
Stability, folding dynamics, and long-range conformational ...
Upload
khangminh22Category
view
2download
0
The Synthesis and Conformational Analysis of
13- and 14-Membered Macro-cyclic Ethers
by
DEAN SUTHERLAND CLYNE
B.Sc, The University of Lethbridge, 1990
A THESIS SUBMITTED IN PARTIAL FULFILLMENT OF
THE REQUIREMENTS FOR THE D E G R E E OF
DOCTOR OF PHILOSOPHY
in
THE FACULTY OF GRADUATE STUDIES
(Department of Chemistry)
We accept this thesis as conforming to the required standard
THE UNIVERSITY OF BRITISH COLUMBIA
January, 1998
© Dean S. Clyne, 1998

In presenting this thesis in partial fulfilment of the requirements for an advanced
degree at the University of British Columbia, I agree that the Library shall make it
freely available for reference and study. I further agree that permission for extensive
copying of this thesis for scholarly purposes may be granted by the head of my
department or by his or her representatives. It is understood that copying or
publication of this thesis for financial gain shall not be allowed without my written
permission.
Department
The University of British Columbia Vancouver, Canada
DE-6 (2/88)

ABSTRACT ii
As part of an ongoing study of the chemistry of macrocyclic compounds in our
laboratory, the 14-membered macrocyclic ethers 90, 92, 103, 104, 116, 119, 137, and
154, and the 13-membered macrocyclic ethers 168, 171, 179, 180, 190, and 193 with
substituents both close to and remote from the oxygen atom were synthesized. The
strategy for the preparation of these macrocyclic ethers involved either the Baeyer-
Villiger ring expansion of a cyclic ketone, or the macrolactonization of a long chain
hydroxy acid to give a lactone. Ultimately, the ether oxygen of the lactone would
become the oxygen of the macrocyclic ether. The lactone was often used to introduce
substituents in the vicinity of the ether oxygen. Once this purpose was served, the
carbonyl of the lactone was removed either via a conversion to an intermediate
thionolactone obtained by reaction with Lawesson's reagent, or reduced directly via a
boron trifluoride etherate mediated sodium borohydride reaction.
The diastereomeric 14-membered ethers 103 and 104, and the 13-membered
ethers 179 and 180 were prepared under both radical reduction and hydrogenation
conditions, and the stereoselectivities of these methods were compared. In general,
the stereoselectivities were low (<18% d. e.). The relative configurations of 103, 104,
179, and 180 were determined through chiral GC analysis.
The unsaturated 14-membered ethers 157, 158, 163, and 164 were prepared via
the ruthenium catalyzed metathesis of an acyclic diene ether. The configuration of the
double bond in these unsaturated ethers was determined with 1H homonuclear
decoupling NMR experiments. The isomerization of the carbon-carbon double bond
using phenyl disulfide under photolysis conditions was studied. The product ratios of
the metathesis cyclization and the isomerization reactions were compared to values
obtained from molecular mechanics calculations.
The conformation of the 13- and 14-membered ethers was analyzed using both
NMR spectroscopy and molecular mechanics calculations. The diamond lattice

Ill
conformations were good starting points in the analysis of the 14-membered rings but
were not suited to the 13-membered rings. The [13333] conformation was found to be
a good model for the analysis of the odd-sized 13-membered rings. Additional 1H-DNMR experiments were performed at low temperatures where the conformational
interconversion rates of the macrocyclic ethers were slowed. The DNMR spectra were
interpreted using predicted A8 values from both anisotropy and van der Waals steric
compression effects. The results from the analysis of the DNMR spectra and the
molecular mechanics calculations were compared. The calculations often gave one or
two preferred low energy conformations with a regular geometry. The alkyl substituents
were found to complicate the conformations of some of the macrocyclic ethers studied.
The transition state energies of the individual macrocyclic ethers were
determined from the DNMR spectra to be approximately 8-10 kcal/mol in the case of
the 14-membered ethers and 6-8 kcal/mol in the case of the 13-membered ethers. The
14-membered ether values were compared to computer calculated values obtained
using a dihedral drive method. The calculated values were in general higher and in the
range of 10-15 kcal/mol.


V
TABLE OF CONTENTS
Abstract ii
Table of Contents v
List of Schemes viii
List of Figures x
List of Tables xiii
Abbreviations xvi
Acknowledgments xix
1 Introduction 1
1.1.1 Synthesis of Macrocyclic Ethers by Intramolecular O-Alkylation 3
1.1.2 Synthesis of Macrocyclic Ethers by Olefin Metathesis 4
1.1.3 Synthesis of Macrocyclic Ethers from Macrocyclic Lactones 8
1.2.1 Conformational Analysis 20
1.2.2 Nuclear Magnetic Resonance in Conformational Analysis 20
1.2.3 Conformational Analysis of 6-Membered Rings 27
1.2.4 Conformational Analysis of Medium and Large Rings 30
1.2.5 Conformational Analysis of 14-Membered Rings 33
1.2.6 Conformational Analysis of 13-Membered Rings 38
1.2.7 Transition State Theory in Large Rings 40
2 14-Membered Macrocyclic Ethers 45
2.0.1 Synthesis of 14-Membered Macrocyclic Ethers 46
2.0.2 Conformational Analysis of 14-Membered Macrocyclic Ethers 48
2.1.1 Synthesis of Oxacyclotetradecane (90) and 2-Methyloxacyclotetra-decane (92) 50
2.1.2 Conformational Analysis of Oxacyclotetradecane (90) 52
2.1.3 Conformational Analysis of 2-Methyloxacyclotetradecane (92) 64
2.2.1 Synthesis of 2,14-Dimethyloxacyclotetradecanes (103) and (104) 73

vi
2.2.2 Conformational Analysis of (2R*, 14R*)-2,14-Dimethyloxacyclotetra-decane (103) 82
2.2.3 Conformational Analysis of (2S*, 14R*)-2,14-Dimethyloxacyclotetra-decane(104) 91
2.3.1 Synthesis of 2,2-Dimethyloxacyclotetradecane (116) 100
2.3.2 Conformational Analysis of 2,2-Dimethyloxacyclotetradecane (116) 108
2.4.1 Synthesis of 3,3-Dimethyloxacyclotetradecane (119) 117
2.4.2 Conformational Analysis of 3,3-Dimethyloxacyclotetradecane (119) 118
2.5.1 Synthesis of 6,6-Dimethyloxacyclotetradecane (137) 128
2.5.2 Conformational Analysis of 6,6-Dimethyloxacyclotetradecane (137) 136
2.6.1 Synthesis of 8,8-Dimethyloxacyclotetradecane (154) 146
2.6.2 Conformational Analysis of 8,8-Dimethyloxacyclotetradecane (154) 154
2.7.1 Conclusion 160
3 14-Membered Macrocyclic Unsaturated Ethers 166
3.1.1 Synthesis of (Z/E)-Oxacyclotetradec-5-enes (157) and (158) 167
3.1.2 Cis-Trans Isomerization of (Z/E)-Oxacyclotetradec-5-ene169 (157) and (158) 169
3.2.1 Synthesis of (Z/E)-14-Methyloxacyclotetradec-5-enes (163) and (164) 172
3.2.2 Cis-Trans Isomerization of (Z/E)-14-Methyloxacyclotetradec-5-enes (163) and (164) 176
3.3.1 Conclusion 178
4 13-Membered Macrocyclic Ethers 180
4.0.1 Synthesis of 13-Membered Macrocyclic Ethers 180
4.0.2 Conformational Analysis of 13-Membered Macrocyclic Ethers 181
4.1.1 Synthesis of Oxacyclotridecane (168) and 2-Methyloxacyclotri-decane(171) 185
4.1.2 Conformational Analysis of Oxacyclotridecane (168) 187
4.1.3 Conformational Analysis of 2-Methyloxacyclotridecane (171) 193
4.2.1 Synthesis of 2,13-Dimethyloxacyclotridecanes (179) and (180) 199
4.2.2 Conformational Analysis of 2,13-Dimethyloxacyclotridecane (179) .. 205

4.2.3 Conformational Analysis of 2,13-Dimethyloxacyclotridecane (180) .. 210
4.3.1 Synthesis of 2,2-Dimethyloxacyclotridecane (190) 216
4.3.2 Conformational Analysis of 2,2-Dimethyloxacyclotridecane (190) .... 220
4.4.1 Synthesis of 3,3-Dimethyloxacyclotridecane (193) 227
4.4.2 Conformational Analysis of 3,3-Dimethyloxacyclotridecane (193) .... 228
4.5.1 Conclusion 236
4.6.1 General Conclusion 236
5 Experimental 239
5.1.1 General 239
5.1.2 Conformational Analysis Methods 242
5.1.3 Chemical Methods 242
References 336
Spectral Appendix 345

LIST OF SCHEMES
Scheme 1. Synthesis of Laurenan (37) 14
Scheme 2. Conversion of Thionocaprolactone (61) into an Oxacyclo-
heptane63 19
Scheme 3. Synthesis of the BCD ring Fragment 67 of Brevetoxin A (1) 19
Scheme 4. Synthetic Strategy for the Preparation of Macrocyclic Ethers 47 Scheme 5. Synthesis of Oxacyclotetradecane (90) and 2-Methyloxacyclotetra-
decane (92) 51
Scheme 6. Retrosynthetic Analysis of 2,14-Dimethyloxacyclotetradecanes (103) and (104) 74
Scheme 7. Synthesis of 2-Methylcyclotridecanone (97) via 1-Dibromomethyl-cyclododecanol (94) 75
Scheme 8. Synthesis of 2,14-Dimethyloxacyclotetradecanes (103) and (104) via Thionolactone 101 77
Scheme 9. Synthesis of 2,14-Dimethyloxacyclotetradecanes (103) and (104)
via Enol Ether 100 78
Scheme 10. Retrosynthetic Analysis of 2,2-Dimethyloxacyclotetradecane (116) ... 101
Scheme 11. Synthesis of 2,2-Dimethylcyclotridecanone (106) 102
Scheme 12. Retrosynthetic Analysis of 13-Methyl-13-tetradecanolide (114) 104
Scheme 13. Synthesis of 13-Methyl-13-tetradecanolide (114) 105
Scheme 14. Synthesis of 3,3-Dimethyloxacyclotetradecane (119) 118
Scheme 15. Retrosynthetic Analysis of 6,6-Dimethyloxacyclotetradecane (137) ... 129
Scheme 16. Synthesis of 8-Bromooctanal ethylene acetal (123) 130
Scheme 17. Synthesis of Bisalkylated Dithiane 127 131
Scheme 18. Synthesis of 9-Methylene-13-hydroxytridecanoic acid (131) 133
Scheme 19. Synthesis of 6,6-Dimethyloxacyclotetradecane (137) 135
Scheme 20. Retrosynthetic Analysis of 8,8-Dimethyloxacyclotetradecane (154) ... 147
Scheme 21. Synthesis of Alkylating Agents 141 and 142 148
Scheme 22. Synthesis of 7-Methylene-13-hydroxytridecanoic acid (149) 151
Scheme 23. Synthesis of 8,8-Dimethyloxacyclotetradecane (154) 153
Scheme 24. Synthesis of Oxacyclotetradec-5-enes (163) and (164) 168

ix
Scheme 25. Retrosynthetic Analysis of 14-Methyloxacyclotetradec-5-enes
(163) and (164) 172
Scheme 26. Synthesis of 14-Methyloxacyclotetradec-5-enes (163) and (164) 174
Scheme 27. Synthesis of Oxacyclotridecane (168) and 2-Methyloxacyclotri-
decane(171) 186
Scheme 28. Synthesis of 2-Methyloxacyclotridecane (171) via Hydrogenation 187
Scheme 29. Retrosynthetic Analysis of 2,13-Dimethyloxacyclotridecanes (179) and (180) 199
Scheme 30. Synthesis of 2,13-Dimethyloxacyclotridecanes (179) and (180) via Radical Reduction 201
Scheme 31. Synthesis of 2,13-Dimethyloxacyclotridecanes (179) and (180) via Hydrogenation 202
Scheme 32. Retrosynthetic Analysis of 2,2-Dimethyloxacyclotridecane (190) 216
Scheme 33. Synthesis of Methyl 11-carbomethoxy-12-oxotridecanoate (186) 218
Scheme 34. Synthesis of 2,2-Dimethyloxacyclotridecane (190) 219
Scheme 35. Synthesis of 3,3-Dimethyloxacyclotridecane (193) 228

LIST OF FIGURES X
Figure 1. Intramolecular Formation of Cyclic Monoethers and Diethers 4
Figure 2. Mechanism for the Intramolecular Metathesis Cyclization of a Diene 5
Figure 3. Synthesis of a Brevetoxin A Subunit 11 via Metathesis Cyclization 7
Figure 4. Synthesis of frans-Fused Oxacycles 15-17 via Metathesis Cyclization ... 7
Figure 5. Mechanism of the Free Radical Reduction of a Lactone with
Trichlorosilane 9
Figure 6. Competitive Pathways in the Trichlorosilane Reaction of Esters 10
Figure 7. Reduction of Steroidal Lactones with Sodium Borohydride 11
Figure 8. Proposed Mechanism of the Reaction of Tebbe Reagent 32 with a
Lactone 12
Figure 9. Comparison of Nucleophilic Attack on Lactones and Thionolactones ... 15
Figure 10. Mechanism of Reaction of Lawesson's Reagent 48 with an Ester 17
Figure 11. Regions of Shielding and Deshielding for a Carbon-Carbon Single Bond as the Result of Diamagnetic Anisotropy 23
Figure 12. Possible Orbital Arrangements for y-Anti and y-Gauche Effects in 3,3-Dimethyloxacyclohexane 27
Figure 13. Shielding of the Axial Proton (Ha) in Cyclohexane as the Result of
the Diamagnetic Anisotropy of a 3 Carbon-Carbon Bond 29
Figure 14. Differences in 8 a e for C-2 and C-5 Geminal Protons in 1,3-Dioxane 30
Figure 15. The Lowest Energy Diamond Lattice Conformation of Cyclotetradecane 34
Figure 16. The Corner and Pseudocorner Positions and the Surrounding
Dihedral Angles 35
Figure 17. Transannular Hydrogen Interactions in Cyclotetradecane 37
Figure 18. Movement of a Corner Atom by One Position with an Accompanying Change in Sign of the Surrounding Gauche Dihedral Angles 42
Figure 19. Conformation Interconversion Pathways for Cyclotetradecane as the Result of the Single Corner Movement Mechanism 43
Figure 20. Variable Temperature 500 MHz 1 H NMR of Oxacyclotetradecane (90) in CHCI 2 F:CHCIF 2 (4:1) 56
Figure 21. Single Corner Movement Transition State for Interconversion of the [3434]-1 90-A and the [3344]-1 90-B Conformations of 90 63

xi
Figure 22. 1H NMR Assignments of the C-2 and C-14 Protons of 2-Methyloxa-cyclotetradecane (92) from COSY and NOEDS Experiments 64
Figure 23. Newman Projections of 92 Showing the Geometry of C-2 in the [3434]-1 and [3434]-4 Conformations 66
Figure 24. Variable Temperature 500 MHz 1H NMR of 2-Methyloxacyclotetra-decane(92) in CHCI2F:CHCIF2 (4:1) 69
Figure 25. Interconversion of Conformations of 92 via Single Corner Movements 73
Figure 26. GC Analysis for 2,14-Dimethyloxacyclotetradecanes (103) and (104) on a Chiral Cyclodex-B Column 80
Figure 27. Variable Temperature 500 MHz 1H NMR of (2R*,14R*)-2,14-Dimethyl-oxacyclotetradecane (103) in CHCI2F:CHCIF2 (4:1) 84
Figure 28. Variable Temperature 500 MHz 1H NMR of (2S*,14R*)-2,14-Dimethyl-oxacyclotetradecane(104)inCHCI2F:CHCIF2(4:1) 94
Figure 29. Interconversion of Conformations of 104 via Single Corner Movements 100
Figure 30. Variable Temperature 500 MHz 1H NMR of 2,2-Dimethyloxacyclotetra-decane(116) in CHCI2F:CHCIF2 (4:1) 110
Figure 31. Interconversion of Conformations of 116 via Single Corner Movements 116
Figure 32. Variable Temperature 500 MHz 1H NMR of 3,3-Dimethyloxacyclotetradecane (119) in CHCI2F:CHCIF2 (4:1) 121
Figure 33. Interconversion of Conformations of 119 via Single Corner Movements 127
Figure 34. Variable Temperature 500 MHz 1H NMR of 6,6-Dimethyloxacyclotetra-decane(137)inCHCI2F:CHCIF2(4:1) 139
Figure 35. Interconversion of Conformations of 137 through the [3344J-1 Conformation 145
Figure 36. Variable Temperature 500 MHz 1H NMR of 8,8-Dimethyloxacyclotetradecane (154) in CHCI2F:CHCIF2 (4:1) 156
Figure 37. Interconversion of Conformations of 154 via Single Corner Movements 163
Figure 38. Variable Temperature 500 MHz 1H NMR of Oxacyclotridecane (154) in CHCI2F:CHCIF2 (4:1) 189
Figure 39. 1H NMR Assignments of the C-2 and C-13 Protons of 2-Methyloxa-cyclotridecane (171) from COSY and NOEDS Experiments 194
Figure 40. Variable Temperature 500 MHz 1H NMR of 2-Methyloxacyclotri-decane(171)inCHCI2F:CHCIF2(4:1) 196

xii
Figure 41. GC Analysis for 2,13-Dimethyloxacyclotridecanes (179) and (180)
on a Chiral Cyclodex-B Column 203
Figure 42. Lowest Energy Conformation of Vinyl Ether 176 205
Figure 43. Variable Temperature 500 MHz 1H NMR of (2R*,13R*)-2,13-Dimethyl-oxacyclotridecane (179) in CHCI2F:CHCIF2 (4:1) 207
Figure 44. Variable Temperature 500 MHz 1H NMR of (2S*,13R*)-2,13-Dimethyl-oxacyclotridecane(180)inCHCI2F:CHCIF2(4:1) 212
Figure 45. Variable Temperature 500 MHz 1H NMR of 2,2-Dimethyloxacyclotridecane (190) in CHCI2F:CHCIF2 (4:1) 222
Figure 46. Variable Temperature 500 MHz 1H NMR of 3,3-Dimethyloxacyclotridecane (193) in CHCI2F:CHCIF2 (4:1) 231

LIST OF TABLES xiii
Table 1. Reagents used in the Thionation of Hexadecanolide (54) 16
Table 2. The Three Lowest Energy Conformations of Cyclotetradecane 36
Table 3. The Two Lowest Energy Conformations of Cyclotridecane 39
Table 4. 1H and 13C NMR Assignments for Oxacyclotetradecane (90) in CDCI3 at Room Temperature 53
Table 5. van der Waals Radii for Some Atom Groups 54
Table 6. Low Energy Conformations of Oxacyclotetradecane (90) 60
Table 7. Thermodynamic Values for the Five Lowest Energy Conformations of 90 61
Table 8. 1H and 13C NMR Assignments for 2-Methyloxacyclotetradecane (92) in CDCI3 at Room Temperature 65
Table 9. Experimental and Calculated Coupling Constants for the Low Energy
Conformations of 92 67
Table 10. Low Energy Conformations of 2-Methyloxacyclotetradecane (92) 71
Table 11. Thermodynamic Values for the Five Lowest Energy Conformations of 92 72
Table 12. Yield and Selectivity in the Preparation of 2,14-Dimethyloxacyclotetradecanes (103) and (104) 82
Table 13. 1H and 13C NMR Assignments for (2R*,14R*)-2,14-Dimethyloxa-cyclotetradecane (103) in CDCI3 at Room Temperature 83
Table 14. Low Energy Conformations of (2R*, 14R*)-2,14-Dimethyloxa-cyclotetradecane (103) 89
Table 15. Thermodynamic Values for the Five Lowest Energy Conformations of 103 90
Table 16. 1H and 13C NMR Assignments for (2S*. 14R*)-2,14-Dimethyloxa-cyclotetradecane (104) in CDCI3 at Room Temperature 92
Table 17. Thermodynamic Values for the Five Lowest Energy Conformations of 104 97
Table 18. Low Energy Conformations of (2S*. 14R*)-2,14-Dimethyloxa-cyclotetradecane (104) 98
Table 19. Reaction Conditions used in the Attempted Baeyer-ViNiger Oxidation of Ketone 106 103

xiv
Table 20. Reaction Conditions used in the Attempted Thionation of Lactone 114 107
Table 21. 1H and 13C NMR Assignments for 2,2-Dimethyloxacyclotetradecane (116) in CDCI3 at Room Temperature 108
Table 22. Low Energy Conformations of 2,2-Dimethyloxacyclotetradecane (116) 114
Table 23. Thermodynamic Values for the Five Lowest Energy Conformations of116 115
Table 24. 1H and 13C NMR Assignments for 3,3-Dimethyloxacyclotetradecane (119) in CDCI3 at Room Temperature 119
Table 25. Low Energy Conformations of 3,3-Dimethyloxacyclotetradecane (119) 125
Table 26. Thermodynamic Values for the Five Lowest Energy Conformations of 119 ' 126
Table 27. 1H and 13C NMR Assignments for 6,6-Dimethyloxacyclotetradecane (137) in CDCI3 at Room Temperature 137
Table 28. Low Energy Conformations of 6,6-Dimethyloxacyclotetradecane (137) 143
Table 29. Thermodynamic Values for the Five Lowest Energy Conformations of 137 144
Table 30. 1H and 13C NMR Assignments for 8,8-Dimethyloxacyclotetradecane (154) in CDCI3 at Room Temperature 155
Table 31. Low Energy Conformations of 8,8-Dimethyloxacyclotetradecane (154) 160
Table 32. Thermodynamic Values for the Five Lowest Energy Conformations of 154 161
Table 33. Experimental and Theoretical Equilibrium Ratios for the Isomerization Reaction of Oxacyclotetradec-5-enes (157) and (158) 171
Table 34. Experimental and Theoretical Equilibrium Ratios for the Isomerization Reaction of 14-Methyloxacyclotetradec-5-enes (163) and (164) 177
Table 35. Relative Energies of Conformations of (Z/E)-14-Methyloxacyclo-tetradec-5-ene (163) and (164) and their Percent Population 178
Table 36. The Oxygen Substituted [13333] Conformations and their Relative Strain Energies 182
Table 37. Other Oxygen Substituted 13-Membered Conformations with Low Strain Energy 184

XV
Table 38. 1H and 13C NMR Assignments for Oxacyclotridecane (168) in
CDCI3 at Room Temperature 188
Table 39. Low Energy Conformations of Oxacyclotridecane (168) 192
Table 40. Thermodynamic Values for the Five Lowest Energy Conformations of 168 193
Table 41. 1H and 13C NMR Assignments for 2-Methyloxacyclotridecane (171) in CDCI3 at Room Temperature 195
Table 42. Thermodynamic Values for the Five Lowest Energy Conformations
of 171 197
Table 43. Low Energy Conformations of 2-Methyloxacyclotridecane (171) 198
Table 44. Yield and Selectivity in the Preparation of 2,13-Dimethyloxa-cyclotridecanes (179) and (180) 205
Table 45. 1H and 13C NMR Assignments for (2R*,13R*)-2,13-Dimethyloxa-cyclotridecane (179) in C D C I 3 at Room Temperature 206
Table 46. Thermodynamic Values for the Five Lowest Energy Conformations of 179 208
Table 47. Low Energy Conformations of (2R*, 13R*)-2,13-Dimethyloxa-cyclotridecane (179) 209
Table 48. 1H and 13C NMR Assignments for (2S*,13R*)-2,13-Dimethyloxa-cyclotridecane (180) in CDCI3 at Room Temperature .• 210
Table 49. Thermodynamic Values for the Five Lowest Energy Conformations of 180 214
Table 50. Low Energy Conformations of (2S*, 13R*)-2,13-Dimethyloxacyclotetradecane (180) 215
Table 51. 1H and 13C NMR Assignments for 2,2-Dimethyloxacyclotridecane (190) in CDCI3 at Room Temperature 220
Table 52. Thermodynamic Values for the Five Lowest Energy Conformations
of 190 225
Table 53. Low Energy Conformations of 2,2-Dimethyloxacyclotridecane (190) .. 226
Table 54. 1H and 13C NMR Assignments for 3,3-Dimethyloxacyclotridecane
(193) in CDCI3 at Room Temperature 229
Table 55. Low Energy Conformations of 3,3-Dimethyloxacyclotridecane (193) .. 235
Table 56. Thermodynamic Values for the Five Lowest Energy Conformations of 193 236

LIST OF ABBREVIATIONS
2-dimensional
acetyl
azobis(isobutyronitrile)
aqueous
boiling point
butyl
chemical ionization
concentrated
correlation spectroscopy
cyclopentadienyl
cyclohexyl
change in chemical shift
change in chemical shift between a geminal pair of
axial and equatorial protons in a cyclohexane system
desorption chemical ionization
diastereomeric excess
dihydropyran
4-dimethylaminopyridine
/V,/V-dimethylformamide
1,3-dimethyl-3,4,5,6-tetrahydro-2(1 H)-pyrimidinone
dimethyl sulfoxide
dynamic nuclear magnetic resonance
activation energy
electron ionization
ethyl
ethyl acetate
Gibbs free energy
gas chromatography
hour

xvii
H enthalpy
HMBC heteronuclear multiple bond connectivity spectroscopy
HMPA hexamethylphosphoramide
HMQC heteronuclear multiple guantum coherence
spectroscopy
HRMS high resolution mass spectrum or spectrometry
/Pr isopropyl
IR infrared (spectroscopy)
J coupling constant
kcal kilocalorie
LAH lithium aluminum hydride
LDA lithium diisopropylamide
LRMS low resolution mass spectrum or spectrometry
LTMP lithium 2,2,6,6-tetramethylpiperidine
M parent mass {mass spectra) or
molar, moles per Litre (concentration)
MABR bis(4-bromo-2,6-di-te/t-butylphenoxide)
mCPBA mefa-chloroperbenzoic acid
Me methyl
mp melting point
m/z mass-to-charge ratio
n normal
NBS /V-bromosuccinimide 1H NMR nuclear magnetic resonance (proton) 13C NMR nuclear magnetic resonance (carbon)
NOE nuclear Overhauser effect
p para
PCC pyridinium chlorochromate
Ph phenyl
ppm parts per million
PPTS pyridinium para-toluenesulfonate

pyr pyridine
Rf retention factor or ratio-to-front
rt room temperature
S entropy
Tc coalescence temperature
Tf triflate
tert tertiary
TFAA trifluroacetic acid
THF tetrahydrofuran
TLC thin-layer chromatography
TTMSH tris(trimethylsilyl)silane
Ts or p-Ts tosyl or para-toluenesulfonyl
UHP urea hydrogen peroxide
v/v volume per volume

ACKNOWLEDGMENTS xix
Firstly, I would like to thank Professor Larry Weiler, my Ph.D. research
supervisor, for his guidance, encouragmenent, insight and patience. I am privileged to
have been a member of the Weiler lab.
I thank the staff of the NMR Laboratory (Liane and Marietta), Mass Spectrometry
Laboratory, Microanalysis Laboratory (Mr. Peter Borda), and Glass Shop (Mr. Steve
Rak) for their assistance. My thanks to Dr. Nick Burlinson and Mr. Ray Syvitski for their
helpful discussions and suggestions regarding various aspects of NMR spectroscopy. I
thank also Professor Thomas Money for reading this thesis prior to its submission.
The assistance and efforts of Mr. Mardy Leibovitch (now Dr. Mardy Leibovitch)
and Mr. Matthew Netherton with the photolysis reactions and chiral GC analysis
performed during the course of this research are gratefully acknowledged.
Special thanks to Dr. Michael Ivery (How are things...), Dr. Anurag Sharadendu
(Where are we going for lunch?), and Dr. Michael Wong (A special thank ewe), for their
helpful suggestions and advice both scientific and otherwise.
I thank Vivienne for her seemingly endless display of patience during the writing
of this thesis. I can not thank you enough for giving me the time and space necessary
to complete this task. Believe.
"There are those that break and bend
I'm the other kind, I'm the other kind"
S. Earle

D7 dedicate tAis ifiesis to myfamily
UAanA youfor your fooe, support,
and encouragement over years.

1
CHAPTER 1
INTRODUCTION
The phenomenon known as red tide is the result of vast blooms of unicellular
algae. The name is derived from the colour of certain blooms which contain the
carotenoid pigment peridinin, however the term is used in a broader sense to describe
blooms of other colours as well as colourless ones. One such algae is Gymnodinium
breve Davis which produce very potent neurotoxins of which the brevetoxins are a
prominent subclass. These algae are responsible for major environmental, economic
and health problems each year. The catastrophic consequences of red tide include
massive fish kills, and mollusk poisoning. Humans can also be affected, as the result
of seafood consumption during outbreaks of red tide.
One of the earliest recorded incidents of red tide poisoning involved Captain
George Vancouver in 1793 when he and his crew suffered poisoning after consuming
seafood in a coastal area of British Columbia.1,2 On the east coast of the United States
in 1987 and 1988, a total of 740 bottlenose dolphins were found washed up along the
Atlantic coast from New Jersey to Florida, also the victims of red tide poisoning.2
Biologically, brevetoxins bind to sodium channels in cell membranes, and thereby keep
the channels open and allow for continuous, and damaging sodium ion influx into the
cell. The symptoms of brevetoxin poisoning in humans include: tingling sensations in
the mouth and digits, disruption of coordination (ataxia), hot-cold reversal of
temperature sensation, dilated pupils, brachyrdia, diarrhea, and respiratory problems.2
Poisoning with these symptoms is commonly known as neurotoxic shellfish poisoning
(NSP), or paralytic shellfish poisoning (PSP).
Cultures of the dinoflagellate Gymnodinium breve Davis (Ptychodiscus brevis
Davis) were extracted to give samples of several brevetoxins including brevetoxin A (1),
the most potent ichthyotoxin of this family. Analysis of this toxin culminated in the
solving of its X-ray crystal structure in 1986 by Shimizu and coworkers.3 Extensive
2
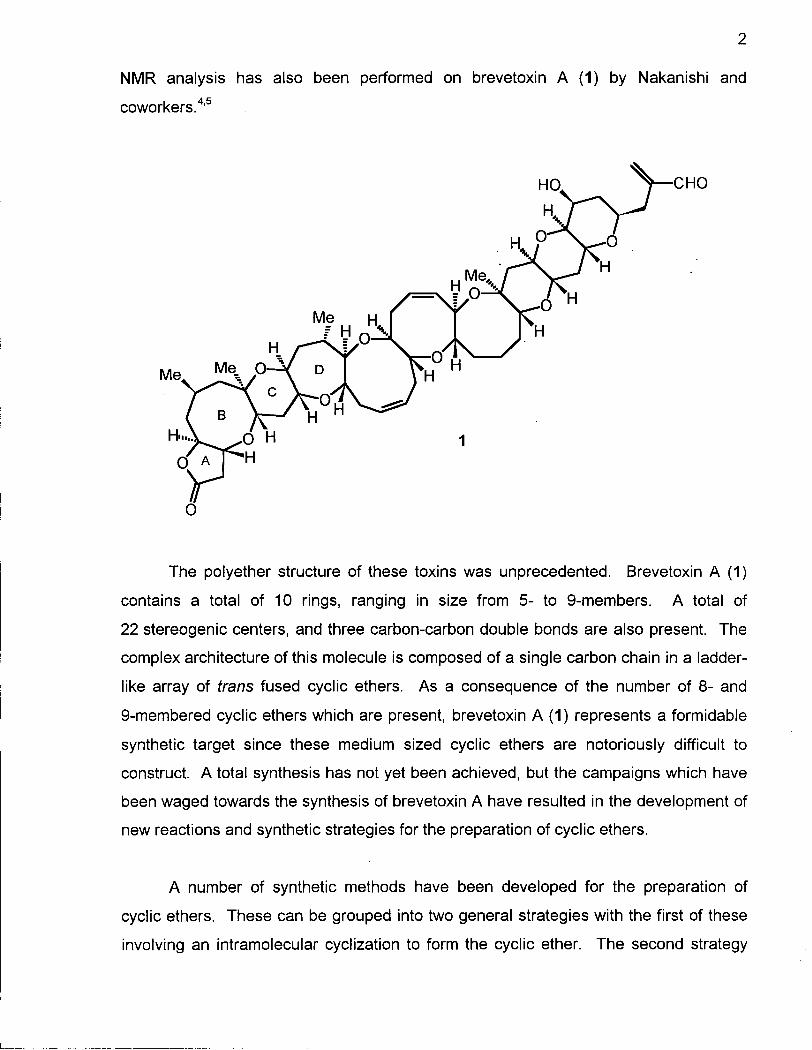
NMR analysis has also been performed on brevetoxin A (1) by Nakanishi and
coworkers.4'5
0
The polyether structure of these toxins was unprecedented. Brevetoxin A (1)
contains a total of 10 rings, ranging in size from 5- to 9-members. A total of
22 stereogenic centers, and three carbon-carbon double bonds are also present. The
complex architecture of this molecule is composed of a single carbon chain in a ladder
like array of trans fused cyclic ethers. As a consequence of the number of 8- and
9-membered cyclic ethers which are present, brevetoxin A (1) represents a formidable
synthetic target since these medium sized cyclic ethers are notoriously difficult to
construct. A total synthesis has not yet been achieved, but the campaigns which have
been waged towards the synthesis of brevetoxin A have resulted in the development of
new reactions and synthetic strategies for the preparation of cyclic ethers.
A number of synthetic methods have been developed for the preparation of
cyclic ethers. These can be grouped into two general strategies with the first of these
involving an intramolecular cyclization to form the cyclic ether. The second strategy

3
involves the modification of an existing ring, such as a lactone, to give the cyclic ether.
Large ring lactones are readily available via a number of hydroxy acid cyclization
methods. Thus, lactones can be viewed as precursors to macrocyclic ethers, since the
difficult issue of ring closure has already been solved in these systems. The problem
of cyclic ether synthesis is thereby reduced to that of converting a lactone into the
desired cyclic ether.
1.1.1 Synthesis of Macrocyclic Ethers by Intramolecular O-Alkylation
The intramolecular cyclization at the ether oxygen is a difficult reaction to
perform successfully in large rings. A study of the cyclization of a series of bromo
alcohols using a variety of base and solvent combinations in our laboratory by Kelly in
1991 met with limited success in the production of the desired 14-membered
macrocyclic ethers.6
In a study of the kinetics of cyclic ether formation, it was shown by llluminati and
coworkers that the rate of the cyclization reaction was dependant on the size of the
formed ring (Figure 1).7 The reactivity was noted to drop off significantly by a factor
greater than 104 as the ring size increased from 6- to 9-membered. A levelling of the
rate was observed for further increases in the ring size, with similar values obtained for
ring sizes 11-16. A comparison of the rates of monoether 4 and diether 5 formation in
general, showed the diether formation rates to be higher. This was attributed to a
reduction of transannular interactions as a result of the substitution of the methylene
groups for oxygen, and is consistent with Dale's view that 1,4- and 1,5-CH-O
interactions are favoured over the corresponding CH-HC interactions.8

4
a0 " 75% (v/v) ag. EtOH A 0^
X(CH2)nBr ^Nc^-J 2 X = CH2,n = 5-16 j X = CH2
3X=0 5 X = °
Figure 1. Intramolecular formation of cyclic monoethers and diethers (from ref. 7).
In such an intramolecular cyclization, an activation at one end of the
hydrocarbon chain, similar to the methods now commonly used to afford lactone
cyclizations might be successful. One such method involves the formation of a
trichloroacetimidate intermediate. This chemistry has been applied to the introduction
of benzyl ether protecting groups.9
1.1.2 Synthesis of Macrocyclic Ethers by Olefin Metathesis
An alternative to cyclization at the ether oxygen involves cyclization at some
point in the hydrocarbon chain via a carbon-carbon bond forming process. The
metathesis reaction which takes a pair of alkenes and couples them in an
intramolecular sense in the presence of a catalyst is suited for such a cyclization.10
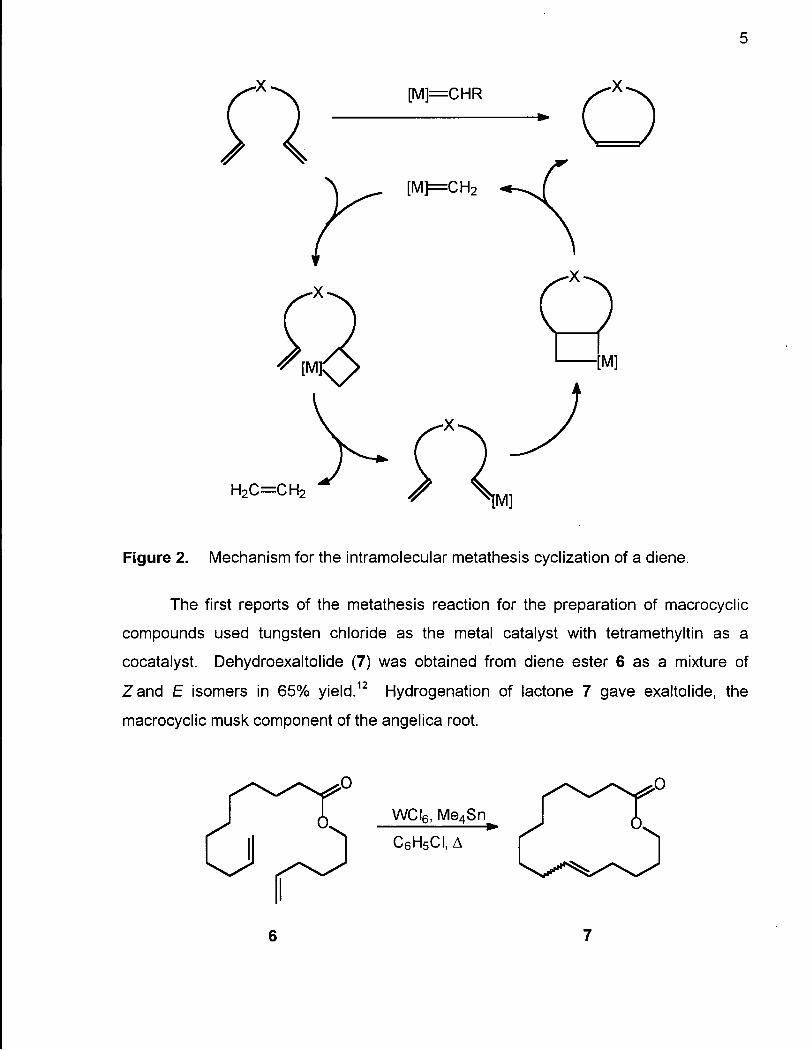
The reaction is believed to proceed through a metallocyclobutane intermediate formed
by reaction of one of the alkenes with the catalyst (Figure 2). This intermediate
undergoes elimination of ethylene to form a new metal carbene which further reacts to
form a fused metallocyclobutane intermediate. Elimination of a metal carbene fragment ** A A
results in the formation of a carbon-carbon double bond in the newly formed ring.
5
Figure 2. Mechanism for the intramolecular metathesis cyclization of a diene.
The first reports of the metathesis reaction for the preparation of macrocyclic
compounds used tungsten chloride as the metal catalyst with tetramethyltin as a
cocatalyst. Dehydroexaltolide (7) was obtained from diene ester 6 as a mixture of
Zand E isomers in 65% yield.12 Hydrogenation of lactone 7 gave exaltolide, the
macrocyclic musk component of the angelica root.
6 7

6
Other catalyst systems have been employed in metathesis reactions including:
WCIe/CpzTiMez and WOCIVCpaTiMez,13,14 Re207/AI203,15 and CH3Re03.16 However, all
of these catalyst systems in general have a low tolerance for the presence of other
functional groups in the metathesis precursors, and the yield of the metathesis products
can be low. In recent years, more complex organometallic catalysts have been
introduced most notably the molybdenum neophylidene complex 8 developed by
Schrock and coworkers.17 The development and application of the molybdenum
neophylidene complex 8 was largely responsible for the recent advances of olefin
metathesis as a synthetically useful carbon-carbon bond forming reaction. However,
owing to the difficulty of preparing 8, and also the sensitivity of this catalyst to oxygen,
water, and polar functional groups, another generation of catalysts with ruthenium at
the core of the organometallic complex has been developed by Grubbs and
coworkers.18 This organometallic ruthenium alkylidene complex 9 is easier to prepare,
essentially air stable as a solid, and still catalytically active without rigorous oxygen and
water exclusion from the reaction system. As a result of the compatibility of these
catalysts with a range of functional groups, a variety of heteroatom containing
compounds have been prepared using olefin metathesis including: ethers,11 crown
ethers,19 lactones,20 ketones,16 amines,21 lactams,22 and sulfides.23
P|Cy3
N Ph C k
(CFafeMeCOJI J>le KRu=^
(CF3)2MeCO- V - f c l ^ C y &
P h
8
As indicated earlier, the marine toxin brevetoxin A (1) contains several medium
sized cyclic ethers. An approach to some medium sized cyclic ether subunits was
recently reported by Clark and Kettle using the olefin metathesis reaction to perform the
cyclization.24 The diene 10 was reacted with the Schrock catalyst 8 to give the
9-membered cyclic ether 11 in 86% yield (Figure 3). This chemistry has also been
7
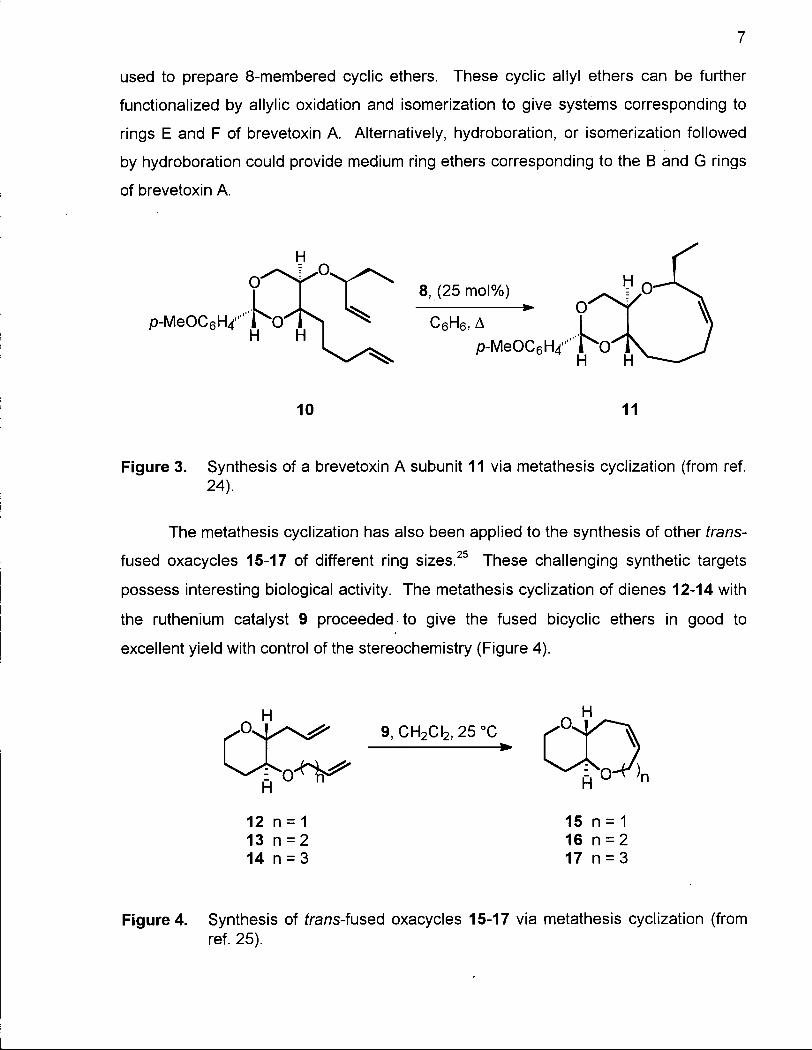
used to prepare 8-membered cyclic ethers. These cyclic allyl ethers can be further
functionalized by allylic oxidation and isomerization to give systems corresponding to
rings E and F of brevetoxin A. Alternatively, hydroboration, or isomerization followed
by hydroboration could provide medium ring ethers corresponding to the B and G rings
of brevetoxin A.
p-MeOC 6 H4
Figure 3. Synthesis of a brevetoxin A subunit 11 via metathesis cyclization (from ref. 24).
The metathesis cyclization has also been applied to the synthesis of other trans
fused oxacycles 15-17 of different ring sizes.25 These challenging synthetic targets
possess interesting biological activity. The metathesis cyclization of dienes 12-14 with
the ruthenium catalyst 9 proceeded to give the fused bicyclic ethers in good to
excellent yield with control of the stereochemistry (Figure 4).
12 n = 1 15 n = 1 13 n = 2 16 n = 2 14 n = 3 17 n = 3
Figure 4. Synthesis of frans-fused oxacycles 15-17 via metathesis cyclization (from ref. 25).

8
1.1.3 Synthesis of Macrocyclic Ethers from Macrocyclic Lactones
As an alternative to the acyclic approaches used in the preparation of
macrocyclic ethers mentioned above, a number of approaches in which a macrocyclic
precursor is converted into the corresponding ether have also been developed. These
methods involve the modification of an existing ring, generally a lactone to give the
macrocyclic ether. Often the lactones are accessible via the cyclization of a hydroxy
acid precursor. A number of methods have been developed for the cyclization of
macrocyclic lactone precursors.26"30
Tsuragi and coworkers have prepared both acyclic and cyclic ethers from
aliphatic esters or lactones via a reduction with trichlorosilane under free radical
conditions.31,32 Ring opened side products as the result of ionic intermediates are
minimized by this free radical process. This reaction can be initiated with either y or uv
radiation or with the photoinduced decomposition of di-te/f-butyl peroxide. This
chemistry has been applied to the reduction of small ring y, 5, and s lactones,32
heptanolide and 3,3,8,8-oVheptanolide33 as well as to the bicyclic lactones 18 and 19. 3 4
Kinetic studies have shown the reduction to proceed via a free radical mechanism with
the addition of trichlorosilane to the carbonyl group of the ester or lactone 20 followed
by further attack of the silane onto the resulting acetal-type intermediate to give the
ether 21 (Figure 5).
19
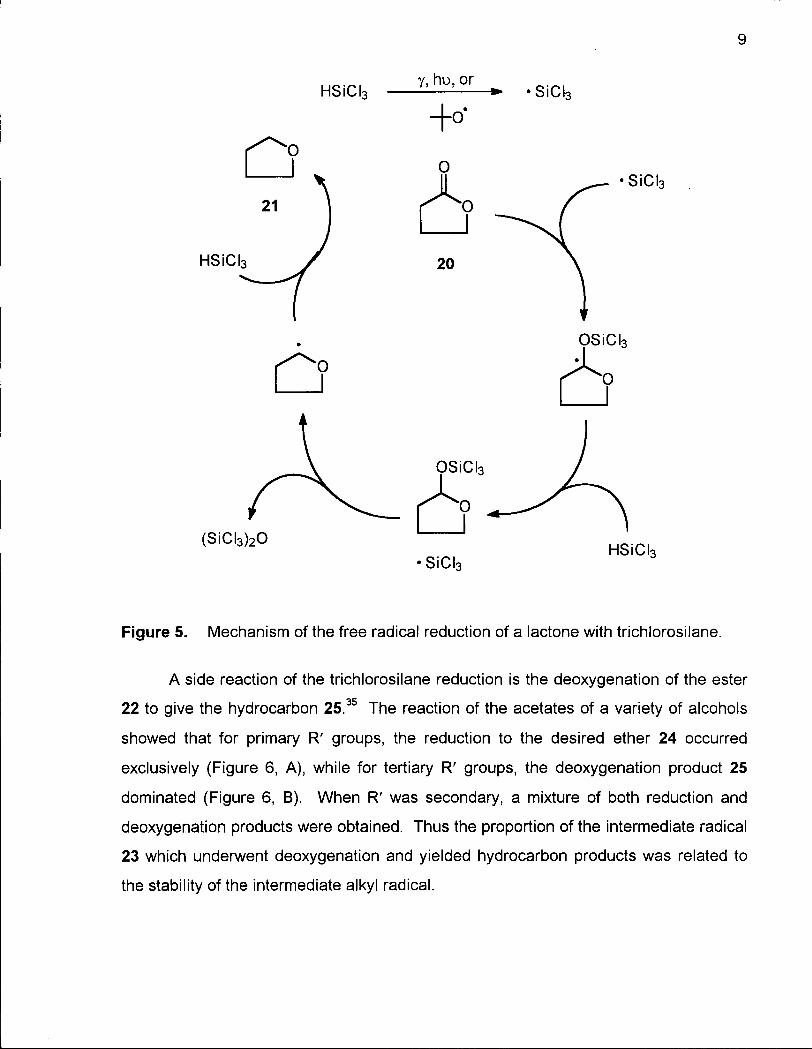
9
HSiCI3
(SiCI3)20
SiCI3
SiCI3
HSiCI3
Figure 5. Mechanism of the free radical reduction of a lactone with trichlorosilane.
A side reaction of the trichlorosilane reduction is the deoxygenation of the ester
22 to give the hydrocarbon 25. 3 5 The reaction of the acetates of a variety of alcohols
showed that for primary R' groups, the reduction to the desired ether 24 occurred
exclusively (Figure 6, A), while for tertiary R' groups, the deoxygenation product 25
dominated (Figure 6, B). When R' was secondary, a mixture of both reduction and
deoxygenation products were obtained. Thus the proportion of the intermediate radical
23 which underwent deoxygenation and yielded hydrocarbon products was related to
the stability of the intermediate alkyl radical.

10
OSiCI3
R- - O R R-
SiCI3H - H
R-0 sicb ?SiC'3
-OR' R—i—OR'
H
H
24
- O R (A)
22 23 \ OSiCI3
R—1=0 +
•R'
SiCI3H R'H
25
(B)
Figure 6. Competitive pathways in the trichlorosilane reaction of esters.
Pettit and coworkers have shown that a lactone can also be directly reduced to
give cyclic ethers using a mixture of sodium borohydride and boron trifluoride
etherate.36 The reducing agent in these reactions was presumed to be diborane formed
in situ. The reduction of lactone 27 under these reaction conditions gave 44% of the
cyclic ether 29, and 42% of diol 31, 3 6 while the reduction of the unsubstituted lactone
26 gave only diol 30, and none of cyclic ether 28 (Figure 7). In contrast to the silane
reduction described above, the presence of alkyl branching adjacent to the ether
oxygen here, results in an increase of the yield of the ether product.37

11
28, R = H 30, R = H 29, R=CH3 31,R=CH3
Figure 7. Reduction of steroidal lactones with sodium borohydride (from ref. 36).
Alkyl substituents can be introduced adjacent to the oxygen of a cyclic ether by
reaction of a lactone with the organotitanium reagent 32 developed by Tebbe and
coworkers.38 The reactive species is thought to be a titanium carbene which reacts with
the carbon-oxygen double bond of the lactone to form intermediate 33 with a four-
membered ring (Figure 8 ) . 3 9 Elimination of a titanium-oxygen species gives the product
34, a cyclic ether with an exocyclic methylene.
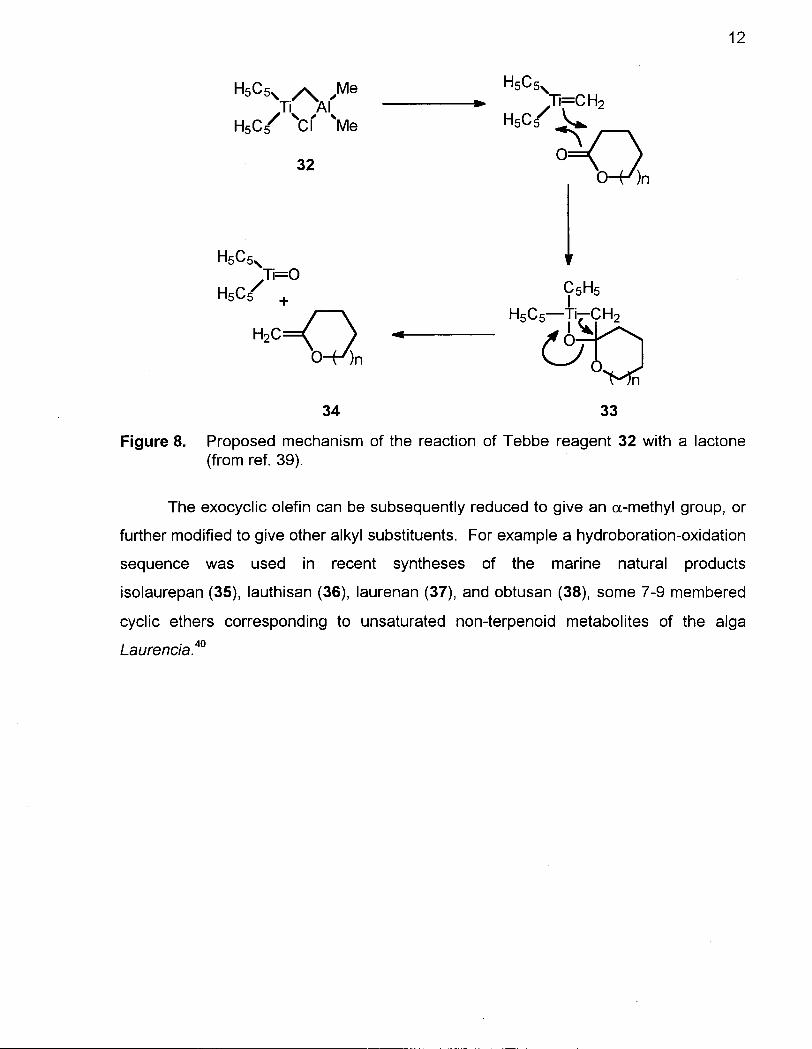
12
H 5 C 5 n / v Me 71 Al
H5C5
/ SCf NMe
32
Ti=CH2
H5C/
. 0-K)n
34 33
Figure 8. Proposed mechanism of the reaction of 7ebbe reagent 32 with a lactone (from ref. 39).
7he exocyclic olefin can be subsequently reduced to give an a-methyl group, or
further modified to give other alkyl substituents. For example a hydroboration-oxidation
sequence was used in recent syntheses of the marine natural products
isolaurepan (35), lauthisan (36), laurenan (37), and obtusan (38), some 7-9 membered
cyclic ethers corresponding to unsaturated non-terpenoid metabolites of the alga
Laurencia.40
13
13
The methylenation of lactone 39 using the Tebbe reagent 32 followed by rapid
chromatographic purification on alumina gave the unstable enol ether 40 (Scheme 1).
This enol ether was subjected to hydroboration with oxidative workup to give the
hydroxymethyl compound 41. Very high selectivity for the desired diastereomer was
obtained when diisoamylborane was used as the hydroborating agent. The hydroxy
methyl group of 41 was oxidized with PCC and chain extended to give alkene 42.
Hydrogenation of the carbon-carbon double bond of 42 gave the C-8 propyl group to
complete the synthesis of laurenan (37).40
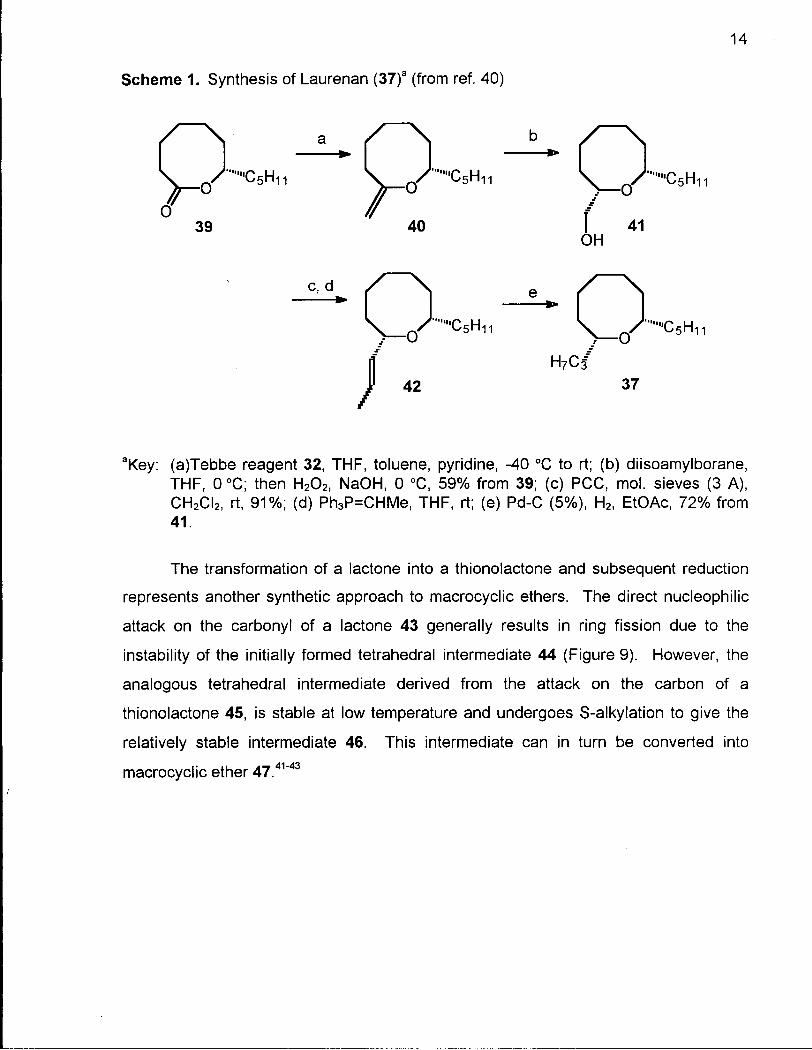
14
Scheme 1. Synthesis of Laurenan (37)a (from ref. 40)
aKey: (a)Tebbe reagent 32, THF, toluene, pyridine, -40 °C to rt; (b) diisoamylborane, THF, 0 °C; then H202, NaOH, 0 °C, 59% from 39; (c) PCC, mol. sieves (3 A), CH2CI2, rt, 91%; (d) Ph3P=CHMe, THF, rt; (e) Pd-C (5%), H2, EtOAc, 72% from 41.
The transformation of a lactone into a thionolactone and subsequent reduction
represents another synthetic approach to macrocyclic ethers. The direct nucleophilic
attack on the carbonyl of a lactone 43 generally results in ring fission due to the
instability of the initially formed tetrahedral intermediate 44 (Figure 9). However, the
analogous tetrahedral intermediate derived from the attack on the carbon of a
thionolactone 45, is stable at low temperature and undergoes S-alkylation to give the
relatively stable intermediate 46. This intermediate can in turn be converted into
macrocyclic ether 47.41"43
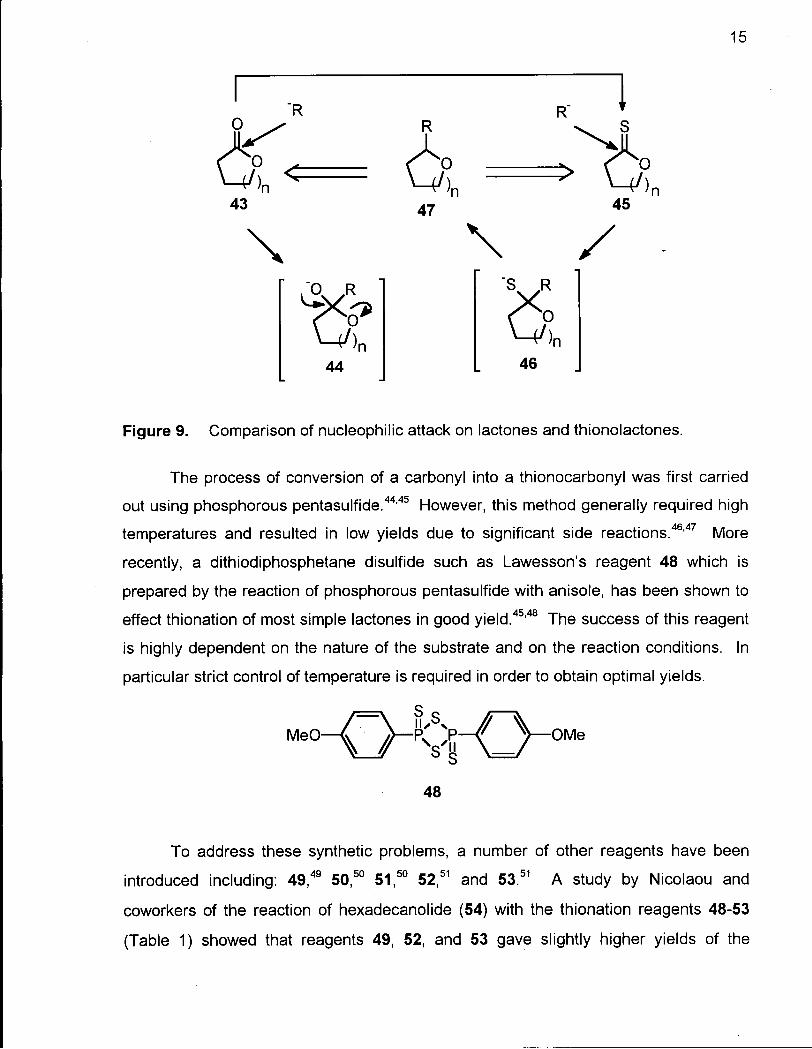
15
Figure 9. Comparison of nucleophilic attack on lactones and thionolactones.
The process of conversion of a carbonyl into a thionocarbonyl was first carried
out using phosphorous pentasulfide.44,45 However, this method generally required high
temperatures and resulted in low yields due to significant side reactions.46,47 More
recently, a dithiodiphosphetane disulfide such as Lawesson's reagent 48 which is
prepared by the reaction of phosphorous pentasulfide with anisole, has been shown to
effect thionation of most simple lactones in good yield.45,48 The success of this reagent
is highly dependent on the nature of the substrate and on the reaction conditions. In
particular strict control of temperature is required in order to obtain optimal yields.
48
To address these synthetic problems, a number of other reagents have been
introduced including: 49,49 50,50 51,50 52,51 and 53.51 A study by Nicolaou and
coworkers of the reaction of hexadecanolide (54) with the thionation reagents 48-53
(Table 1) showed that reagents 49, 52, and 53 gave slightly higher yields of the
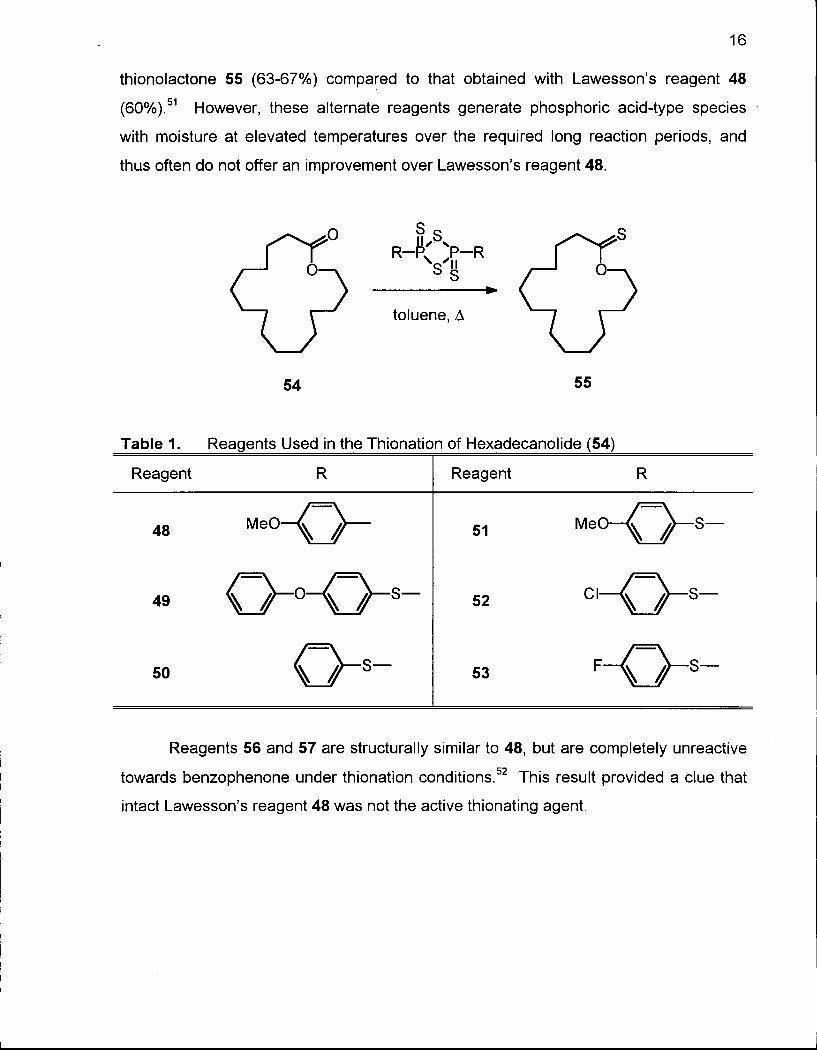
16
thionolactone 55 (63-67%) compared to that obtained with Lawesson's reagent 48
(60%).51 However, these alternate reagents generate phosphoric acid-type species
with moisture at elevated temperatures over the required long reaction periods, and
thus often do not offer an improvement over Lawesson's reagent 48.
R-Pv P-R b S
toluene, A
54 55
Table 1. Reagents Used in the Thionation of Hexadecanolide (54)
Reagent R Reagent R
48 M e 0 - O ~ ~ 51 M e O — S —
49 0 - ° ^ 0 - s - 52 c i - 0 - s _
50 53 F - Q - s -
Reagents 56 and 57 are structurally similar to 48, but are completely unreactive
towards benzophenone under thionation conditions.52 This result provided a clue that
intact Lawesson's reagent 48 was not the active thionating agent.
17
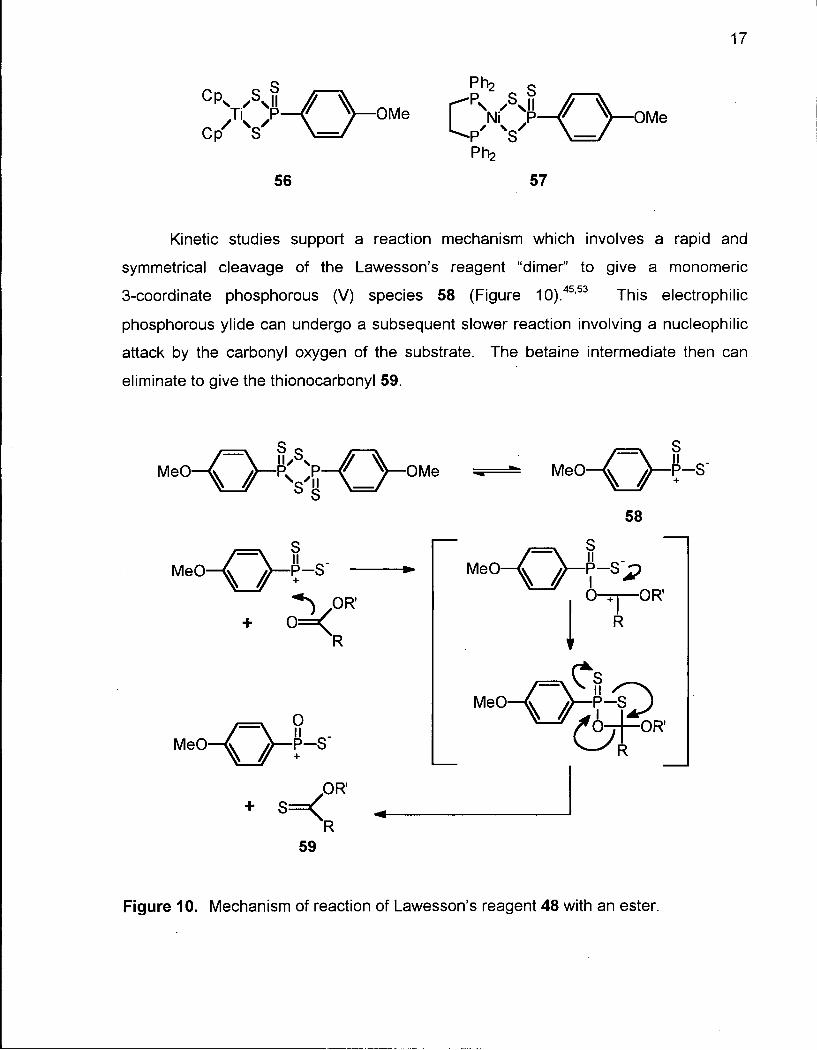
Kinetic studies support a reaction mechanism which involves a rapid and
symmetrical cleavage of the Lawesson's reagent "dimer" to give a monomeric
3-coordinate phosphorous (V) species 58 (Figure 10).45,53 This electrophilic
phosphorous ylide can undergo a subsequent slower reaction involving a nucleophilic
attack by the carbonyl oxygen of the substrate. The betaine intermediate then can
eliminate to give the thionocarbonyl 59.

18
A report by Baxter and Bradshaw in which compounds with electron withdrawing
substituents conjugated to an ester carbonyl failed to react under thionation conditions
while compounds with conjugated electron donating substituents experienced an
increased rate of reaction supports this mechanism.54 Moreover, it was found that
esters containing an ether functionality such as 60 were difficult to thionate. This was
attributed to a competition between the carbonyl oxygen and the more basic ether
oxygen atoms for the electrophilic phosphorous.54
Once formed, the C-1 carbon of a thionolactone such as 61 can be reacted with
a variety of nucleophiles to give after S-alkylation with methyl iodide the mixed thioketal
62 (Scheme 2). A variety of organometallic reagents were examined by Nicolaou and
coworkers, and reagents such as methyl lithium, allyl lithium, and lithium
triethylborohydride gave good yields of the desired mixed thioketals.51 Reductive
desulfurization of these thioketals using triphenyltin hydride, gave the cyclic ether
60
63. 51,55
19
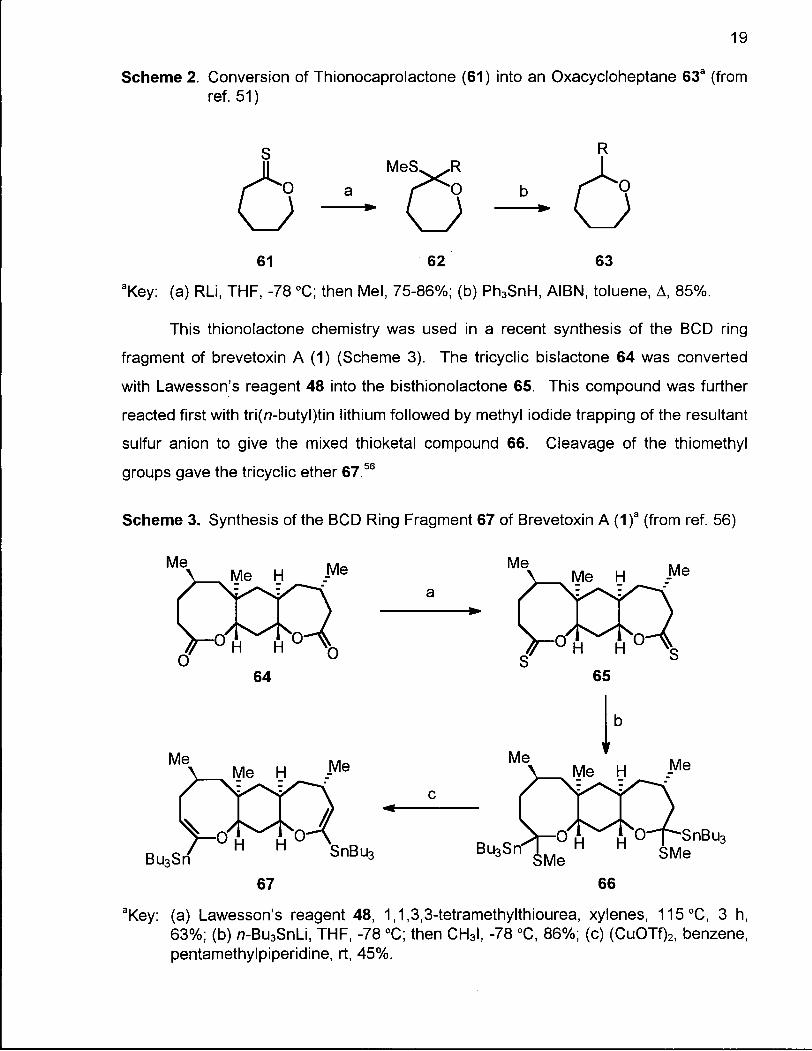
Scheme 2. Conversion of Thionocaprolactone (61) into an Oxacycloheptane 63a (from ref. 51)
61 62 63
aKey: (a) RLi, THF, -78 °C; then Mel, 75-86%; (b) Ph3SnH, AIBN, toluene, A , 85%.
This thionolactone chemistry was used in a recent synthesis of the BCD ring
fragment of brevetoxin A (1) (Scheme 3). The tricyclic bislactone 64 was converted
with Lawesson's reagent 48 into the bisthionolactone 65. This compound was further
reacted first with tri(n-butyl)tin lithium followed by methyl iodide trapping of the resultant
sulfur anion to give the mixed thioketal compound 66. Cleavage of the thiomethyl
groups gave the tricyclic ether 67.56
Scheme 3. Synthesis of the BCD Ring Fragment 67 of Brevetoxin A (1)a (from ref. 56)
67 66
aKey: (a) Lawesson's reagent 48, 1,1,3,3-tetramethylthiourea, xylenes, 115°C, 3 h, 63%; (b) n-Bu3SnLi, THF, -78 °C; then CH3I, -78 °C, 86%; (c) (CuOTf)2, benzene, pentamethylpiperidine, rt, 45%.

2 0
1.2.1 Conformational Analysis
The shape of organic molecules can be specified according to three levels of
increasing precision and sophistication. The first of these levels, constitution,
designates the manner in which the atoms are joined together with chemical bonds.
The next level, configuration, designates which of several possible ways the atoms of a
molecule with a given constitution are spatially connected so that isomeric forms can
be obtained. The final level of sophistication, conformation, designates in which of
several possible ways the atoms of a molecule with a given configuration are arranged
in space. Conformational isomers or conformers usually cannot be isolated since their
interconversion involves a rotation about single bonds within a molecule.
Conformational analysis is the interpretation or prediction of physical or chemical
properties, and of the relative energies of compounds as determined by their
conformation or conformations.
1.2.2 Nuclear Magnetic Resonance in Conformational Analysis
One of the most powerful tools for conformational analysis is dynamic NMR
spectroscopy (DNMR). DNMR can be used for the qualitative and quantitative study of
conformational changes in organic compounds as a function of changes in
temperature.57,58 A classical application of DNMR is the study of rotation about the
carbon-nitrogen bond in dimethylformamide (68).58 At low temperatures the two methyl
groups give two distinct signals in the 1H NMR spectrum (slow exchange rate). As the
temperature is raised the barrier to rotation about the carbon-nitrogen bond which has
significant double bond character is overcome and the methyl groups become
indistinguishable. The signals for the two methyl groups broaden (intermediate
exchange rate), and finally merge into one signal (fast exchange rate). The
temperature at which the signals are broadest and can not be distinguished from each
other is referred to as the coalescence temperature (Tc).

21
68
From the temperature dependence of the spectra, rate constants and activation
parameters can be determined. The rate of exchange (kc) at coalescence can be
calculated using either equation 1 or 2, where Av is the separation of the signals in
hertz measured at a temperature below Tc. Equation 1 is applied in the case of
uncoupled nuclei, and equation 2 is applied when the nuclei are coupled to each other,
and J is the coupling constant in hertz.
The rate of exchange can in turn be used to calculate the free energy of
activation {AG*) for the conformational process using the Eyring equation (3) where: R
is the ideal gas constant, Tc is the coalescence temperature, kB is the Boltzmann
constant, kc is the rate constant, and h is Planck's constant.
Several computer programs have been developed to assist in the analysis of
DNMR data.59 These programs can be used to analyze the line shape of 1H NMR
spectra collected at various temperatures and calculate the rates for the conformational
process at the temperature over which the conformational change occurs. The
activation energy (Ea) can be determined from Arrhenius plots of log k vs. 1/T, and the
enthalpy (AH*) and entropy (AS*) of activation can be determined from Eyring plots of
log (k/T) vs. 1/T.
kc=7c Av/2V2
kc =JL(AV2 - 6 J2)v
2*
(1)
(2)
AG* = RTc In (kBTc/kch)
= R Tc (23.76 + In (Tc / kc))
(3)

22
DNMR studies provide information about the coupling constants, the chemical
shifts, the relaxation time, and the line shape changes of the atoms in a compound as a
function of temperature. The coupling constants can provide information about the
geometry of the compound. For example in cyclic systems, the Karplus equation (4)
can be used to determine the dihedral angles of vicinal protons, and hence the
torsional angles of the ring itself. Here, A and C are constants, and 0 is the H-C-C-H
dihedral angle.
3J = A cos29 + C (4)
The chemical shift (5) gives information about the shielding of protons in the
molecule. For example, the value of 5 a e which is the difference in chemical shift of a
geminal axial and an equatorial proton for a specific carbon in a molecule. A positive
value indicates that the chemical shift of the axial proton is at higher field (shielded)
relative to the equatorial proton. The chemical shift difference of geminal protons in a
molecule is determined by a number of shielding effects including: diamagnetic
anisotropy (O-AN), steric compression (O-ST), and electric field (aE).60
Much of the pioneering work in the determination of the diamagnetic
anisotropies (O-AN) of bonds was performed by ApSimon and coworkers who derived the
anisotropies of carbon-carbon and carbon-hydrogen bonds through a comparison of
the chemical shifts of protons in a series of cycloalkanols.61 It is the bonds located p to
the methylene group of interest which are thought to contribute most to this shielding
effect. The bonds a to the geminal pair of protons are symmetrical with respect to both
the axial and the equatorial protons and therefore do not have a differential affect on
the chemical shift of the geminal protons in a cyclohexane system.

23
The screening contribution is composed of both an angular term, as well as an
anisotropy term as calculated by the McConnell expression (5), where the anisotropy
(Ax) or (XL - XT) is composed of terms parallel and perpendicular to the axis of symmetry
of the atom-hydrogen (X-H) bond, R is the distance between the proton and the centre
of the induced magnetic dipole of the bond, and y is the angle between the direction of
R and the symmetry axis. This leads to regions of shielding and deshielding about the
bond of interest.
Aa = A Y (1 - 3 cos y) 3 R3 (5)
For a carbon-carbon single bond, this equation describes a region shaped like a
double cone with areas of deshielding within the cones, and areas of shielding outside
of the cones (Figure 11).
Shielding
+AX +AX Deshielding
Figure 11. Regions of shielding and deshielding for a carbon-carbon single bond as the result of diamagnetic anisotropy.
When a hydrogen atom is held in close proximity to another atom in a molecule,
at a distance less than the sum of their van der Waals radii,62 the chemical shift of the

24
hydrogen can be shifted downfield as a result of the steric compression effect (O-ST)-63
In the study of tricyclic compounds 69-73, a series of half cage compounds related to
the birdcage hydrocarbon, unusually high shielding and deshielding effects were
observed in the NMR spectra.64,65 The rigid geometry present in these compounds
results in steric repulsion between the endo hydrogen and oxygen groups. The
chemical shift of the endo hydrogen (Hb) in 69 which has an exo hydroxyl group was
2.40 ppm compared to alcohol 71, which has an endo hydroxyl group, and a chemical
shift of 3.55 ppm for the opposing endo hydrogen (Hb). The size of this effect was
found to vary with the nature of the functional group opposite to the sterically
compressed hydrogen. In a series of oxygen substituted compounds 70-73, the
magnitude of the chemical shift change varied in the order 0"Na+ > OH > OMe > OAc.
This chemical shift trend parallels the magnitude of the electron density at the oxygen
atom in each of these compounds.
69 70 R = Na+ 71 R = H 72 R = OMe 73 R = OAc
The change in chemical shift for the sterically compressed hydrogen is attributed
to the electron cloud of the oxygen functional group repelling the bonding electrons in
the C-Hb bond towards the C-Ha bond. This polarization of the methylene electron
cloud accounts for the deshielding of the inside hydrogen mentioned above, and the
shielding of the outside hydrogen atoms (Ha) as well; 1.10 ppm in the case of 69,
compared to 0.88 ppm in 71.

25
The shielding of the proton is caused by a steric repulsion of the electron cloud
in the opposing C-Ha bond, away from the hydrogen nucleus and towards the carbon
nucleus, it follows that the effect should be observed in the 13C NMR spectrum as a
result of this charge polarization as well.66,67 In a study of the bicyclo[3.3.1]nonanes
74-76, C-3 and C-7 are in close spatial proximity to each other.68 In fact, it is the
through space van der Waals interaction of endo groups at C-3 and C-7 which is
thought to be the main driving force for conformational preferences in these
systems.69,70 Substituents at C-7 have an influence on the chemical shift of C-3. For
isomers having an endo hydrogen at C-3, the chemical shift of C-7 is approximately
21 ppm. For example, the chemical shift of C-7 is 21.1 ppm in the unsubstituted 74,
and 20.6 ppm in the exo substituted 75. However in 76 with a C-3 endo substituent, the chemical shift of C-7 is shifted upfield by 5 ppm as a result of steric compression
shielding to 15.5 ppm. In all three compounds, the distance between the C-3 and C-7
endo substituents was determined to be less than 2 A.
The polarization of carbon-carbon and carbon-hydrogen bonds by a dipole or
charge can also influence the shielding and chemical shift of the protons in a molecule.
The magnitude of this electric field effect (aE) is calculated from: the polarizability (P) of
the bond of length (L), the size of the charge (q) at a distance (r) and an angle (0) from
the field gradient to the bond of interest using the Buckingham equation (6).71
CTE = k S q (cos 0) P / (L r2) (6)

26
This shielding effect (aE) is greatest for bonds which are parallel to each other
and perpendicular to their line of centers, and of a lesser magnitude for gauche bonds
or bonds of other skewed geometries. For example, in cyclohexane an axial C-H bond
is affected by the shielding of two parallel y C-H axial bonds and two vicinal gauche
C-H bonds. While an equatorial C-H bond would experience the lesser polarization
shielding effect of four vicinal gauche C-H bonds. The shielding of axial hydrogen
atoms has been noted to increase with the number of axial C-H bonds in other
saturated hydrocarbons,72 and in some steroids studied by Schneider and coworkers as
The effect of introducing a heteroatom into a cyclohexane results in changes to
the 13C NMR spectrum of the molecule. Deshielding of the carbons a and p to the new
heteroatom are largely the result of the increased electronegativity of the heteroatom
relative to that of carbon. This shift varies approximately linearly with the
electronegativity of the heteroatom.75 The heteroatom also has a significant affect on
the chemical shift of the carbon three bonds away. It was first proposed that this effect
resulted from a shielding of the y-carbon by the heteroatom via a polarization of
electrons through the steric compression mechanism described above.66 However,
results from subsequent studies were not explained by a steric effect alone. The
y-effect was found to be of a similar magnitude for a number of substituents which
differed widely in their A value and van der Waals radii.76 This indicated that the
y-effect was controlled by more than simple size considerations, and both steric and
electronic factors were considered. The y-effect was found to depend on both the
electronegativity of the heteroatom, and also on the dihedral angle between the
heteroatom and the y-carbon.76,77
A stereoelectronic interaction is likely responsible for the dihedral dependence
of the y-effect since the steric relationship between the y-carbon and the new
heteroatom orbitals changes with their dihedral angle; The transmission of electronic
information along a molecular chain is associated with the overlap of properly aligned
(parallel) orbitals. This leads to distinct stereoelectronic pathways for arrangements

27
having either an anti or a gauche arrangement between the heteroatom and the
y-carbon.
The effect of the heteroatom in a series of 3,3-dimethyl substituted 6-membered
heterocycles on the y-carbon through an anti relationship gave either a slight shielding
or a deshielding effect of 1-2 ppm depending on the nature of the heteroatom and the
dihedral angle.77 The effect was believed to result from the electronic interaction of
orbitals antiperiplanar with respect to the bond in the y-anti pathway (Figure 12). In
comparison, the affect of the heteroatom on a y-carbon in a gauche relationship was
larger than the y-anti effect, typically shielding the y-carbon by 4-8 ppm. The y-gauche
effect was believed to be the result of the interaction of parallel orbitals on adjacent
atoms (Figure 12).
y-anti y-Qauch©
Figure 12. Possible orbital arrangement for y-anti and y-gauche effects in 3,3-dimethyloxacyclohexane.
1.2.3 Conformational Analysis of 6-Membered Rings
The concepts and ideas of conformational analysis are now widely used in the
interpretation of chemical transformations and reaction mechanisms in organic
chemistry, and also in the explanation of steric and electronic effects in organic
compounds. The conformational analysis of cyclohexane and it's derivatives is one of
the most widely studied topics in organic chemistry. In 1890, Sachse first suggested
that cyclohexane existed in two puckered arrangements which later became known as
the chair and boat conformations.78 Until that time, the prevailing theory depicted
cyclohexane as a regular planar hexagon, rather than a three dimensional structure. In
1925, Hueckel clearly showed that cyclohexanes were in fact not planar structures with

28
the synthesis of the bicyclic cis and trans isomers of decalin/s However, it was not
until 1950 when the analysis of reactions of cyclohexanes and steroids, with their
multiple cyclohexane rings, by Barton that the power of cyclohexane conformational
analysis received the recognition it deserved. By viewing cyclohexane as having a
three dimensional conformation, Barton was able to explain the results of organic
reactions in these systems which had previously puzzled chemists.80 The axial
positions (Ha) in cyclohexane are more hindered than the equatorial positions (He) due
to transannular interactions, and this leads to the conformational preference of
transition states, reaction pathways, and substituents in these systems.
The chair-chair ring interconversion of cyclohexane which converts the ring to its
mirror image via rotation of carbon-carbon single bonds is rapid at rt. This process
interchanges the axial and equatorial substituents, thus making them spectrally
equivalent by NMR analysis. The rate of this process is dependent on the temperature
of the system. As the temperature is lowered, the interconversion of the axial and
equatorial substituents is slowed. At low temperature a particular conformer with either
the axial or the equatorial substituent would predominate. The axial and equatorial
substituents are no longer spectrally equivalent, and accordingly the NMR spectrum
becomes more complex.
A DNMR study of cyclohexane gave a value of 10.3 kcal/mol for the free energy
of activation (AG*) for ring inversion, with a value of 10.8 kcal/mol obtained for the
enthalpy of activation (AH*) for this same process.81 Determination of the vicinal
coupling constants for cyclohexane gave, via the Karplus equation (4), an internal
torsion angle for cyclohexane of 58° which is slightly distorted from 60°, the angle

29
predicted if all the carbons had an ideal tetrahedral geometry.82 The difference in
chemical shift at low temperature between a geminal pair of axial and equatorial
protons (5ae) was found to be 0.48 ppm.83 In cyclohexane, the axial proton lies outside
of the deshielding cone resulting from the diamagnetic anisotropy of the p carbon-
carbon bonds while the equatorial proton lies within the deshielding cone (Figure 13).
Figure 13. Shielding of the axial proton (Ha) in cyclohexane as the result of the diamagnetic anisotropy of a p carbon-carbon bond.
The torsional change resulting from replacing a methylene group with a
heteroatom (X) can result in either a flattening or a puckering of the ring. In a study of
the oxygen heterocycle, tetrahydropyran, the larger C-O-C bond angle, and the shorter
C-0 bond length caused a slight flattening of the chair conformation as compared to
cyclohexane. Changes in bond angle in this heterocycle were of less importance than
changes to the torsion angles in influencing the magnitude of the free energy of
activation of ring inversion (AG 4 ). 6 0 DNMR studies gave a A G 4 of 10.3 kcal/mol for the
ring inversion and a chemical shift difference (5ae) for the protons at C-2 of 0.50 ppm in
tetrahydropyran.84 This chemical shift difference is similar to that obtained for
cyclohexane itself, thus suggesting that the diamagnetic anisotropy of the p carbon-
oxygen single bond is similar to that of a carbon-carbon single bond. Results from a
study of 1,3-dioxanes indicate that the orientation of the carbon-oxygen bond can
influence the value of 5 a e . 8 5 In this study, 8 a e for C-2 was positive denoting shielding of
the axial proton, but 8 a e for C-5 was negative, indicative of a deshielding of the axial
proton at that carbon (Figure 14). The geometry of the C-2 and C-5 protons with
respect to the carbon-oxygen bonds is approximately the same however the orientation

30
of the carbon-oxygen bond at C-2 is different from that of C-5 which may account for
the difference in the shielding observed in this system.
Figure 14. Differences in 8aefor C-2 and C-5 geminal protons in 1,3-dioxane.
To summarize, the introduction of a heteroatom into a ring can result in changes
to the NMR spectrum as a result of differences in the electronegativity of the
heteroatom relative to the methylene group. The lone pairs of the heteroatom can also
introduce new electronic interactions, and the magnitude and sign of the diamagnetic
anisotropy of the C-X bonds can affect the NMR spectrum. Finally, changes in ring
shape as a result of differences in the C-X-C bond angle and C-X bond length relative
to the carbocycle can also affect the NMR spectrum.
1.2.4 Conformational Analysis of Medium and Large Rings
This section begins with a brief historical account of large ring or macrocyclic
compounds. The first macrocyclic compounds were isolated in 1926 by Ruzicka while
investigating the constituents of musk oil.86,87 The structure of the large ring ketones,
civetone (77) and muscone (78) were elucidated using chemical methods only, a
process complicated by the scarcity of functional groups in these compounds. This
research was of twofold importance. First these musklike compounds were of
commercial value in the fragrance industry, and second, little was known about the
physical and chemical properties of large rings compounds leading to a fundamental
interest as well.
Ha
H a
31
O
78 79
Research in the area of macrocyclic chemistry continued through the efforts of
Ruzicka88,89 and Prelog90 and their coworkers who investigated the chemical properties
of medium and large ring hydrocarbons, alcohols, ketones, and lactones. The physical
and chemical properties of these macrocyclic ring compounds showed an interesting
and unexpected dependency on ring size. For example, it was found that the
relationship between melting point and ring size did not rise monotonically as with
aliphatic acyclic hydrocarbons.88"90
Pikromycin (79), the first of the complex macrocycles called the macrolides, was
isolated by Brockmann and Henkel in 1950 from an Actinomyces culture.91 Many of
these large ring lactone macrolides possess interesting biological activity and also
share several characteristic structural features. They contain 12-, 14-, or 16-membered
lactones of secondary alcohols and are composed of an array of hydroxyl and alkyl
substituents characteristically distributed around the ring. Attached to one or more of
the secondary hydroxyl groups are sugars, which are often amino sugars.92 An
understanding of the conformation of these macrolides is important in the
rationalization of the chemical activity and the structure activity relationships of these
antibiotics. This has been an area of extensive research, and a combination of
spectroscopy methods and X-ray crystallography have been employed to determine the
conformation of this and other macrolides in both solution and the solid state.

