
Effective Application of Bicelles for Conformational Analysis of G Protein-Coupled Receptors by...
10
B American Society for Mass Spectrometry, 2015 DOI: 10.1007/s13361-015-1083-4 J. Am. Soc. Mass Spectrom. (2015) 26:808Y817 RESEARCH ARTICLE Effective Application of Bicelles for Conformational Analysis of G Protein-Coupled Receptors by Hydrogen/Deuterium Exchange Mass Spectrometry Nguyen Minh Duc, 1 Yang Du, 2 Thor S. Thorsen, 2 Su Youn Lee, 1 Cheng Zhang, 2,3 Hideaki Kato, 2 Brian K. Kobilka, 2 Ka Young Chung 1 1 School of Pharmacy, Sungkyunkwan University, 2066 Seobu-ro, Jangan-gu, Suwon, 440740, South Korea 2 Department of Molecular and Cellular Physiology, Stanford University Medical School, 297 Campus Drive, Beckman Center, Stanford, CA 94305, USA 3 Present Address: Department of Pharmacology and Chemical Biology, University of Pittsburgh School of Medicine, E 1358 Thomas E. Starzl Biomedical Science Tower, 200 Lothrop St., Pittsburgh, PA 15213, USA Abstract. G protein-coupled receptors (GPCRs) have important roles in physiology and pathology, and 40% of drugs currently on the market target GPCRs for the treatment of various diseases. Because of their therapeutic importance, the structural mechanism of GPCR signaling is of great interest in the field of drug discovery. Hydrogen/deuterium exchange mass spectrometry (HDX-MS) is a useful tool for analyzing ligand binding sites, the protein–protein interaction interface, and confor- mational changes of proteins. However, its application to GPCRs has been limited for various reasons, including the hydrophobic nature of GPCRs and the use of deter- gents in their preparation. In the present study, we tested the application of bicelles as a means of solubilizing GPCRs for HDX-MS studies. GPCRs (e.g., β 2 -adrenergic receptor [β 2 AR], μ-opioid receptor, and protease-activated receptor 1) solubilized in bicelles produced better sequence coverage (greater than 90%) than GPCRs solubilized in n-dodecyl-β-D-maltopyranoside (DDM), suggesting that bicelles are a more effective method of solubilization for HDX-MS studies. The HDX-MS profile of β 2 AR in bicelles showed that transmembrane domains (TMs) undergo lower deuterium uptake than intracel- lular or extracellular regions, which is consistent with the fact that the TMs are highly ordered and embedded in bicelles. The overall HDX-MS profiles of β 2 AR solubilized in bicelles and in DDM were similar except for intracellular loop 3. Interestingly, we detected EX1 kinetics, an important phenomenon in protein dynamics, at the C-terminus of TM6 in β 2 AR. In conclusion, we suggest the application of bicelles as a useful method for solubilizing GPCRs for conformational analysis by HDX-MS. Keywords: GPCR, HDX-MS, Bicelles, Detergent, Conformation, EX1 Received: 16 September 2014/Revised: 15 January 2015/Accepted: 16 January 2015/Published Online: 5 March 2015 Introduction G -protein coupled receptors (GPCRs) are the most impor- tant class of membrane receptors with over 800 identified to date in humans, many of which are involved in diseases, such as oncologic, psychiatric, metabolic, neurodegenerative, cardiovascular, and infectious diseases [1, 2]. Approximately 40%–50% of drugs circulating in the market target GPCRs for the treatment of various diseases [2]. Ligand binding induces conformational changes of GPCRs, which in turn regulate interactions with downstream signaling molecules, such as heterotrimeric G-proteins or β-arrestins [3, 4]. Understanding the conformational changes that are induced in GPCRs upon activation or inactivation would greatly advance our under- standing of the mechanism of activation and inactivation in- duced by endogenous or synthetic ligands, and might ultimate- ly lead to the design of more effective and less toxic drugs. Nguyen Minh Duc and Yang Du contributed equally to this work. Electronic supplementary material The online version of this article (doi:10.1007/s13361-015-1083-4) contains supplementary material, which is available to authorized users. Correspondence to: Brian Kobilka; e-mail: [email protected], Ka Chung; e-mail: [email protected]
Transcript of Effective Application of Bicelles for Conformational Analysis of G Protein-Coupled Receptors by...

B American Society for Mass Spectrometry, 2015DOI: 10.1007/s13361-015-1083-4
J. Am. Soc. Mass Spectrom. (2015) 26:808Y817
RESEARCH ARTICLE
Effective Application of Bicelles for Conformational Analysisof G Protein-Coupled Receptors by Hydrogen/DeuteriumExchange Mass Spectrometry
Nguyen Minh Duc,1 Yang Du,2 Thor S. Thorsen,2 Su Youn Lee,1 Cheng Zhang,2,3
Hideaki Kato,2 Brian K. Kobilka,2 Ka Young Chung1
1School of Pharmacy, Sungkyunkwan University, 2066 Seobu-ro, Jangan-gu, Suwon, 440740, South Korea2Department of Molecular and Cellular Physiology, Stanford University Medical School, 297 Campus Drive, Beckman Center,Stanford, CA 94305, USA3Present Address: Department of Pharmacology and Chemical Biology, University of Pittsburgh School of Medicine, E 1358Thomas E. Starzl Biomedical Science Tower, 200 Lothrop St., Pittsburgh, PA 15213, USA
Abstract. G protein-coupled receptors (GPCRs) have important roles in physiologyand pathology, and 40% of drugs currently on the market target GPCRs for thetreatment of various diseases. Because of their therapeutic importance, the structuralmechanism of GPCR signaling is of great interest in the field of drug discovery.Hydrogen/deuterium exchange mass spectrometry (HDX-MS) is a useful tool foranalyzing ligand binding sites, the protein–protein interaction interface, and confor-mational changes of proteins. However, its application toGPCRs has been limited forvarious reasons, including the hydrophobic nature of GPCRs and the use of deter-gents in their preparation. In the present study, we tested the application of bicelles asa means of solubilizing GPCRs for HDX-MS studies. GPCRs (e.g., β2-adrenergic
receptor [β2AR], μ-opioid receptor, and protease-activated receptor 1) solubilized in bicelles produced bettersequence coverage (greater than 90%) than GPCRs solubilized in n-dodecyl-β-D-maltopyranoside (DDM),suggesting that bicelles are a more effective method of solubilization for HDX-MS studies. The HDX-MS profileof β2AR in bicelles showed that transmembrane domains (TMs) undergo lower deuterium uptake than intracel-lular or extracellular regions, which is consistent with the fact that the TMs are highly ordered and embedded inbicelles. The overall HDX-MS profiles of β2AR solubilized in bicelles and in DDM were similar except forintracellular loop 3. Interestingly, we detected EX1 kinetics, an important phenomenon in protein dynamics, atthe C-terminus of TM6 in β2AR. In conclusion, we suggest the application of bicelles as a useful method forsolubilizing GPCRs for conformational analysis by HDX-MS.Keywords: GPCR, HDX-MS, Bicelles, Detergent, Conformation, EX1
Received: 16 September 2014/Revised: 15 January 2015/Accepted: 16 January 2015/Published Online: 5 March 2015
Introduction
G-protein coupled receptors (GPCRs) are the most impor-tant class of membrane receptors with over 800 identified
to date in humans, many of which are involved in diseases,
such as oncologic, psychiatric, metabolic, neurodegenerative,cardiovascular, and infectious diseases [1, 2]. Approximately40%–50% of drugs circulating in the market target GPCRs forthe treatment of various diseases [2]. Ligand binding inducesconformational changes of GPCRs, which in turn regulateinteractions with downstream signaling molecules, such asheterotrimeric G-proteins or β-arrestins [3, 4]. Understandingthe conformational changes that are induced in GPCRs uponactivation or inactivation would greatly advance our under-standing of the mechanism of activation and inactivation in-duced by endogenous or synthetic ligands, and might ultimate-ly lead to the design of more effective and less toxic drugs.
