
Engineering and optimization of the miR106b cluster for ectopic expression of multiplexed anti-HIV...
27
Downloaded from UvA-DARE, the institutional repository of the University of Amsterdam (UvA) http://dare.uva.nl/document/153040 File ID 153040 Filename 6: Engineering and optimization of the mir-106b-cluster for ectopic expression of multiplexed anti-HIV RNAs SOURCE (OR PART OF THE FOLLOWING SOURCE): Type Dissertation Title RNAi based gene therapy for HIV-1, from bench to bedside Author K.J. von Eije Faculty Faculty of Medicine Year 2009 Pages ix, 180 ISBN 978-94-90371-06-7 FULL BIBLIOGRAPHIC DETAILS: http://dare.uva.nl/record/321556 Copyright It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/or copyright holder(s), other then for strictly personal, individual use. UvA-DARE is a service provided by the library of the University of Amsterdam (http://dare.uva.nl)
-
Upload
independent -
Category
Documents
-
view
3 -
download
0
Transcript of Engineering and optimization of the miR106b cluster for ectopic expression of multiplexed anti-HIV...

Downloaded from UvA-DARE, the institutional repository of the University of Amsterdam (UvA)http://dare.uva.nl/document/153040
File ID 153040Filename 6: Engineering and optimization of the mir-106b-cluster for ectopic expression of multiplexed
anti-HIV RNAs
SOURCE (OR PART OF THE FOLLOWING SOURCE):Type DissertationTitle RNAi based gene therapy for HIV-1, from bench to bedsideAuthor K.J. von EijeFaculty Faculty of MedicineYear 2009Pages ix, 180ISBN 978-94-90371-06-7
FULL BIBLIOGRAPHIC DETAILS: http://dare.uva.nl/record/321556
Copyright It is not permitted to download or to forward/distribute the text or part of it without the consent of the author(s) and/orcopyright holder(s), other then for strictly personal, individual use. UvA-DARE is a service provided by the library of the University of Amsterdam (http://dare.uva.nl)

6Engineering and optimization of the mir-106b-clusterfor ectopic expression of multiplexed anti-HIV RNAs
Lars Aagaard, Karin J. von Eije†, Jane Zhang†, Haitang Li, P̊al Sætrom,Mohammed Amarzguioui, and John J. Rossi† These authors contributed equally to this studyGene Therapy 2008. 15(23):1536-49
6.1 Abstract
Many microRNAs (miRNAs) are encoded within the introns of RNA Pol II tran-scripts, often as polycistronic precursors. Here, we demonstrate the optimizationof an intron encoding three endogenous miRNAs for the ectopic expression of het-erologous anti-HIV-1 small interfering RNAs (siRNAs) processed from a single RNApolymerase II primary transcript. Our expression system, designated as MCM7, isengineered from the intron-embedded, tri-cistronic miR-106b cluster that endoge-nously expresses miR-106b, miR-93 and miR-25. Manipulation of the miR-106b clus-ter demonstrated a strict requirement for maintenance of the native flanking primarymiRNA (pri-miRNA) sequences and key structural features of the native miRNAsfor efficient siRNA processing. As a model for testing the efficacy of this approach,we have replaced the three endogenous miRNAs with siRNAs targeting the tat andrev transcripts of human immunodeficiency virus type 1 (HIV-1). This study hasenabled us to establish guidelines for optimal processing of the engineered miRNAmimics into functional siRNAs. In addition, we demonstrate that the incorporationof a small nucleolar RNA TAR chimeric decoy (snoRNA) inserted within the MCM7intron resulted in a substantial enhancement of HIV suppression in long-term acuteinfectious HIV-1 challenges.
61

CHAPTER 6. ENGINEERING AND OPTIMIZATION OF THEMIR-106B-CLUSTER FOR ECTOPIC EXPRESSION OF MULTIPLEXEDANTI-HIV RNAS
6.2 Introduction
Since its discovery in the worm Caenorhabditis elegans [7], and subsequent demon-stration in essentially all higher organisms, including humans [6], RNA interference(RNAi) has become the method of choice for studying gene function in mammaliancells and is also attracting increasing attention as a therapeutic modality [88, 123, 152].RNAi-mediated knockdown or post-transcriptional gene silencing of the gene of in-terest is achieved either by transfection of synthetic small interfering RNAs (siRNAs)[90], by expression of short hairpin RNAs (shRNA) [50, 228] or by mimics [331] oftheir naturally occurring counterparts, the microRNAs (miRNAs) [28]. A wide rangeof viral diseases have been successfully targeted in cell culture and animal models byeither siRNA or shRNA vector-based approaches [5, 8, 12]. Stable expression systemscan be efficiently delivered to mammalian cells by viral vectors in cell culture and invivo, resulting in long-term silencing of the target gene [184, 249, 271].
Traditional small-molecule-based drug treatments of viral diseases are burdenedwith two related serious limitations: the great genetic diversity of the various strainsand the frequent evolution of drug-resistant strains under the selective pressure oftreatment [88, 109]. This problem was quickly recognized as a limitation that wasalso shared by RNAi for treating RNA viruses with high mutation rates. In one ofthe first published papers on the use of RNAi against a viral target in cell culture,Gitlin et al. [102], demonstrated that although effective inhibition of poliovirus repli-cation could be achieved, escape mutants in the target sequence of the siRNA effectoremerged upon extended culturing of the infected cells. Similar observations were sub-sequently reported by various other researchers for other viruses, including humanimmunodeficiency virus (HIV) [2, 4, 18, 19, 20]. Some researchers have attempted tobypass the problem of escape by using a combination of anti-viral inhibitory agents[5, 9]. The feasibility of this strategy was first demonstrated by Song et al. [273],who observed more pronounced and longer lasting siRNA-mediated protection againstHIV infection upon simultaneous targeting of both CCR5 and CA-p24 than either ofthe individual targets alone. Similar effects were reported for targeting of hepatitis Cvirus by two chemically synthesized siRNAs [20]. Successful incorporation of multipleeffector molecules (including non-RNAi effectors) within the same expression vectorhas subsequently been reported by various researchers for targeting both HIV andhepatitis C virus [18, 61, 128, 183, 287, 290]. In a comprehensive study of siRNAstargeting HIV, ter Brake et al. [290] screened 86 different shRNA constructs targetingconserved sequences within 8 different regions of the HIV genome, before combiningtheir two and three best candidates as separate Pol III expression cassettes. This re-sulted in markedly improved inhibition of virus replication and substantially delayedemergence of escape mutants. A computational model of the propagation of HIVinfection in cells expressing RNAi suggested that simultaneous expression of four op-timally designed RNAi effectors might be sufficient to achieve a complete protectionagainst the emergence of escape mutants [178, 287], although it remains to be seenwhether this prediction bears out experimentally.
The ability to express multiple RNAi effector units from a single Pol II poly-
62

6.2. INTRODUCTION
cistronic transcript is potentially very important for the avoidance of toxicities ob-served when shRNAs are ectopically expressed from strong Pol III promoters [13, 110].This problem of Pol III-expressed shRNA toxicity can be circumvented by taking ad-vantage of endogenous RNAi transcripts, miRNAs [27, 153, 154], for the expressionof transgenes. miRNAs are structurally and functionally very similar to siRNAs,and are incorporated into RNA-induced silencing complex (RISC) [215]. They areprimarily transcribed by RNA Pol II as parts of larger primary transcripts [52, 173]that undergo initial processing in the nucleus by the microprocessor complex, con-sisting of the nuclease Drosha and its specificity binding partner, DGCR8/Pasha[75, 108, 120, 168, 171]. The resulting imperfect hairpin of approximately 65-nt (nu-cleotide), the precursor miRNA (pre-miRNA), is then exported to the cytoplasmthrough Exportin-5 [193, 329, 330], where it is further processed by Dicer. For themajority of miRNAs, one strand is asymmetrically loaded into RISC to function asthe guide strand [63, 107, 153, 172]. Endogenous miRNAs can be expressed as in-tronic clusters from a single polycistronic transcript and the miRNAs simultaneouslydownregulate the expression of multiple mRNAs [7, 126, 155, 245, 286].
Cullen and co-workers [331] pioneered the use of Pol II-based miRNA expressioncassettes through their demonstration that the region of the primary transcript ofmiR-30 corresponding to the mature miRNA may be replaced with a heterologousstem without compromising activity, enabling the generation of an miRNA-based ex-pression cassette with generalizable targeting properties. miR-30-based expressionconstructs have been incorporated into lentiviral vectors and used to generate wholelibraries of validated targeting vectors [60, 269, 275], allowing temporal control of tar-get gene silencing for functional studies [79]. The known localization of many miRNAgenes within introns has prompted researchers to attempt the expression of miRNAsfrom polycistronic transcripts. Zhou et al. [339] expressed an siRNA from an miR-30background within an intron of an enhanced green fluorescent protein expression cas-sette, thus directly coupling miRNA and marker gene expressions. miR-155 has beenused by others in a similar fashion [64, 85], including the simultaneous expression oftwo active miRNAs from the embedded intron, allowing dual targeting [64]. Resultsfrom various studies based on different miRNA backgrounds are somewhat conflict-ing, but an emerging consensus appears to be that proper processing of the chimerictranscript requires maintaining the secondary structure of at least one helical turnimmediately preceding the Drosha processing site, with possible contributions alsofrom the terminal loop [121, 332].
In this study, we have developed a platform for the incorporation of multipleheterologous miRNAs, which allows for the simultaneous expression of up to three in-dividual siRNAs as a means of multiplexing siRNA production. This platform is basedon a naturally occurring miRNA cluster, miR-106b–miR-93–miR-25 [7], encoded en-tirely within intron 13 of the putative protein-encoding gene MCM7 on chromosome7. Within the intron, the three microunits are closely spaced, with only 124 and 145-nt separating the three pre-miRNA hairpins. This spacing makes this system usefulfor incorporation into viral vectors that have spatial limitations for gene insertions.We have cloned the native cluster, including parts of upstream and downstream ex-
63