32
Initially, the shape or conformation of the large ring molecules was poorly
understood. In 1961, Dunitz and coworkers reported an X-ray diffraction study of a
series of cyclodecane derivatives all of which had crystallized in a similar
conformation.93 This was a surprising result at the time as these large rings were
thought to be a flexible chain of atoms capable of existing in many conformations. In
1963, Dale realized that the solid state conformations of the cyclodecane derivatives
closely followed the diamond lattice, an extended tetrahedral array of carbon-carbon
bonds having ideal bond lengths, bond angles, and dihedral angles.94,95 A
conformation which was superimposable on the diamond lattice was therefore predicted
to possess a minimum of angle and torsion strain.
From inspection of space-filling molecular models, Dale proposed diamond
lattice conformations for all even membered rings ranging in size from 6- to
16-membered by maximizing the number of anti dihedral angles and avoiding the
eclipsing of bonds.94 Dale also recognized a tendency for saturated even-membered
large rings to adopt compact conformations consisting of two parallel methylene chains
linked by bridges of minimum length.94 These rectangular conformations were
proposed to be more stable and possess less torsion and angle strain than those with a
large hole in the ring interior. In addition, Dale concluded that conformations of odd-
membered cycloalkanes would not be strain free as they were not superimposable on
the diamond lattice, and that for even-membered rings between C 6 and C i 4 no totally
strain free conformations were possible either since the diamond lattice conformations
would have intraannular interactions between internally oriented hydrogen atoms.94,95
The qualitative recognition of low energy diamond lattice conformations was
followed by exploratory calculations of strain energies in medium and large rings.
Semi-quantitative calculations of the enthalpies of medium and large rings were
performed by Dale using Dreiding models.96 These models have the correct carbon-
carbon bond lengths and tetrahedral bond angles. The dihedral angles of the
macrocycles were manually determined, and compared to a butane potential energy
curve in order to determine the dihedral torsion energies. Subsequently, Anet and

33
coworkers have reported the strain energies of medium and large rings as determined
with molecular mechanics calculations.97,98
1.2.5 Conformational Analysis of 14-Membered Rings
From these analyses, the 14-membered ring was predicted to exist largely in a
quadrangular diamond lattice conformation with two four-bond sides in the anti
configuration joined by two parallel three-bond sides with gauche torsional angles at
the joints. The 14-membered ring in this diamond lattice conformation was the first
large ring in which the transannular interactions were small. This preferred
conformation also contained minimal torsion and bond angle strain, and therefore was
designated as being "strain-free".99 In addition to this lowest energy diamond lattice
conformation of cyclotetradecane, the calculations also suggested the existence of two
low energy non-diamond lattice conformations.96
To determine all of the diamond lattice conformations that were theoretically
possible for cyclotetradecane, Saunders used a ring building program. A total of 13
diamond lattice conformations were found, but as expected, most of these possessed
severe transannular interactions.100 With the exception of the one lowest energy
diamond lattice conformation, the strain energy of the remaining diamond lattice
conformations were calculated to be higher than the two non-diamond lattice
conformations found earlier. The energies of these remaining diamond lattice
conformations ranged from 3-12 kcal/mol above the lowest energy conformation. Thus,
a total of 15 possible conformations were found for cyclotetradecane including 13
diamond lattice conformations and two non-diamond lattice conformations.

34
side view top view
Figure 15. The lowest energy diamond lattice conformation of cyclotetradecane.
The rectangular nature of the lowest energy conformation of cyclotetradecane is
easily recognized from the top view. This shows the conformation to have four atoms
located at the "corners" of the rectangle (Figure 15). These corner atoms are flanked
on either side by gauche dihedral angles that are themselves flanked by anti dihedral
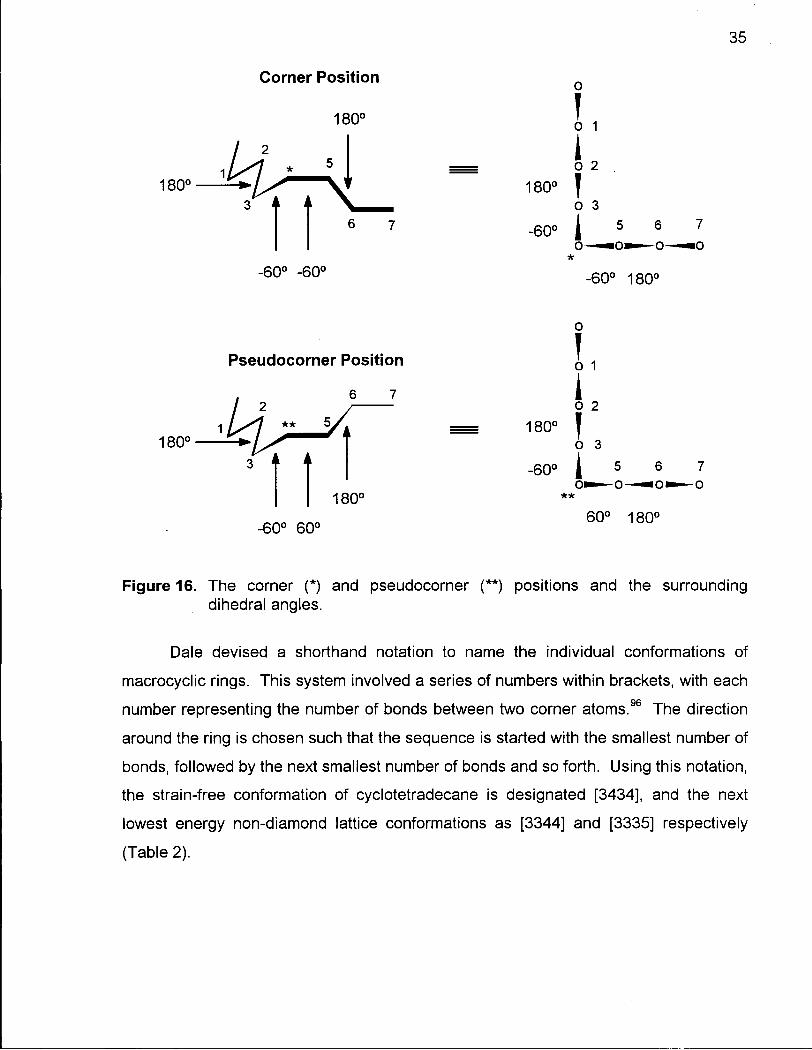
angles (Figure 16). A corner atom is formally defined as an atom flanked by gauche
dihedral angles of the same sign with anti dihedral angles surrounding the gauche
torsions. This is the lowest energy arrangement of dihedral angles about a corner
atom.
Another type of corner has been recognized by Dale and coworkers from an
X-ray crystal study of 1,4,8,11-tetraoxacyclotetradecane,101 and by Neeland during the
study of some 14-membered lactones.102 This involves an atom with gauche dihedral
angles on either side, further flanked by anti dihedral angles, but the gauche dihedral
angles have opposite sign (i.e. 180°, -60°, 60°, 180°) (Figure 16). This arrangement
was termed a pseudocorner102 and is higher in energy than the corner arrangement
described above.
35
180°
Corner Position
180°
-60° -60°
180°
-60°
o I i L ! O 3 | 5 6
k
-60° 180°
7
180°
Pseudocorner Position
6 7
180°
-60°
-60° 60°
o I , i O 2
!, J 5 6 O ^ — O — ^ O i
ft
60° 180°
7 -o
Figure 16. The corner (*) and pseudocorner (**) positions and the surrounding dihedral angles.
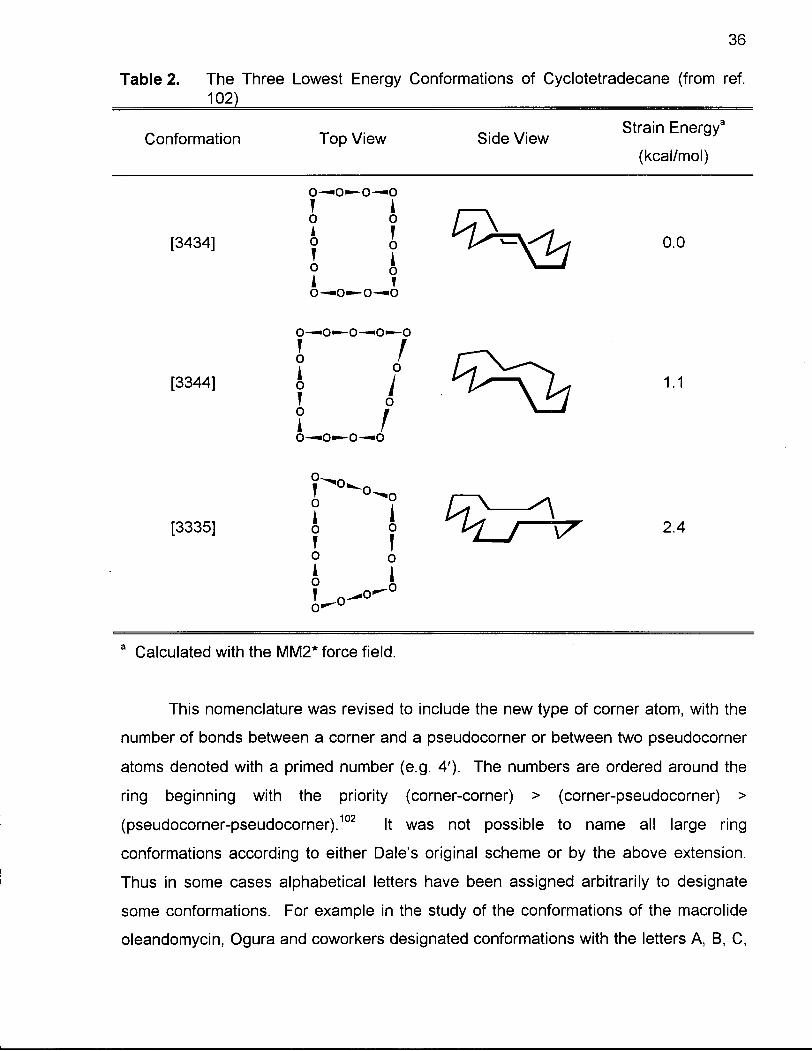
Dale devised a shorthand notation to name the individual conformations of
macrocyclic rings. This system involved a series of numbers within brackets, with each
number representing the number of bonds between two corner atoms.96 The direction
around the ring is chosen such that the sequence is started with the smallest number of
bonds, followed by the next smallest number of bonds and so forth. Using this notation,
the strain-free conformation of cyclotetradecane is designated [3434], and the next
lowest energy non-diamond lattice conformations as [3344] and [3335] respectively
(Table 2).
36
Table 2. The Three Lowest Energy Conformations of Cyclotetradecane (from ref. 102}
Conformation Top View Side View Strain Energy3
(kcal/mol)
o—o—o—o
[3434] o 6 i ^ ^ C l ^ i 0.0
o—o—o—o
o—o—o—o—o
' 1 [3344] o I
1 0—0—0—0
• 1
1.1
[3335] o 6 ' \7 2.4
a Calculated with the MM2* force field.
This nomenclature was revised to include the new type of corner atom, with the
number of bonds between a corner and a pseudocorner or between two pseudocorner
atoms denoted with a primed number (e.g. 4'). The numbers are ordered around the
ring beginning with the priority (corner-corner) > (corner-pseudocorner) >
(pseudocorner-pseudocorner).102 It was not possible to name all large ring
conformations according to either Dale's original scheme or by the above extension.
Thus in some cases alphabetical letters have been assigned arbitrarily to designate
some conformations. For example in the study of the conformations of the macrolide
oleandomycin, Ogura and coworkers designated conformations with the letters A, B, C,

37
and D.103,104 In this case, conformation A has been shown to be the same as the [3434]
conformation of cyclotetradecane.
The [3434] conformation of cyclotetradecane belongs to the C2h symmetry point
group. It contains four diastereotopic methylene groups which experience varying
degrees of transannular steric interactions (Figure 17). With the exception of the
corner methylenes, all other methylenes have at least one hydrogen atom pointed into
the ring, with the endo-hydrogen of the methylene at the centre of the four-bond side
having the most severe steric interaction. In contrast, the hydrogen atoms of the corner
methylenes are both directed to the outside of the ring. Accordingly, these positions
are best able to accommodate geminal substitution without suffering the severe
transannular interaction which would result from geminal substitution at other locations
on the ring.105 In general, there is a preference for a geminally substituted carbon to be
located first at a corner atom, followed next at a pseudocorner atom, and finally at a
non-corner atom.
number of transannular interactions
Figure 17. Transannular hydrogen interactions in cyclotetradecane.
That the preferred conformation of cyclotetradecane in the solid state is actually
the [3434] diamond lattice conformation has been experimentally determined with X-ray
crystallographic studies performed by Groth.106 This study gave carbon-carbon bond
lengths of 1.53 A for cyclotetradecane, and average bond angles of 114.6° for all
angles with the exception of the middle of the four-bond side which had a bond angle of
112.3°. Spectroscopy studies including NMR studies performed by Anet and
coworkers,107 and by Moller and coworkers108 as well as IR and Raman studies
performed by Shannon et al.109 are in agreement with this conformation being the major

38
conformer in solution. The conformation of other 14-membered macrocycles including:
1,3,8,10-tetraoxacyclotetradecane (80),1 1 0 cyclotetradecanone (81),1 1 1 and cyclotetra
decane oxime (82) 1 1 2 have also been determined by X-ray crystallographic studies.
The conformation of the ring was found to be [3434] in all cases with some disorder in
the location of the carbonyl of the macrocyclic ketone.
O
81 82
1.2.6 Conformational Analysis of 13-Membered Rings
In comparison to 14-membered rings, little is known experimentally about the
conformation of 13-membered rings. This ring size falls on the borderline between
medium and large sized rings. The diamond lattice has been used to define idealized
geometries for even-membered macrocyclic rings, but odd-membered rings are not
superimposable on this lattice. As a strain-free diamond lattice geometry is not
accessible, the conformations of the odd-membered rings are predicted to be more
strained as a result of distorted bond lengths, bond angles, and dihedral angles.
However, bond length distortion can be minimized in either 3- or 5-sided conformational
minima. Semi-quantitative calculations on the conformation of 13-membered rings
performed by Dale using molecular models suggested five low energy conformations.96
More accurate values have been reported by Anet and Rawdah from iterative force field
calculations the results of which also indicate five low energy conformations.98
However, the comparative energies and ordering of the minima differ between the two
calculations. Anet and Rawdah concluded that the [13333] conformation was the
global minimum conformation with the [12433] conformation only 1.4 kcal/mol higher in
energy (Table 3). Three triangular conformations were found to have the next lowest
strain energies. The [346] conformation was calculated to have a strain energy of
1.6 kcal/mol with the [445] and [355] conformations at 2.9 kcal/mol, and 3.3 kcal/mol

39
higher in energy relative to the [13333] conformation.98 The conformation set proposed
by Dale had the [12433] conformation as the global minimum conformation.96 A close
geometric relationship exists between the triangular and the quinquangular
conformations in that the sign of the torsion angles around the 1-bond side of the
quinquangular conformations alternate in exactly the same fashion as in the
corresponding near anti bonds of the triangular conformation.113
Table 3. The Two Lowest Energy Conformations of Cyclotridecane (from ref. 98)
Conformation Top View Side View Strain Energy3
(kcal/mol)
[13333]
o
\ ? o o 0—0—0—0
0.0
[12433]
0—0—0—0
T 1 0 o 1 ! 0 0 T i 1 / 0—0—0
1.4
a Calculated with the MOL-BUILD program.
The conformation of some 13-membered compounds have been determined.
The X-ray crystal structures have been reported for three nitrogen containing
compounds 83-85. Thiolactam 83 1 1 4 and the substituted 13-membered amine 84 1 1 5
were both found to have crystallized in the low energy [13333] conformation, although
some disorder was present in portions of the rings. The nitrogen atom and the carbon
of the thionocarbonyl of 83 were on the corners of the 1-bond side, and the nitrogen
atom of the amine 84 was also on the corner of the 1-bond side. The 13-membered
rings of the bisamine 85 were also found to have the [13333] conformation with the
substituted nitrogen occupying the corner of a 3-bond side of the ring.116
40
^ -J 85
1.2.7 Transition State Theory in Large Rings
The interconversion of conformers occurs as the result of rotation about single
bonds. A knowledge of the energy of the molecule as a function of changes to the
molecular geometry is helpful in rationalizing the mechanistic details of such
transformations. In cyclic molecules, a number of conformational processes have been
described by Anet including: ring inversion, local ring inversion, and ring
pseudorotation.117
An example of ring inversion is the change from one chair form to the alternate
chair form in 6-membered rings. In general, this process involves a change in sign of
all the dihedral angles in the ring with the exception of those dihedral angles that are
close to 0° or to 180°. The magnitude of the dihedral angles, the bond lengths and the
internal angles are either unaffected or only slightly changed. The path followed by this

41
inversion process is not specified, and therefore no particular mechanism for the
process is implied.
A local ring inversion is a conformational process which occurs in only part of
the molecule. The conversion of the chair to the boat in a 6-membered ring is an
example of this type of process. Changes to the signs of only two of the dihedral
angles occurs while the values of the remaining four dihedral angles change in
magnitude, but not sign.
Ring pseudorotation is a conformational process that results in a conformation,
which is superimposable on the original. This new conformation may differ from the
original conformation by an apparent rotation about one or more of the molecular axes.
Minor changes in the ring skeleton that may occur as a result of the pseudorotation
process are ignored. This process was first used to describe the conformational
properties of cyclopentane. The atoms of this ring apparently rotate around the ring
with each atom residing in the flap position of the envelope conformation for a portion
of the time.
A conformational isomer or conformer is a structure corresponding to a
conformational energy minima. The transition state between these minima is the lowest
energy "pass" between the pair of conformational minima. Whether a single step or a
sequence of steps are involved in the conformational exchange process is difficult to
determine experimentally, but a knowledge of the geometry and symmetry of the
populated conformations can assist in the suggestion of the interconversion
mechanism. Additional support for the mechanism can be provided by a comparison
with data from qualitative or quantitative calculations of the relative strain energies of
the conformational minima and the transition states that separate them.
A mechanism for the interconversion of cyclic conformations has been proposed
by Dale involving the movement of a single corner atom within the ring.118 This process
can result in the exchange of both ring atoms and ring substituent sites. In a manner
similar to that used for the determination of the geometry and strain energies of

42
possible conformational minima described earlier,96 Dale used molecular models and
calculated butane dihedral torsion energies to calculate the barriers between
conformational minima.118 He proposed the most favourable transition state to have a
0 torsional angle between the new and the old corner atoms which become eclipsed
during the conformational interconversion (Figure 18).
The corner atom which is flanked by two gauche dihedral angles of the same
sign, can be moved by one position in the ring with a resultant change in sign for both
of the gauche dihedral angles about the new corner. This process proceeds through a
transition state with the bond between the new and the old corner atoms eclipsed while
the two adjacent bonds have 120° dihedral angles of opposite sign.118 Further rotation
of the ring bonds gives a new conformation with the old corner atom shifted onto a side
of the conformation, and gauche dihedral angles of opposite sign around the new
corner atom. These local or partial conformational changes can occur without major
geometric changes occurring elsewhere in the molecule.113
Figure 18. Movement of a corner atom by one position with an accompanying change in sign of the surrounding gauche dihedral angles.
The transition state structures for such conformational processes can be
designated in a similar fashion to that used by Dale for conformational minima. The
syn eclipsed bond of the transition state is considered to be a one-bond side, and this
number is written in italics to differentiate the transition state structure from that of
conformational minima.118 In general, n-sided conformations have (n+1)-sided barriers.

43
In cyclotetradecane, the [3434] lowest energy conformation would proceed to the
higher energy [3344] conformation by passing over the [73343] conformational barrier.
After several more repetitions of this process, the atoms are rotated around the ring
and complete site exchange of both ring atoms and substituents can occur (Figure 19).
Dale calculated this barrier to be 13.8 kcal/mol higher in energy than the [3434]
conformation.118 There is also the possibility of an alternate pathway proceeding from
the [3344] conformation over the [73334] barrier to the less stable [3335] intermediate
conformation. This barrier was calculated to be 13.0 kcal/mol higher than the [3434]
conformation.118 Conformational interconversion over this alternate barrier would lead
to exchange of carbon atoms only and not of the substituent hydrogen atoms.113
Passing through the [3434] conformation in the middle of the first interconversion
pathway has the effect of exchanging geminal substituents and after six repetitions, all
hydrogen sites are exchanged.113 These calculated transition state barriers were found
to be too high because of approximations made in the calculations. For example, Anet
and coworkers have reported that the transition state barriers for cyclotetradecane are
approximately 7.0 kcal/mol based on 1H and 13C DNMR studies.107
[3335]
Figure 19. Conformation interconversion pathways for cyclotetradecane as the result of the single corner movement mechanism.

44
The conformational minima of the 13-membered rings were more complex than
that of the 14-membered rings, and the conformational interconversion processes are
also thought to be complicated.116 The interconversion paths for 13-membered rings
have been described as "complex and interwoven".113 The lowest energy [13333] and
[12433] conformational minima can interconvert by passing over the [721333] barrier.
However, if these 5-sided conformers first interconvert to their triangular partners, the
barrier to interconversion is thought to lie even lower in energy.118 The [346]
conformation, which was calculated to have a strain energy of 2.9 kcal/mol, can also
interconvert with the [445] conformation that can in turn interconvert over the [7444]
barrier. This process would lead to complete site exchange in the molecule. Dale has
calculated this conformational barrier at 7.2 kcal/mol.118 However, the barriers
calculated by Dale have been shown to be too high, so the actual value should be
lower. Initial 13C DNMR studies of cyclotridecane gave only a single line at
temperatures as low as -135 °C.98 This indicates a rapid rate of pseudorotation with the
conformational barrier estimated at 6 kcal/mol.

CHAPTER 2 45
RESULTS AND DISCUSSION
Synthesis and Conformational Analysis of 14-Membered Macrocyclic Ethers
Some conformational analyses of macrocyclic compounds have been reported in
the literature, and a few of these were presented in Chapter 1. Large ring monoethers
have received little attention with oxacyclooctane the largest cyclic ether previously
studied.33 As part of an ongoing study of the chemistry of macrocyclic compounds in
our laboratory, methods for the synthesis of 14-membered unsubstituted cyclic ethers,
and 14-membered cyclic ethers with alkyl substituents both adjacent to and remote
from the ether oxygen were examined. Once prepared, the conformational properties
of these macrocyclic ethers were analyzed using NMR spectroscopic techniques, and
molecular mechanics calculations. The conformational preferences of these
macrocyclic ethers, the location of the ether oxygen in the conformation, and the effect
of alkyl substituents, especially gem-dimethyl substituents on the conformation of these
ethers were of key interest. We also hoped to gain an increased understanding of the
conformational interconversion processes of these macrocycles and of the associated
transition state energies.
Replacing a methylene group in a large ring with an oxygen atom is believed to
have a limited affect on the ring conformation. However, the elimination of some
hydrogen atoms as a result of such a substitution can lead to a reduction of the number
of steric interactions, particularly transannular interactions, in the molecule. The effect
of alkyl substitution on the conformation of the ring is also of interest. Whereas an
oxygen atom would be expected to be located at a position with the. most severe
hydrogen interactions in the parent hydrocarbon, an alkyl substituent would be
expected to be found at a position having the fewest hydrogen steric interactions in the
parent hydrocarbon. Since each of the four possible diastereotopic ring sites in the
[3434] conformation of a 14-membered ring has at least one hydrogen pointing outside

46
the ring, monosubstitution should be readily accommodated. However, for the case of
gem-disubstituted molecules, the substituted carbon must be at a corner position.
Severe steric interactions would result from its placement elsewhere in the ring.
2.0.1 Synthesis of 14-Membered Macrocyclic Ethers
The general synthetic strategy for the preparation of the macrocyclic ethers in
this study involved the ring expansion of a cyclic ketone to a lactone, thereby
eliminating the potential problem of forming the macrocycle via a cyclisation reaction.
The endocyclic oxygen of the lactone ultimately became the macrocyclic ether oxygen.
This lactone functionality was used to introduce substitution in the vicinity of the ether
oxygen. Once this role was served, the carbonyl was removed to give the macrocyclic
ether with a procedure developed by Nicolaou and coworkers55 using 2,4-bis(4-
methoxyphenyl)-1,3,2,4-dithiadiphosphetane-2,4-disulfide, Lawesson's reagent 48, 4 8 to
give an intermediate thionolactone. This strategy allowed for the production of a
variety of macrocyclic ethers as the result of variations in the substitution pattern of the
ketone (R1 and R2) and lactone (R4 and R5) from the alkylation reactions, and the
nucleophile (R3) used in the Nicolaou conversion (Scheme 4).
48
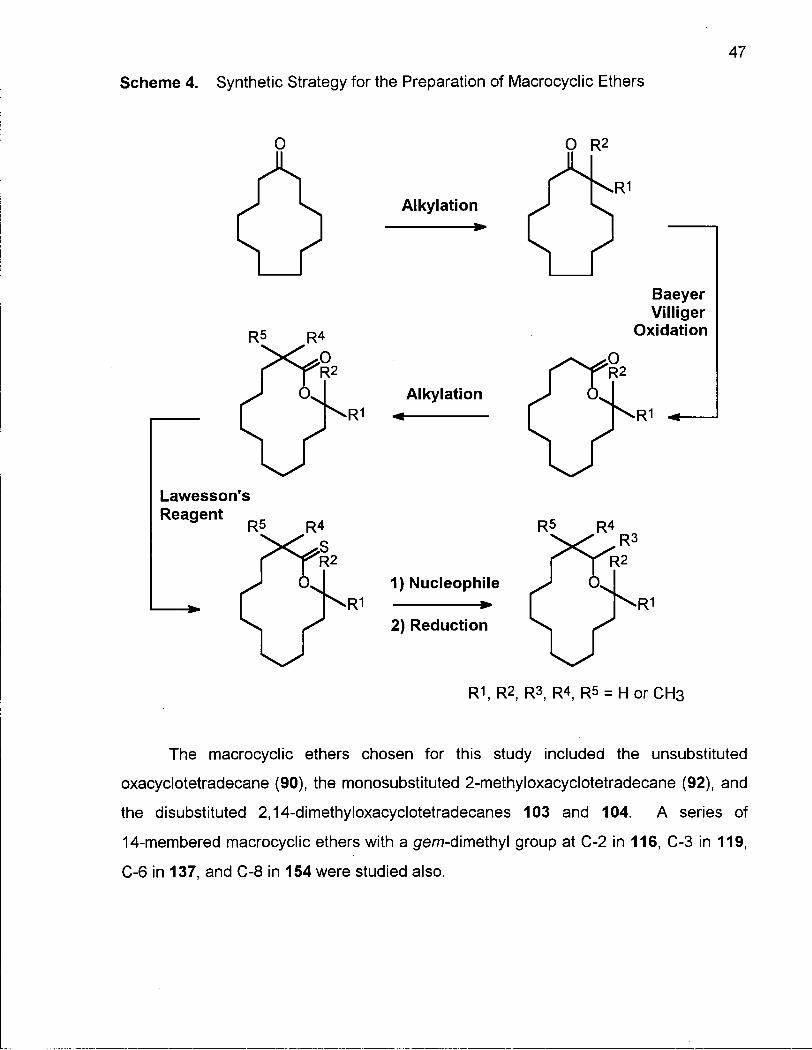
47
Scheme 4. Synthetic Strategy for the Preparation of Macrocyclic Ethers
0 O R 2
Baeyer Villiger
Oxidation
R 1 , R 2 , R3, R 4 R5 = H or C H 3
The macrocyclic ethers chosen for this study included the unsubstituted
oxacyclotetradecane (90), the monosubstituted 2-methyloxacyclotetradecane (92), and
the disubstituted 2,14-dimethyloxacyclotetradecanes 103 and 104. A series of
14-membered macrocyclic ethers with a gem-dimethyl group at C-2 in 116, C-3 in 119,
C-6 in 137, and C-8 in 154 were studied also.

48
104 116 119
137 154
2.0.2 Conformational Analysis of 14-Membered Macrocyclic Ethers
The conformations of these macrocyclic ethers were analyzed using both NMR
spectroscopy and molecular mechanics calculations. The data obtained from a series
of 1- and 2-D NMR experiments (1H, 13C, NOE, COSY, HMQC, HMBC) were used to
assign as much of the spectra as possible. Although the introduction of the oxygen
atom did offer some dispersion in the chemical shifts of the signals of the atoms close
to the ether oxygen, approximately half of the methylenes in each molecule were
remote enough from the ether oxygen to experience very little of this dispersion effect.
Accordingly, the signals of many of the ring methylenes overlapped, and the complete
assignment of the macrocyclic ether NMR spectra was not possible, even at high-field

49
(1H, 500 MHz). This problem was also encountered in the study of the parent
hydrocarbon.107 Once the chemical shifts and coupling constants of a particular ether
were determined, any anomalous values indicative of key conformational features could
be identified.
A series of DNMR experiments were performed to provide further information
about the conformation of these cyclic ethers. Since the molecules undergo rapid site
exchange at rt, these DNMR studies were performed at colder temperatures with 135 K
as an approximate lower temperature limit. This temperature limit was a function of
both the melting point of the solvent system, and the solubility of the cyclic ethers at
these cold temperatures. Experiments could be performed at temperatures as low as
100 K on the spectrometer used, however the solvent systems could not be used to
such low temperatures. These DNMR studies provided information about the
interconversion of the conformations through the processes of ring inversion, local ring
inversion, and pseudorotation, in addition to the thermodynamic barriers for these
processes.
As the temperature is lowered in the DNMR studies, the rates of the
conformational interconversion processes slow, and the signals of individual protons
change as the effects of site exchange slow. The changes in the chemical shift of
various protons as a result of the electronegativity of neighbouring atoms, steric effects
from intramolecular van der Waals repulsions, and diamagnetic anisotropic effects from
both the type and the orientation of the neighbouring chemical bonds is used to
rationalize the conformation of the molecule both at rt and at the lower temperatures.
The results of molecular mechanics calculations are used to assist the rationalization of
the experimental data in an effort to more fully describe the conformational properties
of the compounds studied.
To simplify the comparison of the 14-membered macrocyclic ether
conformations, an extension of the Dale nomenclature was developed to designate the
position of the ether oxygen atom in the conformation. The [3434] conformation of
cyclotetradecane, contains four diastereotopic ring positions. These are numbered

50
starting with position-1 at the middle of a 4-bond side. In the low energy, non-diamond
lattice [3344] conformation, the positions were referred to in a similar manner again
beginning with position-1 at the middle of a 4-bond side and continuing around the ring
through the adjacent 3-bond side. Using this nomenclature, the [3434] conformation of
oxacyclotetradecane (90) with the ether oxygen in the middle of a 4-bond side would be
the [3434]-1 conformation.
[3434] [3344]
2.1.1 Synthesis of Oxacyclotetradecane (90) and 2-Methyloxacyclotetradecane (92)
The first macrocyclic ether in this study, oxacyclotetradecane (90), was prepared
via the Baeyer-Villiger oxidation of cyclotridecanone (86) with trifluoroperacetic acid to
give 13-tridecanolide (87). The peracid was generated by the addition of either 70%
H202 solution119,120 or solid urea hydrogen peroxide (UHP)121 to a solution of
trifluoroacetic anhydride (TFAA) in CH2CI2. The UHP method was superior usually
giving higher yields of the desired lactone.122,123 The UHP reaction was also easier to
perform since the Na2HP04 buffer tended to form a difficult to stir paste with the water
present in the 70% H202 solution. The lactone was converted into thionolactone 88
with Lawesson's reagent 48. The 1H NMR spectrum of the resultant oil showed two-
proton multiplets between 4.46-4.48 ppm and 2.85-2.88 ppm for the C-13 methylene
and the C-2 methylene protons of 88 respectively. The 13C NMR spectrum contained a
signal at 224.66 ppm for the C-1 thionocarbonyl. The HRMS and chemical analysis
results were also consistent with the composition of 88.
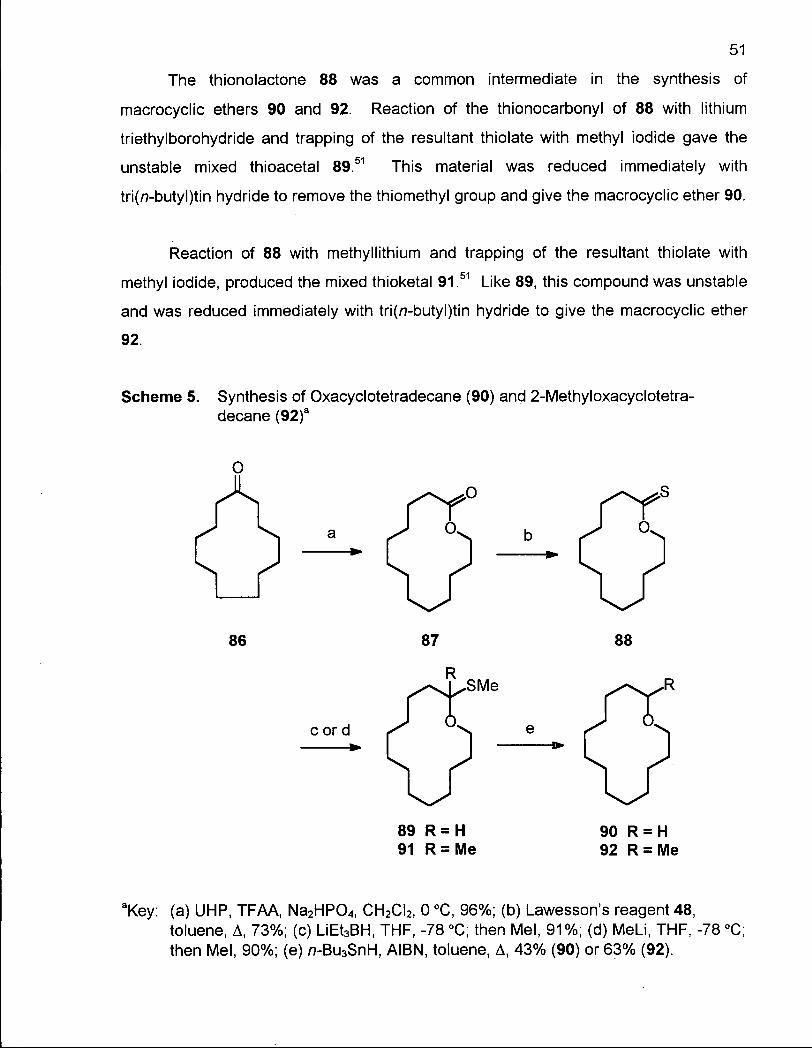
51
The thionolactone 88 was a common intermediate in the synthesis of
macrocyclic ethers 90 and 92. Reaction of the thionocarbonyl of 88 with lithium
triethylborohydride and trapping of the resultant thiolate with methyl iodide gave the
unstable mixed thioacetal 89. 5 1 This material was reduced immediately with
tri(/7-butyl)tin hydride to remove the thiomethyl group and give the macrocyclic ether 90.
Reaction of 88 with methyllithium and trapping of the resultant thiolate with
methyl iodide, produced the mixed thioketal 91. 5 1 Like 89, this compound was unstable
and was reduced immediately with tri(n-butyl)tin hydride to give the macrocyclic ether
92
Scheme 5. Synthesis of Oxacyclotetradecane (90) and 2-Methyloxacyclotetradecane (92)a
O
89 R = H 90 R = H 91 R = Me 92 R = Me
aKey: (a) UHP, TFAA, Na2HP04, CH2CI2, 0 °C, 96%; (b) Lawesson's reagent 48, toluene, A, 73%; (c) LiEt3BH, THF, -78 °C; then Mel, 91%; (d) MeLi, THF, -78 °C; then Mel, 90%; (e) A7-Bu3SnH, AIBN, toluene, A, 43% (90) or 63% (92).

52
2.1.2 Conformational Analysis of Oxacyclotetradecane (90)
The 1H NMR spectrum of oxacyclotetradecane (90) at rt in CDCI3 contained a
four-proton triplet at 3.41 ppm, a four-proton quintet at 1.57 ppm, a 16-proton multiplet
from 1.29-1.43 ppm, and a two-proton multiplet from 1.21-1.27 ppm. The low-field
signals at 3.41 ppm and 1.57 ppm were assigned to the C-2/C-14 and C-3/C-13 protons
based on their proximity to the electronegative ether oxygen. The results of the HRMS
and chemical analysis were also consistent with the composition of 90.
The 13C NMR spectrum of 90 contained seven signals. The assignments of
these signals can be found in Table 4. The simplicity of these spectra indicate that 90
is undergoing rapid exchange on the NMR timescale, and that the exchange results in
a conformation with a plane of symmetry. Each carbon resonance corresponded to a
pair of methylenes in the macrocyclic ether with the exception of the signal at
23.19 ppm that was half the height of the other signals, and was assigned to C-8 on
this basis. The location of this carbon was opposite to the ether oxygen leaving it
without a symmetrical carbon partner. The signal at 68.58 ppm was assigned to
C-2/C-14, the carbons adjacent to the ether oxygen. These carbons were expected to
have the lowest field signal as the result of their proximity to the electronegative ether
oxygen. The remaining 13C and 1H signals were assigned with the aid of COSY and
HMQC 2D-NMR experiments. The chemical shift of the signals for the C-6 and C-7
methylenes were very similar and the unambiguous assignment of these signals was
not possible. The rt NMR spectra of oxacyclotetradecane (90) are consistent with the
[3434]-1 conformation.
[3434]-1
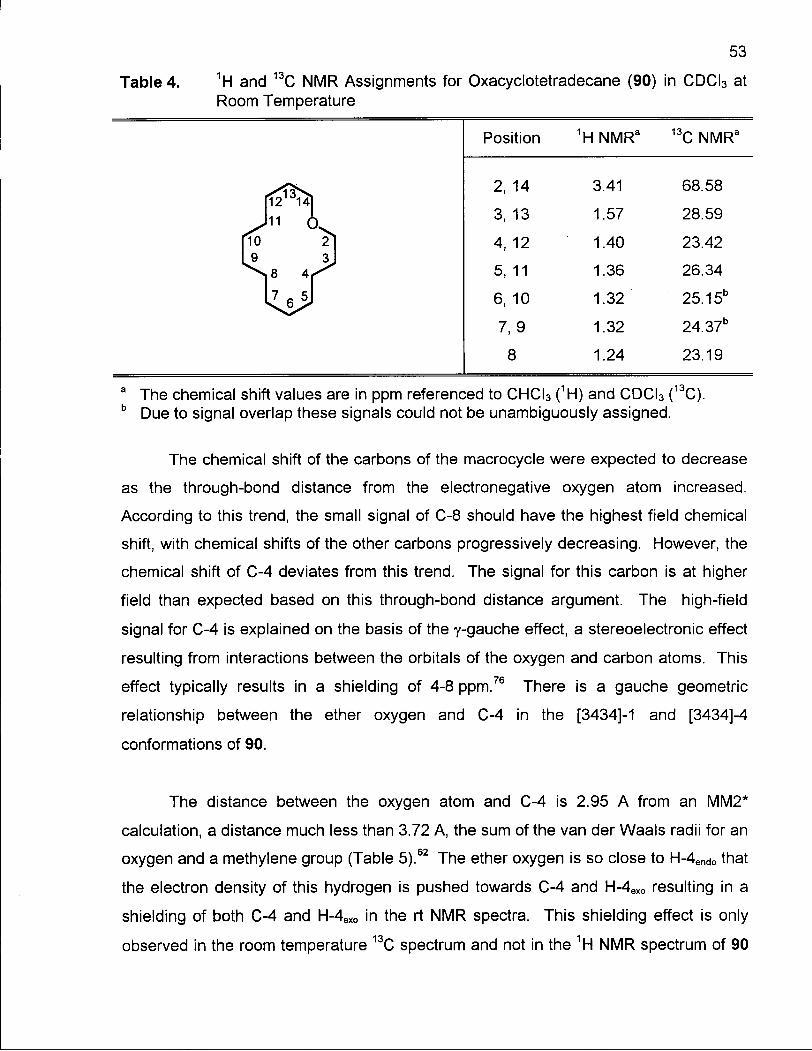
53
Table 4. 1H and 13C NMR Assignments for Oxacyclotetradecane (90) in CDCI3 at Room Temperature
Position 1H NMRa 13C NMRa
3.41 68.58
1.57 28.59
1.40 23.42
1.36 26.34
1.32 25.15b
1.32 24.37b
1.24 23.19
a The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI3 (13C). b Due to signal overlap these signals could not be unambiguously assigned.
The chemical shift of the carbons of the macrocycle were expected to decrease
as the through-bond distance from the electronegative oxygen atom increased.
According to this trend, the small signal of C-8 should have the highest field chemical
shift, with chemical shifts of the other carbons progressively decreasing. However, the
chemical shift of C-4 deviates from this trend. The signal for this carbon is at higher
field than expected based on this through-bond distance argument. The high-field
signal for C-4 is explained on the basis of the y-gauche effect, a stereoelectronic effect
resulting from interactions between the orbitals of the oxygen and carbon atoms. This
effect typically results in a shielding of 4-8 ppm.76 There is a gauche geometric
relationship between the ether oxygen and C-4 in the [3434]-1 and [3434]-4
conformations of 90.
The distance between the oxygen atom and C-4 is 2.95 A from an MM2*
calculation, a distance much less than 3.72 A, the sum of the van der Waals radii for an
oxygen and a methylene group (Table 5).62 The ether oxygen is so close to H-4endo that
the electron density of this hydrogen is pushed towards C-4 and H-4exo resulting in a
shielding of both C-4 and H-4exo in the rt NMR spectra. This shielding effect is only
observed in the room temperature 13C spectrum and not in the 1H NMR spectrum of 90

54
due in part to the overlap of signals in the 1H NMR spectrum in the region of the C-4
proton signals. Also, since the ring is conformationally mobile at rt, any effects
experienced by the C-4 methylene protons are averaged between both H-4exo and
H~4endo-
Table 5. van der Waals Radi for Some Atom Groups3
van der Waals radii
H 1.20 A
0 1.52 A
CH2 1.70 A
3 From ref. 62.
The low temperature spectra of 90 were obtained in a 4:1 mixture of CHCI2F
(Freon21) and CHCIF2 (Freon 22) as solvent. Using this mixed solvent system the
data could be collected over a broader range of temperatures than the more common
NMR solvents such as methanol and methylene chloride-d2 that freeze at 175 K and
178 K respectively. Experiments were performed in the mixed freon solvent at
temperatures as low as 135 K. These freons are protio solvents, and the DNMR
experiments were performed without a deuterium signal lock. The magnitude of the
freon solvent peaks was quite large relative to the macrocyclic ether signals, but the
freon signals had chemical shifts of 7.5 ppm and 7.2 ppm, and were observed in the 1H NMR spectra downfield from the macrocyclic ether signals where they did not
interfere with the analysis.
A series of low temperature 1H NMR experiments were performed on
oxacyclotetradecane (90) (Figure 20). The 1H NMR spectrum of 90 at 220 K contained
four signals of relative integration 4:4:16:2, similar to the rt spectrum with the
multiplicity of the signals lost at the lower temperature. At 200 K the high-field signal
for the C-8 methylene protons was no longer visible, and at 190 K the signal for the C-3
protons coalesced into the methylene envelope, and was no longer distinct. At 180 K,
the signal for the C-2 protons adjacent to the ether oxygen broadened. Some new

55
signals were also visible downfield of the methylene envelope at 1.84 ppm and
1.61 ppm, and upfield of the methylene envelope at 1.04 ppm and 0.57 ppm. At 175 K,
the C-2 methylene signal continued to broaden, and the signals downfield of the
methylene envelope became more distinct. At 165 K, the signal for the C-2 methylenes
split into three signals clustered around 3.4 ppm. The intensities of these partially
overlapping signals were approximately equal. The relative integration of the six
signals visible in the spectrum collected at 165 K was approximately 4:2:4:11:4:1.
Further cooling to 135 K, the lowest temperature in this series of DNMR experiments,
did not produce significant changes in the line shape of the spectrum of 90.
As the temperature was lowered, the 1H NMR spectrum of 90 changed as a
result of the slowing of both ring-site and ring substituent exchange. The
rationalization of these spectral changes began with the protons of C-2 adjacent to the
ether oxygen. This signal progressively broadened until at 165 K, it split into three
signals with chemical shifts of 3.43, 3.40 and 3.38 ppm. The relative intensity of these
signals was 1.2:1:1 based on their peak height. As the temperature was lowered, the
process leading to ring inversion and averaging of the C-2 proton signals was slowed,
and the signals for H-2endo and H-2exo became distinct. In the [3434]-1 conformation,
H-2exo is deshielded by the anisotropy of the C-3/C-4 bond, with a corresponding
shielding of the H-2endo proton. The H-2end0 proton is deshielded by a van der Waals
steric interaction with H-5end0 leading to a shielding of H-2eXo- Here, these steric and
anisotropic shielding effects are opposed, and are expected to partially cancel.
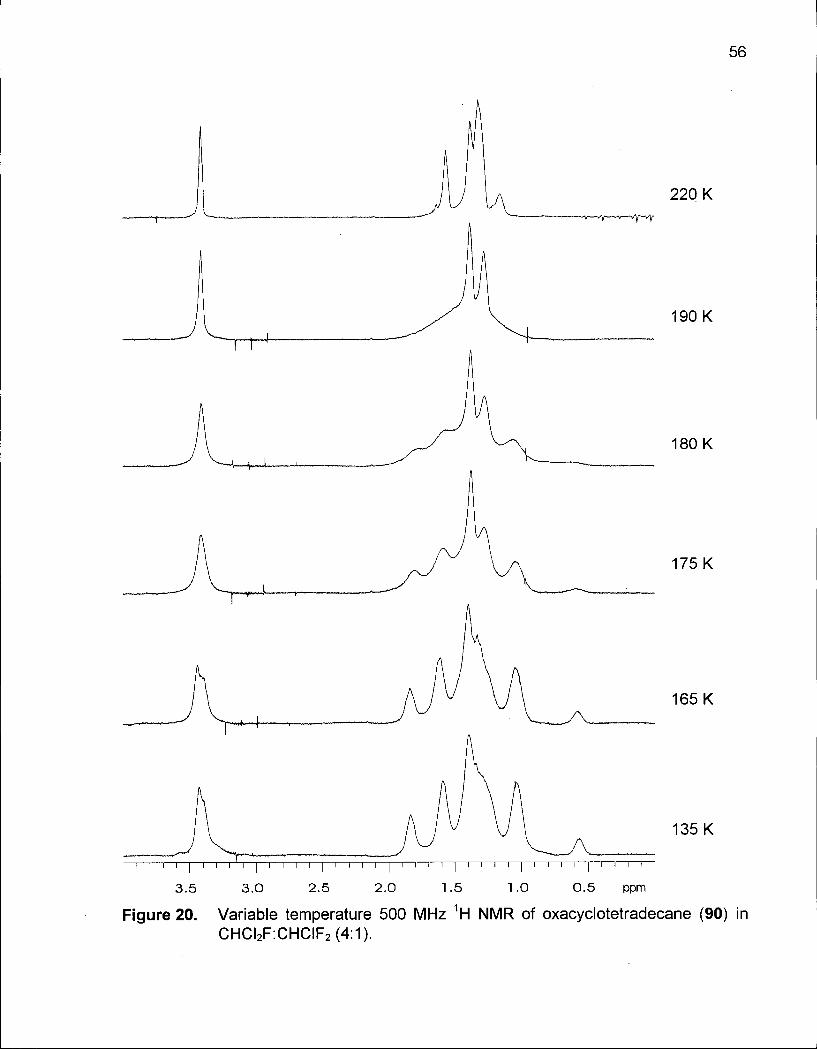

5 7
A large vicinal coupling constant was expected between H-2endo and H-3P in the
[3434]-1 conformation while all other coupling constants for the C-2 protons were
predicted to be small. It was for this reason that the low-field portion of the multiplet at
3.43 ppm was assigned to the H-2exo proton, and the two high-field portions at 3.40 ppm
and 3.38 ppm were assigned to the H-2end0 proton. The presence of several small
coupling constants in the complex pattern, and the broadened line shape at low
temperature contribute to the slightly higher intensity of the H-2exo portion of the
multiplet.
The line shape of the DNMR spectra of 90 indicated the presence of a single
major conformation at low temperature. This conformation was suggested by a
comparison of the predicted line shapes of the signals for the protons adjacent to the
ether oxygen in the low energy conformations of 90 using molecular models and MM2*
calculations.
In the [3434]-4 conformation, the corner C-2 methylene protons, and the C-14
protons are predicted to have different line shapes. The H-2 P proton is deshielded as a
result of the diamagnetic anisotropy of the O/C-14 bond. The H-2 a proton is deshielded
by the anisotropy of the C-3/C-4 bond. These deshielding effects are predicted to be of
a similar magnitude.84 Thus, only a small AS was predicted for the C-2 protons. The
H-14 e x o proton is deshielded by the anisotropy of the C-12/C-13 bond between C-12
and C-13, and shielded by a van der Waals steric interaction between H-14 e nd 0 and
H-11endo- The magnitude of these anisotropy and steric effects is unknown. Since the
C-2 and C-14 protons are in different environments, the line shape in the low
a
[3434]-4

58
temperature spectra for these methylene protons is predicted to be symmetric, but
complex with more lines than are visible here. Therefore, this conformation was not
considered to be a a highly populated conformation of 90.
The low temperature spectra contained a high-field signal at 0.57 ppm of relative
integration 1:4 in comparison to the signals at 3.4 ppm of the protons adjacent to the
ether oxygen. The high-field signal is assigned to H-8exo because of the following
rationalization. In the [3434]-1 conformation, the H-8endo proton is deshielded by van
der Waals steric interactions with the H-5endo/H-11 e n d o protons. This leads to a shielding
of the H-8exo proton. No transannular steric repulsion between H-8end0 and the ether
oxygen appears possible based on the MM2* calculated distance between these atoms
which is 3.10 A. The H-8exo proton is further shielded by electric field effects caused by
the parallel bonds of the C-6a and C-10a protons. The sum of these effects causes an
upfield shift of the H-8exo proton. The signal for H-8end0 is believed to overlap with the
methylene envelope. There are no protons in the [3344]-1 conformation that possess
the correct geometry to give this upfield signal since the distorted geometry of this
conformation does not allow for an alignment of these shielding effects. Thus, the low
temperature spectra of 90 are consistent with the presence of a single conformer; the
[3434]-1 conformation in which both ring inversion and pseudorotation have slowed.
The C-3/C-13 corner protons could be assigned in the low temperature spectra
of 90. The C-3P proton is deshielded by the diamagnetic anisotropy of the C-4/C-5
bond, and by the O/C-2 bond. These effects reinforce each other to give a large A 8
with a chemical shift of 1.84 ppm for the H-3P/H-13P protons and 1.61 ppm for the
H-3a/H-13a protons. The upfield signal of the H-3a/H-13a protons overlapped that of
two other protons, but insufficient information is available to unambiguously identify
these other protons. The signals of the remaining protons overlap between
1.0-1.5 ppm, and can not be unambiguously assigned either.
A molecular mechanics search for the low energy conformations of 90 was
conducted using the Monte Carlo technique and the MM2* force field. The global

59
minimum conformation was the [3434]-1 conformation 90-A with the [3344]-1
conformation 90-B calculated to have the next lowest energy, 0.99 kcal/mol higher.
These calculations suggested the existence of three other low energy conformations
within 2 kcal/mol of the global minimum conformation (Table 6). Higher energy
conformations were ignored as they were not considered to be significantly populated
over the temperature range studied. The relative populations of these conformations at
different temperatures were calculated from enthalpy values (AH°) obtained from the
MM2* calculations, and entropy values (AS°) considering both symmetry and mixing
term contributions (Table 7). The entropic symmetry component takes into account the
principle that conformations with high symmetry have low entropy as calculated by
equation 7 where R is the ideal gas constant, and a is the symmetry number of the
conformer in question. The symmetry of mixing component is applied when the
conformer is chiral. Since both enantiomeric conformations are equally populated, this
increases the entropy value by a factor of R(ln2) or 1.38 cal/mol. The results of these
calculations suggest the [3434]-1 conformation of 90 to be the major conformation over
the temperature range studied in agreement with the DNMR data.
ASsvm = -R In a (7)
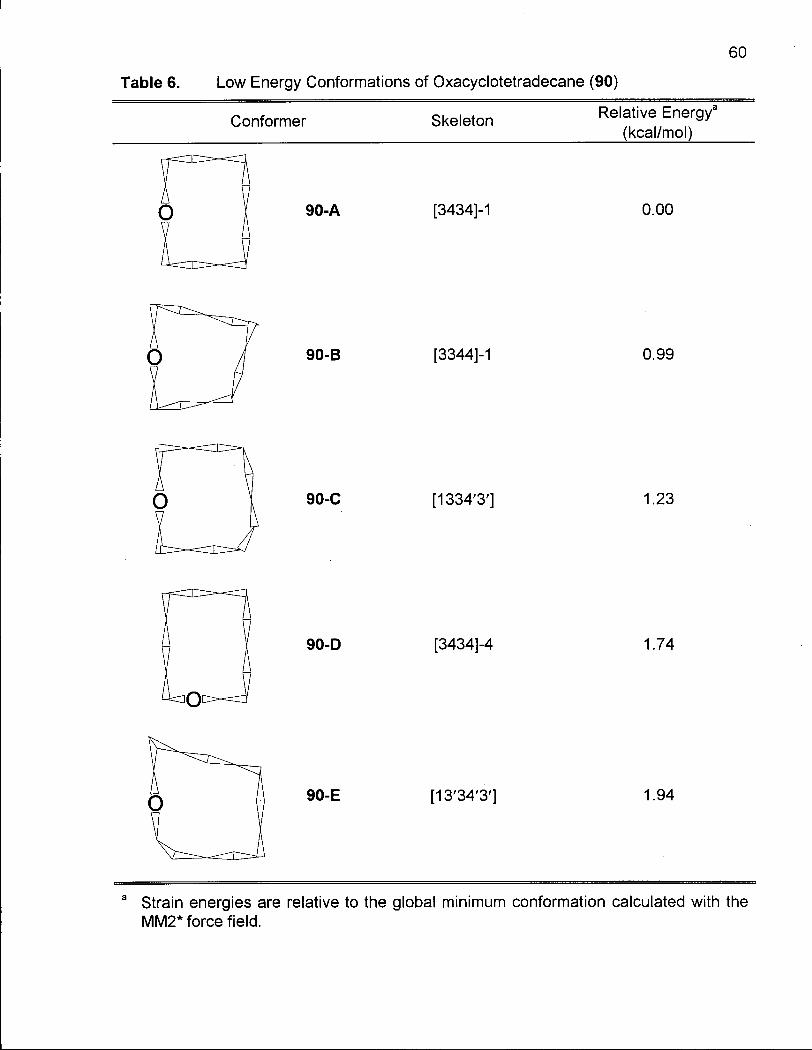
60
Table 6. Low Energy Conformations of Oxacyclotetradecane (90)
Conformer Skeleton Relative Energy (kcal/mol)
90-A [3434]-1 0.00
90-B [3344]-1 0.99
90-C [1334'3'] 1.23
90-D [3434]_4 1.74
90-E [13'34'3'] 1.94
a Strain energies are relative to the global minimum conformation calculated with the MM2* force field.
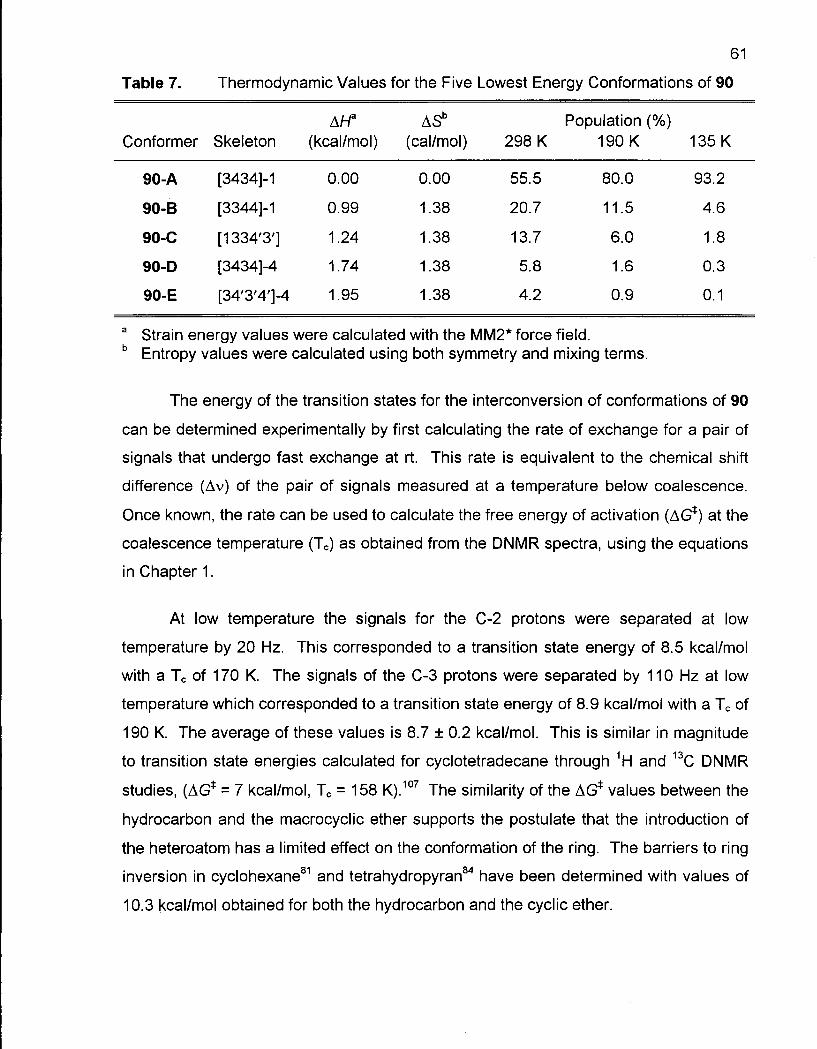
61
Table 7. Thermodynamic Values for the Five Lowest Energy Conformations of 90
ASb Population (%) Conformer Skeleton (kcal/mol) (cal/mol) 298 K 190 K 135 K
90-A [3434]-1 0.00 0.00 55.5 80.0 93.2
90-B [3344]-1 0.99 1.38 20.7 11.5 4.6
90-C [1334'3'] 1.24 1.38 13.7 6.0 1.8
90-D [3434]_4 1.74 1.38 5.8 1.6 0.3
90-E [34'3'4']-4 1.95 1.38 4.2 0.9 0.1
a Strain energy values were calculated with the MM2* force field. b Entropy values were calculated using both symmetry and mixing terms.
The energy of the transition states for the interconversion of conformations of 90
can be determined experimentally by first calculating the rate of exchange for a pair of
signals that undergo fast exchange at rt. This rate is equivalent to the chemical shift
difference (Av) of the pair of signals measured at a temperature below coalescence.
Once known, the rate can be used to calculate the free energy of activation (AG*) at the
coalescence temperature (Tc) as obtained from the DNMR spectra, using the equations
in Chapter 1.
At low temperature the signals for the C-2 protons were separated at low
temperature by 20 Hz. This corresponded to a transition state energy of 8.5 kcal/mol
with a Tc of 170 K. The signals of the C-3 protons were separated by 110 Hz at low
temperature which corresponded to a transition state energy of 8.9 kcal/mol with a Tc of
190 K. The average of these values is 8.7 ± 0.2 kcal/mol. This is similar in magnitude
to transition state energies calculated for cyclotetradecane through 1H and 13C DNMR
studies, (AG* = 7 kcal/mol, Tc = 158 K).107 The similarity of the A G * values between the
hydrocarbon and the macrocyclic ether supports the postulate that the introduction of
the heteroatom has a limited effect on the conformation of the ring. The barriers to ring
inversion in cyclohexane81 and tetrahydropyran84 have been determined with values of
10.3 kcal/mol obtained for both the hydrocarbon and the cyclic ether.

62
Dale has proposed a mechanism for these conformational interconversions that
involves the movement of a single corner atom in the starting conformation. The
transition state has an eclipsed torsional angle between the old-corner atom and an
adjacent non-corner atom. This adjacent atom becomes the corner atom in the new
conformation. The other dihedral angles in the ring undergo a minimum of change
during this interconversion process.113 The repetition of this movement at other corner
positions can lead to site exchange of both ring atoms and substituents. This
mechanism is more complicated for 90 than for a hydrocarbon since more possible
transition state structures exist as a result of the ether oxygen atom. Consequently, the
energies of all possible transition states were not determined. The energy of a [73343]
transition state structure for the interconversion of the [3434]-1 and [3344]-1
conformations 90-A and 90-B was calculated to be 12.9 kcal/mol using the dihedral
drive method124 with 10° increments of the appropriate dihedral angles (Figure 21).
This calculated value was larger than the observed AG* value, however, the difference
between the experimental and calculated transition state energy values may be due to
the inaccuracy of the assumption that the dihedral angles of the 1-bond side and the
adjacent bonds were exactly 120°, 0°, -120°. Also, minimization of the dihedral angles
in the remainder of the ring may have lead to a better agreement of the experimental
and theoretical values.
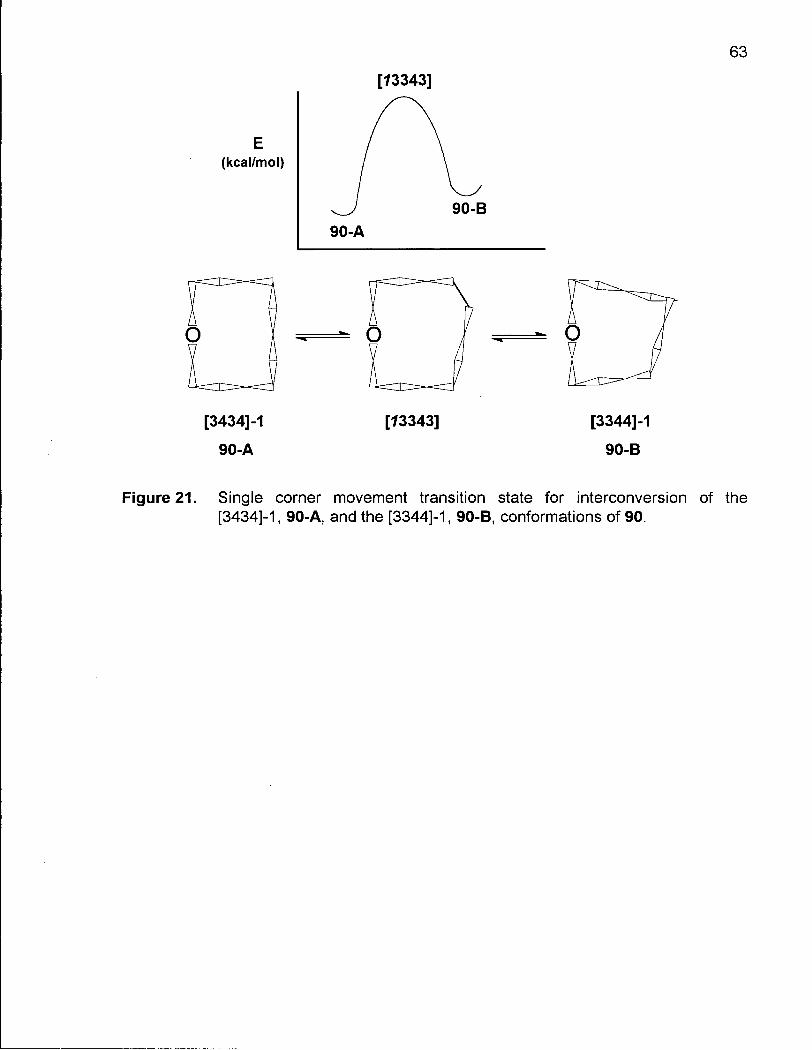
63
[73343]
[3434]-1 [73343] [3344]-1
90-A 90-B
Figure 21. Single corner movement transition state for interconversion of the [3434]-1, 90-A, and the [3344]-1, 90-B, conformations of 90.

64
2.1.3 Conformational Analysis of 2-Methyloxacyclotetradecane (92)
The 1H NMR spectrum of 2-methyloxacyclotetradecane (92) at rt in CDCI3
contained a one-proton doublet of triplets at 3.61 ppm, a one-proton doublet of doublet
of quartets at 3.43 ppm, a one-proton doublet of doublet of doublets at 3.22 ppm, a
22-proton multiplet from 1.10-1.73 ppm, and a three-proton doublet at 1.09 ppm. The
three low-field signals between 3 and 4 ppm were assigned to the C-2 methine and the
C-14 methylene protons. These three protons were unambiguously assigned with a 1H COSY spectrum that showed a correlation between the signals at 3.61 ppm and
3.22 ppm. These signals were assigned to the C-14 methylene, and the remaining
signal at 3.43 ppm was assigned to the C-2 methine proton. A series of 1H NOE
difference experiments were used to differentiate between the C-14 methylene protons.
Irradiation of the signal at 3.61 ppm showed an enhancement of the 3.22 ppm geminal
H-14 signal, while irradiation at 3.22 ppm showed an enhancement of both the geminal
H-14 signal at 3.61 ppm, and the methine H-2 signal at 3.43 ppm. Thus, it was
determined that the 3.22 ppm signal corresponded to the H-14 proton in a conformation
syn to the methine H-2 proton (Figure 22). The high-field doublet at 1.09 ppm was
assigned to the C-15 methyl group. The results of the HRMS and chemical analysis
were also consistent with the composition of 92.
92
Figure 22. 1H NMR assignments of the C-2 and C-14 protons of 2-methyloxacyclotetradecane (92) from COSY and NOEDS experiments.
3.43 ppm
H CH
The 13C NMR spectrum of 92 contained 14 lines, two of which were at low-field,
and were assigned to the C-2 and C-14 carbons. The highest field carbon at

65
19.82 ppm was assigned to the C-15 methyl group. The balance of the 13C signals
were visible around 25 ppm. The assignment of the remaining 13C and 1H signals was
aided with COSY, HMQC, and HMBC 2D-NMR experiments (Table 8). Unfortunately,
due to the overlap of signals in the 1H NMR spectrum, and the small A5 between
several signals in the 13C NMR spectrum, not all of the signals could be assigned.
Table 8. 1H and 13C NMR Assignments for 2-Methyloxacyclotetradecane (92) in CDCI3 at Room Temperature
Position 1H NMRa 13C NMRa
2
3
3.43
1.50, 1.39
73.32
36.42
4-11 not assigned'
12 1.57, 1.23 22.99
13 1.69, 1.45 29.00
14 3.22, 3.61 65.99
15 1.09 19.82
a The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI; > (13C). Due to signal overlap these signals could not be unambiguously assigned.
As in the case of oxacyclotetradecane (90), the chemical shifts of the ring
carbons of 92 were expected to decrease as the through-bond distance from the ether
oxygen increased. The chemical shift of C-12 deviated from this trend as a result of the
y-gauche effect, with an observed upfield chemical shift to 22.99 ppm.
Examination of the coupling constants for the H-2 and H-14 protons provided
some information about the preferred conformation of this macrocyclic ether. The H-2
proton had a large and small coupling constant to the adjacent methylene protons
(Table 9). If the C-15 methyl group is exo and the ether oxygen is in the middle of a
4-bond side, the H-2 proton would be endo to the ring. In this orientation, both large
and small coupling constants are predicted between H-2 and the protons at C-3
(Figure 23). In contrast, if the ether oxygen is adjacent to the corner on a 3-bond side

66
in the [3434]-4 conformation, the C-15 methyl group occupies a corner position. Since
a carbon-oxygen bond is shorter than a carbon-carbon bond, a C-15p methyl group is
preferred as the 1,3-interaction between a C-15a methyl group and H-14exo is greater
than the 1,3-interaction between the C-15p methyl group and H-14exo. In this
conformation, no large coupling constants are expected between H-2 and the C-3
protons (Figure 23).
[3434]-1 [3434]-4
H CH3
H-2 Proton
Figure 23. Newman projections of 92 showing the geometry of C-2 in the [3434]-1 and [3434]-4 conformations.
The coupling constants for the H-2 and H-14 protons in the [3434]-1, [3434]-4,
and [3344]-1 conformations of 92 were calculated and compared to the actual values
(Table 9). The calculated coupling constants for H-2 in the [3434]-4 conformation and
H-14exo in the [3344]-1 conformation with C-14 adjacent to a 4-bond side were in poor
agreement with the observed values. These conformations were not predicted to be
major conformations of 92.
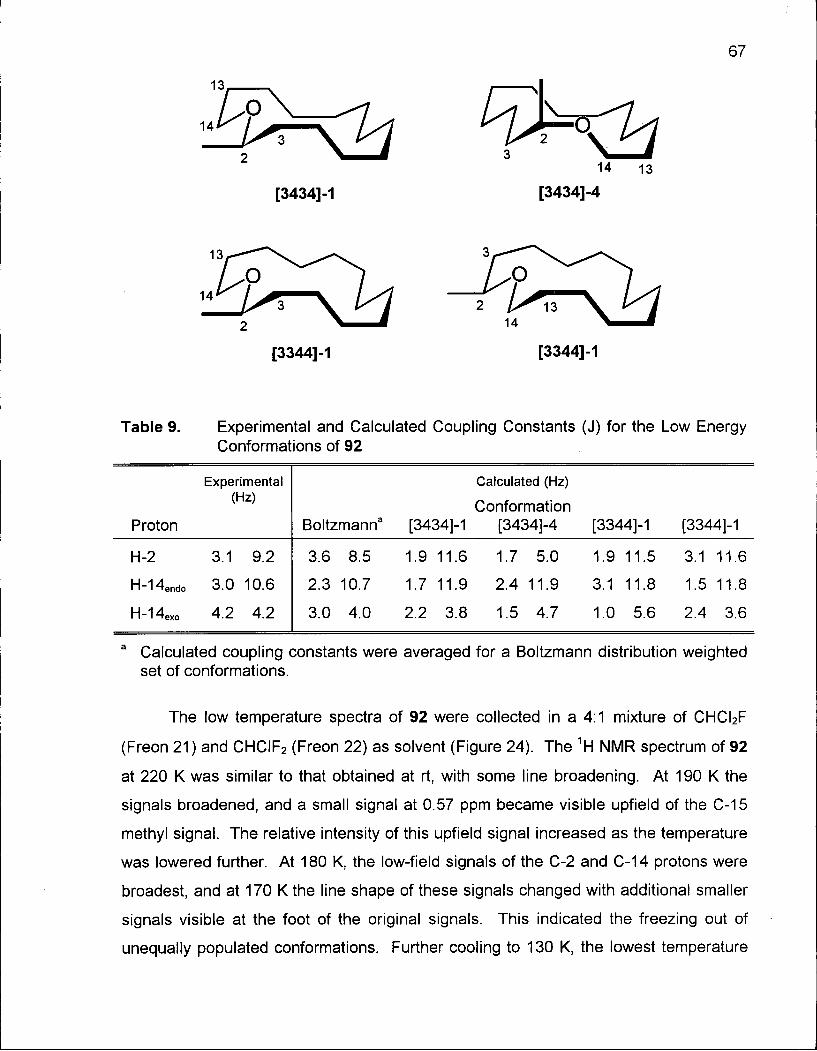
67
[3344]-1 [3344]-1
Table 9. Experimental and Calculated Coupling Constants (J) for the Low Energy Conformations of 92
Proton
Experimental (Hz)
Boltzmann3 [3434]-1
Calculated (Hz)
Conformation [3434]-4 [3344]-1 [3344]-1
H-2 3.1 9.2 3.6 8.5 1.9 11.6 1.7 5.0 1.9 11.5 3.1 11.6
H-14endo 3.0 10.6 2.3 10.7 1.7 11.9 2.4 11.9 3.1 11.8 1.5 11.8
H-14exo 4.2 4.2 3.0 4.0 2.2 3.8 1.5 4.7 1.0 5.6 2.4 3.6
3 Calculated coupling constants were averaged for a Boltzmann distribution weighted set of conformations.
The low temperature spectra of 92 were collected in a 4:1 mixture of CHCI2F
(Freon 21) and CHCIF2 (Freon 22) as solvent (Figure 24). The 1H NMR spectrum of 92
at 220 K was similar to that obtained at rt, with some line broadening. At 190 K the
signals broadened, and a small signal at 0.57 ppm became visible upfield of the C-15
methyl signal. The relative intensity of this upfield signal increased as the temperature
was lowered further. At 180 K, the low-field signals of the C-2 and C-14 protons were
broadest, and at 170 K the line shape of these signals changed with additional smaller
signals visible at the foot of the original signals. This indicated the freezing out of
unequally populated conformations. Further cooling to 130 K, the lowest temperature

68
in this series of DNMR experiments, did not produce further significant changes in the
line shape of spectra of 92.
The additional small signals present in the low-field portion of the spectra at
3.71, 3.34, and 3.03 ppm belong to a minor conformation or conformations of 92. The
similarity of the chemical shifts of the major signals at low-field over the temperature
range studied suggests that the major conformation is the same at both rt and low
temperature. Additional small signals were expected between 1.5 and 2 ppm as well,
but no such signals were observed. Presumably, these were concealed by the signals
of protons in the major conformation also visible in that region. Examination of the
spectra in the region of the C-15 methyl signal at 1.03 ppm at low temperature, showed
other signals at 0.90 and 1.16 ppm. The integration of the signal at 0.90 ppm was 1:3.4
relative to the minor signals at 3.34 and 3.03 ppm, but whether this upfield signal can
be assigned as a C-15 methyl signal of a minor conformer, or to other major conformer
proton signals is unclear.
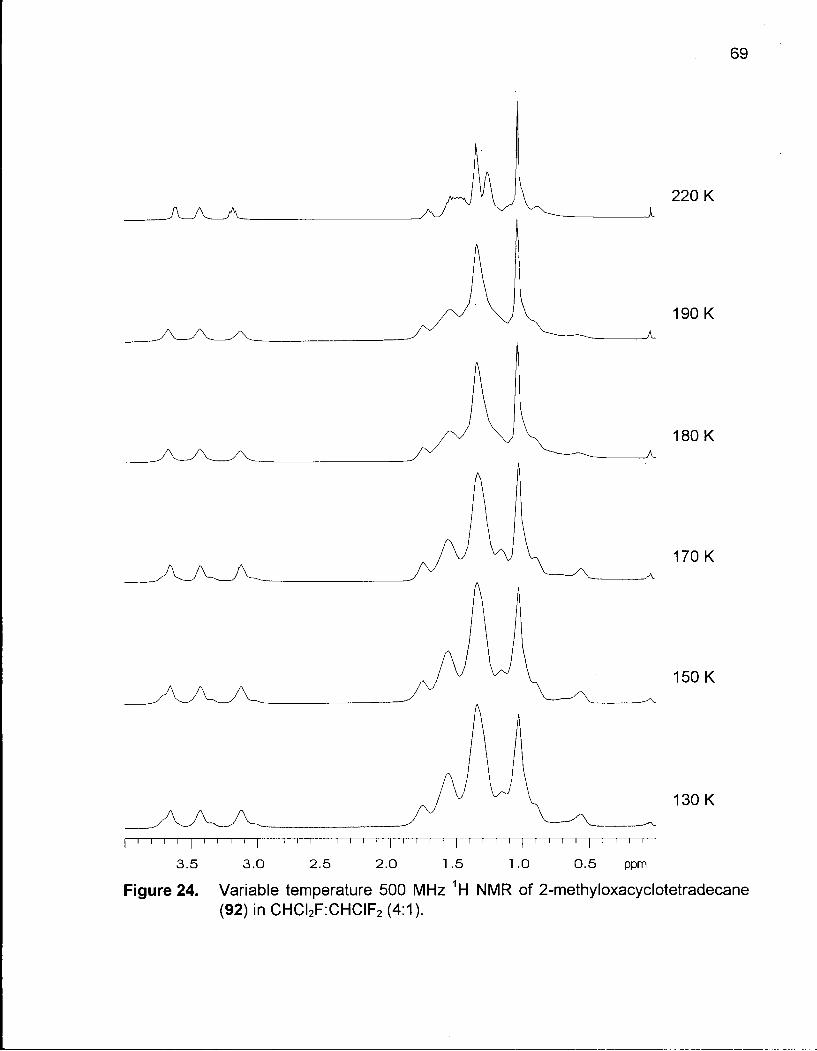
69