Nguyen Minh Duc and Yang Du contributed equally to this work.
Electronic supplementary material The online version of this article(doi:10.1007/s13361-015-1083-4) contains supplementary material, which isavailable to authorized users.
Correspondence to: Brian Kobilka; e-mail: [email protected], Ka Chung;e-mail: [email protected]

Hence, enormous efforts have been invested in the charac-terization of the structure of GPCRs. X-ray crystallography andNMR spectroscopy are standard techniques for obtaining high-resolution structures of proteins. Recent breakthroughs inobtaining high-resolution X-ray crystal structures of GPCRsprovide the most comprehensive insights into the unique func-tional properties of these receptors in both inactive and activestates [5]. Although X-ray crystallography gives an importantthree-dimensional overview of structure, it has certain limita-tions. The X-ray crystal structure represents a single staticconformational state, giving little information about conforma-tional changes or dynamics. Another major limitation is thecrystallization process for GPCR engineering, which requires alot of effort and time, and selection of a ligand and detergent [5,6]. Additionally, the introduction of nonfunctional insertions,truncations, or point mutations into native GPCRs might affectthe endogenous conformation. More importantly, the condi-tions under which proteins function are generally not compat-ible with the conditions required for X-ray diffraction. NMRhas restrictions associated with protein size and sample prepa-ration, such as expression and isotope labeling of proteins [7],and the application of NMR to structural studies of GPCRs iscurrently very limited. Therefore, other techniques are neededin order to study the conformation of GPCRs.
Hydrogen/deuterium exchange mass spectrometry (HDX-MS) measures the exchange rates of peptide amide hydrogenwith deuterium in the solvent. In folded proteins, the exchangerate varies depending on the conformation of the proteins [8, 9];exposed or highly dynamic regions show rapid exchange rateswhereas excluded and rigid regions show slow exchange rates [8,9]. Thus, HDX-MS has been successfully used to study confor-mational changes [10, 11], the protein–protein interaction inter-face, protein–small molecule interaction sites, and protein fold-ing [12, 13]. Previously, the Griffin group presented methodol-ogy for HDX-MS analysis of β2-adrenergic receptor (β2AR) [14]and analyzed ligand-dependent perturbation of the conformation-al ensemble of β2AR by HDX-MS [15]. Their study showedapproximately 71% sequence coverage but the transmembrane(TM) regions were mostly not covered [14, 15]. Other studiesanalyzed the conformational changes of GPCR-interacting mol-ecules (e.g., G proteins and β-arrestin) upon binding to GPCRsby HDX-MS, but conformational information on the GPCRsthemselves is limited [16–18]. The low sequence coverage in theTM regions in the study by the Griffin group and the even lowersequence coverage when analyzed with GPCR-interacting mol-ecules might reflect the technical challenges associated withstudying membrane proteins by mass spectrometry.
GPCRs are insoluble and unstable membrane proteins, andthe use of detergents is obviously required for their extractionand purification from cell systems. The detergents are used notonly to solubilize and stabilize GPCRs but also to keep thepurified GPCRs in functionally folded states in the absence ofa phospholipid membrane. Among many commercially avail-able detergents, n-dodecyl-β-D-maltopyranoside (DDM) is themost widely used for GPCR studies (Figure 1b). A few studieshave analyzed detergent-solubilized GPCRs by mass
spectrometry-based approaches and achieved fairly good se-quence coverage [19–21]. However, the LC-MS conditions ofthese studies are different from those of HDX-MS; for HDX-MS, the separation by LC should be performed at low temper-ature (near 0°C) over a short period of time (less than 10 min).As discussed above, when the Griffin group used DDM forHDX-MS analysis of β2AR, they could not get good sequencecoverage in the TM regions [14, 15].
In addition to conventional detergents, various other ap-proaches to the solubilization and stabilization of GPCRs havebeen investigated [22]. Among those, nanodiscs have beentested for HDX-MS compatibility [23, 24]. Nanodiscs arecomposed of scaffold protein-assisted phospholipid bilayers,which provide more physiological conditions than detergentmicelles [23]. γ-Glutamyl carboxylase, a membrane protein,was reconstituted in nanodiscs and analyzed by HDX-MS withonly approximately 45% sequence coverage overall and nocoverage of most TM regions [23, 24]. Therefore, it is neces-sary to develop a new methodology that can provide reliablesequence coverage for HDX-MS studies of membrane pro-teins, including GPCRs.
Bicelles (bilayered micelles) are lipid-detergent assemblieswith discrete bilayer fragments that are edge-stabilized bycertain detergents (Figure 1a) [25]. They provide a lipid-richmedium that mimics the natural phospholipid bilayer environ-ment and is compatible with structural studies of membraneproteins [26]. Of note, bicelles express the attractive character-istics of both micellar and lipid bilayer systems in structuralstudies of membranous biomolecules [26, 27]. Because ofthese attractive features, a number of studies have used bicellesto maintain functional membrane proteins in EPR [28] andNMR [28–30] studies, as well as X-ray crystallography [31].More recently, there have been several reports of the use ofbicelles to study membrane proteins by mass spectrometry [32,33]. However, to our knowledge, there is no published HDX-MS study using bicelles. In the present study, we tested thefeasibility of using bicelles for structural analysis of GPCRs byHDX-MS. Three GPCRs, β2AR, μ-opioid receptor (μOR), andprotease-activated receptor 1 (PAR-1), were used as GPCRmodels and the HDX-MS profiles were compared betweenβ2AR reconstituted in bicelles and in DDM.