CHAPTER 6. ENGINEERING AND OPTIMIZATION OF THEMIR-106B-CLUSTER FOR ECTOPIC EXPRESSION OF MULTIPLEXEDANTI-HIV RNAS
ons, verified the targeting efficacy and strand selectivity of the original miRNAs, andmodified the original cluster to facilitate independent replacement of each unit withheterologous, trans-targeting siRNAs. As a model system for testing this platform,we have used HIV type 1 (HIV-1) as a target and inserted three different siRNAstargeting different sites within the HIV-1 genome. When the three siRNAs were ex-pressed in T lymphocyte cell lines and challenged with two different strains of HIV,the siRNAs effectively suppressed the replication of HIV-LAI, but were less effective insuppressing the more pathogenic lab strain HIV-IIIB. In an effort to further suppressHIV replication, we incorporated a small nucleolar-localizing TAR-decoy (U16TAR)[184, 209] in three different positions of the MCM7 intron. In each case, a mature-sizedchimeric construct was produced. In acute challenges with HIV-LAI and HIV-IIIB,the inclusion of the TAR-decoy within the intron resulted in a marked enhancement ofanti-HIV function. Our studies provide both a platform unit for multiplexing siRNAsand other small RNAs as well as a novel therapeutic construct for the treatment ofHIV infection.
6.3 Materials and methods
Generation of polycistronic expression constructsWe cloned the entire miR-106b cluster downstream of a CMV promoter in the ex-pression plasmid pcDNA3.1 by PCR of human genomic DNA using primers E-F1 andE-R4 (see details on all oligos in Table 6.1). The resulting PCR product was ini-tially cloned into the TA vector pCR2.1 (Invitrogen, Carlsbad, CA, USA) and thensubcloned as a HindIII/XbaI fragment to pcDNA3.1 to generate pcDNA3-MCM7-E.pcDNA3-MCM7-E contains the miR-106b-bearing intron from the MCM7 gene as wellas parts of the upstream and downstream exon sequences. An miR-106b expressionplasmid without the flanking exon structure, pcDNA3-MCM7-I, was cloned directlyinto pcDNA3.1 by PCR from genomic DNA using HindIII- and XbaI-tagged primers(I-F and I-R, respectively). Next, we built an miR-106b-based scaffold, pcDNA3-MCM7-S, that could be used for insertion of heterologous sequences at the positionsof miR-106b, miR-93 and miR-25. This was accomplished by PCR amplification ofeach of the four regions flanking miR-106b, miR-93 and miR25, and inserting thoseone by one into pBSIISK (region 1 used primers E-F1 and E-R1 and so forth). Eachprimer was tagged with restriction sites corresponding to the order of the multi-ple cloning site in pBSIISK (see Figure 6.1) in a manner that introduced minimalchanges in the native miR-106b cluster sequence. After cloning all four regions intopBSIISK, the entire scaffold was cloned directionally as a KpnI/BssHII fragmentinto KpnI/AscI sites of the pcDNA3.1-modified plasmid pcDNA3-PL4 (in which thepolylinker between HindIII and XbaI in pcDNA3.1 was replaced with a shorter linkercontaining AgeI, KpnI, XhoI, BamH I, AscI and HpaI sites, destroying the originalHindIII and XbaI sites in the cloning process) to generate pcDNA3-MCM7-S.
Using pcDNA3-MCM7-S, heterologous hairpin structures were inserted into theoriginal positions of miR-106b, miR-93 and miR-25 as XhoI/HindIII, EcoRI/BamHI
64

6.3. MATERIALS AND METHODS
or XbaI/SacII fragments, respectively. Inserts were generated by PCR extension ofpartially annealed overlapping 60-nt oligonucleotides, followed by digestion with theappropriate restriction enzymes. The U16TAR-decoy structure was PCR amplifiedwith the appropriate restriction enzymes from the U6-TAR-decoy harboring Shrek3plasmid [236] and cloned into S3B. Restriction sites ClaI and SwaI were introduced70-nt upstream of the miRNA cluster’s 3′ splice site for the insertion of the forwardand reverse U16TAR-decoy. The revU16TAR was cloned with a Pol III U6 promoterfor the expression of the decoy in the reverse orientation.
A splice-deficient version of pcDNA3-MCM7-S1/S2/S3 (called delSplice) withdeleted splice signals was generated by PCR amplification from the full-length con-struct with KpnI-tagged delS-F and BssHII-tagged delS-R primers. The resultingKpnI+BssHII restriction product was cloned into KpnI+AscI sites of pcDNA3-PL4.A minimal hairpin cassette, named miniMCM7, was generated by cloning the S1,S2 and S3 pre-miRNA units into the multiple cloning site of pBSIISK, followed bysubcloning the KpnI/BssHII fragment into KpnI/AscI sites of pcDNA3-PL4.
We created promoter derivatives of pcDNA3-MCM7-S1/S2M/S3B in which theoriginal CMV promoter was replaced with the non-CMV-interfering U1 promoter.The U1 promoter version was generated similarly by replacing the CMV MluI-KpnIpromoter fragment with an SpeI-KpnI fragment from an U1 plasmid (pKS-U1, ourunpublished data). All constructs were verified by restriction analysis and sequencing.
To generate lentiviral vectors, the U1-MCM7-S1/S2M/S3B and U1-MCM7-S1/-S2M/S3B revU16TAR fragments were excised from the pcDNA3 plasmid by MluIand KpnI digestion and inserted into separate pHIV-7-GFP lentiviral vectors.
Cell cultureHEK293, HCT116 and 293T cells were purchased from ATCC (Manassas, VA, USA).The cells were maintained in high glucose (4.5 g/l) Dulbecco’s modified Eagle’smedium supplemented with 2 mM glutamine, 10% FBS (fetal bovine serum), and2 mM penicillin/streptomycin. The human T cell line CEM was cultured in RPMI1640 medium supplemented with 10% FBS. SupT1 cells were cultured in advancedRPMI with 2 mM glutamine, 1% FBS, and 2 mM penicillin/streptomycin.
Construction of psiCHECK reportersDual luciferase (psiCHECK2, Promega, San Luis Obispo, CA, USA) reporters weregenerated for the guide and passenger strands for each of the miRNA and anti-HIV-1siRNA expressed from the various MCM7 constructs. The reporters contained theminimal target site of the respective putative RISC-loaded mi/siRNA strand plusthree additional target-derived sequences at each end to account for slight variationsin processing of miRNAs. Target sequences were cloned into XhoI+NotI sites in the3′ UTR of the Renilla luciferase gene of the psiCHECK2.2 (a version of the commer-cial psiCHECK2 with an extended multiple cloning site). Oligonucleotide cassettesbearing XhoI and NotI compatible 4-nt overhangs at each end were generated by an-nealing two in vitro phosphorylated partially overlapping oligonucleotides. Cleavageof the target site results in the degradation of reporter mRNA, with a concomitant
65