70
The high-field signal at 0.57 ppm in the low temperature spectra of 92, is similar
to that of the high-field signal observed in the DNMR study of oxacyclotetradecane
(90). The relative integration of this high-field signal and the signal at 3.12 ppm for
H-14 e x o of the major conformer is approximately 1:1. In the [3434]-1 conformation of 92,
the C-8 protons are expected to have a geometry similar to that of the C-8 protons in
90. The H-8endo proton is deshielded by steric interactions with H-5endo and H-11 e n d 0 that
are calculated to be separated from H-8endo by 2.22 A and 2.22 A respectively. This
leads to a shielding of H-8 e x o, and results in the upfield shift of this proton signal to
0.57 ppm in the low temperature spectra of 92. In contrast, the geometry of the
[3434]-4 conformation would lead to two high-field signals in the low temperature
spectra. The [3344]-1 conformation does not have the correct geometry to cause the
large shielding effect of a single proton as observed. Thus, the [3434]-1 conformation
is believed to be the major conformation of 92. In this conformation, the A5 of the
H-14endo and H-14 e x o protons can be rationalized as a deshielding of the H-14 e x o proton
by the diamagnetic anisotropy of the C-12/C-13 bond.
A molecular mechanics search for the lowest energy conformations of 92 was
conducted using the Monte Carlo technique and the MM2* force field. These
calculations gave a total of 13 conformations within 2 kcal/mol of the global minimum
conformation, the [3434]-1 conformation 92-A (Table 10). The second lowest energy
conformation was the [3434]-4 conformation 92-B with the methyl substituent at a
corner position. The relative populations of these low energy conformations at different
temperatures were calculated from relative energy values obtained from the MM2*
calculations (Table 11). Since macrocyclic ether 92 is a chiral compound, there were
no symmetry contributions to the entropy. The results of these calculations suggest the
[3434]-1 conformation of 92 to be the major conformation over the temperature range
studied in agreement with the DNMR data.
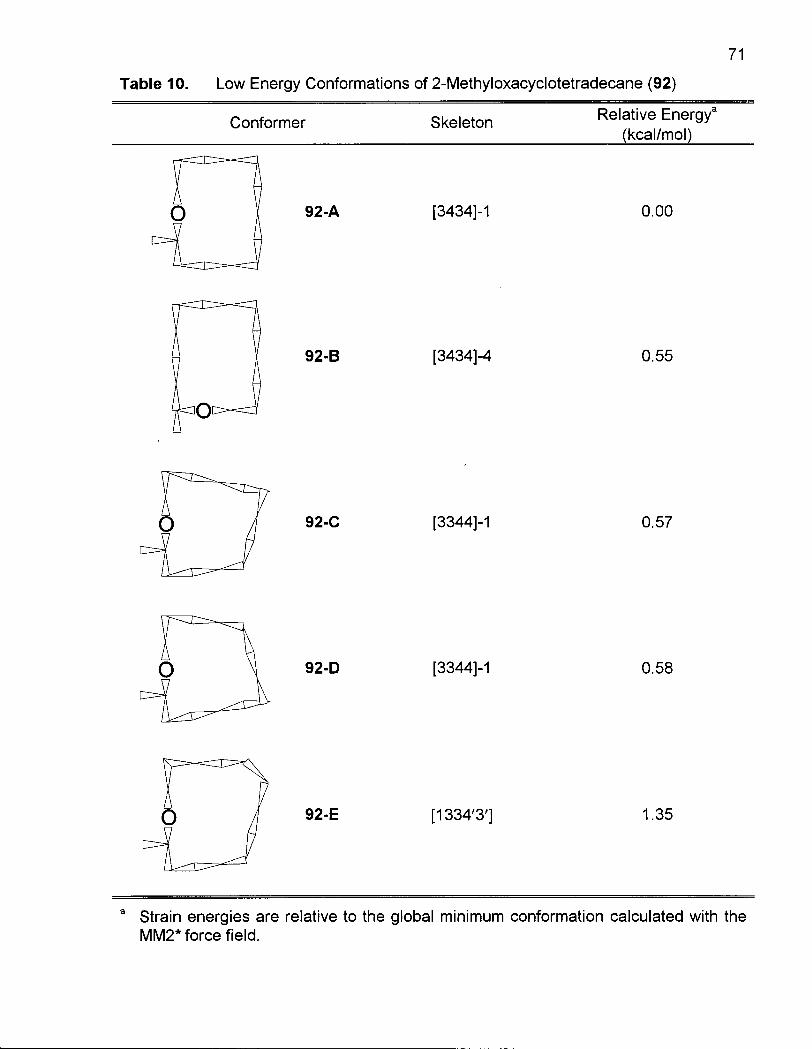
71
Table 10. Low Energy Conformations of 2-Methyloxacyclotetradecane (92)
a Strain energies are relative to the global minimum conformation calculated with the MM2* force field.
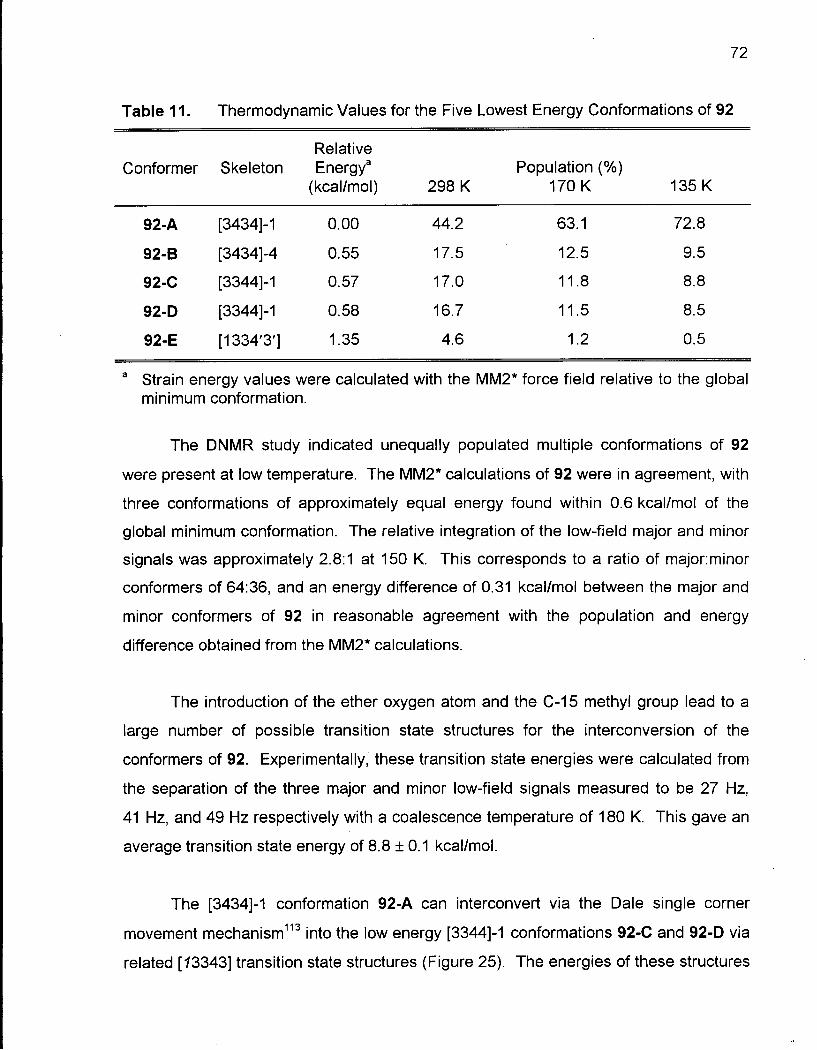
72
Table 11. Thermodynamic Values for the Five Lowest Energy Conformations of 92
Conformer Skeleton Relative Energy3
(kcal/mol) 298 K Population (%)
170 K 135 K
92-A [3434]-1 0.00 44.2 63.1 72.8
92-B [3434J-4 0.55 17.5 12.5 9.5
92-C [3344]-1 0.57 17.0 11.8 8.8
92-D [3344]-1 0.58 16.7 11.5 8.5
92-E [1334'3'] 1.35 4.6 1.2 0.5
a Strain energy values were calculated with the MM2* force field relative to the global minimum conformation.
The DNMR study indicated unequally populated multiple conformations of 92
were present at low temperature. The MM2* calculations of 92 were in agreement, with
three conformations of approximately equal energy found within 0.6 kcal/mol of the
global minimum conformation. The relative integration of the low-field major and minor
signals was approximately 2.8:1 at 150 K. This corresponds to a ratio of majonminor
conformers of 64:36, and an energy difference of 0.31 kcal/mol between the major and
minor conformers of 92 in reasonable agreement with the population and energy
difference obtained from the MM2* calculations.
The introduction of the ether oxygen atom and the C-15 methyl group lead to a
large number of possible transition state structures for the interconversion of the
conformers of 92. Experimentally, these transition state energies were calculated from
the separation of the three major and minor low-field signals measured to be 27 Hz,
41 Hz, and 49 Hz respectively with a coalescence temperature of 180 K. This gave an
average transition state energy of 8.8 ±0.1 kcal/mol.
The [3434]-1 conformation 92-A can interconvert via the Dale single corner
movement mechanism113 into the low energy [3344]-1 conformations 92-C and 92-D via
related [73343] transition state structures (Figure 25). The energies of these structures

73
were calculated with the dihedral drive method124 to be 13.0 kcal/mol and 12.8 kcal/mol
for the interconversion with 92-C and 92-D respectively and involve hydrogen-hydrogen
eclipsing. The interconversion of conformation 92-C with conformation 92-B involves a
[73343] transition state with an eclipsing interaction between the C-15 methyl group
and proton on C-13. The calculated energy of this transition state structure was
9.8 kcal/mol. This structure was expected to be higher in energy than other [73343]
transition states with hydrogen-hydrogen eclipsing only. The calculated transition state
energies were higher than the observed values of 92.
92-D [3434]-4
Figure 25. Interconversion of conformations of 92 via single corner movements.
2.2.1 Synthesis of 2,14-Dimethyloxacyclotetradecanes (103) and (104)
The diastereomeric pair of 2,14-dimethyloxacyclotetradecanes 103 and 104
were the next macrocyclic ethers prepared using the general synthetic strategy
presented earlier. The additional methyl group would be introduced onto the ketone
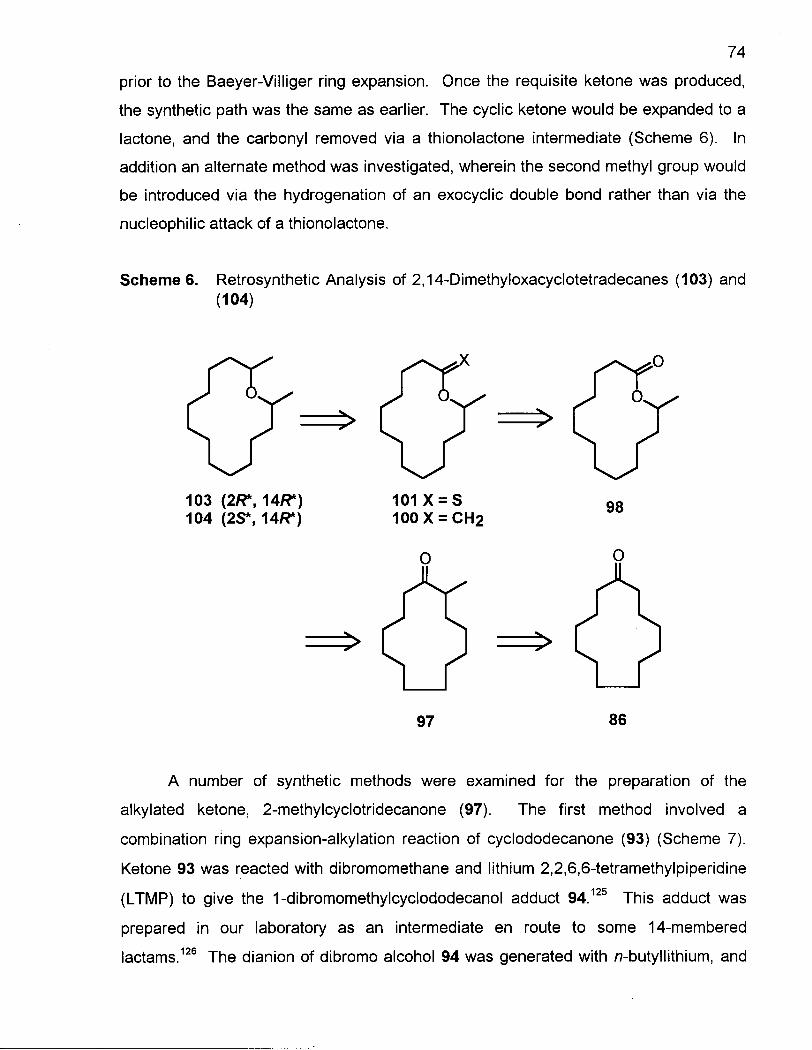
74
prior to the Baeyer-ViNiger ring expansion. Once the requisite ketone was produced,
the synthetic path was the same as earlier. The cyclic ketone would be expanded to a
lactone, and the carbonyl removed via a thionolactone intermediate (Scheme 6). In
addition an alternate method was investigated, wherein the second methyl group would
be introduced via the hydrogenation of an exocyclic double bond rather than via the
nucleophilic attack of a thionolactone.
Scheme 6. Retrosynthetic Analysis of 2,14-Dimethyloxacyclotetradecanes (103) and (104)
104 (2S*, 14/?*) 100X = CH2
97 86
A number of synthetic methods were examined for the preparation of the
alkylated ketone, 2-methylcyclotridecanone (97). The first method involved a
combination ring expansion-alkylation reaction of cyclododecanone (93) (Scheme 7).
Ketone 93 was reacted with dibromomethane and lithium 2,2,6,6-tetramethylpiperidine
(LTMP) to give the 1-dibromomethylcyclododecanol adduct 94. 1 2 5 This adduct was
prepared in our laboratory as an intermediate en route to some 14-membered
lactams.126 The dianion of dibromo alcohol 94 was generated with n-butyllithium, and
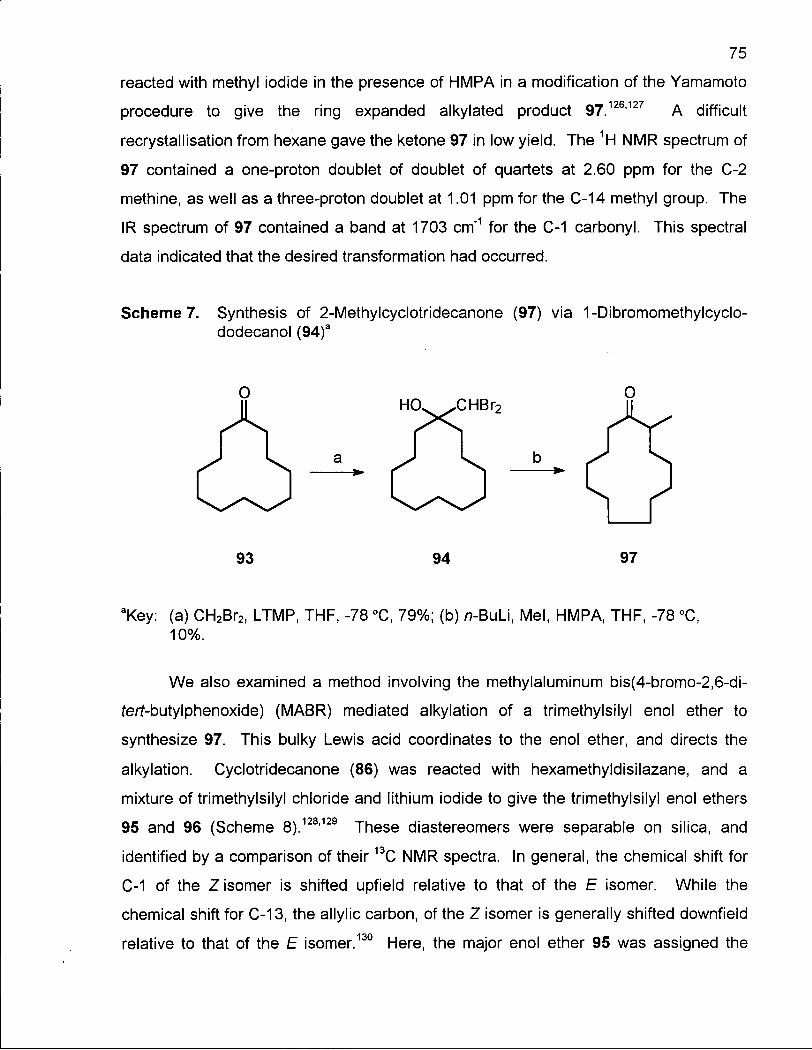
75
reacted with methyl iodide in the presence of HMPA in a modification of the Yamamoto
procedure to give the ring expanded alkylated product 97.126'127 A difficult
recrystallisation from hexane gave the ketone 97 in low yield. The 1H NMR spectrum of
97 contained a one-proton doublet of doublet of quartets at 2.60 ppm for the C-2
methine, as well as a three-proton doublet at 1.01 ppm for the C-14 methyl group. The
IR spectrum of 97 contained a band at 1703 cm"1 for the C-1 carbonyl. This spectral
data indicated that the desired transformation had occurred.
Scheme 7. Synthesis of 2-Methylcyclotridecanone (97) via 1-Dibromomethylcyclo-dodecanol (94)a
93 94 97
aKey: (a) CH2Br2, LTMP, THF, -78 °C, 79%; (b) n-BuLi, Mel, HMPA, THF, -78 °C, 10%.
We also examined a method involving the methylaluminum bis(4-bromo-2,6-di-
fert-butylphenoxide) (MABR) mediated alkylation of a trimethylsilyl enol ether to
synthesize 97. This bulky Lewis acid coordinates to the enol ether, and directs the
alkylation. Cyclotridecanone (86) was reacted with hexamethyldisilazane, and a
mixture of trimethylsilyl chloride and lithium iodide to give the trimethylsilyl enol ethers
95 and 96 (Scheme 8).128,129 These diastereomers were separable on silica, and
identified by a comparison of their 13C NMR spectra. In general, the chemical shift for
C-1 of the Z isomer is shifted upfield relative to that of the E isomer. While the
chemical shift for C-13, the allylic carbon, of the Z isomer is generally shifted downfield
relative to that of the E isomer.130 Here, the major enol ether 95 was assigned the

76
Z configuration based on chemical shifts of 150.17 ppm and 36.11 ppm for C-1 and
C-13 compared to chemical shifts of 151.70 ppm and 29.48 ppm for C-1 and C-13 of
the minor E isomer.
A solution of MABR was generated by the addition of trimethylaluminum in
hexanes to a solution of 4-bromo-2,6-di-fe/f-butylphenol in CH2CI2.131,132 A mixture of
95 and 96 was reacted with an aliquot of this MABR solution and subsequently
alkylated with methyl triflate to give ketone 97.133 This two-step reaction sequence
proceeded in 57% overall yield.
The Baeyer-ViNiger oxidation of ketone 97 was performed with trifluoroperacetic
acid in the presence of Na2HP04 to give 13-tetradecanolide (98). This peracid was
generated by the addition of either 70% H202 solution,119'120 or UHP,122123 to a solution
of trifluoroacetic anhydride in CH2CI2. The reaction with H202 did not go to completion,
and the unreacted ketone was inseparable from the lactone by chromatography. To
obtain pure lactone it was necessary to derivatize the residual ketone into an oxime by
reaction of the mixture of ketone 97 and lactone 18 with hydroxylamine hydrochloride.
The lactone 98 and the oxime 99 were easily separated via column chromatography.
The UHP reaction of 97 did proceed to completion and eliminated the need for this
derivatization step. The lactone 98 was converted into the thionolactone 101 by
reaction with Lawesson's reagent 48.5 1 5 5 The 13C NMR spectrum of 101 contained the
expected 14 lines with a signal at 224.35 ppm indicative of the C-1 thionocarbonyl.
The 1H NMR spectrum contained a one-proton doublet of doublet of quartets at
5.62 ppm for the C-3 methine and a three-proton doublet at 1.30 ppm for the C-14
methyl group. The HRMS and chemical analysis results were also consistent with the
composition of thionolactone 101. The thionolactone 101 was reacted with
methyllithium, and trapping of the resultant sulfur anion with methyl iodide gave the
unstable mixed thioketal 102.5155 This material was reacted immediately under radical
conditions with either tri(n-butyl)tin hydride, or tris(trimethylsilyl)silane (TTMSH),134 to
cleave the thiomethyl group and give the desired macrocyclic ethers 103 and 104. The
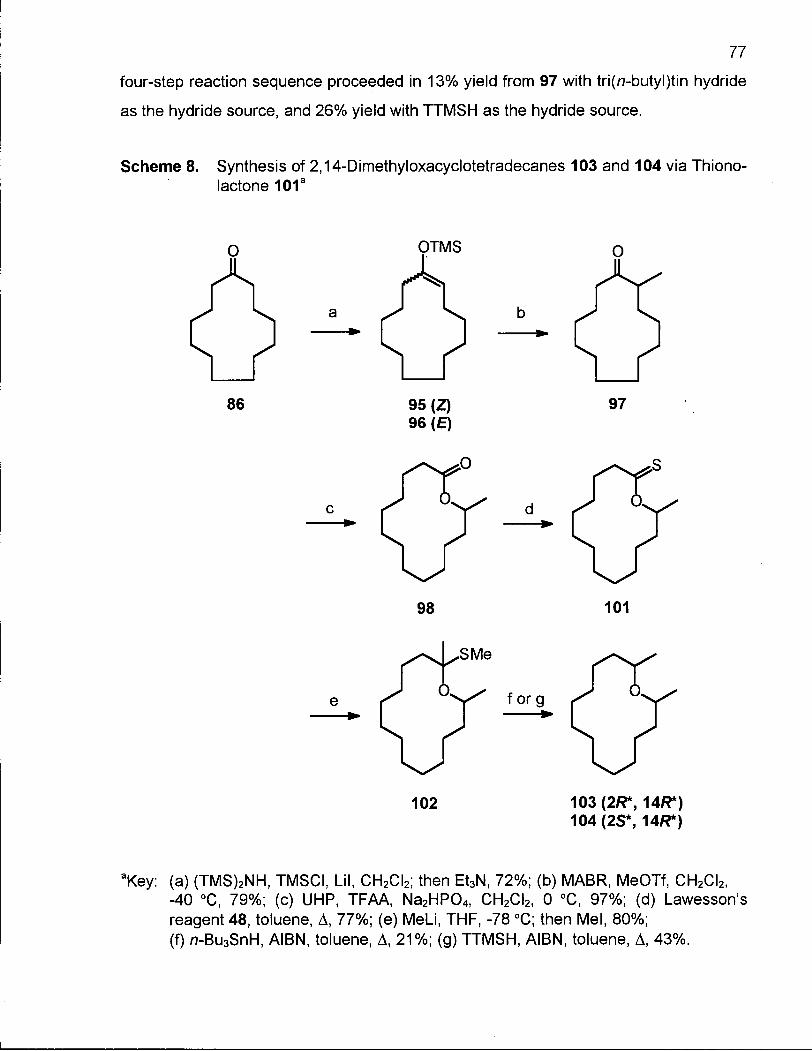
77
four-step reaction sequence proceeded in 13% yield from 97 with tri(/?-butyl)tin hydride
as the hydride source, and 26% yield with TTMSH as the hydride source.
Scheme 8. Synthesis of 2,14-Dimethyloxacyclotetradecanes 103 and 104 via Thionolactone 101a
O ?™S O
86 95 (Z) 97 96 (E)
102 103 {2R*, 14/?*) 104 (2S*, 14/?*)
Key: (a) (TMS)2NH, TMSCI, Lil, CH2CI2; then Et3N, 72%; (b) MABR, MeOTf, CH2CI2, -40 °C, 79%; (c) UHP, TFAA, Na2HP04, CH2CI2, 0 °C, 97%; (d) Lawesson's reagent 48, toluene, A, 77%; (e) MeLi, THF, -78 °C; then Mel, 80%; (f) n-Bu3SnH, AIBN, toluene, A, 21%; (g) TTMSH, AIBN, toluene, A, 43%.

78
A solution of (|i-chloro)(|i-methylene)bis(cyclopentadienyl)(dimethylaluminum)
titanium, Tebbe reagent 32,38 in toluene was generated by the addition of a solution of
trimethylaluminum in toluene to dichlorotitanocene.135 When stored under nitrogen at
0 °C this solution was stable for several months.
C5H5v / v Me Ti Al
C 5 H 5 ' cf vMe
32
The lactone 98 was reacted with this solution of Tebbe reagent 32.38 The
resultant vinyl ether 100 was unstable and was purified by passing the reaction solution
directly through a column of basic alumina with petroleum ether as eluant. The vinyl
ether 100 was immediately hydrogenated to give the macrocyclic ethers 103 and 104.
The reaction sequence proceeded in 22% yield for two-steps.
Scheme 9. Synthesis of 2,14-Dimethyloxacyclotetradecanes 103 and 104 via Enol Ether 100a
98 100 103 (2R*, 14/?*) 104 (2S*. 14/?*)
aKey: (a) Tebbe reagent 32, DMAP, pyridine, THF, -40 °C, 86%; (b) Pt02, H2, Et20, 26%.
The macrocyclic ethers 103 and 104 were separable with silica chromatography,
and each gave a single, distinct peak on GC analysis with a DB-210 column. The

7 9
relative configuration of the C-2 and C-14 methyl substituents of 103 and 104 was
determined through analysis with a chiral Cyclodex-B GC column. The (2R*, 14R*) or
anti isomer of 2,14-dimethyloxacyclotetradecane is a dl pair of enantiomers which
would give rise to two peaks under chiral GC conditions. The (2S*, 14R*) or syn isomer
of the macrocyclic ether is a meso compound which would give rise to only a single
peak under chiral GC conditions. GC analysis of 103, the first macrocyclic ether eluted
on silica, with the Cyclodex-B column resulted in two peaks of equal intensity with
retention times of 45.5 minutes and 46.4 minutes respectively. GC analysis of 104, the
second macrocyclic ether eluted on silica, gave only a single peak with a retention time
of 51.5 minutes (Figure 26). Thus, 103 was identified as the diastereomer with the C-2
and C-14 methyl groups in an anti configuration {2R*, *\4R*) and 104 was identified as
the diastereomer with the C-2 and C-14 methyl groups in a syn configuration
(2S*. 14R*).
103 104
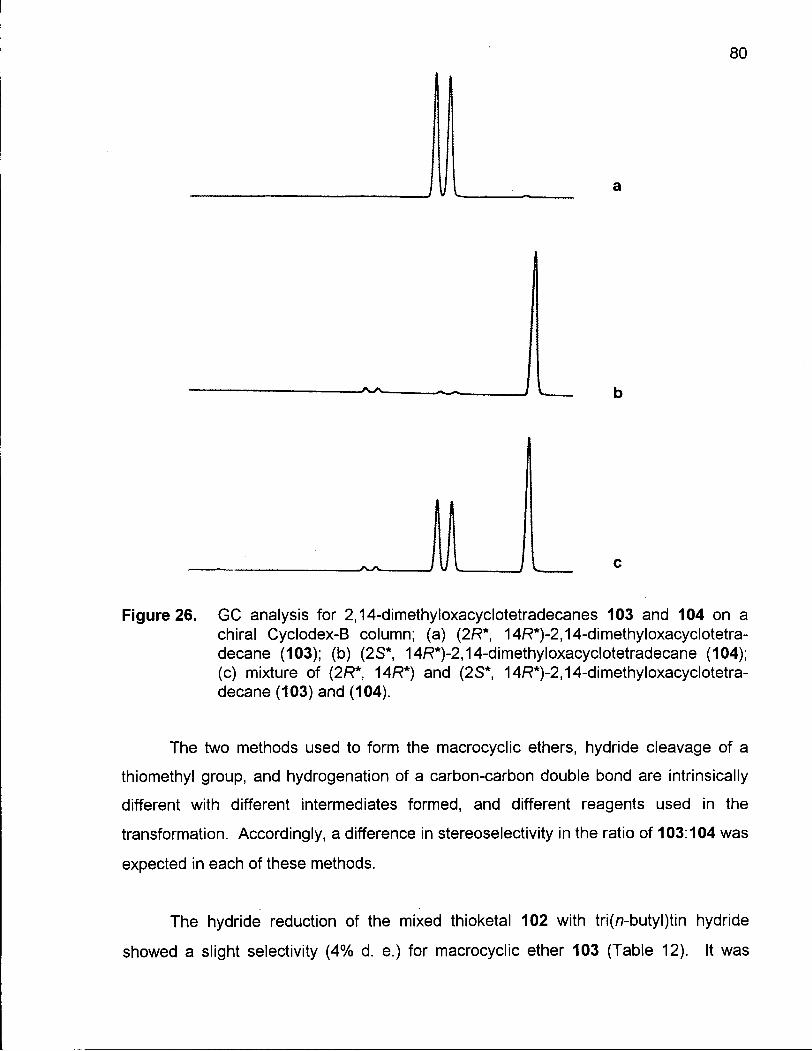
1
80
Figure 26. GC analysis for 2,14-dimethyloxacyclotetradecanes 103 and 104 on a chiral Cyclodex-B column; (a) (2R*, 14R*)-2,14-dimethyloxacyclotetra-decane (103); (b) (2S*, 14R*)-2,14-dimethyloxacyclotetradecane (104); (c) mixture of (2R*, 14R*) and (2S*, 14R*)-2,14-di methyl oxacyclotetradecane (103) and (104).
The two methods used to form the macrocyclic ethers, hydride cleavage of a
thiomethyl group, and hydrogenation of a carbon-carbon double bond are intrinsically
different with different intermediates formed, and different reagents used in the
transformation. Accordingly, a difference in stereoselectivity in the ratio of 103:104 was
expected in each of these methods.
The hydride reduction of the mixed thioketal 102 with tri(n-butyl)tin hydride
showed a slight selectivity (4% d. e.) for macrocyclic ether 103 (Table 12). It was

81
hoped that the different properties of the silane hydride reagent would offer an
improvement in the selectivity of this reduction. Tris(trimethylsilyl)silane is a bulkier
reagent with a greater metal-hydrogen bond strength of 79 kcal/mol compared to
74 kcal/mol in the case of the stannane.134 As well, the metal-hydrogen bond length is
ca. 1.48 A in the case of the silane136 compared to 1.53 A for the tin-hydrogen bond.137
These features make the silane a more selective hydride reagent. Unfortunately, only
a modest improvement in the stereoselectivity of the reduction of 102 was observed
(14% d. e.) with the silane as the hydride source (Table 12). Reaction of pure 103 and
104 with tri(n-butyl)tin hydride under radical conditions showed no isomerisation to the
other macrocyclic ether. Therefore it was assumed that no isomerisation of the
macrocyclic ethers occurred in the hydride reduction of the mixed thioketal.
The reduction of the vinyl ether 100 with Adams' catalyst (Pt02) proceeded with
only a slight stereoselectivity (2% d. e.) (Table 12). The vinyl ether was very
susceptible to hydrolysis and the choice of platinum oxide as the catalyst was important
for the success of the reduction. Palladium on charcoal, another common
hydrogenation catalyst, gave lower yields of the desired macrocycles presumably due
to hydrolysis of the starting material during the hydrogenation. Rhodium on alumina
also gave lower yields of the macrocyclic ethers.
Molecular modeling calculations with the MM3* force field suggested that the
[3434]-1 conformation is the most stable conformation of 100 with the exocyclic double
bond essentially perpendicular to the plane of the ring. The next lowest energy
conformation, 1.84 kcal/mol higher in energy was a [3344]-1 conformation. It was
believed that either the C-14 methyl group flanking the ether oxygen or the macrocyclic
ring itself would have a directing effect on the hydrogenation. However essentially no
stereoselectivity was observed in this reduction, hence the exocyclic double bond must
be blocked to approximately the same degree by the C-14 methyl group on one side
and by the macrocyclic ring on the other.

100
Table 12. Yield and Selectivity in the Preparation of 2,14-Dimethyloxacyclotetra-decanes 103 and 104
Reagent Starting Material 103:104b Total Yield of 103+104 (%)
n-Bu3SnH, AIBN3 102 52:48 21c
TTMSH, AIBN 102 57:43 43d
Pt02, H2 100 49:51 26c
3 A syringe pump was used to slowly add the solution of tri(n-butyl)tin hydride and AIBN in toluene to the reaction solution.
b The ratio of 103:104 was determined by gas chromatography. 0 The diastereomers 103 and 104 were separated via radial chromatography. d The diastereomers 103 and 104 were purified but not separated.
2.2.2 Conformational Analysis of (2A?*,14/?*)-2,14-Dimethyloxacyclotetradecane (103)
The 1H NMR spectrum of 103 at rt in CDCI3 contained a two-proton sextet at
3.65 ppm, another two-proton sextet at 1.63 ppm, a 20-proton multiplet from
1.18-1.43 ppm, and a six-proton doublet at 1.08 ppm. The low-field signal at 3.65 ppm
was assigned to the protons of C-2/C-14, while the signal at 1.63 ppm was assigned to
two of the four protons of C-3/C-13. The doublet at 1.08 ppm was assigned to the C-15
and C-16 methyl groups. The HRMS and chemical analysis data were also consistent
with the composition of 103.
The 13C NMR spectrum contained seven lines indicative of either a plane, or a
symmetry-averaged plane of symmetry in the molecule. Thus, C-2 and C-14 had the
same chemical shift, as did C-3 and C-13, and so forth. The low-field signals at
69.02 ppm and 33.64 ppm were assigned to C-2/C-14 and C-3/C-13 on the basis of

83
their through bond distance from the ether oxygen atom. Three carbons in the
molecule including C-8, were assigned to the peak at 25.15 ppm which was higher than
the other carbon signals. The assignment of the remaining 1H and 13C signals was
aided by COSY, HMQC, and HMBC 2D-NMR experiments (Table 13). The C-4/C-12
signal was shifted to higher field by the y-gauche effect as a result of its geometric
relationship to the ether oxygen atom.
Table 13. 1H and 13C NMR Assignments for (2R*,14R*)-2,14-Dimethyloxacyclo-tetradecane (103) in CDCI3 at Room Temperature
Position 1H NMRa 13C NMRa
2, 14 3.65 69.02
3, 13 1.63, 1.25 33.64 I T J 0. ,
4, 12 1.37 23.13
5, 11 1.40 26.57
6, 10 1.32 25.34b
7,9 1.32 25.15b
8 1.20 25.15
15, 16 1.08 19.63
a The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI3 (13C). b Due to overlap these signals could not be unambiguously assigned.
A series of low temperature spectra of 103 were collected in a 4:1 mixture of
Freon 21 and Freon 22 as solvent (Figure 27). The 1H NMR spectrum of 103 at 220 K
contained four broad signals. As the temperature was lowered, the low-field signal at
3.65 ppm broadened with a coalescence temperature of 195 K. At lower temperatures,
this signal split into two signals of unequal intensity indicative of unequally populated
conformations being present. The chemical shift of the major signal, which remained
fairly broad even at low temperature, was 3.55 ppm. The chemical shift of the minor
signal was 3.87 ppm. The signal at 1.87 ppm, also broadened as the temperature was
lowered, with a coalescence temperature of 195 K. This signal did not split as the
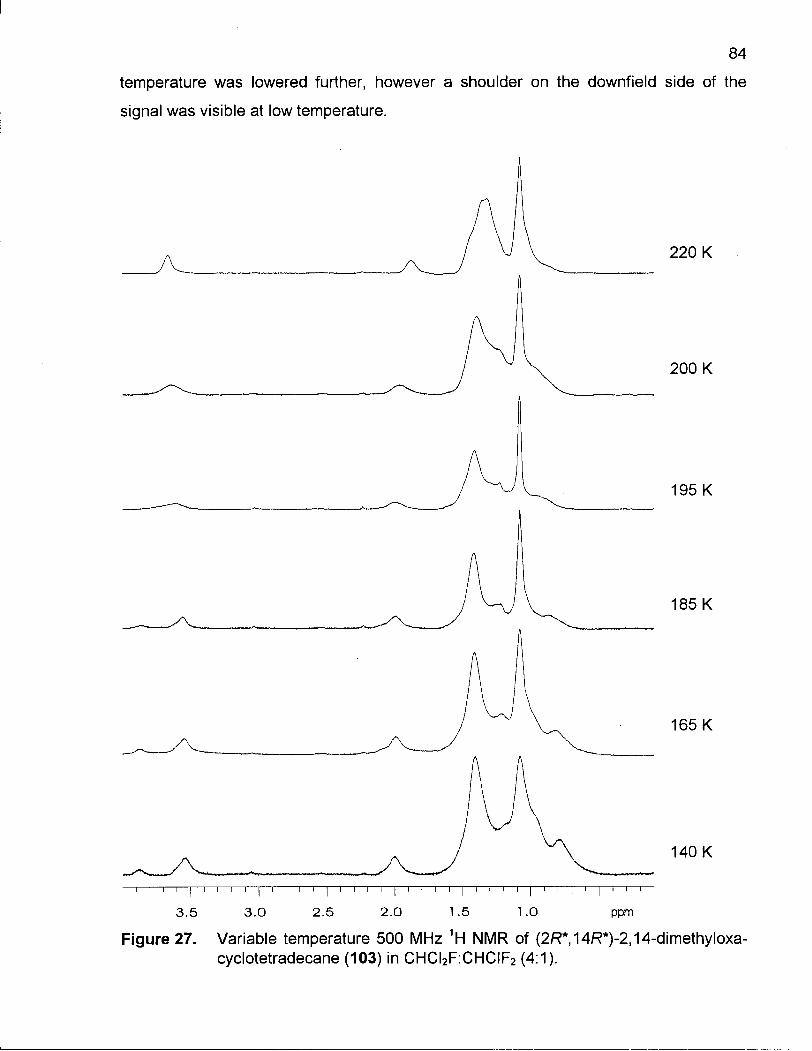
84
temperature was lowered further, however a shoulder on the downfield side of the
signal was visible at low temperature.

85
Analysis of the methylene envelope region of the spectra was complicated by
the overlap of multiple signals. The line shape of the methylene envelope did change
as the temperature was lowered. The line shape of the signal for the C-15 and C-16
methyl groups did not undergo significant changes as the temperature was lowered. A
new signal was visible with a chemical shift of 0.82 ppm at higher field than the methyl
signal as the temperature was lowered in the DNMR spectra.
To begin the analysis of the DNMR data, conformations of 103 likely to have low
energy were sought. Molecular models were used to evaluate some diamond lattice
conformations of this macrocyclic ether. The [3434] conformation has four
diastereotopic methylene groups. In the unsubstituted 14-membered ether, the
[3434]-1 conformation, with the ether oxygen in the middle of a 4-bond side, is the
lowest energy conformation. However, in the case of the 2,14-disubstituted ether 103,
one of the methyl groups would be endo to the ring, thereby raising the energy of this
conformation. The [3434]-4 conformation of the unsubstituted 14-membered ether is
also low in energy, but the methyl groups of 103 would experience a 1,3-diaxial
interaction in the [3434]-4 conformation. There is also a 1,3-diaxial interaction between
the methyl groups of 103 in the [3434]-2 conformation. The methyl groups of 103 are
both exo to the ring in the [3434]-3 conformation, and furthermore no 1,3-diaxial
interactions occur between these groups since they are on different sides of the ring.
Unfortunately, in this conformation the ether oxygen atom is located at a corner position
and no transannular hydrogen interactions are eliminated by the oxygen when at this
position.

86
[3344J-1
None of the [3434] conformations of 103 appeared to be low in strain energy, so
the search was widened to include some non-diamond lattice conformations. In the
[3344]-1 conformation, one of the methyl groups would be endo to the ring as in the
[3434]-1 conformation. Both the [3344]-2 and the [3344]-4 conformations have
1,3-diaxial interactions between the methyl groups, and are therefore not expected to
be low in energy. There are no unfavourable steric interactions between the methyl
groups in the [3344]-3 conformation, but the oxygen atom is at a corner position which
is known to be high in energy. Thus, a priori the identity of the low energy
conformations of 103 was unclear. It is likely that the low energy conformations are
similar to some of these, where the steric repulsions of the methyl groups have been
reduced by small distortions to the appropriate dihedral angles. Alternatively, the low
energy conformations may be other non-diamond lattice conformations. In either case,
the identity of low energy conformation of 103 was not predicted.

87
A regular, low energy conformation of 103 could not be identified Therefore the
DNMR data was analyzed with attention to quantifying the ratio of major and minor
conformations present, rather than specifically trying to identify the low energy
conformations. The signal for the H-2 and H-14 methine protons adjacent to the ether
oxygen of 103 broadened, and gave two signals of unequal intensity at low
temperature. The unequal intensity of the signals eliminated the possibility of the
presence of only one conformation at low temperature. Only one major signal was
observed in the low-field portion of the DNMR spectra at 3.55 ppm, hence the major
conformation of 103 has a small A5 value between the H-2 and H-14 protons. The
methine protons of the minor conformation of 103 could have a large A5 with a signal at
3.87 ppm, and another signal overlapped with the major conformation signal at
3.55 ppm representing the H-2 and H-14 protons. Alternatively, the A5 of the minor
conformation could also be small, and these protons are represented by only the peak
at 3.87 ppm. No shoulder was visible on the major signal at 3.55 ppm to support the
minor conformation, large A5 value argument, and therefore the minor conformation
likely has a small A8 value. The relative intensities of the major and minor downfield
signals was 5.2:1. This corresponded to an 84:16 ratio of conformers with an energy
difference of 0.46 kcal/mol.
Signals for the minor conformation were also expected in other regions of the
spectra, a shoulder was visible at 2.08 ppm, on the downfield side of the signal for two
of the protons p to the ether oxygen. The chemical shift of this major signal drifted
downfield to 1.99 ppm as the temperature was reduced. No obvious signals for the
C-15 and C-16 methyl groups of the minor conformations were identified in the 1 ppm
region of the spectrum.
A molecular mechanics search for the low energy conformations of 103 was
performed with the Monte Carlo technique and the MM2* force field. The global
minimum conformation was a non-diamond lattice [1334'3'] conformation 103-A, with
another [1334'3'] conformation 103-B calculated to have a similar energy, only

88
0.02 kcal/mol higher (Table 14). These calculations suggested the existence of ten
other low energy conformations within 1 kcal/mol of the global minimum conformation.
The results of the calculations were in good agreement with the DNMR data with
respect to the energy difference between the major and minor conformations. The
relative populations of these conformations at different temperatures were calculated
from relative energy values obtained from the MM2* calculations (Table 15).

89
Table 14. Low Energy Conformations of (2R*,14R*)-2,14-Dimethyloxacyclotetra-decane (103)
Conformer Skeleton Relative Energy (kcal/mol)
103-A [1334'3'] 0.00
103-B [1334'3'] 0.02
103-C [3434]-1 0.41
103-D [1334'3'] 0.43
o 103-E [12431'3'] 0.45
a Strain energies are relative to the global minimum conformation calculated with the MM2* force field.
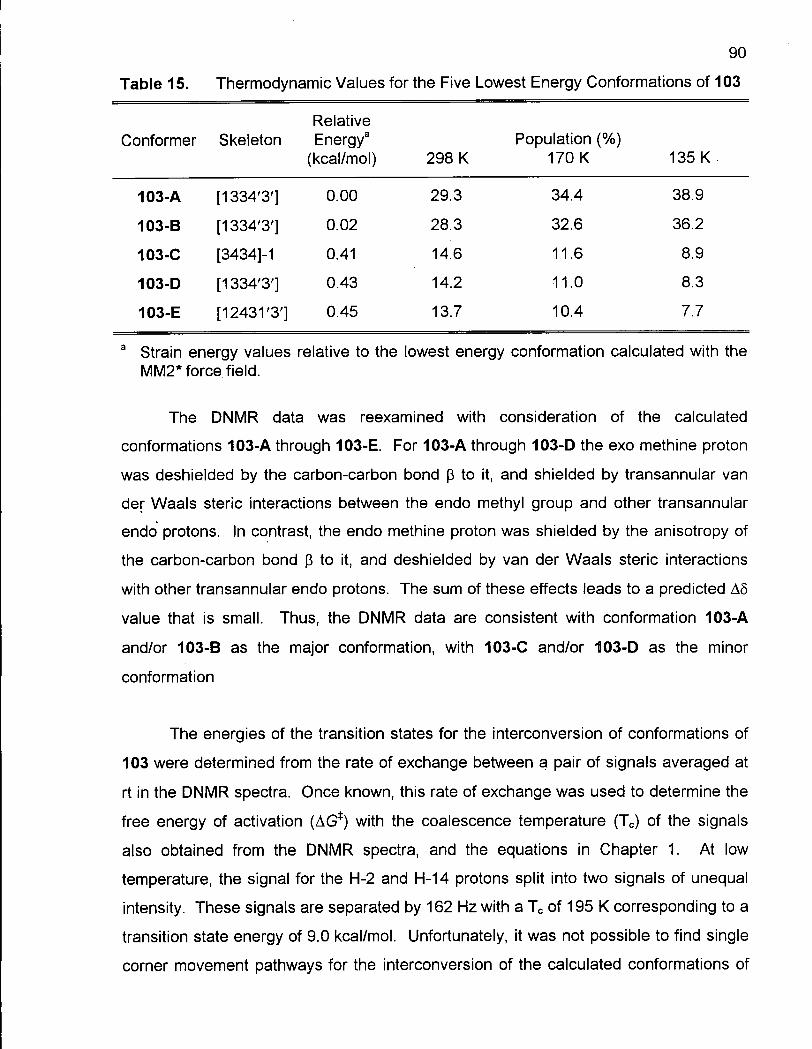
90
Table 15. Thermodynamic Values for the Five Lowest Energy Conformations of 103
Relative Conformer Skeleton Energy3
(kcal/mol) 298 K Population (%)
170 K 135 K .
103-A [1334'3'] 0.00 29.3 34.4 38.9
103-B [1334'3'] 0.02 28.3 32.6 36.2
103-C [3434]-1 0.41 14.6 11.6 8.9
103-D [1334'3'] 0.43 14.2 11.0 8.3
103-E [12431'3'] 0.45 13.7 10.4 7.7
Strain energy values relative to the lowest energy conformation calculated with the MM2* force field.
The DNMR data was reexamined with consideration of the calculated
conformations 103-A through 103-E. For 103-A through 103-D the exo methine proton
was deshielded by the carbon-carbon bond (3 to it, and shielded by transannular van
der Waals steric interactions between the endo methyl group and other transannular
endo protons. In contrast, the endo methine proton was shielded by the anisotropy of
the carbon-carbon bond 3 to it, and deshielded by van der Waals steric interactions
with other transannular endo protons. The sum of these effects leads to a predicted A5
value that is small. Thus, the DNMR data are consistent with conformation 103-A
and/or 103-B as the major conformation, with 103-C and/or 103-D as the minor
conformation
The energies of the transition states for the interconversion of conformations of
103 were determined from the rate of exchange between a pair of signals averaged at
rt in the DNMR spectra. Once known, this rate of exchange was used to determine the
free energy of activation (AG*) with the coalescence temperature (Tc) of the signals
also obtained from the DNMR spectra, and the equations in Chapter 1. At low
temperature, the signal for the H-2 and H-14 protons split into two signals of unequal
intensity. These signals are separated by 162 Hz with a Tc of 195 K corresponding to a
transition state energy of 9.0 kcal/mol. Unfortunately, it was not possible to find single
corner movement pathways for the interconversion of the calculated conformations of

91
103, and the transition state energies could not be estimated by the computer
modelling methods described earlier.
2.2.3 Conformational Analysis of (2S*,14/?*)-2,14-Dimethyloxacyclotetradecane (104)
The 1H NMR spectrum of 104 at rt contained a two-proton doublet of doublet of
quartets at 3.54 ppm, a 22-proton multiplet between 1.18-1.49 ppm, and a six-proton
doublet at 1.10 ppm. The downfield signal was assigned to the protons of C-2/C-14,
and the high-field doublet was assigned to the C-15 and C-16 methyl groups
(Table 16).
The 13C NMR spectrum contained eight signals for this 15-carbon molecule,
indicative of a plane of symmetry or a symmetry-averaged plane of symmetry at rt.
Thus, C-2 and C-14 had the same chemical shift, as did C-3 and C-13, and so forth.
The low-field signals at 71.11 ppm and 36.10 ppm were assigned to C-2/C-14 and
C-3/C-13 on the basis of their through-bond distance to the ether oxygen. The signal
for C-4 was shifted upfield to 22.95 ppm by a y-gauche effect caused by the geometric
relationship of this carbon to the ether oxygen. The highest field signal at 21.18 ppm
was assigned to the methyl groups. The signal at 24.86 ppm was approximately half
the height of the other 13C signals and was assigned to C-8. This carbon is located
across the ring from the ether oxygen atom, and does not have a symmetrical carbon
partner. The assignment of the remaining 1H and 13C signals was assisted with COSY,
HMQC, and HMBC 2D-NMR experiments (Table 16). The HRMS data was also
consistent with the composition of 104.

92
Table 16. 1H and 13C NMR Assignments for (2S*,14R*)-2,14-Dimethyloxacyclotetra-decane (104) in CDCI3 at Room Temperature
Position 1H NMRa 13C NMRa
3.54 71.11
1.46 36.10
1.30b 22.95
1.30b 26.41b
1.30b 26.17b
1.38 25.55
1.20 24.86
1.10 21.18
a The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI3 (13C). b Due to overlap these signals could not be unambiguously assigned.
A series of low temperature spectra of 104 were obtained in a 4:1 mixture of
Freon 21 and Freon 22 as solvent (Figure 28). The 1H NMR spectrum of 104 at 220 K
contained three signals. This spectrum was similar to the rt spectrum, with much of the
signal multiplicity lost. As the temperature was lowered, the downfield signal at
3.74 ppm broadened (Tc = 185 K) to form at low temperature, a pair of sharp signals at
3.82 ppm and 4.03 ppm. At low temperature, a small signal emerged downfield of the
methylene envelope at 1.80 ppm. The signal for the methyl groups also broadened,
and split into two closely spaced signals at 1.13 ppm and 1.09 ppm (Tc = 165 K). At
high-field in the low temperature spectra, signals at 1.00 and 0.76 ppm were visible
with a relative intensity approximately equal to that of the 1.80 ppm signal downfield of
the methylene envelope. No significant line shape changes occurred upon further
cooling to 145 K the lowest temperature of this DNMR study. The series of 1H DNMR
spectra indicate the slowing of a conformational process as the temperature is lowered
that results in a loss of molecular symmetry. Individual signals were obtained at low
temperature for the C-15 and C-16 methyl groups as well as for the H-2 and H-14
methine protons. The sharpness of these signals suggests the presence of only one
conformation at low temperature.
2, 14
8
15, 16

93
The highly symmetric 14-membered [3434] diamond lattice conformation has
four diastereotopic methylene groups with different numbers of transannular steric
interactions. The [3434]-1 conformation with the ether oxygen atom of 104 located in
the middle of a 4-bond side, results in a 1,3-diaxial interaction between the syn methyl
groups. This severe steric interaction raises the energy of the [3434]-1 and [3344]-1
conformations, and these are not considered further. The oxygen atom in the [3434]-4
conformation with the ether oxygen atom located on a 3-bond side adjacent to a corner
atom leads to relief of the second largest number of transannular interactions. In this
conformation, one of the methyl groups is on the corner atom, and is therefore pointing
outside the ring. Secondly, and of greater importance, this corner methyl group is
pointed away from the other methyl group on the side of the ring, thus avoiding a
1,3-diaxial interaction. The DNMR spectra of 104 were analyzed in terms of the
[3434]-4 conformation.
[3434]-4
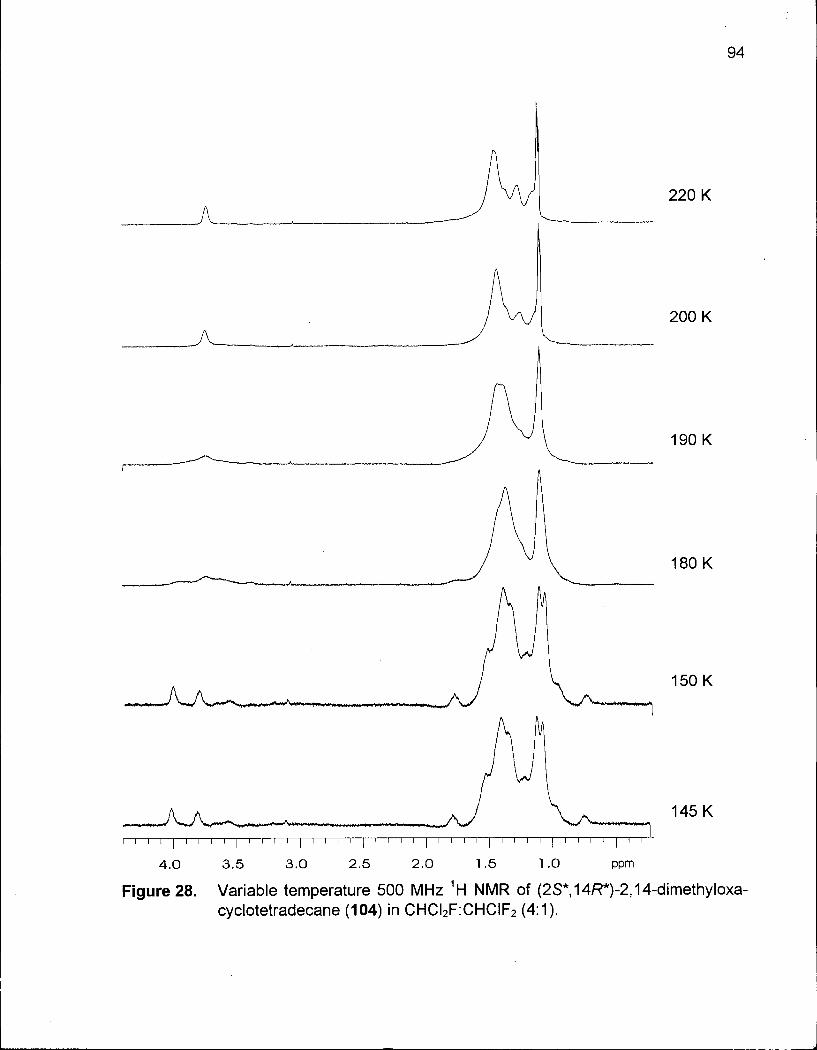
94
180 K
150 K
145 K
4.0 3.5 3.0 2.5 2.0 1.5 1 .0 ppm
Figure 28. Variable temperature 500 MHz 1H NMR of (2S*,14R*)-2,14-dimethyloxa-cyclotetradecane (104) in CHCI2F:CHCIF2 (4:1).

95
In the [3434J-4 conformation, the corner H-2a proton is shielded by the
anisotropy of the O/C-14 bond. The H-14endo proton points to the inside of the ring, and
is shielded by the anisotropy of the C-12/C-13 bond, but is deshielded by van der
Waals steric interactions with H-3endo and H-11endo that are calculated to be 2.17 A and
2.21 A away from H-14endo. Electric field effects play only a small role here with H-2a
slightly shielded as the result of a pair of vicinal gauche carbon-hydrogen bonds. The
H-14endo proton is shielded to a lesser degree by a single vicinal gauche carbon-
hydrogen bond. Thus, the H-14end0 proton is predicted to be more deshielded, and is
assigned to the signal at 4.03 ppm in the low temperature spectra. The other signal at
3.82 ppm was assigned to the corner H-2a proton.
In the rt 1H NMR spectrum of 104, the signals due to the H-3 and H-13 protons p
to the ether oxygen overlap with the signals of the methylene envelope. The new
signal visible downfield of the methylene envelope in the low temperature spectra of
104 had a relative integration of 1:1 in comparison to each of the sharp downfield
signals of H-2a and H-14end0. In the [3434J-4 conformation, the H-3endo proton is
deshielded by the anisotropy of both the O/C-2 and C-2/C-16 bonds. The H-3end0
proton is also deshielded by a van der Waals steric interaction with H-14end0 that is
calculated to be 2.17 A away. The H-13p proton is deshielded by the anisotropy of the
O/C-14 and C-11/C-12 bonds. The corner H-13 protons do not experience any
transannular van der Waals steric interactions since they point to the outside of the
ring. The signals of the other p-protons, H-3exo and H-13a are shifted upfield by these
shielding effects. As a result, the downfield signal at 1.80 ppm is assigned to the most
deshielded of these p-protons, namely the H-3end0 proton.
The signal of the C-15 and C-16 methyl groups split into two signals of
approximately equal intensity at low temperature. The protons of both the C-15 methyl
group and the C-16 methyl group are deshielded to approximately the same extent by
the surrounding bonds. However, the C-15 methyl group is also shielded as a result of
a van der Waals steric repulsion between the H-14end0 proton and the H-11end0 and

96
H-3endo protons. The corner C-16 methyl group does not experience such an effect.
Thus, the low temperature methyl signal at 1.09 ppm is assigned to the C-15 methyl
group, and the lower field signal at 1.13 ppm is assigned to the C-16 methyl group.
The high-field signal at 0.76 ppm in the low temperature spectra had a relative
integration of 1:1 in comparison to the downfield H-2a and H-14end0 proton signals. The
H-4endo and H-11endo protons are deshielded in the [3434]-4 conformation as a result of
van der Waals steric interactions with other endo protons on the ring. This results in
the shielding of the H-4exo and H-11exo protons. Specifically, the H-4endo proton is
deshielded by van der Waals steric interactions with the H-7end0 and H-11endo protons
that are calculated to be 2.19 A and 2.23 A from H-4endo. The H-11endo proton is
deshielded by van der Waals steric repulsions with the H-8end0 and H-14endo protons that
are calculated to be 2.15 A and 2.21 A away from H-11endo- The magnitude of the van
der Waals shielding effect has been found to be proportional to the electronegativity of
the sterically opposing group.64 The van der Waals steric interaction between H-4endo
and the electronegative ether oxygen, calculated to be 2.63 A apart, further shields the
H-4exo proton. Therefore, on the basis of these arguments, the high-field signal at
0.76 ppm is assigned to H-4exo. The signal at 1.00 ppm on the high-field shoulder of the
methyl signals of 104 is assigned to the H-11exo proton.
A molecular mechanics search for low energy conformations of 104 was
conducted using the Monte Carlo technique and the MM2* force field. The global
minimum conformation was the [3434]-4 conformation 104-A, as predicted, with the
[34'3'4']-4 conformation 104-B calculated to have the next lowest energy, 1.21 kcal/mol
higher than the global minimum (Table 18). From these calculations we found a total of
16 conformations within 2 kcal/mol of the global minimum. The fifth lowest energy
conformation 104-E was the [3434]-1 conformation. This was not predicted to be a low
energy conformation due to a steric interaction between the syn methyl groups.
However, a distortion of the dihedral angle of one of the methine carbons resulted in a
reduction of the 1,3-diaxial interaction between the methyl groups, and lowered the
energy of this conformation
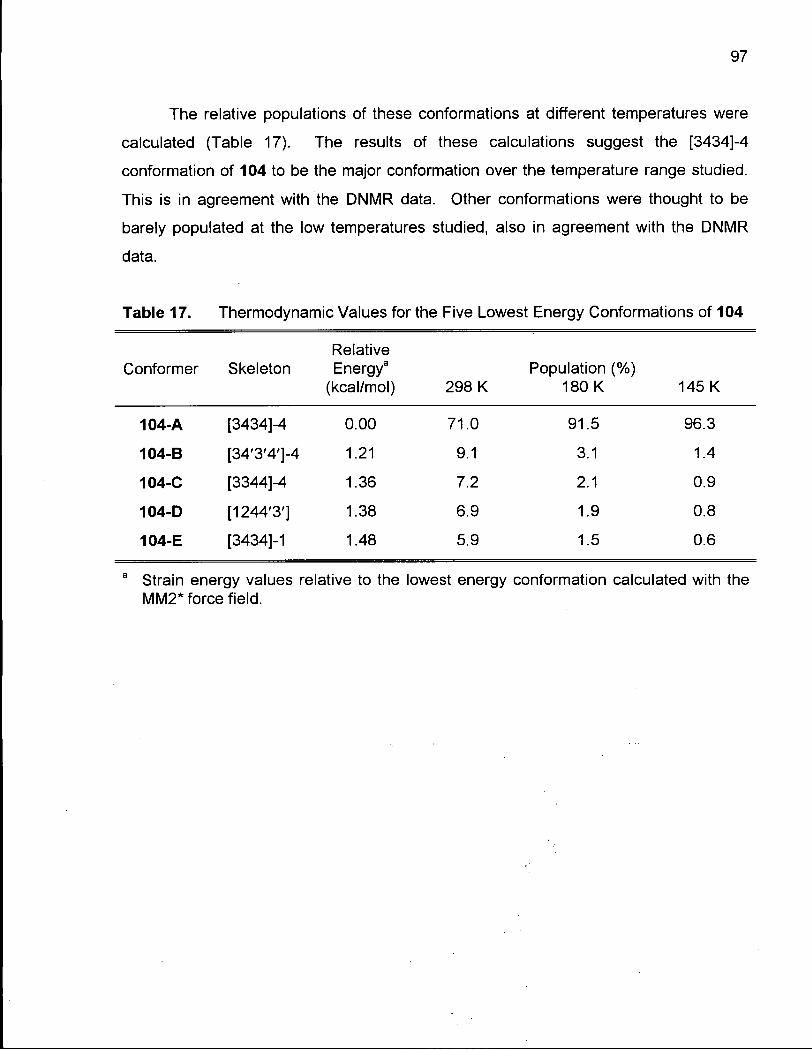
97
The relative populations of these conformations at different temperatures were
calculated (Table 17). The results of these calculations suggest the [3434]-4
conformation of 104 to be the major conformation over the temperature range studied.
This is in agreement with the DNMR data. Other conformations were thought to be
barely populated at the low temperatures studied, also in agreement with the DNMR
data.
Table 17. Thermodynamic Values for the Five Lowest Energy Conformations of 104
Relative Conformer Skeleton Energy3 Population (%)
(kcal/mol) 298 K 180 K 145 K
104-A [3434]_4 0.00 71.0 91.5 96.3
104-B [34'3'4']-4 1.21 9.1 3.1 1.4
104-C [3344]-4 1.36 7.2 2.1 0.9
104-D [1244'3'] 1.38 6.9 1.9 0.8
104-E [3434]-1 1.48 5.9 1.5 0.6
a Strain energy values relative to the lowest energy conformation calculated with the MM2* force field.

98
Table 18. Low Energy Conformations of (2S*,14R*)-2,14-Dimethyloxacyclotetra-decane (104)
Conformer Skeleton Relative Energy (kcal/mol)
104-A [3434]-4 0.00
104-B [34'3'4']-4 1.21
104-C [3344]-4 1.36
[1244'3'] 1.38
104-E [3434J-1 1.48
a Strain energies are relative to the global minimum conformation calculated with the MM2* force field.

99
The energy barriers to the interconversion of conformations of 104 were
calculated by first determining the rate of exchange between a pair of averaged signals
in the DNMR spectra. Once known, this rate of exchange was used to calculate the
free energy of activation (AG*) with the coalescence temperature of the signals (Tc)
also obtained from the DNMR spectra, using the equations in Chapter 1. At low
temperature the signals for the H-2a and H-14endo methine protons were separated by
102 Hz. This corresponded to a transition state energy of 8.7 kcal/mol with a Tc of 185
K. The signals of the C-15 and C-16 methyl groups were separated by 22 Hz. This
corresponded to a transition state energy of 8.2 kcal/mol with a Tc of 165 K. The
average of these values is 8.5 ± 0.3 kcal/mol. These transition state energy values are
approximately that of the unsubstituted macrocycle ether, oxacyclotetradecane (90),
8.7 ± 0.2 kcal/mol.
The single corner movement mechanism proposed by Dale for the
interconversion of cyclic conformations was used to describe the transition states of the
interconversion of low energy conformations of 104. The energies of these transition
state structures were estimated with molecular modeling calculations using the dihedral
drive method124 and the MM2* force field. An incremental step of 10° was used in these
calculations. The global minimum [3434J-4 conformation 104-A can interconvert with
the [3344J-4 conformation 104-C through a [73343] transition state (Figure 29). This
transition state energy was estimated at 12.9 kcal/mol. The [3344]-4 conformation
104-C can also interconvert with the higher energy [3434]-1 conformation 104-E via
another [73343] transition state with an estimated energy of 14.7 kcal/mol. The
calculated transition state energies were larger than the observed values.

100
[3434]-4 [3344]-4 [3434]-1
Figure 29. Interconversion of conformations of 104 via single corner movements.
2.3.1 Synthesis of 2,2-Dimethyloxacyclotetradecane (116)
The first approach to the synthesis of macrocyclic ether 116 followed the general
synthetic strategy presented earlier. The synthetic plan was to ring expand a
dialkylated ketone to give a 14-membered lactone with the gem-dimethyl substituents
already in place adjacent to the ether oxygen. The carbonyl of this lactone would be
removed to give the macrocyclic ether 116 (Scheme 10).
101
Scheme 10. Retrosynthetic Analysis of 2,2-Dimethyloxacyclotetradecane (116)
116 115 114
0 O
106 86
The dialkylated ketone 106 was prepared via a two-step sequence starting with
the monoalkylated ketone 97 which was reacted with hexamethyldisilazane,
trimethylsilyl chloride, and lithium iodide to form the trimethylsilyl enol ether 105. 1 2 8 , 1 2 9
Unfortunately, a mixture of regioisomers was obtained in the enol ether formation step,
and no separation of these isomers was attempted. The MABR mediated alkylation of
this mixture of trimethylsilyl enol ethers with methyl triflate gave ketone 106, and also
(2S*. 13R*)- and (2R*, 13R*)-dimethylcyclotridecanone (107) and (108) resulting from
the alkylation of the regioisomeric trimethylsilyl enol ethers.133

102
0 0
107 108
The alkylation products 107 and 108 were identified by a GC comparison with
authentic samples.126 A smaller proportion of the desired thermodynamic enol ether
105 was obtained than expected. Based on GC analysis of the ketones formed in the
subsequent alkylation step, an approximate ratio of 1:1 kinetic to thermodynamic
product was formed. The two-step sequence proceeded in a modest 31% yield due to
the formation of significant quantities of 107 and 108 resulting from the isomeric kinetic
enol ethers. The 13C NMR spectrum of 106 contained a signal at 216.09 ppm for the
C-1 carbonyl and a signal at 24.62 ppm for the C-2 geminal methyl groups. The 1H NMR spectrum of 106 contained a two-proton multiplet between 2.48-2.51 ppm for
the C-13 methylene, and a six-proton singlet at 1.09 ppm for the C-2 gem-dimethyl
groups consistent with the structure of 106.
Scheme 11. Synthesis of 2,2-Dimethylcyclotridecanone (106)a
O OTMS O
97 105 106
aKey: (a) (TMS)2NH, TMSCI, Lil, CH2CI2; then Et3N, 94%; (b) MABR, MeOTf, CH2CI2, -40 °C, 33%.

103
The Baeyer-ViNiger oxidation of ketone 106 was attempted under a variety of
conditions. The reaction of ketone 106 with trifluoroperacetic acid formed in situ from
trifluoroacetic anhydride and UHP (Table 19, Entries 1-3) was unsuccessful even with a
ten-fold excess of the reagents.122,123 The reaction of m-CPBA in the presence of either
p-TsOH138 (Entry 4) or Li2C03
139 (Entry 5) also failed to yield any of the desired lactone
114.
O
106 114
Table 19. Reaction Conditions used in the Attempted Baeyer-Vi Niger Oxidation of Ketone 106
Entry Reaction Conditions equiv. of oxidant
yield3
1 UHP, TFAA, Na2HP04, CH2CI2, rt 6 0
2 UHP, TFAA, Na2HP04, CH2CI2, rt 10 0
3 UHP, TFAA, Na2HP04, CH2CI2, A 10 0
4 /77-CPBA, p-TsOH, CH2CI2 10 0
5 m-CPBA, Li2C03, CH2CI2 10 0
3 Analysis of the product mixture by gas chromatography showed only starting material to be present.
The synthesis of the gem-dimethyl lactone 114 did not proceed as outlined in
the original synthetic plan. However, the keto acid 112, an intermediate prepared
previously in our laboratory en route to some p-keto lactones, represented an alternate
precursor to lactone 114.140 This keto acid could be reacted to give a tertiary hydroxy
acid, however it was unclear whether this sterically hindered compound would cyclize
104
to give lactone 114 using standard macrolactonisation techniques (Scheme 12). Once
formed the lactone 114 was to be converted into the macrocyclic ether 116.
Scheme 12. Retrosynthetic Analysis of 13-Methyl-13-tetradecanolide (114)
114 113 112
The bromo acid 109 was converted into the methyl ester 110 under Fischer
esterification conditions. This ester was chain extended by alkylation with the anion of
methyl acetoacetate to give diester 111. 1 4 1 The diester 111 was decarboxylated under
strongly acidic conditions to give the keto acid 112. The desired gem-dimethyl group
was introduced using Grignard chemistry to give the hydroxy acid 113. The hydroxy
acid 113 was cyclized with the Yamaguchi procedure with triethylamine and
2,4,6-trichlorobenzoyl chloride, and subsequently reacted with a catalytic amount of
DMAP under high dilution conditions to give the gem-dimethyl lactone 114. 2 8 The five-
step reaction sequence proceeded in 18% yield with the Grignard and cyclisation
reactions having the lowest yields of the sequence. The 1H NMR spectrum of 114
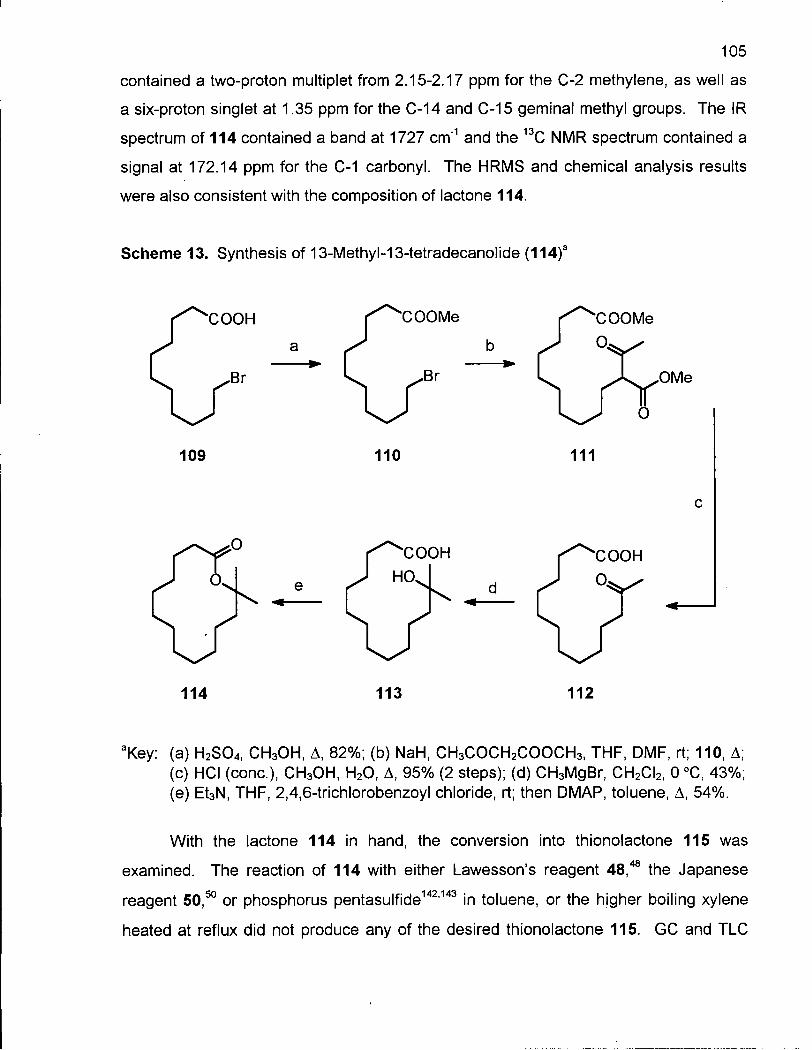
105
contained a two-proton multiplet from 2.15-2.17 ppm for the C-2 methylene, as well as
a six-proton singlet at 1.35 ppm for the C-14 and C-15 geminal methyl groups. The IR
spectrum of 114 contained a band at 1727 cm"1 and the 13C NMR spectrum contained a
signal at 172.14 ppm for the C-1 carbonyl. The HRMS and chemical analysis results
were also consistent with the composition of lactone 114.
Scheme 13. Synthesis of 13-Methyl-13-tetradecanolide (114)a
114 113 112
aKey: (a) H2S04, CH3OH, A , 82%; (b) NaH, CH3COCH2COOCH3, THF, DMF, rt; 110, A; (c) HCI (cone), CH3OH, H20, A , 95% (2 steps); (d) CH3MgBr, CH2CI2, 0 °C, 43%; (e) Et3N, THF, 2,4,6-trichlorobenzoyl chloride, rt; then DMAP, toluene, A , 54%.
With the lactone 114 in hand, the conversion into thionolactone 115 was
examined. The reaction of 114 with either Lawesson's reagent 48, 4 8 the Japanese
reagent 50, 5 0 or phosphorus pentasulfide142,143 in toluene, or the higher boiling xylene
heated at reflux did not produce any of the desired thionolactone 115. GC and TLC

106
analysis of the reaction mixture in all cases showed no evidence of the formation of the
desired thionolactone (Table 20).
50
Decomposition of the starting material was noted, presumably via acid hydrolysis
caused by acidic species formed from the thionation reagents. The reaction of
Lawesson's reagent 48 in xylene heated at reflux with either pyridine or thiourea as a
base to counter this hydrolysis was investigated. GC analysis of these reactions again
showed no formation of the desired thionolactone 115, although the starting material
was still present even after two days reaction time (Table 20, Entry 6-7). The reaction
of the Lawesson's reagent 48 was apparently blocked by the sterically demanding C-13
gem-dimethyl substituents adjacent to the lactone functionality of 114.
114 115

107
Table 20. Reaction Conditions Used in the Attempted Thionation of Lactone 114
Entry Reaction Conditions3 solvent yield/%
1 48 toluene 0b
2 50 toluene 0b
3 P2S5 toluene 0b
4 48 xylene 0b
5 50 xylene 0b
6 48, pyridine (cat.) xylene 0C
7 48, thiourea (cat.) xylene 0C
3 These reactions were performed with the solvent heated at reflux. b Analysis of the product mixture by gas chromatography showed that the starting
material decomposed and no product was present. c Analysis of the product mixture by gas chromatography showed that only starting
material was present.
The effort to synthesize the macrocyclic ether 116 was apparently at an
insurmountable barrier. Preparation of the gem-dimethyl lactone 114 had proven to be
challenging, but ultimately successful. However, all attempts to prepare the
thionolactone 115 had failed. In search of additional alternatives, a more detailed
search of the literature uncovered a report of the reduction of a lactone with sodium
borohydride in the presence of boron trifluoride etherate to directly give a cyclic ether in
a steroidal system.36 The application of this methodology to our system met with
success. The boron trifluoride etherate mediated reaction of sodium borohydride with
lactone 114 gave the macrocyclic ether 116 in 51% yield.
114 116

108
2.3.2 Conformational Analysis of 2,2-Dimethyloxacyclotetradecane (116)
The 1H NMR spectrum of 2,2-dimethyloxacyclotetradecane (116) at rt in CDCI3
contained a two-proton triplet at 3.25 ppm, a two-proton quintet at 1.57 ppm, a
20-proton multiplet from 1.23-1.43 ppm, and a six-proton singlet at 1.13 ppm. The
low-field signal was assigned to the protons of C-14, adjacent to the ether oxygen
atom, and the signal at 1.57 ppm was assigned to the protons of C-13. The singlet was
assigned to the C-2 geminal methyl groups. The remaining proton signals were
overlapped in the methylene envelope region.
The 13C spectrum of this compound contained 14 lines. Due to the overlap of
the signals in the 1H NMR spectrum, even with HMQC and HMBC 2D-NMR
experiments, only a limited number of the carbon signals could be assigned (Table 21).
The low-field signal at 73.88 ppm was assigned to the quaternary C-2 carbon, and the
signal at 37.97 ppm was assigned to the adjacent C-3. The other low-field signal at
58.88 ppm was assigned to the C-14 methylene carbon, and the geminal methyl
carbons C-15 and C-16 were assigned to the signal at 26.66 ppm. The HRMS analysis
was also consistent with the composition of 116.
Table 21. 1H and 13C NMR Assignments for 2,2-Dimethyloxacyclotetradecane (116) in CDCI3 at Room Temperature
Position 1H NMRa 13C NMRa
2 — 73.88
(121314| 15 3 1.46 37.97
J11 c> 4-10 not assigned15
16 assigned15
I 3 11 1.27 24.56
1 4r 12 1.40 24.50
13 1.57 28.34
14 3.25 58.88
15, 16 1.13 26.66
a The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI3 (13C). b Due to signal overlap these signals could not be assigned.

109
A series of DNMR experiments were performed with 116 using a 4:1 mixture of
Freon 21 and Freon 22 as solvent (Figure 30). The 1H NMR spectrum of 116 at 220 K
contained four signals. This was similar to the rt spectrum, however the signals had
broadened at the lower temperature. As the temperature was lowered, the downfield
signal at 3.25 ppm for the C-14 protons broadened to form at low temperature a pair of
signals at 3.31 and 3.15 ppm (Tc = 180 K). The signal for the C-13 protons at 1.57 ppm
also broadened as the temperature was lowered to give at low temperature, signals at
1.69 ppm and 1.50 ppm. The signal for the geminal methyl groups at 1.13 ppm split as
the temperature was lowered to give signals at 1.15 ppm and 1.10 ppm (Tc = 190 K).
As only one pair of major signals were observed for the geminal methyl groups, only
one major conformation was predicted to be present. Also visible in the high-field
region of the low temperature spectra were two signals at 0.93 ppm and 0.67 ppm.
A gem-dimethyl substituted carbon is always restricted to a corner position in
low energy conformations of 14-membered rings. If located at another position on the
ring, one of the methyl groups points into the ring, resulting in a severe transannular
steric interaction.105 In the case of the 14-membered macrocyclic ether 116 that
contains the C-2 gem-dimethyl group adjacent to the ether oxygen, this restriction
prevents the oxygen from occupying the 1-position in the middle of a 4-bond side in the
[3434]-1 conformation. Two possible diamond lattice conformations that have C-2 at a
corner position are the [3434]-4 and [3434]-2 conformations. In the unsubstituted
ether, the [3434]-4 conformation is the preferred conformation of this pair. The
transannular steric interactions eliminated by an oxygen atom at this position are
greater than in the case of the [3434]-2 conformation. These diamond lattice
conformations were both considered as possible major conformations of 116 in the
analysis of the DNMR data.
[3434]-4 [3434]-2
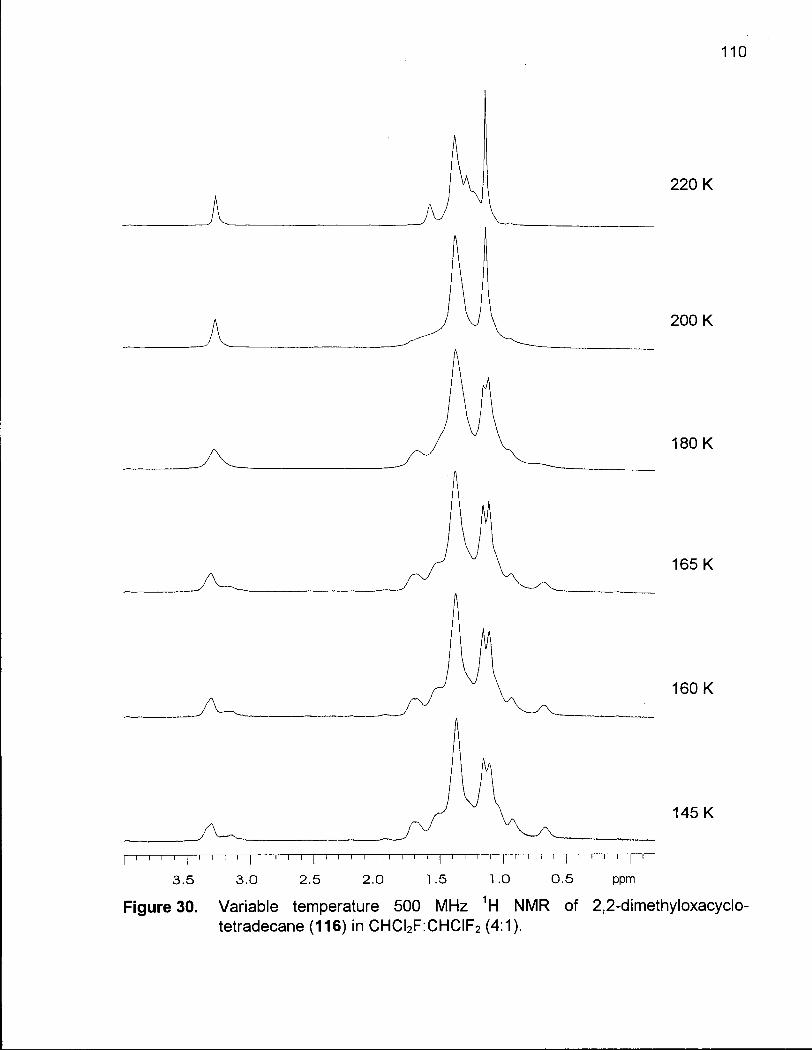

111
The analysis of the DNMR data begins with the downfield signals of the protons
of C-14. In the [3434]-4 conformation, the H-14endo proton is shielded by the
diamagnetic anisotropy of the C-12/C-13 bond. However, van der Waals steric
interactions between this proton and the H-11endo and H-3endo protons results in a
deshielding of the H-14end0 proton. The calculated distances between these protons
and the H-14endo proton are 2.20 A and 2.17 A respectively. Both of these values are
less than the sum of the van der Waals radii for a pair of hydrogens.62 Conversely, the
H-14exo proton is deshielded by the anisotropy of the neighbouring bond, and shielded
by the van der Waals interactions. The relative magnitudes of these effects is
unknown, but the anisotropy contribution is thought to be larger. The H-14endo proton is
expected to have two large coupling constants, a vicinal coupling to H-13p, and a
geminal coupling to H-14exo. In contrast, H-14exo would have only the large Jgem
coupling constant. Thus, the broad signal at 3.15 ppm is assigned to H-14endo, and the
sharper signal at 3.31 ppm to the H-14exo proton based on these chemical shift and
coupling constant arguments.
In the [3434]-2 conformation, C-14 is located at the middle of a 4-bond side of
the ring. Here, H-14exo is shielded as the result of van der Waals interactions between
the H-14endo proton, and the H-3end0 and H-11endo protons. The calculated distances
between H-14end0 and these other protons are 2.10 A and 2.14 A respectively. The
H-14exo proton is further shielded by electric field effects caused by the parallel
alignment of the carbon-hydrogen bonds between C-14 and H-14exo, and C-12 and
H-12p. The reinforcement of these shielding effects would result in a larger A 5 than
observed here in the low temperature spectra of 116. Moreover, in the [3434]-2
conformation, a pair of large coupling constants (3J, Jgem) are expected for BOTH the
H-14endo and H-14exo protons. This predicted lineshape is in poor agreement with that
observed here in the low temperature spectra. Therefore, this conformation was not
considered further as a major conformation in the analysis of the low temperature
spectra of 116.
In the rt 1H NMR spectrum of 116, the C-13 protons p to the ether oxgyen, were
resolved from the methylene envelope while the protons of C-3 overlapped with the

112
methylene envelope. At low temperature however, two signals were visible at 1.69 ppm
and 1.50 ppm which were assigned to protons p to the ether oxygen. In the [3434]-4
conformation, H-3endo is deshielded by the diamagnetic anisotropy of the O/C-2 bond,
and also by the carbon-carbon bond between C-2 and the p-methyl group. A van der
Waals steric interaction with H-14endo further deshields this proton. The H-13a proton is
shielded by the anisotropy of both the C-14/0 bond, and the C-11/C-12 bond. No van
der Waals steric interactions are expected for the C-13 corner protons in the [3434]-4
conformation since both are exo to the ring. This combination of effects lead to the
assignment of the lower field signal at 1.69 ppm to the H-3endo and H-13p protons. The
higher field signal at 1.50 ppm was assigned to the more shielded H-3eXo and H-13a
protons.
The averaging of the C-2 geminal methyl groups of 116 is slow at low
temperature, and a pair of signals of approximately equal intensity at 1.15 and
1.10 ppm are visible at low temperature. The presence of this pair of signals indicates
that a conformational interconversion that results in exchange of the geminal methyl
groups is no longer rapid at the low temperature. The assignment of the signals at 1.15
and 1.10 ppm to the C-2a and C-2P methyl groups is ambiguous at this time.
The signals observed at high-field in the DNMR spectra of 116 are assigned to
the H-4exo and H-11eXo protons. In the [3434]-4 conformation, the H-4endo and H-11endo
protons are deshielded as a result of a series of van der Waals steric interactions. The
H-4endo proton is only 2.17 A from H-7end0, and 2.29 A from the H-11endo proton. The
H-11endo proton is located 2.20 A from H-14end0, and 2.11 A from H-8endo- These
deshielding effects result in the shielding of the H-4exo and H-11exo protons. It is the
distance from the ether oxygen that results in the different chemical shifts of these
protons at low temperature. In a study described in Chapter 1, the magnitude of the van
der Waals shielding effect was found to be proportional to the electronegativity of the
sterically opposing group.64 The electronegative ether oxygen is calculated to be
2.60 A from H-4endo, and 3.25 A from H-11end0, and hence would make a greater
contribution to the shielding of the H-4exo proton. For these reasons the highest field

113
signal at 0.67 ppm is assigned to H-4exo, and the other high-field signal at 0.93 ppm to
the H-11exo proton.
A molecular mechanics search for the lowest energy conformations of 116 was
conducted using the Monte Carlo technique and the MM3* force field. These
calculations gave a total of eight conformations within 2 kcal/mol of the calculated
lowest energy conformation which was the [3434]-4 conformation 116-A. The second
lowest energy conformation was the [3434]-2 conformation 116-B. As expected, the
gem-dimethyl group was situated at a corner position in all of the low energy
conformations (Table 22). These two lowest energy conformations were the same as
those predicted by analysis of transannular interactions in 116. The relative
populations of these conformations at different temperatures were calculated from
relative strain energies obtained from the MM3* calculations (Table 23). These results
suggested that a single conformation, the [3434]-4 conformation 116-A, was the major
conformation over the temperature range studied in agreement with the DNMR data.
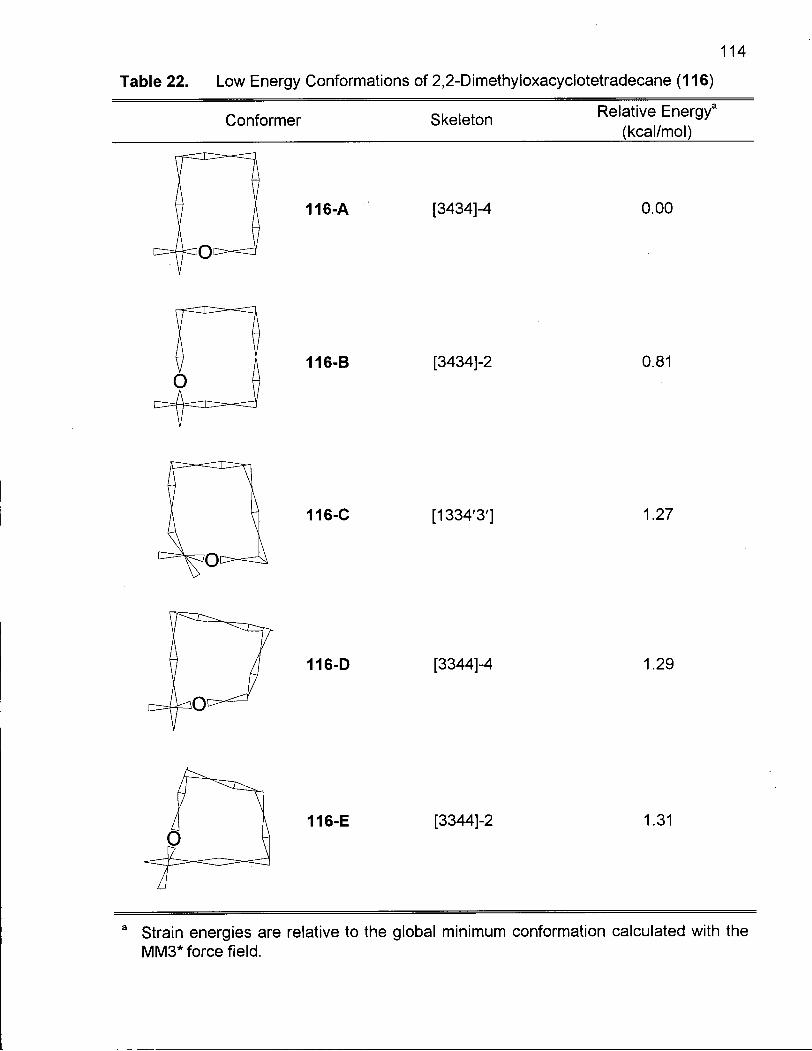
114
Table 22. Low Energy Conformations of 2,2-Dimethyloxacyclotetradecane (116)
Conformer Skeleton Relative Energy (kcal/mol)
116-A ' [3434]-4 0.00
116-B [3434]-2 0.81
116-C [1334'3'] 1.27
116-D [3344]-4 1.29
o \ 116-E [3344]-2 1.31
Strain energies are relative to the global minimum conformation calculated with the MM3* force field.
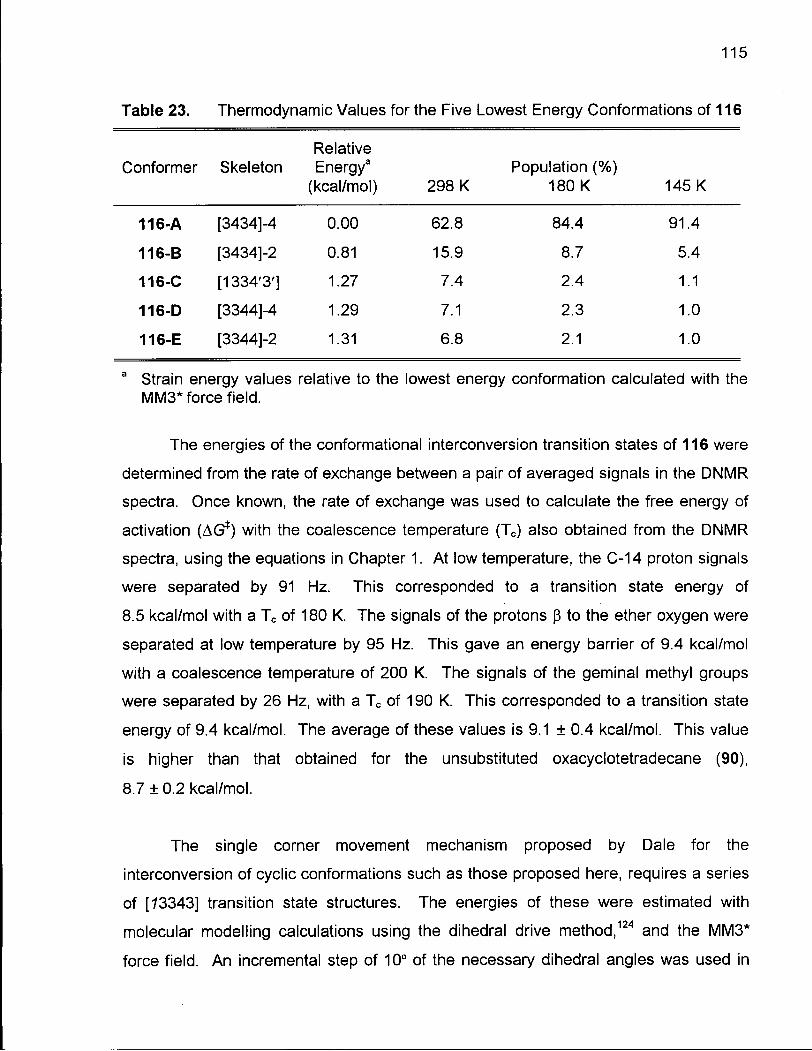
115
Table 23. Thermodynamic Values for the Five Lowest Energy Conformations of 116
Relative Conformer Skeleton Energy3 Population (%)
(kcal/mol) 298 K 180 K 145 K
116-A [3434]-4 0.00 62.8 84.4 91.4
116-B [3434]-2 0.81 15.9 8.7 5.4
116-C [1334'3'] 1.27 7.4 2.4 1.1
116-D [3344]-4 1.29 7.1 2.3 1.0
116-E [3344]-2 1.31 6.8 2.1 1.0
3 Strain energy values relative to the lowest energy conformation calculated with the MM3* force field.
The energies of the conformational interconversion transition states of 116 were
determined from the rate of exchange between a pair of averaged signals in the DNMR
spectra. Once known, the rate of exchange was used to calculate the free energy of
activation (AG 4) with the coalescence temperature (Tc) also obtained from the DNMR
spectra, using the equations in Chapter 1. At low temperature, the C-14 proton signals
were separated by 91 Hz. This corresponded to a transition state energy of
8.5 kcal/mol with a Tc of 180 K. The signals of the protons p to the ether oxygen were
separated at low temperature by 95 Hz. This gave an energy barrier of 9.4 kcal/mol
with a coalescence temperature of 200 K. The signals of the geminal methyl groups
were separated by 26 Hz, with a Tc of 190 K. This corresponded to a transition state
energy of 9.4 kcal/mol. The average of these values is 9.1 ± 0.4 kcal/mol. This value
is higher than that obtained for the unsubstituted oxacyclotetradecane (90),
8.7 ± 0.2 kcal/mol.
The single corner movement mechanism proposed by Dale for the
interconversion of cyclic conformations such as those proposed here, requires a series
of [73343] transition state structures. The energies of these were estimated with
molecular modelling calculations using the dihedral drive method,124 and the MM3*
force field. An incremental step of 10° of the necessary dihedral angles was used in

116
this calculation. The steric requirements of the gem-dimethyl group demand that this
functional group remain at a corner position in the low energy conformations. Similarly,
the transition state structures for the interconversion of the conformations of 116 also
have the gem-dimethyl group at a corner position. The suggested energies of the
[73343] transition state conformations for the interconversion of the global minimum
[3434]_4 conformation 116-A with the [3344]-2 and [3344]-4 conformations 116-E and
116-D were calculated at 13.3 kcal/mol and 12.8 kcal/mol (Figure 31). The energy of
the [73343] transition state conformation involved in the interconversion of the higher
energy [3434]-2 conformation 116-B with the [3344]-2 conformation 116-E was
calculated to be 13.5 kcal/mol. These calculated transition state energies were larger
than the observed values. This may be due to the inaccuracy of the assumption that
the dihedral angles of the 1-bond side and adjacent bonds were 120°, 0°, and 120°
respectively. Any conformational interconversion involving movement of the geminally
substituted carbon atom away from the corner position would also be expected to have
a higher energy.
[3344]-4 [3434]-2
Figure 31. Interconversion of conformations of 116 via single corner movements.