MethodsExpression and Purification of β2AR
Recombinant baculovirus was prepared using the Bestbac ex-pression system (Expression Systems Inc., Davis, CA, USA)with pVL1392 as a vector, and β2AR was extracted and puri-fied as previously described [34]. Briefly, cell pellets werelysed by incubation in lysis buffer (20 mM HEPES pH 7.5,5 mM EDTA, 1 μM alprenolol, 2.5 mg/mL leupeptin, 160 mg/mL benzamindine) with stirring for 20 min. The receptorswere extracted from the cell membrane by dounce ho-mogenization in solubilization buffer (20 mM HEPES,pH 7.5, 100 mM NaCl, 1% DDM, 1 μM alprenolol,
N. M. Duc et al.: Application of Bicelles for GPCR HDX-MS 809

2.5 mg/mL leupeptin, 160 mg/mL benzamindine) for 1 h atroom temperature. After clarification by high-speed centrifu-gation at 18,000×g for 30 min, receptors bearing the N-terminal FLAG epitope were captured by M1 antibody affinitychromatography. The column was extensively washed withHMS-CHS buffer (20 mM HEPES pH 7.5, 350 mM NaCl,0.1% DDM, 0.01% cholesterol hemisuccinate [CHS], 2 mMCaCl2), and the receptors were eluted with HMS-CHS buffersupplemented with 5 mM EDTA and 200 μg/mL free FLAGpeptide. The eluted receptors were further purified by affinitychromatography using alprenolol-Sepharose as previously de-scribed [35] to select functional receptors. Size-exclusion chro-matography (SEC) with Superdex-200 column (GEHealthcare, Pittsburgh, PA, USA) equilibrated in HMS-CHS
buffer was finally used to clean up the receptor. The receptorwas concentrated to 125 μMandmixed with different ligands ifneeded. The purity of the sample was higher than 95% asassessed by SDS-PAGE.
Expression and Purification of μOR
Expression and purification of the construct was performedessentially as described by Manglik et al. [36]. Briefly, theconstruct used was the WT receptor from residue 6 fused toan N-terminal signal FLAG-tag and a C-terminal 6-histidineHis tag. The construct was expressed in sf9 cells.Harvested cells were lysed in lysis buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA, 10 μM naloxone). The cell
Fig. 1. Sequence coverage of β2AR. Illustration of β2AR in bicelle (a) and DDM (b). Sequence coverage of β2ARprepared in bicelles (c) and DDM (d). Blue bars represent the peptic peptides identified in this study. The figures arerepresentative of four independent experiments
810 N. M. Duc et al.: Application of Bicelles for GPCR HDX-MS

pellet was solubilized in solubilization buffer (20 mMHEPES pH 7.5, 0.5 M NaCl, 0.5% DDM, 0.3% 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulphonate,0.03% CHS, 30% v/v glycerol, 10 μM naloxone). Nickel-NTA agarose was added and the mixture was stirred for 1 h at4°C. The beads were recovered by centrifugation and washed inbatches with nickel wash buffer, and then the receptors wereeluted in wash buffer supplemented with 250 mM imidazole.The receptors were further purified by loading onto a M1antibody column. The column was washed with wash bufferand eluted with elution buffer supplemented with 5 mM EDTAand 0.2mg/mL FLAGpeptide. Finally, SECwith Superdex-200column (GE Healthcare) equilibrated in HMS-CHS buffer wasused to clean up the receptor.
Expression and Purification of PAR-1
The human PAR-1 construct was prepared as described previ-ously with slight modification [37]. Briefly, the construct wasexpressed in Sf9 cells at 27°C for 48 h before harvest. To purifythe receptor, infected cells were lysed by osmotic shock inlow-salt buffer containing 10 mM Tris-HCl, pH 7.5, 1 mMEDTA, 100 nM vorapaxar derivative, and 100 μM Tris(2-carboxyethyl)phosphine hydrochloride (TCEP). The vorapaxarderivative was generated by reduction of the non-aromaticcarbon–carbon double bond of vorapaxar and showed a muchfaster dissociation rate than vorapaxar in cell-based assays. Theprotein was further extracted from cell membranes with abuffer containing 20 mM Hepes, pH 7.5, 500 mM NaCl, 1%DDM, 0.03% CHS, 0.2% sodium cholate, 15% glycerol, 100nM vorapaxar derivative, and 100 μM TCEP. Cell debris wasremoved by high-speed centrifugation. From this point on,1 μM vorapaxar derivative was added to all of the buffers usedfor purification except for the buffer used in size exclusionchromatography. Nickel-NTA agarose resin was added to thesupernatant after homogenization and stirred for 1 h at 4°C.The resin was then washed three times in batches with buffercontaining 20 mM HEPES, pH 7.5, 500 mM NaCl, 0.1%DDM, 0.02% CHS, and 1 μM vorapaxar derivative, and trans-ferred to a glass column. The bound receptor was eluted withbuffer containing 300 mM imidazole and loaded onto an anti-Flag M1 affinity column. After extensive washing with buffercontaining 20 mM HEPES, pH 7.5, 500 mM NaCl, 0.1%DDM, 0.02% CHS, 1 μM vorapaxar derivative, and 2 mMCa2+, the receptor was eluted from M1 resin using the samebuffer without Ca2+ but containing 200 μg/mL FLAG peptideand 5 mM EDTA. Size exclusion chromatography was used toobtain the final monodisperse receptor preparation. The run-ning buffer contained 20 mM HEPES, pH 7.5, 100 mM NaCl,0.1% DDM, and 0.02% CHS. The flow rate was set at 0.2 mL/min to give enough time for the vorapaxar derivative to disso-ciate from the receptor.
Reconstitution of Receptors into Bicelles
A 10% bicelle stock was prepared with a 3:1 molar ratio ofDMPC and CHAPSO. One aliquot of the bicelles was thawed
at room temperature until the phase changed to a clear gel andthen transferred to ice to liquefy. The aliquot was vortexedbriefly to reestablish a homogenous phase and placed back onice. One microliter of 10% bicelle stock was added to 9 μLreceptor to give a final 1% working concentration of bicelles.The protein–bicelle mixture was gently pipetted up and downuntil the solution became clear and homogenous. The mixturewas incubated on ice for approximately 30 min to allow com-plete reconstitution of the receptor into bicelles, and was kepton ice until preparation for HDX-MS studies.