CHAPTER 6. ENGINEERING AND OPTIMIZATION OF THEMIR-106B-CLUSTER FOR ECTOPIC EXPRESSION OF MULTIPLEXEDANTI-HIV RNAS
Table6.1:DNAoligosused
forclo
ning.
Nam
eSequence
E-F
1G
AAG
TG
GTACCTG
ACTACATCACAG
CAG
CATACG
E-R
1G
ATATTctC
gagcgggaggtg
gggag
E-F
2G
TTCTA
AG
CTTCCG
TG
GAG
GG
AA
AG
GC
E-R
2G
AA
ACG
AATTCCA
AG
ACG
GG
AG
GACAG
E-F
3G
AAG
TG
GATCCG
TTCTCTCTG
CCA
ATT
GT
cE-R
3ccagtT
cTA
gagttc
agctg
tcctg
tgagg
E-F
4TA
AG
ACCG
CG
GATG
CTG
GG
GG
GE-R
4G
AATCG
AG
CTCTG
TCTG
CCCC
TTG
TCT
CC
I-FG
aagtA
AG
CTTG
TA
AG
TG
CCCA
AATTG
CT
GI-R
gaatc
AG
ATCTCTA
AG
GG
GA
AG
GTAG
GG
GG
S1-A
agaggaCTCG
AG
ccctg
ccggggcG
CG
GAG
ACAG
CG
ACG
AAG
AG
Ctttg
tgta
gG
CT
CT
TS1-B
gaagA
AG
CTTtg
cgtg
ccctg
ctg
gagcG
CG
GAG
ACAG
CG
ACG
AAG
AG
Ccta
cacaaaG
CS2-A
agaggaG
AATTCagtc
ctg
ggggctc
GCCTG
TG
CCTCTTCAG
CTACCtttg
tgta
gG
GTA
S2-B
agaggaG
GATCCcgggggctc
GCCTG
TG
CCTCTTCAG
CTACCcta
cacaaaG
GTAG
S3-A
agagTCTAG
Aactg
gccagtg
ttgCATCTCCTATG
GCAG
GA
AG
AAtttg
tgta
gTTC
TT
CS3-B
agagCCG
CG
Gtg
ttgggccggcactg
CATCTCCTATG
GCAG
GA
AG
AA
cta
cacaaaTTCT
min
iS1-A
TCG
AG
GCG
GAG
ACAG
CG
ACG
AAG
AG
Ctttg
tgta
gG
CTC
TT
CG
TCG
CTG
TCT
CC
GCA
min
iS1-B
AG
CTTG
CG
GAG
ACAG
CG
ACG
AAG
AG
Ccta
cacaaaG
CTC
TTC
GT
CG
CTG
TCT
CCG
CC
min
iS2-A
AATTCG
CCTG
TG
CCTCTTCAG
CTAC
Ctttg
tgta
gG
GTAG
CTG
AAG
AG
GC
ACAG
GC
Gm
iniS
2-B
GATCCG
CCTG
TG
CCTCTTCA
GCTACCcta
cacaaaG
GTAG
CTG
AAG
AG
GC
ACA
GG
CG
min
iS3-A
CTAG
ACATCTCCTATG
GCAG
GA
AG
AAtttg
tgta
gTT
CT
TCC
TG
CCATAG
GAG
ATG
CC
GC
min
iS3-B
GG
CATCTCCTATG
GCAG
GA
AG
AA
cta
cacaaaTTCTTC
CTG
CCATAG
GAG
ATG
TD
elS
-FG
AAG
TG
GTACCagggccatc
tgttttg
accc
DelS
-RG
AAG
TG
CG
CG
Caaagatg
ggaacgggaggag
S2M
-Acttg
GA
ATTCagtc
ctg
ggggctg
gTAG
CTG
AAG
AG
GCACAG
GCagugugauuacccaac
S2M
-BagaacG
GATCCcgggggcta
tcgAG
CTG
AA
aG
AG
GCCAG
GCaggttg
ggta
atc
acac
S3M
-AaacTCTAG
Aactg
gccagtg
ttgaggCATgTCCTA
aaG
GAG
GA
AaG
ggtttg
tgta
gctc
S3M
-BcatC
CG
CG
Gtg
ttgggccggcactg
tcgCATCTCCTATG
GCAG
GA
AG
Agcta
cacaaacc
S3M
2-A
aacTCTAG
Aactg
gccagtg
ttgaggCATgTCCTA
aaG
aAG
GA
aaG
ggcgacgctg
cact
S3M
2-B
catC
CG
CG
Gtg
ttgggccggcactg
tcgCATCTCCTATG
GCAG
GA
AG
agcagtg
cagcgtc
gcc
S3A
-Atg
aacTCTAG
Aagccctg
ccggggcCATCTCCTATG
GCAG
GG
AG
AAtttg
tgta
gTTCTT
S3A
-BcatC
CG
CG
Gtg
tgccctg
ctg
gagcCATCTCCTATG
GCAG
GA
AG
AA
cta
cacaaaTTCTC
S3B-A
gaacTCTAG
Aagtc
ctg
ggggctc
gCTTCCTG
CCATAG
GAG
ATG
cgtg
tgatta
cccaac
S3B-B
agcatC
CG
CG
Gtg
tgTCCcgggggctc
ttgTTCCTG
CtC
ATAG
AG
ATG
cggttg
ggta
atc
acac
S4-A
aacTCTAG
Aactg
gccagtg
ttgtc
tGCA
cCATTA
ccAtC
ACCttA
ggcgacgctg
cact
S4-B
catC
CG
CG
Gtg
ttgggccggcactg
agtG
CAG
CATTA
ATA
ACACCA
Aagcagtg
cagcgtc
gcc
U3R
-FG
TTA
AG
CG
GCCG
CacgcgtT
ggaagggcta
atttg
gtc
ccaa
U3R
-RG
TTA
Aggta
ccATCG
ATCG
TACG
tgggttc
ccta
gtta
gccagaga
66

6.3. MATERIALS AND METHODS
decrease in translated product, which can be detected by a luminescence-based assaysystem. Firefly luciferase expressed from the same vector serves as an internal nor-malization control.
Reporter co-transfection experimentsHEK293 or HCT116 cells, seeded 1 day before, were transfected in duplicate in 24-well plates at 80% confluency with a Lipofectamine 2000 (Invitrogen, Carlsbad, CA,USA)-complexed mixture of 25 ng psiCHECK reporter plasmid and 250 ng MCM7effector plasmid DNA per well. Twenty-four hours post-transfection, the cells werelysed with 120 µl Passive Lysis Buffer (Promega, San Luis Obispo, CA, USA) andluciferase levels were analyzed from 10 µl lysate using the Dual Luciferase reporterassay (50 µl of each substrate reagents, Promega) on a Veritas Microplate Luminome-ter (Turner BioSystems, Sunnyvale, CA, USA). Changes in the expression of Renillaluciferase (target) were calculated relative to firefly luciferase (internal control) andnormalized to levels in cells transfected with “empty” pcDNA3 control plasmid.
Northern blot analysisHCT116 or HEK293 cells were transfected in six-well plates at 80% confluency witha Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA)-complexed mixture of 2 µgMCM7 effector plasmid DNA and/or 400 ng enhanced green fluorescent protein con-trol plasmid DNA per well. Total RNA was isolated 40 h after transfection usingSTAT-60 (Tel-Test Inc., Friendswood, TX, USA) and resuspended in nuclease-freewater.
For detection of siRNA or miRNA expression, 10–20 µg of total RNA was elec-trophoresed in 10% denaturing polyacrylamide gel electrophoresis in 1 × TBE bufferfor 1–2 h at 300 V together with a RNA size marker (Decade Marker, Ambion, Austin,TX, USA). The RNA was transferred to a positively charged nitrocellulose mem-brane, Hybond-N+ (Amersham Biosciences, Piscataway, NJ, USA) by electroblotting.Ultraviolet-fixed membranes were hybridized overnight in PerfectHyb solution (Sigma-Aldrich, St Louis, MA, USA) at 37 ∘C with purified 𝛾-32P-labeled oligos complemen-tary to the predicted mature miRNA or siRNA strand. The oligo probes (2 pmol) wereend-labeled with PNK (10 units, NEB) and 𝛾-32P-ATP (3 pmol) in 20 µl reactions for30 min, and purified with G-25 Sephadex spin-columns (GE Healthcare, Waukesha,WI, USA). Hybridized membranes were washed once with 6 × SSC/0.1% SDS, thentwice with 2 × SCC/0.1% SDS, before exposure to film (Biomax MS, Kodak, NewHaven, CT, USA) or PhosphorImager analysis. miR-106b probe: 5′-ATCTGCAC-TGTCAGCACTTTA-3′, miR-93 probe: 5′-CTACCTGCACGAACAGCACTTT-3′,miR-25 probe: 5′-TCAGACCGAGACAAGTGCAATG-3′, S1 probe: 5′-GCGGAG-ACAGCGACGAAGAGC-3′, S2 probe: 5′-GCCTGTGCCTCTTCAGCTACC-3′, S3probe: 5′-CATCTCCTATGGCAGGAAGAA-3′, TAR probe: 5′-CCAGAGAGCTC-CCAGGCTCAG-3′, miR-21 probe: 5′-ATCGAATAGTCTGACTACAACT, U6 probe:5′-TATGGAACGCTTCTCGAATT-3′.
67