117
2.4.1 Synthesis of 3,3-Dimethyloxacyclotetradecane (119)
The Baeyer Villiger oxidation of cyclotridecanone (86) gave lactone 87. The
gem-dimethyl substituents were introduced via a sequential alkylation with LDA and
methyl iodide to give ultimately the gem-dimethyl lactone 118 (Scheme 14). The
1H NMR spectrum of lactone 118 contained a two-proton multiplet from 4.04-4.07 ppm
for the C-13 methylene and a six-proton singlet at 1.15 ppm for the C-2 geminal methyl
groups. The 13C NMR spectrum contained 14 lines with a signal at 178.21 ppm for the
C-1 carbonyl. The HRMS and chemical analysis results were also consistent with the
composition of lactone 118. Formation of a thionolactone with Lawesson's reagent 48
is problematic in the case of sterically hindered lactones such as 118. 1 4 4 Hence, the
conversion of this lactone to the macrocyclic ether 119 was performed via the direct
reduction with sodium borohydride in the presence of boron trifluoride etherate in THF
heated at reflux to give the macrocyclic ether 119 in a low yield of 11%.36 This
reduction did not proceed at room temperature. The four-step reaction sequence
proceeded in an overall yield of 8%.
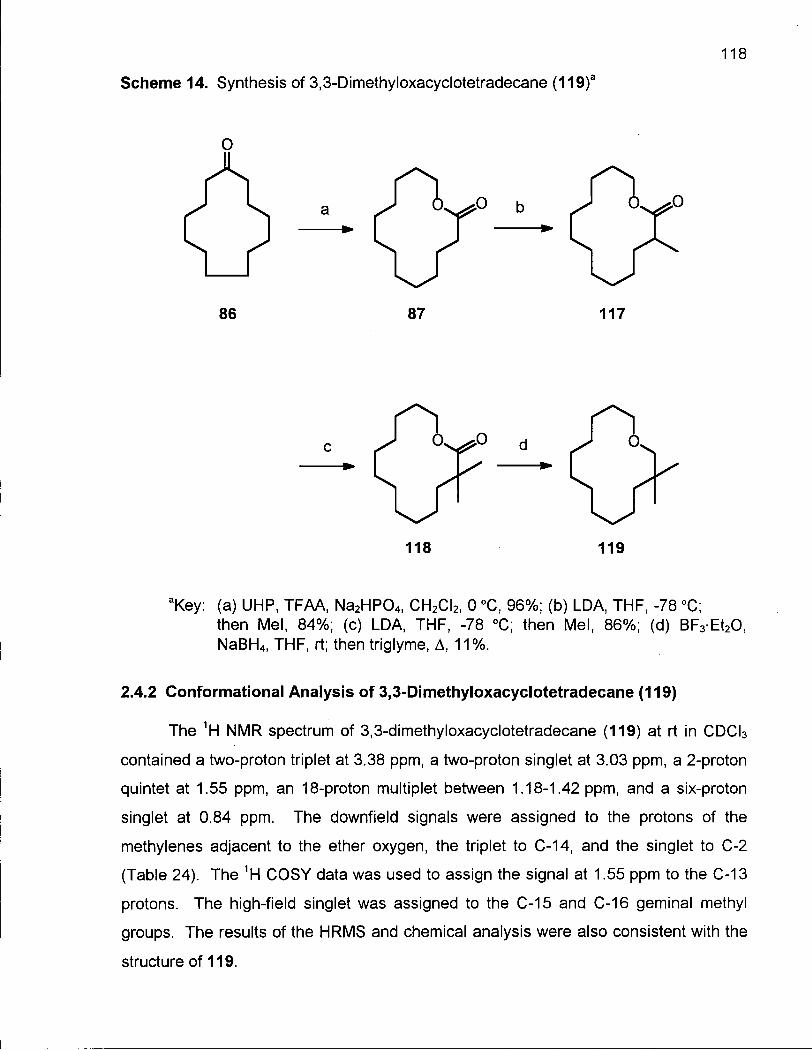
Scheme 14. Synthesis of 3,3-Dimethyloxacyclotetradecane (119)1
118
0
86 87 117
118 119
aKey: (a) UHP, TFAA, Na2HP04, CH2CI2, 0 °C, 96%; (b) LDA, THF, -78 °C; then Mel, 84%; (c) LDA, THF, -78 °C; then Mel, 86%; (d) BF3Et20, NaBH4, THF, rt; then triglyme, A , 11 %.
2.4.2 Conformational Analysis of 3,3-Dimethyloxacyclotetradecane (119)
The 1H NMR spectrum of 3,3-dimethyloxacyclotetradecane (119) at rt in CDCI3
contained a two-proton triplet at 3.38 ppm, a two-proton singlet at 3.03 ppm, a 2-proton
quintet at 1.55 ppm, an 18-proton multiplet between 1.18-1.42 ppm, and a six-proton
singlet at 0.84 ppm. The downfield signals were assigned to the protons of the
methylenes adjacent to the ether oxygen, the triplet to C-14, and the singlet to C-2
(Table 24). The 1H COSY data was used to assign the signal at 1.55 ppm to the C-13
protons. The high-field singlet was assigned to the C-15 and C-16 geminal methyl
groups. The results of the HRMS and chemical analysis were also consistent with the
structure of 119.

119
The 13C spectrum of 119 contained 14 lines. The two lowest field signals at
77.38 and 68.81 ppm were assigned to C-2 and C-14 respectively. The assignment of
other 13C and 1H signals was aided with COSY, HMQC, and HMBC 2D-NMR
experiments (Table 24). The signal at 34.09 ppm was assigned to the C-3 quaternary
carbon, and the signal at 37.43 ppm to the adjacent C-4 methylene. The chemical shift
of the C-3 geminal methyl groups was 26.12 ppm.
Table 24. 1H and 13C NMR Assignments for 3,3-Dimethyloxacyclotetradecane (119) in CDCI3 at Room Temperature3
Position 1H NMRb 13C NMRb
3.03 77.38
34.09
1.22 37.43
not assigned0
1.36 22.84
1.55 28.81
3.38 68.81
0.84 26.12
3 Arrows show HMBC correlations. b The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI3 (13C). c Due to signal overlap these signals could not be assigned.
The low temperature 1H NMR spectra of 119 were obtained in a 4:1 mixture of
Freon 21 and Freon 22 as solvent (Figure 32). The spectrum of 119 at 220 K was
similar to the rt spectrum however the signals had broadened at the lower temperature.
At 200 K, the C-14 methylene signal broadened, and the signal for the C-2 protons was
extremely broad. At this same temperature, the signal for the C-13 protons, p to the
ether oxygen, was unresolved on the low-field shoulder of the methylene envelope, and
the C-3 geminal methyl signal had broadened to a significant degree. At 190 K, the
signals for the C-2 and C-14 protons broadened further, and the signal for the C-13
15, 16

120
protons downfield from the methylene envelope also was poorly resolved. In the
upfield portion of the spectrum, the C-3 geminal methyl signal split into a pair of equally
intense signals at 0.93 ppm and 0.77 ppm.
At lower temperatures, the line shape of the a-proton signals became distinct
with a total of seven peaks visible in this region from 2.5 to 3.7 ppm at 140 K, the
lowest temperature examined in this DNMR series. This is indicative of multiple
conformations being present even at low temperature. Three signals at 1.61, 1.75, and
1.84 ppm were visible in the region where the C-13 proton signals were expected. The
geminal methyl signals were quite broad, with no additional peaks visible in that region
to indicate the presence of minor conformations. Also, at temperatures below 175 K an
unresolved, broad peak at 0.62 ppm on the high-field shoulder of the methyl signals
was visible.
The low energy conformations of 119 are predicted to have the C-3
gem-dimethyl substituted carbon at a corner position of the ring. This configuration
allows for the ether oxygen to be located in the middle of a 4-bond side in the [3434]-1
conformation, thereby removing the transannular hydrogen interactions present at that
location in the parent hydrocarbon. The ether oxygen can also be located on the
3-bond side in the [3434]-4 conformation where transannular hydrogen interactions are
present in cyclotetradecane as well. The non-diamond lattice [3344]-1 conformations
of 119 with the gem-dimethyl group at a corner position flanked by either two 4-bond
sides, or a 3- and a 4-bond side were also expected to have low strain energy. These
conformations were considered as likely low-energy conformations in the analysis of
the DNMR spectra of 119.

1 2 1

122
13 12
14 13
2 14
[3434]-1 [3434]-4
14
[3344]-1 [3344]-1
The downfield portion of the low temperature spectra of 119 contained seven
peaks. The three lowest field peaks were triplet-like with relative heights of 1:2:1.
However the A8 values of these peaks are approximately 50 Hz, and therefore too large
to have been the result of vicinal coupling. The downfield peaks of the DNMR spectra
therefore must result from a number of unequally populated conformations present at
the low temperature. The four higher field signals at 3.27, 3.12, 2.99, and 2.65 ppm
were assigned to the C-2 protons of 119. The downfield pair of signals in this group
are assigned to the major conformation and are more intense than the upfield pair of
signals that are assigned to the minor conformation, with an approximate relative
intensity of 2.2:1. This corresponds to an energy difference of 0.21 kcal/mol between
the major and minor conformations. The A5 value of the C-2 protons in the major
conformation is small, while in the minor conformation it is large. The predicted A5
values for the C-2 protons in the low energy conformations suggested above were
compared in an effort to identify the major and minor conformations of 119.
In the [3434]-1 conformation, the H-2exo proton is deshielded by the anisotropy of
the C-3/C-4 bond, and shielded by the anisotropy of the carbon-carbon bond between
C-3 and the C-3P methyl group. Also, this proton is shielded by a van der Waals steric
interaction between the H-2endo and the H-5endo protons that are calculated to be 2.20 A

123
apart. The result of these effects is a predicted small A5 for the C-2 protons in this
conformation with the H-2exo proton at higher field. In the [3434]-4 conformation, the
H-2exo proton is deshielded by the anisotropy of the C-3/C-4 bond, but shielded by the
anisotropy of the carbon-carbon bond between C-3 and the C-3a methyl group. Also,
this proton is shielded by van der Waals steric interactions between the H-2endo proton
and the H-5endo and H-13endo protons that are calculated to be 2.20A and 2.19 A from
H-2endo- The combination of these effects is a predicted A8 value that is large for the
C-2 protons in this conformation with the H-2exo proton at higher field.
In the [3344]-1 conformation with the gem-dimethyl substituted corner atom
flanked by a pair of 4-bond sides, The H-2exo proton is deshielded by the anisotropy of
the C-3/C-4 bond, but shielded by the anisotropy of the carbon-carbon bond between
C-3 and the C-3P methyl group. The magnitude of these effects is unequal as a result
of the distorted geometry of this non-diamond lattice conformation. Also, the H-2exo
proton is shielded by a van der Waals steric interaction between the H-2end0 and H-5endo
protons that are calculated to be 2.27 A apart. A large A5 value is predicted for the C-2
protons in this conformation as a result of these effects. In the other [3344J-1
conformation where the substituted corner atom is between a 3- and a 4-bond side, the
environment of the C-2 protons is similar to that of the C-2 protons in the [3434]-1
conformation, with a small A8 value predicted.
In summary, the chemical shift differences between the C-2 protons is small in
the [3434]-1 conformation and in the [3344]-1 conformation with the gem-disubstituted
corner atom flanked by a 3- and a 4-bond side. These are possible candidates for the
major conformation of 119. The A8 value was predicted to be large for the [3434]-4 and
the [3344]-1 conformation where the gem-disubstituted corner atom is flanked by a pair
of 4-bond sides. These are possible candidates for the minor conformation of 119.
A similar analysis was performed for the three signals of the C-14 protons at
3.61, 3.53, and 3.42 ppm. The triplet-like pattern here was the result of the unequally
intense overlapping doublets of the major and minor conformations of 119. Although
the heights of the peaks at 3.61 and 3.42 ppm are approximately equal, the 3.61 ppm

124
peak is partially overlapped with the middle 3.53 ppm peak, artificially increasing the
height of this signal. Thus, the two signals for the minor conformation are the peaks at
3.61 and 3.53 ppm, with the major conformation signals at 3.42 ppm and 3.53 ppm.
The observed A8 values of the C-14 protons in the major and minor conformations of
119 are both small. In all cases, a comparison of the shielding effects experienced by
the C-14 protons in the four low energy conformations gave small predicted A5 values
and hence did not assist in determining which conformation might be the major or minor
one observed.
The signal of the C-3 gem-dimethyl groups of 119 split as the temperature was
lowered to give a pair of signals at 0.93 and 0.77 ppm. Unfortunately, no signals for
the methyl groups of the minor conformation could be identified. However, these
signals may be hidden by the major conformation proton signals in this region, and also
by the somewhat broad signals of the major conformation methyl groups.
A molecular mechanics search for low energy conformations of 119 was
conducted with the Monte Carlo technique and the MM3* force field. The calculated
global minimum was the [3434]-1 conformation 119-A with the [3344]-1 conformation
119-B calculated to have the next lowest energy, 0.49 kcal/mol higher. These
calculations suggested the existence of five other conformations within 2 kcal/mol of
the global minimum conformation (Table 25). Higher energy conformations were
ignored since they were not considered to be significantly populated over the
temperature range examined. The relative populations of these low energy
conformations at different temperatures were calculated from the relative energies
obtained from the MM3* calculations (Table 26). The results of these calculations
suggest the [3434]-1 conformation 119-A to be the major conformation over the
temperature range studied with the second most populated conformation, the [3344]-1
conformation 119-B, also significantly populated. These assignments are consistent
with the DNMR data and above proposals. The calculated relative energies between
the major and minor conformations is 0.49 kcal/mol, a value higher than the
0.21 kcal/mol observed in the DNMR spectra.
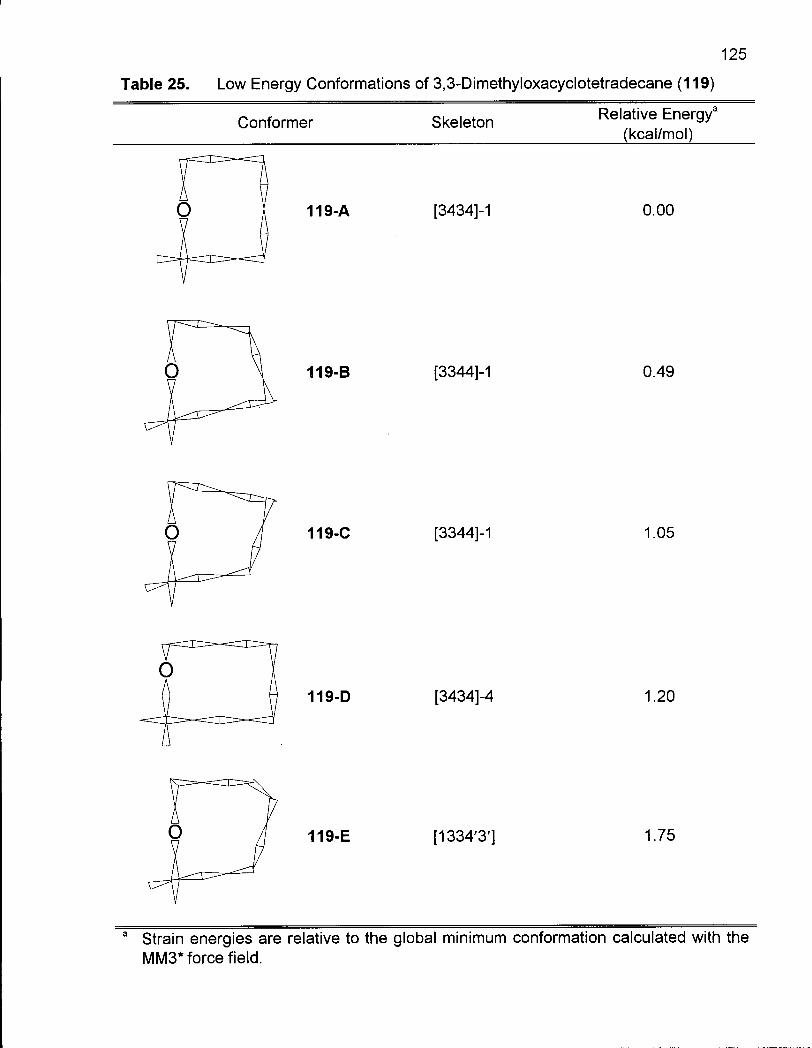
125
Table 25. Low Energy Conformations of 3,3-Dimethyloxacyclotetradecane (119)
Conformer Skeleton Relative Energy (kcal/mol)
119-A [3434]-1 0.00
119-B [3344]-1 0.49
119-C [3344]-1 1.05
119-D [3434]-4 1.20
119-E [1334'3'] 1.75
Strain energies are relative to the global minimum conformation calculated with the MM3* force field.
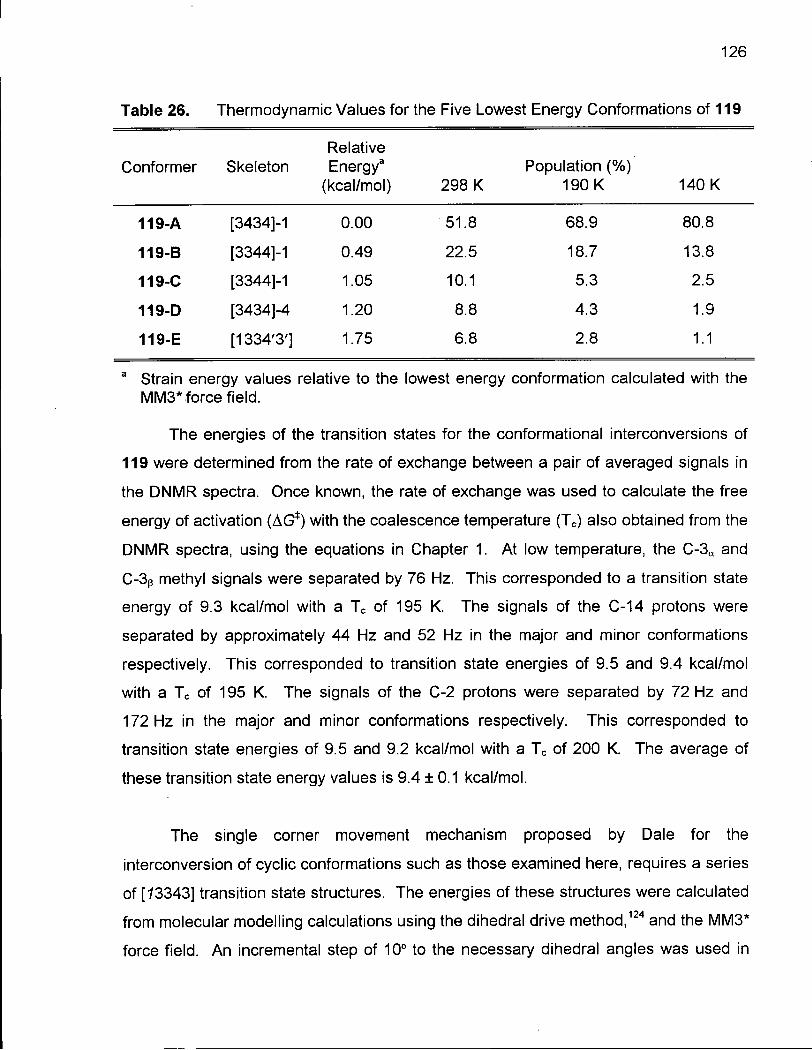
126
Table 26. Thermodynamic Values for the Five Lowest Energy Conformations of 119
Relative Conformer Skeleton Energy3 Population (%)
(kcal/mol) 298 K 190 K 140 K
119-A [3434]-1 0.00 51.8 68.9 80.8
119-B [3344]-1 0.49 22.5 18.7 13.8
119-C [3344J-1 1.05 10.1 5.3 2.5
119-D [3434]-4 1.20 8.8 4.3 1.9
119-E [1334'3'] 1.75 6.8 2.8 1.1
3 Strain energy values relative to the lowest energy conformation calculated with the MM3* force field.
The energies of the transition states for the conformational interconversions of
119 were determined from the rate of exchange between a pair of averaged signals in
the DNMR spectra. Once known, the rate of exchange was used to calculate the free
energy of activation (AG*) with the coalescence temperature (Tc) also obtained from the
DNMR spectra, using the equations in Chapter 1. At low temperature, the C-3a and
C-3P methyl signals were separated by 76 Hz. This corresponded to a transition state
energy of 9.3 kcal/mol with a Tc of 195 K. The signals of the C-14 protons were
separated by approximately 44 Hz and 52 Hz in the major and minor conformations
respectively. This corresponded to transition state energies of 9.5 and 9.4 kcal/mol
with a Tc of 195 K. The signals of the C-2 protons were separated by 72 Hz and
172 Hz in the major and minor conformations respectively. This corresponded to
transition state energies of 9.5 and 9.2 kcal/mol with a Tc of 200 K. The average of
these transition state energy values is 9.4 ± 0.1 kcal/mol.
The single corner movement mechanism proposed by Dale for the
interconversion of cyclic conformations such as those examined here, requires a series
of [73343] transition state structures. The energies of these structures were calculated
from molecular modelling calculations using the dihedral drive method,124 and the MM3*
force field. An incremental step of 10° to the necessary dihedral angles was used in
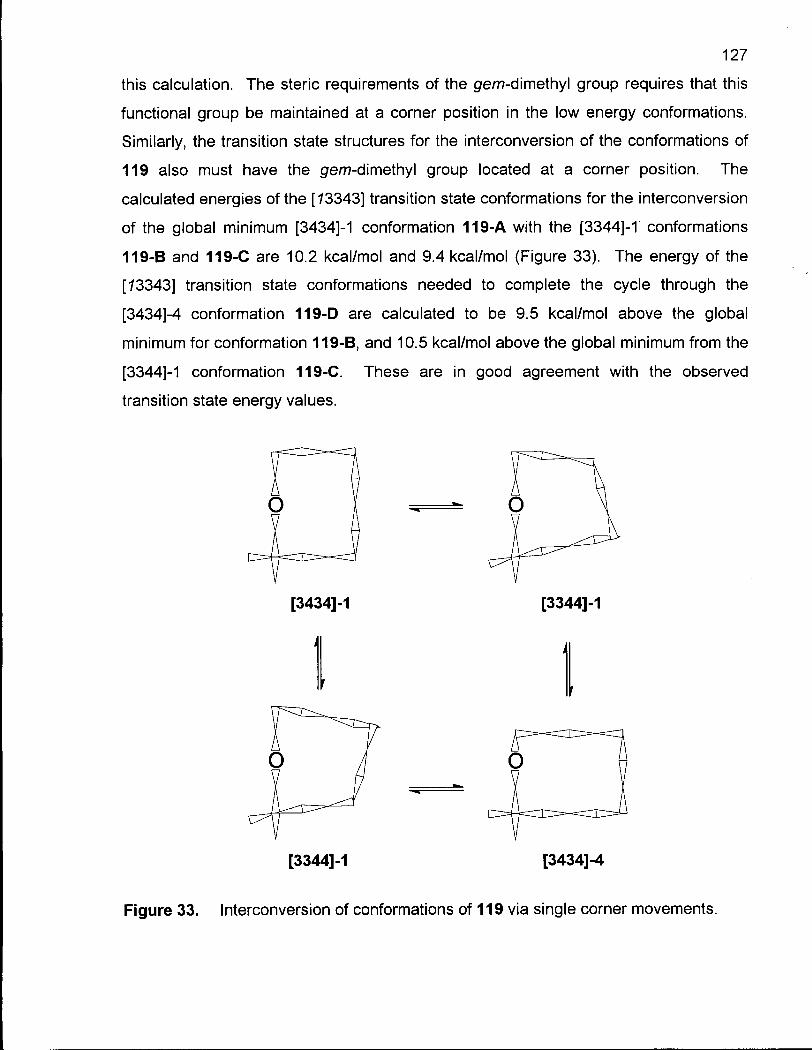
127
this calculation. The steric requirements of the gem-dimethyl group requires that this
functional group be maintained at a corner position in the low energy conformations.
Similarly, the transition state structures for the interconversion of the conformations of
119 also must have the gem-dimethyl group located at a corner position. The
calculated energies of the [73343] transition state conformations for the interconversion
of the global minimum [3434J-1 conformation 119-A with the [3344]-T conformations
119-B and 119-C are 10.2 kcal/mol and 9.4 kcal/mol (Figure 33). The energy of the
[73343] transition state conformations needed to complete the cycle through the
[3434]-4 conformation 119-D are calculated to be 9.5 kcal/mol above the global
minimum for conformation 119-B, and 10.5 kcal/mol above the global minimum from the
[3344J-1 conformation 119-C. These are in good agreement with the observed
transition state energy values.
[3434]-1 [3344]-1
[3344]-1 [3434]-4
Figure 33. Interconversion of conformations of 119 via single corner movements.

128
2.5.1 Synthesis of 6,6-Dimethyloxacyclotetradecane (137)
The preparation of the macrocyclic ether 137 required a different approach than
that used to synthesize the macrocyclic ethers presented above. The introduction of a
gem-dimethyl group at a position remote from the oxygen atom of the macrocyclic ether
meant that these substituents could not be introduced through the alkylation of the
intermediate lactone or ketone. Instead, the synthetic strategy involved the preparation
of the molecule from two parts, which were coupled together using a dithiane ring as
the central unit of the acyclic molecule (Scheme 15). This dithiane ring was eventually
converted into the desired gem-dimethyl group. The macrocycle was formed via the
cyclisation of a hydroxy acid, and the resultant lactone was transformed into the
macrocyclic ether 137.
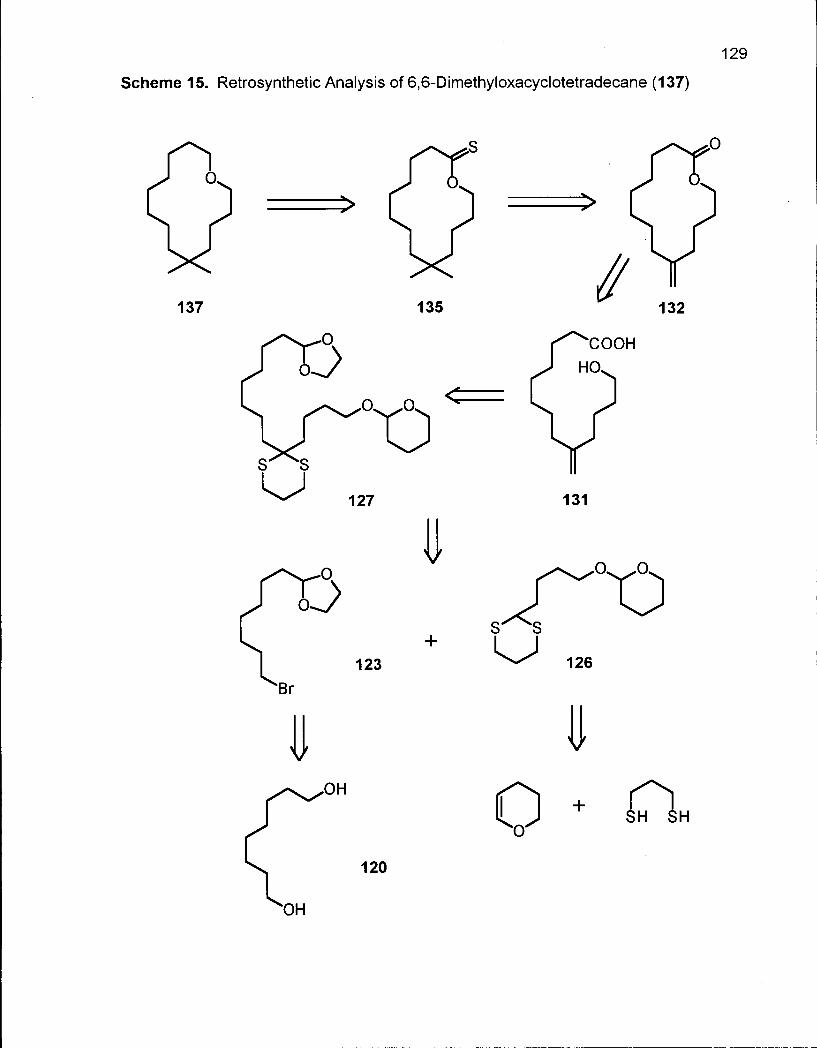

130
The symmetric 1,8-octanediol (120) was treated with 48% hydrobromic acid to
give the monobrominated alcohol 121.145 This bromo alcohol was oxidized under
Swern conditions to give the bromo aldehyde 122 which was subsequently protected by
reaction with ethylene glycol to give the ethylene acetal 123 (Scheme 16). This
three-step reaction sequence proceeded in an overall yield of 77%. The 1H NMR
spectrum of 123 contained a one-proton triplet at 4.78 ppm for the C-1 methine proton
of the acetal. As well, a pair of multiplets between 3.77-3.92 ppm for the C-1' and C-2'
methylenes of the acetal were observed. In the IR spectrum, the band at 1711 cm"1
from the C-1 carbonyl of the penultimate aldehyde 122 was absent.
Scheme 16. Synthesis of 8-bromooctanal ethylene acetal (123)a
123 122
aKey: (a) 48% HBr, C6H6, A, 92%; (b) (COCI)2, DMSO, Et3N, CH2CI2, -78 °C, 90%; (c) PPTS, HOCH2CH2OH, C6H6, A, 93%.
A solution of 1,3-propanedithiol and dihydropyran in CH2CI2 was treated with
boron trifluoride etherate to give the hydroxy dithiane 125,146 which was protected as a
tetrahydropyranyl ether to give 126 in a yield of 81 % for two-steps (Scheme 17).147 The
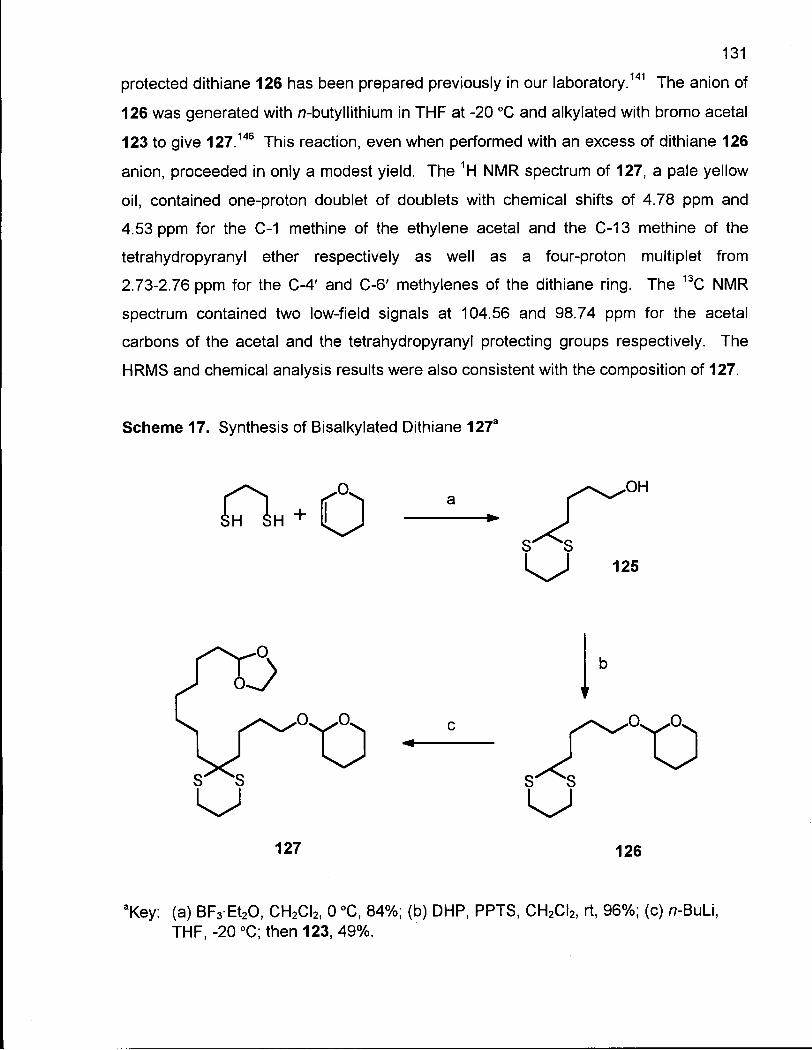
131
protected dithiane 126 has been prepared previously in our laboratory.141 The anion of
126 was generated with n-butyllithium in THF at -20 °C and alkylated with bromo acetal
123 to give 127.146 This reaction, even when performed with an excess of dithiane 126
anion, proceeded in only a modest yield. The 1H NMR spectrum of 127, a pale yellow
oil, contained one-proton doublet of doublets with chemical shifts of 4.78 ppm and
4.53 ppm for the C-1 methine of the ethylene acetal and the C-13 methine of the
tetrahydropyranyl ether respectively as well as a four-proton multiplet from
2.73-2.76 ppm for the C-4' and C-6' methylenes of the dithiane ring. The 13C NMR
spectrum contained two low-field signals at 104.56 and 98.74 ppm for the acetal
carbons of the acetal and the tetrahydropyranyl protecting groups respectively. The
HRMS and chemical analysis results were also consistent with the composition of 127.
Scheme 17. Synthesis of Bisalkylated Dithiane 127a
S S 125
.0. 0 c
•
b
127 126
aKey: (a) BF3Et20, CH2CI2, 0 °C, 84%; (b) DHP, PPTS, CH2CI2, rt, 96%; (c) n-BuLi, THF,-20 °C; then 123, 49%.

132
With the dithiane 127 in hand, the next task was to transform the dithiane ring
into the desired gem-d\methyl group. Difficulties were encountered with the unwanted
cleavage of the protecting groups when this conversion was attempted on the protected
compound, and hence the conversion was performed in two stages. The dithiane ring
of 127 was hydrolyzed into the ketone 128 (Scheme 18). This was first attempted with
NBS under standard conditions148,149 but difficulty with the cleavage of the acetal
protecting groups was encountered. To overcome this problem, an alternative
cleavage involving the use of mercuric perchlorate with calcium carbonate was
investigated.150 This reaction proceeded rapidly to give the ketone 128 with both
acetals intact. The carbonyl of 128 was converted into the exo-methylene group of 129
using the Tebbe reagent 3 2 . 3 8 1 5 1 " 1 5 3 Removal of the acetal protecting groups gave the
hydroxy aldehyde 130. 1 4 7
The oxidation of the aldehyde in the presence of the primary alcohol of 130
required chemoselective conditions. Too powerful an oxidant could have also oxidized
the primary hydroxyl group. The method chosen was a silver oxide oxidation with the
oxidant generated in situ from silver nitrate and sodium hydroxide.154 This reaction was
performed in the absence of light to minimize the photoreduction of Ag+. Unfortunately,
this oxidation step proceeded in only a modest 30% yield to give the hydroxy acid 131.
The 1H NMR spectrum of this oil contained a two-proton singlet at 4.68 ppm for the
C-14 exo-methylene and two-proton triplets at 3.64 ppm and 2.32 ppm for the C-2 and
the C-13 methylenes respectively. The 13C NMR spectrum contained a signal at
179.13 ppm for the C-1 carbonyl of the carboxylic acid. The IR spectrum of 131
contained bands at 3639 cm'1 and 1712 cm"1 for the carboxylic acid and 1644 cm"1 for
the C-9 double bond consistent with the structure 131.

133
Scheme 18. Synthesis of 9-Methylene-13-hydroxytridecanoic acid (131)
b
131 130 129
aKey: (a) Hg(CI04)2, CaC03, THF, H20, 80%; (b) Tebbe reagent 32, 3 8 DMAP, pyr, THF, -40 °C, 53%; (c) PPTS, acetone, H20, A, 90%; (d) AgN03, NaOH, THF, H20, 30%.
The Yamaguchi procedure wherein the hydroxy acid is first activated as a mixed
anhydride with triethylamine and 2,4,6-trichlorobenzoyl chloride was used to cyclize the
hydroxy acid 131. 2 8 This activated anhydride was cyclized under high dilution
conditions to give the lactone 132 (Scheme 19).28,155 The exocyclic methylene of 132
then was converted into a cyclopropyl group using diethylzinc and chloroiodomethane
via a procedure similar to that developed by Denmark.156"158 This reaction was found to
be superior to the traditional Simmons-Smith procedure159,160 since the
bis(chloromethyl)zinc reagent is more reactive than the bis(iodomethyl)zinc reagent

134
used in the Simmons-Smith conditions.156,157 The cyclopropyl group of 133 was ring
opened under hydrogenolysis conditions with Adams catalyst (Pt02) in acetic acid to
give the gem-dimethyl lactone 134 . 1 6 1 Hydrogenolysis of cyclopropyl rings occurs
preferentially at the least substituted carbon-carbon bond leading to the desired
gem-dimethyl product.162. The 1H NMR spectrum of 134 contained a six-proton singlet
at 0.82 ppm for the geminal methyl groups, as well as a two-proton multiplet from
4.10-4.12 ppm for the C-13 methylene group. The 1 3 C NMR contained 14 lines, with
one low-field signal at 173.70 ppm for the C-1 carbonyl. The HRMS data was also
consistent with the composition of lactone 134.
The remainder of the synthesis involved the conversion of the lactone into the
desired macrocyclic ether. The lactone 134 was reacted with Lawesson's reagent 48 to
give the thionolactone 1 3 5 . 5 1 5 5 This thionolactone was reduced with lithium
triethylborohydride and trapped with methyl iodide to give the mixed thioacetal 1 3 6 . 5 1 5 5
This compound was reacted immediately with a solution of tri(n-butyl)tin hydride to
remove the thiomethyl group of 136 under radical conditions to give the desired
macrocyclic ether 137. This six-step reaction sequence proceeded in an overall yield
of 10%.
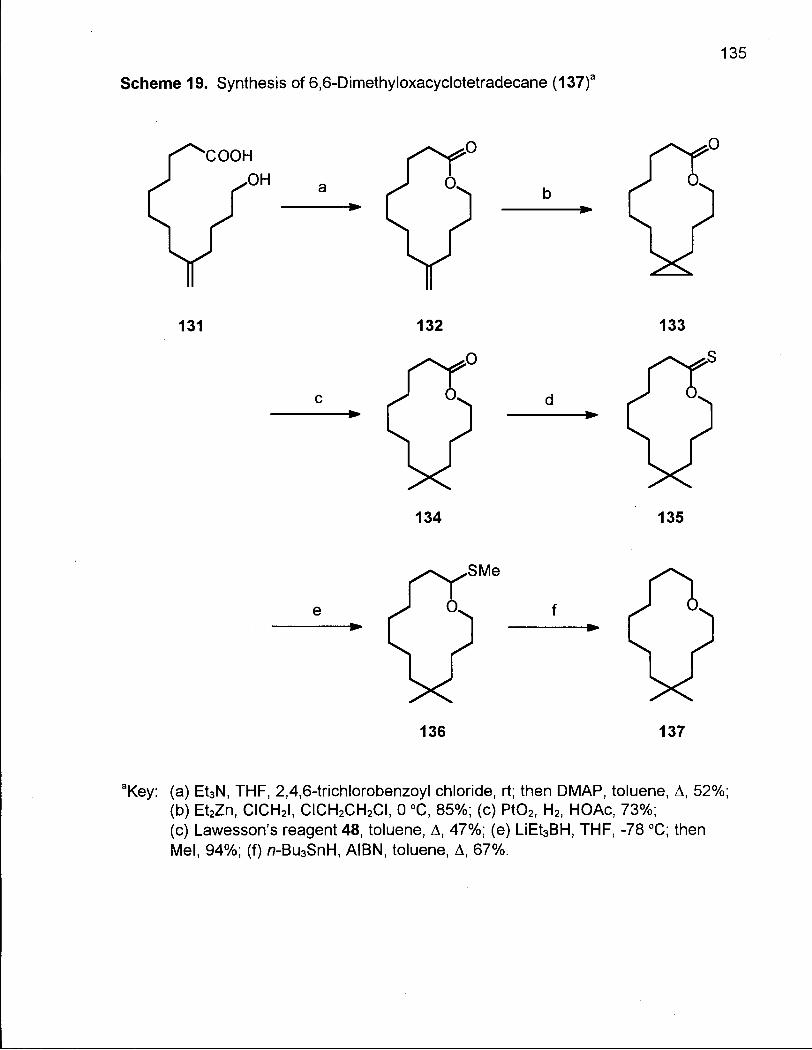
Scheme 19. Synthesis of 6,6-Dimethyloxacyclotetradecane (137)a
135
136 137
aKey: (a) Et3N, THF, 2,4,6-trichlorobenzoyl chloride, rt; then DMAP, toluene, A, 52%; (b) Et2Zn, CICH2I, CICH2CH2CI, 0 °C, 85%; (c) Pt02, H2, HOAc, 73%; (c) Lawesson's reagent 48, toluene, A, 47%; (e) LiEt3BH, THF, -78 °C; then Mel, 94%; (f) n-Bu3SnH, AIBN, toluene, A, 67%.

136
2.5.2 Conformational Analysis of 6,6-Dimethyloxacyclotetradecane (137)
The 1H NMR spectrum of 6,6-dimethyloxacyclotetradecane (137) at rt in CDCI3
contained a two-proton triplet at 3.43 ppm, a two-proton triplet at 3.42 ppm, a
two-proton quintet at 1.60 ppm, a two-proton triplet at 1.54 ppm, a ten-proton multiplet
from 1.29-1.42 ppm, a six-proton multiplet between 1.11-1.17 ppm, and a six-proton
singlet at 0.84 ppm. The low-field signals at 3.43 and 3.42 ppm were assigned to the
a-methylene protons of C-2 and C-14 respectively. The signals at 1.54 and 1.60 ppm
were assigned to C-3 and C-13, the methylenes 3 to the ether oxygen. The signal at
0.84 ppm was assigned to the C-15 and C-16 geminal methyl groups. The 13C NMR
spectrum contained 14 lines. The assignment of these signals was aided with COSY,
HMQC, and HMBC 2D-NMR experiments (Table 27). The long-range 1H-13C NMR data
was integral in distinguishing between the signals in the region of C-2 and C-14. The
chemical shifts of the C-2 to C-4 and the C-14 to C-12 portions of this macrocyclic ether
were very similar. However a correlation between one of the carbon atoms adjacent to
the quaternary C-6, and the protons of one of the methylenes y to the ether oxygen
made these assignments possible. The downfield signals at 68.17 and 67.70 ppm were
assigned to C-2 and C-14 adjacent to the ether oxygen atom. The C-6 quaternary
carbon had a chemical shift of 32.39 ppm. The chemical shifts of C-5 and C-7, the
carbons that flanked the quaternary carbon were 37.76 and 38.88 ppm respectively.
The HRMS data was also consistent with the structure of 137.

137
Table 27. 1H and 13C NMR Assignments for 6,6-Dimethyloxacyclotetradecane (137) in CDCI3 at Room Temperature3
Position 1H NMRb 13C NMRb
2 3.43 68.17
3 1.54 29.20
4 1.34 22.48
5 1.17 37.76
6 . ~ 32.39
7 1.12 38.88
8-11 not assigned0
12 1.40 23.55
13 1.60 28.74
14 3.42 67.70
15, 16 0.84 29.32
3 Arrows show HMBC correlations. b The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI3 (13C). c Due to signal overlap these signals could not be unambiguously assigned.
The low temperature NMR spectra of 137 were obtained in a 4:1 mixture of
Freon 21 and Freon 22 as solvent (Figure 34). The 1H NMR spectrum of 137 at 220 K
had already broadened in comparison to the rt spectrum. The signals of the
a-methylenes, C-2 and C-14, at 3.4 ppm continued to broaden with a coalescence
temperature of 190 K. Below this temperature, two closely spaced signals were visible
at 3.46 and 3.42 ppm. No small signals for minor conformations were visible in this
region, suggesting the presence of only a single conformation at low temperature. The
p-proton signals between 1.5 and 1.6 ppm in the rt spectrum coalesced above 220 K.
This was the highest temperature of the DNMR series, and the signals for these
protons were already broad. At lower temperatures, signals at 1.86 ppm, 1.59 ppm,
and 1.52 ppm were visible for these protons. The signal at 0.88 ppm, downfield of the
methyl singlet is an impurity. The signal for the C-6 geminal methyl groups at 0.84 ppm

138
did broaden somewhat as the temperature was lowered, but it remained averaged over
the temperature range examined. The remaining signals for other methylene protons in
137 were overlapped at rt, and although line shape changes did occur in this region as
the temperature was lowered, signal overlap prevented a detailed analysis.
The combination of a number of factors including, a general preference of
14-membered rings for the [3434] diamond lattice conformation over the non-diamond
lattice [3344] conformation, the placement of the ether oxygen at the middle of a 4-bond
side, and the placement of the gem-dimethyl group at a corner position suggested
some likely low energy conformations for 137. These included the diamond lattice
[3434]-1 and [3434]-4 conformations, and the non-diamond lattice [3344]-1
conformation. In the analysis of the DNMR study of 137, these conformations were
considered as candidates for the major conformation of 137.
[3344J-1
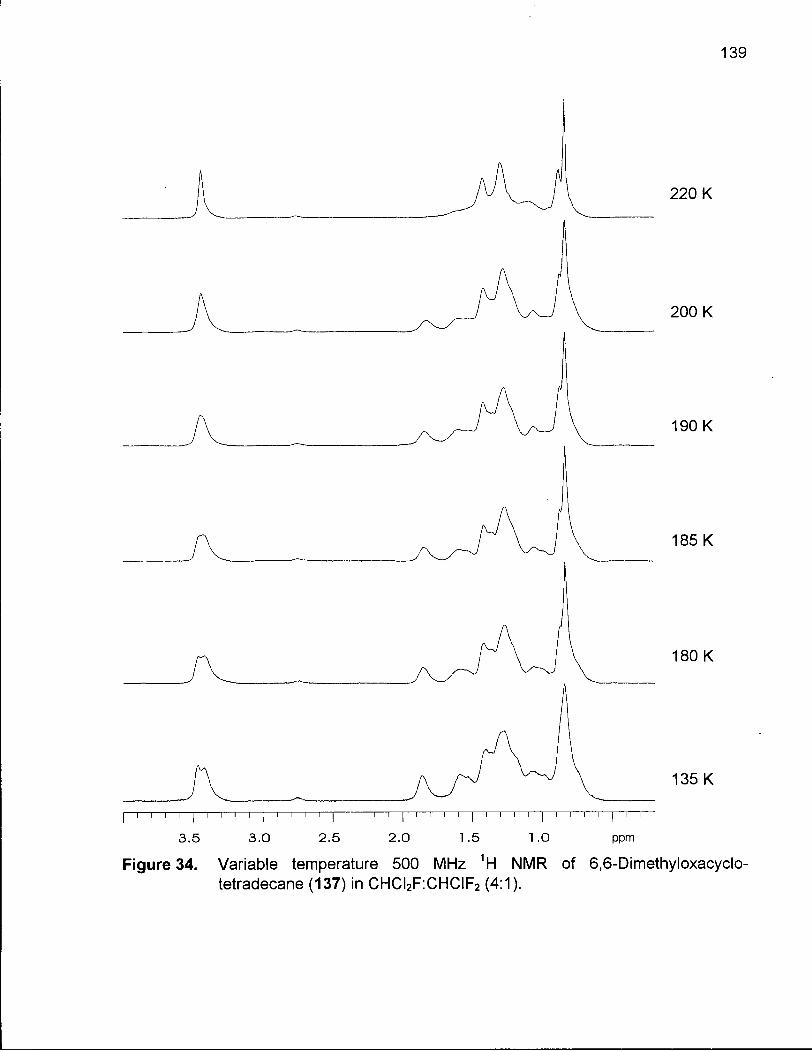
139

140
In the [3434]-4 conformation, the ct-methylene protons are not in similar
environments. One set occupies a corner position, while the other is on a 3-bond side.
The H-2a proton is deshielded by the anisotropy of the C-3/C-4 bond, while the H-2P
proton is deshielded by the anisotropy of the O/C-14 bond. These effects are of a
similar magnitude, and a small A6 value is predicted. In the case of the C-14 protons,
the H-14exo proton is deshielded by the anisotropy of the C-12/C-13 bond, but shielded
by van der Waals steric interactions between H-14end0 and the H-3endo and H-11endo
protons that are calculated to be 2.18 A and 2.23 A away from H-14endo- These van der
Waals steric shielding effects oppose the larger anisotropic effect, and a smaller A5 is
expected than for the anisotropic shielding alone. Thus, since the C-2 and C-14
methylene protons occupy different environments in this conformation, the line shape
for these protons is predicted to be more complex than observed here.
The non-diamond lattice geometry of the [3344]-1 conformation is distorted and
places the C-2 and C-14 protons in slightly different environments. The C-12 dihedral
angle is calculated to be 16° less than in the [3434]-1 conformation. This changes the
anisotropic shielding contribution to the chemical shift of the C-14 protons. The
calculated distances between the H-2end0 and H-5endo, and the H-11endo and H-14endo
protons are 2.27 A and 2.29 A. These distances are less than the sum of the van der
Waals radii for a pair of hydrogens,62 and a small van der Waals steric repulsion
contribution to the A5 of the a-methylene protons is expected. The sum of these effects
could lead to four separate signals for the a-methylene protons. However, depending
on the magnitude of the chemical shift changes caused by the dihedral angle distortion,
the signals may be partially overlapped.
In the [3434]-1 conformation of 137, the C-2 and C-14 protons adjacent to the
ether oxygen are on a 4-bond side. The local environment of these methylenes is
essentially equivalent, and any effects experienced by the C-2 protons are also
experienced by the C-14 protons. The H-2exo proton is deshielded by the anisotropy of
the C-3/C-4 bond, and the H-2endo proton is deshielded by a steric interaction with

141
H-5endo, calculated to be 2.22 A away. This van der Waals steric repulsion results in a
shielding of H-2ex0. The anisotropic shielding effect opposes the van der Waals
shielding effect and the magnitude of the overall shielding is reduced. Thus, the
expected A8 for the a-methylene protons in the [3434]-1 conformation is small, in
agreement with the low temperature 1H NMR spectra, and the [3434]-1 conformation is
likely to be a major conformation of 137. The downfield portion of the signal at
3.46 ppm was assigned to H-2exo and H-14exo, while the upfield portion at 3.42 ppm was
assigned to the endo protons.
The p-methylene protons of the [3434]-4 conformation are expected to give a
symmetric line shape as the result of anisotropy and van der Waals shielding effects.
The H-3endo proton is deshielded by the anisotropy of the C-2/0 bond, and deshielded
by a van der Waals steric repulsion with H-14endo. The reverse effects are experienced
by H-3exo, and a normal A5 value is predicted. The H-13p proton is deshielded by the
anisotropy of both the C-11/C-12 bond and the O/C-14 bond. No van der Waals steric
repulsions are expected since these protons occupy a corner position in this
conformation. The anisotropy shielding effects are additive giving a large predicted A5
value for C-13. Four signals are expected for the p-methylene protons in this
conformation as a result of these effects with the signals of the C-3 protons flanked by
the signals of the C-13 protons. The lowest field p-methylene proton signal at
1.86 ppm integrates to two-protons, where a one-proton signal at low-field for the H-13p
proton is expected for this conformation. Therefore, the [3434]-4 conformation is not a
major conformation of 137.
In the [3434]-1 conformation, the C-3 and C-13 methylenes are both located at
corner positions, and similar chemical shifts for each methylene are expected. The
H-3P proton is deshielded by the anisotropy of both the C-4/C-5 bond, and the O/C-2
bond. These effects are additive and a large A5 value is predicted. The relative
integration of the p-methylene signals at 1.86, 1.59, and 1.52 ppm compared to the
a-methylene signals at 3.4 ppm is approximately 2:1:1:4. The downfield signal is
assigned to H-3P and H-13p, and the upfield signals at 1.59 and 1.52 ppm are assigned

142
to H-3a and H-13a. The chemical shift difference of the upfield signals at low
temperature is equal to the chemical shift difference of the C-3 and C-13 methylene
protons at rt. It is unclear why this chemical shift difference is not also observed in the
downfield signal. The observed DNMR data is still consistent with the [3434]-1
conformation of 137.
A 13C DNMR study of 137 was also carried out in a 4:1 mixture of Freon 21 and
Freon 22 as solvent. One signal was observed for the C-6 quaternary carbon through
the 145-220 K temperature range examined. Only one signal was observed at low
temperatures for each of the C-5 and C-7 carbons adjacent to the quaternary carbon.
These results are consistent with one conformation being present at low temperature,
or alternatively a case where one conformation is considerably more populated than all
others present. The C-15 and C-16 geminal methyl groups gave one signal at high
temperature (220 K), a broad signal at 200 K, and two signals as the temperature was
lowered to 145 K. At low temperature these signals are separated by 50 Hz. This
result is consistent with a single conformation present at low temperature where the
process of ring inversion is slow, and the C-15 and C-16 methyl groups are no longer
averaged as in the [3434]-1 conformation.
A molecular mechanics search for low energy conformations of 137 was
conducted with the Monte Carlo technique and the MM3* force field. The global
minimum conformation was the [3434]-1 conformation 137-A with the [3434]-4
conformation 137-B calculated to have the next lowest energy, 1.15 kcal/mol higher.
These calculations suggested the existence of four other low energy conformations
within 2 kcal/mol of the global minimum conformation (Table 28). Higher energy
conformations were ignored as they were not considered to be significantly populated.
The relative populations of these conformations at different temperatures were
calculated from relative energies obtained from the MM3* calculations (Table 29). The
results of these calculations suggested the [3434]-1 conformation of 137 to be the
major conformation over the temperature range studied in agreement with the DNMR
data.
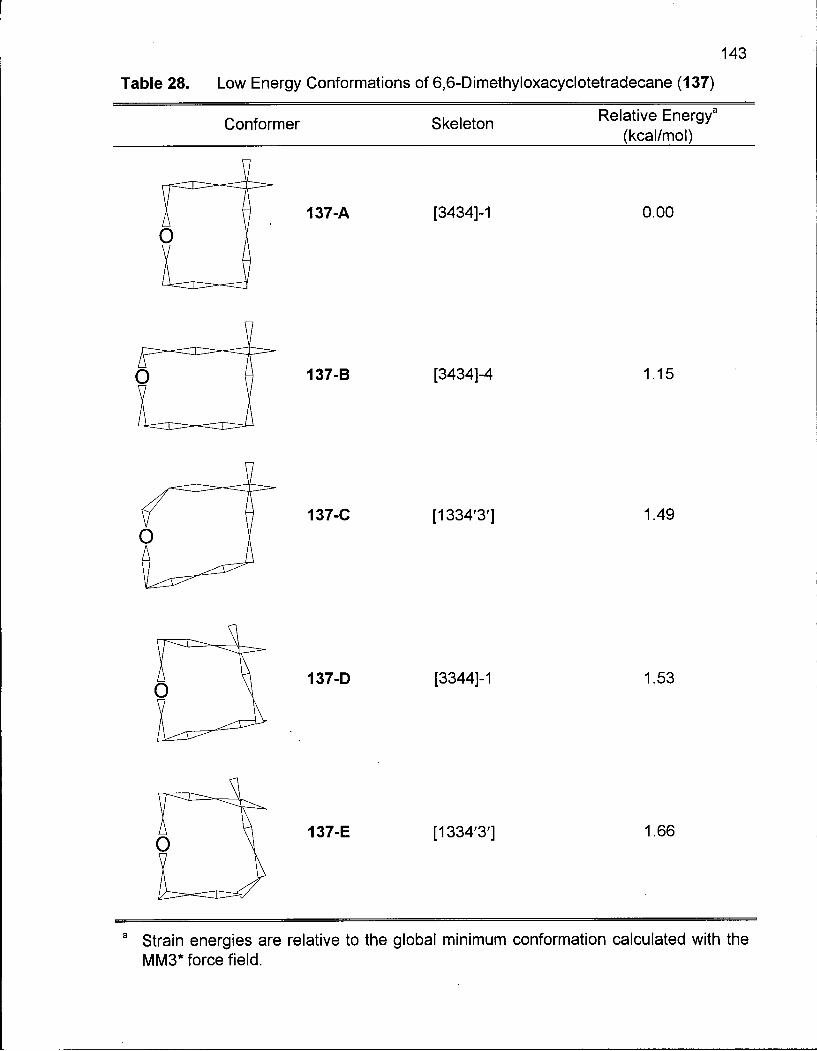
143
Table 28. Low Energy Conformations of 6,6-Dimethyloxacyclotetradecane (137)
Conformer Skeleton Relative Energy (kcal/mol)
137-A [3434]-1 0.00
137-B [3434J-4 1.15
137-C [1334'3'] 1.49
137-D [3344]-1 1.53
o 137-E [1334'3'] 1.66
a Strain energies are relative to the global minimum conformation calculated with the MM3* force field.
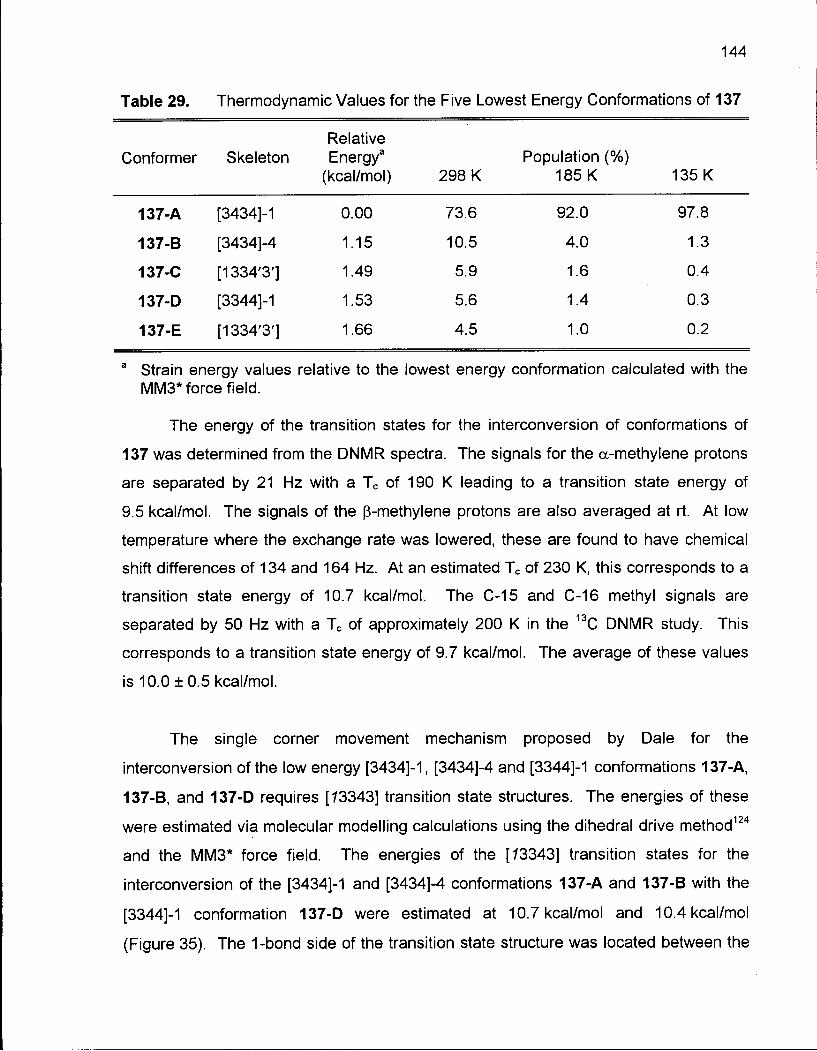
144
Table 29. Thermodynamic Values for the Five Lowest Energy Conformations of 137
Relative Conformer Skeleton Energy3 Population (%)
(kcal/mol) 298 K 185 K 135 K
137-A [3434J-1 0.00 73.6 92.0 97.8
137-B [3434]-4 1.15 10.5 4.0 1.3
137-C [1334'3'] 1.49 5.9 1.6 0.4
137-D [3344J-1 1.53 5.6 1.4 0.3
137-E [1334'3'1 1.66 4.5 1.0 0.2
Strain energy values relative to the lowest energy conformation calculated with the MM3* force field.
The energy of the transition states for the interconversion of conformations of
137 was determined from the DNMR spectra. The signals for the a-methylene protons
are separated by 21 Hz with a Tc of 190 K leading to a transition state energy of
9.5 kcal/mol. The signals of the p-methylene protons are also averaged at rt. At low
temperature where the exchange rate was lowered, these are found to have chemical
shift differences of 134 and 164 Hz. At an estimated Tc of 230 K, this corresponds to a
transition state energy of 10.7 kcal/mol. The C-15 and C-16 methyl signals are
separated by 50 Hz with a Tc of approximately 200 K in the 13C DNMR study. This
corresponds to a transition state energy of 9.7 kcal/mol. The average of these values
is 10.0 ±0.5 kcal/mol.
The single corner movement mechanism proposed by Dale for the
interconversion of the low energy [3434]-1, [3434]-4 and [3344]-1 conformations 137-A,
137-B, and 137-D requires [73343] transition state structures. The energies of these
were estimated via molecular modelling calculations using the dihedral drive method124
and the MM3* force field. The energies of the [73343] transition states for the
interconversion of the [3434]-1 and [3434]-4 conformations 137-A and 137-B with the
[3344]-1 conformation 137-D were estimated at 10.7 kcal/mol and 10.4 kcal/mol
(Figure 35). The 1-bond side of the transition state structure was located between the

145
"moving" corner atoms of the interconverting conformers. The conformations with
1-bond sides can be interconverted with the 4-sided conformers through the [3344]-1
conformation. These conformations do not interconvert via the single corner movement
mechanism, but rather via the rotation of dihedral angles on the side of the
conformation. The energy of the transition state for the interconversion of the [1334'3']
conformation 137-E with the [3344]-1 conformation 137-D was estimated at
10.4 kcal/mol. This value was obtained by driving the C-10 and C-12 dihedral angles.
The calculated and observed transition state energies are in good agreement. These
values are both more than 1 kcal/mol higher than the observed transition state energy
of oxacyclotetradecane (90). Transition states involving movement of the geminally
substituted carbon away from the corner position, as would occur in the pseudorotation
of 137, are expected to be higher in energy.
[3434]-1 [3344]-1
[1334'3'] [3434]-4
Figure 35. Interconversion of conformations of 137 through the [3344]-1 conformation.

146
2.6.1 Synthesis of 8,8-Dimethyloxacyclotetradecane (154)
The synthetic plan for the preparation of the macrocyclic ether 154 was
developed according to the same strategy used for the preparation of
6,6-dimethyloxacyclotetradecane (137). Again the molecule was prepared in two parts
and coupled using a dithiane ring as the central unit (Scheme 20). In this case, both
the left and right synthetic fragments of 154 were prepared from 1,6-hexanediol (138).
The central dithiane ring was subsequently converted into the gem-dimethyl group
while the ring of the macrocycle was formed via the cyclisation of a hydroxy acid to give
a lactone that would be converted into macrocyclic ether 154.
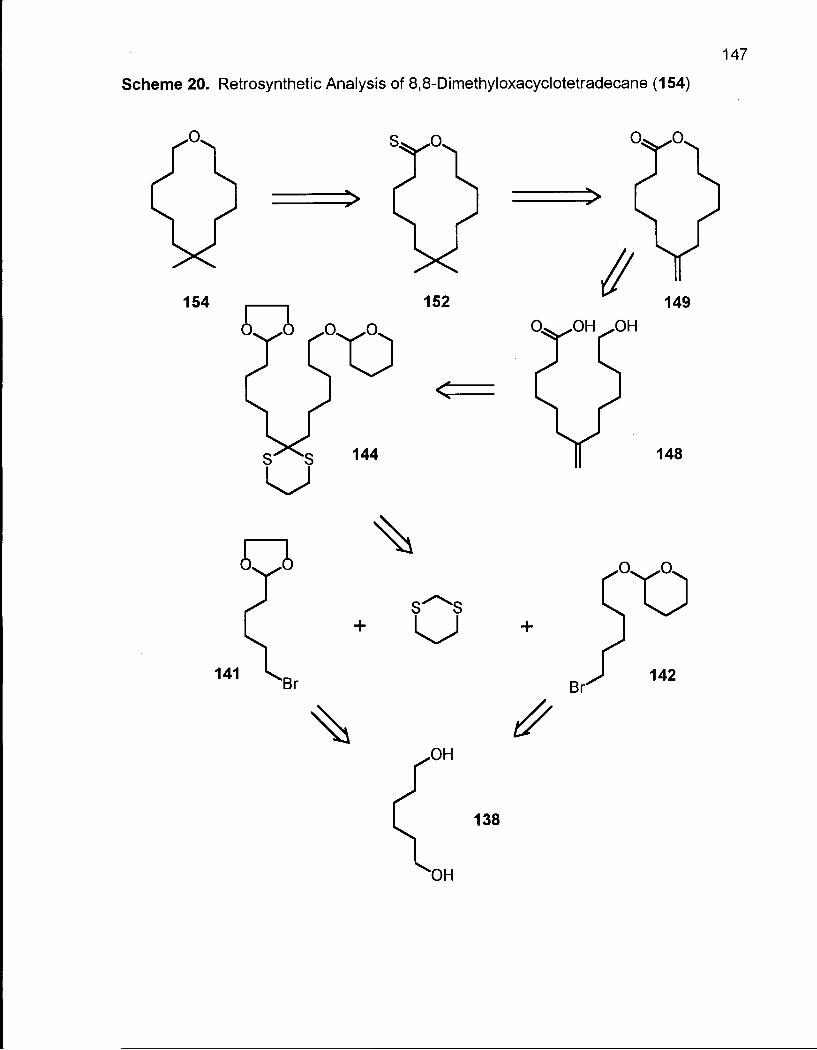
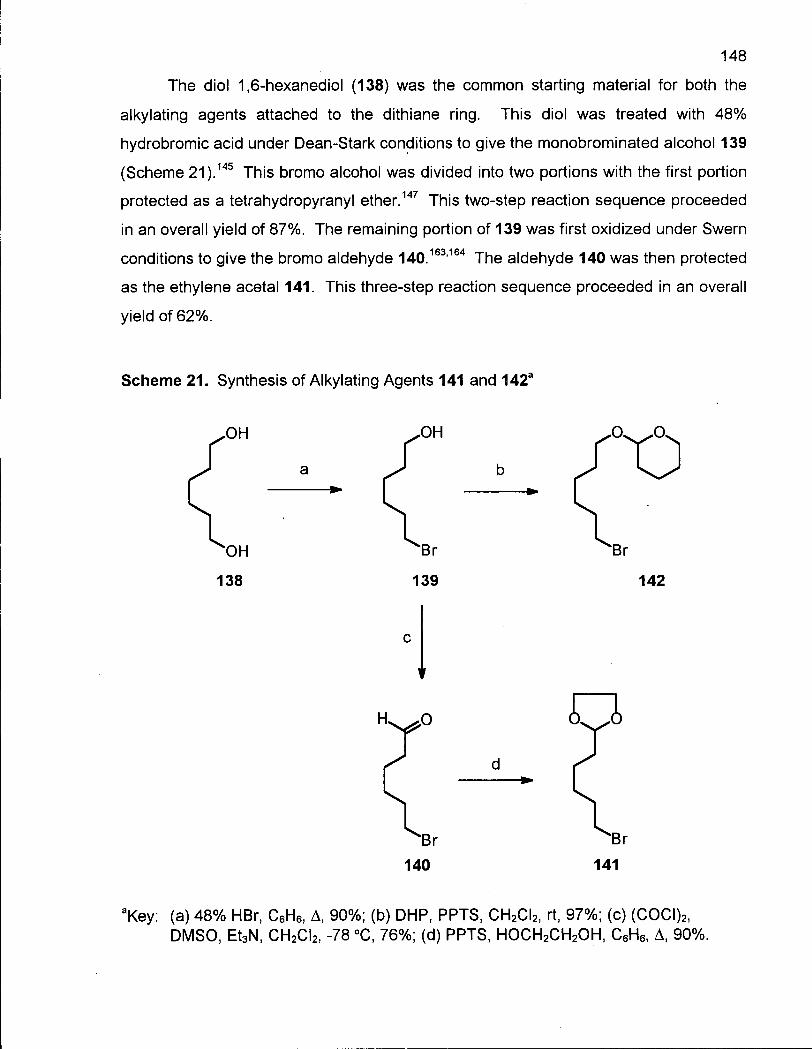
148
The diol 1,6-hexanediol (138) was the common starting material for both the
alkylating agents attached to the dithiane ring. This diol was treated with 48%
hydrobromic acid under Dean-Stark conditions to give the monobrominated alcohol 139
(Scheme 21 ).145 This bromo alcohol was divided into two portions with the first portion
protected as a tetrahydropyranyl ether.147 This two-step reaction sequence proceeded
in an overall yield of 87%. The remaining portion of 139 was first oxidized under Swern
conditions to give the bromo aldehyde 140. 1 6 3 , 1 6 4 The aldehyde 140 was then protected
as the ethylene acetal 141. This three-step reaction sequence proceeded in an overall
yield of 62%.
Scheme 21. Synthesis of Alkylating Agents 141 and 142a
140 141
aKey: (a) 48% HBr, C6H6, A, 90%; (b) DHP, PPTS, CH2CI2, rt, 97%; (c) (COCI)2, DMSO, Et3N, CH2CI2, -78 °C, 76%; (d) PPTS, HOCH2CH2OH, C6H6, A, 90%.

149
The bisalkylated dithiane 144 was prepared via a two-step reaction sequence
(Scheme 22). The anion of 1,3-dithiane was generated with n-butyllithium and
alkylated with 0.66 equivalents of bromo acetal 141 to give the heptane 143.146 This
monoalkylated product was reacted further with n-butyllithium to generate the anion of
143 which was alkylated with 1.2 equivalents of bromide 142 to give 144.146 Hydrolysis
of the dithiane ring gave ketone 145.150 This reaction sequence proceeded in 16%
yield for three-steps with the second alkylation giving the lowest yield. The 1H NMR
spectrum of 145 contained a one-proton triplet at 4.81 ppm for the C-1 methine of the
acetal, and a one-proton doublet of doublets at 4.54 ppm for the C-13 methine of the
tetrahydropyranyl ether. The 13C NMR spectrum of 145 showed a signal at 211.32 ppm
and the IR spectrum showed a sharp band at 1716 cm"1 for the C-7 carbonyl.
Conversion of the ketone into the gem-dimethyl group was accomplished in two-
parts as in the case of 6,6-dimethyloxacyclotetradecane (137). The small scale
reaction of ketone 145 with Tebbe reagent 3238 proceeded to give, after column
chromatography with basic alumina, alkene 146 in 70% yield.151,152 Unfortunately,
these reaction conditions gave low yields on the required larger scale. The crude
reaction mixture was filtered directly through basic alumina which quenched the excess
Tebbe reagent as well as removed unwanted titanium compounds from the alkene
product. This filtration step was problematic when performed on a large scale. To
overcome this, the methylenation of the ketone with a Wittig reagent was investigated.
The anion of methyl triphenylphosphonium iodide was generated and reacted with
ketone 145 to give the alkene 146 in 66% yield (Scheme 22). This Wittig chemistry
was found to be better suited to the larger reaction scale. The 1H NMR spectrum of
146 contained a two-proton singlet at 4.64 ppm and the IR spectrum contained a band
at 1643 cm"1 for the carbon-carbon double bond. The HRMS and chemical analysis
results were also consistent with the composition of alkene 146.
Removal of the protecting groups under weakly acidic conditions gave the
hydroxy aldehyde 147.147 The chemoselective oxidation of the aldehyde of 147 was
performed with sodium chlorite to give hydroxy acid 148.165 This reaction sequence

150
proceeded in 36% yield for the three-steps beginning with the Wittig reaction. The
1H NMR spectrum of this colourless oil contained a singlet at 4.65 ppm for the C-14
methylene of the double bond as well as a triplet at 2.30 ppm for the C-2 methylene
adjacent to the acid group of 148. The IR spectrum contained bands at 3637 cm"1 and
1712 cm"1 for the carboxylic acid terminus of the hydrocarbon and at 1644 cm"1 for the
double bond of 148.
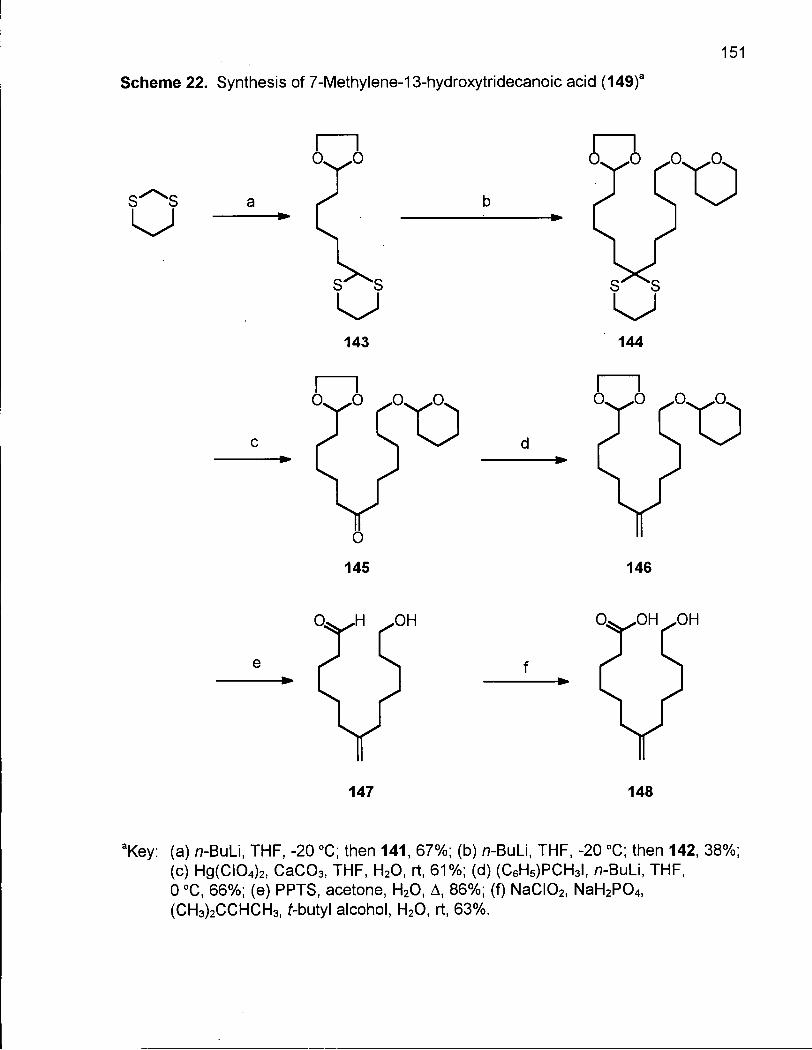
151
Scheme 22. Synthesis of 7-Methylene-13-hydroxytridecanoic acid (149)a
147 148
aKey: (a) n-BuLi, THF, -20 °C; then 141, 67%; (b) n-BuLi, THF, -20 °C; then 142, 38%; (c) Hg(CI04)2, CaC03, THF, H20, rt, 61%; (d) (C6H5)PCH3I, n-BuLi, THF, 0 °C, 66%; (e) PPTS, acetone, H20, A, 86%; (f) NaCI02, NaH2P04, (CH3)2CCHCH3, f-butyl alcohol, H20, rt, 63%.

152
The cyclisation of the hydroxy acid 148 was achieved with the Yamaguchi
procedure with the hydroxy acid first activated as a mixed anhydride with
2,4,6-trichlorobenzoyl chloride28 and then cyclized under high dilution conditions to give
the lactone 149 in 42% yield (Scheme 23).28 The exocyclic methylene of 149 was
converted into a cyclopropyl group.159,160 This reaction was sluggish and further
addition of the zinc complex precursors was necessary to optimize the yield. The
cyclopropyl group of 150 was hydrogenolyzed with Adams' catalyst to give the
ge/77-dimethyl lactone 151. 1 6 1 The 1H NMR spectrum of 151 contained a two-proton
multiplet between 4.14-4.16 ppm for the C-13 methylene adjacent to the ether oxygen
as well as a six-proton singlet at 0.81 ppm for the new geminal methyl groups. The 13C NMR spectrum contained 14 lines with a low-field signal at 173.56 ppm for the C-1
carbonyl. The IR spectrum contained a band at 1736 cm"1 also for the C-1 carbonyl.
The HRMS results were also consistent with the composition of lactone 151.
The lactone 151 was reacted with Lawesson's reagent 48 to give the
thionolactone 152. 5 1 The thionolactone was further reacted with lithium
triethylborohydride followed by trapping of the resultant sulfur anion with methyl iodide
to give the mixed thioacetal 153. 5 1 This compound was reacted immediately with a
solution of tri(/7-butyl)tin hydride under radical conditions to reduce the thiomethyl
group of 153 to give the macrocyclic ether 154. This reaction sequence proceeded in
2% yield for the six-steps with the Simmons-Smith cyclopropanation and the hydride
reduction steps having the lowest yields.

Scheme 23. Synthesis of 8,8-Dimethyloxacyclotetradecane (154)a
153
153 154
aKey: (a) Et3N, THF, 2,4,6-trichlorobenzoyl chloride, rt; then DMAP, toluene, A, 42%; (b) Zn-Cu, CH2I2, l2, Et20, A, 34%; (c) Pt02, H2, HOAc, rt, 78%; (d) Lawesson's reagent 48, toluene, A, 54%; (e) LiEt3BH, THF, -78 °C; then Mel, 98%; (f) /7-Bu3SnH, AIBN, tol, A, 40%.

154
2.6.2 Conformational Analysis of 8,8-Dimethyloxacyclotetradecane (154)
The 1H NMR spectrum of 8,8-dimethyloxacyclotetradecane (154) in CDCI3 at rt
contained a four-proton triplet at 3.40 ppm, three four-proton multiplets between
1.53-1.58 ppm, 1.41-1.47 ppm, and 1.28-1.35 ppm, an eight-proton multiplet between
1.14-1.24 ppm, and a six-proton singlet at 0.81 ppm. The downfield triplet was
assigned to the protons of C-2/C-14. The singlet at 0.81 ppm was assigned to the
geminal methyl groups of C-8 (Table 30). The HRMS analysis was also consistent with
the composition of 154.
The 13C NMR spectrum contains eight lines indicating that either 154 has a
plane of symmetry, or is undergoing site exchange that is rapid on the NMR timescale.
The downfield signal at 69.27 ppm was assigned to C-2/C-14, and the signal at
29.09 ppm was assigned to the C-8 geminal methyl groups. The signal at 32.80 ppm
was assigned to the quaternary C-8 carbon since this signal was approximately half the
height of the other 13C signals. The remaining 13C and 1H signals were assigned with
data from COSY, HMQC, and HMBC 2D-NMR experiments. The signal for C-4/C-12
was shifted to higher field than expected based on its through-bond distance from the
ether oxygen. This upfield shift was caused by a y-gauche effect with the ether oxygen
as a result of the gauche dihedral relationship between C-4 and the ether oxygen atom.