Hydrogen/Deuterium Exchange and MassSpectrometry
The purified proteins were prepared at a concentration of 125pmol/μL. Hydrogen/deuterium exchange was initiated bymixing 2 μL of protein with 28 μL of D2O buffer (20 mMHEPES, pH 7.4, 100 mM NaCl, and 0.1% DDM or bicelles inD2O), and the mixtures were incubated for various time periods(10, 100, 1000, and 10,000 s) on ice. At the indicated timepoints, the mixtures were quenched by addition of 30 μL of ice-cold quench buffer (100 mM KH2PO4, 20 mM TCEP,pH 2.01) and immediately placed on dry ice to freeze. Fornon-deuterated samples, 2 μL of purified protein was mixedwith 28 μL of H2O buffer (20 mM HEPES, pH 7.4, 100 mMNaCl, and 0.1% DDM or bicelles in H2O), and 30 μL of ice-cold quench buffer was added. The quenched samples weredigested online by passing through an immobilized pepsincolumn (2.1×30 mm) (Life Technologies, Carlsbad, CA,USA) at a flow rate of 100 μL/min with 0.05% formic acid inH2O at 10°C. Peptide fragments were collected on a C18VanGuard trap column (1.7 μm×30 mm) (Waters, Milford,MA, USA) for desalting with 0.05% formic acid in H2O andthen separated by ultra-pressure liquid chromatography usingan Acquity UPLC C18 column (1.7 μm, 1.0×100 mm)(Waters) at a flow rate of 40 μL/min with an acetonitrilegradient using two pumps, starting with 8% B and increasingto 85% B over the next 8.5 min. The mobile phase A was 0.1%formic acid in H2O, and solvent B was acetonitrile containing0.1% formic acid. To minimize back-exchange of deuterium tohydrogen, the sample, solvents, trap, and UPLC column weremaintained at pH 2.5 and 0.5°C during analysis. Mass spectralanalyses were performed with a Xevo G2 Qtof equipped with astandard ESI source in MSE mode (Waters). The mass spectrawere acquired in the range of m/z 100–2000 for 10 min in thepositive ion mode.
Peptide Identification and HDX-MS DataProcessing
Peptic peptides in non-deuterated samples were identified withProteinLynx Global Server 2.4 (Waters). Searches were runwith variable methionine oxidation modification, and the pep-tides were filtered on a peptide score of 6. To process the HDX-MS data, the amount of deuterium in each peptide was deter-mined by measuring the centroid of the isotopic distributionusing DynamX software (Waters). EX1 kinetics were
N. M. Duc et al.: Application of Bicelles for GPCR HDX-MS 811

determined by visual inspection of the distribution of the twoisotopes. All experiments were conducted in at least triplicate.
Statistical Analysis
Results were expressed as means±S.E.M. Statistical analysiswas performed using Graph Prism 5.0 software. Statisticalsignificance of differences between bicelles and DDM wasdetermined by Student's t-test. Differences between data wereconsidered statistically significant at PPG0.05.
ResultsSuccessful HDX-MS analysis is often determined by the se-quence coverage derived from the identified peptic peptides ofnon-deuterated samples. It is, therefore, necessary to optimizebuffers, digestion conditions, and LC-MS conditions to achievethe highest sequence coverage. We tested quench buffer(100 mM KH2PO4, pH 2.01) supplemented with various con-centrations of denaturant (guanidine-HCl, 0.0–4.0 M) and re-ductant (TCEP, 0.01–0.2 M). Addition of 20 mM TCEP wasoptimal but addition of guanidine-HCl with 20 mM TCEP hada deleterious effect, resulting in an average 23% decrease insequence coverage compared with 20 mM TCEP alone (datanot shown). Based on this result, we selected quench buffercomposed of 100 mM KH2PO4, pH 2.01, and 20 mM TCEP.
The feasibility of using bicelles for mass spectrometry wasinvestigated and compared with the use of DDM. Sequencecoverage was analyzed for the selected GPCRs (β2AR, μOR,and PAR-1) prepared in either bicelles or DDM. High redun-dancy is also an important parameter because we can use morepeptides for the analysis of a single region, thus increasing ourconfidence in evaluation of the deuterium uptake level of thisregion. All three GPCRs showed improved sequence coveragewith higher redundancy when prepared in bicelles than inDDM (average sequence coverage of 93.6% versus 79.5%and average redundancy of 4.87 versus 3.28; Table 1,Figure 1, Supplementary Figure 1, and SupplementaryFigure 2). Interestingly, worse sequence coverage (64.43±0.44% with redundancy of 2.02±0.07) was obtained whenβ2AR was solubilized in maltose neopentyl glycol-3(MNG-3), a detergent that was recently introduced forGPCR structural studies [38] (Supplementary Figure 3).
The sequence coverage maps of β2AR are shown inFigure 1c and d, and the sequence coverage maps of μORand PAR-1 are provided in Supplementary Figure 1 andSupplementary Figure 2. Hydrophobic peptides are generallymore difficult to be analyzed by mass spectrometry thanhydrophilic peptides [39]. As expected, the intracellularor extracellular regions are well covered, whereas thetransmembrane (TM) regions are poorly covered for bothbicelle and DDM samples (Figure 1, Supplementary Figure 1,and Supplementary Figure 2). Interestingly, bicelle-preparedsamples yielded increased identification for peptic peptides inboth intracellular/extracellular regions and TM regions and,therefore, showed enhanced sequence coverage in bothintracellular/extracellular and TM regions compared toDDM-prepared samples (Figure 1, Supplementary Figure 1,and Supplementary Figure 2).
To test the compatibility of bicelles and DDM for HDX-MS, we analyzed the HDX profile of all three GPCRs preparedin bicelles and in DDM (Figure 2). The average standarddeviation of each deuterium exchange time point was 2.2%with a range of 0.1% to 5.3%. The thermodynamic stability ofall GPCRs in bicelles was not significantly different from thatin DDM, and the stability of GPCRs over time of D2O bufferincubation was tested and showed no significant change inGPCR stability (Supplementary Figure 4). As shown inFigure 2, Supplementary Figure 5, and SupplementaryFigure 6, the TM regions showed lower deuterium uptake thanintracellular or extracellular regions for both bicelle- andDDM-prepared GPCRs. This is expected because the TMregions are highly ordered and relatively protected by bicellesor DDM. These results suggest that both bicelles and DDM arecompatible with HDX-MS. However, because bicellesgave better sequence coverage than DDM, we were ableto obtain HDX-MS information for more regions fromGPCRs prepared in bicelles than from those prepared inDDM (Figure 2). In particular, we could get new infor-mation from TM regions (e.g., TM1, TM3, and TM7 ofβ2AR) in only the bicelle samples. These results suggest thatbicelles are a better solubilization method than DDM for HDX-MS analysis of GPCRs.
As bicelles appeared to be a good system for HDX-MSanalysis of GPCRs, we next compared the conformation ofβ2AR prepared in bicelles with that in DDM. The HDX profile
Table 1. Comparison of Sequence Coverage Between GPCRs Prepared in Bicelles or DDM
β2AR μOR PAR-1
Bicelles DDM Bicelles DDM Bicelles DDM
Sequence coverage (%) 92.77±1.21 74.87±1.34a 94.4±1.82 78.6±3.10b 93.67±3.39 85.23±3.60c
Number of peptic peptides 116±9.71 62.33±3.93a 152.67±17.57 76.67±9.94b 89.67±7.42 69±11.53Redundancy* 4.27±0.11 3.66±0.187a 6.67±1.26 3.36±0.39c 3.28±0.24 2.81±0.23
*Number of peptides covering the same amino acids.aPG0.001.bPG0.01.cPG0.05 compared to bicelles.