CHAPTER 6. ENGINEERING AND OPTIMIZATION OF THEMIR-106B-CLUSTER FOR ECTOPIC EXPRESSION OF MULTIPLEXEDANTI-HIV RNAS
Lentiviral vector production and transduction of target cellsLentiviral vector production, transduction and generation of stable cell lines weregenerated as described earlier [290].
HIV-1 challenge assaysHEK 293 cells were co-transfected in triplicate with the various vectors of interest andHIV-1 NL4-3 at a ratio of 4:1. Culture supernatants were collected at 24, 48 and 72 htime points, and analyzed by a bDNA assay using the QuantiGene Reagent System(Panomics, Fremont, CA, USA). The bDNA assay values for relative luciferase lightunits (RLU) were divided by the viable cells for each time point to give RLU per cell.
One million CEM-T cells were infected with wild-type HIV-1 strain IIIB at amultiplicity of infection of 0.001 in duplicate. After overnight incubation, cells werewashed three times with Hank’s balanced salt solution and cultured in RPMI 1640with 10% FBS. The SupT1-T cells (2 × 105 cells in 1 ml of medium) were challengedwith 0.006 ng of measured CA-p24 of HIV-1 strain LAI in triplicate. At designatedtime points, culture supernatants were collected and analyzed for HIV replication bya CA-p24 ELISA assay.
6.4 Results
6.4.1 Generation of an miR-106b-based expression system
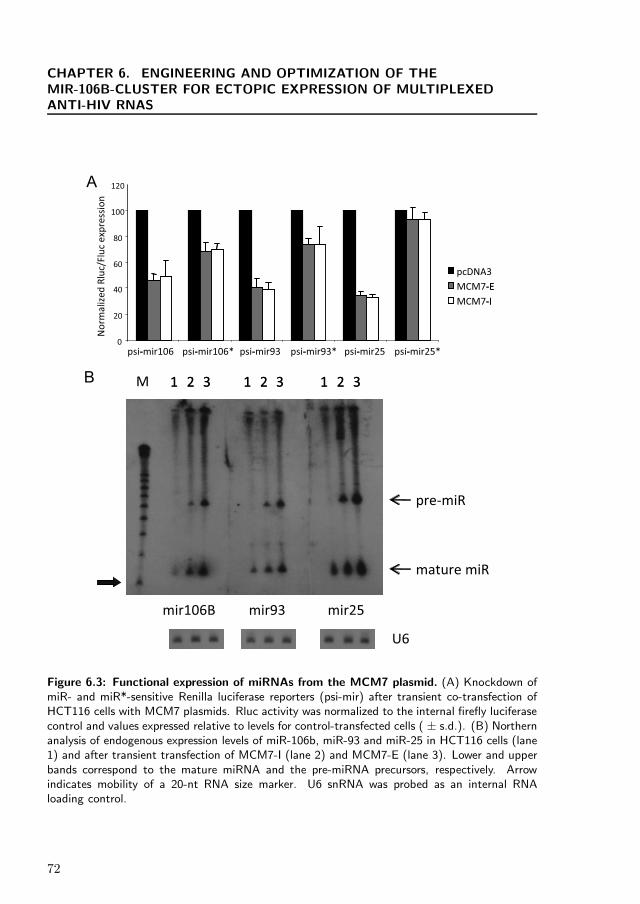
With the aim of generating a siRNA multiplexing expression system within a singletranscript, we sought to use a naturally occurring polycistronic miRNA as a scaffold.We based our work on the tri-cistronic miR-106b cluster located in intron 13 of theprotein encoding MCM7 gene on chromosome 7 [7]. This cluster, which consists ofmiR106b, miR-93 and miR-25, is relatively compact, and the entire intron is less than800 bp long. We initially cloned the miR-106b cluster into a cytomegalovirus (CMV)-driven expression plasmid, pcDNA3.1, by PCR amplification of human genomic DNA.Two versions were cloned: harboring the intron sequence only (MCM7-I) or incor-porating an additional 100-nt of flanking exon sequences on either side (MCM7-E)(Figure 6.1). Functional processing of mature miRNAs and their predicted passengerstrands (miR*) from our expression plasmid was then tested by co-transfecting var-ious expression plasmids with dual luciferase psiCHECK reporter constructs, whichharbored targets specific for each of the two strands of the miRNAs. Each psiCHECK2.2-based reporter (derived from psiCHECK 2 by extending the polylinker for cloning)contains a short stretch of target sequence with perfect complementarity to a specificmiR (or its miR* partner) inserted into the 3′ UTR (untranslated region) of Re-nilla luciferase. Normalized reporter expression is a measure of productive RISCloading and reporter mRNA cleavage by miR (or miR*). As shown in Figure 6.3A,co-transfections with the two versions of the MCM7 plasmid resulted in equivalentlevels of silencing for all three mature miRNAs and their passengers strands, sug-gesting that the inclusion of exonic regions did not substantially enhance activity.
68

6.4. RESULTS
The greatest level of silencing, 65%, was observed for miR-25, whereas slightly lowerlevels were observed for miR-93 (60%) and miR-106b (50%). This order of efficacywas reversed for the passenger strand-specific reporters, with negligible knockdownobserved for miR-25*, 25% for miR-93* and 30% for miR-106b* (Figure 6.3A). Theinverse relationship between the levels of silencing mediated by the two strands ofthe miRNAs is consistent with the asymmetric nature of strand selection, in whichone strand is preferentially utilized by RISC at the expense of the other [129, 293].Our observation that silencing by the designated mature miRNA strand was in allcases superior to that by the designated passenger strand is consistent with assignedasymmetries of strand utilization for the three miRNAs of this cluster, which is basedon cloning of recoverable short RNAs.
Mature miRNA expression from our plasmids was also evaluated using Northernblot analyses of transfected human HCT116 cells. All three mature miRNAs and theirprecursors were easily detectable by 5′-radiolabeled complementary oligonucleotideprobes (Figure 6.3B). Variable background levels of mature miRNAs were detectedin control-transfected cells, indicative of endogenous miRNA expression in HCT116cells. Our target inhibition data suggested that the exon-deficient version of themiRNA cluster was as effective as the exon-inclusive version, but Northern blot anal-yses indicated that the exon-inclusive construct consistently produced higher levels ofmature miRNA. On the basis of the Northern analyses, we decided to proceed withthe exon-inclusive version of our expression construct as a scaffold for heterologousmiRNA expression.
6.4.2 Expression of heterologous siRNAs from the miR-106bscaffold
We next sought to modify the MCM7-E plasmid in a manner that would allow usto replace the individual pre-miRNA stemloop structures with any given sequenceor hairpin structure. To accomplish this, we introduced unique restriction sites onboth sides and as close as possible to the three pre-miRNA sequences, using a mini-mal number of nucleotide substitutions. Importantly, secondary structure predictionsdid not indicate that the substitutions would disrupt the endogenous miRNA stem-loop structure (data not shown). An miRNA-depleted scaffold of the MCM7 intronwas generated by PCR amplification of each of the flanking and intervening pri-miR“spacer” sequences with restriction site-tagged primers followed by re-assembly ofthe individual units in a suitable vector in the native sequence order. We next sub-cloned this MCM7 scaffold into a pcDNA3.1-derived plasmid to generate MCM7-S(Figure 6.1). In MCM7-S, the miR-106b stemloops (and a few flanking nucleotides)are essentially replaced with small linkers, which serve as stuffers for insertions of anysequence using the introduced restriction sites. This platform itself should be use-ful to investigators wishing to insert their own miRNA clusters. We next introducedstemloop structures, S1S3, targeting three conserved sites in the HIV-1 genome calledsites 1, 2 and 3. S1 targets the common exon of early HIV-1 tat and rev transcripts,whereas S2 and S3 target rev and tat, respectively.
69

CHAPTER 6. ENGINEERING AND OPTIMIZATION OF THEMIR-106B-CLUSTER FOR ECTOPIC EXPRESSION OF MULTIPLEXEDANTI-HIV RNAS
miR-106b miR-93 miR-25
MCM7-E
MCM7-I
XhoI
MCM7-S
HindIII EcoRI BamHI XbaI SacII
MCM7-
S1/S2M/S3B
si-S1 si-S2M si-S3B
SD SA
XhoI HindIII EcoRI BamHI XbaI SacIIKpnI
121 nt 94 nt 128 nt 116 nt 150 nt 111 nt
771 nt
MCM7-
S1/S2M-TAR-
si-S1 si-S2M TAR
XhoI HindIII EcoRI BamHI XbaI SacIIKpnI
exon
intron
miRNA
shRNA
linker
miniS2
miniMCM7
delSplice
MCM7
-S1/S2M/S3B
forTARd
MCM7
-S1/S2M/S3BrevTARd
TARd
si-S1 si-S2M si-S3B
XhoI HindIII EcoRI BamHI XbaI SacIIKpnI
si-S1 si-S2M si-S3B
XhoI HindIII EcoRI BamHI XbaI SacII
U6-TARd
KpnI
Figure 6.1: Overview of miR-106b polycistron-based expression plasmids. Schematic diagramof the miR-106b polycistronic miRNA expression plasmids and the derived siRNA expressionplasmid used in this study. The ’MCM7’ name was used to indicate that the miR-106b clusteris located in intron 13 of the protein encoding gene MCM7 on chromosome 7q22.1. MCM7-E,MCM7-I and MCM7-S, refers to exon, intron and scaffold, respectively. MCM7-S1/S2/S3 refersto targeting of three independent targets sites, S1S3, in the viral HIV-1 genome. U16TAR-decoy refers to the nucleolar RNA TAR-decoy, with forU16TAR being U16TAR expressed in theforward direction and revU16TAR in the opposite orientation. The constructs delSplice, miniS2and miniMCM7, represent deletion variants of MCM7-S1/S2/S3.
70