155
Table 30. 1H and 13C NMR Assignments for 8,8-Dimethyloxacyclotetradecane (154) in CDCI3 at Room Temperature
Position 1H NMRa 13C NMRa
2, 14 3.40 69.27
12 / \ l 4 3, 13 1.55 27.31
4, 12 1.44 24.80 1 0 j ^ 15 V
J 5, 11 1.31 27.90
8 6, 10 1.20 21.56
7,9 1.18 38.61 6
8 ~ 32.80
15, 16 0.81 29.09
The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI3 (13C).
A series of DNMR experiments were carried out with 154 using a mixture of
Freon 21 and Freon 22 as solvent (Figure 36). The 1H NMR spectrum of 154 at 220 K
contained six signals similar to the rt spectrum however at the lower temperature, the
signals had broadened. At lower temperatures, the downfield signal at 3.40 ppm for the
C-2/C-14 protons broadened to form, at intermediate temperatures, a pair of signals at
3.55 and 3.27 ppm. The upfield signal at 3.27 ppm broadened as the temperature was
lowered further. The signal at 1.55 ppm for the C-3 protons broadened to give at
temperatures below 185 K a downfield signal at 1.80 ppm, with another signal
presumably concealed by the signals of the methylene envelope. The C-8 geminal
methyl group signal did not change significantly over the temperature range studied.
This data is consistent with one major conformation of 154 being present at low
temperature.
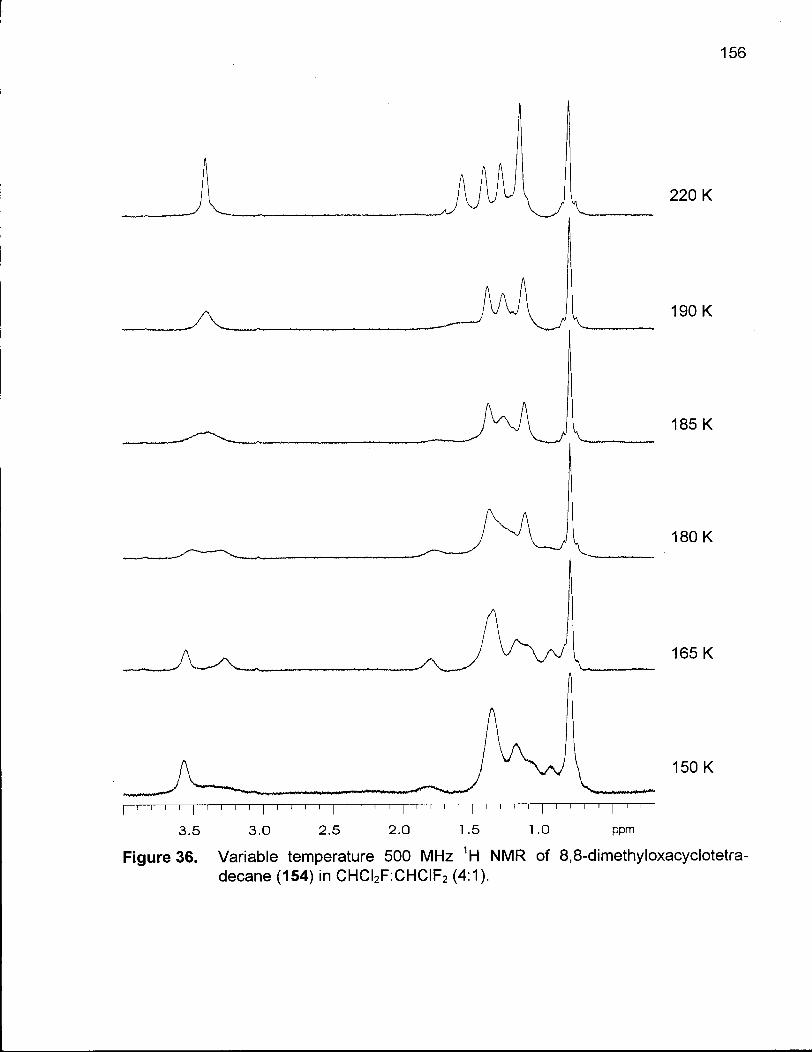
156
decane (154) in CHCI2F:CHCIF2 (4:1).

157
It is significant that the signal for the C-8 geminal methyls remains averaged
even at low temperature while the signal for the methylene protons adjacent to the
ether oxygen does not. One possibile explanation is that the major conformation of 154
is symmetric with a C 2 axis running through the C-8 corner atom. This C 2 axis would
interconvert the equivalent C-8 geminal methyl groups. However the protons adjacent
to the ether oxygen would be inequivalent since they are in different environments.
The [3434]-1 and [3434]-4 conformations were low energy conformations in the
unsubstituted 14-membered ether, but these conformations would have transannular
steric interactions involving an endo methyl group in the macrocyclic ether 154. The
low energy conformations of 154 would have the C-8 gem-dimethyl group at a corner
position of the ring. Therefore, the diamond lattice [3434]-3 conformation, and the non-
diamond lattice [3344]-2, [3344]-3 and [3344]-6 conformations of 154, where the C-8
gem-dimethyl group is located at a corner position are possible low energy
conformations of the macrocyclic ether 154. The ether oxygen atom is located at a
corner position in each of these conformations with the exception of the [3344]-2
conformation. An oxygen atom does not eliminate any transannular hydrogen
interactions when located at a corner position, and in general such conformations are
unfavoured. Non-corner oxygen atoms do however eliminate some transannular
hydrogen interactions, and the [3344]-2 conformation with its non-corner oxygen atom
is likely to have a low strain energy. The [3344]-2 conformation with a C 2 axis of
symmetry is a likely major conformation for 154.

158
6
3 4 4 5
[3434]-3 [3344]-2
2
o
2
[3344]-3 [3344]-6
The two peaks observed in the DNMR spectra of 154 for the protons adjacent to
the ether oxygen were of approximately equal intensity. In the [3344]-2 conformation,
the C-2 corner protons do not experience any van der Waals steric interactions since
both protons are exo to the ring. The H-2 a proton is deshielded by the anisotropy of the
O/C-14 bond, but shielded by the anisotropy of the C-3/C-4 bond. These opposing
effects are of a similar magnitude, and a small A8 value is expected for the C-2 protons.
The H-14endo proton is deshielded by van der Waals steric interactions with the H-3endo
and H-11endo protons calculated to be 2.08 A and 2.19 A away from H-14endo. This
results in a shielding of the H-14 e x o proton, and a larger A5 value than that of the C-2
protons. The C-2 and C-14 methylene protons are in different environments in the
[3344]-2 conformation, and different lineshapes are predicted for the two methylene
groups. The line shape of the C-2 and C-14 protons in this conformation is predicted to
be more complex than that observed in the DNMR spectra. The C-2 and C-14 protons
are also in different environments in the diamond lattice [3434]-3 conformation with the
oxygen atom at a corner position. The line shape is again predicted to be more
complex than that observed here.
In the symmetric [3344]-3 and [3344]-6 conformations, the geometry of the C-2
and C-14 protons is similar in each conformation. The H-2eXo proton of the [3344]-3

159
conformation is deshielded by the anisotropy of the O/C-14 bond, but shielded by a van
der Waals steric interaction between H-2endo and H-13endo which are calculated to be
2.12 A apart. The predicted A5 value for the H-2endo and H-2exo protons in this
conformation is small. The H-2exo proton in the [3344]-6 conformation experiences both
of these effects, but is further shielded by a van der Waals steric interaction between
H-2endo and H-5endo calculated to be 2.18 A apart. The A8 between H-2endo and H-2exo in
this latter conformation is predicted to be larger than that of the [3344]-3 conformation.
The A5 value for the C-2 and C-14 protons averaged over both these conformations is
consistent with the observed low temperature spectra.
A molecular mechanics search for low energy conformations of 154 was
conducted with the Monte Carlo technique, and the MM3* force field. The global
minimum conformation was found to be the non-diamond lattice conformation 154-A
(Table 31). The second lowest energy conformation 154-B, was also non-diamond
lattice, but symmetric and 0.41 kcal/mol higher in energy. The C-8 gem-dimethyl group
is at a corner position in all of the low energy conformations found. The first diamond
lattice conformation found was 154-E. This was a [3434]-3 conformation with a corner
C-8 gem-dimethyl group, and the ether oxygen at the opposite corner position. Higher
energy conformations were not considered to be significantly populated over the
temperature range studied. The relative populations of these conformations at different
temperatures were calculated from enthalpy values (AH0) and entropy values (AS°) with
both symmetry and mixing contributions (Table 32). These calculations suggest the
global minimum non-diamond lattice conformation 154-A is the major conformation over
the temperature range examined.
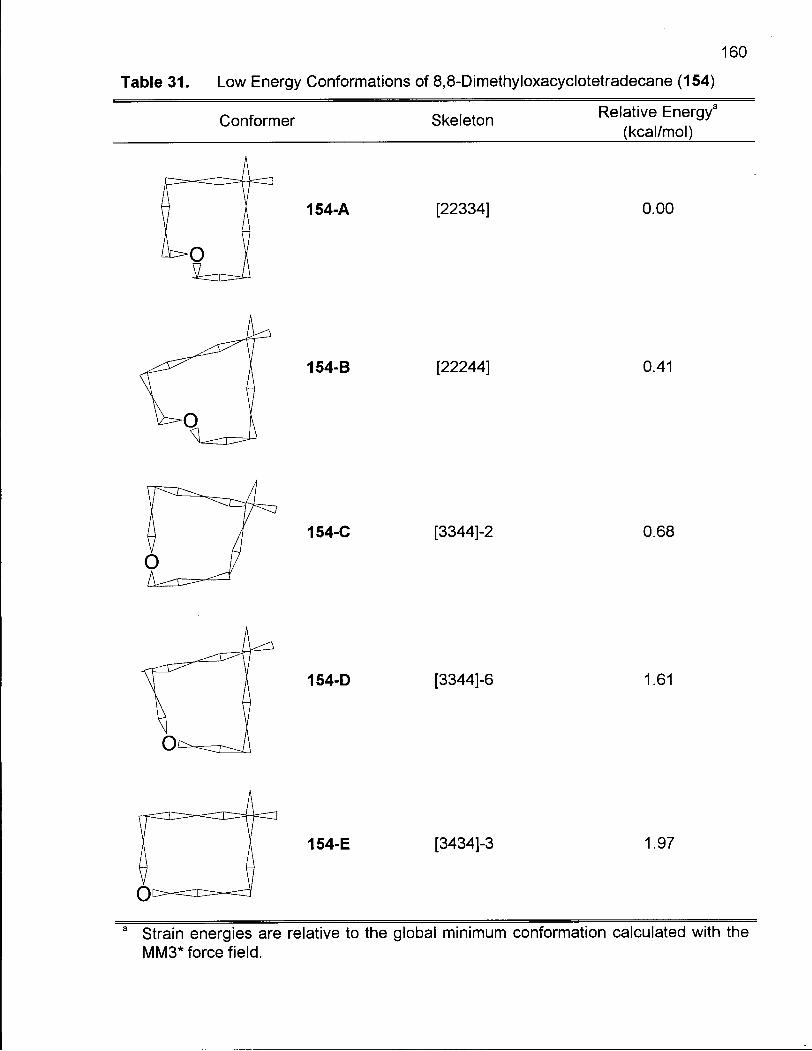
160
Table 31. Low Energy Conformations of 8,8-Dimethyloxacyclotetradecane (154)
Conformer Skeleton Relative Energy (kcal/mol)
154-A [22334] 0.00
154-B [22244] 0.41
154-C [3344]-2 0.68
154-D [3344]-6 1.61
a Strain energies are relative to the global minimum conformation calculated with the MM3* force field.

161
Table 32. Thermodynamic Values for the Five Lowest Energy Conformations of 154
Relative Conformer Skeleton Energy3 ASb Population (%)
(kcal/mol) (cal/mol) 298 K 190 K 150 K
154-A [22334] 0.00 1.38 61.0 74.2 81.0
154-B [22244] 0.41 0.00 15.3 12.6 10.3
154-C [3344]-2 0.68 1.38 19.5 12.4 8.4
154-D [3344]-6 1.61 0.00 2.0 0.5 0.2
154-E [3434]-3 1.97 1.38 2.2 0.4 0.1
3 Strain energy values are relative to the global minimum conformation calculated with the MM3* force field.
b Entropy values were calculated with both symmetry and mixing terms.
The DNMR data was reexamined with the non-diamond lattice conformations
154-A and 154-B in mind. Conformation 154-B has a C2 axis through the C-8 corner
position, and one signal was expected for the geminal methyl groups. No transannular
van der Waals shielding effects are experienced by the C-2 and C-14 protons in these
two conformations since they are all exo to the ring. The H-2P and H-14a protons are
both deshielded by the anisotropy of the p-carbon-carbon bond in conformation 154-A,
while the H-2P and H-14p protons are deshielded in conformation 154-B. The predicted
A 5 value between these protons in conformation 154-A and conformation 154-B is
consistent with that observed in the DNMR spectra here. Overall, the DNMR data is
consistent either with conformation 154-B, or with conformation 154-A where a local
inversion to conformation 154-B still occurs at low temperature.
The transition state energies for the interconversion of conformations of the
macrocyclic ether 154 were calculated by first determining the rate of exchange
between a pair of averaged signals in the DNMR spectra. The rate of exchange was
used to calculate the free energy of activation ( A G * ) with the coalescence temperature
(Tc) also obtained from the DNMR spectra, and the equations in Chapter 1. At low
temperature, the C-2 proton signals of 154 are separated by 139 Hz. This

\
162
corresponded to a transition state energy of 8.8 kcal/mol at the coalescence
temperature of 190 K. This was the only averaged set of signals in the DNMR spectra
of 154 from which a rate of exchange could be determined.
The single corner movement mechanism proposed by Dale for the
interconversion of large ring conformations such as the [3344]-2 154-C, [3344]-6
154-D, and [3434]-3 154-E conformations involve [73343] transition state structures
with a 1-bond side between the moving corner atoms (Figure 37). The energies of
these transition states were calculated via molecular modelling calculations using the
dihedral drive method124 and the MM3* force field. The necessary dihedral angles were
incremented by 10° during these calculations. The energies of the [73343] transition
state structures involved in the interconversion of the diamond lattice [3434J-3
conformation 154-E with the non-diamond lattice [3344]-2 and [3344]-6 conformations
154-C and 154-D were estimated at 13.4 kcal/mol and 14.2 kcal/mol respectively. The
low energy, non-diamond lattice conformations 154-A and 154-B can interconvert via
the single corner movement mechanism as well (Figure 37). The energy of the
transition state structure was estimated at 10.8 kcal/mol using the dihedral drive
method. These conformations are not interconvertable with the other low energy
conformations 154-C through 154-E via this same mechanism. This latter transition
state energy was in good agreement with the value derived from the DNMR data,
whereas the transition state barriers of the interconversion through the diamond lattice
conformation 154-E were higher. The observed transition state energy is in better
agreement with that of the interconversion of the non-diamond lattice conformations of
154, in support of the presence of these conformations.
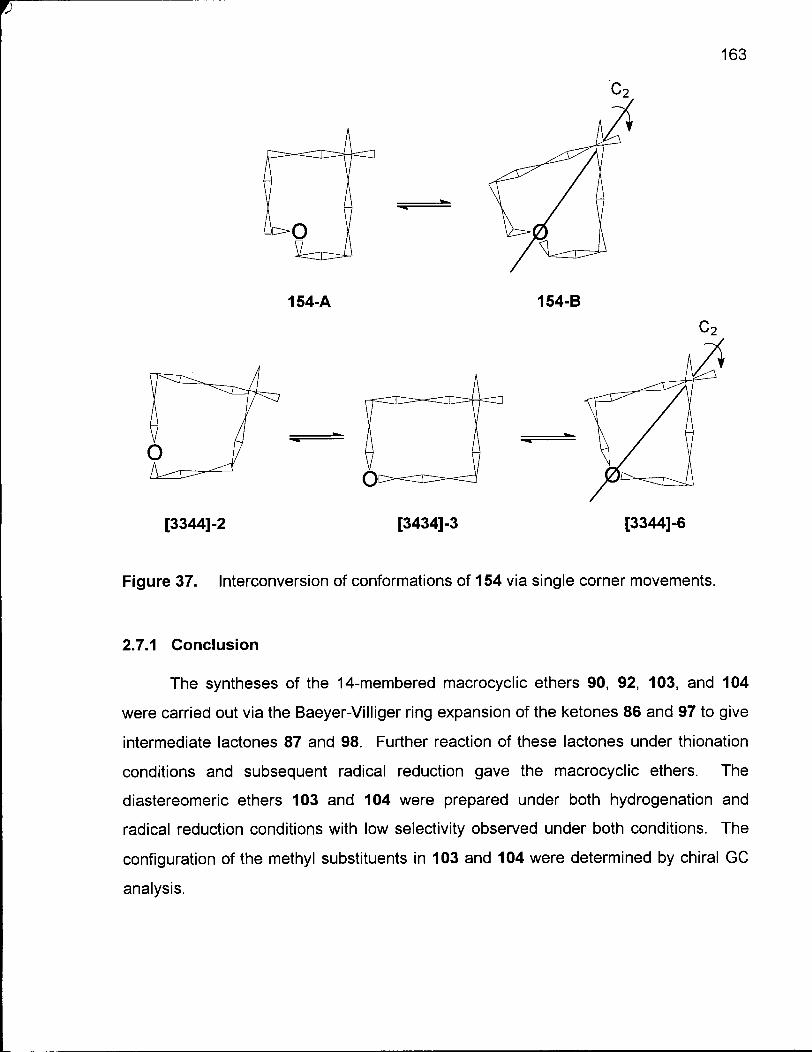
163
C2
[3344]-2 [3434]-3 [3344]-6
Figure 37. Interconversion of conformations of 154 via single corner movements.
2.7.1 Conclusion
The syntheses of the 14-membered macrocyclic ethers 90, 92, 103, and 104
were carried out via the Baeyer-Vi Niger ring expansion of the ketones 86 and 97 to give
intermediate lactones 87 and 98. Further reaction of these lactones under thionation
conditions and subsequent radical reduction gave the macrocyclic ethers. The
diastereomeric ethers 103 and 104 were prepared under both hydrogenation and
radical reduction conditions with low selectivity observed under both conditions. The
configuration of the methyl substituents in 103 and 104 were determined by chiral GC
analysis.

164
The Baeyer-Vi Niger ring expansion of ketone 106 did not proceed. The required
lactone 114 was instead prepared via the cyclisation of hydroxy acid 113 with the
Yamaguchi reagent. The thionation of this lactone also failed, and a direct reduction of
the lactone with sodium borohydride in the presence of boron trifluoride etherate was
employed to give macrocyclic ether 116.
The macrocyclic ether 119 was prepared via the reduction of lactone 118.
However, even under refluxing conditions, the boron trifluoride mediated sodium
borohydride reduction of this lactone proceeded in low yield.
Macrocyclic ethers 137 and 154 were prepared from hydroxy acid intermediates
131 and 148. The cyclisation of these intermediates with the Yamaguchi reagent gave
lactones 132 and 149. Further reaction of these lactones under thionation conditions
and subsequent radical reduction gave the desired macrocyclic ethers.
The conformation of these 14-membered ethers was analyzed with data from 1H-DNMR experiments. The low-temperature chemical shift difference of protons with
signals that were averaged at rt, were generally in agreement with predictions based on
anisotropy and van der Waals shielding effects in the low energy conformations.
Although many different possible conformations for these large ring compounds exist,
only a few conformations were found to be appreciably populated at room temperature
and below. The conformations were consistent with the substituents generally located
exo to the ring, with geminal substituted carbon atoms occupying corner positions
exclusively. These results were consistent with the molecular mechanics calculations.
In general, the diamond lattice [3434] conformation was preferred with the oxygen atom
at either the 1-position or the 4-position. Thus the introduction of the oxygen atom in
these macrocyclic ethers did not have a significant effect on the conformation of the
ring.
The transition state energies for the conformational interconversion were
determined from the 1H DNMR experiments to be in the range of 8.5 to 9.6 kcal/mol.

165
The interconversion barriers of the gem-dimethyl substituted macrocyclic ethers 116,
119, and 137 were found to be higher than those of the other macrocyclic ethers
studied. The calculated single corner movement transition state energies of the
macrocyclic ethers were between 10 and 15 kcal/mol and higher than the observed
values. Both of these values were larger than those previously obtained for the
hydrocarbon cyclotetradecane.

C H A P T E R 3
166
Synthesis and Isomerization of Unsaturated 14-Membered Macrocyclic Ethers
Macrocyclic compounds are commonly formed via the modification of an existing
ring, or through the cyclization of an acyclic precursor. Both of these methods were
used to good advantage during the study of a series of 14-membered macrocyclic
ethers, the results of which were discussed in Chapter 2. A cyclization method that is
currently receiving much attention in the literature is the olefin metathesis cyclization of
an acyclic diene. This reaction uses an organometallic catalyst to give a cyclic
compound with a carbon-carbon double bond in the ring at the location of the ring
closure.10 An organometallic catalyst shown to be quite useful for this chemistry is the
ruthenium alkylidene 9 prepared by Grubbs and coworkers.18,166
PCy3
X= CH2, COO, CON, NH, 0, S
The metathesis cyclization is a general reaction for the formation of cyclic
compounds with a variety of ring sizes. The method is compatible with a range of other
functional groups which allows for the formation of not only cyclic hydrocarbons, but
also heterocyclic compounds containing oxygen,11,19 sulfur,167 and nitrogen21,22 atoms.
The product of the cyclization contains a new synthetic handle, the newly formed
carbon-carbon double bond, that can be further modified to introduce additional
substituents into the cyclic system. To evaluate this reaction, the synthesis of some
14-membered macrocyclic ethers was undertaken. The target ethers contained
different alkyl substitution and also different configurations of the carbon-carbon double

167
bond as a result of the metathesis cyclization. What the preferred configuration of the
double bond would be in each case, as well as, what affect if any the C-2 methyl group
would have on the cyclization were questions of interest.
157 (Z) R = H, 163 (Z) R = CH3
158(E) R = H, 164(E) R = CH3
3.1.1 Synthesis of Oxacyclotetradec-5-enes (157) and (158)
The macrocyclic alkenes 157 and 158 were prepared using the metathesis
reaction to form the macrocyclic ring. This required as the cyclization precursor an
acyclic diene ether which was produced via the O-alkylation of a primary alcohol. The
anion of 9-decenol (155) was generated with potassium hydride and reacted with
5-bromo-1-pentene in the presence of DMPU to give the acyclic diene ether 156. This
diene was cyclized under metathesis conditions with the Grubbs catalyst 9. 1 6 6
Solutions of the diene 156 and the metathesis catalyst 9, both in toluene, were slowly
combined under high dilution conditions using a syringe pump. This two-step reaction
sequence proceeded to give the macrocyclic ethers 157 and 158 in a ratio of 59:41 with
an overall yield of 35% (Scheme 24). The macrocyclic ethers 157 and 158 had slightly
different Rf values on silica, as well as different retention times on a DB-210 GC
column. This allowed for the separation of the isomeric products.

168
Scheme 24. Synthesis of Oxacyclotetradec-5-enes (157) and (158)a
155 156 157 (Z) 158(E)
aKey: (a) KH, THF, 0 °C; then 5-bromo-1-pentene, DMPU, 84%; (b) Grubbs catalyst 9,166 toluene, 42%.
The 1H NMR spectrum of 157 contained one-proton signals at 5.26 and
5.51 ppm for the C-5 and C-6 methine protons of the double bond. Signals were also
present at 3.41 and 3.38 ppm for the protons of the C-2 and C-14 methylenes adjacent
to the ether oxygen. The 13C NMR spectrum contained 13 lines with the C-5 and C-6
methine carbon signals at 129.59 and 130.87 ppm, and the C-2 and C-14 methylene
carbons at 68.81 and 68.69 ppm. Unfortunately the C-2 and C-14 signals could not be
unambiguously assigned even with an HMQC experiment due to the similarity of their
chemical shifts. The IR spectrum of 157 contained a weak band at 1649 cm"1 for the
carbon-carbon stretch of the double bond. In addition, the HRMS and chemical
analysis results were consistent with the composition of ether 157.
The 1H NMR spectrum of ether 158 contained overlapping signals for the C-5
and C-6 methine protons of the double bond at 5.39 and 5.34 ppm respectively. The
signals for the C-2 and C-14 methylene protons had chemical shifts of 3.37 and
3.48 ppm respectively. The 13C NMR spectrum of 158 contained 13 lines with the C-5
and C-6 methine carbon signals at 130.61 and 131.80 ppm. The C-2 and C-14
methylene carbon signals had chemical shifts of 67.01 and 69.54 ppm respectively.
The HRMS and chemical analysis data were also consistent with the composition of
ether 158. A band for the carbon-carbon stretch of the double bond was not visible In
the IR spectrum of 158. In order for a molecular vibration to give rise to an IR

169
absorption, the molecular motion must result in a change of the dipole moment of the
molecule. The change in dipole moment for the stretching of the carbon-carbon double
bond of 158 is either very small or zero, and hence no band is visible in the IR
spectrum at ca. 1600 cm"1 for the carbon-carbon double bond stretch.
In order to determine whether the major isomer of oxacyclotetradec-5-ene had
the E or the Z configuration, a series of 1H homonuclear decoupling NMR experiments
were performed. The coupling constant for olefinic protons of a trans double bond (E)
are typically in the range of 12-18 Hz, while olefinic protons of a cis double bond (Z)
typically have a smaller coupling constant in the range of 6-12 Hz. 1 6 8 From the 1H NMR decoupling experiments, it was determined that the olefinic protons of the
major isomer 157 had a coupling constant of 10.7 Hz and was correspondingly
assigned the Z configuration. The double bond of the minor isomer 158 was
determined to have the E configuration based on a coupling constant of 15.2 Hz
between the olefinic protons. Additional 1H NOE difference experiments on isomer 157
showed an enhancement of the signal of one olefinic proton upon irradiation of the
other olefinic proton thus confirming the cis configuration of the double bond of 158.
The trans isomer would not have been expected to show such an enhancement.
Once the configuration of the double bond in 157 and 158 was determined, the
above IR results were clear. The IR spectrum of 157 contained a weak absorption for
the carbon-carbon stretch of the double bond. Typically, the corresponding absorption
in the trans isomer is weaker.169 In the case of the trans isomer 158 this absorption is
not visible in the IR spectrum at all.
3.1.2 Cis-Trans Isomerization of (Z/£)-Oxacyclotetradec-5-ene (157) and (158)
Once prepared, the cis-trans isomerization of the carbon-carbon double bond in
157 and 158 was examined. This reaction is expected to give an equilibrium mixture of
the isomers that is dependent on the relative energies of the low energy conformations
of each isomer. Isomerization of carbon-carbon double bonds can be performed with a
variety of reagents including: tri(n-butyl)tin hydride,170 tris(trimethylsilyl)silane,171,172

170
iodine,173 nitrous acid,174 and phenyl disulfide.175,176 Of these possible reagents, the
reaction of phenyl disulfide under photolysis conditions, to generate benzene thiyl
radicals, was the method chosen. This method is less likely to produce positional
isomers, a problem commonly encountered with the isomerization of non-conjugated
alkenes such as 157 and 158 using iodine or acid reagents.175
A solution of ether 157 and phenyl disulfide immersed in a 0.0014 M K2Cr04
solution was photolysed with an Hanovia 450W medium pressure mercury lamp. A
combination of pyrex glass and the chromate solution gave an irradiation window from
290-340 nm.177 When photolysed, the ether 157 gave a mixture of ethers 157 and 158
(GC ratio, 41:59). Using equation 8, this corresponded to an energy difference of
0.22 kcal/mol at 25 °C (Table 33, Entry 1). When reacted under similar conditions,
ether 158 also gave a mixture of ethers 157 and 158 (GC ratio, 39:61) which
corresponded to an energy difference of 0.26 kcal/mol (Table 33, Entry 2).
AG° = - RTInK (8)
These experimental results were compared to values obtained from molecular
mechanics calculations using the MM3* force field. The global minimum conformation
of the cis alkene 157 was a distorted [3434] conformation with an energy of
15.74 kcal/mol. The next conformation was 1.08 kcal/mol higher in energy. The global
minimum conformation for the trans isomer 158 was a non-diamond lattice
conformation with an energy of 15.51 kcal/mol. The second lowest energy
conformation of 158 was 0.92 kcal/mol higher in energy. Using these calculated
enthalpy values as an estimation of the relative energies of 157 and 158 (AS = 0), the
energy difference of 0.23 kcal/mol corresponded to a 40:60 Boltzmann distribution of
ethers 157 and 158 at 25 °C (Table 33, Entry 3). This was in good agreement with the
experimental data.
171
Table 33. Experimental and Theoretical Equilibrium Ratios for the Isomerization Reaction of Oxacyclotetradec-5-enes (157) and (158)
Entry Ratio 157:158a AG° (kcal/mol)b
1 157 (Z) 41:59 0.22
2 158(E) 39:61 0.26
3 MM3* 40:60 0.23c
a The equilibrium ratio of 157:158 was determined by analysis with a DB-210 GC column.
b AG0 values were calculated using equation 8 at 25 °C. c Calculated relative strain energies were taken as an approximation of AG0.
It is interesting to note, that the trans isomer 158 was the favoured isomer in the
isomerization and hence lower in energy. However, cis isomer 157 was the major
isomer produced in the metathesis cyclization. This indicates that even though the
metathesis reaction is believed to proceed via a series equilibrium processes
(Chapter 1),11 it does not necessarily yield an equilibrium ratio of products. The
metathesis reaction is believed to proceed through a metallocyclobutane intermediate
(Chapter 1). The trans isomer of this intermediate is in principle lower in energy and
predicted to lead to the trans isomer of the product. However, the results of molecular
mechanics calculations showed the lowest energy conformation of the trans isomer 158
is distorted in order to accommodate the carbon-carbon double bond. This deviation
from the diamond lattice conformations for the trans alkene 158 could result in a
preference for the cis isomer. Therefore, it may be the conformation of the rest of the
ring that determines the course of this reaction giving the cis metallocyclobutane
intermediate as the preferred intermediate, and 157 as the major isomer with the cis
configuration of the double bond although the selectivity was not very high (18% d. e.).
The observed selectivity difference between the metathesis products and the
isomerization products could also result from a solvent effect caused by the difference
in polarity between toluene (metathesis solvent) and cyclohexane (isomerization
solvent).
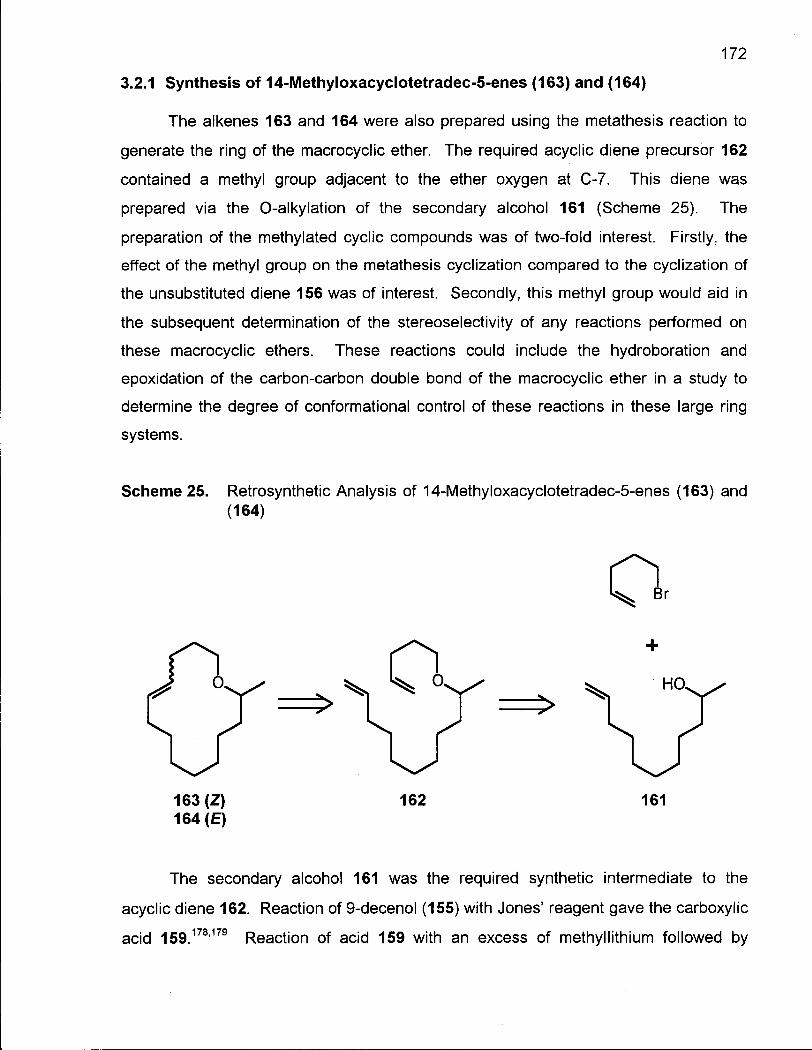
172
3.2.1 Synthesis of 14-Methyloxacyclotetradec-5-enes (163) and (164)
The alkenes 163 and 164 were also prepared using the metathesis reaction to
generate the ring of the macrocyclic ether. The required acyclic diene precursor 162
contained a methyl group adjacent to the ether oxygen at C-7. This diene was
prepared via the O-alkylation of the secondary alcohol 161 (Scheme 25). The
preparation of the methylated cyclic compounds was of two-fold interest. Firstly, the
effect of the methyl group on the metathesis cyclization compared to the cyclization of
the unsubstituted diene 156 was of interest. Secondly, this methyl group would aid in
the subsequent determination of the stereoselectivity of any reactions performed on
these macrocyclic ethers. These reactions could include the hydroboration and
epoxidation of the carbon-carbon double bond of the macrocyclic ether in a study to
determine the degree of conformational control of these reactions in these large ring
systems.
Scheme 25. Retrosynthetic Analysis of 14-Methyloxacyclotetradec-5-enes (163) and (164)
163(Z) 162 161 164 (E)
The secondary alcohol 161 was the required synthetic intermediate to the
acyclic diene 162. Reaction of 9-decenol (155) with Jones' reagent gave the carboxylic
acid 159. 1 7 8 , 1 7 9 Reaction of acid 159 with an excess of methyllithium followed by

173
trimethylsilyl chloride gave the methyl ketone 160. 1 8 0 The addition of trimethylsilyl
chloride minimizes the formation of the undesired tertiary alcohol side-product.
Reduction of ketone 160 with lithium aluminum hydride gave the desired secondary
alcohol 161. The anion of 161 was generated with potassium hydride and reacted with
5-bromo-1 -pentene to give the metathesis precursor, diene ether 162. The 13C NMR of
162 contained 16 lines with four lines between 100 and 140 ppm for the four olefinic
carbons. The 1H NMR spectrum contained a doublet at 1.09 ppm for the C-7 methyl
group. This four-step reaction sequence proceeded in an overall yield of 49%. Slow
addition of a solution of the Grubbs catalyst 9 1 6 6 in CH2CI2 to a solution of diene 162
under high dilution conditions gave a mixture of the cis and trans isomers of 163 and
164 in 63% yield (Scheme 26).
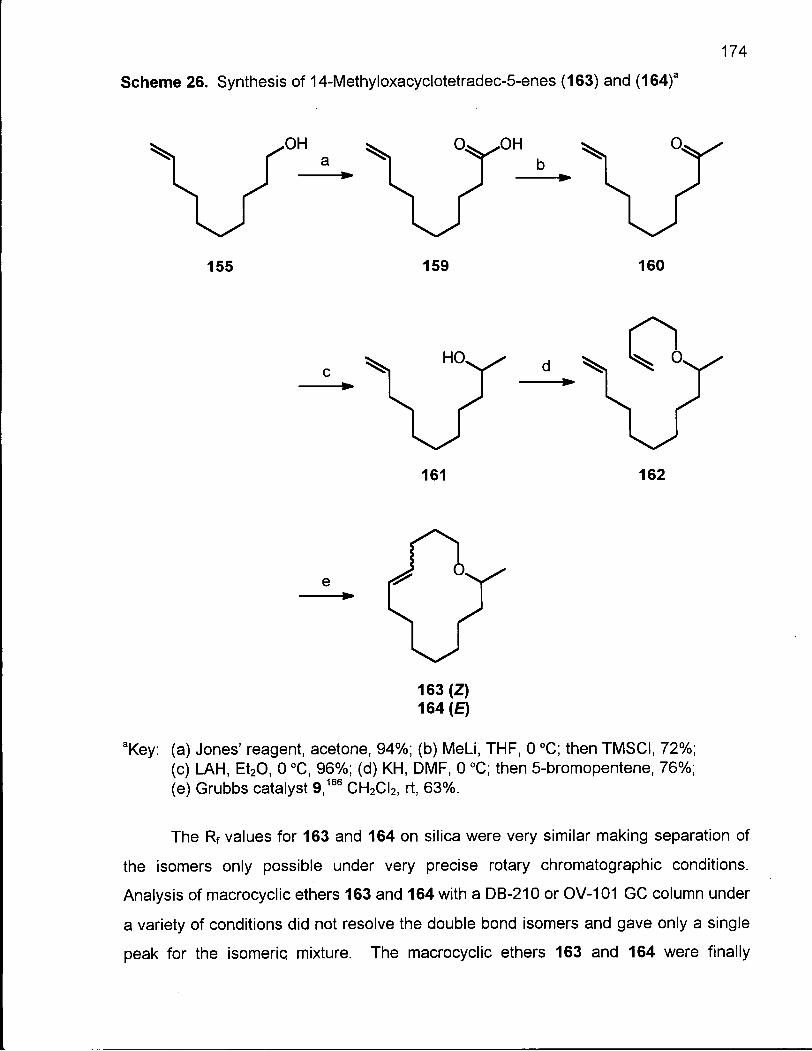
174
Scheme 26. Synthesis of 14-Methyloxacyclotetradec-5-enes (163) and (164)a
163 (Z) 164 (£)
aKey: (a) Jones' reagent, acetone, 94%; (b) MeLi, THF, 0 °C; then TMSCI, 72%; (c) LAH, Et20, 0 °C, 96%; (d) KH, DMF, 0 °C; then 5-bromopentene, 76%; (e) Grubbs catalyst 9,166 CH2CI2, rt, 63%.
The Rf values for 163 and 164 on silica were very similar making separation of
the isomers only possible under very precise rotary chromatographic conditions.
Analysis of macrocyclic ethers 163 and 164 with a DB-210 or OV-101 GC column under
a variety of conditions did not resolve the double bond isomers and gave only a single
peak for the isomeriq mixture. The macrocyclic ethers 163 and 164 were finally

175
resolved when analyzed with a chiral p-Dex 360 GC column (Supelco). This analysis
showed that the cyclization reaction proceeded to give 163 and 164 in a 43:57 ratio. It
should also be noted that under these GC conditions, each isomer gave rise to a
distinct pair of peaks due to each double bond isomer itself being composed of a pair of
enantiomers.
The 1H NMR spectrum of 163 contained one-proton signals at 5.46 and
5.26 ppm for the C-5 and C-6 methine protons of the carbon-carbon double bond. Also
visible was a three-proton doublet with a chemical shift of 1.09 ppm for the C-15 methyl
group. The C-2 methine and the C-14 methylene protons were resolved and had
chemical shifts of 3.41, 3.59 and 3.23 ppm respectively. The 13C NMR spectrum of 163
contained 14 lines with the olefinic carbons C-5 and C-6 having chemical shifts of
130.80 and 129.88 ppm respectively. The C-14 methine carbon and the C-2 methylene
carbon adjacent to the ether oxygen had chemical shifts of 73.35 and 66.24 ppm. The
HRMS and chemical analysis data was also consistent with the composition of 163.
The 1H NMR spectrum of 164 contained overlapping one-proton signals at 5.34
and 5.37 ppm for the C-5 and C-6 methine protons of the carbon-carbon double bond.
Also visible was a three-proton doublet with a chemical shift of 1.11 ppm for the C-15
methyl group. The C-14 methine and the C-2 methylene protons were resolved with
chemical shifts of 3.48, 3.41 and 3.34 ppm respectively. The 13C NMR spectrum of 164
contained 14 lines with the olefinic carbons C-5 and C-6 having chemical shifts of
131.87 and 130.77 ppm. The unambiguous assignment of these olefinic signals was
not possible due to the overlap of the olefinic proton signals in the HMQC experiment.
The C-14 methine carbon and the C-2 methylene carbon adjacent to the ether oxygen
had chemical shifts of 73.71 and 65.29 ppm respectively.
The carbon-carbon double bond of the major isomer produced in the metathesis
cyclization had the trans geometry as in 164. This was determined through a series of
1H homonuclear decoupling experiments which showed the coupling constant between
the olefinic protons to be 15.2 Hz. The double bond of the minor isomer 163 was

176
determined to have the Z configuration (c/'s) based on a coupling constant of 10.2 Hz
between the olefinic protons.
163 (Z) 164 (E)
3.2.2 Cis-Trans Isomerization of (27£)-14-Methyloxacyclotetradec-5-enes (163) and (164)
Treatment of the alkenes 163 and 164 with phenyl disulfide under photolysis
conditions was the method chosen to study alkene isomerization. A solution of ether
163 and phenyl disulfide was photolysed with a Hanovia 450W medium pressure
mercury lamp through pyrex glass and a 0.0014 M K2Cr04 solution resulting in an
irradiation window from 290-340 nm.177 The ether 163 gave a mixture of ethers 163
and 164 (GC ratio, 29:71). This corresponded to an energy difference of 0.53 kcal/mol
at 25 °C (Table 34, Entry 1). When reacted under similar conditions, ether 164 gave
the same mixture of ethers 163 and 164 (GC ratio, 29:71) (Table 34, Entry 2).
These experimental results were compared to values obtained from molecular
mechanics calculations using the MM3* force field. The global minimum conformation
of the cis alkene 163 is a distorted [3434] conformation with an energy of
21.67 kcal/mol. The next conformation is 0.25 kcal/mol higher in energy. The global
minimum conformation of 164, the trans isomer, was the [34'3'4'] conformation with an
energy of 20.69 kcal/mol. The second conformation of 164 is 0.89 kcal/mol higher in
energy. Using these calculated relative energies of 163 and 164, the energy difference
of 0.98 kcal/mol corresponded to a 16:84 equilibrium ratio of ethers 163 and 164 at
25 °C (Table 34, Entry 3). The agreement between the calculated and the

177
experimentally derived data was not as good as in the case of the unsubstituted ethers
157 and 158. Although the calculations did suggest that the trans isomer was in fact
lower in energy than the cis isomer 163, the calculated energy difference was greater
than observed.
Table 34. Experimental and Theoretical Equilibrium Ratios for the Isomerization Reaction of 2-Methyloxacyclotetradec-10-enes (163) and (164)
Entry Ratio 163:164a AG° (kcal/mol)b
1 163 (Z) 29:71 0.53
2 164(E) 29:71 0.53
3 MM3* 16:84 0.98c
a The equilibrium ratio of 163:164 was determined by analysis with a (3-DEX 360 chiral GC column (Supelco).
b AG° values were calculated using equation 8 at 25 °C. c Calculated relative strain energies were taken as an approximation of AG0.
Since the theoretical and experimental equilibrium ratios for the isomerization
reaction of ethers 163 and 164 were not in close agreement, a more detailed
calculation involving several of the lowest energy conformations of the ethers together
as an ensemble was performed (Table 35). This was in contrast to merely comparing
the energies of the global minimum conformations of each isomer. Using the
Boltzmann distribution expression and equation (9) to weight the contributions of the
individual conformations a weighted enthalpy value of 21.06 kcal/mol for 164 the
E isomer, and a weighted enthalpy value of 21.99 kcal/mol for 163 the Z isomer was
obtained. This corresponded to an estimated AG0 of 0.93 kcal/mol and an equilibrium
ratio of 17:83 (Z:E) for ethers 163 and 164. This more detailed calculation gave a
similar equilibrium ratio to that obtained from the simple comparison of the lowest
energy conformations of 163 and 164. Thus, either the calculated enthalpy values are
over estimated, or the AS term for the isomerization is relevant, and cannot be ignored
(AS*0).
AH = SniAHi (9)
178
Table 35. Relative Energies of Conformations of (Z/E)-14-Methyloxacyclotetradec-5-enes (163) and (164) and their Percent Population
— — = Ether 163 (Z) Ether 164(E)
Conformation Relative Energy3 Population' Relative Energyc Population'
(kcal/mol) (%) (kcal/mol) (%)
1 0.00 44 0.00 51
2 0.25 29 0.46 23
3 0.85 11 0.89 11
4 0.88 10 0.98 10
5 1.15 6 1.38 5
3 Estimated from relative energies at 25 °C calculated for 163 with the MM3* force field.
' Calculated with equation 8. c Estimated from relative energies at 25 °C calculated for 164 with the MM3* force
field.
Like the unsubstituted diene 156, the metathesis cyclization of diene 162, did
not produce an equilibrium ratio of the ether products. The unsubstituted diene gave
the cis isomer 157 as the major product, whereas the introduction of the C-2 methyl
group in the diene cyclization precursor resulted in the trans isomer 164 being the
major isomer in the metathesis cyclization. The trans isomer is in general the preferred
product in these macrocyclization reactions.19,181 The C-2 methyl group is several
carbons removed from the reacting carbon-carbon double bonds and accordingly the
influence of this stereocenter on the transition state of the reaction was not expected to
be large.182 However, this C-2 methyl group must play a role in the conformation of the
transition state and influence the selectivity of ethers 163 and 164 in the metathesis
cyclization.
3.3.1 Conclusion
The unsaturated 14-membered macrocyclic ethers 157, 158, 163, and 164 were
prepared via the ruthenium catalyzed metathesis reaction. These cyclizations
proceeded with low stereoselectivity giving macrocyclic ethers 157 and 158 in a ratio of
59:41, and macrocyclic ethers 163 and 164 in a ratio of 43:57. The geometry of the

179
carbon-carbon double bond in these macrocycles was determined by 1H homonuclear
decoupling NMR experiments. The double bond was isomerized with phenyl disulfide
under photolysis conditions to give in the case of ethers 157 and 158, a 40:60 mixture
of isomers. This corresponded to an energy difference of 0.23 kcal/mol in excellent
agreement with the value calculated from the energies of the global minimum
conformations of both 157 and 158. The isomerization of ethers 163 and 164 gave a
29:71 mixture of isomers. This corresponded to an energy difference of 0.53 kcal/mol
in reasonable agreement with 0.98 kcal/mol, the molecular mechanics calculated
energy difference between the global minimum conformations of 163 and 164.

CHAPTER 4 180
Synthesis and Conformational Analysis of 13-Membered Macrocyclic Ethers
The synthetic strategy for the preparation of the 13-membered macrocyclic
ethers in this study was the same as that employed in the preparation of the
14-membered macrocyclic ethers (Chapter 2). The 13-membered macrocyclic ether
precursors are less expensive than those used in the preparation of the 14-membered
analogues. Therefore, the chemistry of these smaller-ring analogues was of interest
not only for the synthetic and conformational data that could be collected from these
odd-numbered large ring systems, but also as a 'testing ground' for the chemical
reactions used here. Many of the synthetic problems were resolved in the study of the
13- membered ring systems prior to their application to the 14-membered ring systems.
This allowed for conservation of the more expensive 14-membered macrocyclic ether
precursors. For example, the cost of cyclododecanone (93) a common starting material
for many of the 13-membered macrocyclic ethers is $0.23/gram183 compared to
$125/gram184 for cyclotridecanone (86) used in the preparation of several of the
14- membered macrocyclic ethers.
4.0.1 Synthesis of 13-Membered Macrocyclic Ethers
The synthetic strategy involved the ring expansion of a cyclic ketone to a
lactone, thereby eliminating the potential problem of attempting to form the macrocycle
via a cyclization reaction. The ether oxygen of the lactone would ultimately become the
oxygen of the macrocyclic ether. This lactone functionality was also used to introduce
substituents in the region of the ether oxygen. Once this role was served, the carbonyl
was removed to give the macrocyclic ether with a procedure developed by Nicolaou
and coworkers55 using Lawesson's reagent 4848 to give an intermediate thionolactone.
This strategy allowed for a range of macrocyclic ethers to be produced as the result of
variations in the substitution pattern of the intermediate ketone and lactone resulting
from alkylation reactions, and the nucleophile used in the Nicolaou conversion.

181
The 13-membered macrocyclic ethers prepared in this study included the
unsubstituted oxacyclotridecane (168), the monosubstituted 2-methyloxacyclotridecane
(171), and the disubstituted 2,13-dimethyloxacyclotridecanes 179 and 180. The
13-membered macrocyclic ethers with a gem-dimethyl group at C-2 as in 190, and at
C-3 as in 193 were also prepared.
168 171 179
180 190 193
4.0.2 Conformational Analysis of 13-Membered Macrocyclic Ethers
The conformations of odd-membered rings such as the 13-membered cyclic
ethers are not superimposable on the diamond lattice. The strain energy of the
distorted conformations of cyclotridecane were shown to be lowest for either 3- or
5-sided conformations.98 The replacement of a ring carbon atom with an oxygen atom
should not influence the conformation of the ring since no new angular strain is
introduced. However, since some hydrogen atoms are eliminated by this substitution,
the number of transannular hydrogen interactions can be reduced. The introduction of
the ether oxygen atom, and of alkyl substituents further increases the possible number
of conformations of these 13-membered rings. For example, in the case of the
unsubstituted oxacyclotridecane (168), the introduction of the oxygen atom gives
13 possible [13333] conformations, 13 possible [12433] conformations, 13 possible
[346] conformations, and so forth.

182
A molecular mechanics search for low energy conformations of cyclotridecane
with the MM3* force field found a total of seven conformations within 2 kcal/mol of the
global minimum, [13333] conformation. The [13333] conformation is unsymmetric, and
all of the ring carbon positions are unique. The strain energies of the [13333]
conformations of the unsubstituted 13-membered ether resulting from the systematic
substitution of oxygen for each carbon atom were calculated with the MM3* force field
(Table 36). Some of these conformations were higher in strain energy, and need not
be considered in detail. For example, the conformations with the oxygen atom at a
corner position were all found to have high strain energies. Substitution of an oxygen
atom for C-2, C-11, or C-12 (Table 36 numbering) gave the lowest energy [13333]
conformations for this 13-membered ring.
Table 36. The Oxygen Substituted [13333] Conformations and Their Relative Strain Energies
Oxygen Position
Relative Energy3
(kcal/mol)
1 2.68
2 0.00
1 3 3.15
1 4 4.76
13 ^ ^ ^ J ^ ^ 5 1.31
A V 6 1.87
v / 5 7 4.18 11 l H 8 1.58
9 7 9
10
1.60
3.75
11 0.79
12 0.76
13 3.18
3 Strain energies are relative to the lowest energy [13333] conformation calculated with the MM3* force field.

183
To simplify the comparison of the 13-membered ether conformations, an
extension of the Dale nomenclature was developed to designate the position of the
ether oxygen atom in the conformation. The 13 positions of the [13333] conformation
are numbered starting with the 1-position at a 1-bond corner and increasing in a
clockwise fashion as shown in Table 36. Using this nomenclature, the low energy
[13333] conformation with the ether oxygen at the 2-position would be the [13333]-2
conformation.
Similar calculations were performed with the other low energy conformations of
cyclotridecane. The strain energy of the conformations resulting from the systematic
substitution of each carbon with an oxygen atom was also calculated. This exploration
gave three other 13-membered ether conformations likely to have low strain energy.
These are the [12334], [12433], and [13324] conformations with oxygen substitution as
indicated in Table 37. These calculations gave the [12334] conformation of a
13-membered cyclic ether the lowest strain energy and it was comparable to the lowest
energy [13333] conformation found above. This set of six conformations was used as a
starting point in the conformational analysis of the 13-membered macrocyclic ethers
examined in this study.

184
Table 37. Other Oxygen Substituted 13-Membered Conformations with Low Strain Energy
Conformer Skeleton Relative Energy (kcal/mol)
[12334] 0.01
[13324] 0.60
[12433] 1.00
a Strain energies are relative to the lowest energy, oxygen substituted [13333] conformation calculated with the MM3* force field.
The transition state interconversions of 13-membered rings are more complex
than those of the 14-membered rings. Both single corner movements, and
interconversions between 5- and 3-sided conformations are believed to occur, with
interconversions over the latter pathway thought to be lower in energy.118 Therefore, in
this study the transition state energies for the interconversion of the 13-membered
ethers were not calculated by driving the dihedral angles of corner atoms in low energy
conformations.

185
4.1.1 Synthesis of Oxacyclotridecane (168) and 2-Methyloxacyclotridecane (171)
The first 13-membered macrocyclic ether in this study, oxacyclotridecane (168),
was prepared via the Baeyer-Villiger oxidation of cyclododecanone (93) with
trifluoroperacetic acid to give 12-dodecanolide (165). The peracid was generated by
the addition of solid UHP to a solution of trifluoroacetic anhydride in CH2CI2.121 The
lactone 165 was converted into thionolactone 166 with Lawesson's reagent 48. 5 1 This
two-step sequence proceeded in 83% yield. The 1H NMR spectrum of the resultant oil
showed two-proton signals at 4.51 ppm and 2.87 ppm for the C-3 and C-13 methylene
protons of 166 respectively. The 13C NMR spectrum contained a signal at 225.20 ppm
for the C-1 thionocarbonyl, consistent with the structure of 166.
The thionolactone 166 was a common intermediate in the synthesis of the
macrocyclic ethers 168 and 171. Reaction of the thionocarbonyl of 166 with lithium
triethylborohydride and trapping of the resultant sulfur anion with methyl iodide gave
the unstable mixed thioacetal 167, 5 1 which was reduced immediately with tri(A7-butyl)tin
hydride to give the macrocyclic ether 168. The two-step reaction sequence proceeded
in 52% yield (Scheme 27).
Reaction of 166 with methyllithium and trapping of the resultant sulfur anion with
methyl iodide, produced the mixed thioketal 169. 5 1 Like 167, this compound was
unstable and was reduced immediately under radical conditions with tri(n-butyl)tin
hydride and AIBN to give the macrocyclic ether 171 in an overall yield of 30% for the
two-steps.
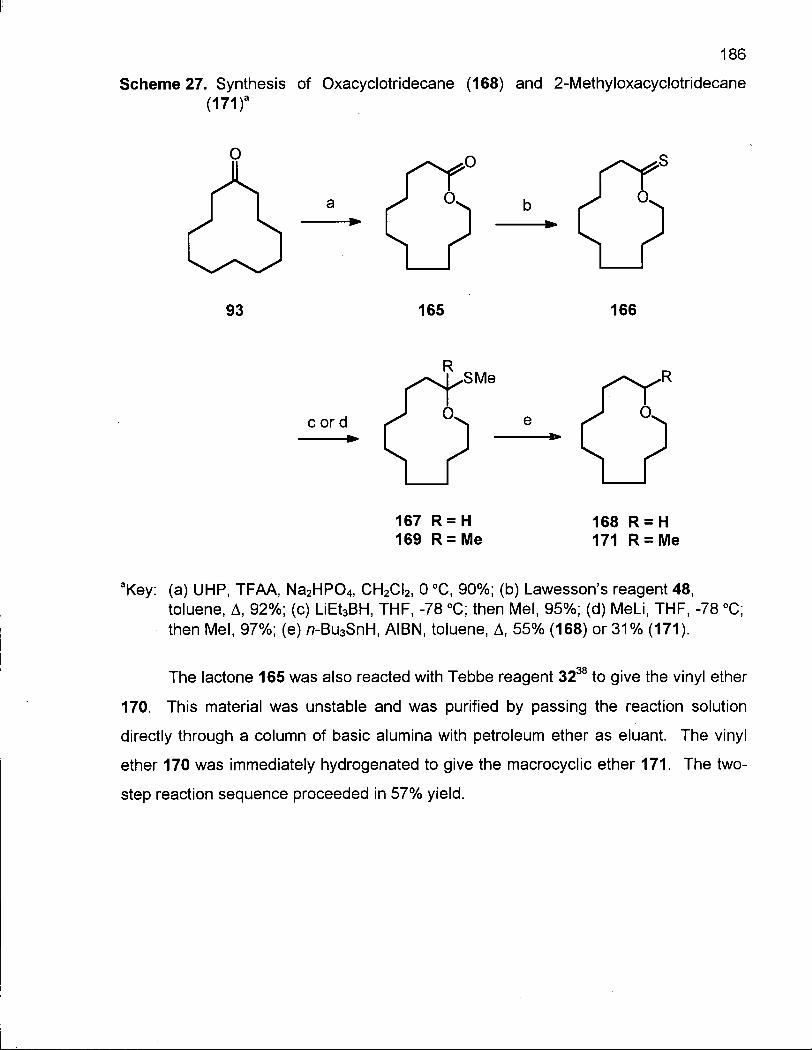
186
Scheme 27. Synthesis of Oxacyclotridecane (168) and 2-Methyloxacyclotridecane (171)a
167 R = H 168 R = H 169 R = Me 171 R = Me
aKey: (a) UHP, TFAA, Na2HP04, CH2CI2, 0 °C, 90%; (b) Lawesson's reagent 48, toluene, A, 92%; (c) LiEt3BH, THF, -78 °C; then Mel, 95%; (d) MeLi, THF, -78 °C; then Mel, 97%; (e) n-Bu3SnH, AIBN, toluene, A, 55% (168) or 31% (171).
The lactone 165 was also reacted with Tebbe reagent 32 3 8 to give the vinyl ether
170. This material was unstable and was purified by passing the reaction solution
directly through a column of basic alumina with petroleum ether as eluant. The vinyl
ether 170 was immediately hydrogenated to give the macrocyclic ether 171. The two-
step reaction sequence proceeded in 57% yield.

187
Scheme 28. Synthesis of 2-Methyloxacyclotridecane (171) via Hydrogenation3
aKey: (a) Tebbe reagent 32 , DMAP, pyridine, THF, -40 °C, 67%; (b) Pt02, H2, Et20, 85%.
4.1.2 Conformational Analysis of Oxacyclotridecane (168)
The 1H NMR spectrum of 168 at rt in CDCI3 contained a four-proton triplet at
3.42 ppm, a four-proton quintet at 1.54 ppm, a four-proton multiplet between
1.41-1.46 ppm, and a 12-proton multiplet between 1.30-1.38 ppm. The downfield
signal was assigned to the protons on C-2/C-13 adjacent to the ether oxygen
(Table 38). The signal at 1.54 ppm was assigned to the protons on C-3/C-12. The
chemical shifts of the remaining signals were very similar and could not be
unambiguously assigned. The 13C NMR spectrum contained six signals indicating that
the conformation of 168 either has a plane of symmetry, or site exchange processes
that are rapid at rt. The downfield signals at 70.33 and 28.54 ppm were assigned to the
C-2/C-13 and C-3/C-12 methylene carbons. The HRMS and chemical analysis data
were also consistent with the composition of 168.
165 170 171

188
Table 38. 1H and 13C NMR Assignments for Oxacyclotridecane (168) in CDCI3 at Room Temperature
Position 1H NMRa 13C NMRa
[11 13]
/ °>» 2, 13 3.42 70.33
3, 12 1.54 28.54
4-11 not assigned15
a The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI3 (13C). b Due to signal overlap these signals could not be unambiguously assigned.
The low temperature NMR spectra of 168 were obtained in a 4:1 mixture of
Freon 21 and Freon 22 as solvent (Figure 38). The 1H NMR spectrum of 168 at 220 K
was essentially the same as the rt spectrum, but with broader signals. The signals
continued to broaden as the temperature was lowered. At 148 K, the signal for the
C-2/C-13 protons split into two signals of approximate equal intensity. At temperatures
down to 130 K, these signals were resolved with chemical shifts of 3.61 and 3.23 ppm.
At 125 K, the lowest temperature reached in this DNMR experiment, the upfield signal
at 3.23 ppm broadened again. The sample froze before the coalescence temperature
of this second process could be determined.
The broadness of the signals in the DNMR study of 168, is consistent with the
presence of multiple conformations, even at low temperatures. The signals are
insufficiently resolved to determine the ratio of the individual conformations present.
The substitution of an oxygen atom into low energy conformations of cyclotridecane
suggested that the [13333], [12334], and [13324] conformations of 168 were likely to
have low strain energy. These were the first conformations considered in the analysis
of the DNMR spectra of this macrocyclic ether.
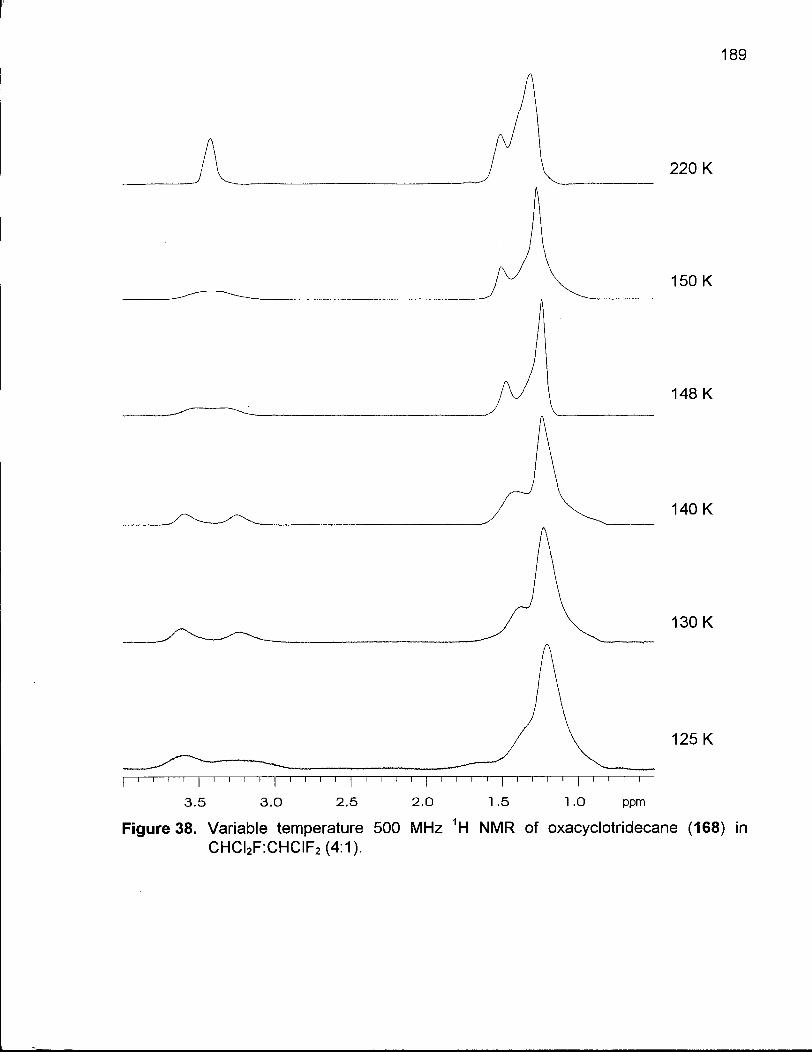

190
[13333]-2 [12334] [13324]
The signal for the C-2 and C-13 protons adjacent to the ether oxygen split into
two signals as the temperature was lowered. In the [13333]-2 conformation, the H-2exo
proton is deshielded by the anisotropy of the C-3/C-4 bond, and the H-13a proton is
deshielded by the anisotropy of the C-11/C-12 bond. No van der Waals steric
interactions are predicted for the C-13 protons since both are pointing to the outside of
the ring. The closest transannular hydrogen to the H-2endo proton is H-5endo calculated
to be 2.32 A away. This distance is only slightly less than the sum of the van der
Waals radii for a pair of protons, and at best a small contribution from this effect is
expected. The A 5 value is predicted to be similar in magnitude for both the C-2 and
C-13 protons, with a large value expected.
In the [12334] and [13324] conformations, the A 5 value for the C-2 and C-13
protons is predicted to be of a similar magnitude to that of the [13333]-2 conformation.
In each case, one of the H-2 protons, and one of the H-13 protons is deshielded by the
anisotropy of a neighbouring carbon-carbon bond. No van der Waals steric
interactions are expected here, as a result of the distorted geometry of the
13-membered ring. The DNMR spectra of 168 are consistent with each of these
conformations, and the likely low energy conformations cannot be narrowed any
further. For the case of the [13333]-2 conformation, the downfield signal at 3.61 ppm in
the low temperature 1H NMR spectrum could be assigned to the H-2exo and H-13a
protons, and the upfield signal at 3.23 ppm could be assigned to the H-2endo and H-13p
protons.

191
A molecular mechanics search for low energy conformations of 168 was
performed with the Monte Carlo technique and the MM2* force field. The global
minimum conformation was the non-diamond lattice [13333]-2 conformation 168-A as
suggested above. The second conformation 168-B was 0.15 kcal/mol higher in energy.
A total of six conformations were found within 2 kcal/mol of the global minimum. The
relative populations of these conformations at different temperatures were calculated
from the relative energies obtained from the MM2* calculations (Table 39). The results
of the calculations suggest that conformations 168-A and 168-B are both significantly
populated over the temperature range studied. This is in agreement with the results of
the DNMR study which first were consistent with both of these conformations, and
second indicated more than one conformation to be present at low temperatures.

Table 39. Low Energy Conformations of Oxacyclotridecane (168)
192
a Strain energies are relative to the global minimum conformation calculated with the MM2* force field.
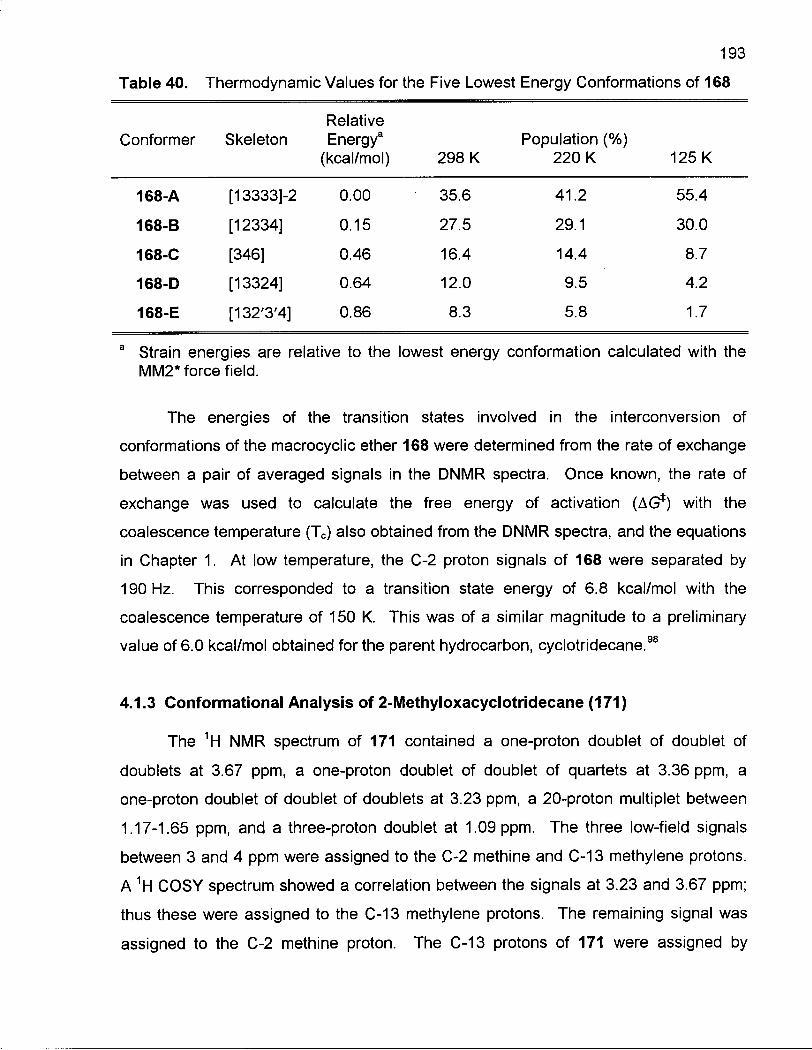
193
Table 40. Thermodynamic Values for the Five Lowest Energy Conformations of 168
Relative Conformer Skeleton Energy3
(kcal/mol) 298 K Population (%)
220 K 125 K
168-A [13333]-2 0.00 35.6 41.2 55.4
168-B [12334] 0.15 27.5 29.1 30.0
168-C [346] 0.46 16.4 14.4 8.7
168-D [13324] 0.64 12.0 9.5 4.2
168-E [132'3'4] 0.86 8.3 5.8 1.7
3 Strain energies are relative to the lowest energy conformation calculated with the MM2* force field.
The energies of the transition states involved in the interconversion of
conformations of the macrocyclic ether 168 were determined from the rate of exchange
between a pair of averaged signals in the DNMR spectra. Once known, the rate of
exchange was used to calculate the free energy of activation (AG*) with the
coalescence temperature (Tc) also obtained from the DNMR spectra, and the equations
in Chapter 1. At low temperature, the C-2 proton signals of 168 were separated by
190 Hz. This corresponded to a transition state energy of 6.8 kcal/mol with the
coalescence temperature of 150 K. This was of a similar magnitude to a preliminary
value of 6.0 kcal/mol obtained for the parent hydrocarbon, cyclotridecane.98
4.1.3 Conformational Analysis of 2-Methyloxacyclotridecane (171)
The 1H NMR spectrum of 171 contained a one-proton doublet of doublet of
doublets at 3.67 ppm, a one-proton doublet of doublet of quartets at 3.36 ppm, a
one-proton doublet of doublet of doublets at 3.23 ppm, a 20-proton multiplet between
1.17-1.65 ppm, and a three-proton doublet at 1.09 ppm. The three low-field signals
between 3 and 4 ppm were assigned to the C-2 methine and C-13 methylene protons.
A 1H COSY spectrum showed a correlation between the signals at 3.23 and 3.67 ppm;
thus these were assigned to the C-13 methylene protons. The remaining signal was
assigned to the C-2 methine proton. The C-13 protons of 171 were assigned by

194
comparing them to the 1H NMR data of the 14-membered ether,
2-methyloxacyclotetradecane (92). In 92, the C-14 proton syn to the C-2 methine
proton had the higher field chemical shift of the C-14 pair of protons. On this basis, the
signal at 3.23 ppm in the 1H NMR of 171 was assigned to the H-13 proton syn to H-2,
and the signal at 3.67 ppm was assigned to the H-13 proton syn to the C-14 methyl
group (Figure 39).
82
Figure 39. 1H NMR assignments of the C-2 and C-13 protons of 2-methyloxacyclo-tridecane (171).
The 13C NMR spectrum of 171 contained 13 lines. Two of these signals were at
low-field, and were assigned to the C-2 and C-13 carbons adjacent to the ether oxygen.
The highest field carbon at 20.20 ppm was assigned to the C-14 methyl group. The
remaining 13C signals occurred around 25 ppm. The assignment of these signals was
aided with data from COSY and HMQC 2D-NMR experiments (Table 41). The results
of the HRMS and chemical analysis were also consistent with the composition of 171.
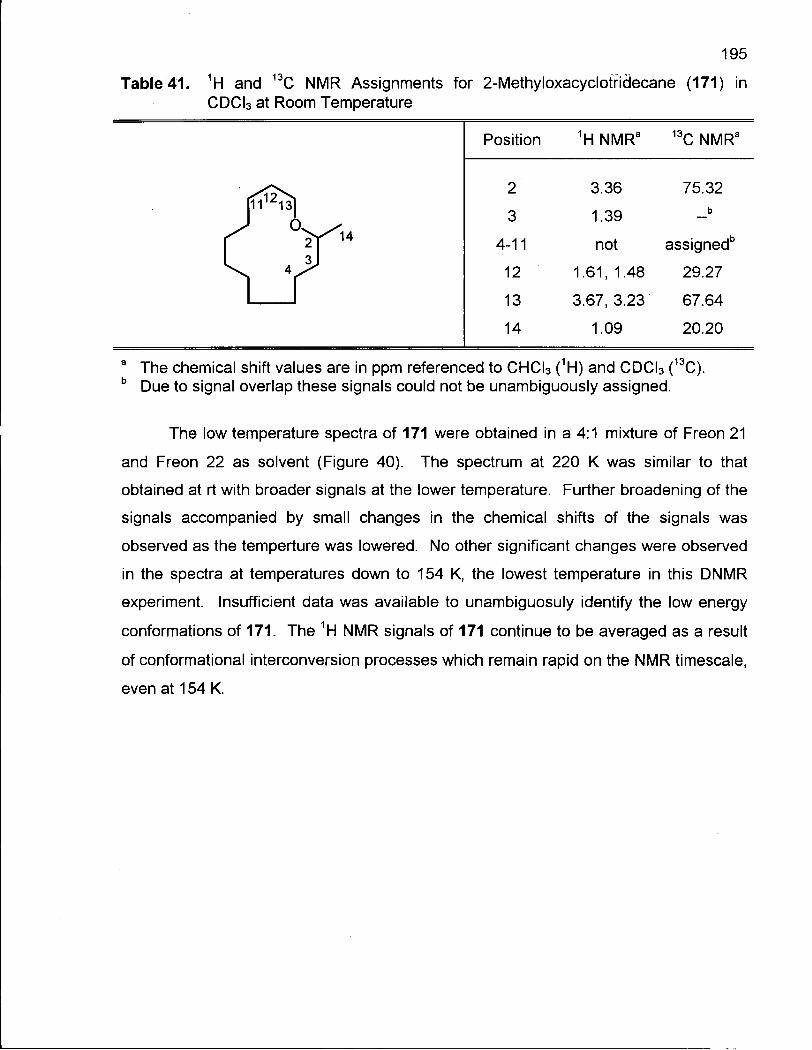
195
Table"41 . 1H and 13C NMR Assignments for 2-Methyloxacyclofridecane (171) in CDCI3 at Room Temperature
Position 1H NMRa 13C NMRa
r i i 1 2 i 3 | 2 3.36 75.32
r i i 1 2 i 3 | u
3 1.39 D
4-11 not assigned6
I * 3J 1 r 12 1.61, 1.48 29.27
1 1 13 3.67, 3.23 67.64
14 1.09 20.20
a The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI3 (13C). b Due to signal overlap these signals could not be unambiguously assigned.
The low temperature spectra of 171 were obtained in a 4:1 mixture of Freon 21
and Freon 22 as solvent (Figure 40). The spectrum at 220 K was similar to that
obtained at rt with broader signals at the lower temperature. Further broadening of the
signals accompanied by small changes in the chemical shifts of the signals was
observed as the temperture was lowered. No other significant changes were observed
in the spectra at temperatures down to 154 K, the lowest temperature in this DNMR
experiment. Insufficient data was available to unambiguosuly identify the low energy
conformations of 171. The 1H NMR signals of 171 continue to be averaged as a result
of conformational interconversion processes which remain rapid on the NMR timescale,
even at 154 K.

196
220 K
200 K
168 K
162 K
156 K
154 K
n — i — i — r ~ i — i — i — r
3.5 3.0 2.5 2.0 1.5 1.0 ppm
Figure 40. Variable temperature 500 MHz 1H NMR of 2-methyloxacyclotridecane (171) in CHCI2F:CHCIF2 (4:1).
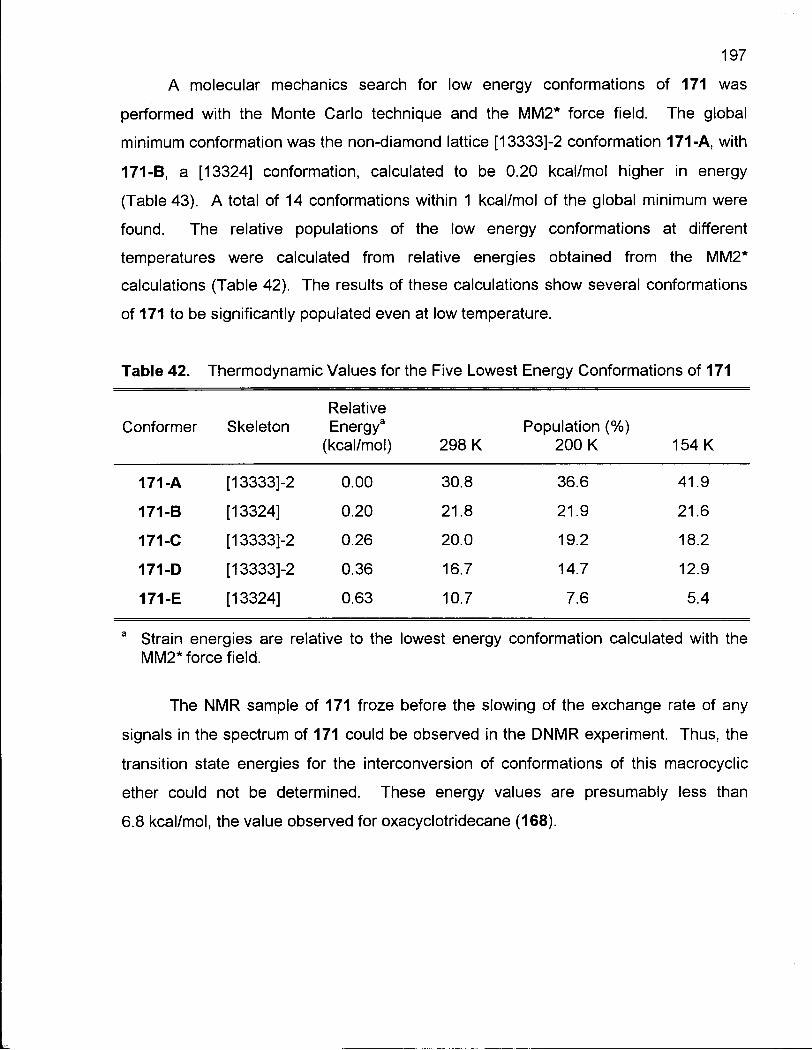
197
A molecular mechanics search for low energy conformations of 171 was
performed with the Monte Carlo technique and the MM2* force field. The global
minimum conformation was the non-diamond lattice [13333]-2 conformation 171-A, with
171-B, a [13324] conformation, calculated to be 0.20 kcal/mol higher in energy
(Table 43). A total of 14 conformations within 1 kcal/mol of the global minimum were
found. The relative populations of the low energy conformations at different
temperatures were calculated from relative energies obtained from the MM2*
calculations (Table 42). The results of these calculations show several conformations
of 171 to be significantly populated even at low temperature.
Table 42. Thermodynamic Values for the Five Lowest Energy Conformations of 171
Relative Conformer Skeleton Energy3 Population (%)
(kcal/mol) 298 K 200 K 154 K
171-A [13333]-2 0.00 30.8 36.6 41.9
171-B [13324] 0.20 21.8 21.9 21.6
171-C [13333J-2 0.26 20.0 19.2 18.2
171-D [13333J-2 0.36 16.7 14.7 12.9
171-E [13324] 0.63 10.7 7.6 5.4
Strain energies are relative to the lowest energy conformation calculated with the MM2* force field.
The NMR sample of 171 froze before the slowing of the exchange rate of any
signals in the spectrum of 171 could be observed in the DNMR experiment. Thus, the
transition state energies for the interconversion of conformations of this macrocyclic
ether could not be determined. These energy values are presumably less than
6.8 kcal/mol, the value observed for oxacyclotridecane (168).
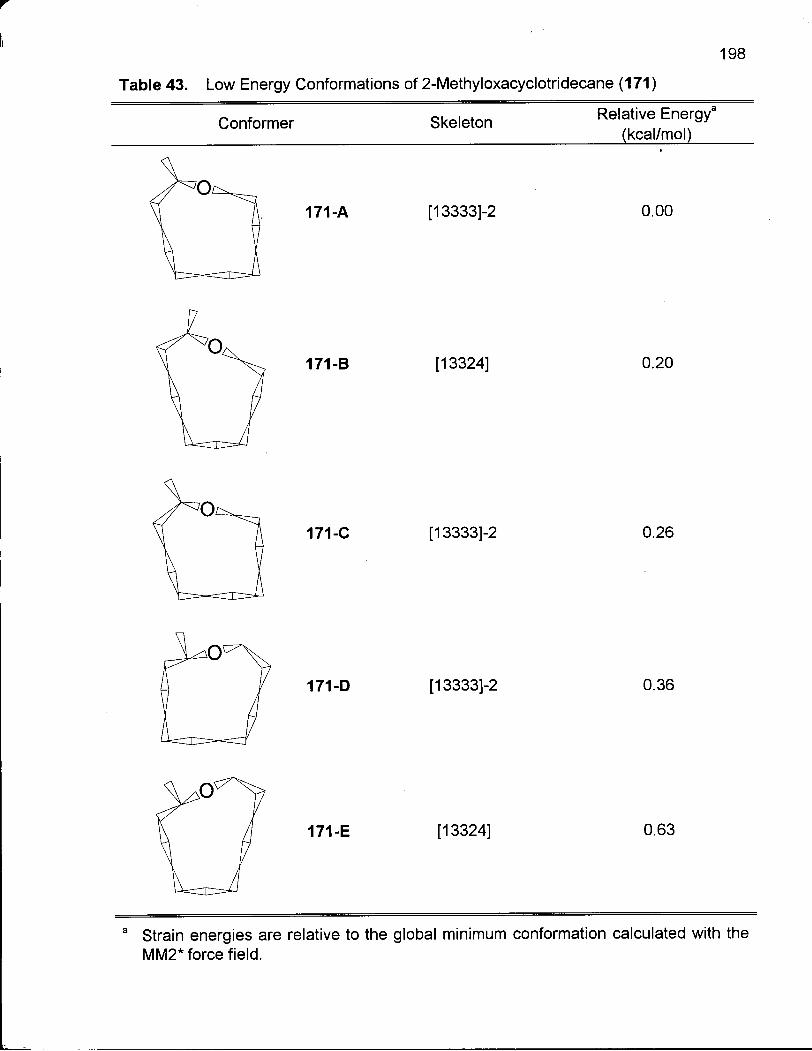
198
Table 43. Low Energy Conformations of 2-Methyloxacyclotridecane (171)
Conformer Skeleton Relative Energy (kcal/mol)
171-A [13333]-2 0.00
171-B [13324] 0.20
171-C [13333]-2 0.26
171-D [13333]-2 0.36
171-E [13324] 0.63
a Strain energies are relative to the global minimum conformation calculated with the MM2* force field.

199
4.2.1 Synthesis of 2,13-Dimethyloxacyclotridecanes (179) and (180)
The next 13-membered macrocyclic ethers examined were the diastereomeric
pair of 2,13-dimethyloxacyclotridecanes, 179 and 180. The C-2 methyl group was
introduced adjacent to the ketone prior to the Baeyer-Villiger ring expansion. Once the
desired ketone was produced, the synthetic path was the same as earlier. The cyclic
ketone was expanded to a lactone, and the carbonyl was removed via a thionolactone
intermediate (Scheme 29). Another method where the second methyl group was
introduced via the hydrogenation of an exocyclic double bond rather than via the
nucleophilic attack of a thionolactone was also investigated.
Scheme 29. Retrosynthetic Analysis of 2,13-Dimethyloxacyclotridecanes (179) and (180)
179 (2R*, 13/?*) 177X = S 180 (2SM3/?*) 176X = CH2
174 93
A method involving the methylaluminum bis(4-bromo-2,6-di-terf-butylphenoxide)
(MABR) mediated alkylation of a trimethylsilyl enol ether was employed in order to
prepare the intermediate ketone 174. Cyclododecanone (93) was reacted with
hexamethyldisilazane, and a mixture of trimethylsilyl chloride and lithium iodide to give

200
the trimethylsilyl enol ethers 172 and 173 in a 48:52 ratio as determined from GC
analysis (Scheme 30).128,129 Another method involving reaction of ketone 93 with
triethylamine and trimethylsilyl chloride gave lower yields of the enol ethers. These
diastereomers were separable on silica, and were identified by a comparison of their 13C NMR spectra. In general, the chemical shift for C-1 of the Z isomer is upfield
relative to that of the E isomer. While, the chemical shift for C-12, the allylic carbon, of
the Z isomer is generally downfield relative to that of the E isomer.130 Here, enol ether
172 was assigned the Z configuration based on chemical shifts of 149.76 ppm and
36.44 ppm for C-1 and C-12 compared to chemical shifts of 151.87 ppm and 28.42 ppm
for C-1 and C-12 of 173, the E isomer.
A solution of MABR was generated by the addition of trimethylaluminum in
hexanes to a solution of 4-bromo-2,6-di-te/?-butylphenol in CH2CI2.131,132 A mixture of
enol ethers 172 and 173 was reacted with an aliquot of this MABR solution and
subsequently alkylated with methyl triflate to give ketone 174.133 The bulky Lewis acid
coordinated to the enol ether and directed the alkylation with methyl triflate. The
BaeyerA/illiger oxidation of ketone 174 was performed with trifluoroperacetic acid in the
presence of Na2HP04 to give 12-tridecanolide (175). This peracid was generated by
the addition of UHP122,123 to a solution of trifluoroacetic anhydride in CH2CI2. The
lactone 175 was converted into the thionolactone 177 by reaction with Lawesson's
reagent 48.51 5 5 The 1H NMR spectrum of 177 contained a one-proton signal at
5.61 ppm for the C-3 methine, as well as a three-proton doublet at 1.27 ppm for the
C-14 methyl group. The 13C NMR spectrum of 177 contained 13 lines with the C-1
thionocarbonyl signal at 224.35 ppm, consistent with the structure of 177.
Subsequent reaction of thionolactone 177 with methyllithium and trapping of the
resultant sulfur anion with methyl iodide gave the unstable mixed thioketal 178.51,55
This material was reduced immediately under radical conditions with either
tri(/7-butyl)tin hydride or tris(trimethylsilyl)silane (TTMSH)134 to give the desired
macrocyclic ethers 179 and 180. The four-step reaction sequence proceeded in 13%

201
yield from 174 with tri(/7-butyl)tin hydride as the hydride source, and 23% yield with
TTMSH as the hydride source.
Scheme 30. Synthesis of 2,13-Dimethyloxacyclotridecanes (179) and (180) via Radical Reduction3
93 172 (Z) 174 173 (£)
175 177
178 179 (2R*, 13fl*) 180 (2S*, 13/?*)
aKey: (a) (TMS)2NH, TMSCI, Lil, CH2CI2; then Et3N, 92%; (b) MABR, MeOTf, CH2CI2, -40 °C, 71%; (c) UHP, TFAA, Na2HP04, CH2CI2, 0 °C, 92%; (d) Lawesson's reagent 48, toluene, A, 75%; (e) MeLi, THF, -78 °C; then Mel, 71%; (f) n-Bu3SnH, AIBN, toluene, A, 26%; (g) TTMSH, AIBN, toluene, A, 46%.