812 N. M. Duc et al.: Application of Bicelles for GPCR HDX-MS

is dependent on the peptic peptides used for the analysis, andanalyzing the same region with different peptides could resultin slightly different HDX-MS profiles even though the sampleswere in the same conformation. Bicelle- and DDM-preparedsamples produce dissimilar peptic peptide patterns (Figure 1,Supplementary Figure 1, and Supplementary Figure 2). For thisreason, it is not possible to directly compare the HDX-MSprofile of bicelle- and DDM-prepared samples. For example,in Figure 3, TM5 and TM6 of β2AR show different deuteriumuptake levels in bicelles compared with DDM; this mightreflect either the different conformation or the effect of usingdifferent peptides. Therefore, we chose peptides commonlyfound in both bicelle and DDM samples for side-by-side com-parison. New heat maps were generated using these common
peptides (Figure 3a, Supplementary Figure 5c, andSupplementary Figure 6c), and differences in deuterium uptakeof β2AR between bicelle and DDM samples at the 1000 s timepoint are illustrated in the snake map (Figure 3b). The deuteri-um uptake levels were not statistically different betweenbicelle- and DDM-prepared samples for μOR and PAR-1(Supplementary Figure 5c and Supplementary Figure 6c). Forβ2AR, most regions showed statistically similar deuteriumuptake except for ICL3, which showed slightly, but statisticallysignificant, lower deuterium uptake in bicelles than in DDM(Figure 3a and b red boxes, and 3c). These results indicate thatthe conformation of β2AR is similar between bicelle- andDDM-prepared samples in most regions, but ICL3 is moredynamic in DDM.
Fig. 2. Deuterium uptake profile of β2AR. Heat map of β2AR prepared in bicelles (a) and DDM (b). Incorporation of deuterium at 10,100, 1000, and 10,000 s is indicated by color-coded blocks underlining the amino acid sequence. The color legend shows deuteriumuptake levels. Snake maps showing deuterium uptake at 1000 s for β2AR prepared in bicelles (c) and DDM (d). The figures arerepresentative of four independent experiments
N. M. Duc et al.: Application of Bicelles for GPCR HDX-MS 813

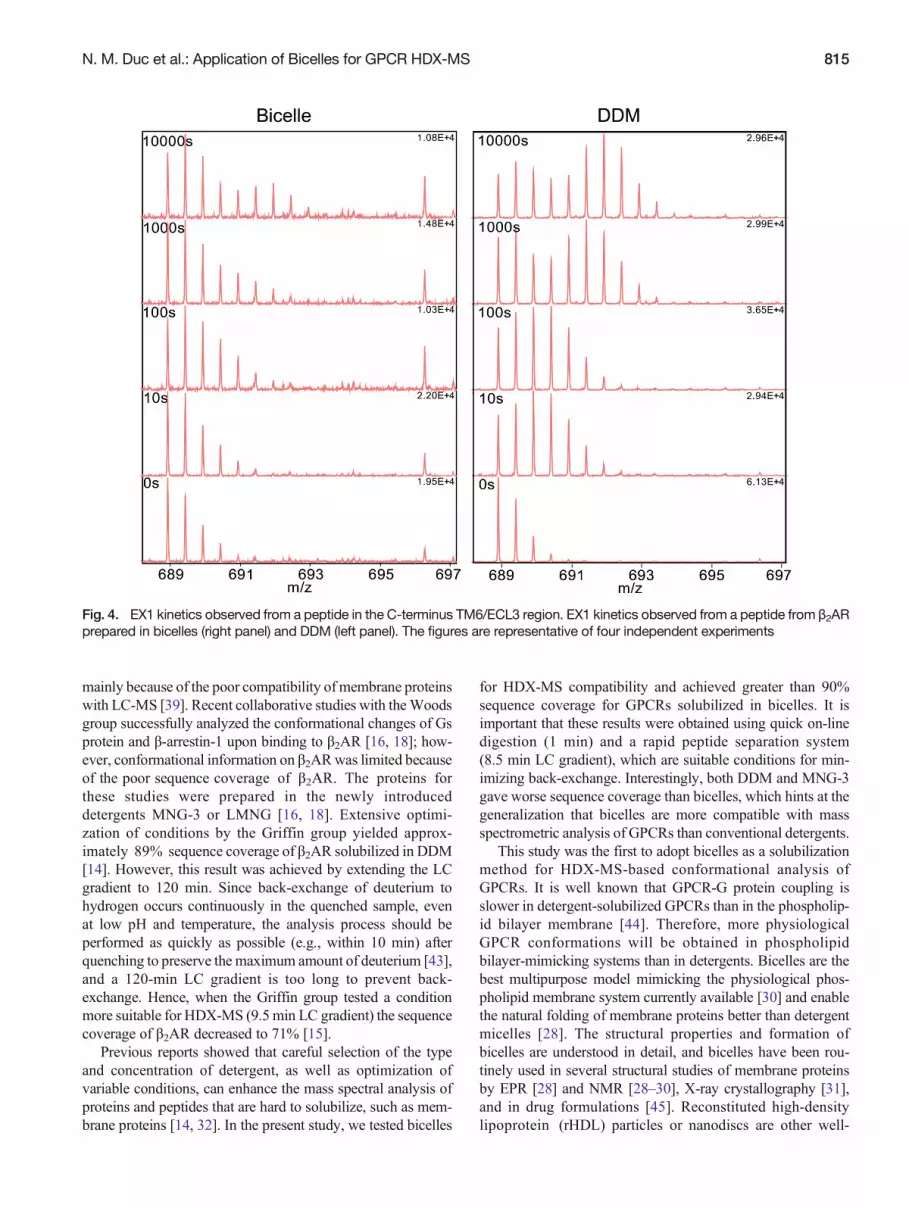
The kinetics (i.e., EX1 and EX2 kinetics) of the HDXprofile provide information on the folding status of aprotein [40]. EX2 kinetics occur when the rate of HDXis much slower than the rate of protein unfolding, andmight represent local conformational fluctuations close tothe surface of the protein [41]. In contrast, EX1 kineticsoccur when the rate of protein unfolding reaction is slowerthan the rate of HDX, which might represent the globalunfolding/refolding of proteins [41, 42]. In physiologicalconditions, EX1 kinetics are rare but very interesting be-cause they help us understand the dynamics of a protein[42]. We did not find EX1 kinetics in μOR or PAR-1.Interestingly, we identified intermediate EX1 and EX2 kinetics(i.e., the existence of both bimodal isotopic distribution [EX1]and time-dependent mass shifts [EX2]) from a peptide inthe C-terminus of TM6/ECL3 (IVNIVHVIQDNL) ofβ2AR prepared both in bicelles (Figure 4, left) and DDM(Figure 4, right). The ratio between folded and unfolded stateswas not statistically different in bicelle samples compared withDDM samples.
DiscussionIn this study, we could achieve greater than 90% sequencecoverage of GPCRs in a HDX-MS experimental setup bysolubilizing GPCRs with bicelles. Importantly, this was ob-served for three unrelated GPCRs—PAR-1, μOR, andβ2AR—which gave 93.7%, 94.4%, and 92.7% sequence cov-erage respectively. Of note, not only the sequence coverage butalso the redundancy covering an amino acid improved signif-icantly with bicelles. Redundancy is an important parameterbecause a greater number of peptides covering a single regionwill provide a higher quality and more reliable HDX-MSprofile. Good sequence coverage and increased redundancyled to successful analysis of GPCRs by HDX-MS, indicatingthat bicelles are a good solubilization method for HDX-MSstudies on GPCRs.