6.4. RESULTS
miR106b
--- a c ua- g aga -- ucgcu cG gcccugc ggggc aagugcu acagugc uagu gg ccga gc cgggacg ccucg uucaugg ugucacg aucg cc u
uU Aac a a ucg g cc- ug uc
S1--- a c UG
gcu cG gcccugc ggggcGCGGAGACAGCGACGAAGAGCUU \cga gc cgggacg ccuCGCCCUCUGUCGCUGCUUCUCGGA U
uU Aac a a UG
ua-aagugcu
gacagugc
aga u
gg
ucgg uucaugg ugucacg
g cc
CGCCCUCUGUCGCUGCUUCUGCGGAGACAGCGACGAAGAGC
Figure 6.2: Example of replacing the native miR stemloop with an HIV-1 targeting shRNAsequence. For replacement of the native miR stemloop with an HIV-1 targeting shRNA sequencewe introduced restriction sites in MCM7-S, here illustrated with miR-106b and S1 using XhoIand Hind III. Mature miRNA/guide strand in light grey, predicted passenger strand (miR*) indark grey, heterologous sequences in uppercase, restriction sites in bold.
The heterologous hairpins were cloned as cassettes generated by PCR extensionof overlapping oligonucleotides followed by restriction endonuclease digestion. We de-signed our HIV-1 targeting hairpins by maintaining the natural pri-miR sequences atthe base of the stems up to the final closing base pair proximal to the mature miRNAand then simply replaced the upper part of the structure with an artificial, fully paired23 bp stem and an artificial 5-base loop (Figure 6.2). The upper part of this hybridstructure closely resembles a classical Pol III-promoted shRNA construct. The finalconstruct was designated as MCM7-S1/S2/S3. We tested this construct for process-ing of each of the siRNA species by co-transfection assays with individual S1, S2 andS3 target site-specific dual luciferase reporters (psi-Sx). As shown in Figure 6.4A, thetwo first units S1 and S2 worked efficiently and reduced luciferase reporter activityby approximately 75%. The third unit, S3, exhibited reduced knockdown ability (lessthan 50%). Importantly, the intrinsic activity of the S3 target, in the context of anshRNA construct, does not differ significantly from S1 or S2 (data not shown). Hence,the diminished activity suggested that si-S3 was processed less efficiently than si-S1and si-S2. Consistent with this, a Northern blot analysis demonstrated lower levels ofmature si-S3 than si-S1 or si-S2 (Figure 6.4B). Note that for the Northern analysis,the MCM7-S1/S2/S3 was transcribed by the Pol-II U1 snRNA promoter to avoidinterference with the CMV-driven green fluorescent protein (GFP) marker used asa transfection control. Our data suggest that the restriction site-modified miR-106bexpression system can be used to direct Pol II-promoted expression of three differentsiRNAs and that at least two of these siRNAs had comparable target knockdownefficacy relative to the native miRNAs.
71
CHAPTER 6. ENGINEERING AND OPTIMIZATION OF THEMIR-106B-CLUSTER FOR ECTOPIC EXPRESSION OF MULTIPLEXEDANTI-HIV RNAS
0
20
40
60
80
100
120
psi-mir25 psi-mir25*psi-mir93 psi-mir93*psi-mir106 psi-mir106*
No
rma
lize
d R
luc/
Flu
c e
xpre
ssio
n
pcDNA3
MCM7-E
MCM7-I
- -- -- -
-E
-I
mir25 mir93mir106B
MB
pre-miR
mature miR
U6
1 2 31 2 3 1 2 31 2 3 1 2 31 2 3
A
Figure 6.3: Functional expression of miRNAs from the MCM7 plasmid. (A) Knockdown ofmiR- and miR*-sensitive Renilla luciferase reporters (psi-mir) after transient co-transfection ofHCT116 cells with MCM7 plasmids. Rluc activity was normalized to the internal firefly luciferasecontrol and values expressed relative to levels for control-transfected cells ( ± s.d.). (B) Northernanalysis of endogenous expression levels of miR-106b, miR-93 and miR-25 in HCT116 cells (lane1) and after transient transfection of MCM7-I (lane 2) and MCM7-E (lane 3). Lower and upperbands correspond to the mature miRNA and the pre-miRNA precursors, respectively. Arrowindicates mobility of a 20-nt RNA size marker. U6 snRNA was probed as an internal RNAloading control.
72

6.4. RESULTS
A
0
20
40
60
80
100
120
psi-S1 psi-S2 psi-S3
pcDNA3
MCM7-E
MCM7-S1/S2/S3
No
rma
lize
d R
luc/
Flu
c e
xpre
ssio
n
B
S1 S2 S3
U6
Figure 6.4: Functional expression of siRNAs from the MCM7-S1/S2/S3 plasmid. (A)Knockdown of HIV-1 siRNA guide strand-sensitive RLuc reporters (psi-Sx) after transient co-transfection of HEK293 cells with MCM7 plasmids. Rluc activity was normalized to the inter-nal firefly luciferase control and values expressed relative to levels for control-transfected cells(± s.d.). (B) Northern analysis of siRNA guide strand expression levels after transient trans-fection of HCT116 cells with the MCM7-S1/S2/S3 construct. Arrow indicates mobility of a20-nt RNA size marker. Note that for the Northern blot analysis, MCM7-S1/S2/S3 expressionwas driven from a Pol-II U1-promoter plasmid to avoid interference from the CMV-driven greenfluorescent protein marker, which was used as a transfection control. U6 snRNA was probed asan internal RNA loading control.
73

CHAPTER 6. ENGINEERING AND OPTIMIZATION OF THEMIR-106B-CLUSTER FOR ECTOPIC EXPRESSION OF MULTIPLEXEDANTI-HIV RNAS
6.4.3 Removal of flanking pri-miRNA sequences abolishes siRNAfunctionality
Using the established knockdown efficiency of MCM7-S1/S2/S3 as measured by dualluciferase reporters, we set out to determine whether the flanking primary miRNA(pri-miRNA) sequences are crucial for functional processing of siRNAs, or whetherwe could configure the MCM7 expression platform in a simpler manner. First, weindividually replaced the longer mi/shRNA hybrid stemloops with a simple 23-nt-stem shRNA. We did this by direct cloning of oligo cassettes that encompassed therestriction sites and shRNA sequences only, thereby deleting the pre-miR leader se-quences as well as a few flanking nucleotides. Reporter co-transfections with theseminimal constructs, designated miniSx’s (see Figure 6.1 for a diagram of miniS2),demonstrated that the removal of pre-miR leader sequences completely abolished theactivity of the siRNA (Figure 6.5A). It can be noted that the removal of the leadersequence of one pre-miRNA unit had little effect on the functional release of siRNAsfrom the two remaining units (Figure 6.5A), which suggests that each unit is pro-cessed independently. Next, we generated a pcDNA3-derived expression construct,miniMCM7 (Figure 6.1), in which all sequences flanking each of the three mi/shRNAunits were removed and replaced by short, unstructured 6-nt linkers. This constructexhibited only marginal silencing activity (Figure 6.5B and data not shown). Thus,structural features within the miR-106b scaffold are critical for efficient siRNA pro-cessing. Our results are consistent with recently published data, indicating that effec-tive Drosha/DGCR8-mediated nuclear processing of the primary transcript dependson a base-paired stem of about one full helical turn flanked by unstructured regionsof the sequence outside of the pre-miRNA [155].
6.4.4 Splicing enhances siRNA functionality in the miR-106bsystem
Among the various features missing in miniMCM7 is the exon-intron-exon structure.To test whether splicing affected siRNA functionality, we cloned a shorter versionof MCM7-S1/S2/S3, delSplice, in which flanking MCM7-derived exons and all splic-ing signals within the introns were removed (Figure 6.1). This construct exhibiteda twofold reduction in silencing of psi-S1 in co-transfection assays (Figure 6.5B).Reduced silencing potential was also observed for the other two siRNAs expressedfrom the tri-cistronic construct (data not shown). These data are similar to our ob-servation that the inclusion of flanking exons enhances the expression of the nativemiRNAs from this cluster. Clearly, however, removal of splicing potential does notin itself abolish the ability of intron-embedded polycistronic miRNAs to be processedby Drosha/DGCR8. This is consistent with recent observations by others in whichdeletion of splice sites only marginally affected pre-miRNA generation from an intron[155].
74

6.4. RESULTS
0
20
40
60
80
100
120
psi-S1 psi-S2 psi-S3
No
rma
lize
d R
luc/
Flu
c e
xpre
ssio
n
0
pcDNA3
MCM7-S1/S2/S3-
miniS1
miniS2
miniS3
A B
0
20
40
60
80
100
120
psi-S1
pcDNA3
MCM7-S1/S2/S3
delSplice
miniMCM7
No
rma
lize
d R
luc/
Flu
c e
xpre
ssio
nFigure 6.5: Limited efficacy of siRNAs expressed from MCM7-S1/S2/S3 deletion mu-tants. (A) Knockdown of siRNA-sensitive RLuc reporters after transient co-transfection ofHEK293 cells with MCM7-S1/S2/S3 plasmids lacking the lower part of the miRNA stemloopstructure (miniSx). (B) Knockdown of siRNA-sensitive Renilla luciferase reporters in MCM7-S1/S2/S3 plasmids lacking the flanking exon and the splice sites (delSplice) or missing thepri-miR-derived sequences between the stemloop structures (miniMCM7). See Figure 6.1 forconstruct diagrams. Rluc activity was normalized to the internal firefly luciferase control andvalues expressed relative to levels for control-transfected cells (± s.d.).
6.4.5 Strand selectivity of the S2 unit is improved by a fullmiR-93 mimic
When expressed as standard shRNAs from U6 promoters, all three RNAi effectorsdisplay favorable asymmetry in strand utilization (data not shown). We decidedto investigate whether or not asymmetry was retained in the polycistronic miRNAexpression background. Co-transfection experiments with strand-specific reportersdemonstrated highly variable strand selectivity for the various units. The reportersfor the presumptive guide and passenger strands of si-S2 were silenced almost equallyby MCM7-S1/S2/S3, indicating little discrimination between strands in RISC incor-poration (Figure 6.6A). In contrast, si-S1 and si-S3 demonstrated negligible andmarginal passenger strand activities, respectively (data not shown). The poor strandselectivity of S2 is clearly undesirable as this increases the likelihood of passengerstrand-mediated off-target effects. The poor strand selectivity of si-S2 compared withits parent miR-93 in the context of the polycistronic transcript may result from skewedprocessing of the hybrid transcript due to changes in secondary structure. In an at-tempt to control for such an effect, we changed our “simple” S2 mi/shRNA hybridto a version, designated as S2M, in which the passenger strand was selectively mu-
75