202
The lactone 175 was also reacted with Tebbe reagent 3238 to give the vinyl
ether 176. This material was unstable, however it could be purified by passing the
reaction solution directly through a column of basic alumina with petroleum ether as
eluant. The vinyl ether 176 was immediately hydrogenated to give the macrocyclic
ethers 179 and 180. This two-step reaction sequence proceeded in 36% yield.
Scheme 31. Synthesis of 2,13-Dimethyloxacyclotridecanes (179) and (180) via Hydrogenation3
175 176 179 (2R*, 13/?*) 180 (2S*, 13/?*)
3Key: (a) Tebbe reagent 32 , DMAP, pyridine, THF, -40 °C, 70%; (b) Pt02, H2, Et20, 52%.
The relative configuration of the C-2 and C-13 methyl substituents of 179 and
180 was determined through their analysis with a chiral Cyclodex-B GC column. The
macrocyclic ethers 179 and 180 were separable with silica chromatography and each
gave a single, distinct peak on GC analysis with a DB-210 column. The (2R*,13R*) or
anti isomer of 2,13-dimethyloxacyclotridecane is a dl pair of enantiomers which would
give rise to two peaks under chiral GC conditions. The (2S*,13R*) or syn isomer of the
macrocyclic ether is a meso compound which would give rise to only a single peak
under chiral GC conditions. GC analysis of 179, the first macrocyclic ether eluted on
silica, with the Cyclodex-B column resulted in two peaks of equal intensity with
retention times of 45.2 minutes and 46.0 minutes respectively. GC analysis of 180, the
second macrocyclic ether eluted on silica, gave only a single peak with a retention time
of 48.5 minutes (Figure 41). Thus, 179 was identified as the diastereomer with the C-2
and C-13 methyl groups in an anti configuration (2R*,13R*) and 180 was identified as

203
the diastereomer with the C-2 and C-13 methyl groups in a syn configuration
(2S*,13R*).
179 180
II
Figure41. GC analysis of 2,13-dimethyloxacyclotridecanes (179) and (180) on a chiral Cyclodex-B column; (a) (2R*,13R*)-2,13-dimethyloxacyclotridecane (179) ; (b) (2S*,13R*)-2,13-dimethyloxacyclotridecane (180); (c) mixture of (2R*,13R*) and (2S*,13R*)-2,13-dimethyloxacyclotridecane (179) and (180) .

204
The two methods used to form the macrocyclic ethers, hydride reduction of the
thiomethyl group, and hydrogenation of a carbon-carbon double bond are intrinsically
different with different intermediates and different reagents used in the transformation.
Accordingly, a difference in stereoselectivity in the ratio of 179:180 was expected for
each of these methods.
The hydride reduction of the mixed thioketal 178 with tri(n-butyl)tin hydride
showed no selectivity for either macrocyclic ether 179 or 180 (Table 44). It was hoped
that the different properties of the silane hydride reagent would offer an improvement in
the stereoselectivity of this reduction. The tris(trimethylsilyl)silane is a bulkier reagent
with a greater metal-hydrogen bond strength and a shorter metal-hydrogen bond
length. These features make the silane a more selective hydride reagent.
Unfortunately, only a very small stereoselectivity for macrocyclic ether 179 was
observed (8% d. e.) with the silane as the hydride source (Table 44). Reactions
performed with pure 2,14-dimethyloxacyclotetradecanes (103) and (104) the analogous
14-membered compounds with tri(n-butyl)tin hydride under radical conditions showed
no evidence of isomerization to the other macrocyclic ether. Therefore it was assumed
here that no isomerization of the macrocyclic ethers occurred in the hydride reduction
of the 13-membered ring system either.
The reduction of the vinyl ether 176 with Adams' catalyst (Pt02) proceeded with
low stereoselectivity (18% d. e.) (Table 44). The choice of platinum oxide as the
catalyst was important for the success of the reduction. Palladium on charcoal, and
rhodium on alumina, other common hydrogenation catalysts, gave lower yields of the
desired macrocycles in the hydrogenation reaction.
Molecular modeling calculations with the MM3* force field suggested that the
[13333] conformation is the most stable conformation of vinyl ether 176 with the
exocyclic double bond in an orientation essentially perpendicular to the plane of the
ring (Figure 42). The next lowest energy conformation was 0.22 kcal/mol higher in

205
energy and also a [13333] conformation. The local conformation in the region of the
double bond is similar in both of these conformations. It was believed that either the
methyl group flanking the ether oxygen or the macrocyclic ring itself would have a
directing effect on the hydrogenation. Modest stereoselectivity for 180 was observed in
this reduction, hence the exocyclic double bond must be blocked by the macrocyclic
ring on one face, and by a slightly lesser degree by the methyl group on the other face.
Figure 42. Lowest energy conformation of vinyl ether 176.
Table 44. Yield and Selectivity in the Preparation of 2,13-Dimethyloxacyclotridecanes (179) and (180)
Reagent Starting Material 1791803 Total Yield of 179+180
(%)
n-Bu3SnH, AIBN 178 50:50 26b
TTMSH, AIBN 178 54:46 46b
Pt02, H2 176 41:59 26c
a The ratio of 179:180 was determined by gas chromatography. b The diastereomers 179 and 180 were purified but not separated. c The diastereomers 179 and 180 were separated via radial chromatography.
4.2.2 Conformational Analysis of (2/?*,13R*)-2,13-Dimethyloxacyclotridecane (179)
The 1H NMR spectrum of 179 at rt in CDCI3 contained a two-proton sextet at
3.69 ppm, a 20-proton multiplet between 1.16-1.58 ppm, and a six-proton doublet at

206
1.08 ppm. The low-field signal at 3.69 ppm was assigned to the protons of C-2/C-13,
and the doublet at 1.08 ppm was assigned to the C-14 and C 15 methyl groups.
The 13C NMR spectrum at rt contained seven lines indicative of a either a plane
of symmetry, or a rapid site exchange process leading to symmetry in this molecule.
Thus, C-2 and C-13 had the same chemical shift as did C-3 and C-12, and so forth.
The low-field signals at 69.11 and 34.82 ppm were assigned to C-2/C-13 and C-3/C-12.
The signal at 19.56 ppm was assigned to the C-14 and C-15 methyl groups (Table 45).
The HRMS data was also consistent with the composition of 179.
Table 45. 1H and 13C NMR Assignments for (2R*,13R*)-2,13-Dimethyloxacyclo-tridecane (179) in CDCI3 at Room Temperature
Position 1H NMRa 13C NMRa
2, 13
3, 12
3.69
1.49-1.57
69.11
34.82
4-11 not assigned'
1 I 14, 15 1.08 19.56
a The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI3 (13C). b Due to signal overlap these signals could not be unambiguously assigned.
The low temperature spectra of 179 were obtained in a 4:1 mixture of Freon 21
and Freon 22 as solvent (Figure 43). The spectrum of 179 at 220 K was similar to that
obtained at rt with broadening of the signals at the lower temperature. Further
broadening of the signals accompanied by small changes in the chemical shifts of the
signals was observed as the temperature was lowered. No other significant changes
were observed in the spectra at temperatures down to 150 K, the lowest temperature in
this DNMR experiment. Insufficient data was available to unambiguosuly identify the
low energy conformations of 179. The 1H NMR signals of 179 continue to be averaged
as a result of conformational interconversion processes which remain rapid on the NMR
timescale, even at 150 K.
207
220 K
190 K
180 K
170 K
165 K
150 K
~7—I—I—r ~i i i r 1 i i r
3 .5 3 .0 2 .5 2 .0 1.5 1.0 0 . 5 ppm
Figure 43. Variable temperature 500 MHz 1H NMR of (2R*,13R*)-2,13-dimethyloxa-cyclotridecane (179) in CHCI2F:CHCIF2 (4:1).
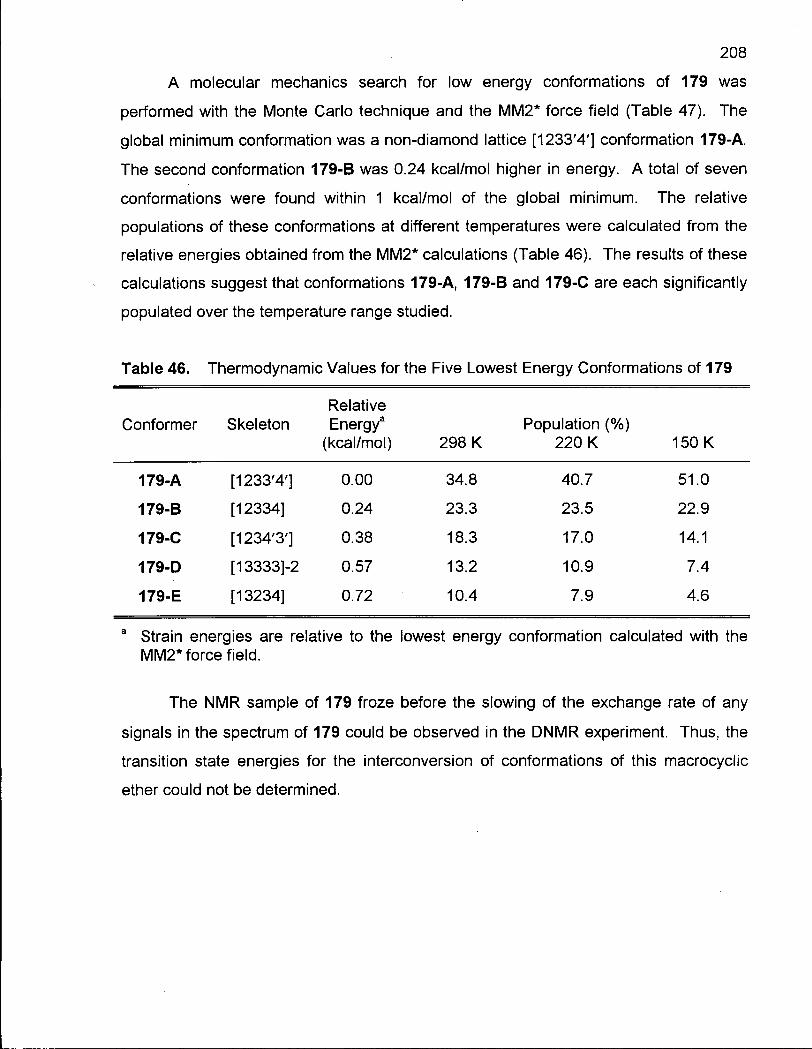
208
A molecular mechanics search for low energy conformations of 179 was
performed with the Monte Carlo technique and the MM2* force field (Table 47). The
global minimum conformation was a non-diamond lattice [1233'4'] conformation 179-A.
The second conformation 179-B was 0.24 kcal/mol higher in energy. A total of seven
conformations were found within 1 kcal/mol of the global minimum. The relative
populations of these conformations at different temperatures were calculated from the
relative energies obtained from the MM2* calculations (Table 46). The results of these
calculations suggest that conformations 179-A, 179-B and 179-C are each significantly
populated over the temperature range studied.
Table 46. Thermodynamic Values for the Five Lowest Energy Conformations of 179
Relative Conformer Skeleton Energy3 Population (%)
(kcal/mol) 298 K 220 K 150 K
179-A [1233'4'] 0.00 34.8 40.7 51.0
179-B [12334] 0.24 23.3 23.5 22.9
179-C [1234'3'] 0.38 18.3 17.0 14.1
179-D [13333]-2 0.57 13.2 10.9 7.4
179-E [13234] 0.72 10.4 7.9 4.6
3 Strain energies are relative to the lowest energy conformation calculated with the MM2* force field.
The NMR sample of 179 froze before the slowing of the exchange rate of any
signals in the spectrum of 179 could be observed in the DNMR experiment. Thus, the
transition state energies for the interconversion of conformations of this macrocyclic
ether could not be determined.

209
Table 47. Low Energy Conformations of (2R*,13R*)-2,13-Dimethyloxacyclotridecane (179)
Conformer Skeleton Relative Energy (kcal/mol)
179-A [1233'4'] 0.00
179-B [12334] 0.24
179-C [1234'3'] 0.38
179-D [13333]-2 0.57
179-E [13234] 0.72
a Strain energies are relative to the global minimum conformation calculated with the MM2* force field.

210
4.2.3 Conformational Analysis of (2S*,13/?*)-2>13-Dimethyloxacyclotridecane (180)
The 1H NMR spectrum of 180 contained a two-proton doublet of doublet of
quartets at 3.43 ppm, a 20-proton multiplet between 1.23-1.48 ppm, and a six-proton
doublet at 1.10 ppm. The low-field signal at 3.43 ppm was assigned to the protons of
C-2/C-13, and the doublet at 1.10 ppm was assigned to the C-14 and C-15 methyl
groups (Table 48).
The 13C NMR spectrum at rt contained seven lines indicative of a either a plane
of symmetry, or a rapid site exchange process leading to symmetry in this molecule.
Thus, C-2 and C-13 had the same chemical shift as did C-3 and C-12, and so forth.
The low-field signals at 74.23 and 37.67 ppm were assigned to C-2/C-13 and C-3/C-12.
The signal at 22.31 ppm was assigned to the C-14 and C-15 methyl groups (Table 48).
The HRMS and chemical analysis data were also consistent with the composition of
180.
Table 48. 1H and 13C NMR Assignments for (2S*,13R*)-2,13-Dimethyloxa-cyclotridecane (180) in CDCI3 at Room Temperature
Position 1H NMRa 13C NMRa
J CL ^ [ 21 1 5
2, 13
3, 12
3.43 b
74.23
37.67
r 4-11 not assigned" r 14, 15 1.10 22.31
a The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI3 (13C). b Due to signal overlap these signals could not be unambiguously assigned.
The low temperature spectra of 180 were obtained in a 4:1 mixture of Freon 21
and Freon 22 as solvent (Figure 44). The spectrum at 220 K was similar to that
obtained at rt with the signals broadened at the lower temperature. The signal for the
a-protons was extremely broad at 210 K, and split into a pair of equally intense signals

211
with chemical shifts of 3.64 and 3.28 ppm at low temperature. The signal for the C-3
and C-12 protons p to the ether oxygen also changed as the temperature was lowered.
At temperatures below 210 K, a signal of equally intensity to either the H-2 or H-13
protons became visible at 1.85 ppm and was assigned to one of the p-protons. The
signal for the C-14 and C-15 methyl groups broadened as the temperature was
lowered, but even at 150 K, the lowest temperature in this DNMR experiment, only one
signal was observed for these protons. Also visible in the low temperature spectra was
a signal at higher field than the signal of the methyl groups; at 0.96 ppm. The intensity
of this upfield signal was equal to one proton.
The sharpness of the low temperature signals in the DNMR study of 180
suggests the presence of a single conformation at low temperature. The substitution of
an oxygen atom into low energy conformations of cyclotridecane suggested that the
[13333] conformations with the oxygen atom located at the 2-, 5-, 11-, and 12-positidn
(page 182, Table 36 numbering) were likely to have low strain energy. The ring
skeletons of the [12334] and [13324] conformations (page 184, Table 37) were also
considered. In each of these six conformations, one of the methyl groups is at a corner
position, and is thus pointing away from the other methyl group thereby avoiding a
1,3-diaxial interaction between the C-14 and C-15 methyl groups.
[13333] [12334] [13324]

212
220 K
210 K
205 K
200 K
180 K
150 K
I — i — i — i — i — | — i — i — i — i — | — i — i — i — i — | — i — i — i — i — | — i — i — i — i | i i i i | i i i i | i i
3.5 3.0 2.5 2.0 1.5 1.0 ppm
Figure 44. Variable temperature 500 MHz 1H NMR of (2S*,13R*)-2,13-dimethyl-oxacyclotridecane (180) in CHCI2F:CHCIF2 (4:1).

213
The observed A8 value for the H-2 and H-13 protons in the low temperature
spectra of 180 is of an intermediate value. In the [13333]-5 conformation of 180, the
H-13 proton is deshielded by the anisotropy of the C-11/C-12 bond, while the H-2
proton is shielded by the anisotropy of the C-3/C-4 bond and by a van der Waals steric
interaction between H-2 and H-12endo. These protons were calculated to be only 2.27 A
apart. The A5 for these protons in this conformation is predicted to be large. A large
A5 value is also predicted between the H-2 and H-13 protons in the [13333]
conformation with the oxygen atom at the 11-position.
In the [13333]-2 conformation, the H-2 proton is shielded by the anisotropy of the
C-3/C-4 bond and also by a van der Waals steric interaction with the H-5end0 proton
calculated to be 2.27 A away. An intermediate A5 value is predicted between the H-2
and H-13 protons in this conformation. Similarily, intermediate A6 values are predicted
for the a-protons in the [13333]-12 conformation, and also in the [13324] conformation.
The A5 value for the H-2 and H-13 protons in the case of the [12334] conformation is
predicted to be small.
The observed chemical shift difference of the H-2 and H-13 protons in the
DNMR spectra of 180 is consistent with the value predicted for the above three
conformations. The p-proton signals in the DNMR spectra were examined in terms of
these conformations. The DNMR spectra show one of the p-protons of 180 to be
deshielded relative to the others. Unfortunately, in each of these three conformations,
one of the p-protons is predicted to be deshielded relative to the others. In the
[13333]-2 [13333]-12 [13324]
214
[13333]-2 conformation, the H-3a proton is deshielded by the anisotropy of both the
C-4/C-5 and the O/C-2 bonds. The three remaining p-protons are predicted to be more
shielded and give signals at higher field. Thus, the data for the p-protons did not allow
the likely low-energy conformation of 180 to be narrowed further.
A molecular mechanics search for the low energy conformations of 180 was
conducted with the Monte Carlo technique and the MM2* force field. The global
minimum conformation was the [13333]-2 conformation 180-A (Table 50). The second
conformation found was 180-B, 1.55 kcal/mol higher in energy. A total of six
conformations were found within 2 kcal/mol of the global minimum. The relative
populations of the low energy conformations at different temperatures were calculated
from relative energies obtained from the MM2* calculations (Table 49). The results of
these calculations suggest that conformation 180-A is the major conformation over the
temperature range studied. This is in agreement with the results of the DNMR study
where the results were consistent with conformation 180-A.
Table 49. Thermodynamic Values for the Five Lowest Energy Conformations of 180
Relative Conformer Skeleton Energy3 Population (%)
(kcal/mol) 298 K 210 K 150 K
180-A [13333]-2 0.00 82.5 94.1 98.8
180-B [13333]-5 1.55 6.0 2.3 0.5
180-C [1234'3'] 1.72 4.5 1.5 0.3
180-D [13324] 1.86 3.6 1.1 0.2
180-E [12433] 1.89 3.4 1.0 0.2
Strain energies are relative to the lowest energy conformation calculated with the MM2* force field.

215
Table 50. Low Energy Conformations of (2S*,13R*)-2,13-Dimethyloxacyclotridecane (180)
Conformer Skeleton Relative Energy (kcal/mol)
180-A [13333]-2 0.00
180-B [13333]-5 1.55
180-C [1234'3'] 1.72
180-D [13324] 1.86
180-E [12433] 1.89
Strain energies are relative to the global minimum conformation calculated with the MM2* force field.

216
The energies of the transition states involved in the interconversion of
conformations of the macrocyclic ether 180 were determined from the rate of exchange
between a pair of averaged signals in the DNMR spectra. Once known, the rate of
exchange was used to calculate the free energy of activation (AG*) with the
coalescence temperature (Tc) also obtained from the DNMR spectra and the equations
in Chapter 1. At low temperature, the signals for the H-2 and H-13 protons were
separated by 175 Hz. This corresponded to a transition state energy of 9.7 kcal/mol at
a coalescence temperature of 210 K. The transition state energy for this macrocyclic
ether is significantly larger than the 6.8 kcal/mol obtained for the unsubstituted
oxacyclotridecane (168). The methyl substituents in 180 must prevent conformations of
this macrocyclic ether from interconverting via lower energy pathways.
4.3.1 Synthesis of 2,2-Dimethyloxacyclotridecane (190)
The synthesis of macrocyclic ether 190 followed the general synthetic strategy
presented earlier. The synthetic plan was to ring expand a dialkylated ketone to give a
13-membered lactone with the gem-dimethyl substituents already in place adjacent to
the ether oxygen. The carbonyl of this lactone would be removed to give the
macrocyclic ether 190 (Scheme 32).
Scheme 32. Retrosynthetic Analysis of 2,2-Dimethyloxacyclotridecane (190)
O
190 189 181
Unfortunately, difficulties were encountered with both the dialkylation of the
ketone and the Baeyer-Vi Niger oxidation of the ketone to the proposed lactone 189, and
this synthetic route was abandoned. Earlier, it was shown that the cyclization of the

217
tertiary hydroxy acid 113 gave the C-13 grem-dimethyl lactone 114 in the preparation of
the 14-membered macrocyclic ether 116, the 14-membered analogue to ether 190.
This synthetic route in which the desired ring was formed as the result of a cyclization
reaction rather than through the expansion of an existing ring was viewed as a
promising alternative synthetic path.
113 114 116
The preparation of the desired tertiary hydroxy acid, the acyclic precursor to
lactone 189, began with the reaction of 1,10-decanediol (182) with 48% hydrobromic
acid to give the bromo alcohol 183 (Scheme 33).145 Oxidation of this alcohol with the
Jones' reagent gave the bromo acid 184. 1 7 8 , 1 7 9 Reaction of the bromo acid 184 under
Fischer esterification conditions gave the methyl ester 185. This ester was chain
extended by alkylation with the anion of methyl acetoacetate to give diester 186. 1 4 1 The
four-step reaction sequence proceeded in an overall yield of 72%. The 1H NMR
spectrum of 186 contained three singlets for the three methyl groups, the ester methyl
groups had chemical shifts of 3.70 and 3.60 ppm, while the chemical shift of the C-13
methyl group was 2.19 ppm. Three carbonyl signals were visible in the 13C NMR
spectrum of 186 at 203.18, 174.19 and 170.13 ppm for the C-12 ketone and the two
ester carbonyls respectively. This spectral data as well as the HRMS and the chemical
analysis results were all consistent with the structure of 186.
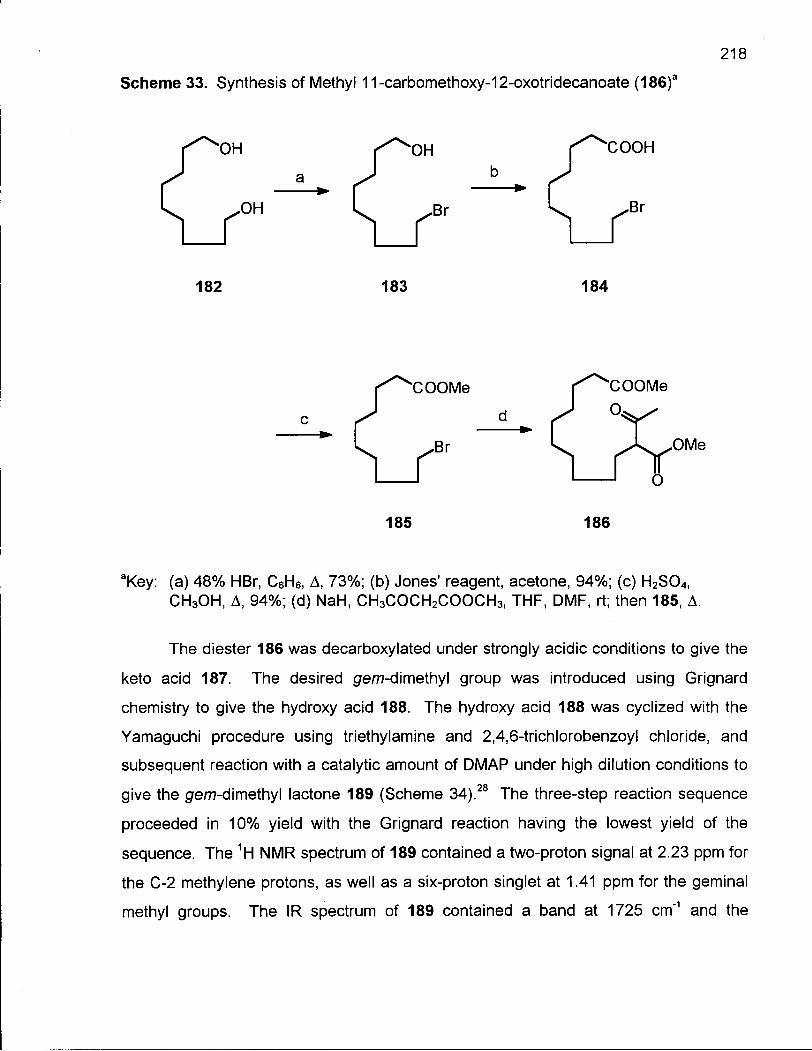
218
Scheme 33. Synthesis of Methyl 11-carbomethoxy-12-oxotridecanoate (186)a
185 186
aKey: (a) 48% HBr, C6H6, A, 73%; (b) Jones' reagent, acetone, 94%; (c) H2S04, CH3OH, A, 94%; (d) NaH, CH3COCH2COOCH3, THF, DMF, rt; then 185, A.
The diester 186 was decarboxylated under strongly acidic conditions to give the
keto acid 187. The desired gem-dimethyl group was introduced using Grignard
chemistry to give the hydroxy acid 188. The hydroxy acid 188 was cyclized with the
Yamaguchi procedure using triethylamine and 2,4,6-trichlorobenzoyl chloride, and
subsequent reaction with a catalytic amount of DMAP under high dilution conditions to
give the gem-dimethyl lactone 189 (Scheme 34).28 The three-step reaction sequence
proceeded in 10% yield with the Grignard reaction having the lowest yield of the
sequence. The 1H NMR spectrum of 189 contained a two-proton signal at 2.23 ppm for
the C-2 methylene protons, as well as a six-proton singlet at 1.41 ppm for the geminal
methyl groups. The IR spectrum of 189 contained a band at 1725 cm"1 and the
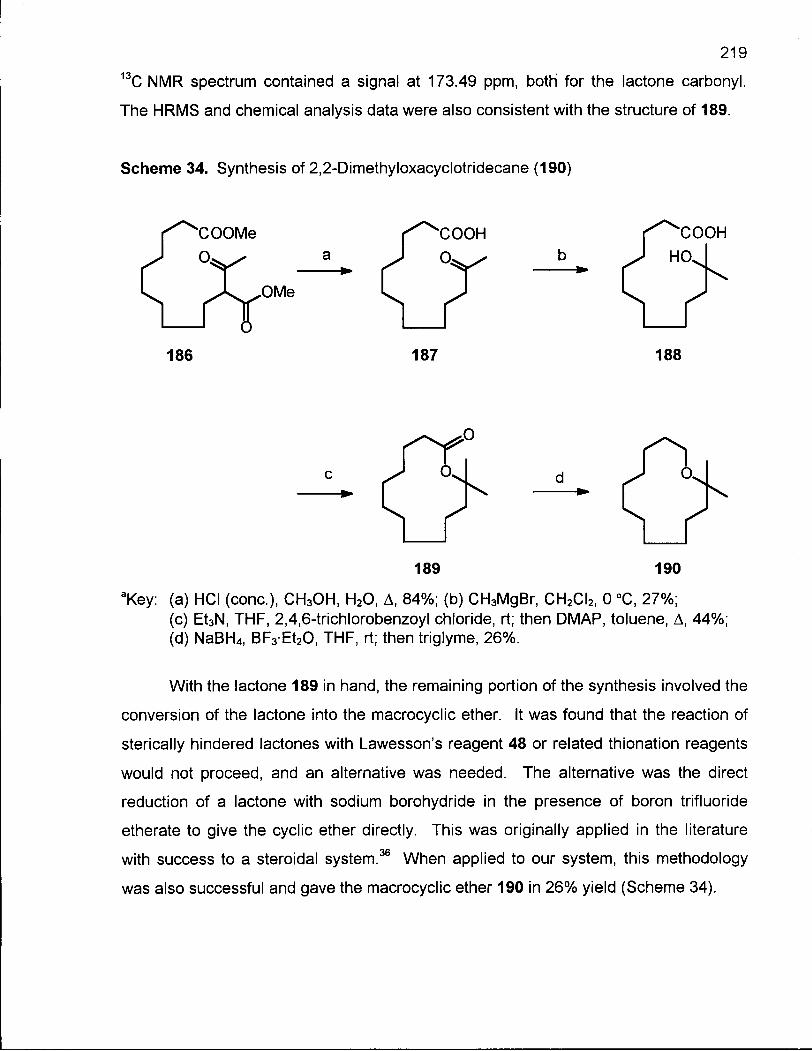
219 13C NMR spectrum contained a signal at 173.49 ppm, both for the lactone carbonyl.
The HRMS and chemical analysis data were also consistent with the structure of 189.
Scheme 34. Synthesis of 2,2-Dimethyloxacyclotridecane (190)
189 190
aKey: (a) HCI (cone), CH3OH, H20, A, 84%; (b) CH3MgBr, CH2CI2, 0 °C, 27%; (c) Et3N, THF, 2,4,6-trichlorobenzoyl chloride, rt; then DMAP, toluene, A, 44%; (d) NaBH4, BF3Et20, THF, rt; then triglyme, 26%.
With the lactone 189 in hand, the remaining portion of the synthesis involved the
conversion of the lactone into the macrocyclic ether. It was found that the reaction of
sterically hindered lactones with Lawesson's reagent 48 or related thionation reagents
would not proceed, and an alternative was needed. The alternative was the direct
reduction of a lactone with sodium borohydride in the presence of boron trifluoride
etherate to give the cyclic ether directly. This was originally applied in the literature
with success to a steroidal system.36 When applied to our system, this methodology
was also successful and gave the macrocyclic ether 190 in 26% yield (Scheme 34).

220
4.3.2 Conformational Analysis of 2,2-Dimethyloxacyclotridecane (190)
The 1H NMR spectrum of 190 at rt in CDCI3 contained a two-proton triplet at
3.31 ppm, a two-proton multiplet between 1.50-1.55 ppm, an 18-proton multiplet
between 1.29-1.44 ppm, and a six-proton singlet at 1.12 ppm. The downfield signal at
3.31 ppm was assigned to the C-13 protons, and the singlet at 1.12 ppm was assigned
to the C-14 and C-15 geminal methyl groups. The 13C NMR spectrum contained
13 lines. The low-field signals at 74.19 and 59.96 ppm were assigned to the C-2 and
C-13 carbons. The signal at 39.27 ppm was assigned to C-3 adjacent to the
quaternary carbon C-2, while the signal at 26.16 ppm was assigned to the C-14 and
C-15 geminal methyl groups. The assignment of the remaining 1H and 13C signals was
aided by COSY, HMQC, and HMBC 2D-NMR experiments (Table 51). The overlap of
some signals in these spectra prevented the complete assignment of the NMR data.
The HRMS data was also consistent with the composition of 190.
Table 51. 1H and 13C NMR Assignments for 2,2-Dimethyloxacyclotridecane (190) in CDCI3 at Room Temperature
Position 1H NMRa 13C NMRa
74.19
1.40 39.27
not assigned"
1.42 24.32
1.53 29.26
3.31 59.96
1.12 26.16
a The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI3 (13C). b Due to signal overlap these signals could not be unambiguously assigned.
A series of DNMR experiments were carried out on 190 in a 4:1 mixture of
Freon 21 and Freon 22 as solvent (Figure 45). The 1H NMR spectrum at 220 K
contained four signals and was similar to the rt spectrum with broadening of the signals
at the lower temperature. The signal for the C-13 protons broadened at intermediate
14,15

221
temperatures to give a pair of signals with chemical shift 3.35 and 3.23 ppm at low
temperature. The upfield signal of this pair was slightly broader at low temperatures.
Since a pair of signals was observed for these protons at low temperature, it is believed
that a single major conformation of 190 is present at low temperature. The signal for
the C-12 protons p to the ether oxygen also broadened as the temperature was
lowered. The coalescence temperature for both the C-13 and the C-12 proton signals
was 160 K. At low temperature, the signal for the C-12 protons split to give signals at
1.68 and 1.59 ppm. The lineshape of the signals in the methylene envelope region of
the spectra also changed as the temperature was lowered, however overlap of these
signals prevented a detailed analysis. The signal for the C-2 geminal methyl groups
broadened as the temperature was lowered, but even at 125 K, the lowest temperature
in this DNMR experiment, only one signal was observed for these protons.
A gem-dimethyl substituted carbon is restricted to a corner position in
13-membered conformations. If located at any other position in the ring, one of the
methyl groups is directed to the interior of the ring and a severe transannular steric
interaction results.105 There are eight possible [13333] conformations where the
oxygen atom is adjacent to a corner position. These are the conformations with the
oxygen atom at the 2- 3- 5- 6-, 8- 9- 11- or 12-positions (page 182, Table 36
numbering). Three other conformations were also considered as possible low energy
conformations of 190 (page 184, Table 37). The [12433] conformation from Table 37
was immediately disqualified from this set of conformations since the C-2 gem-dimethyl
group cannot be located at a corner position in such a conformation.
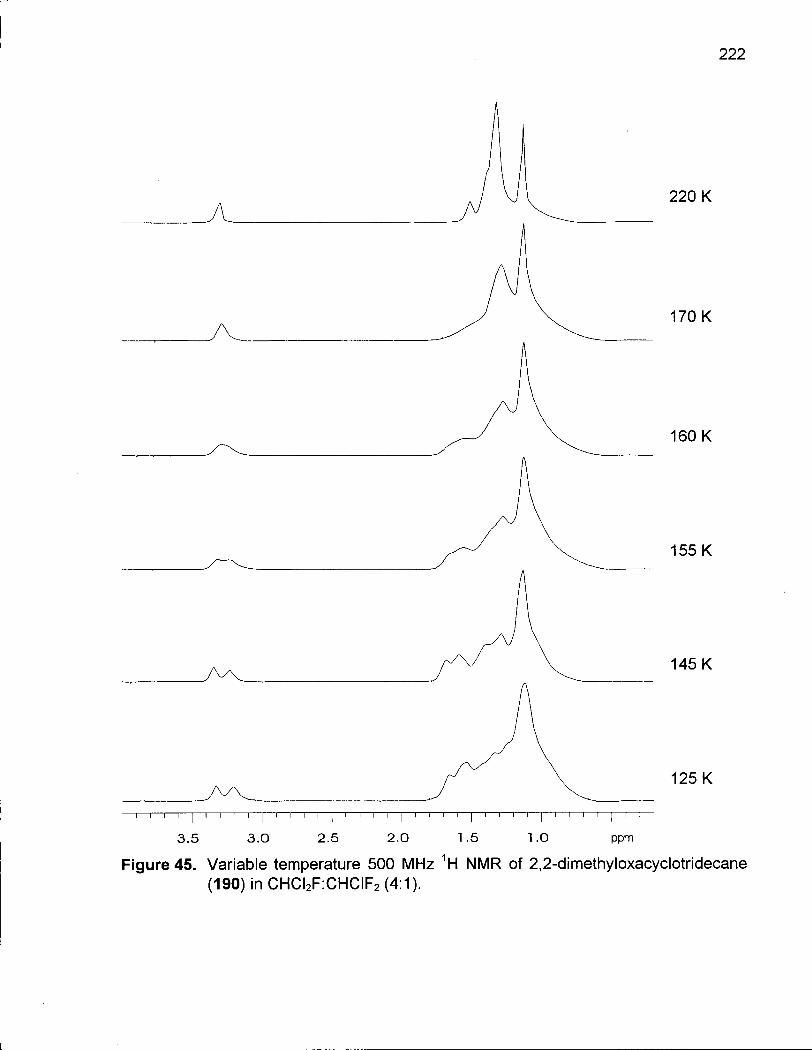
222

223
[13333] [12334] [13324]
The A5 value for the C-13 protons in the low temperature spectra of 190 is small.
In the [13333]-2 conformation of 190, the H-13exo proton is deshielded by the anisotropy
of the C-11/C-12 bond. This proton is also shielded by a van der Waals steric
repulsion between H-13endo and H-10endo- The A5 value for the H-13 protons in this
conformation is predicted to be of an intermediate magnitude. The A6 value for the
C-13 protons is predicted to be of an intermediate value in the [13333] conformations
with the ether oxygen atom at the 3- 5- 11- and 12-positions as well. In the [13333]
conformation with the oxygen atom at the 6-position, the H-13exo proton is deshielded
by the anisotropy of the C-11/C-12 bond. It is also shielded as a result of van der
Waals steric interactions between both H-10endo and H-3endo with H-13endo- The two van
der Waals shielding effects are in opposition to the anisotropic deshielding effect and a
small A5 value is predicted. A similarly small A5 value is predicted for the [13333]
conformations with the oxygen atom at the 8- and 9-positions. The A5 values of the
C-13 protons in the [12334] and [13324] conformations are predicted to be of an
intermediate value as the result of a deshielding of the H-13exo proton by the anisotropy
of the C-11/C-12 bond, and one opposing van der Waals shielding effect.

224
[13333]-6 [13333]-8 [13333]-9
On the basis of the observed chemical shift difference of the C-13 protons in
macrocyclic ether 190, the likely low energy conformations were narrowed to the above
three [13333] conformations. These are all consistent with the DNMR data where at
low temperature, the H-13exo proton is assigned to the downfield signal at 3.35 ppm,
and the H-13end0 proton to the signal at 3.23 ppm. The signal for H-13end0 was broader
than that of H-13exo. The H-13endo proton is expected to have two large coupling
constants ( 3J, J g e m ) , while the H-13exo proton has only one large coupling constant
(Jgem)-
The A8 value for the signals of the C-12 protons also was small in the low
temperature DNMR spectra. In each of the above three remaining conformations, the
C-12 methylene carbon is at a corner position. The H-12a proton is deshielded by the
anisotropy of the C-10/C-11 bond, but shielded by the anisotropy of the O/C-13 bond.
Thus, the DNMR data is in agreement with the predicted A5 of the C-12 protons in each
of these three conformations, and the identity of the low energy conformation cannot be
narrowed further with the data available.
A molecular mechanics search for low energy conformations of 190 was
performed with the Monte Carlo technique and the MM3* force field. The global
minimum conformation was the [13333]-9 conformation 190-A (Table 53). The second
conformation found was 190-B, 0.41 kcal/mol higher in energy. A total of six
conformations were found within 1 kcal/mol of the global minimum. The relative
populations of these conformations at different temperatures were calculated from the
relative energies obtained from the MM3* calculations (Table 52). The results of these
225
calculations suggest that conformation 190-A is the major conformation over the
temperature range studied. This is in agreement with the results of the DNMR study
which gave data that was consistent with conformation 190-A.
Table 52. Thermodynamic Values for the Five Lowest Energy Conformations of 190
Relative Conformer Skeleton Energy3 Population (%)
(kcal/mol) 298 K 170 K 125 K
190-A [13333]-9 0.00 41.3 59.2 72.1
190-B [12334] 0.41 20.8 17.8 14.1
190-C [13333]-5 0.55 16.3 11.6 7.9
190-D [1234'3'] 0.79 10.9 5.8 3.0
190-E [13333]-2 0.80 10.7 5.5 2.9
3 Strain energies are relative to the lowest energy conformation calculated with the MM3* force field.
226
Table 53. Low Energy Conformations of 2,2-Dimethyloxacyclotridecane (190)
Conformer Skeleton Relative Energy (kcal/mol)
190-A [13333]-9 0.00
190-B [12334] 0.41
190-C [13333J-5 0.55
190-D [1234'3'] 0.79
190-E [13333]-2 0.80
a Strain energies are relative to the global minimum conformation calculated with the MM3* force field.

227
The energies of the transition states involved in the interconversion of
conformations of the macrocyclic ether 190 were determined from the rate of exchange
between a pair of averaged signals in the DNMR spectra. Once known, the rate of
exchange was used to calculate the free energy of activation (AG*) with the
coalescence temperature (Tc) also obtained from the DNMR spectra and the equations
in Chapter 1. At low temperature, the signals of the C-13 protons of 190 were
separated by 58 Hz. This corresponded to a transition state barrier of 7.6 kcal/mol with
the coalescence temperature of 160 K. The signals for the C-12 protons were
separated by 48 Hz. The coalescence temperature for the signals of these protons was
also 160 K for a transition state energy of 7.7 kcal/mol. The average of these values is
7.7 ± 0.1 kcal/mol. Since movement of the gem-dimethyl group from the corner position
would result in transannular steric interactions involving an endo methyl group rather
than the lower energy hydrogen-hydrogen transannular interactions of the
unsubstituted ether, the transition state energy for the interconversion of conformations
of this gem-dimethyl ether is larger than that of the unsubstituted 13-membered ether
168 (6.8 kcal/mol).
4.4.1 Synthesis of 3,3-Dimethyloxacyclotridecane (193)
The final 13-membered macrocyclic ether prepared here was
3,3-dimethyloxacyclotridecane (193). The gem-dimethyl substituents of 193 could be
introduced through alkylation at C-2 of lactone 165. First, the Baeyer-Villiger oxidation
of cyclododecanone (93) was performed with trifluoroperacetic acid to give lactone 165.
The gem-dimethyl substituents were introduced individually via a sequential double
alkylation with LDA to generate the anion at C-2 of the lactone, and alkylation with
methyl iodide to give ultimately the gem-dimethyl lactone 192 (Scheme 35). The three-
step reaction sequence proceeded in 27% yield overall. The 1H NMR spectrum of 192
contained a signal at 4.11 ppm for the C-12 methylene protons and a six-proton singlet
at 1.13 ppm for the C-13 and C-14 geminal methyl groups. The IR spectrum contained
an absorption at 1715 cm'1 for the lactone carbonyl. Formation of a thionolactone with
Lawesson's reagent 48 was not attempted as this reaction was known to fail in the case
of sterically hindered lactones.144 The conversion of this lactone to the macrocyclic
228
ether 193 was performed via the direct reduction with sodium borohydride in the
presence of boron trifluoride etherate to give the macrocyclic ether 193 in a very
modest yield of 6%.36 Unlike the reduction of lactone 189 with a gem-dimethyl group at
C-12, the reduction of the C-2 gem-dimethyl lactone 192 was more difficult and would
not proceed at room temperature. Refluxing conditions were found to be necessary.
Scheme 35. Synthesis of 3,3-Dimethyloxacyclotridecane (193)a
O
93 165 191
192 193
aKey: (a) UHP, TFAA, Na2HP04, CH2CI2, 0 °C, 90%; (b) LDA, THF, -78 °C; then Mel, 43%; (c) LDA, THF, -78 °C; then Mel, 70%; (d) BF3Et20, NaBH4, THF, rt; then triglyme, A, 6%.
4.4.2 Conformational Analysis of 3,3-Dimethyloxacyclotridecane (193)
The 1H NMR spectrum of 193 contained a two-proton triplet at 3.40 ppm, a
two-proton singlet at 3.05 ppm, a two-proton broad quintet between 1.49-1.53 ppm, a
16-proton multiplet between 1.19-1.44 ppm, and a six-proton singlet at 0.84 ppm. The
downfield signals at 3.40 and 3.05 ppm were assigned to the protons of C-13 and C-2
respectively (Table 54). The data from a COSY experiment was used to assign the
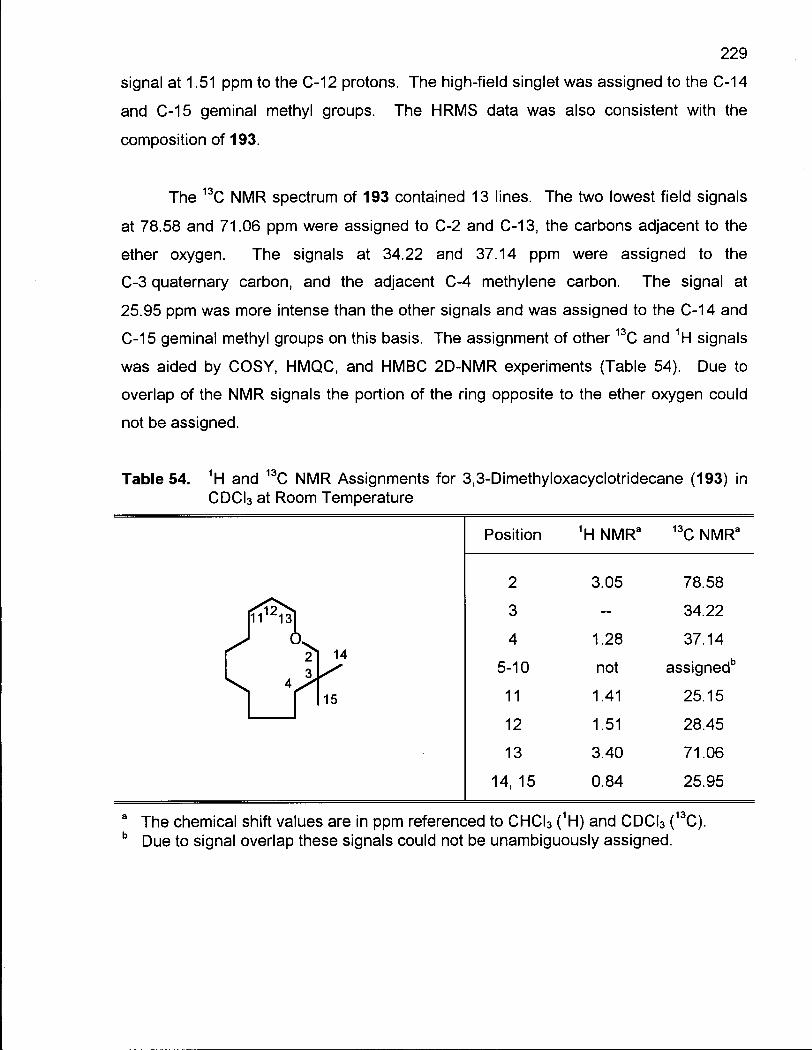
229
signal at 1.51 ppm to the C-12 protons. The high-field singlet was assigned to the C-14
and C-15 geminal methyl groups. The HRMS data was also consistent with the
composition of 193.
The 13C NMR spectrum of 193 contained 13 lines. The two lowest field signals
at 78.58 and 71.06 ppm were assigned to C-2 and C-13, the carbons adjacent to the
ether oxygen. The signals at 34.22 and 37.14 ppm were assigned to the
C-3 quaternary carbon, and the adjacent C-4 methylene carbon. The signal at
25.95 ppm was more intense than the other signals and was assigned to the C-14 and
C-15 geminal methyl groups on this basis. The assignment of other 13C and 1H signals
was aided by COSY, HMQC, and HMBC 2D-NMR experiments (Table 54). Due to
overlap of the NMR signals the portion of the ring opposite to the ether oxygen could
not be assigned.
Table 54. 1H and 13C NMR Assignments for 3,3-Dimethyloxacyclotridecane (193) in CDCI3 at Room Temperature
Position 1H NMRa 13C NMRa
3.05 78.58
34.22
1.28 37.14
not assigned"
1.41 25.15
1.51 28.45
3.40 71.06
0.84 25.95
2
13
14, 15
a The chemical shift values are in ppm referenced to CHCI3 (1H) and CDCI3 (13C). b Due to signal overlap these signals could not be unambiguously assigned.

230
The low temperature 1H NMR spectra of 193 were obtained in a 4:1 mixture of
Freon 21 and Freon 22 as solvent (Figure 46). The spectrum of 193 at 220 K was
similar to the rt spectrum with broadening of the signals at the lower temperature. The
signals of the C-13 protons broadened at intermediate temperature to give a pair of
signals at 3.75 and 3.25 ppm at low temperature. In contrast, the signal for the C-2
protons was essentially unchanged at all but the lowest temperatures examined in this
DNMR study. Broadening of the C-2 proton signal below 150 K was observed.
Similarily, the signal for the C-12 protons was largely unchanged over the temperature
range examined with the signal broadening at the lowest temperatures. The signal for
the geminal methyl groups also broadened at intermediate temperatures with a
coalescence temperature of 155 K. At 130 K this signal was split into two equally
intense signals at 0.91 and 0.77 ppm. Since one pair of intense signals were observed
for the geminal methyl groups, only one major conformation of 193 is present at low
temperature.
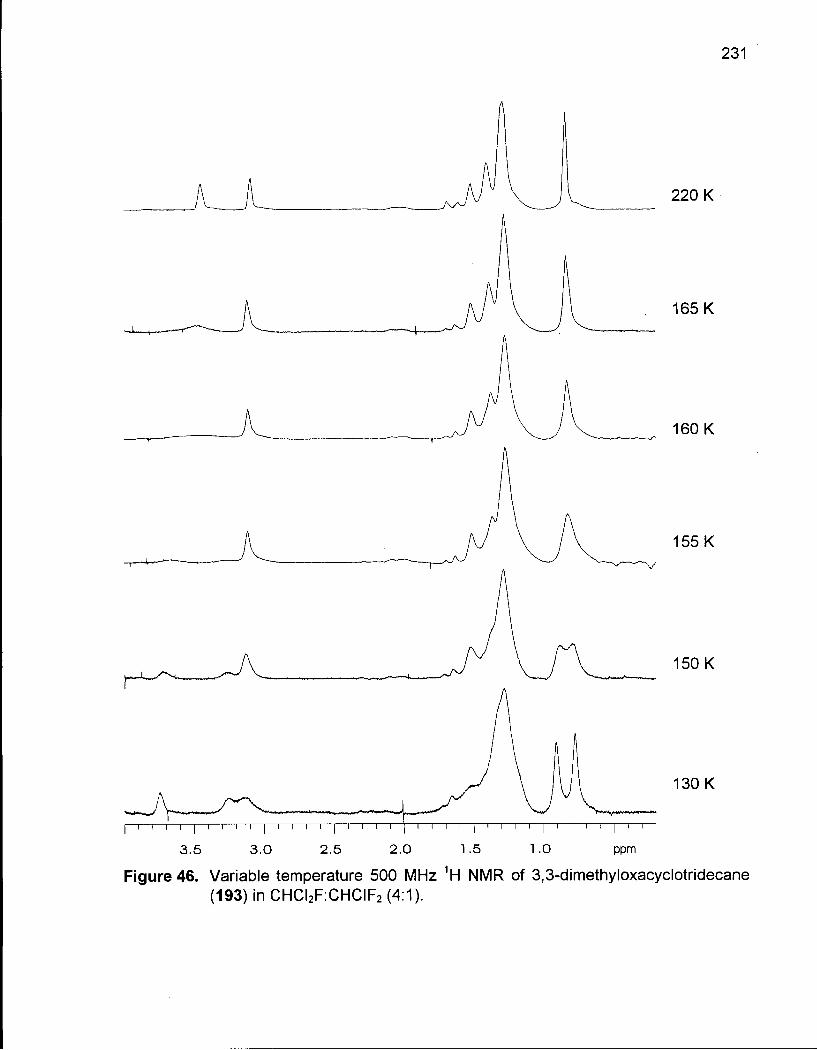
231

232
A gem-dimethyl substituted carbon is restricted to a corner position in
13-membered conformations. If located at any other position in the ring, one of the
methyl groups is pointing into the ring and a severe transannular steric interaction
results.105 There are nine possible [13333] conformations where the oxygen atom is
located p to a corner position. These are the conformations with the oxygen atom at
the 2- 3- 5- 6-, 8- 9- 11- or 12-positions (page 182, Table 36 numbering). The other
conformations in Table 37 (page 184) were also considered as ring skeletons for low
energy conformations of 193.
[13324] [12433] [12433]
The A8 value for the C-13 protons in the low temperature spectra of 193 is large.
In the [13333] conformation of 193 with the oxygen atom at the 2-position, the H-136Xo
proton is deshielded by the anisotropy of the C-11/C-12 bond. The A8 value for the
H-13 protons in this conformation is predicted to be large. The H-13p proton in the
[13333] conformation of 193 with the oxygen atom at the 3-position, is at a corner
position, and is deshielded by the anisotropy of the C-11/C-12 bond, but shielded by

233
the anisotropy of the O/C-2 bond. The A5 value for the C-13 protons is predicted to be
small in this conformation, as well as for the [13333] conformations with the ether
oxygen at the 5- 6- 8- 9-, 11- and 12-positions.
The A5 value of the C-13 protons in the [12334] conformation is predicted to be
large as the result of a deshielding of the H-13exo proton by the anisotropy of the
C-11/C-12 bond. In the [13324] conformations presented above, the A5 is predicted to
be small as the result of a deshielding of the H-13exo proton by the anisotropy of the
C-11/C-12 bond, with either an opposing additional anisotropic shielding effect, or an
opposing van der Waals shielding effect. Small A8 values are also predicted for the
C-13 protons in the [12433] conformations.
On the basis of the observed chemical shift differences of the C-13 protons in
macrocyclic ether 193, the number of likely low energy conformations was narrowed to
the above two conformations. These are both consistent with the observed DNMR data
for the C-13 protons where at low temperature, the H-13exo proton is assigned to the
downfield signal at 3.75 ppm, and the H-13end0 proton to the signal at 3.25 ppm
(Figure 46).
The A8 value of the low temperature signals of the C-2 protons in 193 was either
zero, or very small. In the above [13333]-2 conformation, the C-2 methylene carbon is
adjacent to a corner position. Thus, the H-2exo proton is deshielded by the anisotropy of
the C-3/C-4 bond and shielded by a van der Waals steric interaction between H-2endo
[13333] [12334]

234
and H-5end0. These opposing effects sum to a small A5 value. The H-2P proton in the
[12334] conformation is deshielded by the anisotropy of the C-3/C-4 bond. This results
in a large A5 value for the C-2 protons in this conformation. Thus, of the two remaining
conformations, the [13333]-2 conformation is the best fit to the DNMR signals of the
C-2 and C-13 protons of 193.
The averaging of the C-3 geminal methyl groups of 193 is slow at low
temperature and a pair of signals of equal intensity at 0.91 and 0.77 ppm are visible in
the low temperature DNMR spectra. The presence of this pair of signals indicates that
a conformational interconversion process that results in exchange of the geminal
methyl groups is slow at the low temperature. The unambiguous assignment of the
signals at 0.91 and 0.77 ppm to the C-3a and C-3P methyl groups cannot be made from
this data.
A molecular mechanics search for the low energy conformations of 193 was
carried out with the Monte Carlo technique and the MM3* force field. The global
minimum conformation was the [13333]-2 conformation 193-A (Table 55). The second
conformation 193-B was 0.69 kcal/mol higher in energy. A total of six conformations
were found within 1 kcal/mol of the global minimum. The relative populations of these
conformations at different temperatures were calculated from the relative energies
obtained from the MM3* calculations (Table 56). The results of these calculations
suggest that conformation 193-A is the major conformation over the temperature range
studied. This is in agreement with the above analysis of the DNMR data.
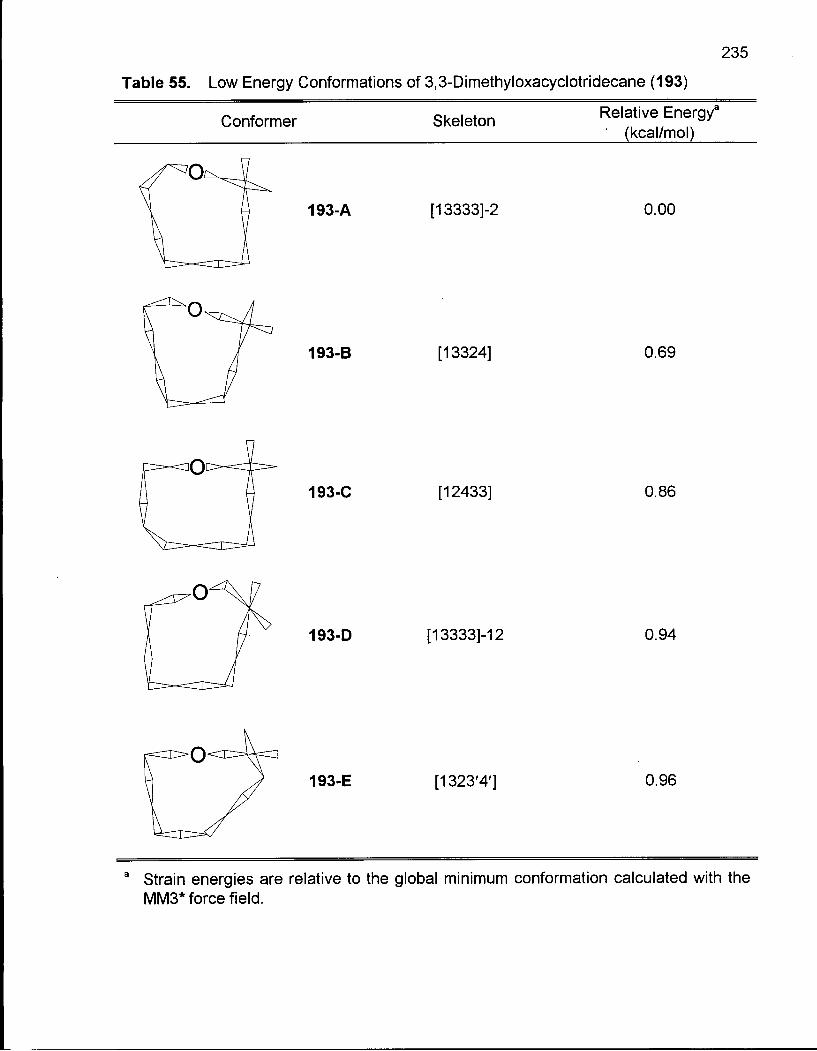
235
Table 55. Low Energy Conformations of 3,3-Dimethyloxacyclotridecane (193)
Conformer Skeleton Relative Energy (kcal/mol)
193-A [13333]-2 0.00
193-B [13324] 0.69
193-C [12433] 0.86
193-D [13333J-12 0.94
a Strain energies are relative to the global minimum conformation calculated with the MM3* force field.
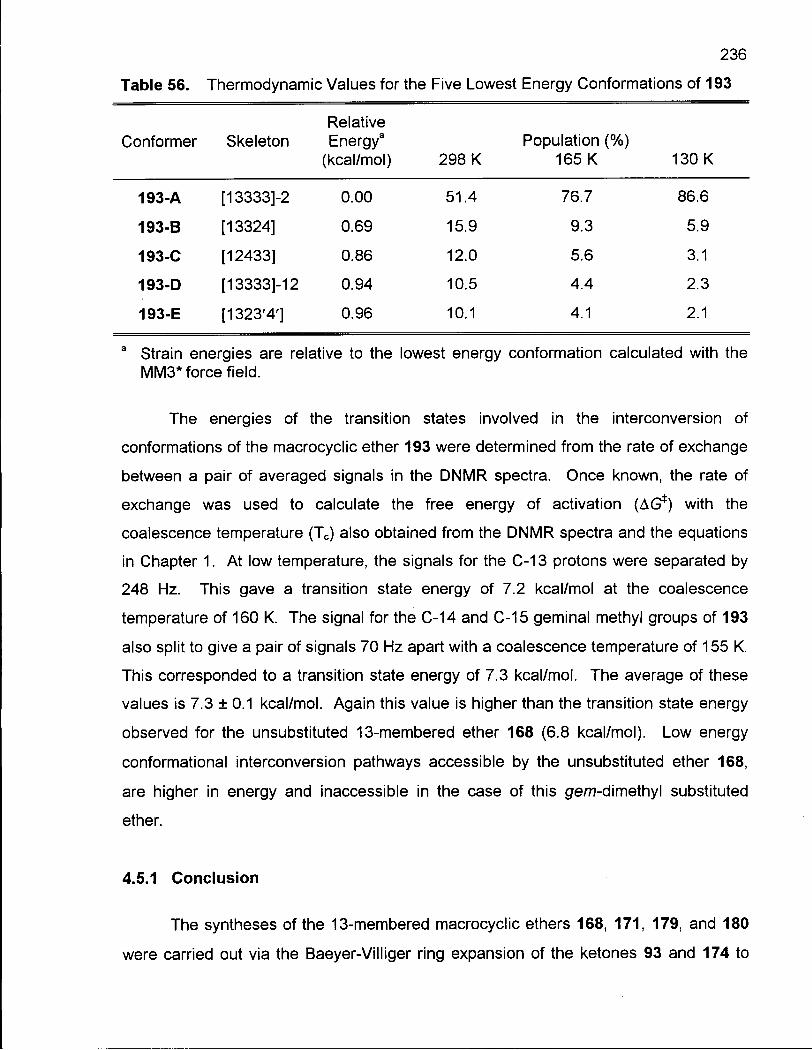
236
Table 56. Thermodynamic Values for the Five Lowest Energy Conformations of 193
Relative Conformer Skeleton Energy3
(kcal/mol) 298 K Population (%)
165 K 130 K
193-A [13333]-2 0.00 51.4 76.7 86.6
193-B [13324] 0.69 15.9 9.3 5.9
193-C [12433] 0.86 12.0 5.6 3.1
193-D [13333]-12 0.94 10.5 4.4 2.3
193-E [1323'4'] 0.96 10.1 4.1 2.1
a Strain energies are relative to the lowest energy conformation calculated with the MM3* force field.
The energies of the transition states involved in the interconversion of
conformations of the macrocyclic ether 193 were determined from the rate of exchange
between a pair of averaged signals in the DNMR spectra. Once known, the rate of
exchange was used to calculate the free energy of activation (AG*) with the
coalescence temperature (Tc) also obtained from the DNMR spectra and the equations
in Chapter 1. At low temperature, the signals for the C-13 protons were separated by
248 Hz. This gave a transition state energy of 7.2 kcal/mol at the coalescence
temperature of 160 K. The signal for the C-14 and C-15 geminal methyl groups of 193
also split to give a pair of signals 70 Hz apart with a coalescence temperature of 155 K.
This corresponded to a transition state energy of 7.3 kcal/mol. The average of these
values is 7.3 ±0.1 kcal/mol. Again this value is higher than the transition state energy
observed for the unsubstituted 13-membered ether 168 (6.8 kcal/mol). Low energy
conformational interconversion pathways accessible by the unsubstituted ether 168,
are higher in energy and inaccessible in the case of this gem-dimethyl substituted
ether.
4.5.1 Conclusion
The syntheses of the 13-membered macrocyclic ethers 168, 171, 179, and 180
were carried out via the Baeyer-ViNiger ring expansion of the ketones 93 and 174 to

237
give intermediate lactones 165 and 175. Further reaction of these lactones under
thionation conditions and subsequent radical reduction gave the macrocyclic ethers.
The diastereomeric ethers 179 and 180 were prepared under both hydrogenation and
radical reduction conditions with low stereoselectivity observed under both conditions.
The configuration of the methyl substituents in 179 and 180 was determined by chiral
GC analysis.
The macrocyclic ether 190 was prepared via the cyclization of hydroxy acid 188
with the Yamaguchi reagent to give lactone 189. The direct reduction of the lactone
with sodium borohydride in the presence of boron trifluoride etherate was employed to
give macrocyclic ether 190.
The macrocyclic ether 193 was prepared via the reduction of lactone 192.
However, even under refluxing conditions, the boron trifluoride mediated sodium
borohydride reduction of this lactone proceeded in low yield.
The conformation of these 13-membered ethers was analyzed with data from 1H-DNMR experiments. The low-temperature chemical shift difference of protons with
signals that were averaged at rt, were generally in agreement with predictions based on
anisotropy and van der Waals shielding effects in the low energy conformations.
Although many different possible conformations for these large ring compounds exist,
only a few conformations were found to be appreciably populated at room temperature
and below. Generally the conformations were consistent with the substituents located
exo to the ring, with geminal substituted carbon atoms occupying corner positions
exclusively. These results were consistent with the molecular mechanics calculations.
In general, the non-diamond lattice [13333] conformation was preferred just as in the
case of cyclotridecane. Thus, the introduction of the oxygen atom in these macrocyclic
ethers did not have a significant effect on the conformation of the ring.
The transition state energies for the conformational interconversion were
determined from the 1H DNMR experiments to be in the range of 6.8 to 9.7 kcal/mol.

238
The transition state energies of the gem-disubstituted ethers 190 and 193 were both
larger than that of the unsubstituted 13-membered ether 168. As expected, the
transition state energy values obtained for these macrocyclic compounds with an odd-
numbered ring size were in general smaller than those obtained for the 14-membered
ethers. This is in agreement with the greater conformational mobility of the
13-membered rings and their non-diamond lattice based conformations. These
conformations are in general more distorted and higher in strain energy than the
diamond lattice conformations.
4.6.1 General Conclusion
The reduction of a lactone was shown to be an effective method for the
preparation of 13- and 14-membered macrocyclic ethers. In the case of sterically
hindered lactones, the method involving a thionolactone intermediate failed, and a
direct reduction of the lactone was employed instead. The observed stereoselectivity
of the reductions leading to the dialkylated ethers 103, 104, 179, and 180 was low
under both radical and hydrogenation conditions.
The 14-membered ethers were in general found to have conformations which
were superimposable on the diamond lattice, while the 13-membered ether were
conformationally less regular, and not superimposable on the diamond lattice.
Comparison of the conformations of the 13- and 14-membered ethers showed the alkyl
substituents to have a similar impact for both ring sizes. In the case of the structurally
similar ethers 103 and 179 (R* R*) vs. 104 and 180 (S*, R*), the (S*, R*) isomers were
predominantly one conformation, while the (R*, R*) isomers had multiple low-energy
conformations. Entropy contributions were largely not considered in the calculations
performed here. However, it is likely that in the case of the (R*, R*) isomers described
above and in other macrocycles having several conformations with low energies there
may be significant entropic contributions due to the interconversion of those low-lying
conformations.

C H A P T E R 5
239
E X P E R I M E N T A L
5.1.1 General
Unless otherwise stated, all reactions were performed under a nitrogen
atmosphere in flame- or oven-dried glassware. Elevated temperature reactions were
performed in either a silicone oil bath or with a Glas-Col heating mantle heated to the
desired temperature. Low temperature reactions were performed in a cold bath
prepared as follows: -78 °C (dry ice, acetone), -40 °C (dry ice, acetonitrile), -20 °C (dry
ice, carbon tetrachloride), 0 °C (ice, water).
Anhydrous solvents were obtained by distillation. Diethyl ether, tetrahydrofuran
(THF) and toluene were distilled from sodium. Methylene chloride was distilled from
calcium hydride. Dimethyl formamide and dimethyl sulphoxide were distilled at reduced
pressure from calcium hydride. The low boiling fraction of petroleum ether
(bp 35-60 °C) was used. Toluene and 1,2-dichloroethane were deoxygenated with the
freeze-pump-thaw method. Methylene chloride and cyclohexane were deoxygenated
by sparging with nitrogen for 30 minutes. Otherwise the solvent was used as received
from the supplier.
Reagents were purified according to the procedure given in the literature.185
Unless otherwise noted, reagents were purchased from the Aldrich Chemical Co.
Alkyllithium reagents were standardized by titration with 2,5-dimethoxybenzyl alcohol in
THF at 0 °C to a faint red colour indicative of the endpoint.186 Urea hydrogen peroxide
(UHP) was either purchased from Aldrich or prepared by the method of Lu, Hughes,
and Giguere.187 Tri(n-butyl)tin hydride was either purchased from Aldrich or prepared
by the method of Kuivila and Beumel.188 The Tebbe reagent 32 was prepared
according to the method of Cannizzo and Grubbs.153 Adams' Catalyst (Pt02) was
purchased from BDH Chemicals Ltd. Pyridinium p-toluenesulfonate was prepared by
the method of Miyashita, Yoshikoshi, and Grieco.147 Zinc-copper couple was prepared

240
according to the method of Shank and Shechter.189 The Grubbs' catalyst 9 was
prepared by Mr. Andre Hodder according to the method of Schwab, Grubbs, and
Ziller.166 The Jones reagent was prepared via the method of Eisenbraun.179
Analytical gas-liquid chromatography (GC) was performed on a Hewlett-Packard
model 5880A gas chromatograph, equipped with a split mode capillary injection system
and a flame ionization detector. The stationary phase consisted of a either an OV-101
or a DB-210 capillary column of dimensions 0.22 mm x 12 m. Chiral GC columns
Cyclodex-B (Chromatographic Specialties Inc.) and |3-Dex 360 (Supelco) both having
dimensions of 0.25 mm x 30 m were also employed. Helium was used as the carrier
gas in all cases.
The concentration or evaporation of solvents under reduced pressure refers to
the use of a Buchi rotary evaporator. A brine solution refers to a saturated NaCl
solution. Thin layer chromatography (TLC) was performed on commercially available
aluminum backed plates of silica gel 60 (Merck 5554, 0.2 mm thickness). TLC plates
were visualized with ultraviolet light (254 nm) or 1% p-anisaldehyde spray. Flash
chromatography190 was performed using silica gel 60, 230-400 mesh, supplied by
E. Merck Co. In most cases a solvent system was chosen such that the desired
product had an Rf of approximately 0.30-0.35 on TLC. Radial chromatography was
performed using a Harrison Chromatotron model 8924. The adsorbant used was silica
gel 60, PF254 with gypsum binder supplied by EM Science. In most cases a solvent
system was chosen such that the desired product had an Rf of approximately 0.10-0.20
on TLC.
Melting points were performed using a Mel-Temp II apparatus (Lab Devices
USA) and are uncorrected. Isomerization reactions were performed under photolysis
conditions with the sample immersed in a 0.0014 M K2Cr04 solution and a 450 W
Hanovia medium pressure mercury vapour lamp.
Infrared (IR) spectra were recorded on a Bomem Michelson 100 FT-IR
spectrometer with internal calibration. IR spectra were taken on either

241
deuteriochloroform or carbon tetrachloride solutions held between two NaCl plates of
4 mm thickness with an internal well of 0.2 mm thickness.
Proton nuclear magnetic resonance (1H NMR) spectra were recorded on either
deuteriochloroform or benzene-cf6 solutions using a Bruker WH-400 (400 MHz), or a
Bruker AMX-500 (500 MHz) spectometer. Chemical shifts are given in parts per million
(ppm) on the 8 scale, referenced to chloroform (8 7.24) or benzene (8 7.15) as internal
standard. Signal multiplicity, coupling constants, and integration ratios are indicated in
parentheses. The abbreviations used to denote NMR signal multiplicities are as
follows: br (broad), s (singlet), d (doublet), t (triplet), q (quartet), quint (quintet), sext
(sextet), m (multiplet), dd (doublet of doublets), dt (doublet of triplets), ddd (doublet of
doublet of doublets), etc.
Proton and carbon dynamic nuclear magnetic resonance spectra were recorded
on Freon 21 (CHCI2F) and Freon 22 (CHCIF2) (4:1) solutions using a Bruker AMX-500
(1H, 500 MHz, 13C, 125 MHz) spectrometer. Carbon nuclear magnetic resonance
(13C NMR) spectra were recorded on either deuteriochloroform or benzene-d6 solutions
using a Bruker AMX-500 (125 MHz) spectrometer. Chemical shifts are given in parts
per million (ppm) on the 8 scale, using deuteriochloroform (8 77.0) or benzene-d6
(8 128.0) as internal standard.
Low resolution mass spectra (LRMS) in electron ionization (El) mode were
recorded on a Kratos-AEI model MS 50 spectrometer. LRMS in chemical ionization
(CI) mode were recorded on either a Kratos MS 80 spectrometer or a Kratos Concept II
HQ spectrometer. LRMS in desorption chemical ionization (DCI) mode were recorded
on a Delsi Nermag R10-10 C spectrometer. Only peaks with greater than 20% relative
intensity or those which were analytically useful are reported.
High resolution mass spectra (HRMS) in El mode were recorded on a Kratos-AEI
model MS 50 spectrometer. HRMS in CI mode were recorded on either a Kratos MS 80
spectrometer or a Kratos Concept II HQ spectrometer.

242
Microanalyses were performed by Mr. Peter. Borda in the Microanalytical
Laboratory at the University of British Columbia on a Carlo Erba Elemental Analyzer
Model 1106 or a Fisons CHN-0 Elemental Analyzer Model 1108.
5.1.2 Conformational Analysis Methods
BATCHMIN, a part of the MACROMODEL molecular modelling program
developed by Still and coworkers.191 was used to calculate the global minimum
conformations of the macrocyclic ethers studied in this work. A starting structure was
chosen, random variations to internal coordinates were applied (torsional angles), the
new structure was minimized using either the MM2* or the MM3* force field parameters,
and the result was compared with conformations found during previous conformational
search steps. After this resulting structure had been either stored as a new unique
conformer or rejected as a duplicate, the cycle was repeated. This method is known as
the Monte Carlo Multiple Minimum Search (MCMM). The MM2* and MM3* force fields
are based on the MM2192 and MM3193 parameter sets developed by Allinger and
coworkers.
5.1.3 Chemical Methods
13-Tridecanolide (87)
(a) Baever-Villiaer Oxidation of Cvclotridecanone (86) with 70% Hydrogen Peroxide
Trifluoroacetic anhydride (0.44 mL, 3.1 mmol), was added to a solution of 70% H202
(0.10 mL, 3.1 mmol) in CH2CI2 (6 mL) at -10 °C and the reaction was stirred for

243
45 minutes. A solution of cyclotridecanone (86) (61 mg, 0.31 mmol) in CH2CI2 (3 mL)
was added dropwise via syringe followed by solid Na2HP04 (0.22 g, 1.6 mmol), and the
resultant mixture was stirred for three hours. The reaction was diluted with CH2CI2,
sequentially washed with 10% KOH solution, 10% Na2S03 solution and brine, and dried
over anhydrous MgS04. The extracts were filtered and the solvent was removed under
reduced pressure. Column chromatography of the residue with 2% ethyl acetate in
petroleum ether as eluant gave lactone 87 (24 mg, 39%) as a pale yellow oil.
IR(CDCI3): 2934, 2861, 1719, 1447, 1251, 1051 cm'1; 1H NMR (500 MHz, CDCI3): 5 4.11-4.13 (m, 2 H), 2.34-2.36 (m, 2 H), 1.60-1.66 (m, 4 H),
1.22-1.44 (m, 16 H);
13C NMR (125 MHz, CDCI3): 5 173.92, 63.25, 34.36, 27.64, 26.22, 26.04, 25.86, 25.66,
24.72, 24.63, 24.03, 23.70, 22.77;
LRMS (El) m/z (relative intensity): 212 (M+, 32), 194 (39), 152 (79), 137 (32), 124 (64),
110 (100), 98 (94), 83 (44);
HRMS (El) m/z calculated for Ci3H2402: 212.1776, found: 212.1775;
Analysis calculated for Ci3H2402: C, 73.54; H, 11.39. Found: C, 73.40; H, 11.43.
(b) Baeyer-ViNiger Oxidation of Cyclotridecanone (86) with UHP
Trifluoroacetic anhydride (1.9 mL, 14 mmol) was added via syringe to a mixture of
cyclotridecanone (86) (0.42 g, 2.1 mmol), urea hydrogen peroxide (1.20 g, 12.8 mmol),
and Na2HP04 (2.09 g, 14.7 mmol) in CH2CI2 (20 mL) stirred at 0 °C and the reaction
was stirred for 18 hours with slow warming to rt. The reaction was diluted with CH2CI2,
sequentially washed with saturated NaHC03 solution, saturated Na2S203 solution,
water and brine, and dried over anhydrous MgS04. The extracts were filtered and the
solvent was removed under reduced pressure to give lactone 87 (0.43 g, 96%) as a
pale yellow oil with spectral data in agreement with that reported above. This material
was used in subsequent reactions without further purification.

244
2-Oxacyclotetradecanethione (88)
A solution of lactone 87 (0.21 g, 0.97 mmol) in toluene (5 mL) was added via cannula to
a suspension of Lawesson's reagent 48 (0.87 g, 2.2 mmol) in toluene (5 mL) and the
reaction was heated at reflux for 4.5 days. The reaction was cooled to rt, filtered
through cotton, and the solid residue was rinsed with diethyl ether. The organic layers
were combined, and the solvent was removed under reduced pressure. Column
chromatography of the residue with 2% ethyl acetate in petroleum ether as eluant gave
thionolactone 88 (0.16 g, 73%) as a yellow oil.
IR(CDCI3): 3154, 2908, 1445, 1273, 1198, 1019, 829 cm"1; 1H NMR (500 MHz, CDCI 3): 5 4.46-4.48 (m, 2 H), 2.85-2.88 (m, 2 H), 1.74-1.82 (m, 2 H),
1.67-1.73 (m, 2 H), 1.43-1.48 (m, 2 H), 1.16-1.39 (m, 14 H);
1 3 C NMR (125 MHz, CDCI 3): 5 224.66, 71.37, 47.14, 27.25, 27.00, 26.09, 25.97, 25.84,
25.03, 24.38 (2), 23.39, 23.19;
LRMS (El) m/z (relative intensity): 228 (M+, 4), 195 (21), 95 (48), 83 (47), 69 (68), 55
(100);
HRMS (El) m/z calculated for Ci3H24OS: 228.1548, found: 228.1547;
Analysis calculated for Ci3H24OS: C, 68.37; H, 10.59. Found: C, 68.12; H, 10.54.

245
2-(Methylthio)oxacyclotetradecane (89)
A solution of lithium triethylborohydride in THF (1.8 mL, 1.8 mmol) was added to a
solution of thionolactone 88 (81 mg, 0.36 mmol) in THF (5 mL) at -78 °C and the
reaction was stirred for 30 minutes. Methyl iodide (0.14 mL, 2.2 mmol) was added, and
the reaction was stirred for a further 30 minutes at -78 °C, and then allowed to slowly
warm to rt. The solution was diluted with diethyl ether and cooled to -78 °C. Aqueous
3M NaOH solution (3 mL) and 30% H202 (1.5 mL) were added sequentially. The
solution was stirred for 20 minutes at -78 °C and then allowed to warm to rt. The
reaction was sequentially washed with saturated Na2S203 solution, water, and brine,
and dried over anhydrous MgS04. The extracts were filtered and the solvent was
removed under reduced pressure to give mixed thioacetal 89 (79 mg, 91%) as an oil.
Thioacetal 89 was unstable and was used immediately without further purification.
LRMS (DCI(+), ammonia) m/z: 262 (M++18), 245 (M++1);
HRMS (El) m/z calculated for C14H28OS: 244.1861, found: 244.1856.

246
Oxacyclotetradecane (90)
A deoxygenated solution of tri(n-butyl)tin hydride (0.38 mL, 1.4 mmol) and AIBN
(10 mg) in toluene (2.6 mL) was added over three hours via syringe pump to a
deoxygenated solution of mixed thioacetal 89 (57 mg, 0.23 mmol) and AIBN (5 mg) in
toluene (15 mL) heated at reflux. After the addition of the tri(n-butyl)tin hydride, the
solvent was removed under reduced pressure. The tin compounds were removed by
column chromatography of the residue with 2% ethyl acetate in petroleum ether.
Further column chromatography using AgN03 impregnated silica with petroleum ether
as eluant gave ether 90 (20 mg, 43%) as a pale yellow oil.
IR(CDCI3): 3932, 2860, 1442, 1351, 1266, 1119, 1038 cm"1;
1H NMR (500 MHz, CDCI3): 5 3.41 (t, J = 5.5 Hz, 4 H), 1.57 (quint, J = 5.5 Hz, 4 H),
1.29-1.43 (m, 16 H), 1.21-127 (m, 2 H);
13C NMR (125 MHz, CDCI3): 5 68.58 (2), 28.59 (2), 26.34 (2), 25.15 (2), 24.37 (2),
23.42 (2), 23.19;
LRMS (DCI(+), ammonia) m/z (relative intensity): 216 (M++18, 97), 199 (M++1, 100);
HRMS (El) m/z calculated for Ci3H260: 198.1984, found: 198.1991;
Analysis calculated for Ci3H260: C, 78.72; H, 13.21. Found: C, 79.08; H, 13.18.

247
2-Methyl-2-(methylthio)oxacyclotetradecane (91)
Methyllithium in diethyl ether (1.7 mL, 1.7 mmol) was added to a solution of
thionolactone 88 (73 mg, 0.32 mmol) in THF (5 mL) at -78 ° C and the reaction was
stirred for 40 minutes at -78 ° C . Methyl iodide (0.12 mL, 1.9 mmol) was added, the
reaction was stirred for an additional 20 minutes at -78 ° C , and was finally allowed to
warm to rt. The reaction was diluted with diethyl ether, and sequentially washed with
water and brine, and dried over anhydrous MgS04. The extracts were filtered and the
solvent was removed under reduced pressure to give the mixed thioketal 91 (74 mg,
90%) as a pale yellow oil. Thioketal 91 was unstable and was used immediately
without further purification.
LRMS (El) m/z (relative intensity): 258 (M+, 1), 211 (26), 97 (22), 83 (30), 71 (35), 59
(100), 43 (65);
HRMS (El) m/z calculated for Ci5H30OS: 258.2018, found: 258.2027.

248
2-Methyloxacyclotetradecane (92)
A deoxygenated solution of tri(/7-butyl)tin hydride (2.2 mL, 8.2 mmol) and AIBN (10 mg)
in toluene (7.8 mL) was added over ten hours via syringe pump to a deoxygenated
solution of mixed thioketal 91 (0.21 g, 0.82 mmol) and AIBN (5 mg) in toluene (10 mL)
heated at reflux. After addition of the tri(n-butyl)tin hydride solution, the solvent was
removed under reduced pressure. Column chromatography of the residue with 2%
ethyl acetate in petroleum ether as eluant removed the tin compounds. Further column
chromatography using AgN03 impregnated silica with petroleum ether as eluant gave
ether 92 (0.11 g, 63%) as a pale yellow oil.
IR(CDCI3): 2929, 2859, 1459, 1372, 1340, 1130, 1098, 1039 cm"1; 1H NMR (500 MHz, CDCI3): 6 3.61 (dt, J = 9.2, 4.2 Hz, 1 H), 3.43 (ddq, J = 3.1, 9.2, 6.2
Hz, 1 H), 3.22 (ddd, J = 3.0, 9.2, 10.6 Hz, 1 H), 1.65-1.73 (m, 1 H), 1.10-1.61 (m,
21 H), 1.09 (d, J = 6.2 Hz, 3 H); 13C NMR (125 MHz, CDCI3): 5 73.32, 65.99, 36.42, 29.00, 26.48, 26.22, 25.46, 25.27,
24.93, 24.66, 23.84, 23.19, 22.99, 19.82;
LRMS (El) m/z (relative intensity): 212 (M+, 4), 197 (23), 109 (25), 97 (73), 82 (100), 69
(96), 55 (97), 43 (37);
HRMS (El) m/z calculated for Ci4H280: 212.2140, found: 212.2140;
Analysis calculated for C14H280: C, 79.18; H, 13.29. Found: C, 79.23; H, 13.70.

249
(Z/E)-1-(Trimethylsiloxy)cyclotridecene (95) and (96)
OTMS
1,1,1,3,3,3-Hexamethyldisilazane (0.21 mL, 1.0 mmol) and trimethylsilyl chloride
(0.13 mL, 1.0 mmol) were added sequentially via syringe to a mixture of
cyclotridecanone (86) (0.10 g, 0.51 mmol) and lithium iodide (0.13 g, 1.0 mmol) in
CH2CI2 (5 mL), and the reaction was stirred for 19 hours in the dark at rt. Triethylamine
(0.14 mL, 1.0 mmol) was added to the reaction mixture, and it was stirred for an
additional 30 minutes. The reaction was diluted with diethyl ether, and sequentially
washed with saturated NaHC03 solution, brine and dried over anhydrous MgS04. The
extracts were filtered and the solvent was removed under reduced pressure. Column
chromatography of the residue (GC ratio 95:96, 83:17) with petroleum ether as eluant
gave silyl enol ethers 95 (86 mg, 63%) and 96 (12 mg, 9%) both as colourless oils.
95 (Z)
IR (CDCI3): 2929, 2857, 1670, 1451, 1362, 1252, 1164, 1047, 947, 850 cm"1;
1H NMR (500 MHz, C6D6): 5 4.44 (t, J = 7.3 Hz, 1 H), 2.10 (dt, J = 7.3, 6.7 Hz, 2 H),
2.01-2.03 (m, 2 H), 1.50-1.55 (m, 2 H), 1.33-1.46 (m, 16 H), 0.14 (s, 9 H);
13C NMR (125 MHz, C6D6): 6 150.17, 110.67, 36.11, 28.32, 26.82, 26.70, 26.69, 26.09,
25.94, 25.91, 25.74, 25.12, 24.87, 0.63 (3);
LRMS (El) m/z (relative intensity): 268 (M+, 15), 143 (72), 130 (92), 73 (100);
HRMS (El) m/z calculated for Ci6H32OSi: 268.2222, found: 268.2215;
Analysis calculated for Ci6H32OSi: C, 71.57; H, 12.01. Found: C, 71.71; H, 11.89.

250
96 (£)
IR (CDCI3): 2929, 2859, 2353, 1659, 1452, 1252, 1135, 859 cm'1; 1H NMR (500 MHz, C 6D 6): 5 4.66 (t, J = 7.3 Hz, 1 H), 2.17 (t, J = 6.6 Hz, 2 H), 2.04 (dt,
J = 7.3, 6.7 Hz, 2 H), 1.64-1.68 (m, 2 H), 1.32-1.45 (m, 16 H), 0.21 (s, 9 H); 1 3 C NMR (125 MHz, C 6D 6): 8 151.70, 108.47, 29.48, 29.33, 28.76 (2), 27.58, 27.22,
26.54, 25.66, 25.23, 25.21, 24.50, 0.50 (3);
LRMS (El) m/z (relative intensity): 268 (M+, 17), 143 (75), 130 (100), 73 (97);
HRMS (El) m/z calculated for Ci6H32OSi: 268.2222, found: 268.2222.
2-Methylcyclotridecanone (97)
0
(a) Ring Expansion/Alkvlation of 1-Dibromomethvlcyclododecanol (94)
A solution of n-butyllithium in hexanes (4.5 mL, 6.3 mmol) was added over 30 minutes
via syringe pump to a solution of dibromoalcohol 94 (1.07 g, 2.99 mmol) in THF (10 mL)
at -78 °C. The reaction was stirred for 30 minutes at -78 °C, warmed to 0 °C, and
stirred for 10 minutes at 0 °C. The reaction was cooled to -78 °C and HMPA (1.0 mL)
and methyl iodide (0.56 mL, 9.0 mmol) were added simultaneously. The reaction was
stirred for 30 minutes, warmed to rt, and stirred for an additional two hours at rt. The
reaction was quenched with 1 M HCI, diluted with diethyl ether, and the organic layer
was sequentially washed with saturated CuS04 solution, saturated Na2S203 solution,
brine, and dried over anhydrous MgS04. The extracts were filtered, and the solvent
was removed under reduced pressure. Column chromatography of the residue with 2%
ethyl acetate in petroleum ether as eluant, followed by recrystallization from hexanes
gave ketone 97 (65 mg, 10%) as white needles.