With recent developments in instrumentation and technolo-gy, HDX-MS has become a powerful technique for studyingthe dynamic structures of various proteins. However, analysisof membrane proteins such as GPCRs remains challenging,
Fig. 3. Comparison of deuterium uptake profile of β2AR prepared in bicelles andDDM.(a) Heatmap of β2AR prepared in bicelles andDDManalyzed from common peptides. Incorporation of deuterium at 10, 100, 1000, and 10,000 s is indicated by color-coded blocksunderlining the amino acid sequence. The color legend shows deuterium uptake levels. (b) Snake maps showing the difference indeuterium uptake between β2AR prepared in bicelles and DDM. The color legend shows the difference in deuterium uptake levels,where blue indicates less deuterium uptake and red indicates more deuterium uptake in DDM than in bicelles samples. Regions thatshow differences between bicelles and DDM samples are highlighted with red boxes. (c) Deuterium uptake plot. Error bars arestandard error of four independent experiments. * PG0.05
814 N. M. Duc et al.: Application of Bicelles for GPCR HDX-MS
mainly because of the poor compatibility of membrane proteinswith LC-MS [39]. Recent collaborative studies with theWoodsgroup successfully analyzed the conformational changes of Gsprotein and β-arrestin-1 upon binding to β2AR [16, 18]; how-ever, conformational information on β2ARwas limited becauseof the poor sequence coverage of β2AR. The proteins forthese studies were prepared in the newly introduceddetergents MNG-3 or LMNG [16, 18]. Extensive optimi-zation of conditions by the Griffin group yielded approx-imately 89% sequence coverage of β2AR solubilized in DDM[14]. However, this result was achieved by extending the LCgradient to 120 min. Since back-exchange of deuterium tohydrogen occurs continuously in the quenched sample, evenat low pH and temperature, the analysis process should beperformed as quickly as possible (e.g., within 10 min) afterquenching to preserve the maximum amount of deuterium [43],and a 120-min LC gradient is too long to prevent back-exchange. Hence, when the Griffin group tested a conditionmore suitable for HDX-MS (9.5min LC gradient) the sequencecoverage of β2AR decreased to 71% [15].
Previous reports showed that careful selection of the typeand concentration of detergent, as well as optimization ofvariable conditions, can enhance the mass spectral analysis ofproteins and peptides that are hard to solubilize, such as mem-brane proteins [14, 32]. In the present study, we tested bicelles
for HDX-MS compatibility and achieved greater than 90%sequence coverage for GPCRs solubilized in bicelles. It isimportant that these results were obtained using quick on-linedigestion (1 min) and a rapid peptide separation system(8.5 min LC gradient), which are suitable conditions for min-imizing back-exchange. Interestingly, both DDM and MNG-3gave worse sequence coverage than bicelles, which hints at thegeneralization that bicelles are more compatible with massspectrometric analysis of GPCRs than conventional detergents.
This study was the first to adopt bicelles as a solubilizationmethod for HDX-MS-based conformational analysis ofGPCRs. It is well known that GPCR-G protein coupling isslower in detergent-solubilized GPCRs than in the phospholip-id bilayer membrane [44]. Therefore, more physiologicalGPCR conformations will be obtained in phospholipidbilayer-mimicking systems than in detergents. Bicelles are thebest multipurpose model mimicking the physiological phos-pholipid membrane system currently available [30] and enablethe natural folding of membrane proteins better than detergentmicelles [28]. The structural properties and formation ofbicelles are understood in detail, and bicelles have been rou-tinely used in several structural studies of membrane proteinsby EPR [28] and NMR [28–30], X-ray crystallography [31],and in drug formulations [45]. Reconstituted high-densitylipoprotein (rHDL) particles or nanodiscs are other well-
Fig. 4. EX1 kinetics observed from a peptide in the C-terminus TM6/ECL3 region. EX1 kinetics observed from a peptide from β2ARprepared in bicelles (right panel) and DDM (left panel). The figures are representative of four independent experiments
N. M. Duc et al.: Application of Bicelles for GPCR HDX-MS 815

characterized GPCR solubilization methods that mimic thephospholipid bilayer [46]. However, certain properties ofrHDL particles make them less attractive than bicelles. Thereconstitution process of rHDL particles is more complex thanthat of bicelles, and rHDL particles use a dimer of ApoA-I as ascaffolding protein to surround the lipid bilayer, which mightadd complexity to the HDX-MS data analysis [46]. The prop-erties of bicelles make them well suited for investigating theconformation of GPCRs by HDX-MS.
The HDX-MS profile of all three GPCRs tested showedhigher deuterium uptake in intracellular and extracellular re-gions and lower deuterium uptake in TM regions, which cor-relates well with the overall seven-transmembrane structure ofGPCRs [14]. Interestingly, we observed EX1 kinetics atthe C-terminus of the TM6/ECL3 region of β2AR but notμOR or PAR-1, suggesting that this region in β2AR slowlyexchanges the folded and unfolded conformations. To ourknowledge, this is the first report of EX1 kinetics in GPCRs.TM6 undergoes outward movement upon GPCR activationthat leads to G protein coupling and plays an important rolein GPCR oligomerization [34, 47–50]. The importance of EX1kinetics for β2AR function will be an interesting field toexplore.
ConclusionThe present study suggests that bicelles are a better GPCRsolubilization tool for HDX-MS studies than conventionaldetergents such as DDM or MNG-3. It will be interesting toinvestigate why bicelles are more compatible with HDX-MSthan detergents such as DDM or MNG-3. Elucidation of theunderlyingmechanismmight lead to the development of HDX-MS-compatible detergents for GPCRs and other membraneproteins.
AcknowledgmentsThe authors acknowledge support for this work by the BasicScience Research Program through the National ResearchFoundation of Korea funded by the Ministry of Education,Science, and Technology (2012R1A1A1039220) and un-der the framework of an international cooperation pro-gram managed by the National Research Foundation ofKorea (2012K2A1A2031900).