CHAPTER 6. ENGINEERING AND OPTIMIZATION OF THEMIR-106B-CLUSTER FOR ECTOPIC EXPRESSION OF MULTIPLEXEDANTI-HIV RNAS
tated to closely mimic the secondary structure of miR-93 while retaining the desiredS2 guide strand (Figure 6.6B). This miR-93 mimic exhibited no passenger strandactivity (in co-transfection experiments with a new reporter specific for the mutatedpassenger strand, psi-S2M-AS) while still silencing the psi-S2 reporter more than 70%(Figure 6.6A). Importantly, the MCM7-S1/S2M/S3 construct maintained S1 and S3targeting activities (data not shown). These observations suggest that to achieve thedesired combination of efficacy and specificity, polycistronic miRNA-based expressioncassettes should maintain the general secondary structures of the pre-miRNA leadersequences and heterologous stems.
6.4.6 Increased expression of si-S3 from an miR-93 mimic butneither an miR-106 nor an miR-25 mimic
Having achieved efficient knockdown and good strand selectivity for the S1 and S2units, we next addressed the issue of poor functionality from the S3 unit. To im-prove S3, we decided to investigate whether the secondary structures of the first twounits might be transferable to the third unit in a heterologous tri-cistron. The S3siRNA was therefore incorporated into the third position as an S1/miR-106b, anS2M/miR-93 or an miR25 structural mimic, generating the hybrids S3A, S3B andS3C, respectively. Whereas the S3A hairpin resulted in only a modest improvementof psi-S3 knockdown and S3C resulted in no improvement (data not shown), the S3Bvariant improved silencing to 80% (Figure 6.7A). Concomitant with this improvementin functionality, Northern blot analyses demonstrated a marked increase in the levelsof mature si-S3 from S3B as compared with S3 (Figure 6.7C). It can be noted thatMCM7-S1/S2M/S3B maintained knockdown efficacy of the S1 and S2 units (Fig-ure 6.7A) while exhibiting good strand selectivity for S3 (Figure 6.7B). We are thusconfident that this final configuration of the MCM7 intron is appropriate for use inother applications besides HIV-1 infection.
6.4.7 Incorporation of U16 snoRNA-TAR-decoy hybrid RNAto increase the functionality of the MCM7 system
In targeting HIV-1, it is important to use a combinatorial inhibitory approach andit seems wise not to depend on a single inhibitory modality, such as RNAi. Priorresults from our lab demonstrated a nucleolar localizing RNA TAR-decoy (U16TAR)served as a good inhibitory agent of HIV-1 replication [209]. We therefore decidedto create combinations of the anti-HIV siRNAs with the U16TAR-decoy. In the firstconstruct, S3B was replaced with the U16TAR element. Two additional constructsincorporating U16TAR were also made. One of these incorporated an U6 Pol IIIpromoter-driven U16TAR-decoy downstream of the siRNAs and transcribed in theopposite orientation with respect to the siRNAs (revU16TAR). We also inserted theU16TAR-decoy downstream of the siRNAs in an intron position shown to be favorableto snoRNA processing [129, 130] and designated this as forU16TAR. The forU16TARis co-transcribed with the siRNAs from the upstream Pol II promoter (Figure 6.1).
76

6.4. RESULTS
A
0
20
40
60
80
100
120
psi-S2-AS psi-S2M-AS psi-S2
pcDNA3
MCM7-S1/S2/S3
MCM7-S1/S2M/S3
No
rma
lize
d R
luc/
Flu
c e
xpre
ssio
n
B
S2Muca gU- - A ug au
gaAU guccugggggcug AGCUG AAGAGGC CAGGCag ug ug g
cuug Uagggcccccgau UCGAC UuCUCCG GUCCGuc ac acC- agc U - ca cc
ggg g
S2uca UG
gaAU guccugggggcucGCCUGUGCCUCUUCAGCUACCUU \
cuug UagggcccccgagCGGACACGGAGAAGUCGAUGGGA UcC- UG
uca ca- - u g ug au gaAU guccugggggcuc aagugcu guucg gcag uag ug ucuug Uagggcccccgag uucacga cgagu cguc auc ac a
cC- ccc u - - ca cc
miR93
a- u g aagugcu guucg gcag
guag
cccuucacga cgagu cguc auc
u
GCCUGUGCCUCUUCAGCUACC
agCGGACACGGAGAAGUCGAU
AGCUG AAGAGGCA CAGGCag
UCGAC UuCUCCG GUCCGU agc
U
Figure 6.6: Improved asymmetry of S2 siRNA RISC loading by an miR-93 structuralmimic. (A) Knockdown of passenger strand-sensitive RLuc reporters (psi-S2-AS or psi-S2M-AS) after transient co-transfection of HEK293 cells with MCM7-S1/S2/S3 plasmid or an miR-93mimic (S2M). Rluc activity was normalized to the internal firefly luciferase control and values ex-pressed relative to levels for control-transfected cells (± s.d.). (B) Predicted stemloop structureof miR-93, the fully base-paired S2 hairpin and its miR-93 mimic, S2M. Mature miRNA/guidestrand in light grey, predicted star/passenger strand in dark grey, heterologous sequences inuppercase, restriction sites (EcoR I and BamHI) in bold.
77

CHAPTER 6. ENGINEERING AND OPTIMIZATION OF THEMIR-106B-CLUSTER FOR ECTOPIC EXPRESSION OF MULTIPLEXEDANTI-HIV RNAS
0
20
40
60
80
100
120
S1 S2M S3
No
rmali
zed
Rlu
c/F
luc e
xp
ressio
n
0
20
40
60
80
100
120
No
rma
lize
d R
luc
/Flu
c e
xp
res
sio
n
0
20
40
60
80
100
120
S1 S2M S3
No
rmali
zed
Rlu
c/F
luc e
xp
ressio
n
pcDNA
MCM7S1S2MS3
MCM7S1S2MS3B
pcDNA
MCM7S1S2MS3
MCM7S1S2MS3B
0
20
40
60
80
100
120
psi-S3as
No
rma
lize
d R
luc
/Flu
c e
xp
res
sio
n
pcDNA
MCM7S1S2MS3B
pcDNA
MCM7S1S2MS3B
S1 S2 S3
U6
1 2 1 2 1 2A
B
C
Figure 6.7: Optimized S3B containing cluster maintains the knockdown efficiencies ofS1 and S2M, but has greatly improved S3 efficacy. Plasmids containing the three anti-HIVmiRNA mimics were co-transfected with psiCHECK vectors harboring the targets for each ofthe three siRNAs targeting S1, S2 and S3. The S3 unit was tested in the context of S1 andS2M in its original configuration (MCM7-S1/S2M/S3) or in the optimized configuration (MCM7-S1/S2M/S3B). The MCM7-S1/S2M/S3B cluster provides comparable levels of knockdown forsites S1 and S2, but demonstrated enhanced knockdown of the S3 target (A). Good strandselectivity is maintained for MCM7-S1/S2M/S3B when the target is inserted in the antisenseorientation (B). Northern analysis of siRNA guide strand expression levels after transient trans-fection of HCT116 cells (C). RNA from MCM7-S1/S2/S3 (lane 1)- and MCM7-S1/S2M/S3B(lane 2)-transfected cells, respectively. Arrow indicates mobility of a 20-nt RNA size marker. U6snRNA was probed as an internal RNA loading control.
78

6.4. RESULTS
Expression analyses of the siRNAs as well as the U16TAR-decoy by Northern blottingrevealed good expression of the siRNAs and the U16TAR RNA, with the most robustU16TAR expression deriving from the revTARd (Figure 6.8).
S3
S2
S1
S1S
2MS3B
forU
16TA
R
pcDNA
S1S
3MS3B
S1S
2MS3B
revU
16TA
R
shS1
shS2
shS3
S1S
2MU16
TAR
Shr
ek 3
U16TAR
Figure 6.8: Expression of the RNAs in the optimized MCM7 constructs. HEK293 cells weretransiently transfected with each of the optimized MCM7 constructs and controls. Forty-eighthours after transfection, total RNA was collected, electrophoresed in an 8% polyacrylamide gelwith 8 M urea, blotted onto a nylon membrane and hybridized with the corresponding 32P-labeled probes. Hybridization to U6-driven short hairpin siRNAs S1, S2, S3 and the U16TAR-decoy expressed from Shrek3 [236] were used as positive controls. RNA prepared from an emptyvector (pcDNA)-transfected cell line was used as a negative control. S1, S2 and S3 siRNAs areapproximately 21 nucleotides. The U16TAR-decoy is 132 nucleotides.
We reasoned that the forU16TAR should be processed from the intron in a fashion
79