251
mp: 31-33 °C;
IR (CDCI3): 2933, 2862, 1703, 1595, 1491, 1214, 1017, 792 cm"1;
1H NMR (500 MHz, CDCI3): 5 2.60 (ddq, J = 3.6, 7.1, 6.9 Hz, 1 H), 2.57 (ddd, J = 3.8,
9.5, 16.4 Hz, 1 H), 2.30 (ddd, J = 3.8, 7.6, 16.4 Hz, 1 H), 1.72-1.79 (m, 1 H),
1.60-1.67 (m, 1 H), 1.46-1.53 (m, 1 H), 1.09-1.37 (m, 17 H), 1.01 (d, J = 6.9 Hz,
3H);
13C NMR (125 MHz, CDCI3): 5 215.52, 46.19, 40.17, 32.86, 26.55, 26.26, 26.09, 25.57,
25.24, 24.93, 24.36, 24.30, 22.61, 16.94;
LRMS (El) m/z (relative intensity): 210 (M+, 22), 111 (20), 98 (36), 83 (42), 69 (58), 55
(100), 41 (54);
HRMS (El) m/z calculated for Ci4H260: 210.1984, found: 210.1985;
Analysis calculated for Ci4H260: C, 79.94; H, 12.46. Found: C, 79.69; H, 12.30.
(b) MABR Mediated Alkylation of (Z/E)-1-(Trimethvlsiloxv)cvclotridecene (95) and (96)
A solution of MABR was generated by the addition of trimethylaluminum in hexanes
(6.0 mL, 12 mmol) to a solution of 4-bromo-2,6-di-fe/if-butylphenol (3.42 g, 12.0 mmol)
in CH2CI2 (24 mL) and the reaction was stirred for 2.5 hours at rt. An aliquot of the
MABR solution (33 mL, 6.6 mmol) was added to a solution of silyl enol ethers 95 and
96 (1.27 g, 4.73 mmol) in CH2CI2 (50 mL) at -40 °C and the reaction was stirred for
20 minutes. Methyl triflate (1.1 mL, 9.5 mmol) was added, and the reaction was stirred
with slow warming to rt over 15 hours. The reaction was diluted with CH2CI2, and
sequentially washed with 1 M HCI, saturated NaHC03 solution, brine, and dried over
anhydrous MgS04. The extracts were filtered and the solvent was removed under
reduced pressure. Column chromatography of the residue with 2% ethyl acetate in
petroleum ether as eluant gave ketone 97 (0.79 g, 79%) as a pale yellow oil with
spectral data in agreement with that reported above.

252
13-Tetradecanolide (98)
(a) Baever-Villiger Oxidation of 2-Methvlcvclotridecanone (97) with Hydrogen Peroxide
Trifluoroperacetic acid was generated from 70% H202 (0.50 mL, 11 mmol) and
trifluoroacetic anhydride (1.8 mL, 13 mmol) in CH2CI2 (1.3 mL) at 0 °C. An aliquot of
this peracid (0.47 mL, 1.5 mmol) was added to a mixture of ketone 97 (0.10 g,
0.49 mmol) and Na2HP04 (0.43 g, 3.1 mmol) in CH2CI2 (0.50 mL), and the reaction was
stirred for five hours at 0 °C. An additional aliquot of trifluoroperacetic acid (0.55 mL,
1.7 mmol) was added, and the reaction was stirred for a further two hours at 0 °C. The
reaction mixture was poured into water, neutralized with saturated NaHC03 solution,
and the organic layer was dried over anhydrous MgS04. The extracts were filtered and
the solvent was removed under reduced pressure. Column chromatography of the
residue with 2% ethyl acetate in petroleum ether as eluant gave impure lactone 98
(74 mg, 66%; GC ratio 98:97, 94:6) as a pale yellow oil. The unreacted ketone 97 was
inseparable from lactone 98 using either column chromatography or HPLC.
(b) Derivatization of 2-Methvlcvclotridecanone (97) into 2-Methylcvclotridecane oxime
(99)
Sodium acetate (0.19 g, 2.2 mmol) and hydroxylamine hydrochloride (0.14 g,
2.0 mmol), were added to a mixture of ketone 97 and lactone 98 (90 mg; GC ratio
98:97, 86:14) in methanol (3 mL) and the reaction was stirred for 17.5 hours at rt. The
reaction was poured into water, extracted with diethyl ether, and dried over anhydrous
MgS04. The extracts were filtered and the solvent was removed under reduced
pressure. The oxime 99 was identified by comparison to an authentic sample using

253
TLC analysis.126 Column chromatography of the residue with 2% ethyl acetate in
petroleum ether as eluant gave lactone 98 (46 mg) as a pale yellow oil.
IR(CDCI3): 2933, 2861, 1715, 1448, 1345, 1253, 1179, 1128, 1037 cm"1;
1H NMR (500 MHz, CDCI3): 8 4.98 (sext, J = 6.3 Hz, 1 H), 2.39 (ddd, J = 3.4, 9.2, 14.4
Hz, 1 H), 2.24 (ddd, J = 3.4, 8.6, 14.4 Hz, 1 H), 1.52-1.72 (m, 4 H), 1.17-1.41 (m,
16 H), 1.19 (d, J = 6.3Hz, 3 H); 13C NMR (125 MHz, CDCI3): 8 173.62, 69.93, 35.03, 34.51, 26.32, 26.19, 25.88,
25.55 (2), 24.82, 23.97, 23.87, 22.11, 20.28;
LRMS (El) m/z (relative intensity): 226 (M+, 2), 208 (14), 182 (15), 111 (29), 98 (54), 83
(49), 69 (58), 55(100), 41 (76);
HRMS (El) m/z calculated for C14H2602: 226.1933, found: 226.1927;
Analysis calculated for Ci4H2602: C, 74.29; H, 11.58. Found: C, 74.20; H, 11.45.
(c) Baeyer-Vi Niger Oxidation of 2-Methvlcyclotridecanone (97) with UHP
Trifluoroacetic anhydride (2.4 mL, 17 mmol) was added via syringe to a mixture of
ketone 97 (0.55 g, 2.6 mmol), UHP (1.47 g, 15.6 mmol) and Na2HP04 (2.58 g,
18.2 mmol) in CH2CI2 (30 mL) at 0 °C and the reaction was stirred with slow warming to
rt over 12 hours. The reaction was diluted with CH2CI2, and sequentially washed with
water, saturated Na2S203 solution, saturated NaHC03 solution, brine, and dried over
anhydrous MgS04. The extracts were filtered, and the solvent was removed under
reduced pressure. Column chromatography of the residue with 1 % ethyl acetate in
petroleum ether gave lactone 98 (0.57 g, 97%) as a pale yellow oil with spectral data in
agreement with that reported above.

2-Methylene-14-methyloxacyclotetradecane (100)
254
A solution of Tebbe reagent 3 2 3 8 1 5 3 in toluene (0.22 mL, 0.22 mmol) was added to a
solution of lactone 98 (25 mg, 0.11 mmol), DMAP (20 mg, 0.13 mmol), and pyridine
(10 pL, 1.3 pmol) stirred in THF (2 mL) at -40 °C and the reaction was warmed slowly to
rt overnight. The reaction mixture was filtered through basic alumina with petroleum
ether as eluant, and the solvent was removed under reduced pressure to give alkene
100 (21 mg, 86%) as a pale yellow oil. Enol ether 100 was unstable and was used
immediately without further purification.
IR (CDCI3): 2930, 2859, 1647, 1455, 1375, 1274, 1132 cm"1;
LRMS (El) m/z (relative intensity): 224 (M+, 100), 166 (24), 125 (20), 96 (25), 71 (24);
HRMS (El) m/z calculated for Ci5H280: 224.2140, found: 224.2138.
3-Methyl-2-oxacyclotetradecanethione (101)
A solution of lactone 98 (0.41 g, 1.8 mmol) in toluene (10 mL) was added via cannula to
a suspension of Lawesson's reagent 48 (1.46 g, 3.62 mmol) in toluene (10 mL) and the

255
reaction was heated at reflux for five days. The reaction was cooled to rt, filtered, and
the solid residue was rinsed with diethyl ether. The organic layers were combined, and
the solvent was removed under reduced pressure. Column chromatography of the
residue with petroleum ether as eluant gave thionolactone 101 (0.34 g, 77%) as a
yellow oil. Further column chromatography with 2% ethyl acetate in petroleum ether as
eluant gave recovered lactone 98 (0.07 g, 17%).
IR (CDCI3): 2930, 2860, 1455, 1357, 1289, 1181', 1094, 773 cm"1;
1H NMR (500 MHz, CDCI3): 5 5.62 (ddq, J = 7.4, 3.7, 6.3 Hz, 1 H), 2.86 (ddd, J = 5.1,
8.0, 13.0 Hz, 1 H), 2.73 (ddd, J = 4.6, 7.4, 13.0 Hz, 1 H), 1.67-1.75 (m, 4 H),
1.17-1.42 (m, 16 H), 1.30 (d, J = 6.3 Hz, 3 H); 13C NMR (125 MHz, CDCI3): 6 224.35, 78.27, 47.50, 34.83, 27.70, 26.16, 26.01, 25.80,
25.76, 25.22, 24.08, 23.77, 22.27, 19.06;
LRMS (El) m/z (relative intensity): 242 (M+, 3), 209 (30), 109 (24), 98 (38), 83 (35), 69
(66), 55(100), 41 (85);
HRMS (El) m/z calculated for Ci4H26OS: 242.1704, found: 242.1704;
Analysis calculated for Ci4H26OS: C, 69.36; H, 10.81. Found: C, 69.23; H, 10.67.
2-Methyl-2-(methylthio)-14-methyloxacyclotetradecane (102)
Methyllithium in diethyl ether (0.39 mL, 0.54 mmol) was added to a solution of
thionolactone 101 (44 mg, 0.18 mmol) in THF (5 mL) stirred at -78 °C and the reaction
was stirred for 30 minutes. Methyl iodide (36 uL, 0.58 mmol) was added and the
reaction was stirred for 15 minutes at -78 °C, warmed to rt, and stirred for an additional
15 minutes at rt. The reaction was diluted with diethyl ether, and sequentially washed

256
with water, brine, and dried over anhydrous MgS04. The extracts were filtered and the
solvent was removed under reduced pressure to give mixed thioketal 102 (39 mg, 80%)
as a pale yellow oil. Thioketal 102 was unstable and was used immediately without
further purification.
LRMS (El) m/z (relative intensity): 272 (M+, 3), 225 (61), 209 (32), 123 (26), 109 (56),
95 (70), 83 (69), 69 (85), 55 (98), 43 (100);
HRMS (El) m/z calculated for Ci 6H 3 2OS: 272.2174, found: 272.2169.
(2R*, 14/?*) and (2S*, 14/?*)-Dimethyloxacyclotetradecane (103) and (104)
(a) Reduction of 2-Methylene-14-methyloxacvclotetradecane (100) with Adams'
Catalyst
Adams' catalyst was added to a solution of alkene 100 (62 mg, 0.28 mmol) in diethyl
ether (5 mL) and the mixture was stirred under H2 overnight at rt. The reaction was
filtered through silica with diethyl ether as eluant, and the solvent was removed under
reduced pressure. Radial chromatography of the residue (GC ratio 103:104, 49:51)
with petroleum ether as eluant gave ethers 103 (8.0 mg, 13%) and 104 (7.7 mg, 13%)
both as oils.
103(2/?*, 14/?*)
IR (CDCI3): 2928, 2859, 1457, 1374, 1135, 1059 cm"1;
1H NMR (500 MHz, CDCI3): 5 3.65 (sext, J = 6.2 Hz, 2 H), 1.63 (sext, J = 6.2 Hz, 2 H),
1.18-1.43 (m, 20 H), 1.08 (d, J = 6.2 Hz, 6 H);

257
1 3 C NMR (125 MHz, CDCI 3): 8 69.02 (2), 33.64 (2), 26.56 (2), 25.34 (2), 25.15 (3),
23.13 (2), 19.63 (2);
LRMS (El) m/z (relative intensity): 226 (M+, 14), 211 (14), 182 (23), 111 (37), 97 (69),
83 (80), 69 (86), 55 (100), 41 (73);
HRMS (El) m/z calculated for Ci5H30O: 226.2297, found: 226.2294;
Analysis calculated for Ci5H30O: C, 79.58; H, 13.36. Found: C, 79.42; H, 13.37.
104 (2S*, 14/?*)
IR (CDCI3): 2928, 2860, 1458, 1371, 1330, 1123, 1051 cm"1; 1H NMR (500 MHz, CDCI 3): 8 3.54 (ddq, J = 4.2, 5.7, 6.2 Hz, 2 H), 1.18-1.49 (m, 22 H),
1.10 (d, J = 6.2 Hz, 6H); 1 3 C NMR (125 MHz, CDCI 3): 8 71.77 (2), 36.10 (2), 26.41 (2), 26.17 (2), 25.55 (2),
24.86, 22.95 (2), 21.18 (2);
LRMS (El) m/z (relative intensity): 226 (M+, 11), 211 (9), 111 (31), 97 (61), 83 (87), 69
(87), 55(100), 41 (74);
HRMS (El) m/z calculated for Ci5H30O: 226.2297, found: 226.2294.
(b) Reduction of 2-Methvl-2-(methylthio)-14-methvloxacvclotetradecane (102) with
Tri(/7-butvl)tin Hydride
A deoxygenated solution of tri(/?-butyl)tin hydride (1.5 mL, 5.5 mmol) and AIBN (cat.) in
toluene (8.5 mL) was added over ten hours via syringe pump to a deoxygenated
solution of mixed thioketal 102 (0.15 g, 0.55 mmol) and AIBN (cat.) in toluene (20 mL)
heated at reflux. The solvent was removed under reduced pressure, and column
chromatography of the residue (GC ratio 103:104, 52:48) with 1% ethyl acetate in
petroleum ether as eluant removed the tin compounds. Further radial chromatography
with petroleum ether as eluant gave ethers 103 (13.0 mg, 10%) and 104 (13.3 mg,
11%) both as pale yellow oils with spectral data in agreement with that reported above.

258
(c) Reduction of 2-Methvl-2-(methvlthio)-14-methyloxacvclotetradecane (102) with
Tris(trimethvlsilvl)silane (TTMSH)
AIBN (cat.) and TTMSH (0.54 mL, 0.17 mmol) were added to a deoxygenated solution
of mixed thioketal 102 (47 mg, 0.17 mmol) in toluene (20 mL) and the reaction was
heated at reflux for 24 hours. The solvent was removed under reduced pressure, and
column chromatography of the residue with petroleum ether as eluant followed by 1%
ethyl acetate in petroleum ether as eluant gave ethers 103 and 104 (17 mg, 43%; GC
ratio 103:104, 57:43) as a pale yellow oil with spectral data in agreement with that
reported above.
(Z/£)-1-(Trimethylsi loxy)-2-methylcyclotridecene (105)
OTMS
1,1,1,3,3,3-Hexamethyldisilazane (0.68 mL, 3.2 mmol) and trimethylsilyl chloride
(0.41 mL, 3.2 mmol) were added sequentially via syringe to a mixture of
2-methylcyclotridecanone (97) (0.33 g, 1.6 mmol) and lithium iodide (0.43 g, 3.2 mmol)
in CH2CI2 (10 mL) at rt, and the reaction was stirred for three days in the dark.
Triethylamine (0.45 mL, 3.2 mmol) was added and the reaction was stirred for
30 minutes. The reaction was diluted with diethyl ether, and sequentially washed with
saturated NaHC03 solution, brine, and dried over anhydrous MgS04. The extracts
were filtered and the solvent was removed under reduced pressure to give a mixture of
silyl enol ethers (0.43 g, 94%) as a pale yellow oil. This mixture was used without
purification in the subsequent reaction.

259
LRMS (El) m/z (relative intensity): 282 (M\ 9), 157 (22), 144 (65), 129 (8), 73 (100), 41
(21);
HRMS (El) m/z calculated for Ci7H34OSi: 282.2379, found: 282.2376.
2,2-Dimethylcyclotridecanone (106)
(0.43 g, 1.5 mmol) in CH2CI2 (10 mL) at -40 °C and the reaction was stirred for
20 minutes. Methyl triflate (0.34 mL, 3.0 mmol) was added, and the reaction was
stirred with slow warming to rt overnight. The reaction was diluted with diethyl ether,
and sequentially washed with 1 M HCI, water, saturated NaHC03 solution, brine, and
dried over anhydrous MgS04. The extracts were filtered and the solvent was removed
under reduced pressure. Column chromatography of the residue with 2% ethyl acetate
in petroleum ether as eluant followed by radial chromatography with petroleum ether as
eluant gave ketone 106 (0.11 g, 33%) as a pale yellow oil.
1H NMR (500 MHz, CDCI3): 8 2.48-2.51 (m, 2 H), 1.61-1.66 (m, 2 H), 1.47-1.51 (m, 2 H),
1.20-1.34 (m, 16 H), 1.09 (s, 6 H);
13C NMR (125 MHz, CDCI3): 8 216.09, 47.77, 40.75, 35.63, 26.83, 26.61, 26.52, 25.30,
25.12, 24.62 (2), 24.38, 24.33, 22.14, 21.76;
LRMS (El) m/z (relative intensity): 224 (M+, 19), 111 (17), 97 (29), 83 (32), 69 (67), 56
(83), 55 (80), 41 (100);
HRMS (El) m/z calculated for Ci5H280: 224.2140, found: 224.2142.
O
A solution of MABR was generated by the addition of trimethylaluminum in hexanes
(3.0 mL, 6.0 mmol) to a solution of 4-bromo-2,6-di-fe/t-butylphenol (1.71 g, 6.00 mmol)
in CH2CI2 (12 mL) and the reaction was stirred for one hour at rt. An aliquot of the
MABR solution (12 mL, 2.3 mmol) was added to a solution of silyl enol ethers 105

260
Methyl 11-bromoundecanoate (110)
Concentrated sulfuric acid (3 mL) was added to a solution of 11-bromoundecanoic acid
109 (19.27 g, 76.72 mmol) in methanol (100 mL) and the solution was heated at reflux
for nine hours. The solvent was removed under reduced pressure, and the resultant oil
was diluted with diethyl ether. The ether solution was washed sequentially with
saturated NaHC03 solution, water, brine, and dried over anhydrous MgS04. The
extracts were filtered and the solvent was removed under reduced pressure. Column
chromatography of the residue with diethyl ether as eluant gave ester 110 (25.98 g,
82%) as a pale yellow oil. This material was used in subsequent reactions without
further purification. Column chromatography of a sample of 110 (ca. 100 mg) with
2% ethyl acetate in petroleum ether as eluant gave pure 110 with spectral data in
agreement with that reported earlier in our laboratory.140
IR(CCU): 2931, 2857, 1741, 1461, 1437, 1360, 1174 cm"1;
1H NMR (500 MHz, CDCI3): 5 3.62 (s, 3 H), 3.36 (t, J = 7.0 Hz, 2 H), 2.25 (t, J = 7.5 Hz,
2 H), 1.80 (quint, J = 7.0 Hz, 2 H), 1.54-1.60 (m, 2 H), 1.35-1.40 (m, 2 H), 1.22-
1.29 (m, 10 H);
13C NMR (125 MHz, CDCI3): 5 174.15, 51.32, 34.01, 33.85, 32.76, 29.27, 29.23, 29.11,
29.04, 28.65, 28.08, 24.86;
LRMS (El) m/z (relative intensity): 280 (81Br, M+, 1), 278 (79Br, M+, 1), 249 (2), 247 (2),
199 (7), 87 (45), 74 (100), 55 (23), 41 (20);
HRMS (El) m/z calculated for; calculated for Ci2H2302
81Br: 280.0861, found: 280.0855;
Ci2H2302
79Br: 278.0881, found: 278.0875.

261
Methyl 12-carbomethoxy-13-oxotetradecanoate (111)
Methyl acetoacetate (20.1 mL, 186 mmol) was added dropwise to a suspension of
sodium hydride (7.44 g, 186 mmol) in a mixture of THF and DMF (3:1, 400 mL) at rt.
After the effervescence had subsided, ester 110 (25.98 g, 93.04 mmol) was added to
the reaction over three hours, and the mixture was heated at reflux for two days. The
resultant solution was concentrated under reduced pressure, diluted with CH2CI2, and
sequentially washed with 1 M HCI, water, brine, and dried over anhydrous MgS04. The
extracts were filtered and the solvent was removed under reduced pressure to give
crude diester 111 (32 g, 109%). This material was used in subsequent reactions
without further purification. Column chromatography of a sample of 111 (ca. 100 mg)
with 5% ethyl acetate in petroleum ether as eluant gave pure 111 as a white solid for
analysis.
mp: 42-43 °C;
IR (CCI4): 2932, 2857, 1743, 1721, 1436, 1357, 1273, 1171 cm'1;
1H NMR (500 MHz, CDCI3): 6 3.68 (s, 3 H), 3.61 (s, 3 H), 3.37 (t, J = 7.3 Hz, 1 H), 2.25
(t, J = 7.5 Hz, 2 H), 2.17 (s, 3 H), 1.78 (quint, J = 7.3 Hz, 2 H), 1.56 (quint, J =
7.5 Hz, 2 H), 1.11-1.29 (m, 14 H);
13C NMR (125 MHz, CDCI3): 5 203.15, 174.20, 170.34, 59.66, 52.22, 51.32, 34.01,
29.32, 29.27, 29.21, 29.17, 29.12, 29.04, 28.67;
LRMS (El) m/z (relative intensity): 314 (M+, 1), 283 (8), 251 (5), 129 (18), 116 (100), 98
(97), 87 (36), 69 (24), 55 (55), 43 (49);
HRMS (El) m/z calculated for Ci7H3o05: 314.2093, found: 314.2090;
Analysis calculated for C17H30O5: C, 64.94; H, 9.62. Found: C, 64.82; H, 9.66.

262
13-Oxotetradecanoic acid (112)
A solution of diester 111 (10.06 g, 32.00 mmol) in a mixture of concentrated HCI,
methanol, and water (3:1:1, 112 mL) was heated at reflux for nine hours. The reaction
was cooled, diluted with water, and extracted with diethyl ether. The organics were
combined, washed with brine, and dried over anhydrous MgS04. The extracts were
filtered and the solvent was removed under reduced pressure to give keto acid 112
(6.71 g, 87%) as a white solid. This material was used in subsequent reactions without
further purification. Column chromatography of a sample of 112 (ca. 100 mg) with
4% methanol in CH2CI2 gave pure 112 with spectral data in agreement with that
reported earlier in our laboratory.140
mp: 64-66 °C;
IR(CCI4): 3045, 2929, 2856, 1713, 1433, 1359, 1289, 1166 cm"1;
1H NMR (500 MHz, CDCI3): 6 2.38 (t, J = 7.3 Hz, 2 H), 2.30 (t, J = 7.5 Hz, 2 H), 2.10 (s,
3 H), 1.49-1.62 (m, 4 H), 1.13-1.32 (m, 14 H);
13C NMR (125 MHz, CDCI3): 5 209.52, 179.76, 43.76, 34.01, 29.74, 29.42, 29.34, 29.30
(2), 29.14, 29.11, 28.99, 24.64, 23.83;
LRMS (El) m/z (relative intensity): 242 (M+, 1), 224 (2), 98 (18), 83 (21), 81 (13), 69
(33), 67 (18), 58 (100), 43 (88), 41 (26);
HRMS (El) m/z calculated for Ci4H2603: 242.1882, found: 242.1885.

263
13-Hydroxy-13-methyltetradecanoic acid (113)
A solution of methylmagnesium bromide in diethyl ether (5.2 mL, 16 mmol) was added
to a solution of keto acid 112 (1.27 g, 5.24 mmol) in diethyl ether (20 mL) at 0 °C and
the reaction was stirred with slow warming to rt overnight. The reaction mixture was
diluted with diethyl ether, and acidified with 1 M HCI. The organic layer was
sequentially washed with water and brine, and dried over anhydrous MgS04. The
extracts were filtered and the solvent was removed under reduced pressure. Column
chromatography of the residue with 20% ethyl acetate in petroleum ether as eluant
gave hydroxy acid 113 (0.58 g, 43%) as a white solid.
mp: 50-52 °C;
IR (CDCI3): 3607, 2930, 2856, 1709, 1195, cm"1;
1H NMR (500 MHz, CDCI3): 6 2.31 (t, J = 7.5 Hz, 2 H), 1.61 (quint, J = 7.5 Hz, 2 H),
1.44 (t, J = 6.1 Hz, 2H), 1.23-1.34 (m, 16 H), 1.19 (s, 6 H); 13C NMR (125 MHz, CDCI3): 5 179.16, 71.31, 43.92, 34.02, 30.10, 29.45 (3), 29.32,
29.13 (2), 28.99, 24.69, 24.27;
LRMS (DCI(+), ammonia) m/z (relative intensity): 276 (M++18, 100), 259 (M++1, 32),
258 (M+, 100);
HRMS (Cl(+), isobutane) m/z calculated for C 1 5 H 3 1 O 3 (M++1) 259.2273, found:
259.2272;
Analysis calculated for Ci5H3o03: C, 69.72; H, 11.70. Found: C, 69.58; H, 11.58.

264
13-Methyl-13-tetradecanolide (114)
Triethylamine (0.28 mL, 2.0 mmol) was added to a solution of hydroxy acid 113 (0.46 g,
1.8 mmol) in THF (20 mL) at rt and the reaction was stirred for 15 minutes.
2,4,6-Trichlorobenzoyl chloride (0.28 mL, 1.8 mmol) was added and the reaction was
stirred for a further two hours. The reaction mixture was filtered and concentrated
under reduced pressure. Trace amounts of solvent were removed under high vacuum
over one hour. A solution of the resultant mixed anhydride in toluene (100 mL) was
divided into two portions and simultaneously added via syringe pump to two solutions
of DMAP (0.88 g, 7.2 mmol) in toluene (600 mL) at reflux over 40 hours. The reaction
was concentrated under reduced pressure, diluted with diethyl ether, and sequentially
washed with water, 1 M HCI, saturated NaHC03 solution, brine, and dried over
anhydrous MgS04. The extracts were filtered and the solvent was removed under
reduced pressure. Column chromatography of the residue with 1 % ethyl acetate in
petroleum ether gave lactone 114 (0.16 g, 54%) as a colourless oil.
IR(CCU): 2932, 2861, 1727, 1462, 1385, 1368, 1200, 1174, 1150, 1082 cm"1;
1H NMR (500 MHz, CDCI3): 5 2.15-2.17 (m, 2 H), 1.76-1.79 (m, 2 H), 1.48-1.53 (m, 2 H),
1.21-1.40 (m, 16 H), 1.35 (s, 6 H);
13C NMR (125 MHz, CDCI3): 6 172.14, 81.89, 38.56, 34.55, 27.16 (2), 26.98, 26.72,
26.64, 26.07, 25.55, 24.86, 24.03, 23.65;
LRMS (El) m/z (relative intensity): 240 (M+, 51), 225 (39), 182 (69), 167 (19), 125 (20),
111 (39), 98 (52), 83 (54), 69 (100), 55 (83), 41 (63);
HRMS (El) m/z calculated for Ci5H2802: 240.2089, found: 240.2084;
Analysis calculated for Ci5H2802: C, 74.95; H, 11.74. Found: C, 75.13; H, 11.74.

265
2,2-Dimethyloxacyclotetradecane (116)
Boron trifluoride etherate (0.88 mL, 7.0 mmol) and sodium borohydride (0.06 g,
1.6 mmol) were added to a solution of lactone 114 (56.1 mg, 0.233 mmol) in THF
(2 mL) at rt and the reaction was stirred for 45 minutes. Triglyme (1.0 mL) was added
and the reaction was stirred for 16 hours at rt. The reaction was quenched with
saturated NaHC03 solution, and diluted with diethyl ether. The ether layer was
separated and was sequentially washed with saturated NaHC03 solution, water, brine,
and dried over anhydrous MgS04. The extracts were filtered and the solvent was
removed under reduced pressure to give crude ether 116 (37.1 mg, 70%). This
material was combined with additional crude 116 (29.2 mg, 64%) obtained from lactone
114 (48.7 mg, 0.203 mmol) in a second reaction carried out under similar conditions.
Radial chromatography of the combined residue with 0.5% ethyl acetate as eluant gave
ether 116 (50.6 mg, 51%) as a colourless oil.
IR (CCI4): 2928, 2860, 1462, 1381, 1363, 1276, 1202, 1179, 1088, 1031 cm"1;
1H NMR (500 MHz, CDCI3): 5 3.25 (t, J = 6.4 Hz, 2 H), 1.57 (quint, J = 6.4 Hz, 2 H),
1.23-1.43 (m, 20 H), 1.13 (s, 6 H);
13C NMR (125 MHz, CDCI3): 5 73.88, 58.88, 37.97, 28.34, 26.69, 26.66 (2), 26.39,
26.15, 25.15, 24.56, 24.50, 23.52, 23.38, 20.33;
LRMS (El) m/z (relative intensity): 226 (M+, 1), 211 (22), 97 (12), 83 (15), 69 (19), 59
(100), 55 (21);
HRMS (El) m/z calculated for C15H30O: 226.2297, found: 226.2295.

266
2-Methyl-13-tridecanolide (117)
A solution of n-butyllithium in hexanes (8.0 mL, 20 mmol) was added to a solution of
diisopropylamine (3.0 mL, 23 mmol) in THF (9.0 mL) at -78 °C and the reaction was
stirred for 15 minutes, warmed to 0 °C, and stirred for an additional 15 minutes. An
aliquot of this LDA solution (2.6 mL, 2.6 mmol) was added to a solution of lactone 87
(0.43 g, 2.0 mmol) in THF (5 mL) and the reaction was stirred for four hours at -78 °C.
Methyl iodide (0.25 mL, 3.0 mmol) was added, the reaction was stirred for 15 minutes
at -78 °C, warmed to rt, and stirred for an additional 15 minutes at rt. The reaction was
diluted with diethyl ether, and sequentially washed with water, brine, and dried over
anhydrous MgS04. The extracts were filtered and the solvent was removed under
reduced pressure. Column chromatography of the residue with 2% ethyl acetate in
petroleum ether gave lactone 117 (0.38 g, 84%) as a pale yellow oil.
IR(CCU): 2934, 2860, 1732, 1461, 1349, 1170, 1088 cm"1;
1H NMR (500 MHz, CDCI3): 5 4.24 (dt, J = 11.0, 5.5 Hz, 1 H), 3.97 (dt, J = 11.0, 5.3 Hz,
1 H), 2.51 (ddq, J = 3.2, 9.8, 7.0 Hz, 1 H), 1.55-1.65 (m, 4 H), 1.15-1.48 (m, 16
H), 1.12 (d, J = 7.0Hz, 3H);
13C NMR (125 MHz, CDCI3): 8 176.87, 63.16, 39.70, 33.79, 27.73, 26.17, 26.05, 26.04,
24.65, 24.47, 24.00, 23.59, 22.82, 17.61;
LRMS (El) m/z (relative intensity): 226 (M+, 5), 208 (4), 117 (37), 97 (45), 87 (23), 83
(53), 74 (81), 69 (68), 55 (100), 42 (82);
HRMS (El) m/z calculated for Ci4H2602: 226.1933, found: 226.1930.

267
2,2-Dimethyl-13-tridecanolide (118)
An aliquot of LDA solution (3.0 mL, 3.0 mmol) (see 117) was added to a solution of
lactone 117 (0.34 g, 1.5 mmol) in THF (5 mL) at -78 °C and the reaction was stirred for
nine hours at -78 °C. Methyl iodide (0.28 mL, 4.5 mmol) was added and the reaction
was stirred for 15 minutes at -78 °C, warmed to rt, and stirred for an additional
15 minutes. The reaction was diluted with diethyl ether, and was washed with water,
brine, and dried over anhydrous MgS04. The extracts were filtered and the solvent was
removed under reduced pressure. Column chromatography of the residue with 2%
ethyl acetate in petroleum ether gave lactone 118 (0.31 g, 86%) as a pale yellow oil.
IR(CCU): 2934, 2861, 1728, 1464, 1390, 1321, 1162, 1136 cm"1;
1H NMR (500 MHz, CDCI3): 6 4.04-4.07 (m, 2 H), 1.62-1.66 (m, 2 H), 1.45-1.48 (m, 2 H),
1.26-1.41 (m, 12 H), 1.13-1.22 (m, 4 H), 1.15 (s, 6 H); 13C NMR (125 MHz, CDCI3): 5 178.21, 63.20, 42.72, 40.68, 28.03, 26.53, 26.18, 25.92,
25.68 (2), 24.32, 23.69, 22.71, 22.62, 22.47;
LRMS (El) m/z (relative intensity): 240 (M+, 62), 222 (15), 153 (42), 97 (29), 88 (100),
83 (31), 69 (38), 55 (32);
HRMS (El) m/z calculated for Ci5H 2 80 2: 240.2089, found: 240.2086;
Analysis calculated for C15H2802: C, 74.95; H, 11.74. Found: C, 74.93; H, 11.92.

268
3,3-Dimethyloxacyclotetradecane (119)
Boron trifluoride etherate (2.0 mL, 16 mmol) and sodium borohydride (0.14 g, 3.6 mmol)
were added to a solution of lactone 118 (125 mg, 0.520 mmol) in THF (5.0 mL) and the
reaction was stirred for 40 minutes at rt. Triglyme (2.0 mL) was added and the reaction
was heated at reflux for three hours. The reaction was diluted with diethyl ether, and
was quenched with saturated NaHC03 solution. The ether layer was sequentially
washed with saturated NaHC03 solution, water, brine, and dried over anhydrous
MgS04. The extracts were filtered and the solvent was removed under reduced
pressure. Column chromatography of the residue with 0.5% ethyl acetate in petroleum
ether as eluant gave ether 119 (25 mg, 11%) as a pale yellow oil.
IR (CCI4): 2935, 2859, 1463, 1382, 1118 cm"1;
1H NMR (500 MHz, CDCI3): 8 3.38 (t, J = 5.4 Hz, 2 H), 3.03 (s, 2 H), 1.55 (quint, J =
5.4 Hz, 2 H), 1.18-1.42 (m, 18 H), 0.84 (s, 6 H);
13C NMR (125 MHz, CDCI3): 8 77.38, 68.81, 37.43, 34.09, 28.81, 26.79, 26.61, 26.12
(2), 25.79, 24.17, 24.07, 22.84, 22.81, 20.39;
LRMS (Cl(+), ammonia) m/z (relative intensity): 244 (M++18, 30), 227 (M++1, 100);
HRMS (Cl(+), isobutane) m/z calculated for Ci5H310 (M++1): 227.2375, found:
227.2374;
Analysis calculated for C15H30O: C, 79.58; H, 13.36. Found: C, 79.51; H, 13.39.

269
8-Bromo-1-octanol (121)
48% HBr (12.1 mL, 107 mmol) was added to a solution of 1,8-octanediol (120) (10.41 g,
71.19 mmol) in benzene (300 mL) and the solution was heated at reflux under Dean-
Stark conditions for 48 hours. The organic layer was collected and concentrated under
reduced pressure. The residue was diluted with diethyl ether, and sequentially washed
with saturated NaHC03 solution, brine, and dried over anhydrous MgS04. The extracts
were filtered and the solvent was removed under reduced pressure. Column
chromatography of the residue with 20% ethyl acetate in petroleum ether as eluant
gave alcohol 121 (13.65 g, 92%) as a pale yellow oil with spectral data in agreement
with that reported in the literature.145
IR (CCI4): 3635, 3378, 2932, 2858, 1453, 1050 cm"1;
1H NMR (500 MHz, CDCI3): 5 3.57 (t, J = 6.7 Hz, 2 H), 3.35 (t, J = 6.9 Hz, 2 H), 1.85
(br s, 1 H), 1.80 (quint, J = 6.9 Hz, 2 H), 1.51 (quint, J = 6.7 Hz, 2 H), 1.35-1.41
(m, 2 H), 1.26-1.32 (m, 6 H);
13C NMR (125 MHz, CDCI3): 5 62.74, 33.89, 32.67, 32.57, 29.11, 28.60, 27.98, 25.54;
LRMS (DCI(+), ammonia) m/z (relative intensity): 228 (81Br, M++18, 100), 226 (79Br,
M++18, 98);
HRMS (Cl(+), isobutane) m/z calculated for C8H18081Br (M++1): 211.0521, found:
211.0529; calculated for C8H18079Br (M++1): 209.0541, found: 209.0537.

270
8-Bromooctanal (122)
A solution of dimethylsulfoxide (3.0 mL, 42 mmol) in CH2CI2 (10 mL) was added via
cannula to a solution of oxalyl chloride (1.8 mL, 21 mmol) in CH2CI2 (50 mL) stirred at
-78 °C. The resulting solution was stirred for two minutes and alcohol 121 (2.21 g,
10.6 mmol) in CH2CI2 (10 mL) was added via cannula and the mixture was stirred for
40 minutes. Triethylamine (7.4 mL, 53 mmol) was added and the mixture was stirred
for an additional 10 minutes then warmed to rt. The reaction was quenched with water,
and the organic layer was collected. The aqueous layer was extracted with CH2CI2.
The organic layers were combined, sequentially washed with water and brine, and
dried over anhydrous MgS04. The extracts were filtered and the solvent was removed
under reduced pressure. Column chromatography of the residue with 10% ethyl
acetate in petroleum ether as eluant gave aldehyde 122 (1.98 g, 90%) as a pale yellow
oil.
IR (CCI4): 2934, 2859, 1711, 1436, 1288, 937 cm"1;
1H NMR (500 MHz, CDCI3): 5 9.72 (t, J = 1.7 Hz, 1 H), 3.36 (t, J = 6.8 Hz, 2 H), 2.38 (dt,
J = 1.7, 7.4 Hz, 2 H), 1.81 (quint, J = 6.8 Hz, 2 H), 1.56-1.62 (m, 2 H), 1.37-1.42
(m, 2 H), 1.28-1.31 (m, 4 H);
13C NMR (125 MHz, CDCI3): 5 202.57, 43.72, 33.76, 32.58, 28.84, 28.39, 27.83, 21.83;
LRMS (DCI(+), ammonia) m/z (relative intensity): 226 (81Br, M++18, 86), 224 (79Br,
M++18, 100);
HRMS (Cl(+), isobutane) m/z calculated for C8H16081Br (M++1): 209.0364, found:
209.0373; calculated for C8Hi6079Br (M++1): 207.0385, found: 207.0377;
Analysis calculated for C8H15OBr: C, 46.39; H, 7.30. Found: C, 46.64; H, 7.25.

271
8-Bromooctanal ethylene acetal (123)
A solution of aldehyde 122 (1.82 g, 8.79 mmol), ethylene glycol (2.5 mL, 44 mmol), and
PPTS (0.45 g, 1.8 mmol) in benzene (100 mL) was heated at reflux under Dean-Stark
conditions for 12 hours. The solvent was removed under reduced pressure, and the
resultant oil was diluted with diethyl ether, sequentially washed with saturated NaHC03
solution, water, and brine, and dried over anhydrous MgS04. The extracts were filtered
and the solvent was removed under reduced pressure. Column chromatography of the
residue with 10% ethyl acetate in petroleum ether as eluant gave acetal 123 (2.06 g,
93%) as a pale yellow oil.
IR(CCU): 2936, 2861, 1461, 1407, 1136, 1039, 942 cm-1;
1H NMR (500 MHz, CDCI3): 5 4.78 (t, J = 5.0 Hz, 1 H), 3.87-3.92 (m, 2 H), 3.77-3.82 (m,
2 H), 3.34 (t, J = 6.9 Hz, 2 H), 1.80 (quint, J = 6.9 Hz, 2 H), 1.59 (ddd, J = 6.9,
9.6, 5.9 Hz, 2 H), 1.34-1.41 (m, 4 H), 1.26-1.32 (m, 4 H);
13C NMR (125 MHz, CDCI3): 5 104.48, 64.72 (2), 33.80, 33.71, 32.67, 29.20, 28.54,
27.93, 23.82;
LRMS (DCI(+), ammonia) m/z (relative intensity): 270 (81Br, M++18, 7), 268 (79Br,
M++18, 7);
HRMS (Cl(+), isobutane) m/z calculated for Ci0H2o02
81Br (M++1): 253.0626, found:
253.0627; calculated for C10H20O2
79Br (M++1): 251.0647, found: 251.0638.

272
5-(1 ',3'-Dithian-2'-yl)-1 -pentanol (125)
Boron trifluoride etherate (10.0 mL, 81.3 mmol) was added dropwise to a solution of
1,3-propanedithiol (124) (5.4 mL, 54 mmol) and dihydropyran (6.0 mL, 66 mmol) in
CH2CI2 (100 mL) at 0 °C, and the reaction was stirred for 19.5 hours with slow warming
to rt. The reaction was quenched with water, and sequentially washed with 3 M NaOH
solution, water, brine and dried over anhydrous MgS04. The extracts were filtered and
the solvent was removed under reduced pressure. Column chromatography of the
residue with 30% ethyl acetate in petroleum ether as eluant gave alcohol 125 (8.74 g,
84%) as a pale yellow oil.
IR (CCU): 3635, 2939, 2902, 1457, 1423, 1276, 1051, 909 cm"1; 1H NMR (500 MHz, CDCI3): 5 4.01 (t, J = 7.0 Hz, 1 H), 3.58-3.61 (m, 2 H), 2.75-2.86 (m,
4 H), 2.04-2.10 (m, 1 H), 1.52-1.85 (m, 7 H); 13C NMR (125 MHz, CDCI3): 5 62.43, 47.39, 35.08, 32.14, 30.35 (2), 25.91, 22.82;
LRMS (El) m/z (relative intensity): 192 (M+, 31), 119 (100), 85 (26), 45 (30), 42 (29);
HRMS (El) m/z calculated for C8Hi60S2: 192.0643, found: 192.0641.

5-(1',3'-Dithian-2'-yl)-1-(2"-tetrahydropyranyloxy)pentane (126)
273
A solution of alcohol 125 (8.73 g, 45.4 mmol), dihydropyran (5.0 mL, 55 mmol), and
PPTS (2.28 g, 9.08 mmol) in CH2CI2 (100 mL) was stirred at rt for 23 hours. The
resultant solution was sequentially washed with saturated NaHC03 solution, brine and
dried over anhydrous MgS04. The extracts were filtered and the solvent was removed
under reduced pressure. Column chromatography of the residue with 10% ethyl
acetate in petroleum ether as eluant gave dithiane 126 (12.02 g, 96%) as a pale yellow
oil with spectral data in agreement with that reported earlier in our laboratory.141
IR(CCI4): 2912, 1454, 1441, 1423, 1351, 1323, 1276, 1241, 1200, 1182, 1137, 1122,
1076, 1033, 970, 908, 869 cm"1;
1H NMR (500 MHz, CDCI3): 5 4.51 (dd, J = 4.1, 3.0 Hz, 1 H), 3.99 (t, J = 6.9 Hz, 1 H),
3.79 (ddd, J = 2.7, 7.6, 10.9 Hz, 1 H), 3.68 (dt, J = 9.7, 6.2 Hz, 1 H), 3.43 (ddd,
3.6, 5.2, 10.9 Hz, 1 H), 3.32 (dt, J = 9.7, 6.2 Hz, 1 H), 2.73-2.84 (m, 4 H), 1.43-
2.08 (m, 14 H);
13C NMR (125 MHz, CDCI3): 5 98.69, 67.02, 62.13, 47.37, 35.14, 30.60, 30.32 (2),
29.18, 25.91, 25.36, 23.31, 19.48;
LRMS (El) m/z (relative intensity): 276 (M+, 4), 191 (100), 119 (26), 85 (66), 42 (23);
HRMS (El) m/z calculated for Ci3H2402S2: 276.1218, found: 276.1211;
Analysis calculated for Ci3H2402S2: C, 56.48; H, 8.75. Found: C, 56.68; H, 8.90.

274
9-(1',3'-Dithian-2'-yl)-13-(2"-tetrahyclropyranyloxy)-tridecanal ethylene acetal (127)
A solution of n-butyllithium in hexanes (31 mL, 31 mmol) was added to a solution of
dithiane 126 (8.64 g, 31.3 mmol) in THF (50 mL) at -20 °C and the reaction was stirred
at -20 °C for five hours. A solution of bromide 123 (3.13 g, 12.5 mmol) in THF (10 mL)
was added via cannula. This reaction was stirred for one hour at -20 °C, warmed to rt,
and stirred for an additional hour at rt. The reaction was quenched with saturated
NH4CI solution and diluted with diethyl ether. The ether layer was sequentially washed
with water, brine, and dried over anhydrous MgS04. The extracts were filtered and the
solvent was removed under reduced pressure. Column chromatography of the residue
with 10% ethyl acetate in petroleum ether as eluant gave dithiane 127 (2.75 g, 49%) as
a pale yellow oil.
IR (CCU): 2917, 2863, 1458, 1354, 1276, 1132, 1077, 1034, 945, 908 cm"1;
1H NMR (500 MHz, CDCI3): 5 4.78 (dd, J = 4.8, 5.0 Hz, 1 H), 4.53 (dd, J = 3.1, 2.7 Hz, 1
H), 3.77-3.92 (m, 5 H), 3.70 (dt, J = 9.7, 6.6 Hz, 1 H), 3.42-3.47 (m, 1 H), 3.35
(dt, J = 9.7, 6.7 Hz, 1 H), 2.73-2.76 (m, 4 H), 1.76-1.91 (m, 6 H), 1.23-1.68 (m,
13C NMR (125 MHz, CDCI3): 5 104.56, 98.74, 67.14, 64.71 (2), 62.20, 53.22, 38.12,
37.90, 33.77, 30.67, 29.74, 29.60, 29.39, 29.28, 25.91 (2), 25.47, 25.41, 23.92,
23.83,20.79,19.54;
22 H);

275
LRMS (El) m/z (relative intensity): 446 (M+, 33), 361 (27), 289 (45), 275 (22), 85 (100),
73 (70);
HRMS (El) m/z calculated for C23H42O4S2: 446.2524, found: 446.2523;
Analysis calculated for C23H42O4S2: C, 61.84; H, 9.48. Found: C, 62.12; H, 9.60.
9-Oxo-13-(2'-tetrahydropyranyloxy)-tridecanal ethylene acetal (128)
Mercuric perchlorate (2.96 g, 6.52 mmol) in water (2 mL) was added to a mixture of
dithiane 127 (2.65 g, 5.93 mmol) and calcium carbonate (0.71 g, 7.1 mmol) in THF
(40 mL) and the reaction was stirred for 20 minutes at rt. The reaction was diluted with
diethyl ether and filtered. The filtrate was washed with brine, and dried over anhydrous
MgS04. The extracts were filtered and the solvent was removed under reduced
pressure. Column chromatography of the residue with 15% ethyl acetate in petroleum
ether as eluant gave ketone 128 (1.70 g, 80%) as a pale yellow oil.
IR (CCU): 2939, 2866, 1716, 1458, 1410, 1358, 1130, 1078, 1035 cm"1;
1H NMR (500 MHz, CDCI3): 8 4.78 (t, J = 4.8 Hz, 1 H), 4.51 (dd, J = 2.7, 4.0 Hz, 1 H),
3.77-3.92 (m, 5 H), 3.69 (dt, J = 9.7, 6.4 Hz, 1 H), 3.42-3.47 (m, 1 H), 3.33 (dt, J
= 9.7, 6.3 Hz, 1 H), 2.38 (t, J = 7.2 Hz, 2 H), 2.33 (t, J = 7.5 Hz, 2 H), 1.21-1.80
(m, 22 H);
13C NMR (125 MHz, CDCI3): 5 211.11, 104.57, 98.78, 67.09, 64.74 (2), 62.23, 42.70,
42.38, 33.78, 30.66, 29.25, 29.20, 29.18, 29.03, 25.40, 23.90, 23.74, 20.61,
19.56;

276
LRMS (El) m/z (relative intensity): 355 (M+, 1), 255 (18), 98 (13), 85 (71), 73 (100), 55
(17);
HRMS (El) m/z calculated for C2oH3505: 355.2484, found: 355.2488.
9-Methylene-13-(2'-tetrahydropyranyloxy)-tridecanal ethylene acetal (129)
A solution of Tebbe reagent 32 3 8 1 5 3 in toluene (18 mL, 9.3 mmol) was added via syringe
to a solution of ketone 128 (1.66 g, 4.66 mmol), DMAP (0.68 g, 5.6 mmol) and pyridine
(0.20 mL, 2.5 mmol) in THF (100 mL) stirred at -40 °C and the reaction was stirred with
slow warming to rt over 20 hours. The reaction mixture was filtered through basic
alumina with petroleum ether as eluant and the filtrate was collected. The solvent was
removed under reduced pressure and column chromatography of the residue with 5%
ethyl acetate in petroleum ether as eluant gave alkene 129 (0.88 g, 53%) as a pale
yellow oil.
IR (CCU): 2909, 1644, 1451, 1354, 1323, 1132, 1132, 1035, 945, 892 cm-1;
1H NMR (500 MHz, CDCI3): 5 4.81 (t, J = 4.8 Hz, 1 H), 4.66 (s, 2 H), 4.55 (dd, J = 2.9,
4.0 Hz, 1 H), 3.80-3.94 (m, 5 H), 3.71 (dt, J = 9.5, 6.7 Hz, 1 H), 3.45-3.49 (m, 1
H), 3.36 (dt, J = 9.5, 6.5 Hz, 1 H), 2.00 (t, J = 7.6 Hz, 2 H), 1.95 (t, J = 7.5 Hz, 2
H), 1.22-1.83 (m, 22 H);
13C NMR (125 MHz, CDCI3): 6 149.85, 108.68, 104.66, 98.76, 67.41, 64.78 (2), 62.23,
35.94, 35.76, 33.87, 30.73, 29.47, 29.42, 29.40, 29.26, 27.73, 25.48, 24.37,
24.03, 19.61;

277
LRMS (El) m/z (relative intensity): 354 (M+, 3), 269 (10), 208 (5), 155 (6), 85 (100), 73
(47), 55 (8);
HRMS (El) m/z calculated for C 2 iH 3 80 4: 354.2770, found: 354.2768;
Analysis calculated for C2iH3804: C, 71.15; H, 10.80. Found: C, 71.42; H, 10.89.
13-Hydroxy-9-methylenetridecanal (130)
A solution of alkene 129 (0.84 g, 2.4 mmol) and PPTS (0.12 g, 0.47 mmol) in acetone
and water (10:1, 50 mL) was heated at reflux for 20 hours. The acetone was removed
under reduced pressure, and the reaction mixture was diluted with diethyl ether. The
organic layer was sequentially washed with saturated NaHC03 solution, brine, and
dried over anhydrous MgS04. The extracts were filtered and the solvent was removed
under reduced pressure. Column chromatography of the residue with 30% ethyl
acetate in petroleum ether as eluant gave hydroxy aldehyde 130 (0.48 g, 90%) as a
colourless oil.
IR (CCI4): 3635, 3437, 3076, 2932, 2859, 2716, 1729, 1644, 1427, 1052, 891 cm"1;
1H NMR (500 MHz, CDCI3): 5 9.73 (t, J = 1.7 Hz, 1 H), 4.68 (s, 2 H), 3.64 (t, J = 6.4 Hz,
2 H), 2.40 (dt, J = 1.7, 7.4 Hz, 2 H), 2.01 (dt, J = 1.0, 7.5 Hz, 2 H), 1.97 (dt, J =
1.0, 7.6 Hz, 2H), 1.52-1.64 (m, 4 H), 1.25-1.51 (m, 10 H);
13C NMR (125 MHz, CDCI3): 5 202.90, 149.64, 108.87, 62.90, 43.88, 35.90, 35.72,
32.48, 29.21, 29.13, 29.09, 27.65, 23.87, 22.04;

278
LRMS (DCI(+), ammonia) m/z (relative intensity): 244 (M++18, 100), 227 (M++1, 47);
HRMS (Cl(+), isobutane) m/z calculated for Ci4H2702(M++1): 227.2011, found:
227.2012.
13-Hydroxy-9-methylenetridecanoic acid (131)
Silver nitrate (3.50 g, 20.3 mmol) and sodium hydroxide (1.64 g, 41.0 mmol) were
added to a solution of hydroxy aldehyde 130 (0.46 g, 2.03 mmol) in THF and water
(1:1, 50 mL) and the mixture was stirred at rt for six hours in the dark. The reaction
mixture was filtered and the solid residue was washed with ethyl acetate. The aqueous
layer was acidified with 1 M HCI and extracted with ethyl acetate. The organic layers
were combined, washed with brine, and dried over anhydrous MgS04. The extracts
were filtered and the solvent was removed under reduced pressure. Column
chromatography of the residue with 4% methanol in CH2CI2 gave hydroxy acid 131
(0.15 g, 30%) as a colourless oil.
IR (CCI4): 3639, 2933, 2858, 1712, 1644, 1434, 1054, 891 cm"1;
1H NMR (500 MHz, CDCI3): 6 4.68 (s, 2 H), 3.64 (t, J = 6.3 Hz, 2 H), 2.32 (t, J = 7.4 Hz,
2 H), 2.01 (t, J = 7.5 Hz, 2 H), 1.97 (t, J = 7.6 Hz, 2 H), 1.61 (quint, J = 7.4 Hz,
2 H), 1.45-1.57 (m, 4 H), 1.23-1.41 (m, 8 H);
13C NMR (125 MHz, CDCI3): 5 179.13, 149.67, 108.89, 62.84, 35.84, 35.71, 32.34,
29.03, 28.96 (2), 28.87, 27.56, 24.65, 23.88;
LRMS (DCI(+), ammonia) m/z (relative intensity): 260 (M++18, 93), 243 (M++1, 100);

279
HRMS (Cl(+), isobutane) m/z calculated for Ci4H2703 (M++1): 243.1960, found:
243.1961;
Analysis calculated for Ci4H2603. C, 69.38; H, 10.81. Found: C, 69.52; H, 11.00.
9-Methylene-13-tridecanolide (132)
Triethylamine (49 uL, 0.35 mmol) was added to a solution of hydroxy acid 131 (76 mg,
0.31 mmol) in THF (31 mL) and the reaction was stirred for 15 minutes at rt.
2,4,6-Trichlorobenzoyl chloride (48 uL, 0.31 mmol) was added and the reaction was
stirred for an additional two hours. The reaction was filtered and concentrated under
reduced pressure. Trace amounts of solvent were removed under high vacuum over
two hours. A solution of the resultant mixed anhydride in toluene (150 mL) was added
via syringe pump to a solution of DMAP (0.23 g, 1.9 mmol) in toluene (31 mL) heated at
reflux over six hours. The reaction was concentrated under reduced pressure, diluted
with diethyl ether, sequentially washed with 1 M HCI, saturated NaHC03 solution, brine,
and dried over anhydrous MgS04. The extracts were filtered and the solvent was
removed under reduced pressure. Radial chromatography of the residue with 1 % ethyl
acetate in petroleum ether as eluant gave lactone 132 (37 mg, 52%) as a pale yellow
oil.
IR (CCI4): 2935, 2862, 1734, 1643, 1452, 1243, 1138, 1084, 892 cm"1;
1H NMR (500 MHz, CDCI3): 8 4.67 (s, 2 H), 4.11-4.13 (m, 2 H), 2.32-2.35 (m, 2 H), 2.05
(br t, J = 7.6 Hz, 2 H), 1.96 (br t, J = 7.8 Hz, 2 H), 1.58-1.64 (m, 6 H), 1.26-1.56
(m, 8 H);

280
13C NMR (125 MHz, CDCI3): 5 173.63, 149.28, 110.37, 63.09, 34.81, 34.25, 33.14,
27.62, 26.18, 25.73, 25.56, 24.66, 24.40, 23.51;
LRMS (El) m/z (relative intensity): 224 (M+, 20), 109 (45), 96 (75), 95 (62), 81 (100), 67
(69), 55 (56), 41 (56);
HRMS (El) m/z calculated for C 1 4 H 2 4 O 2 : 224.1776, found: 224.1776.
9-Cyclopropyl-13-tridecanolide (133)
Chloroiodomethane (80 uL, 1.1 mmol) was added to a solution of diethylzinc (54 u,L,
0.53 mmol) in deoxygenated CICH2CH2CI (2 mL) at 0 °C and the reaction was stirred for
seven minutes. A solution of lactone 132 (59 mg, 0.26 mmol) in deoxygenated
CICH2CH2CI (1 mL) was added via cannula and the reaction was stirred for an
additional ten minutes at 0 °C. The reaction was quenched with a 1:1 mixture of
saturated Na2S203 solution and saturated NH4CI solution, slowly warmed to rt, and
diluted with CH2CI2. The aqueous layer was extracted with CH2CI2, the organic layers
were combined, sequentially washed with water and brine, and dried over anhydrous
MgS04. The extracts were filtered and the solvent was removed under reduced
pressure. Column chromatography of the residue with 1 % ethyl acetate in petroleum
ether as eluant gave lactone 133 (54 mg, 85%) as a pale yellow oil.
IR (CCI4): 3069, 2933, 2860, 1733, 1458, 1247, 1171, 1137, 1086 cm'1;
1H NMR (500 MHz, CDCI3): 8 4.09-4.14 (m, 2 H), 2.34-2.37 (m, 2 H), 1.60-1.67 (m, 4 H),
1.46-1.52 (m, 2 H), 1.10-1.37 (m, 12 H), 0.16 (dd, J = 1.9, 7.4 Hz, 2 H), 0.13 (dd,
J = 1.9, 7.4 Hz, 2 H);

281
13C NMR (125 MHz, CDCI3): 8 173.87, 63.03, 34.69, 34.37, 33.73, 27.91, 26.31, 25.75,
25.70, 24.63, 21.92, 21.73, 18.24, 12.26 (2);
LRMS (El) m/z (relative intensity): 238 (M+, 12), 209 (33), 123 (25), 110 (77), 95 (98),
81 (100), 67 (95), 55 (68), 41 (74);
HRMS (El) m/z calculated for Ci5H2602: 238.1933, found: 238.1927.
9,9-Dimethyl-13-tridecanolide (134)
Adams' catalyst was added to a solution of lactone 133 (51 mg, 0.21 mmol) in acetic
acid (2 mL) and the mixture was stirred under H2 for 22 hours at rt. The reaction was
diluted with diethyl ether and filtered. The solid residue was rinsed with diethyl ether
and the organic layers were combined. The solution was washed with saturated
NaHC03 solution, water and brine, and dried over anhydrous MgS04. The extracts
were filtered and the solvent was removed under reduced pressure. Column
chromatography of the residue with 2% ethyl acetate in petroleum ether as eluant gave
lactone 134 (37 mg, 73%) as a pale yellow oil.
IR(CCU): 2941, 2861, 1733, 1464, 1364, 1250, 1145, 1086 cm"1;
1H NMR (500 MHz, CDCI3): 6 4.10-4.12 (m, 2 H), 2.33-2.35 (m, 2 H), 1.56-1.65 (m, 4 H),
1.05-1.38 (m, 14 H), 0.82 (s, 6 H);
13C NMR (125 MHz, CDCI3): 5 173.70, 62.99, 37.87, 37.84, 35.11, 32.37, 29.16 (2),
28.82, 26.65, 25.96, 25.82, 24.30, 19.66, 19.22;

282
LRMS (El) m/z (relative intensity): 240 (M+, 5), 166 (23), 96 (100), 81 (50), 69 (65), 55
(94), 41 (63);
HRMS (El) m/z calculated for CisHzsOz: 240.2089, found: 240.2087.
7,7-Dimethyl-2-oxacyclotetradecanethione (135)
A solution of lactone 134 (36 mg, 0.15 mmol) in toluene (5 mL) was added to a
suspension of Lawesson's reagent 48 (0.12 g, 0.30 mmol) in toluene (10 mL) and the
reaction was heated at reflux for five days. The reaction was cooled to rt, filtered, and
the solid residue was rinsed with diethyl ether. The filtrate was concentrated under
reduced pressure, and column chromatography of the residue with petroleum ether as
eluant gave thionolactone 135 (18 mg, 47%) as a yellow oil.
IR(CCI4): 2942, 2861, 1463, 1383, 1362, 1293, 1262, 1199, 1139, 1119, 1085 cm"1;
1H NMR (500 MHz, CDCI3): 8 4.46-4.50 (m, 2 H), 2.87-2.91 (m, 2 H), 1.75-1.80 (m, 2 H),
1.60-1.66 (m, 2 H), 1.27-1.39 (m, 8 H), 1.02-1.16 (m, 6 H), 0.83 (s, 6 H);
13C NMR (125 MHz, CDCI3): 5 224.28, 70.86, 47.85, 38.23, 37.45, 32.34, 29.17 (2),
28.14, 26.56, 25.96 (2), 24.79, 19.64, 19.13;
LRMS (El) m/z (relative intensity): 256 (M+, 34), 223 (34), 201 (100), 167 (50), 149 (28);
HRMS (El) m/z calculated for C15H28OS: 256.1861, found: 256.1856.

283
2-(Methylthio)-10,10-dimethyloxacyclotetradecane (136)
A solution of lithium triethylborohydride in THF (0.28 mL, 0.28 mmol) was added to a
solution of thionolactone 135 (14 mg, 0.056 mmol) in THF (5 mL) at -78 °C and the
reaction was stirred for 30 minutes at -78 °C. Methyl iodide (21 pL, 0.33 mmol) was
added and the reaction was stirred for 30 minutes, warmed to rt, and stirred for an
additional 30 minutes. The reaction was diluted with diethyl ether and cooled to -78 °C.
Aqueous 3 M NaOH solution (ca. 1 mL) and 30% H202 (ca. 0.5 mL) were added and the
solution was stirred for 15 minutes at -78 °C, and warmed to rt. The reaction was
sequentially washed with saturated Na2S203 solution, water, and brine, and dried over
anhydrous MgS04. The extracts were filtered and the solvent was removed under
reduced pressure to give mixed thioacetal 136 (14 mg, 94%) as an oil. Thioacetal 136
was unstable and was used immediately without further purification.
LRMS (Cl(+), isobutane) m/z (relative intensity): 273 (M++1, 16), 225 (100);
HRMS (Cl(+), isobutane) m/z calculated for Ci6H33OS (M++1): 273.2252, found:
273.2252.

284
6,6-Dimethyloxacyclotetradecane (137)
A deoxygenated solution of tri(n-butyl)tin hydride (0.14 mL, 0.52 mmol) and AIBN (cat.)
in toluene (9.8 mL) was added over ten hours via syringe pump to a deoxygenated
solution of mixed thioacetal 136 (14 mg, 0.052 mmol) and AIBN (cat.) in toluene
(10 mL) heated at reflux. The solvent was removed under reduced pressure. Column
chromatography of the residue with petroleum ether as eluant removed the tin
compounds. Further chromatography using AgN03 impregnated silica with petroleum
ether as eluant gave ether 136 (7.9 mg, 67%) as an oil.
IR(CDCI3): 2937, 2861, 1451, 1357, 1116 cm-1;
1H NMR (500 MHz, CDCI3): 5 3.43 (t, J = 5.3 Hz, 2 H), 3.42 (t, J = 5.4 Hz, 2 H), 1.60
(quint, J = 5.3 Hz, 2 H), 1.54 (quint, J = 5.4 Hz, 2 H), 1.29-1.42 (m, 10 H), 1.11-
1.17 (m, 6 H), 0.84 (s, 6 H);
13C NMR (125 MHz, CDCI3): 5 68.17, 67.70, 38.88, 37.76, 32.39, 29.32 (2), 29.19,
28.74, 26.48, 26.22, 23.54, 22.47, 19.84, 18.46;
LRMS (El) m/z (relative intensity): 226 (M+, 16), 211 (4), 152 (13), 115 (13), 109 (16),
96(100), 82(15), 69(16);
HRMS (El) m/z calculated for Ci5H30O: 226.2297, found: 226.2296.

285
6-Bromo-1-hexanol (139)
48% HBr (30 mL, 0.27 mol) was added to a solution of 1,6-hexanediol (138) (21.10 g,
178.5 mmol) in benzene (400 mL) and the solution was heated at reflux under Dean-
Stark conditions for 68 hours. The organic layer was collected and concentrated under
reduced pressure. The resultant oil was diluted with diethyl ether, and the ether
solution was sequentially washed with saturated NaHC03 solution, water and brine,
and dried over anhydrous MgS04. The extracts were filtered and the solvent was
removed under reduced pressure to give alcohol 139 (29.23 g, 90%) as a yellow oil.
This material was used in subsequent reactions without further purification. Column
chromatography of a small sample of 139 (ca. 100 mg) with 20% ethyl acetate in
petroleum ether as eluant gave pure 139 with spectral data in agreement with that
reported earlier in our laboratory.141
IR (CCI4): 3634, 3353, 2935, 2862, 1459, 1435, 1279, 1052, 952 cm"1;
1H NMR (500 MHz, CDCI3): 5 3.58 (t, J = 6.7 Hz, 2 H), 3.36 (t, J = 6.9 Hz, 2 H), 1.93
(br s, 1 H), 1.82 (quint, J = 6.9 Hz, 2 H), 1.52 (quint, J = 6.7 Hz, 2 H), 1.39-1.45
(m, 2 H), 1.30-1.36 (m, 2 H);
13C NMR (125 MHz, CDCI3): 6 62.51, 33.72, 32.59, 32.35, 27.81, 24.82;
LRMS (DCI(+), ammonia) m/z (relative intensity): 200 (81Br, M++18, 100), 198 (79Br,
M++18, 99);
HRMS (Cl(+), isobutane) m/z calculated for C6Hi4081Br (M++1): 183.0208, found:
183.0202; calculated for C6Hi4079Br (M++1): 181.0228, found: 181.0226.

286
6-Bromohexanal (140)
A solution of dimethylsulfoxide (11.4 mL, 160 mmol) in CH2CI2 (17 mL) was added via
cannula to a solution of oxalyl chloride (7.0 mL, 80 mmol) in CH2CI2 (90 mL) at -78 °C.
The solution was stirred for two minutes and alcohol 139 (7.20 g, 39.8 mmol) in CH2CI2
(40 mL) was added via cannula. This mixture was stirred for 15 minutes at -78 °C.
Triethylamine (28 mL, 0.20 mol) was added, the mixture was stirred for 5 minutes, and
warmed to rt. The reaction was quenched with water, and the organic layer was
collected. The aqueous layer was extracted with CH2CI2. The organic layers were
combined, sequentially washed with water and brine, and dried over anhydrous MgS04.
The extracts were filtered and the solvent was removed under reduced pressure.
Column chromatography of the residue with 10% ethyl acetate in petroleum ether as
eluant gave aldehyde 140 (5.41 g, 76%) as a pale yellow oil with spectral data in
agreement with that reported earlier in our laboratory.141
IR (CDCI3): 2939, 2863, 2728, 1724, 1430, 1257 cm'1;
1H NMR (500 MHz, CDCI3): 5 9.76 (t, J = 1.7 Hz, 1 H), 3.39 (t, J = 6.7 Hz, 2 H), 2.45 (dt,
J = 1.7, 7.2 Hz, 2 H), 1.86 (quint, J = 6.7 Hz, 2 H), 1.61-1.67 (m, 2 H), 1.43-1.50
(m, 2 H);
13C NMR (125 MHz, CDCI3): 5 202.13, 43.65, 33.34, 32.43, 27.65, 21.17;
LRMS (DCI(+), ammonia) m/z (relative intensity): 198 (81Br, M++18, 56), 196 (79Br,
M++18, 61), 178 (79Br, M+, 100);
HRMS (Cl(+), isobutane) m/z calculated for C6H12079Br (M++1): 179.0072, found:
179.0070.

287
6-Bromohexanal ethylene acetal (141)
A solution of aldehyde 140 (4.95 g, 27.6 mmol), ethylene glycol (7.7 mL, 0.14 mol), and
PPTS (1.39 g, 5.52 mmol) in benzene (200 mL) was heated at reflux under Dean-Stark
conditions for 21 hours. The solvent was removed under reduced pressure and the
residue was diluted with diethyl ether. The ether solution was sequentially washed with
saturated NaHC03 solution, water and brine, and dried over anhydrous MgS04. The
extracts were filtered and the solvent was removed under reduced pressure to give
acetal 141 (5.50 g, 90%) as a pale yellow oil. This material was used in subsequent
reactions without further purification. Column chromatography of a small sample of 141
(ca. 100 mg) with 5% ethyl acetate in petroleum ether as eluant gave pure 141 with
spectral data in agreement with that reported earlier in our laboratory.141
IR (CDCI3): 2947, 2875, 1460, 1435, 1407, 1360, 1237, 1136, 1041, 946 cm"1;
1H NMR (500 MHz, CDCI3): 5 4.83 (t, J = 5.0 Hz, 1 H), 3.90-3.97 (m, 2 H), 3.79-3.86 (m,
2 H), 3.38 (t, J = 6.8 Hz, 2 H), 1.82-1.88 (m, 2 H), 1.61-1.67 (m, 2 H), 1.41-1.49
(m, 4 H);
13C NMR (125 MHz, CDCI3): 5 104.39, 64.84 (2), 33.67, 33.63, 32.68, 28.04, 23.13;
LRMS (El) m/z (relative intensity): 223 (81Br, M+, 18), 221 (79Br, M+, 17), 83 (27), 73
(100), 45 (22);
HRMS (El) m/z calculated for C8H1402
81Br (M+-1): 223.0157, found: 223.0156;
calculated for C8H1402
79Br (M+-1): 221.0177, found: 221.0183.
6-Bromo-1-(2'-tetrahydropyranyloxy)-hexane (142)
288
A solution of alcohol 139 (5.00 g, 27.6 mmol), dihydropyran (3.0 mL, 33 mmol), and
PPTS (1.38 g, 5.52 mmol) in CH2CI2 (100 mL) was stirred at rt for 15 hours. The
resultant solution was diluted with CH2CI2, sequentially washed with saturated NaHC03
solution and brine, and dried over anhydrous MgS04. The extracts were filtered and
the solvent was removed under reduced pressure to give bromide 142 (7.08 g, 97%) as
a pale yellow oil. This material was used in subsequent reactions without further
purification. Column chromatography of a small sample of 142 (ca. 100 mg) with
5% ethyl acetate in petroleum ether as eluant gave pure 142 with spectral data in
agreement with that reported earlier in our laboratory.141
IR(CCI4): 2940, 2865, 1454, 1440, 1351, 1323, 1273, 1201, 1135, 1121, 1077, 1032,
980, 906, 870 cm"1;
1H NMR (500 MHz, CDCI3): 5 4.51 (dd, J = 2.8, 4.4 Hz, 1 H), 3.80 (ddd, J = 11.1, 7.9,
3.3 Hz, 1 H), 3.68 (dt, J = 9.5, 6.9 Hz, 1 H), 3.44 (ddd, J = 11.1, 2.5, 4.0 Hz, 1 H),
3.31-3.37 (m, 3 H), 1.31-1.81 (m, 14 H);
13C NMR (125 MHz, CDCI3): 5 98.75, 67.26, 62.22, 33.68, 32.64, 30.66, 29.44, 27.88,
25.39, 25.36, 19.57;
LRMS (El) m/z (relative intensity): 265 (81Br, M+-1, 31), 263 (79Br, M+-1, 31), 165 (31),
163 (33), 115 (35), 101 (33), 85 (100), 67 (30), 55 (80), 41 (63);
HRMS (El) m/z calculated for CnH20O2
81Br (M+-1): 265.0626, found: 265.0638;
calculated for CnH20O2
79Br (M+-1): 263.0647, found: 263.0649.

7-(1\3'-Dithian-2'-yl)-heptanal ethylene acetal (143)
289
A solution of n-butyllithium in hexanes (22.5 mL, 33.8 mmol) was added to a solution of
1,3-dithiane (4.06 g, 33.8 mmol) in THF (60 mL) at -20 °C and the reaction was stirred
for two hours. A solution of bromo acetal 141 (5.00 g, 22.5 mmol) in THF (50 mL) was
added and the solution was stirred for one hour, warmed to rt, and stirred for an
additional hour at rt. The reaction was quenched with saturated NH4CI solution, and
diluted with diethyl ether. The ether solution was sequentially washed with water and
brine, and dried over anhydrous MgS04. The extracts were filtered and the solvent was
removed under reduced pressure. Column chromatography of the residue with 10%
ethyl acetate in petroleum ether as eluant gave dithiane 143 (3.95 g, 67%) as a pale
yellow oil.
IR(CCU): 2912, 1457, 1423, 1276, 1137, 1038, 942, 909 cm-1;
1H NMR (500 MHz, CDCI3): 5 4.79 (t, J = 5.4 Hz, 1 H), 3.99 (t, J = 7.1 Hz, 1 H), 3.87-
3.92 (m, 2 H), 3.78-3.82 (m, 2 H), 2.75-2.85 (m, 4 H), 1.29-2.08 (m, 12 H);
13C NMR (125 MHz, CDCI3): 8 104.46, 64.73 (2), 47.47, 35.23, 33.67, 30.37 (2), 29.02,
26.42, 25.96, 23.68;
LRMS (El) m/z (relative intensity): 262 (M+, 81), 155 (21), 119 (63), 73 (100);
HRMS (El) m/z calculated for Ci2H2202S2: 262.1061, found: 262.1065;
Analysis calculated for Ci2H2202S2: C, 54.92; H, 8.45. Found: C, 54.99; H, 8.60.

290
7-(1',3'-Dithian-2'-yl)-13-(2"-tetrahydropyranyloxy)-tridecanal ethylene acetal (144)
A solution of n-butyllithium in hexanes (12.6 mL, 16.4 mmol) was added to a solution of
dithiane 143 (3.59 g, 13.7 mmol) in THF (50 mL) at -20 °C and the reaction was stirred
for two hours. A solution of bromide 142 (4.51 g, 17.0 mmol) in THF (10 mL) was
added via cannula and the reaction was stirred for one hour at -20 °C, warmed to rt,
and stirred for an additional hour at rt. The reaction was quenched with saturated
NH4CI solution and diluted with diethyl ether. The ether solution was sequentially
washed with water and brine, and dried over anhydrous MgS04. The extracts were
filtered and the solvent was removed under reduced pressure. Column
chromatography of the residue with 10% ethyl acetate in petroleum ether as eluant
gave dithiane 144 (1.38 g, 38%) as a pale yellow oil.
IR (CCU): 2940, 2865, 1459, 1353, 1275, 1133, 1078, 1033, 907 cm"1;
1H NMR (500 MHz, CDCI3): 6 4.80 (t, J = 4.8 Hz, 1 H), 4.53 (dd, J = 2.9, 4.2 Hz, 1 H),
3.88-3.94 (m, 2 H), 3.78-3.84 (m, 3 H), 3.68 (dt, J = 9.7, 6.9 Hz, 1 H), 3.45 (ddd,
J = 3.8, 5.0, 10.9, 1 H), 3.33 (dt, J = 9.7, 6.7 Hz, 1 H), 2.73-2.76 (m, 4 H), 1.77-
1.92 (m, 6 H), 1.27-1.69 (m, 22 H);
13C NMR (125 MHz, CDCI3): 5 104.50, 98.73, 67.46, 64.75 (2), 62.20, 53.24, 38.12,
38.00, 33.78, 30.69, 29.66, 29.64, 29.61, 26.07, 25.92 (2), 25.49, 25.43, 23.96,
23.93, 23.82, 19.59;

291
LRMS (El) m/z (relative intensity): 446 (M+, 13), 261 (31), 85 (100), 73 (49);
HRMS (El) m/z calculated for C23H42O4S2: 446.2524, found: 446.2518;
Analysis calculated for C23H42O4S2: C, 61.48; H, 9.48. Found: C, 61.62; H, 9.62.
7-Oxo-13-(2'-tetrahydropyranyloxy)-tridecanal ethylene acetal (145)
Mercuric perchlorate (3.42 g, 7.54 mmol) in water (2 mL) was added to a mixture of
dithiane 144 (3.06 g, 6.85 mmol) and calcium carbonate (0.82 g, 8.2 mmol) in THF
(40 mL) at rt and the reaction was stirred for 20 minutes. The reaction was diluted with
diethyl ether and filtered. The filtrate was washed with brine, and dried over anhydrous
MgS04. The extracts were filtered and the solvent was removed under reduced
pressure. Column chromatography of the residue with 20% ethyl acetate in petroleum
ether as eluant gave ketone 145 (1.50 g, 61%) as a colourless oil.
IR (CCU): 2940, 2866, 1716, 1454, 1409, 1358, 1133, 1078, 1033 cm"1;
1H NMR (500 MHz, CDCI3): 6 4.81 (t, J = 4.8 Hz, 1 H), 4.54 (dd, J = 2.7, 4.2 Hz, 1 H),
3.89-3.94 (m, 2 H), 3.80-3.85 (m, 2 H), 3.69 (dt, J = 9.5, 6.8 Hz, 1 H), 3.42-3.49
(m, 1 H), 3.34 (dt, J = 9.5, 6.6 Hz, 1 H), 2.36 (t, J = 7.4 Hz, 2 H), 2.35 (t, J = 7.4
Hz, 2 H), 1.47-1.82 (m, 15 H), 1.26-1.42 (m, 8 H);
13C NMR (125 MHz, CDCI3): 5 211.32, 104.51, 98.84, 67.49, 64.80 (2), 62.32, 42.69,
42.58, 33.67, 30.75, 29.55, 29.10, 29.06, 26.04, 25.47, 23.77 (2), 23.70, 19.66;

292
LRMS (El) m/z (relative intensity): 356 (M+, 6), 271 (14), 211 (37), 85 (66), 73 (100);
HRMS (El) m/z calculated for C20H36O5: 356.2563, found: 356.2559;
Analysis calculated for C2oH3605: C, 67.38; H, 10.18. Found: C, 67.67; H, 10.03.
7-Methylene-13-(2'-tetrahydropyranyloxy)-tridecanal ethylene acetal (146)
(a) Reaction of Ketone 145 with Tebbe Reagent 3 2 j e 1 5 3
A solution of Tebbe reagent 32 in toluene (12.6 mL, 8.42 mmol) was added via
syringe to a stirred solution of ketone 145 (1.50 g, 4.21 mmol), DMAP (0.62 g,
5.1 mmol) and pyridine (0.20 mL, 2.5 mmol) in THF (100 mL) at -40 °C and the reaction
was slowly warmed to rt over 20 hours. The reaction mixture was filtered through basic
alumina with petroleum ether as eluant and the filtrate was collected. The solvent was
removed under reduced pressure, and column chromatography of the residue with
5% ethyl acetate in petroleum ether as eluant gave alkene 146 (1.05 g, 70%) as a pale
yellow oil.
IR (CCU): 2936, 2862, 1643, 1459, 1354, 1132, 1078, 1033, 892 cm-1;
1H NMR (500 MHz, CDCI3): 5 4.81 (t, J = 4.9 Hz, 1 H), 4.64 (s, 2 H), 4.54 (dd, J = 3.2,
4.2 Hz, 1 H), 3.89-3.94 (m, 2 H), 3.79-3.85 (m, 3 H), 3.69 (dt, J = 9.5, 6.9 Hz, 1
H), 3.44-3.48 (m, 1 H), 3.35 (dt, J = 9.5, 6.7 Hz, 1 H), 1.96 (t, J = 7.6 Hz, 2 H),
1.95 (t, J = 7.6 Hz, 2 H), 1.26-1.83 (m, 22 H);

293
13C NMR (125 MHz, CDCI3): 6 150.01, 108.48, 104.60, 98.78, 67.58, 64.77 (2), 62.26,
35.93, 35.85, 33.82, 30.73, 29.67, 29.26, 29.20, 27.68, 27.63, 26.10, 25.46,
23.92, 19.64;
LRMS (El) m/z (relative intensity): 354 (M+, 4), 269 (14), 208 (16), 85 (100), 73 (51);
HRMS (El) m/z calculated for C2iH3804: 354.2770, found: 354.2764;
Analysis calculated for C2iH3804: C, 71.15; H, 10.80. Found: C, 71.35; H, 11.00.
(b) Reaction of Ketone 145 with Wittig Reagent
A solution of n-butyllithium in hexanes (100 mL, 160 mmol) was added to a suspension
of triphenylphosphonium iodide (65.26 g, 161.4 mmol) in THF (350 mL) at 0 °C and the
reaction was stirred for one hour at 0 °C. A solution of ketone 145 (14.39 g,
40.36 mmol) in THF (100 mL) was added via cannula and the reaction was stirred for
16 hours at 0 °C. The reaction was concentrated under reduced pressure and diluted
with diethyl ether. The ether solution was sequentially washed with water and brine,
and dried over anhydrous MgS04. The extracts were filtered and the solvent was
removed under reduced pressure. Column chromatography of the residue with 10%
ethyl acetate as eluant gave alkene 146 (9.41 g, 66%) as a pale yellow oil with spectral
data in agreement with that reported above.
13-Hydroxy-7-methylenetridecanal (147)
A solution of alkene 146 (0.95 g, 2.7 mmol) and PPTS (0.14 g, 0.54 mmol) in acetone
and water (10:1, 50 mL) was heated at reflux for 20 hours. The acetone was removed
under reduced pressure, and the reaction mixture was diluted with diethyl ether. The

294
ether solution was washed with saturated NaHC03 solution, brine, and dried over
anhydrous MgS04. The extracts were filtered and the solvent was removed under
reduced pressure. Column chromatography of the residue with 30% ethyl acetate in
petroleum ether as eluant gave hydroxy aldehyde 147 (0.52 g, 86%) as a colourless oil.
IR (CCI4): 3635, 2933, 2859, 2715, 1730, 1643, 1455, 1048, 892 cm-1;
1H NMR (500 MHz, CDCI3): 6 9.73 (t, J = 1.7 Hz, 1 H), 4.66 (br d, J = 3.4 Hz, 2 H), 3.61
(t, J = 6.6 Hz, 2 H), 2.40 (dt, J = 1.7, 7.4 Hz, 2 H), 1.95-1.99 (m, 4 H), 1.62 (quint,
J = 7.4, 2 H), 1.54 (quint, J = 6.6 Hz, 2 H), 1.27-1.44 (m, 10 H); 13C NMR (125 MHz, CDCI3): 5 202.81, 149.66, 108.76, 62.95, 43.82, 35.85, 35.70,
32.70, 29.11, 28.82, 27.67, 27.41, 25.59, 21.92;
LRMS (DCI(+), ammonia) m/z (relative intensity): 244 (M++18, 37), 227 (M++1, 15);
HRMS (Cl(+), isobutane) m/z calculated for C14H2702 (M++1): 227.2011, found:
227.2011.
13-Hydroxy-7-methylenetridecanoic acid (148)
A solution of NaCI02 (21.26 g, 235.1 mmol) and NaH2P04 (21.51 g, 179.3 mmol) in
water (100 mL) was added over four hours to a solution of hydroxy aldehyde 147
(5.06 g, 22.4 mmol) and 2-methyl-2-butene (60 mL) in f-butyl alcohol (250 mL), and the
reaction was stirred at rt overnight. The reaction was concentrated under reduced
pressure, diluted with water, and extracted with diethyl ether. The organic layer was
sequentially washed with water and brine, and dried over anhydrous MgS04. The
extracts were filtered and the solvent was removed under reduced pressure. Column

295
chromatography of the residue with 4% methanol in CH2CI2 as eluant gave hydroxy
acid 148 (3.41 g, 63%) as a colourless oil.
IR (CCU): 3637, 3373, 2977, 2933, 2862, 1712, 1644, 1382, 1350, 1120, 891 cm"1;
1 H NMR (500 MHz, CDCI3): 6 6.14 (br s, 1 H), 4.65 (s, 2 H), 3.60 (t, J = 6.7 Hz, 2 H),
2.30 (t, J = 7.5 Hz, 2 H), 1.94-1.98 (m, 4 H), 1.61 (quint, J = 7.5 Hz, 2 H), 1.53
(quint, J = 6.7 Hz, 2 H), 1.25-1.43 (m, 10 H); 1 3 C NMR (125 MHz, CDCI3): 5 178.81, 149.64, 108.66, 62.76, 35.75, 35.65, 33.90,
32.42, 28.99, 28.66, 27.56, 27.23, 25.47, 24.51;
LRMS (DCI(+), ammonia) m/z (relative intensity): 260 (M++18, 93), 243 (M++1, 100);
HRMS (Cl(+), ammonia/methane) m/z calculated for C14H27O3 (M++1): 243.1960, found:
243.1954;
Analysis calculated for Ci 4 H 2 60 3 : C, 69.38; H, 10.81. Found: C, 69.63; H, 10.87.
7-Methylene-13-tridecanolide (149)
Triethylamine (0.32 mL, 2.3 mmol) was added to a solution of hydroxy acid 148 (0.50 g,
2.1 mmol) in THF (20 mL) at rt and the reaction was stirred for 15 minutes.
2,4,6-Trichlorobenzoyl chloride (0.33 mL, 2.1 mmol) was added and the reaction was
stirred for an additional two hours. The reaction was filtered and concentrated under
reduced pressure. Trace amounts of solvent were removed under high vacuum over
two hours. A solution of the resultant mixed anhydride in toluene (100 mL) was divided
into two portions and simultaneously added via syringe pump to two solutions of DMAP
(0.73 g, 6.0 mmol) in toluene (600 mL) heated at reflux over 40 hours. The reaction
was concentrated under reduced pressure and diluted with diethyl ether. The ether

296
solution was sequentially washed with 1 M HCI, saturated NaHC03 solution, and brine,
and dried over anhydrous MgS04. The extracts were filtered and the solvent was
removed under reduced pressure. Column chromatography of the residue with 1%
ethyl acetate in petroleum ether as eluant gave lactone 149 (0.20 g, 42%) as a
colourless oil.
IR (CCU): 2935, 2861, 1734, 1643, 1453, 1252, 1149, 1074, 892 cm'1; 1H NMR (500 MHz, CDCI3): 5 4.72-4.73 (m, 1 H), 4.70-4.71 (m, 1 H), 4.06-4.08 (m, 2 H),
2.30-2.33 (m, 2 H), 2.03 (br t, J = 6.4 Hz, 2 H), 1.98 (br t, J = 6.6 Hz, 2 H), 1.51 -
1.66 (m, 6 H), 1.18-1.46 (m, 8 H); 13C NMR (125 MHz, CDCI3): 8 173.80, 147.36, 109.83, 63.22, 36.50, 34.16, 31.59,
27.50, 27.09, 26.36, 25.24, 25.22, 25.06, 24.13;
LRMS (El) m/z (relative intensity): 224 (M+, 74), 206 (100), 195 (26), 151 (21), 137 (36),
123 (41), 109 (88), 95 (48), 81 (50), 67 (38), 55 (33), 41 (24);
HRMS (El) m/z calculated for C 1 4 H 2 4 O 2 . 224.1776, found: 224.1778;
Analysis calculated for C14H2402: C, 74.95; H, 10.78. Found: C, 75.12; H, 10.83.
7-Cyclopropyl-13-tridecanolide (150)
A catalytic amount of iodine was added to a suspension of zinc-copper couple (0.44 g,
6.7 mmol) in diethyl ether (50 mL) and the mixture was stirred at rt for 15 minutes.
Diiodomethane (0.54 mL, 6.7 mmol) was added and the mixture was stirred for an
additional 15 minutes. A solution of lactone 149 (0.30 g, 1.3 mmol) in diethyl ether
(2 mL) was added, and the mixture was heated at reflux for 19 hours. Additional zinc-
copper couple (0.44 g, 6.7 mmol), iodine (cat.) and diiodomethane (0.54 mL, 6.7 mmol)

297
were added and the reaction was heated at reflux for a further 14 hours. The reaction
was quenched with saturated NH4CI solution and filtered. The solid residue was rinsed
with diethyl ether. The organic layers were combined, and sequentially washed with
saturated NaHC03 solution, water, and brine, and dried over anhydrous MgS04. The
extracts were filtered and the solvent was removed under reduced pressure. Radial
chromatography of the residue with 0.5% ethyl acetate in petroleum ether as eluant
gave lactone 150 (0.11 g, 34%) as a pale yellow oil.
IR (CCI4): 3070, 2934, 2859, 1724, 1580, 1550, 1448, 1347, 1276, 1206, 1159, 1062,
1011, 822 cm"1;
1H NMR (500 MHz, CDCI3): 5 4.13-4.15 (m, 2 H), 2.35-2.37 (m, 2 H), 1.46-1.72 (m, 6 H),
1.14-1.37 (m, 12 H), 0.16 (dd, J = 2.9, 6.6 Hz, 2 H), 0.12 (dd, J = 2.9, 6.6 Hz, 2
H); 13C NMR (125 MHz, CDCI3): 5 173.80, 63.77, 36.20, 33.28, 32.87, 27.86, 27.67, 26.90,
24.59, 24.35, 23.85, 23.27, 18.87, 12.22 (2);
LRMS (El) m/z (relative intensity): 238 (M+, 9), 220 (17), 209 (39), 137 (24), 123 (63),
109 (49), 95 (74), 81 (100), 67 (97), 55 (78), 41 (79);
HRMS (El) m/z calculated for C15H2602: 238.1933, found: 238.1927.
7,7-Dimethyl-13-tridecanolide (151)
Adams' catalyst was added to a solution of lactone 150 (0.11 g, 0.46 mmol) in acetic
acid (10 mL) and the mixture was stirred at rt under H2 for five hours. The reaction was
diluted with diethyl ether and filtered. The solid residue was rinsed with diethyl ether

298
and the organic layers were combined. The solution was neutralized with saturated
NaHC03 solution, washed with brine, and dried over anhydrous MgS04. The extracts
were filtered and the solvent was removed under reduced pressure. Column
chromatography of the residue with 1% ethyl acetate in petroleum ether as eluant gave
lactone 151 (87 mg, 78%) as a pale yellow oil.
IR (CCI4): 2940, 2860, 1736, 1459, 1383, 1277, 1191, 1162, .1124 cm"1;
1H NMR (500 MHz, CDCI3): 5 4.14-4.16 (m, 2 H), 2.34-2.37 (m, 2 H), 1.68-1.73 (m, 2 H),
1.28-1.56 (m, 4H), 1.27-1.35 (m, 4 H), 1.11-1.18 (m, 8 H), 0.81 (s, 6 H);
13C NMR (125 MHz, CDCI3): 8 173.56, 64.15, 39.23, 37.70, 32.68, 32.21, 29.07 (2),
28.09, 27.88, 27.59, 24.47, 24.24, 21.73, 20.88;
LRMS (El) m/z (relative intensity): 240 (M+, 2), 225 (31), 207 (37), 138 (73), 124 (69),
109 (30), 95 (27), 83 (27), 69 (100), 55 (58), 41 (57);
HRMS (El) m/z calculated for Ci5H2802: 240.2089, found: 240.2088.
9,9-Dimethyl-2-oxacyclotetradecanethione (152)
A solution of lactone 151 (87 mg, 0.36 mmol) in toluene (5 mL) was added to a
suspension of Lawesson's reagent 48 (0.29 g, 0.72 mmol) in toluene (15 mL) and the
reaction was heated at reflux for 6.5 days. The reaction was cooled to rt and filtered.
The solid residue was rinsed with diethyl ether and the organic layers were combined.
The solvent was removed under reduced pressure. Column chromatography of the
residue with petroleum ether as eluant gave thionolactone 152 (50 mg, 54%) as a
yellow oil.

