Sci. U. S. A. 97 (2000)
5978–5983.
[5] E. Kmiec, W.K. Holloman, Beta protein of bacteriophage lambda pro-
motes renaturation of DNA, J. Biol. Chem. 256 (1981) 12636–12639.
[6] L.J. Maher III, Prospects for the therapeutic use of antigene
oligonucleotides, Cancer Invest. 14 (1996) 66–82.
[7] H.E. Moser, P.B. Dervan, Sequence-specific cleavage of double
helical DNA by triple helix formation, Science 238 (1987) 645–650.
[8] K.M. Vasquez, J.H. Wilson, Triplex-directed site-specific genome
modification, Methods Mol. Biol. 133 (2000) 183–200.
[9] O. Igoucheva, K. Yoon, Targeted single-base correction by RNA–
DNA oligonucleotides, Hum. Gene Ther. 11 (2000) 2307–2312.
[10] K. Kunzelmann, J.Y. Legendre, D.L. Knoell, L.C. Escobar, Z. Xu,
D.C. Gruenert, Gene targeting of CFTR DNA in CF epithelial cells,
Gene Ther. 3 (1996) 859–867.
[11] L. Liu, S. Cheng, A.J. van Brabant, E.B. Kmiec, Rad51p and Rad54p,
but not Rad52p, elevate gene repair in Saccharomyces cerevisiae
directed by modified single-stranded oligonucleotide vectors, Nucleic
Acids Res. 30 (2002) 2742–2750.
[12] P.H. Thorpe, B.J. Stevenson, D.J. Porteous, Functional correction of
episomal mutations with short DNA fragments and RNA–DNA
oligonucleotides, J. Gene Med. 4 (2002) 195–204.
[13] X.T. Li, N. Costantino, L.Y. Lu, D.P. Liu, R.M. Watt, K.S. Cheah,
D.L. Court, J.D. Huang, Identification of factors influencing strand

L. Ferrara et al. / Experimental Cell Research 300 (2004) 170–179 179
bias in oligonucleotide-mediated recombination in Escherichia coli,
Nucleic Acids Res. 31 (2003) 6674–6687.
[14] L. Krejci, J. Damborsky, B. Thomsen, M. Duno, C. Bendixen,
Molecular dissection of interactions between Rad51 and members
of the recombination-repair group, Mol. Cell. Biol. 21 (2001)
966–976.
[15] C. Lundin, K. Erixon, C. Arnaudeau, N. Schultz, D. Jenssen, M.
Meuth, T. Helleday, Different roles for nonhomologous end joining
and homologous recombination following replication arrest in
mammalian cells, Mol. Cell. Biol. 22 (2002) 5869–5878.
[16] C. Lundin, N. Schultz, C. Arnaudeau, A. Mohindra, L.T. Hansen, T.
Helleday, RAD51 is involved in repair of damage associated with
DNA replication in mammalian cells, J. Mol. Biol. 328 (2003)
521–535.
[17] B. Michel, M.J. Flores, E. Viguera, G. Grompone, M. Seigneur,
V. Bidnenko, Rescue of arrested replication forks by homolo-
gous recombination, Proc. Natl. Acad. Sci. U. S. A. 98 (2001)
8181–8188.
[18] Y. Saintigny, F. Delacote, G. Vares, F. Petitot, S. Lambert, D.
Averbeck, B.S. Lopez, Characterization of homologous recombination
induced by replication inhibition in mammalian cells, EMBO J. 20
(2001) 3861–3870.
[19] E. Sonoda, M.S. Sasaki, J.M. Buerstedde, O. Bezzubova, A.
Shinohara, H. Ogawa, M. Takata, Y. Yamaguchi-Iwai, S. Takeda,
Rad51-deficient vertebrate cells accumulate chromosomal breaks
prior to cell death, EMBO J. 17 (1998) 598–608.
[20] A. Majumdar, N. Puri, B. Cuenoud, F. Natt, P. Martin, A. Khorlin, N.
Dyatkina, A.J. George, P.S. Miller, M.M. Seidman, Cell cycle
modulation of gene targeting by a triple helix-forming oligonucleo-
tide, J. Biol. Chem. 278 (2003) 11072–11077.
[21] K. Rothkamm, I. Kruger, L.H. Thompson, M. Lobrich, Pathways of
DNA double-strand break repair during the mammalian cell cycle,
Mol. Cell. Biol. 23 (2003) 5706–5715.
[22] Y. Saintigny, B.S. Lopez, Homologous recombination induced by
replication inhibition, is stimulated by expression of mutant p53,
Oncogene 21 (2002) 488–492.
[23] L. Liu, M. Usher, Y. Hu, E.B. Kmiec, Nuclease activity of Sac-
charomyces cerevisiae Mre11 functions in targeted nucleotide
alteration, Appl. Environ. Microbiol. 69 (2003) 6216–6224.
[24] H. Parekh-Olmedo, J. Engstrom, E.B. Kmiec, The effect of hydrox-
yurea and trichostatin A on targeted nucleotide exchange in yeast and
mammalian cells, Ann. N. Y. Acad. Sci. 1002 (2003) 43–56.
[25] A. Mohindra, L.E. Hays, E.N. Phillips, B.D. Preston, T. Helleday, M.
Meuth, Defects in homologous recombination repair in mismatch-
repair-deficient tumour cell lines, Hum. Mol. Genet. 11 (2002)
2189–2200.
[26] D.L. Dexter, E.N. Spremulli, Z. Fligiel, J.A. Barbosa, R. Vogel, A.
VanVoorhees, P. Calabresi, Heterogeneity of cancer cells from a single
human colon carcinoma, Am. J. Med. 71 (1981) 949–956.