CHAPTER 6. ENGINEERING AND OPTIMIZATION OF THEMIR-106B-CLUSTER FOR ECTOPIC EXPRESSION OF MULTIPLEXEDANTI-HIV RNAS
similar to endogenous snoRNA processing [129], whereas the U16TAR inserted inplace of an miRNA unit could potentially be processed by the miRNA processingenzyme, Drosha. To address this possibility, we took advantage of a Tet-inducibleanti-Drosha shRNA construct [2]. Following the addition of Doxycycline to induce theanti-Drosha shRNA, we monitored processing of the siRNAs as well as the U16TAR byNorthern analyses. Although the processing of the S1 and S2 siRNAs and endogenousmiR21 RNA was inhibited by Drosha knockdown, there was no inhibition of theU16TAR-decoy (Figure 6.9). Interestingly, the steady-state level of the U6-drivenrevU16TAR was somewhat elevated following Drosha knockdown. We conclude fromthese results that the processing of the forU16TAR and U16TAR takes place throughthe snoRNA processing mechanism.
6.4.8 Optimized multi-cistronic expression units suppresses HIV-1 replication
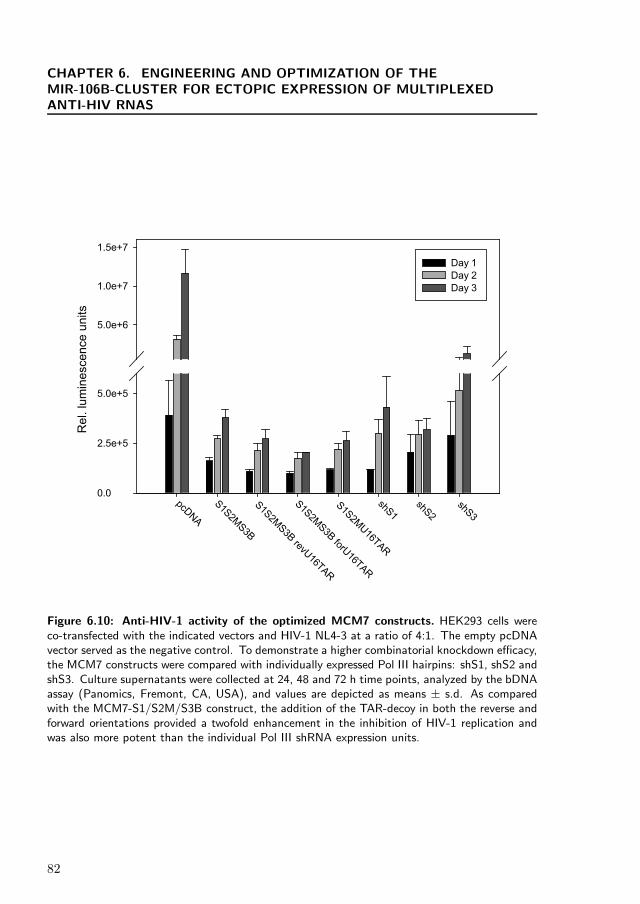
To test the functionality of the MCM7-S1/S2M/S3B/U16TAR constructs, we tran-siently co-transfected HEK293 cells with the various vectors and HIV pNL4-3 proviralDNA. On the days indicated, supernatants were collected and assayed for HIV replica-tion using a branched DNA (bDNA) assay, which measures viral RNA (Figure 6.10).All of the constructs provided two or more logs of inhibition of HIV RNA production,but the revU16TAR and forU16TAR containing MCM7 constructs provided the mostpotent knockdown over the course of the assay and were at least twice as inhibitoryas the MCM7-S1/S2M/S3B construct.
6.4.9 The U16TAR-decoy enhances viral inhibition of the MCM7--S1/S2M/S3B cluster
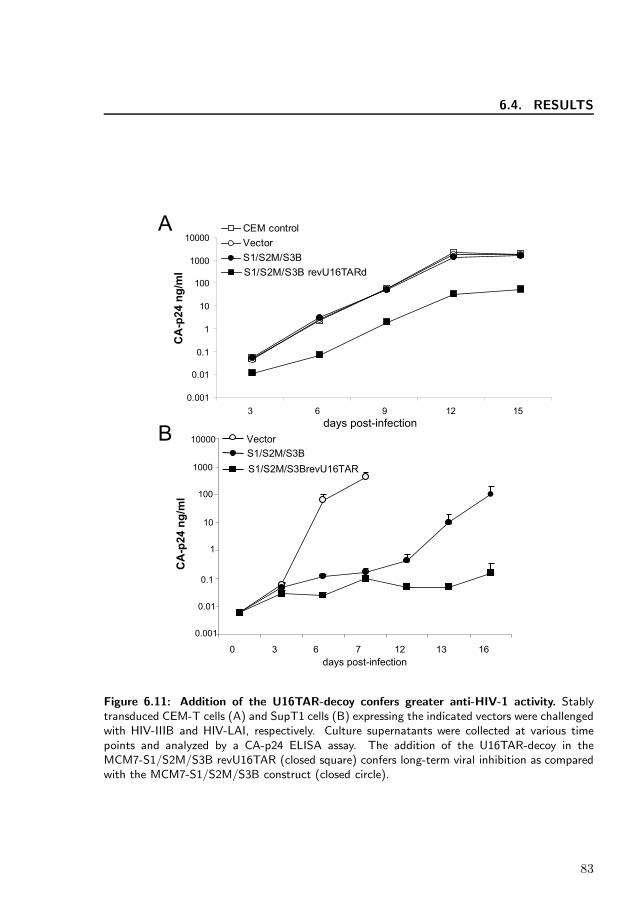
The most robust U16TAR-decoy expression was obtained from the revU16TAR inser-tion that did not compromise the expression of the siRNAs. Therefore, we incorpo-rated this configuration into the pHIV7 lentiviral vector that was used to transducehuman T cell lines for testing in HIV acute challenge assays. We also replaced theCMV promoter with the human U1 snRNA promoter and SV40 poly-A sequence.The entire unit was inserted into pHIV7 in an orientation opposite to that of theCMV packaging promoter to prevent splicing of the MCM7 intron during packag-ing. CEM and SupT1 cells were stably transduced with the control pHIV7 vector,MCM7-S1/S2M/S3B or MCM7-S1/S2M/S3B revU16TAR. The transduced cells weresorted for enhanced green fluorescent protein expression and subsequently challengedwith HIV-1. For the CEM cells, the challenge virus was the HIV-IIIB, whereas theSupT1 cells were challenged with HIV-LAI. Supernatants were collected at varioustime points and levels of viral CA-p24 were measured by ELISA (Figure 6.11). Al-though the MCI7-S1/S2M/S3B construct provided good inhibition with HIV-LAI,it was less protective when challenged with HIV-IIIB. Inclusion of the revU16TARresulted in substantially better inhibition of both strains of HIV-1.
80

6.4. RESULTS
U6
shS1
U16TAR
S1S
2MS3B
U16
S1S
2MS3B
rev
U16
TAR
d
S1S
2MU
16TAR
shS1
+ Dox - Dox
S1S
2MS3B
U16
S1S
2MU
16TAR
shS1
S1S
2MS3B
rev
U16
TAR
d
S1S
2MS3B
U16
S1S
2MU
16TAR
shS1
S1S
2MS3B
rev
U16
TAR
d
miR-21
Figure 6.9: Transfection of MCM7 constructs into a Doxycyline inducible anti-DroshashRNA harboring cell line. Doxycycline (Dox) inducible Drosha knock-out 293 cells were culturedin high glucose (4.5 g/l) DMEM supplemented with 2 mM glutamine and 10% FBS withoutantibiotics. Drosha shRNA expression was induced with 200 ng/ml Dox and replenished every2 days with fresh medium and Dox for continual Drosha shRNA expression. One week afterinitial Dox induction, cells were seeded with or without inducer in 6-well plates. The next day,at 80% confluency, cells were transfected with a Lipofectamine 2000-complexed mixture of 4 µgMCM7 effector plasmid DNA per well. Cells were cultured with the Lipofectamine mixture for 48hours prior to RNA harvesting. RNAs were electrophoresed in a 8% denaturing polyacrylamide gel,transferred onto a nitrocellulose membrane and probed with radioactively labeled oligonucleotidescomplementary to the mature S1 siRNA or the TAR sequence. As controls, U6 snRNA and miR-21 were also probed and S1 was expressed from the U6 Pol III promoter as an shRNA (S1).Drosha knockdown resulted in reductions in the levels of S1, S2 and S3 siRNAs as well as miR-21. In contrast, no reductions in the levels of forU16TAR, revU16TAR or U16TAR were observed,but instead an increase in the level of revU16TAR can be seen.
81
CHAPTER 6. ENGINEERING AND OPTIMIZATION OF THEMIR-106B-CLUSTER FOR ECTOPIC EXPRESSION OF MULTIPLEXEDANTI-HIV RNAS
pcDNA
S1S2MS3B
S1S
2MS3B
revU16TA
R
S1S
2MS3B
forU16TA
R
S1S
2MU16TA
R
shS1
shS2
shS3
Re
l. lu
min
esce
nce
un
its
0.0
2.5e+5
5.0e+5
5.0e+6
1.0e+7
1.5e+7
Day 1
Day 2
Day 3
Figure 6.10: Anti-HIV-1 activity of the optimized MCM7 constructs. HEK293 cells wereco-transfected with the indicated vectors and HIV-1 NL4-3 at a ratio of 4:1. The empty pcDNAvector served as the negative control. To demonstrate a higher combinatorial knockdown efficacy,the MCM7 constructs were compared with individually expressed Pol III hairpins: shS1, shS2 andshS3. Culture supernatants were collected at 24, 48 and 72 h time points, analyzed by the bDNAassay (Panomics, Fremont, CA, USA), and values are depicted as means ± s.d. As comparedwith the MCM7-S1/S2M/S3B construct, the addition of the TAR-decoy in both the reverse andforward orientations provided a twofold enhancement in the inhibition of HIV-1 replication andwas also more potent than the individual Pol III shRNA expression units.
82
6.4. RESULTS
A
B
0.001
0.01
0.1
1
10
100
1000
10000
3 6 9 12 15
CEM control
Vector
S1/S2M/S3B
S1/S2M/S3B revU16TARd
0.001
0.01
0.1
1
10
100
1000
10000
0 3 6 7 12 13 16
Vector
S1/S2M/S3BrevU16TAR
S1/S2M/S3B
days post-infection
days post-infection
CA
-p24 n
g/m
lC
A-p
24 n
g/m
l
Figure 6.11: Addition of the U16TAR-decoy confers greater anti-HIV-1 activity. Stablytransduced CEM-T cells (A) and SupT1 cells (B) expressing the indicated vectors were challengedwith HIV-IIIB and HIV-LAI, respectively. Culture supernatants were collected at various timepoints and analyzed by a CA-p24 ELISA assay. The addition of the U16TAR-decoy in theMCM7-S1/S2M/S3B revU16TAR (closed square) confers long-term viral inhibition as comparedwith the MCM7-S1/S2M/S3B construct (closed circle).
83