References1. Lundstrom, K.: An overview on GPCRs and drug discovery: structure-
based drug design and structural biology on GPCRs. Methods Mol. Biol.552, 51–66 (2009)
2. Lagerström, M.C., Schiöth, H.B.: Structural diversity of G protein-coupledreceptors and significance for drug discovery. Nat. Rev. Drug Discov. 7(4),339–357 (2008)
3. Rajagopal, S., Rajagopal, K., Lefkowitz, R.J.: Teaching old receptors newtricks: biasing seven-transmembrane receptors. Nat. Rev. Drug Discov.9(5), 373–386 (2010)
4. Kahsai, A.W., Xiao, K., Rajagopal, S., Ahn, S., Shukla, A.K., Sun, J., Oas,T.G., Lefkowitz, R.J.: Multiple ligand-specific conformations of the β2-adrenergic receptor. Nat. Chem. Biol. 7(10), 692–700 (2011)
5. Venkatakrishnan, A.J., Deupi, X., Lebon, G., Tate, C.G., Schertler, G.F.,Babu, M.M.: Molecular signatures of G-protein-coupled receptors. Nature494, 185–194 (2013)
6. Kobilka, B.: The structural basis of g-protein-coupled receptor signaling(Nobel Lecture). Angew. Chem. Int. Ed. 52(25), 6380–6388 (2013)
7. Granier, S., Kobilka, B.: A new era of GPCR structural and chemicalbiology. Nat. Chem. Biol. 8(8), 670–673 (2012)
8. Percy, A.J., Rey, M., Burns, K.M., Schriemer, D.C.: Probing proteininteractions with hydrogen/deuterium exchange and mass spectrometry—areview. Anal. Chim. Acta 721, 7–21 (2012)
9. Marcsisin, S.R., Engen, J.R.: Hydrogen exchange mass spectrometry: whatis it and what can it tell us? Anal. Bioanal. Chem. 397(3), 967–972 (2010)
10. Resing, K.A., Ahn, N.G.: Deuterium exchange mass spectrometry as aprobe of protein kinase activation. Analysis of wild-type and constitutivelyactive mutants of MAP kinase kinase-1. Biochemistry 37(2), 463–475(1998)
11. Engen, J.R., Smithgall, T.E., Gmeiner, W.H., Smith, D.L.: Comparison ofSH3 and SH2 domain dynamics when expressed alone or in an SH(3+2)construct: the role of protein dynamics in functional regulation. J. Mol.Biol. 287(3), 645–656 (1999)
12. Heidary, D.K., Gross, L.A., Roy, M., Jennings, P.A.: Evidence for anobligatory intermediate in the folding of interleukin-1 beta. Nat. Struct.Biol. 4(9), 725–731 (1997)
13. Yang, H., Smith, D.L.: Kinetics of cytochrome c folding examined byhydrogen exchange and mass spectrometry. Biochemistry 36(48), 14992–14999 (1997)
14. Zhang, X., Chien, E.Y., Chalmers, M.J., Pascal, B.D., Gatchalian, J.,Stevens, R.C., Griffin, P.R.: Dynamics of the β2 -adrenergic G-proteincoupled receptor revealed by hydrogen-deuterium exchange. Anal. Chem.82(3), 1100–1108 (2010)
15. West, G.M., Chien, E.Y., Katritch, V., Gatchalian, J., Chalmers, M.J.,Stevens, R.C., Griffin, P.R.: Ligand-dependent perturbation of the confor-mational ensemble for the GPCR β2 adrenergic receptor revealed by HDX.Structure 19(10), 1424–1432 (2011)
16. Chung, K.Y., Rasmussen, S.G.F., Liu, T., Li, S., DeVree, B.T., Chae, P.S.,Calinski, D., Kobilka, B.K., Woods, V.L., Sunahara, R.K.: Conformationalchanges in the G protein Gs induced by the β2 adrenergic receptor. Nature477(7366), 611–615 (2011)
17. Orban, T., Jastrzebska, B., Gupta, S.,Wang, B., Miyagi, M., Chance, M.R.,Palczewski, K.: Conformational dynamics of activation for the pentamericcomplex of dimeric G protein-coupled receptor and heterotrimeric G pro-tein. Structure 20(5), 826–840 (2012)
18. Shukla, A.K., Westfield, G.H., Xiao, K., Reis, R.I., Huang, L.Y., Tripathi-Shukla, P., Qian, J., Li, S., Blanc, A., Oleskie, A.N., Dosey, A.M., Su, M.,Liang, C.R., Gu, L.L., Shan, J.M., Chen, X., Hanna, R., Choi, M., Yao,X.J., Klink, B.U., Kahsai, A.W., Sidhu, S.S., Koide, S., Penczek, P.A.,Kossiakoff, A.A.,Woods, V.L. Jr., Kobilka, B.K., Skiniotis, G., Lefkowitz,R.J.: Visualization of arrestin recruitment by a G-protein-coupled receptor.Nature 512(7513), 218–222 (2014)
19. Zvonok, N., Xu, W., Williams, J., Janero, D.R., Krishnan, S.C.,Makriyannis, A.: Mass spectrometry-based GPCR proteomics: compre-hensive characterization of the human cannabinoid 1 receptor. J.Proteome Res. 9(4), 1746–1753 (2010)
20. Kahsai, A.W., Xiao, K., Rajagopal, S., Ahn, S., Shukla, A.K., Sun, J., Oas,T.G., Lefkowitz, R.J.: Multiple ligand-specific conformations of the beta2-adrenergic receptor. Nat. Chem. Biol. 7(10), 692–700 (2011)
21. Angel, T.E., Gupta, S., Jastrzebska, B., Palczewski, K., Chance, M.R.:Structural waters define a functional channel mediating activation of theGPCR, rhodopsin. Proc. Natl. Acad. Sci. U. S. A. 106(34), 14367–14372(2009)
22. Zhao, Q., Wu, B.L.: Ice breaking in GPCR structural biology. ActaPharmacol. Sin. 33(3), 324–334 (2012)
23. Hebling, C.M., Morgan, C.R., Stafford, D.W., Jorgenson, J.W., Rand,K.D., Engen, J.R.: Conformational analysis of membrane proteins in phos-pholipid bilayer nanodiscs by hydrogen exchangemass spectrometry. Anal.Chem. 82(13), 5415–5419 (2010)
24. Parker, C.H., Morgan, C.R., Rand, K.D., Engen, J.R., Jorgenson,J.W., Stafford, D.W.: A conformational investigation of propeptidebinding to the integral membrane protein gamma-glutamyl carbox-ylase using nanodisc hydrogen exchange mass spectrometry.Biochemistry 53(9), 1511–1520 (2014)
816 N. M. Duc et al.: Application of Bicelles for GPCR HDX-MS

25. Vold, R.R., Prosser, R.S., Deese, A.J.: Isotropic solutions of phospholipidbicelles: a new membrane mimetic for high-resolution NMR studies ofpolypeptides. J. Biomol. NMR. 9(3), 329–335 (1997)
26. Sanders, C.R., Prosser, R.S.: Bicelles: a model membrane system for allseasons? Structure 6(10), 1227–1234 (1998)