[27] J.T. Leith, D.L. Dexter, J.K. DeWyngaert, E.M. Zeman, M.Y. Chu, P.
Calabresi, A.S. Glicksman, Differential responses to x-irradiation of
subpopulations of two heterogeneous human carcinomas in vitro,
Cancer Res. 42 (1982) 2556–2561.
[28] V. Bianchi, E. Pontis, P. Reichard, Changes of deoxyribonucleoside
triphosphate pools induced by hydroxyurea and their relation to DNA
synthesis, J. Biol. Chem. 261 (1986) 16037–16042.
[29] L. Thelander, P. Reichard, Reduction of ribonucleotides, Annu. Rev.
Biochem. 48 (1979) 133–158.
[30] R.D. Anderson, N.A. Berger, International commission for protection
against environmental mutagens and carcinogens. Mutagenicity and
carcinogenicity of topoisomerase-interactive agents, Mutat. Res. 309
(1994) 109–142.
[31] J. Thacker, A surfeit of RAD51-like genes? Trends Genet. 15 (1999)
166–168.
[32] L.H. Thompson, D. Schild, The contribution of homologous
recombination in preserving genome integrity in mammalian cells,
Biochimie 81 (1999) 87–105.
[33] L.H. Thompson, D. Schild, Homologous recombinational repair of
DNA ensures mammalian chromosome stability, Mutat. Res. 477
(2001) 131–153.
[34] M.D. Drury, E.B. Kmiec, DNA pairing is an important step in the
process of targeted nucleotide exchange, Nucleic Acids Res. 31
(2003) 899–910.
[35] B.B. Hasinoff, T.I. Kuschak, A.M. Creighton, C.L. Fattman, W.P.
Allan, P. Thampatty, J.C. Yalowich, Characterization of a Chinese
hamster ovary cell line with acquired resistance to the bisdioxopiper-
azine dexrazoxane (ICRF-187) catalytic inhibitor of topoisomerase II,
Biochem. Pharmacol. 53 (1997) 1843–1853.
[36] L.H. Jensen, K.C. Nitiss, A. Rose, J. Dong, J. Zhou, T. Hu, N.
Osheroff, P.B. Jensen, M. Sehested, J.L. Nitiss, A novel mechanism of
cell killing by anti-topoisomerase II bisdioxopiperazines, J. Biol.
Chem. 275 (2000) 2137–2146.
[37] M. Kobayashi, N. Adachi, Y. Aratani, A. Kikuchi, H. Koyama,
Decreased topoisomerase IIalpha expression confers increased resist-
ance to ICRF-193 as well as VP-16 in mouse embryonic stem cells,
Cancer Lett. 166 (2001) 71–77.
[38] L.F. Liu, DNA topoisomerase poisons as antitumor drugs, Annu. Rev.
Biochem. 58 (1989) 351–375.
[39] J.L. Nitiss, W.T. Beck, Antitopoisomerase drug action and resistance,
Eur. J. Cancer 32A (1996) 958–966.
[40] B. van Hille, X. Clerc, A.M. Creighton, B.T. Hill, Differential
expression of topoisomerase I and RAD52 protein in yeast reveals
new facets of the mechanism of action of bisdioxopiperazine
compounds, Br. J. Cancer 81 (1999) 800–807.
[41] B. van Hille, B.T. Hill, Yeast cells expressing differential levels of
human or yeast DNA topoisomerase II: a potent tool for identification
and characterization of topoisomerase II-targeting antitumour agents,
Cancer Chemother. Pharmacol. 42 (1998) 345–356.
[42] A. Kumari, N. Schultz, T. Helleday, p53 protects from replication-
associated DNA double-strand breaks in mammalian cells, Oncogene
23 (2004) 2324–2329.
[43] E.E. Brachman, E.B. Kmiec, The dbiasedT evolution of targeted gene
repair, Curr. Opin. Mol. Ther. 4 (2002) 171–176.
[44] O. Igoucheva, V. Alexeev, M. Pryce, K. Yoon, Transcription affects
formation and processing of intermediates in oligonucleotide-medi-
ated gene alteration, Nucleic Acids Res. 31 (2003) 2659–2670.
[45] J.E. Haber, DNA repair. Gatekeepers of recombination, Nature 398
(1999) 665–667.
[46] F. Paques, G.F. Richard, J.E. Haber, Expansions and contractions in
36-bp minisatellites by gene conversion in yeast, Genetics 158 (2001)
155–166.
[47] E. Van Dyck, A.Z. Stasiak, A. Stasiak, S.C. West, Binding of double-
strand breaks in DNA by human Rad52 protein, Nature 398 (1999)
728–731.
[48] S.E. Golding, E. Rosenberg, A. Khalil, A. McEwen, M. Holmes, S.
Neill, L. Povrik, K. Valerie, Double-strand break repair by homolo-
gous recombination is regulated by cell cycle-independent signaling
via ATM in human glioma cells, J. Biol. Chem. (2004) 1–43
(Electronic publication ahead of print).
[49] J.N. Sarkaria, E.C. Busby, R.S. Tibbetts, P. Roos, Y. Taya, L.M.
Karnitz, R.T. Abraham, Inhibition of ATM and ATR kinase
activities by the radiosensitizing agent, caffeine, Cancer Res. 59
(1999) 4375–4382.
[50] B.B. Zhou, P. Chaturvedi, K. Spring, S.P. Scott, R.A. Johanson, R.
Mishra, M.R. Mattern, J.D. Winkler, K.K. Khanna, Caffeine abolishes
the mammalian G(2)/M DNA damage checkpoint by inhibiting ataxia-
telangiectasia-mutated kinase activity, J. Biol. Chem. 275 (2000)
10342–10348.
Copyright © 2022 FDOKUMEN




