CHAPTER 6. ENGINEERING AND OPTIMIZATION OF THEMIR-106B-CLUSTER FOR ECTOPIC EXPRESSION OF MULTIPLEXEDANTI-HIV RNAS
6.5 Discussion
We have engineered a system for functional co-expression of three different heterolo-gous siRNAs from a polycistronic, intronic, miRNA platform. This system is basedon the miR-106b cluster that is located in a 0.8 kb intron in the host gene MCM7. Ac-cordingly, we have called the system MCM7 (Figure 6.1). The native cluster was ini-tially cloned into a CMV-promoted expression plasmid vector and each of the encodedmiRNAs; miR-106b, miR-93 and miR-25 were shown to be functionally expressedfrom this backbone (Figure 6.3). By introducing a small number of substitutions, wecreated suitable restriction sites flanking each of the three miRNA stem-loops thatallowed for the insertion of any given stemloop at each of the three positions. As atest of this platform, we inserted three different anti-HIV sequences and tested eachunit for function. The HIV-1 targeting sequences were initially introduced as fullybase-paired shRNA-like structures that essentially replaced the pre-miRNA parts ofthe primary transcript, above the Drosha cleavage site [155, 236]. Knockdown ef-ficiencies of the cognate targets of the heterologous siRNAs were comparable withthose observed for the parental miRNAs of the wild-type polycistron (Figure 6.4). Ofimportance, we noticed a strict requirement for the flanking pri-miRNA sequences forfunctionality. Knockdown efficacy was abolished by the removal of leader sequencesbelow the Drosha cleavage site of the extended stem structure, or when the longintervening sequences between the different miRNA units were replaced with shortunstructured linkers (Figure 6.5). Moreover, siRNA expression levels and functional-ity were also reduced when we removed the flanking exon structure and splice sites,suggesting that splicing may enhance processing of polycistronic miRNAs.
Despite the efficient processing of at least two siRNAs from our miR/shRNAchimera (MCM7-S1/S2/S3), we observed two undesirable properties of the initialMCM7 tri-cistronic construct. First, the third unit, S3, was expressed at lower levelsthan the first and second units and concomitantly less active than the other two.Second, S2 displayed equivalent RISC incorporation of both strands. This may be adesirable property for some applications where both strands are intentionally designedto be functional. In our case, it indicated that our design had poor strand asymmetry,which increases the potential for passenger strand-mediated off-target effects. We thussought to correct both deficiencies without altering the HIV-1 targeting properties.Our approach was to introduce targeted mutations in the passenger strand in such away as to convert the original miR/shRNA hybrids to structures that better resembledthe native miRNAs of this cluster. Such a structural mimic of miR-93, S2M, improvedthe strand selectivity of the S2 siRNA to a level at which functional passenger strandactivity was negligible while still maintaining guide strand-mediated functionality(Figure 6.6). We suspect that the initial poor strand selectivity for this unit mightstem from skewed Drosha processing of the primary transcript. Algorithms thatpredict Drosha processing sites based on conserved primary and secondary structuremotifs [127], in fact, predicted the intended cleavage site for S1 while suggestingmultiple cleavages sites for S2 (data not shown). If these predictions are accurate,the resulting siRNAs would most likely differ in strand loading based on differences
84

6.5. DISCUSSION
in thermodynamic end stabilities [151, 265].
Once our MCM7 construct was optimized for efficient processing of the siRNAs,we sought to increase the construct’s multifunctionality by inserting a nucleolar lo-calizing TAR-decoy within the intron. This U16TAR RNA mimic binds Tat andsubsequently sequesters it from the virus and inhibits HIV-1 replication [209]. Here,we demonstrate the processing and expression of the U16TAR-decoy in three dif-ferent configurations within the modified MCM7 intron (revU16TAR, for U16TAR,U16TAR) (Figure 6.8). When these various combinations of the MCM7-S1/S2M/S3Band U16TAR-decoy were tested for anti-viral activity, we observed better inhibitionof HIV-1 as opposed to the parental MCM7-S1/S2M/S3B construct (Figure 6.10). Asmore robust expression of the U16TAR was observed from the MCM7-S1/S2M/S3BrevU16TAR than the forU16TARd MCM7 cluster (Figure 6.8), we decided to utilizethe revU16TAR configuration for insertion into the pHIV7 lentiviral vector. From thisassay, the MCM7-S1/S2M/S3B revU16TAR construct provided a marked reductionin HIV CA-p24 antigen levels compared with the MCM7 cluster without the U16TAR-decoy (Figure 6.11). The HIV-LAI isolate was inhibited by the MCM7-S1/S2M/S3construct alone, but the addition of the U16TAR-decoy enhanced the inhibition (Fig-ure 6.11B) in similarity to the HIV-IIIB challenges. However, the lack of significantinhibition by the MCM7-S1/S2M/S3B when challenged with HIV-IIIB (Figure 6.11A)was somewhat surprising and contrasted with our HIV pNL4-3 co-transfection results.In our hands, HIV-IIIB is one of the more difficult lab isolates of HIV-1 to inhibit incell culture challenges. The relatively low expression levels of the siRNAs from theintegrated lentiviral vector construct, although important for minimizing toxicities,may allow HIV to overwhelm the RNAi effectors. We have previously observed thatshRNAs can be overwhelmed by HIV in acute challenges, but when they are combinedwith other RNA-based inhibitors, such as the TAR-decoy and a ribozyme, virtuallycomplete inhibition can be obtained [184]. It is important to note that on its own theU16TAR-decoy is not a potent inhibitor of HIV replication, but in combination withone or more other anti-HIV RNAs there is an apparent synergy [184, 185]. Bindingof the U16TAR to Tat may subsequently slow the progression of viral transcriptionto reach a critically low level that allows each of the S1, S2M and S3B hairpins tofunction more effectively in downregulating the tat/rev transcripts.
It can be noted that our U16TAR-decoy is a chimeric snoRNA that has beenshown to exclusively localize within the nucleoli of human cells [209]. When encodedin an intron, the assembly of snoRNA particles depends on the position within theintron or the stability of a stem structure flanking the snoRNA [129]. Here, wedemonstrate expression of the U16TAR-decoy configured in three different ways. Inone configuration it is expressed from its own U6 promoter in an orientation oppositeof the MCM7-S1/S2M/S3B cluster (revU16TAR). In the second configuration, it isco-expressed with the MCM7-S1/S2M/S3B miRNAs via its insertion approximately70 bases upstream of the 3′ splice signal (forU16TAR). Finally, we have inserted theU16TAR-decoy unit in place of S3B within a structured stem (U16TAR). Althoughall three variants are expressed and processed, the strongest expression was obtainedfrom the revU16TAR (Figure 6.8). Drosha knockdown experiments did not diminish
85

CHAPTER 6. ENGINEERING AND OPTIMIZATION OF THEMIR-106B-CLUSTER FOR ECTOPIC EXPRESSION OF MULTIPLEXEDANTI-HIV RNAS
the levels of mature U16TAR in any of the constructs despite the reduction in matureS1 and miR21 levels (Figure 6.9).
In nature, multi-cistronic miRNA clusters are common, and the miRNAs producedfrom these often regulate the expression of many different transcripts simultaneously.The use of an miRNA cluster for concurrent expression of multiple siRNAs has sev-eral advantages. First, targeting of viral pathogens at multiple positions should, intheory, reduce the likelihood of viral escape. For HIV-1, it is well established thatthe potency of RNAi rapidly selects for escape mutants [41, 70, 102, 308, 318], andexpression of multiple siRNAs by various means has been shown to improve bothinitial functionality and delay the emergence of resistance [287, 290]. This strategyis analogous to the use of multiple drugs in the current highly active anti-retroviraltherapy (HAART) treatments. This new expression system also has the advantagethat the choice of a promoter is quite flexible, and constitutive, tissue-specific or evenHIV Tat-inducible promoters [299] can be used. Thus, levels of expression can be con-trolled to avert potential toxicities caused by high levels of expression of the miRNAsand resultant siRNAs [110]. As the processing signals for the miRNAs are encodedwithin the miRNAs themselves as well as sequences immediately flanking the hairpinsand there are no specific structural requirements for the 5′ and 3′ ends of the primarytranscripts, this system should prove to be generally useful for expression of multiplehairpins from a variety of promoters.
In summary, we have engineered a novel miR-106b cluster-based expression sys-tem, MCM7, for the delivery of multiple siRNAs from a single RNA Pol II promotedtranscript. We have optimized the designs of the miRNA mimics for efficient func-tional release of all three of the ectopic siRNAs. In addition, we have included withinthe intron a nucleolar U16TAR-decoy, which is co-expressed with the siRNAs. Thecombined siRNAs and U16TAR-decoy demonstrate potent inhibition of HIV-1 repli-cation. Our miRNA-based siRNA expression system may also be useful as a generaltool for silencing of multiple cellular targets and could find applications in both basicexperimentation and in therapeutic applications where multiple siRNA or miRNAexpression is required.
6.6 Acknowledgements
This work was funded by a fellowship to LAA from the Alfred Benzon Foundation,a post-doctoral fellowship to MA from the Norwegian Research Council and NIHAI42552. AI29329 and HL07470 grants to JJR. KJE is supported by ZonMw througha VICI grant. We thank A. Ehsani for his suggestions about the positions for insertingthe TAR-decoy and members of the Rossi lab for their support.
86




