27. Czerski, L., Sanders, C.R.: Functionality of a membrane protein in bicelles.Anal. Biochem. 284, 327–333 (2000)
28. Dürr, U.H., Soong, R., Ramamoorthy, A.: When detergent meets bilayer:Birth and coming of age of lipid bicelles. Prog. Nucl. Magn. Reson.Spectrosc. 69, 1–22 (2013)
29. Park, S.H., Prytulla, S., De Angelis, A.A., Brown, J.M., Kiefer, H., Opella,S.J.: High-resolution NMR spectroscopy of a GPCR in aligned bicelles. J.Am. Chem. Soc. 128(23), 7402–7403 (2006)
30. Dürr, U.H., Gildenberg, M., Ramamoorthy, A.: The magic of bicelles lightsup membrane protein structure. Chem. Rev. 112, 6054–6074 (2012)
31. Ujwal, R., Bowie, J.U.: Crystallizing membrane proteins using lipidicbicelles. Methods 55, 337–341 (2011)
32. Hopper, J.T.S., Yu, Y.T.-C., Li, D., Raymond, A., Bostock, M., Liko, I.,Mikhailov, V., Laganowsky, A., Benesch, J.L.P., Caffrey, M., Nietlispach,D., Robinson, C.V.: Detergent-free mass spectrometry of membrane pro-tein complexes. Nat. Methods 10(12), 1206–1208 (2013)
33. Pan, Y., Brown, L., Konermann, L.: Kinetic folding mechanism of anintegral membrane protein examined by pulsed oxidative labeling andmassspectrometry. J. Mol. Biol. 410(1), 146–158 (2011)
34. Rasmussen, S.G., DeVree, B.T., Zou, Y., Kruse, A.C., Chung, K.Y.,Kobilka, T.S., Thian, F.S., Chae, P.S., Pardon, E., Calinski, D.,Mathiesen, J.M., Shah, S.T., Lyons, J.A., Caffrey, M., Gellman, S.H.,Steyaert, J., Skiniotis, G., Weis, W.I., Sunahara, R.K., Kobilka, B.K.:Crystal structure of the β2 adrenergic receptor-Gs protein complex.Nature 477(7366), 549–555 (2011)
35. Rosenbaum, D.M., Cherezov, V., Hanson, M.A., Rasmussen, S.G., Thian,F.S., Kobilka, T.S., Choi, H.J., Yao, X.J., Weis, W.I., Stevens, R.C.,Kobilka, B.K.: GPCR engineering yields high-resolution structural insightsinto beta2-adrenergic receptor function. Science 318(5854), 1266–1273(2007)
36. Manglik, A., Kruse, A.C., Kobilka, T.S., Thian, F.S., Mathiesen, J.M.,Sunahara, R.K., Pardo, L., Weis, W.I., Kobilka, B.K., Granier, S.: Crystalstructure of the μ-opioid receptor bound to a morphinan antagonist. Nature485(7398), 321–326 (2012)
37. Zhang, C., Srinivasan, Y., Arlow, D.H., Fung, J.J., Palmer, D., Zheng, Y.,Green, H.F., Pandey, A., Dror, R.O., Shaw, D.E., Weis, W.I., Coughlin,S.R., Kobilka, B.K.: High-resolution crystal structure of human protease-activated receptor 1. Nature 492(7429), 387–392 (2012)
38. Chae, P.S., Rasmussen, S.G., Rana, R.R., Gotfryd, K., Chandra, R., Goren,M.A., Kruse, A.C., Nurva, S., Loland, C.J., Pierre, Y., Drew, D., Popot,
J.L., Picot, D., Fox, B.G., Guan, L., Gether, U., Byrne, B., Kobilka, B.,Gellman, S.H.: Maltose-neopentyl glycol (MNG) amphiphiles for solubili-zation, stabilization and crystallization of membrane proteins. Nat. Methods7(12), 1003–1008 (2010)
39. Rabilloud, T.: Membrane proteins and proteomics: love is possible, but sodifficult. Electrophoresis 30(Suppl 1), S174–S180 (2009)
40. Raschke, T.M., Marqusee, S.: Hydrogen exchange studies of protein struc-ture. Curr. Opin. Biotechnol. 9(1), 80–86 (1998)
41. Konermann, L., Tong, X., Pan, Y.: Protein structure and dynamics studiedby mass spectrometry: H/D exchange, hydroxyl radical labeling, and relat-ed approaches. J. Mass Spectrom. 43(8), 1021–1036 (2008)
42. Weis, D.D., Wales, T.E., Engen, J.R., Hotchko, M., Lynn, F., Ten, E.:Identification and characterization of EX1 kinetics in H/D exchange massspectrometry by peak width analysis. J. Am. Soc. Mass Spectrom. 17(11),1498–1509 (2006)
43. Wu, Y., Kaveti, S., Engen, J.R.: Extensive deuterium back-exchange incertain immobilized pepsin columns used for H/D exchange mass spec-trometry. Anal. Chem. 78(5), 1719–1723 (2006)
44. Jastrzebska, B., Debinski, A., Filipek, S., Palczewski, K.: Role of mem-brane integrity on G protein-coupled receptors: rhodopsin stability andfunction. Prog. Lipid Res. 50(3), 267–277 (2011)
45. Rubio, L., Alonso, C., Rodríguez, G., Barbosa-Barros, L., Coderch, L., Dela Maza, A., Parra, J.L., Lopez, O.: Bicellar systems for in vitro percutane-ous absorption of diclofenac. Int. J. Pharmaceut 386(1/2), 108–113 (2010)
46. Whorton, M.R., Bokoch, M.P., Rasmussen, S.G., Huang, B., Zare, R.N.,Kobilka, B., Sunahara, R.K.: A monomeric G protein-coupled receptorisolated in a high-density lipoprotein particle efficiently activates its Gprotein. Proc. Natl. Acad. Sci. U. S. A. 104(18), 7682–7687 (2007)
47. Rasmussen, S.G., Chio, H.J., Fung, J.J., Pardon, E., Casarosa, P., Chae,P.S., Devree, B.T., Rosenbaum, D.M., Thian, F.S., Kobilka, T.S., Schnapp,A., Konetzki, I., Sunahara, R.K., Gellman, S.H., Pautsch, A., Steyaert, J.,Weis, W.I., Kobilka, B.K.: Structure of a nanobody-stabilized active stateof the β2 adrenoceptor. Nature 469(7329), 175–180 (2011)
48. Hebert, T.E., Moffett, S., Morello, J.P., Loisel, T.P., Bichet, D.G., Barret,C., Bouvier, M.: A peptide derived from a beta2-adrenergic receptortransmembrane domain inhibits both receptor dimerization and activation.J. Biol. Chem. 271(27), 16384–16392 (1996)
49. Fung, J.J., Deupi, X., Pardo, L., Yao, X.J., Velez-Ruiz, G.A., Devree, B.T.,Sunahara, R.K., Kobilka, B.K.: Ligand-regulated oligomerization of be-ta(2)-adrenoceptors in a model lipid bilayer. EMBO J. 28(21), 3315–3328(2009)
50. Ghosh, A., Sonavane, U., Joshi, R.: Multiscale modeling to understand theself-assembly mechanism of human beta2-adrenergic receptor in lipidbilayer. Comput. Biol. Chem. 48, 29–39 (2014)
N. M. Duc et al.: Application of Bicelles for GPCR HDX-MS 817




