299
IR(CCU): 2941,2859, 1461, 1366, 1293, 1192, 1134, 1054 cm"1;
1 H NMR (500 MHz, CDCI3): 6 4.51-4.53 (m, 2 H), 2.77-2.80 (m, 2 H), 1.80-1.85 (m, 2 H),
1.66-1.71 (m, 2 H), 1.51-1.57 (m, 2 H), 1.23-1.36 (m, 4 H), 1.09-1.21 (m, 8 H),
0.80 (s, 6 H); 1 3 C NMR (125 MHz, CDCI3): 5 224.77, 72.23, 44.72, 38.80, 37.41, 32.71, 29.03 (2),
28.08, 27.62, 27.10, 26.99, 24.84, 21.50, 21.09;
LRMS (El) m/z (relative intensity): 256 (M+, 9), 223 (28), 173 (46), 139 (100), 97 (28),
83 (24), 69 (31);
HRMS (El) m/z calculated for C i 5 H 2 8 O S : 256.1861, found: 256.1853.
2-(Methylthio)-8,8-dimethyloxacyclotetradecane (153)
Lithium triethylborohydride in THF (0.89 mL, 0.89 mmol) was added to a solution of
thionolactone 152 (45 mg, 0.18 mmol) in THF (5 mL) at -78 °C and the reaction was
stirred for 30 minutes at -78 °C. Methyl iodide (68 uL, 1.1 mmol) was added and the
reaction was stirred for 30 minutes at -78 °C, warmed to rt, and stirred for an additional
30 minutes at rt. The reaction was diluted with diethyl ether and cooled to -78 °C.
Aqueous 3 M NaOH solution (ca. 1 mL) and 30% H202 (ca. 1 mL) were added. The
solution was stirred for 20 minutes at -78 °C, and then warmed to rt. The reaction was
sequentially washed with saturated Na2S203 solution, water, brine, and dried over
anhydrous MgS04. The extracts were filtered and the solvent was removed under
reduced pressure to give mixed thioacetal 153 (47 mg, 98%) as an oil. Thioacetal 153
was unstable and was used immediately without further purification.

300
LRMS (DCI(+), ammonia) m/z (relative intensity): 290 (M++18, 21), 273 (M++1, 100);
HRMS (Cl(+), ammonia/methane) m/z calculated for Ci6H33OS (M++1): 273.2252,
found: 273.2255.
8,8-Dimethyloxacyclotetradecane (154)
A deoxygenated solution of tri(n-butyl)tin hydride (0.47 mL, 1.7 mmol) and AIBN (cat.)
in toluene (9.5 mL) was added over ten hours via syringe pump to a deoxygenated
solution of mixed thioacetal 153 (47 mg, 0.17 mmol) and AIBN (cat.) in toluene (10 mL)
heated at reflux. The solvent was removed under reduced pressure. Column
chromatography of the residue with petroleum ether as eluant removed the tin
compounds. Radial chromatography with 0.5% ethyl acetate in petroleum ether as
eluant gave ether 154 (16 mg, 40%) as an oil.
IR(CDCI3): 2936, 2859, 1457, 1361, 1115 cm"1;
1H NMR (500 MHz, CDCI3): 5 3.40 (t, J = 5.7 Hz, 4 H), 1.53-1.58 (m, 4 H), 1.41-1.47 (m,
4 H), 1.28-1.35 (m, 4 H), 1.14-1.24 (m, 8 H), 0.81 (s, 6 H);
13C NMR (125 MHz, CDCI3): 6 69.27 (2), 38.61 (2), 32.80, 29.09 (2), 27.90 (2), 27.31
(2), 24.80 (2), 21.56 (2);
LRMS (El) m/z (relative intensity): 226 (M+, 4), 143 (16), 124 (100), 109 (48), 97 (13),
95(16), 83(13), 81 (16), 69 (26);
HRMS (El) m/z calculated for Ci5H30O: 226.2297, found: 226.2294.

301
6-Oxahexadeca-1,15-diene (156)
9-Decenol (155) (0.18 mL, 1.0 mol) was added to a suspension of potassium hydride
(0.14 g, 1.2 mmol) in THF (2 mL) at 0 °C and the reaction was stirred for two hours.
DMPU (0.15 ml, 1.2 mmol) and 5-bromo-1 -pentene (0.20 mL, 1.2 mmol) were added
sequentially via syringe, and the reaction was stirred with slow warming to rt overnight.
The reaction was diluted with diethyl ether. The organic layer was washed with water
and brine, and dried over anhydrous MgS04. The extracts were filtered and the solvent
was removed under reduced pressure. Column chromatography of the residue with 2%
ethyl acetate in petroleum ether gave diene 156 (0.19 g, 84%) as a colourless oil.
IR(CCU): 3078, 2929, 2857, 1641, 1451, 1366, 1116, 993, 913 cm-1;
1H NMR (500 MHz, CDCI3): 8 5.80 (ddt, J = 16.6, 10.1, 6.7 Hz, 1 H), 5.79 (ddt, J = 17.0,
10.5, 6.5 Hz, 1 H), 5.00 (ddt, J = 16.6, 1.9, 1.7, 1 H), 4.97 (br dd, J = 17.0, 2.1
Hz, 1 H), 4.94 (br dd, J = 10.1, 1.9 Hz, 1 H), 4.91 (ddt, J = 10.5, 2.1, 1.1 Hz, 1
H), 3.39 (t, J = 6.7 Hz, 2 H), 3.37 (t, J = 6.7 Hz, 2 H), 2.10 (br ddt, J = 6.7, 1.7,
7.1 Hz, 2H), 2.02 (br ddt, J = 6.5, 1.1,7.2 Hz, 2 H), 1.62-1.68 (m, 2 H), 1.51-
1.57 (m, 2 H), 1.24-1.38 (m, 10 H);
13C NMR (125 MHz, CDCI3): 8 139.21, 138.41, 114.60, 114.09, 70.79, 70.13, 33.78,
30.34, 29.76, 29.42 (2), 29.07, 28.95, 28.91, 26.18;
LRMS (El) m/z (relative intensity): 224 (M+, 1), 154 (4), 99 (35), 95 (24), 83 (34), 69
(74), 68(100), 55 (74), 41 (79);
HRMS (El) m/z calculated for Ci5H280: 224.2140, found: 224.2135;
Analysis calculated for Ci5H280: C, 80.29; H, 12.58. Found: C, 80.02; H, 12.55.

(Z/£)-Oxacyclotetradec-5-ene (157) and (158)
302
A deoxygenated solution of diene 156 (103 mg, 0.448 mol) in toluene (50 mL) and a
deoxygenated solution of Grubbs' catalyst 9 1 6 6 (19 mg, 0.24 mmol, 5.3 mol%) in toluene
(50 mL) were added simultaneously using a syringe pump to deoxygenated toluene
(20 mL) stirred at rt over 24 hours. The receiver toluene flask was gently sparged with
N2 gas during the addition. After the addition, the solution was stirred for a further
24 hours, and a spatula of silica was added. The solvent was removed under reduced
pressure, and column chromatography of the silica absorbed residue with 2% ethyl
acetate in petroleum ether as eluant removed ruthenium compounds. Radial
chromatography of the residue (43 mg, 49%, GC ratio 157:158, 59:41) with 0.5% ethyl
acetate in petroleum ether gave ethers 157 (22 mg, 25%) and 158 (15 mg, 17%) both
as pale yellow oils.
157 (Z)
IR (CCU): 3004, 2927, 2859, 2794, 1649, 1483, 1452, 1359, 1291, 1117, 1039, 909,
860 cm-1;
1H NMR (500 MHz, CDCI3): 5 5.51 (dt, J = 10.7, 7.9 Hz, 1 H), 5.26 (dt, J = 10.7, 7.4 Hz,
1 H), 3.41 (t, J = 5.5 Hz, 2 H), 3.38 (t, J = 5.2 Hz, 2 H), 2.22 (dt, J = 7.4, 7.0 Hz,
2 H), 1.97 (dt, J = 7.1, 7.9 Hz, 2 H), 1.55-1.63 (m, 4 H), 1.24-1.45 (m, 10 H);
13C NMR (125 MHz, CDCI3): 5 130.87, 129.59, 68.81, 68.69, 29.56, 28.30, 27.35, 26.29,
26.23, 25.76, 24.23, 23.86, 23.38;

303
LRMS (Cl(+), ammonia) m/z (relative intensity): 197 (M++1, 100);
HRMS (Cl(+), isobutane) m/z calculated for Ci3H250 (M++1): 197.1905, found:
197.1905;
Analysis calculated for Ci3H240: C, 79.53; H, 12.32. Found: C, 79.74; H, 12.47.
158 (£ )
IR (CCU): 2929, 2856, 1447, 1359, 1118, 969 cm-1; 1H NMR (500 MHz, CDCI3): 8 5.39 (dt, J = 15.2, 7.1 Hz, 1 H), 5.33 (dt, J = 15.2, 6.9 Hz,
1 H), 3.48 (t, J = 5.3 Hz, 2 H), 3.37 (t, J = 6.1 Hz, 2 H), 2.11 (ddd, J = 6.9, 6.9,
5.9 Hz, 2 H), 1.96-2.02 (m, 2 H), 1.63-1.68 (m, 2 H), 1.24-1.50 (m, 10 H); 13C NMR (125 MHz, CDCI3): 8 131.80, 130.61, 69.54, 67.01, 31.64, 29.09, 28.60, 26.57,
26.50, 25.30, 25.16, 23.84, 22.04;
LRMS (DCI(+), ammonia) m/z (relative intensity): 214 (M++18, 67), 197 (M++1, 100);
HRMS (Cl(+), isobutane) m/z calculated for C13H250 (M++1): 197.1905, found:
197.1907;
Analysis calculated for Ci3H240: C, 79.53; H, 12.32. Found: C, 79.74; H, 12.46.
(a) Isomerization of (Z)-Oxacvclotetradec-5-ene (157) with Phenyl Disulfide
A catalytic amount of phenyl disulfide was added to a solution of ether 157 (ca. 0.5 mg)
in cyclohexane (1 mL), and the mixture was sparged with N2 for 15 minutes. The
reaction was photolysed with a 450 W medium pressure Hanovia mercury vapour lamp
for six hours. This gave a mixture of ethers 157 and 158 (GC ratio 157:158, 41:59).
(b) Isomerization of (E)-Oxacvclotetradec-5-ene (158) with Phenyl Disulfide
A catalytic amount of phenyl disulfide was added to a solution of ether 158 (ca. 0.5 mg)
in cyclohexane (1 mL), and the mixture was sparged with N2 for 15 minutes. The
reaction was photolysed with a 450 W medium pressure Hanovia mercury vapour lamp
for six hours. This gave a mixture of ethers 157 and 158 (GC ratio 157:158, 39:61).

304
9-Decenoic acid (159)
A solution of Jones' reagent (ca. 8 mL) was added to a solution of 9-decenol (155)
(3.00 g, 19.2 mmol) in acetone (100 mL) stirred at rt until an orange colour persisted.
The reaction was quenched with 2-propanol, and neutralized with solid NaHC03. The
mixture was filtered through silica, and the solid residue was rinsed with diethyl ether.
The organic layers were combined, washed with water and brine, and dried over
anhydrous MgS04. The extracts were filtered and the solvent was removed under
reduced pressure to give acid 159 (3.08 g, 94%) as a pale yellow oil. This material was
used in subsequent reactions without further purification. Column chromatography of a
sample of 159 (ca. 50 mg) with 4% methanol in CH2CI2 as eluant gave pure 159 for
analysis.
IR(CCI4): 3080, 2929, 2857, 1711, 1641, 1420, 1289, 913 cm"1;
1H NMR (500 MHz, CDCI3): 8 5.78 (ddt, J = 10.2, 17.2, 6.8 Hz, 1 H), 4.96 (ddt, J = 17.2,
2.0, 1.7, 1 H), 4.91 (ddt, J = 10.2, 2.0, 1.1 Hz, 1 H), 2.32 (t, J = 7.5 Hz, 2 H), 2.01
(dddt, J = 6.8, 1.1, 1.7, 7.1 Hz, 2 H), 1.61 (quint, J = 7.5 Hz, 2 H), 1.27-1.38 (m, 8
H); 13C NMR (125 MHz, CDCI3): 5 180.54, 139.02, 114.17, 34.08, 33.70, 29.03, 28.96,
28.85, 28.80, 24.60;
LRMS (El) m/z (relative intensity): 170 (M+, 1), 152 (14), 110 (39), 96 (25), 84 (40), 83
(34), 69(85), 55(100), 41 (76);
HRMS (El) m/z calculated for Ci0H18O2: 170.1307, found: 170.1300;
Analysis calculated for Ci0H18O2: C, 70.55; H, 10.66. Found: C, 70.93; H, 10.89.

305
10-Undecen-2-one (160)
Methyllithium in diethyl ether (39 mL, 52 mmol) was added to a solution of acid 159
(2.96 g, 17.4 mmol) in THF (50 mL) stirred at 0 °C and the reaction was stirred for two
hours at 0 °C. Trimethylsilyl chloride (33 mL, 0.26 mol) was added via syringe, and the
reaction was stirred with warming to rt over 30 minutes. The reaction was quenched
with saturated NH4CI solution, and stirred for a further one hour. The reaction mixture
was extracted with diethyl ether and the organic layers were combined, washed with
water, and dried over anhydrous MgS04. The extracts were filtered and the solvent
was removed under reduced pressure. Column chromatography of the residue with 5%
ethyl acetate in petroleum ether gave ketone 160 (2.11 g, 72%) as a pale yellow oil.
IR(CCI4): 3078, 2929, 2856, 1718, 1641, 1436, 1360, 1163, 1120, 993, 912 cm"1;
1H NMR (500 MHz, CDCI3): 6 5.76 (ddt, J = 17.1, 10.2, 6.7 Hz, 1 H), 4.94 (ddt, J = 17.1,
1.9, 1.6 Hz, 1 H), 4.88 (ddt, J = 10.2, 1.9, 1.1 Hz, 1 H), 2.37 (t, J = 7.5 Hz, 2 H),
2.09 (s, 3 H), 1.99 (dddt, J = 1.6, 1.1, 6.7, 7.5 Hz, 2 H), 1.53 (quint, J = 7.5 Hz, 2
H), 1.30-1.36 (m, 2 H), 1.22-1.27 (m, 6 H);
13C NMR (125 MHz, CDCI3): 5 209.13, 139.03, 114.12, 43.71, 33.68, 29.76, 29.16,
29.06, 28.86, 28.79, 23.78;
LRMS (El) m/z (relative intensity): 168 (M+, 1), 150 (2), 125(10), 111 (12) 110 (21), 97
(13), 81 (20), 71 (43), 58 (86), 43 (100);
HRMS (El) m/z calculated for CnH20O: 168.1514, found: 168.1517.

306
2-Hydroxy-10-undecene (161)
A solution of ketone 160 (1.36 g, 8.08 mmol) in diethyl ether (10 mL) was added
dropwise over one hour to a suspension of lithium aluminum hydride (0.31 g, 8.1 mmol)
stirred in diethyl ether (25 mL) at 0 °C. The stirred solution was warmed slowly to rt
overnight. Water (ca. 1.5 mL) and 3 M NaOH solution (ca. 0.5 mL) were sequentially
added dropwise. The reaction was filtered through celite, and the solid residue was
rinsed with diethyl ether. The organic layers were combined and the solvent was
removed under reduced pressure to give alcohol 161 (1.32 g, 96%) as a colourless oil.
This material was used in subsequent reactions without further purification. Column
chromatography of a sample of 161 (ca. 50 mg) with 4% methanol in CH2CI2 as eluant
gave pure 161 for analysis.
IR (CCI4): 3626, 3078, 2928, 2856, 1641, 1458, 1376, 1091, 912 cm"1;
1H NMR (500 MHz, CDCI3): 5 5.78 (ddt, J = 17.1, 10.1 Hz, 6.7 Hz, 1 H), 4.96 (ddt, J =
17.1, 1.5, 1.6 Hz, 1 H), 4.90 (ddt, J = 10.1, 1.5, 1.0 Hz, 1 H), 3.76 (sext, J = 6.1
Hz, 1 H), 2.01 (dddt, J = 6.7, 1.6, 1.0, 7.1 Hz, 2 H), 1.24-1.47 (m, 12 H), 1.15 (d,
J =6.1 Hz, 3 H);
13C NMR (125 MHz, CDCI3): 8 139.14, 114.10, 68.10, 39.33, 33.74, 29.56, 29.41, 29.02,
28.88, 25.71, 23.45;
LRMS (El) m/z (relative intensity): 170 (M+, 1), 152 (2), 110 (27), 96 (28), 95 (40), 82
(56), 81 (67), 69 (52), 68 (49), 67 (52) 55 (62), 45 (100), 41 (47);
HRMS (El) m/z calculated for CnH220: 170.1671, found: 170.1671;
Analysis calculated for CnH220: C, 77.58; H, 13.02. Found: C, 77.31; H, 13.05.

307
7-Methyl-6-oxahexadeca-1,15-diene (162)
Alcohol 161 (0.59 mL, 3.0 mmol) was added via syringe to a suspension of potassium
hydride (1.72 g, 15.0 mmol) in DMF (10 mL) at 0 °C and the reaction was stirred for
three hours. 5-Bromo-1-pentene (1.8 mL, 15 mmol) was added and the reaction was
warmed slowly to rt with stirring overnight. The reaction was diluted with diethyl ether,
washed with water and brine, and dried over anhydrous MgS04. The extracts were
filtered and the solvent was removed under reduced pressure. Column chromato
graphy of the residue with 2% ethyl acetate in petroleum ether gave ether 162 (0.54 g,
76%) as a colourless oil.
IR (CCI4): 3078, 2974, 2929, 2857, 1641, 1451, 1373, 1340, 1108, 993, 912 cm"1;
1H NMR (500 MHz, CDCI3): 5 5.79 (ddt, J = 10.4, 17.0, 6.8 Hz, 1 H), 5.78 (ddt, J = 10.4,
17.1,6.6 Hz, 1 H), 4.99 (ddt, J = 17.0, 1.8, 1.5 Hz, 1 H), 4.96 (br d, J = 17.1 Hz,
1 H), 4.93 (br dd, J = 10.4, 1.8 Hz, 1 H), 4.89 (br d, J = 10.4 Hz, 1 H), 3.46 (dt, J
= 9.3, 6.5 Hz, 1 H), 3.29-3.34 (m, 2 H), 2.09 (ddt, J = 6.8, 1.5, 7.8 Hz, 2 H), 2.01
(br dt, J = 6.6, 7.4 Hz, 2 H), 1.62 (m, 2 H), 1.45-1.52 (m, 2 H), 1.22-1.38 (m, 10
H), 1.09 (d, J = 6.1 Hz, 3H);
13C NMR (125 MHz, CDCI3): 8 139.14, 138.43, 114.54, 114.06, 75.41, 67.61, 36.69,
33.75, 30.38, 29.64, 29.43, 29.31, 29.05, 28.89, 25.57, 19.68;
LRMS (El) m/z (relative intensity): 238 (M+, 1), 113 (21), 95 (23), 71 (49), 69 (100), 55
(27), 41 (50);
HRMS (El) m/z calculated for C16H30O: 238.2297, found: 238.2295;
Analysis calculated for Ci6H30O: C, 80.61; H, 12.68. Found: C, 80.81; H, 12.74.

308
(Z/E)-14-Methyloxacyclotetradec-5-ene (163) and (164)
0 A deoxygenated solution of Grubbs' catalyst 9 1 6 6 (18 mg, 0.022 mmol, 4.9 mol%) in
CH2CI2 (50 mL) was added over three hours via syringe pump to a deoxygenated
solution of ether 162 (108 mg, 0.455 mmol) in CH2CI2 (250 mL) stirred at rt. The ether
162 solution was gently sparged with N2 gas during the addition. The reaction solution
was stirred for a further 17 hours, and was quenched with Et3N (ca. 1 mL). A spatula of
silica was added, and the solvent was removed under reduced pressure. Column
chromatography of the silica absorbed residue with 1% ethyl acetate in petroleum ether
as eluant removed ruthenium compounds. Radial chromatography of the residue
(61 mg, 63%, GC ratio 163:164, 43:57) with 0.3% ethyl acetate in petroleum ether gave
ethers 163 (11 mg, 12%) and 164 (8.0 mg, 8%) both as pale yellow oils.
163 (Z)
IR (CCU): 2928, 2858, 1455, 1372, 1338, 1094, 912 cm"1;
1H NMR (500 MHz, CDCI3): 5 5.46 (dt, J = 4.8, 10.2 Hz, 1 H), 5.26 (dt, J = 5.2, 10.2 Hz,
1 H), 3.59 (ddd, J = 9.4, 6.3, 2.8 Hz, 1 H), 3.41 (ddq, J = 8.5, 3.5, 6.1 Hz, 1 H),
3.23 (dt, J = 2.3, 9.4 Hz, 1 H), 2.47-2.55 (m, 1 H), 2.17-2.25 (m, 1 H), 1.90-1.96
(m, 1 H), 1.74-1.81 (m, 1 H), 1.63-1.70 (m, 1 H), 1.48-1.60 (m, 3 H), 1.23-1.42
(m, 10 H), 1.09 (d, J = 10.6 Hz, 3 H);
13C NMR (125 MHz, CDCI3): 5 130.80, 129.88, 73.35, 66.24, 36.09, 30.07, 27.44, 26.42,
25.86, 25.80, 25.49, 24.26, 23.66, 19.73;
LRMS (DCI(+), ammonia) m/z (relative intensity): 211 (M++1, 68), 210 (M+, 76);
HRMS (Cl(+), methane/ammonia) m/z calculated for Ci4H270 (M++1): 211.2062, found:
211.2052;
Analysis calculated for d4H260: C, 79.94; H, 12.46. Found: C, 80.12; H, 12.52.

309
164 (E)
IR(CCU): 2928, 2857, 1450, 1371, 1342, 1107, 970 cm-1; 1H NMR (500 MHz, CDCI3) 5: 5.38 (dt, J = 15.5, 6.5 Hz, 1 H), 5.33 (dt, J = 15.5, 6.5 Hz,
1 H), 3.48 (ddq, J = 10.2, 2.0, 6.2 Hz, 1 H), 3.41 (dt, J = 8.9, 7.3 Hz, 1 H), 3.34
(dt, J = 8.9, 6.2 Hz, 1 H), 2.14-2.21 (m, 1 H), 1.91-2.05 (m, 3 H), 1.62-1.68 (m, 2
H), 1.47-1.54 (m, 2 H), 1.22-1.41 (m, 10 H), 1.11 (d, J = 6.2 Hz, 3 H);
13C NMR (125 MHz, CDCI3) 5: 131.87, 130.77, 73.71, 65.29, 33.02, 31.58, 28.82, 28.71,
27.01, 25.86, 25.77, 24.26, 20.43, 20.13;
LRMS (DCI(+), ammonia) m/z (relative intensity): 228 (M++18, 2), 211 (M++1, 86), 210
(M+, 64);
HRMS (El) m/z calculated for Ci4H260: 210.1984, found: 210.1991;
Analysis calculated for Ci4H260: C, 79.94; H, 12.46. Found: C, 80.12; H, 12.47.
(a) Isomerization of (Z)-2-Methvloxacvclotetradec-10-ene (163) with Phenyl Disulfide
A catalytic amount of phenyl disulfide was added to a solution of ether 163 (ca. 0.5 mg)
in cyclohexane (1 mL) and the mixture was sparged with N2 for 15 minutes. The
reaction was photolysed with a 450 W medium pressure Hanovia mercury vapour lamp
for seven hours. This gave a mixture of ethers 163 and 164 (GC ratio 163:164, 29:71).
(b) Isomerization of (E)-2-Methvloxacvclotetradec-10-ene (164) with Phenyl Disulfide
A catalytic amount of phenyl disulfide was added to a solution of ether 164 (ca. 0.5 mg)
in cyclohexane (1 mL) and the mixture was sparged with N2 for 15 minutes. The
reaction was photolysed with a 450 W medium pressure Hanovia mercury vapour lamp
for seven hours. This gave a mixture of ethers 163 and 164 (GC ratio 163:164, 29:71).

310
12-Dodecanolide (165)
Trifluoroacetic anhydride (5.0 mL, 36 mmol) was added via syringe to a mixture of
cyclododecanone (93) (1.00 g, 5.49 mmol), urea hydrogen peroxide (3.09 g,
32.9 mmol), and Na2HP04 (5.45 g, 38.4 mmol) in CH2CI2 (50 mL) at 0 °C and the
reaction was stirred for 21 hours with slow warming to rt. The reaction was diluted with
CH2CI2, and was sequentially washed with water, saturated Na2S203 solution, saturated
NaHC03 solution, brine, and dried over anhydrous MgS04. The extracts were filtered
and the solvent was removed under reduced pressure. Column chromatography of the
residue with 2% ethyl acetate in petroleum ether as eluant gave lactone 165 (0.98 g,
90%) as a pale yellow oil.
IR(CDCI3): 2934, 2861, 1718, 1446, 1334, 1252, 1147, 1051,836 cm"1;
1H NMR (500 MHz, CDCI3): 5 4.10-4.12 (m, 2 H), 2.30-2.32 (m, 2 H), 1.59-1.66 (m, 4 H),
1.21-1.41 (m, 14 H);
13C NMR (125 MHz, CDCI3): 5 174.21, 64.50, 34.59, 27.38, 26.57, 26.38 (2), 25.42,
25.37, 24.95, 24.44, 24.20;
LRMS (El) m/z (relative intensity): 198 (M+, 3), 180 (6), 162 (6), 155 (3), 138 (18), 110
(24), 98 (63), 84 (57), 69 (62), 55 (100), 41 (44);
HRMS (El) m/z calculated for Ci2H2202: 198.1620, found: 198.1617;
Analysis calculated for Ci2H2202: C, 72.68; H, 11.18. Found: C, 73.11; H, 11.28.

2-Oxacyclotridecanethione (166)
311
A solution of lactone 165 (0.42 g, 2.1 mmol) in toluene (5 mL) was added via cannula to
a suspension of Lawesson's reagent 48 (1.60 g, 3.96 mmol) in toluene (5 mL) and the
reaction was heated at reflux for 56 hours. The reaction was cooled to rt, filtered
through cotton, and the solvent was removed under reduced pressure. Column
chromatography of the residue with petroleum ether as eluant gave thionolactone 166
(0.42 g, 92%) as a yellow oil.
IR (CDCI3): 2935, 2860, 1448, 1277, 1199, 1137, 1046 cm-1; 1H NMR (500 MHz, CDCI3): 8 4.49-4.52 (m, 2 H), 2.86-2.88 (m, 2 H), 1.78-1.83 (m, 2 H),
1.72-1.77 (m, 2 H), 1.43-1.48 (m, 2 H), 1.30-1.37 (m, 12 H);
13C NMR (125 MHz, CDCI3): 5 225.20, 72.91, 47.00, 27.30, 26.96, 26.44, 26.11, 25.73,
25.57, 25.39, 24.87, 24.50;
LRMS (El) m/z (relative intensity): 214 (M+, 4), 181 (41), 163(12), 111 (12), 97 (34), 83
(40), 69(52), 55(100), 41 (29);
HRMS (El) m/z calculated for C12H22OS: 214.1391, found: 214.1394;
Analysis calculated for Ci2H22OS: C, 67.24; H, 10.34. Found: C, 67.43; H, 10.55.

2-(Methylthio)oxacyclotridecane (167)
312
Lithium triethylborohydride in THF (2.3 ml, 2.3 mmol) was added to a solution of
thionolactone 166 (98 mg, 0.46 mmol) in THF (5 mL) at -78 °C and the reaction was
stirred for 20 minutes. Methyl iodide (0.17 mL, 2.8 mmol) was added and the reaction
was stirred for 30 minutes followed by slow warming to rt. The solution was diluted with
diethyl ether and cooled to -78 °C. Aqueous 3 M NaOH solution (5 mL) and 30% H202
(1 mL) were added sequentially and the solution was stirred for 10 minutes, followed by
warming to rt. The organic layer was sequentially washed with saturated Na2S203
solution, water, brine, and dried over anhydrous MgS04. The extracts were filtered and
the solvent was removed under reduced pressure to give mixed thioacetal 167 (0.10 g,
95%) as an oil. Thioacetal 167 was unstable and was used immediately without further
purification.
LRMS (DCI(+), ammonia) m/z (relative intensity): 248 (M++18, 10), 231 (M++1, 82), 200
(57), 183(100);
HRMS (El) m/z calculated for Ci3H260S: 230.1704, found: 230.1707.

313
Oxacyclotridecane (168)
A deoxygenated solution of tri(n-butyl)tin hydride (1.23 mL, 4.62 mmol) and AIBN (cat.)
was added in four portions over 26 hours to a deoxygenated solution of thioacetal 167
(75 mg, 0.33 mmol) and AIBN (cat.) in toluene (20 mL) heated at reflux. The reaction
was heated at reflux for an additional 22 hours. The solvent was removed under
reduced pressure, and column chromatography of the residue first with petroleum ether
as eluant removed the tin compounds. This was followed by 2% ethyl acetate in
petroleum ether as eluant to give crude ether 168. Further column chromatography
using AgN03 impregnated silica with petroleum ether as eluant gave ether 168 (33 mg,
55%) as a pale yellow oil.
IR(CDCI3): 2935, 2863, 1441, 1352, 1266, 1124, 1067, 1031 cm"1;
1H NMR (500 MHz, CDCI3): 5 3.42 (t, J = 5.2 Hz, 4 H), 1.54 (quint, J = 5.2 Hz, 4 H),
1.41 -1.46 (m, 4 H), 1.30-1.38 (m, 12 H);
13C NMR (125 MHz, CDCI3): 5 70.33 (2), 28.54 (2), 26.57 (2), 25.90 (2), 25.04 (2),
24.71 (2);
LRMS (El) m/z (relative intensity): 184 (M+, 0.8), 166 (2), 137 (5), 123 (10), 109 (24), 95
(44), 82 (100), 68 (59), 55 (66), 41 (18);
HRMS (El) m/z calculated for Ci2H240: 184.1827, found: 184.1830;
Analysis calculated for Ci2H240: C, 78.20; H, 13.12. Found: C, 77.98; H, 12.93.

2-Methyl-2-(methylthio)oxacyclotridecane (169)
314
Methyllithium in diethyl ether (0.39 mL, 0.47 mmol) was added to a solution of
thionolactone 166 (62 mg, 0.29 mmol) in THF (5.0 mL) at -78 °C and the reaction was
stirred for ten minutes. Methyl iodide (36 at, 0.58 mmol) was added, the reaction was
stirred for 10 minutes, and warmed to rt. The reaction was diluted with diethyl ether,
sequentially washed with saturated NaHC03 solution and brine, and dried over
anhydrous MgS04. The extracts were filtered and the solvent was removed under
reduced pressure to give mixed thioketal 169 (68 mg, 97%) as a pale yellow oil.
Thioketal 169 was unstable and was used immediately without further purification.
LRMS (DCI(+), ammonia) m/z (relative intensity): 245 (M++1, 59), 229 (20), 197 (100);
HRMS (El) m/z calculated for Ci4H28OS: 244.1860, found: 244.1856.
2-Methyleneoxacyclotridecane (170)
A solution of Tebbe reagent 32 3 8 , 1 5 3 in toluene (1.5 mL, 1.0 mmol), was added via
syringe to a solution of lactone 165 (0.10 g, 0.50 mmol), DMAP (0.07 g, 0.6 mmol) and
pyridine (20 \xL, 0.25 mmol) in THF (10 mL) stirred at -40 °C and the reaction was

315
stirred for 20 hours with slow warming to rt. The reaction mixture was filtered through
basic alumina with petroleum ether as eluant, and the organic layers were combined.
The solvent was removed under reduced pressure to give alkene 170 (70 mg, 67%) as
a pale yellow oil. Alkene 170 was unstable and was used immediately without further
purification.
LRMS (El) m/z (relative intensity): 196 (M+, 27), 125 (9), 97 (22), 71 (100), 55 (75), 43
(70), 41 (63);
HRMS (El) m/z calculated for C13H24O: 196.1827, found: 196.1826.
2-Methyloxacyclotridecane (171)
(a) Reduction of 2-Methvl-2-(methvlthio)oxacvclotridecane (169) with Trifn-butvDtin
Hydride
Th(/?-butyl)tin hydride (0.15 mL, 0.56 mmol) was added via syringe to a deoxygenated
solution of mixed thioketal 169 (68 mg, 0.28 mmol) and AIBN (cat.) in toluene (20 mL)
and the solution was heated at reflux for 2.5 hours. The solvent was removed under
reduced pressure. Column chromatography of the residue with petroleum ether as
eluant removed the tin compounds. Further column chromatography using AgN03
impregnated silica with petroleum ether as eluant gave ether 171 (17 mg, 31%) as a
pale yellow oil.

316
IR(CDCI3): 2927, 2859, 1456, 1372, 1340, 1139, 1091 cm'1;
1H NMR (500 MHz, CDCI3): 5 3.67 (ddd, J = 3.6, 4.2, 9.5 Hz, 1 H), 3.36 (ddq, J = 3.2,
9.2, 6.1 Hz, 1 H), 3.23 (ddd, J = 2.6, 9.5, 10.3 Hz, 1 H), 1.17-1.65 (m, 20 H), 1.09
(d, J = 6.1 Hz, 3H); 13C NMR (125 MHz, CDCI3): 5 75.32, 67.64, 36.56, 29.27, 26.69, 26.64, 26.45, 25.46,
24.52, 24.31, 23.85, 23.61, 20.20;
LRMS (El) m/z (relative intensity): 198 (M+, 4), 183 (12), 152 (13), 109 (20), 97 (37), 85
(15), 83 (54), 69 (57), 55(100);
HRMS (El) m/z calculated for Ci3H260: 198.1984, found: 198.1986;
Analysis calculated for C13H260: C, 78.72; H, 13.21. Found: C, 79.00; H, 13.36.
(b) Reduction of 2-Methyleneoxacvclotridecane (170) with Adams' Catalyst
Adams' catalyst was added to a solution of alkene 170 (70 mg, 0.36 mmol) in diethyl
ether (5 mL) and the mixture was stirred at rt under H2 overnight. The reaction was
filtered through silica with diethyl ether as eluant and the solvent was removed under
reduced pressure to give ether 171 (60 mg, 85%) as a pale yellow oil with spectral data
in agreement with that reported above.
(27£)-1-(Trimethylsi loxy)cyclododecene (172) and (173)
OTMS
(a) Reaction of Cvclododecanone (93) with Hexamethvldisilazane and Trimethylsilyl
Iodide
1,1,1,3,3,3-Hexamethyldisilazane (0.23 mL, 1.1 mmol) and trimethylsilyl chloride
(0.14 mL, 1.1 mmol) were added sequentially via syringe to a mixture of
cyclododecanone (93) (0.10 g, 0.55 mmol) and lithium iodide (0.15 g, 1.1 mmol) in

317
CH2CI2 (5 mL) stirred at rt. The reaction was stirred for 20 hours in the dark.
Triethylamine (0.15 mL, 1.1 mmol) was added and the reaction was stirred for an
additional 30 minutes. The reaction was diluted with diethyl ether, sequentially washed
with saturated NaHC03 solution and brine, and dried over anhydrous MgS04. The
extracts were filtered and the solvent was removed under reduced pressure. Column
chromatography of the residue (GC ratio 172:173, 48:52) with petroleum ether as
eluant gave silyl enol ethers 172 (61 mg, 44%) and 173 (64 mg, 46%) both as
colourless oils.
172 (Z)
IR(CCU): 2929, 2857, 1668, 1451, 1362, 1252, 1168, 1134, 1081, 1031,850 cm-1; 1H NMR (500 MHz, C6D6): 5 4.52 (t, J = 7.4 Hz, 1 H), 2.14 (dt, J = 7.4, 4.6 Hz, 2 H), 2.02
(t, J = 6.0 Hz, 2 H), 1.47-1.55 (m, 4 H), 1.35-1.45 (m, 12 H), 0.14 (s, 9 H); 13C NMR (125 MHz, C6D6): 5 149.76, 111.39, 36.44, 26.88, 26.67, 26.53, 26.22, 25.30,
25.16, 25.14, 25.09, 24.03, 0.59;
LRMS (El) m/z (relative intensity): 254 (M+, 15), 239 (7), 197 (11), 183 (23), 169 (12),
155 (12), 143 (59), 130 (59), 75 (53), 73 (100);
HRMS (El) m/z calculated for Ci5H30OSi: 254.2066, found: 254.2064;
Analysis calculated for Ci5H30OSi: C, 70.80; H, 11.88. Found: C, 71.06; H, 11.88.
1 7 3 ( E )
IR(CCI4): 2930, 2858, 1659, 1467, 1446, 1252, 1234, 1181, 1133, 1108, 942, 878,
850 cm"1;
1H NMR (500 MHz, C6D6): 5 4.66 (t, J = 7.9 Hz, 1 H), 2.16 (t, J = 6.7 Hz, 2 H), 2.01 (dt,
J = 7.9, 5.7 Hz, 2 H), 1.63-1.68 (m, 2 H), 1.26-1.42 (m, 14 H), 0.20 (s, 9 H);
13C NMR (125 MHz, C6D6): 5 151.87, 108.53, 28.42, 27.81, 25.06, 25.03, 24.81, 24.60,
24.37, 24.34, 22.77, 22.61, 0.52;
LRMS (El) m/z (relative intensity): 254 (M+, 13), 239 (5), 211 (8), 197 (10),-183 (22),
169 (12), 155 (13), 143 (60), 130 (58), 115 (14) 75 (52), 73 (100);
HRMS (El) m/z calculated for Ci5H30OSi: 254.2066, found: 254.2060;
Analysis calculated for C15H30OSi: C, 70.80; H, 11.88. Found: C, 70.82; H, 11.97.

318
(b) Reaction of Cvclododecanone (93) with Triethylamine and Trimethylsilyl Chloride
A solution of cyclododecanone (93) (3.65 g, 20.0 mmol) in DMF (10 mL) was added via
syringe to a solution of triethylamine (5.6 mL, 40 mmol) and trimethylsilyl chloride
(3.1 mL, 24 mmol) in DMF (30 mL) and the reaction was heated at reflux for six hours.
The reaction was cooled to rt, diluted with hexane, washed with saturated NaHC03
solution and brine, and dried over anhydrous MgS04. The extracts were filtered and
the solvent was removed under reduced pressure. Column chromatography of the
residue with petroleum ether as eluant gave silyl enol ethers 172 and 173 (2.60 g, 52%;
GC ratio 172:173, 63:37) as a pale yellow oil with spectral data in agreement with that
reported above.
2-Methylcyclododecanone (174)
A solution of MABR was generated by the addition of trimethylaluminum in hexanes
(3.0 mL, 6.0 mmol) to a solution of 4-bromo-2,6-di-terf-butylphenol (1.71 g, 6.00 mmol)
in CH2CI2 (12 mL) and the reaction was stirred for one hour at rt. An aliquot of the
MABR solution (13.0 mL, 2.60 mmol) was added to a solution of silyl enol ethers 172
and 173 (0.43 g, 1.7 mmol) in CH2CI2 (10 mL) at -40 °C, and the reaction was stirred for
20 minutes. Methyl triflate (0.38 mL, 3.4 mmol) was added and the reaction was stirred
with slow warming to rt over 20 hours. The reaction was diluted with CH2CI2 and
sequentially washed with 1 M HCI, saturated NaHC03 solution, brine, and dried over
anhydrous MgS04. The extracts were filtered and the solvent was removed under
reduced pressure. Column chromatography of the residue with 2% ethyl acetate in
petroleum ether as eluant gave ketone 174 (0.24 g, 71 %) as a pale yellow oil.

319
IR (CDCI3): 2934, 2864, 1709, 1469, 1445, 1371, 1133, 1028 cm"1;
1H NMR (500 MHz, CDCI3): 6 2.68 (ddq, J = 3.6, 9.8, 7.0 Hz, 1 H), 2.58 (ddd, J = 4.8,
6.9, 16.2 Hz, 1 H), 2.32 (ddd, J = 5.2, 7.7, 16.2 Hz, 1 H), 1.62-1.71 (m, 3 H), 1.47
(dddd, J = 3.4, 8.0, 8.0, 13.8 Hz, 1 H), 1.10-1.31 (m, 14 H), 1.02 (d, J = 7.0 Hz, 3
H); 13C NMR (125 MHz, CDCI3): 6 215.33, 45.49, 37.05, 31.51, 25.82, 25.40, 24.14, 24.08,
23.84, 22.71, 22.32, 22.06, 16.68;
LRMS (El) m/z (relative intensity): 196 (M+, 30), 167 (9), 149 (12), 139 (12), 125 (17),
111 (19), 98 (55), 83 (39), 72 (66), 55 (100), 41 (81);
HRMS (El) m/z calculated for C13H24O: 196.1827, found: 196.1829;
Analysis calculated for C13H240: C, 79.53; H, 12.32. Found: C, 79.57; H, 12.23.
12-Tridecanolide (175)
Trifluoroacetic anhydride (1.8 mL, 13 mmol) was added via syringe to a mixture of
ketone 174 (0.40 g, 2.0 mmol), urea hydrogen peroxide (1.13 g, 12.0 mmol) and
Na2HP04 (1.99 g, 14.0 mmol) in CH2CI2 (20 mL) stirred at 0 °C. The reaction was
stirred with slow warming to rt over 12 hours. The reaction was diluted with CH2CI2,
and sequentially washed with water, saturated Na2S203 solution, saturated NaHC03
solution, brine, and dried over anhydrous MgS04. The extracts were filtered and the
solvent was removed under reduced pressure to give lactone 175 (0.40 g, 92%) as a
pale yellow oil. This material was used in subsequent reactions without further
purification. Radial chromatography of a sample of 175 (ca. 50 mg) with 1% ethyl
acetate in petroleum ether as eluant gave pure 175 for analysis.

320
IR (CDCI3): 2933, 2862, 1729, 1457, 1368, 1249, 1134 cm'1;
1H NMR (500 MHz, CDCI3): 5 4.96 (ddq, J = 2.3, 8.8, 6.3 Hz, 1 H), 2.40 (ddd, J = 3.1,
8.4, 13.9 Hz, 1 H), 2.23 (ddd, J = 3.2, 9.7, 13.9 Hz, 1 H), 1.54-1.75 (m, 2 H),
1.22-1.44 (m, 16 H), 1.18 (d, J = 6.3 Hz, 3 H); 13C NMR (125 MHz, CDCI3): 5 173.75, 71.01, 35.10, 35.03, 26.78, 26.46, 26.15, 25.44,
25.14, 24.59, 24.52, 23.01, 20.65;
LRMS (El) m/z (relative intensity): 212 (M+, 2), 194 (14), 176 (9), 168 (20), 150 (12),
134 (7), 125 (27), 111 (37), 98 (72), 83 (40), 69 (61), 55 (100), 41 (89);
HRMS (El) m/z calculated for C13H24O2: 212.1776, found: 212.1770;
Analysis calculated for C13H24O2: C, 73.54; H, 11.39. Found: C, 73.48; H, 11.21.
2-Methylene-13-methyloxacyclotridecane (176)
A solution of Tebbe reagent 32 3 8 , 1 5 3 in toluene (1.9 mL, 1.9 mmol) was added to a
solution of lactone 175 (101 mg, 0.473 mmol), DMAP (0.07 g, 0.6 mmol) and pyridine
(20 pL, 0.25 mmol) in THF (10 mL) stirred at -40 °C. The reaction was stirred with slow
warming to rt over 23 hours. The reaction was filtered through basic alumina with
petroleum ether as eluant and the solvent was removed under reduced pressure to give
alkene 176 (70 mg, 70%) as a pale yellow oil. Alkene 176 was unstable and was used
immediately without further purification.
LRMS (El) m/z (relative intensity): 210 (M+, 14), 194(11), 176 (7), 168(14), 150(14),
135 (10), 125 (27), 98 (48), 82 (57), 71 (74), 55 (100), 41 (79);
HRMS (El) m/z calculated for C14H26O: 210.1984, found: 210.1980.

321
3-Methyl-2-oxacyclotridecanethione (177)
A solution of lactone 175 (0.62 g, 2.9 mmol) in toluene (10 mL) was added via cannula
to a suspension of Lawesson's reagent 48 (2.37 g, 5.86 mmol) in toluene (10 mL) and
the reaction was heated at reflux for two days. The reaction was cooled to rt, filtered
through silica, and the solid residue was rinsed with diethyl ether. The organic layers
were combined and the solvent was removed under reduced pressure. Column
chromatography of the residue with petroleum ether as eluant gave thionolactone 177
(0.50 g, 75%) as a yellow oil.
IR(CDCI3): 2932, 2861, 1457, 1356, 1279, 1191, 1125, 1048 cm'1;
1H NMR (500 MHz, CDCI3): 5 5.61 (ddq, J = 1.9, 9.5, 6.3 Hz, 1 H), 2.90 (ddd, J = 3.2,
9.7, 13.2 Hz, 1 H), 2.75 (ddd, J = 3.2, 8.2, 13.2 Hz, 1 H), 1.72-1.88 (m, 2 H),
1.57-1.67 (m, 2 H), 1.21-1.45 (m, 14 H), 1.27 (d, J = 6.3 Hz, 3 H);
1 3 C NMR (125 MHz, CDCI3): 5 224.35, 79.43, 47.81, 35.08, 26.98, 26.75, 26.35, 25.14,
25.07, 24.72, 24.39, 22.86, 19.42;
LRMS (El) m/z (relative intensity): 228 (M+, 4), 195 (48), 177 (17), 111 (22), 97 (37), 83
(36), 69(61), 55(100), 41 (85);
HRMS (El) m/z calculated for Ci3H24OS: 228.1548, found: 228.1548;
Analysis calculated for Ci3H24OS: C, 68.37; H, 10.59. Found: C, 68.70; H, 10.71.

322
2-Methyl-2-(methylthio)-13-methyloxacyclotridecane (178)
Methyllithium in diethyl ether (0.45 mL, 0.63 mmol) was added to a solution of
thionolactone 177 (47 mg, 0.21 mmol) in THF (5 mL) at -78 °C and the reaction was
stirred for 30 minutes. Methyl iodide (42 uL, 0.67 mmol) was added. The reaction was
stirred for an additional 15 minutes followed by warming to rt and further stirred for
15 minutes. The reaction was diluted with diethyl ether, sequentially washed with water
and brine, and dried over anhydrous MgS04. The extracts were filtered and the solvent
was removed under reduced pressure to give mixed thioketal 178 (38 mg, 71%) as an
oil. Thioketal 178 was unstable and was used immediately without further purification.
LRMS (El) m/z (relative intensity): 258 (M+, 0.4), 242 (2), 211 (20), 195 (83), 177 (38),
152 (8), 121 (30), 103 (75), 71 (73), 55 (100), 41 (68);
HRMS (El) m/z calculated for Ci5H30OS: 258.2018, found: 258.2011.

323
{2R*, 13/?*) and (2S*. 13/?*)-Dimethyloxacyclotridecane (179) and (180)
(a) Reduction of 2-Methvlene-13-methvloxacvclotridecane (176) with Adams' catalyst
Adams' catalyst was added to a solution of alkene 176 (60 mg, 0.29 mmol) in diethyl
ether (10 mL) and the reaction was stirred under H2 for 24 hours at rt. The reaction
was filtered through basic alumina with diethyl ether as eluant and the solvent was
removed under reduced pressure. Radial chromatography of the residue (GC ratio
179:180, 41:59) with petroleum ether as eluant gave ethers 179 (13 mg, 22%) and 180
(19 mg, 30%) both as oils.
179 (2/?*, 13/?*)
IR (CDCI3): 2968, 2928, 2858, 1455, 1374, 1132, 1081, 1048 cm-1; 1H NMR (500 MHz, CDCI3): 5 3.69 (sext, J = 6.1 Hz, 2 H), 1.49-1.57 (m, 4 H), 1.16-1.48
(m, 16 H), 1.08 (d, J = 6.1 Hz, 6 H); 13C NMR (125 MHz, CDCI3): 5 69.11 (2), 34.82 (2), 27.00 (2), 25.84 (2), 25.43 (2),
23.30 (2), 19.56 (2);
LRMS (El) m/z (relative intensity): 212 (M+, 12), 197 (12), 179 (4), 168 (27), 140 (10),
125 (15), 111 (33), 97 (61), 83 (80), 69 (88), 55 (100), 41 (75);
HRMS (El) m/z calculated for d4H280: 212.2140, found: 212.2140.
180 (2S*, 13/?*)
IR(CDCI3): 2966, 2928, 2861, 1456, 1370, 1329, 1140, 1114, 1062 cm"1;
1H NMR (500 MHz, CDCI3): 5 3.43 (ddq, J = 4.2, 6.3, 6.1 Hz, 2 H), 1.23-1.48 (m, 20 H),
1.10 (d, J = 6.1 Hz, 6H);

324
13C NMR (125 MHz, CDCI3): 5 74.23 (2), 37.67 (2), 26.28 (2), 25.78 (2), 22.92 (2),
22.80 (2), 22.31 (2);
LRMS (El) m/z (relative intensity): 212 (M+, 10), 197 (6), 179 (4), 168 (17), 140 (9), 125
(15), 111 (27), 97 (57), 83 (72), 69 (82), 55 (100), 41 (78);
HRMS (El) m/z calculated for C14H280: 212.2140, found: 212.2133;
Analysis calculated for Ci4H280: C, 79.18; H, 13.29. Found: C, 79.01; H, 13.10.
(b) Reduction of 2-Methvl-2-(methvlthio)-13-methyloxacvclotridecane (178) with
Tri(n-butvl)tin Hydride
A deoxygenated solution of tri(n-butyl)tin hydride (0.51 mL, 1.9 mmol) and AIBN (cat.)
in toluene (9.5 mL) was added over ten hours via syringe pump to a deoxygenated
solution of mixed thioketal 178 (50 mg, 0.19 mmol) and AIBN (cat.) in toluene (20 mL)
heated at reflux. The solvent was removed under reduced pressure. Column
chromatography of the residue first with petroleum ether as eluant to remove the tin
compounds followed by 1% ethyl acetate in petroleum ether as eluant to give ethers
179 and 180 (11 mg, 26%; GC ratio 179:180, 50:50) as an oil with spectral data in
agreement with that reported above.
(c) Reduction of 2-Methvl-2-(methvlthio)-13-methvloxacvclotridecane (178) with TTMSH
TTMSH (0.57 mL, 1.8 mmol) was added to a deoxygenated solution of mixed thioketal
178 (48 mg, 0.18 mmol) and AIBN (cat.) in toluene (10 mL) and the reaction was
heated at reflux overnight. The solvent was removed under reduced pressure. Column
chromatography of the residue first with petroleum ether as eluant followed by 1 % ethyl
acetate in petroleum ether as eluant gave ethers 179 and 180 (18 mg, 46%; GC ratio
179:180, 54:46) as an oil with spectral data in agreement with that reported above.

325
10-Bromo-1-decanol (183)
0 48% HBr (11.7 mL, 0.103 mol) was added to a solution of 1,10-decanediol (182)
(12.00 g, 68.85 mmol) in benzene (300 mL) and the solution was heated at reflux under
Dean-Stark conditions for two days. The organic layer was collected and concentrated
under reduced pressure. The resultant oil was diluted with diethyl ether, sequentially
washed with saturated NaHC03 solution, water, brine, and dried over anhydrous
MgS04. The extracts were filtered and the solvent was removed under reduced
pressure. Column chromatography of the residue with 10% ethyl acetate in petroleum
ether as eluant gave alcohol 183 (11.86 g, 73%) as a pale yellow oil with spectral data
in agreement with that reported in the literature.145
IR (CCI4): 3635, 2930, 2857, 1456, 1048, 909 cm-1;
1H NMR (500 MHz, CDCI3): 5 3.59 (t, 6.7 Hz, 2 H), 3.37 (t, J = 6.9 Hz, 2 H), 1.81 (quint,
J = 6.9 Hz, 2 H), 1.52 (quint, J = 6.7 Hz, 2 H), 1.36-1.41 (m, 2 H), 1.22-1.32 (m,
7H);
13C NMR (125 MHz, CDCI3): 6 62.94, 33.97, 32.76, 32.70, 29.41, 29.31, 29.29, 28.67,
28.09, 25.66;
LRMS (DCI(+), ammonia) m/z (relative intensity): 256 (81Br, M++18, 81), 254 (79Br,
M++18, 100);
HRMS (Cl(+), methane, ammonia) m/z calculated for Ci0H25NO81Br (M++18): 256.1099,
found: 256.1103; calculated for Ci0H25NO79Br (M++18): 254.1120, found:
254.1119.

326
10-Bromo-1-decanoic acid (184)
A solution of Jones reagent (ca. 35 mL) was added dropwise to a solution of alcohol
183 (19.63 g, 82.76 mmol) in acetone (300 mL) stirred at rt until an orange colour
persisted. The reaction was quenched with 2-propanol (2 mL) and neutralized with
solid NaHC03. The reaction mixture was filtered through silica and the solid residue
was rinsed with diethyl ether. The organic layers were combined and the solvent was
removed under reduced pressure. The resultant oil was diluted with diethyl ether,
sequentially washed with water and brine, and dried over anhydrous MgS04. The
extracts were filtered and the solvent was removed under reduced pressure to give acid
184 (19.49 g, 94%) as a white solid. This material was used in subsequent reactions
without further purification. Column chromatography of a sample of 184 (ca. 100 mg)
with 4% methanol in CH2CI2 as eluant gave pure 184 for analysis.
mp: 40-41 °C;
IR(CCU): 3541, 3072, 2931, 2858, 1711, 1432, 1287, 934 cm"1;
1H NMR (500 MHz, CDCI3): 5 3.37 (t, J = 6.9 Hz, 2 H), 2.32 (t, J = 7.5 Hz, 2 H), 1.82
(quint, J = 6.9 Hz, 2 H), 1.60 (quint, J = 7.5 Hz, 2 H), 1.36-1.42 (m, 2 H), 1.26-
1.32 (m, 8 H);
13C NMR (125 MHz, CDCI3): 6 180.38, 34.03, 33.92, 32.74, 29.14, 29.05, 28.92, 28.62,
28.07, 24.57;
LRMS (El) m/z (relative intensity): 252 (81Br, M+, 17), 250 (79Br, M+, 17), 209 (7), 207
(8), 73 (80), 60 (100), 55 (54), 41 (46);
HRMS (El) m/z calculated for Ci0Hi9O2
81Br: 252.0548, found: 252.0550; calculated for
Ci0H19O2
79Br: 250.0568, found: 250.0568;
Analysis calculated for C10Hi9OBr: C, 47.82; H, 7.62. Found: C, 47.92; H, 7.73.

327
Methyl 10-bromodecanoate (185)
Concentrated sulfuric acid (3 mL) was added to a solution of acid 184 (19.27 g,
76.72 mmol) in methanol (100 mL) and the solution was heated at reflux overnight. The
solvent was removed under reduced pressure and the residue was diluted with diethyl
ether. The organic phase was sequentially washed with saturated NaHC03 solution,
water, brine, and dried over anhydrous MgS04. The extracts were filtered and the
solvent was removed under reduced pressure to give ester 185 (19.22 g, 94%) as a
pale yellow oil. This material was used in subsequent reactions without further
purification. Radial chromatography of a sample of 185 (ca. 100 mg) with 2% ethyl
acetate in petroleum ether as eluant gave pure 185 for analysis.
IR (CCI4): 2932, 2857, 1742, 1437, 1176 cm-1;
1H NMR (500 MHz, CDCI3): 5 3.59 (s, 3 H), 3.32 (t, J = 6.9 Hz, 2 H), 2.23 (t, J = 7.5 Hz,
2 H), 1.77 (quint, J = 7.5 Hz, 2 H), 1.51-1.57 (m, 2 H), 1.32-1.37 (m, 2 H), 1.21-
1.25 (m, 8 H);
13C NMR (125 MHz, CDCI3): 5 174.04, 51.25, 33.89, 33.76, 32.65, 29.06, 28.97, 28.92,
28.52, 27.97, 24.75;
LRMS (El) m/z (relative intensity): 266 (81Br, M+, 6), 264 (79Br, M+, 6), 235 (5), 233 (6),
87 (44), 74 (100), 55 (21), 41 (21);
HRMS (El) m/z calculated for CnH2i02
81Br: 266.0704, found: 266.0699; calculated for
CnH2i02
79Br: 264.0725, found: 264.0715;
Analysis calculated for CnH2i02Br: C, 49.82; H, 7.98. Found: C, 50.11; H, 8.15.

328
Methyl 11-carbomethoxy-12-oxotridecanoate (186)
COOMe
J OMe
Methyl acetoacetate (15.4 mL, 143 mmol) was added dropwise to a suspension of
sodium hydride (5.72 g, 143 mmol) in a mixture of THF and DMF (3:1, 400 mL) stirred
at rt. After the effervescence had subsided, ester 185 (18.92 g, 71.35 mmol) was
slowly added to the reaction over three hours. The reaction was heated at reflux
overnight. The resultant solution was concentrated under reduced pressure, diluted
with CH2CI2, and sequentially washed with 1 M HCI, water, brine, and dried over
anhydrous MgS04. The extracts were filtered and the solvent was removed under
reduced pressure to give diester 186 (23.76 g) as a pale yellow oil. This material was
used in subsequent reactions without further purification. Column chromatography of a
sample of 186 (ca. 100 mg) with 5% ethyl acetate in petroleum ether as eluant gave
pure 186 for analysis.
IR(CCI4): 2933, 2857, 1743, 1721, 1437, 1357, 1273, 1196, 1167 cm"1;
1H NMR (500 MHz, CDCI3): 5 3.70 (s, 3 H), 3.60 (s, 3 H), 3.38 (t, J = 7.4 Hz, 1 H), 2.26
(t, J = 7.5 Hz, 2 H), 2.19 (s, 3 H), 1.76-1.86 (m, 2 H), 1.54-1.60 (m, 2 H), 1.17-
1.29 (m, 12 H);
13C NMR (125 MHz, CDCI3): 6 203.18, 174.19, 170.13, 59.60, 52.21, 51.31, 49.71,
33.96, 29.16, 29.09, 29.05, 28.97, 28.66, 28.14, 27.28, 24.81;
LRMS (El) m/z (relative intensity): 300 (M+, 2), 129 (17), 116 (99), 98 (100), 87 (36), 84
(23), 74 (24), 69 (21), 55 (29), 43 (92);
HRMS (El) m/z calculated for Ci6H2805: 300.1937, found: 300.1946;
Analysis calculated for Ci6H2805: C, 63.97; H, 9.40. Found: C, 63.79; H, 9.49.

329
12-Oxotridecanoic acid (187)
COOH
7 P
A solution of diester 186 (23.49 g, 78.20 mmol) in a mixture of concentrated HCI,
methanol and water (5:1:1, 224 mL) was heated at reflux for 14.5 hours. The reaction
was cooled and extracted with diethyl ether. The organic layers were combined,
sequentially washed with water and brine, and dried over anhydrous MgS04. The
extracts were filtered and the solvent was removed under reduced pressure to give
keto acid 187 (14.99 g, 84%) as a white solid. This material was used in subsequent
reactions without further purification. Column chromatography of a sample of 187 (ca.
100 mg) with 4% methanol in CH2CI2 as eluant gave pure 187 for analysis.
mp: 59-60 °C;
IR (CCI4): 3084, 2930, 2856, 1713, 1416, 1359, 909 cm"1;
1H NMR (500 MHz, CDCI3): 5 2.39 (t, J = 7.4 Hz, 2 H), 2.32 (t, J = 7.4 Hz, 2 H), 2.11 (s,
3 H), 1.61 (quint, J = 7.4 Hz, 2 H), 1.54 (quint, J = 7.4 Hz, 2 H), 1.21-1.34 (m,
12 H);
13C NMR (125 MHz, CDCI3): 5 209.50, 179.20, 43.80, 33.90, 29.31 (4), 29.14, 29.13,
29.00, 24.65, 23.83;
LRMS (El) m/z (relative intensity): 228 (M+, 1), 210 (2), 152 (18), 135 (18), 112 (17), 98
(30), 83 (18), 71 (24), 69 (31), 58 (100), 43 (86);
HRMS (El) m/z calculated for Ci3H2403: 228.1726, found: 228.1724;
Analysis calculated for Ci3H2403: C, 68.38; H, 10.59. Found: C, 68.21; H, 10.65.

330
12-Hydroxy-12-methyltridecanoic acid (188)
A solution of methylmagnesium bromide in diethyl ether (13.0 mL, 39.0 mmol) was
added to a solution of keto acid 187 (3.00 g, 13.1 mmol) in CH2CI2 (100 mL) at 0 °C and
the reaction was stirred with slow warming to rt overnight. The reaction was diluted
with CH2CI2, acidified with 1 M HCI, sequentially washed with water and brine, and
dried over anhydrous MgS04. The extracts were filtered and the solvent was removed
under reduced pressure. Column chromatography of the residue with 10% ethyl
acetate in petroleum ether as eluant followed by 50% ethyl acetate in petroleum ether
as eluant gave hydroxy acid 188 (0.85 g, 27%) as a white solid.
mp: 62-64 °C;
IR (CDCI3): 3607, 2931, 2857, 1709, 1416, 1264, 1143 cm-1;
1H NMR (500 MHz, CDCI3): 6 2.32 (t, J = 7.4 Hz, 2 H), 1.61 (quint, J = 7.1 Hz, 2 H),
1.42-1.45 (m, 2 H), 1.24-1.35 (m, 14 H), 1.19 (s, 6 H);
13C NMR (125 MHz, CDCI3): 5 178.42, 71.24, 43.91, 33.85, 30.06, 29.45, 29.32, 29.26,
29.19 (2), 29.10, 28.94, 24.68, 24.24;
LRMS (El) m/z (relative intensity): 244 (M+, 1), 226 (2), 211 (22), 186 (11), 69 (15), 59
(100), 43(16);
HRMS (El) m/z calculated for C14H2803: 244.2038, found: 244.2039;
Analysis calculated for Ci4H2803: C, 68.81; H, 11.55. Found: C, 68.97; H, 11.55.

331
12-Methyl-12-tridecanolide (189)
Triethylamine (0.31 mL, 2.2 mmol) was added to a solution of hydroxy acid 188 (0.50 g,
2.0 mmol) in THF (20 mL) stirred at rt and the reaction was stirred for 15 minutes.
2,4,6-Trichlorobenzoyl chloride (0.31 mL, 2.0 mmol) was added and the reaction was
stirred for two hours. The reaction was filtered and concentrated under reduced
pressure. Trace amounts of solvent were further removed under high vacuum over two
hours. A solution of the resultant mixed anhydride in toluene (100 mL) was divided into
two portions and simultaneously added via syringe pump to two solutions of DMAP
(0.73 g, 6.0 mmol) in toluene (600 mL) heated at reflux over 40 hours. The reaction
was concentrated under reduced pressure, diluted with diethyl ether, sequentially
washed with water, 1 M HCI, saturated NaHC03 solution, and brine, and dried over
anhydrous MgS04. The extracts were filtered and the solvent was removed under
reduced pressure. Column chromatography of the residue with 2% ethyl acetate in
petroleum ether gave lactone 189 (0.20 g, 44%) as a colourless oil.
IR(CCU): 2930, 2861, 1725, 1457, 1385, 1368, 1267, 1147, 1085 cm'1;
1H NMR (500 MHz, CDCI3): 8 2.22-2.24 (m, 2 H), 1.69-1.72 (m, 2 H), 1.56-1.61 (m, 2 H),
1.41 (s, 6H), 1.27-1.38 (m, 14 H);
13C NMR (125 MHz, CDCI3): 5 173.49, 82.92, 39.49, 35.94, 27.41, 26.72, 26.53,
26.46 (2), 25.43, 24.98 (2), 24.33, 21.03;
LRMS (El) m/z (relative intensity): 226 (M+, 6), 211 (11), 168(15), 153(11), 125(15),
111 (21), 98 (33), 83 (29), 69 (89), 56 (100), 43 (45), 41 (95);
HRMS (El) m/z calculated for Ci4H2602: 226.1933, found: 226.1930;
Analysis calculated for Ci4H2602: C, 74.29; H, 11.58. Found: C, 74.60; H, 11.52.

332
2,2-Dimethyloxacyclotridecane (190)
Boron trifluoride etherate (1.9 mL, 15 mmol) and sodium borohydride (0.13 g, 3.4 mmol)
were added to a solution of lactone 189 (0.11 g, 0.49 mmol) in THF (5 mL) at rt and the
reaction was stirred for 40 minutes. Triglyme (1.9 mL) was added, and the reaction
was stirred overnight at rt. The reaction was diluted with diethyl ether, and was
quenched with saturated NaHC03 solution (ca. 5 mL). The organic layer was washed
with saturated NaHC03 solution, water and brine, and dried over anhydrous MgS04.
The extracts were filtered and the solvent was removed under reduced pressure.
Column chromatography of the residue with 0.5% ethyl acetate in petroleum ether as
eluant gave ether 190 (27 mg, 26%) as a colourless oil.
IR (CCI4): 2926, 2858, 1459, 1382, 1362, 1136, 1086 cm"1;
1H NMR (500 MHz, CDCI3): 6 3.31 (t, J = 5.4 Hz, 2 H), 1.50-1.55 (m, 2 H), 1.29-1.44 (m,
18 H), 1.12 (s, 6 H);
13C NMR (125 MHz, CDCI3): 5 74.19, 59.96, 39.27, 29.26, 28.22, 26.66, 26.16 (2),
25.88, 25.77, 25.62, 25.49, 24.32, 21.18;
LRMS (El) m/z (relative intensity): 212 (M+, 1), 197 (11), 97 (12), 83 (18), 69 (25), 59
(100), 43(18), 41 (31);
HRMS (El) m/z calculated for C14H280: 212.2140, found: 212.2131.

333
2-Methyl-12-dodecanolide (191)
An LDA solution was generated by the addition of n-butyllithium in hexanes (17.1 mL,
20.0 mmol) to a solution of diisopropylamine (3.4 mL, 24 mmol) in THF (3.9 mL). The
solution was stirred at -78 °C for 15 minutes, warmed to 0 °C, and stirred for a further
15 minutes. An aliquot of this LDA solution (5.4 mL, 4.3 mmol) was added to a solution
of lactone 165 (0.65 g, 3.3 mmol) in THF (5 mL), and the reaction was stirred for four
hours at -78 °C. Methyl iodide (0.41 mL, 6.6 mmol) was added. The reaction was
stirred for 15 minutes, warmed to rt, and stirred for an additional 15 minutes. The
reaction was diluted with diethyl ether, sequentially washed with saturated NaHC03
solution, water, and brine, and dried over anhydrous MgS04. The extracts were filtered
and the solvent was removed under reduced pressure. Column chromatography of the
residue with 2% ethyl acetate in petroleum ether gave lactone 191 (0.30 g, 43%) as a
pale yellow oil.
IR(CDCI3): 2936, 2861, 1718, 1451, 1265, 1187, 1052, 829 cm-1;
1H NMR (500 MHz, CDCI3): 5 4.27 (ddd, J = 4.2, 6.6, 10.9 Hz, 1 H), 4.00 (ddd, J = 3.9,
5.8, 10.9 Hz, 1 H), 2.50 (ddq, J = 3.3, 9.9, 6.9 Hz, 1 H), 1.44-1.66 (m, 4 H), 1.24-
1.37 (m, 14 H), 1.12 (d, J = 6.9 Hz, 3 H);
13C NMR (125 MHz, CDCI3): 5 177.09, 64.42, 39.71, 33.75, 27.55, 26.58, 26.54, 25.49,
25.25, 24.83 (2), 24.16, 17.62;
LRMS (El) m/z (relative intensity): 212 (M+, 9), 194 (8), 138 (37), 112 (31), 110 (28), 98
(56), 96 (38), 83 (100), 69 (85), 55 (79);
HRMS (El) m/z calculated for Ci3H2402: 212.1776, found: 212.1776.

334
2,2-Dimethyl-12-dodecanolide (192)
An aliquot of an LDA solution (3.0 mL, 2.4 mmol) (see 191) was added to a solution of
lactone 191 (0.26 g, 1.2 mmol) in THF (10 mL) at -78 °C and the reaction was stirred for
nine hours. Methyl iodide (0.22 mL, 3.6 mmol) was added, and the reaction was stirred
for 15 minutes, warmed to rt, and stirred for an additional 15 minutes. The reaction was
diluted with diethyl ether, sequentially washed with saturated NaHC03 solution, water,
and brine, and dried over anhydrous MgS04. The extracts were filtered and the solvent
was removed under reduced pressure. Column chromatography of the residue with 2%
ethyl acetate in petroleum ether gave lactone 192 (0.19 g, 70%) as a pale yellow oil.
IR(CDCI3): 2936, 2861, 1715, 1451, 1257, 1180, 1027, 829 cm"1;
1H NMR (500 MHz, CDCI3): 8 4.10-4.12 (m, 2 H), 1.57-1.62 (m, 2 H), 1.49-1.52 (m, 2 H),
1.21-1.39 (m, 14 H), 1.13 (s, 6 H);
13C NMR (125 MHz, CDCI3): 5 178.14, 64.55, 42.77, 40.13, 27.75, 27.08, 26.59,
25.62 (2), 25.17, 24.76, 24.61, 23.91, 22.54;
LRMS (El) m/z (relative intensity): 226 (14), 208 (4), 139 (31), 124 (20), 97 (45), 88
(100), 83 (77), 69 (89), 55 (55);
HRMS (El) m/z calculated for Ci4H2602: 226.1933, found: 226.1927;
Analysis calculated for C14H2602: C, 74.29; H, 11.58. Found: C, 74.60; H, 11.66.

335
3,3-Dimethyloxacyclotridecane (193)
Boron trifluoride etherate (0.65 mL, 5.1 mmol) and sodium borohydride (45 mg,
1.2 mmol) were added to a solution of lactone 192 (39.0 mg, 0.17 mmol) in THF (5 mL)
stirred at rt. The reaction was stirred for 40 minutes at rt. Triglyme (0.65 mL) was
added and the reaction was heated at reflux for four hours. The reaction was diluted
with diethyl ether and was quenched with saturated NaHC03 solution (ca. 1 mL). The
organic layer was sequentially washed with saturated NaHC03 solution, water, and
brine, and dried over anhydrous MgS04. The extracts were filtered and the solvent was
removed under reduced pressure. Column chromatography of the residue with 0.5%
ethyl acetate in petroleum ether as eluant gave ether 193 (2.3 mg, 6%) as a pale yellow
oil.
IR(CDCI3): 2931, 2859, 1452, 1361, 1116 cm"1;
1H NMR (500 MHz, CDCI3): 5 3.40 (t, J = 5.3 Hz, 2 H), 3.05 (s, 2 H), 1.49-1.53 (br quint,
J = 5.3 Hz, 2 H), 1.19-1.44 (m, 16 H), 0.84 (s, 6 H);
13C NMR (125 MHz, CDCI3): 5 78.58, 71.06, 37.14, 34.22, 28.45, 27.72, 27.12, 26.42,
25.95 (2), 25.15, 25.08, 24.90, 20.96;
LRMS (Cl(+), ammonia) m/z (relative intensity): 230 (M++18, 3), 213 (M++1, 100);
HRMS (Cl(+), isobutane) m/z calculated for C14H290: 213.2219 (M++1), found:
213.2218.

R E F E R E N C E S
336
(1) Anderson, D. M.; White, A. W. Oceanus 1992, 35, 55 and references cited
therein.
(2) Nicolaou, K. C. Angew. Chem. Int. Ed. Engl. 1996, 35, 589.
(3) Shimizu, Y.; Choe, H.-N.; Bando, H.; Van Duyne, G.; Clardy, J. C. J. Am. Chem.
Soc. 1986, 708, 514.
(4) Pawlak, J.; Tempesta, M. S.; Golik, J.; Zagorski, M. G.; Lee, M. S.; Nakanishi, K.;
Iwashita, T.; Gross, M. L; Tomer, K. B. J. Am. Chem. Soc. 1987, 709, 1144.
(5) Zagorski, M. G.; Nakanishi, K.; Qin, G.-W.; Lee, M. S. J. Org. Chem. 1988, 53,
4156.
(6) Kelly, S. 1991, unpublished results.
(7) llluminati, G.; Mandolini, L. Acc. Chem. Res. 1981, 14, 95.
(8) Dale, J. Tetrahedron 1974, 30, 1683.
(9) Eckenberg, P.; Groth, U.; Huhn, T.; Richter, N.; Schmeck, C. Tetrahedron 1993,
49, 1619.
(10) Grubbs, R. H.; Miller, S. J.; Fu, G. C. Acc. Chem. Res. 1995, 28, 446.
(11) Fu, G. C; Grubbs, R. H. J. Am. Chem. Soc. 1992, 774, 5426.
(12) Villemin, D. Tetrahedron Lett. 1980, 27, 1715.
(13) Tsuji, J.; Hashiguchi, S. Tetrahedron Lett. 1980, 27, 2955.
(14) Tsuji, J.; Hashiguchi, S. J. Organomet. Chem. 1981, 278, 69.
(15) Mol, J. C; Woerlee, E. F. G. J. Chem. Soc. Chem. Commun. 1979, 330.
(16) Junga, H.; Blechert, S. Tetrahedron Lett. 1993, 34, 3731.
(17) Schrock, R. R.; Murdzek, J. S.; Bazan, G. C; Robbins, J.; DiMare, M.; O'Regan,
M. J. Am. Chem. Soc. 1990, 772, 3875.
(18) Schwab, P.; France, M. B.; Ziller, J. W.; Grubbs, R. H. Angew. Chem. Int. Ed.
Engl. 1995, 34, 2039.
(19) Koenig, B.; Horn, C. SyA?/etf1996, 1013.
(20) Yang, Z.; He, Y.; Vourloumis, D.; Vallberg, H.; Nicolaou, K. C. Angew. Chem. Int.
Ed. Engl. 1997, 36, 166.

337
(21) Fu, G. C.; Grubbs, R. H. J. Am. Chem. Soc. 1992, 114, 7324.
(22) Xu, Z.; Johannes, C. W.; Salman, S. S.; Hoveyda, A. H. J. Am. Chem. Soc.
1996, 118, 10926.
(23) Shon, Y.-S.; Lee, T. R. Tetrahedron Lett. 1997, 38, 1283.
(24) Clark, J. S.; Kettle, J. G. Tetrahedron Lett. 1997, 38, 127.
(25) Delgado, M.; Martin, J. D. Tetrahedron Lett. 1997, 38, 6299.
(26) Corey, E. J.; Nicolaou, K. C. J. Am. Chem. Soc. 1974, 96, 5614.
(27) Mukaiyama, T.; Usui, M.; Saigo, K. Chem. Lett. 1976, 49.
(28) Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. Bull. Chem. Soc.
Jpn. 1979, 52, 1989.
(29) Mitsunobu, O. Synthesis 1981, 1.
(30) Schmidt, U.; Dietsche, M. Angew. Chem. Int. Ed. Engl. 1981, 20, 771.
(31) Nakao, R.; Fukumoto, T.; Tsurugi, J. J. Am. Chem. Soc. 1969, 91, 4587.
(32) Nakao, R.; Fukumoto, T.; Tsurugi, J. J. Org. Chem. 1972, 37, 76.
(33) Meyer, W. L; Taylor, P. W.; Reed, S. A.; Leister, M. C; Schneider, H.-J.;
Schmidt, G.; Evans, F. E.; Levine, R. A. J. Org. Chem. 1992, 57, 291.
(34) Baldwin, S. W.; Doll , R. J.; Haut, S. A. J. Org. Chem. 1974, 39, 2470.
(35) Baldwin, S. W.; Haut, S. A. J. Org. Chem. 1975, 40, 3885.
(36) Dias, J. R.; Pettit, G. R. J. Org. Chem. 1971, 36, 3485.
(37) Pettit, G. R.; Evers, W. J. Can. J. Chem. 1966, 44, 1293.
(38) Tebbe, F. N.; Parshall, G. W.; Reddy, G. S. J.Am. Chem. Soc. 1978, 700, 3611.
(39) Pine, S. H.; Pettit, R. J.; Geib, G. D.; Cruz, S. G.; Gallego, C. H.; Tijerina, T.;
Pine, R. D. J. Org. Chem. 1985, 50, 1212.
(40) Carling, R. W.; Clark, J. S.; Holmes, A. B. J. Chem. Soc. Perkin Trans. 1 1992,
83.
(41) Corey, E. J.; Shimoji, K. J. J. Am. Chem. Soc. 1983, 705, 1662.
(42) Kraus, G. A.; Molina, M. T. J. Org. Chem. 1988, 53, 752.
(43) Nicolaou, K. C; Hwang, C. K.; Duggan, M. E.; Reddy, K. B.; Marron, B. E.;
McGarry, D. J. Am. Chem. Soc. 1986, 708, 6800.
(44) Henry, L. Ann. Chem. Pharm. 1869, 752, 148.
(45) Cava, M. P.; Levinson, M. I. Tetrahedron 1985, 41, 5061.

338
(46) Scheeren, J. W.; Ooms, P. H. J.; Nivard, R. J. F. Synthesis 1973, 149.
(47) Trebaul, C; Teste, J. Bull. Soc. Chim. Fr. 1970, 2272.
(48) Scheibye, S.; Kristensen, J.; Lawesson, S.-O. Tetrahedron 1979, 35, 1339.
(49) Lajoie, G.; Lepine, F.; Maziak, L; Belleau, B. Tetrahedron Lett. 1983, 24, 3815.
(50) Yokoyama, M.; Hasegawa, Y.; Hatanaka, H.; Kawazoe, Y.; Imamoto, T.
Synthesis 1984, 827.
(51) Nicolaou, K. C; McGarry, D. G.; Somers, P. K.; Kim, B. H.; Ogilvie, W. W.;
Yiannikouros, G.; Prasad, C. V. C; Veale, C. A.; Hark, R. R. J. Am. Chem. Soc.
1990, 112, 6263.
(52) Rauchfuss, T. B.; Zank, G. A. Tetrahedron Lett. 1986, 27, 3445.
(53) Perregaard, J.; Thomsen, I.; Lawesson, S.-O. Bull. Soc. Chim. Belg. 1977, 86,
321.
(54) Baxter, S. L; Bradshaw, J. S. J. Org. Chem. 1981, 46, 831.
(55) Nicolaou, K. C; McGarry, D. G.; Somers, P. K.; Vaele, C. A.; Furst, G. T. J. Am.
Chem. Soc. 1987, 109, 2504.
(56) Nicolaou, K. C; McGarry, D. G.; Sommers, P. K. J. Am. Chem. Soc. 1990, 112,
3696.
(57) Dynamic Nuclear Magnetic Resonance Spectroscopy; Jackman, L. M.; Cotton, F.
A. Ed.; Academic Press, New York, 1975; .
(58) Friebolin, H. Basic One- and Two-Dimensional NMR Spectroscopy; VCH, New
York, 1991; Chapter 11.
(59) Friebolin, H. Basic One- and Two-Dimensional NMR Spectroscopy; VCH, New
York, 1991; p268.
(60) Lambert, J. B.; Featherman, S. I. Chem. Rev. 1975, 75, 611.
(61) ApSimon, J. W.; Craig, W. G.; Demarco, P. V.; Mathieson, D. W.; Saunders, L;
Whalley, W. B. Tetrahedron 1967, 23, 2339.
(62) Bondi, A. J. Phys. Chem. 1964, 68, 441.
(63) Cheney, B. V. J. Am. Chem. Soc. 1968, 90, 5386.
(64) Winstein, S.; Carter, P.; Anet, F. A. L; Bourn, A. J. R. J. Am. Chem. Soc. 1965,
87, 5247.
(65) Carter, P.; Winstein, S. J. Am. Chem. Soc. 1972, 94, 2171.

339
(65) Carter, P.; Winstein, S. J. Am. Chem. Soc. 1972, 94, 2171.
(66) Grant, D. M.; Cheney, B. V. J. Am. Chem. Soc. 1967, 89, 5315.
(67) Cheney, B. V.; Grant, D. M. J. Am. Chem. Soc. 1967, 89, 5319.
(68) Aranda, G.; Bernassau, J.-M.; Fetizon, M.; Hanna, I. J. Org. Chem. 1985, 50,
1156.
(69) Murray-Rust, J.; Murray-Rust, P.; Parker, W. C; Tranter, R. L; Watt, C. I. F. J.
Chem. Soc. Perkin Trans. 2 1979, 1496.
(70) Ishiyama, J.; Senda, Y.; Imaizumi, S. Chem. Lett. 1983, 771.
(71) Buckingham, A. D. Can. J. Chem. 1960, 38, 300.
(72) Boaz, H. Tetrahedron Lett. 1973, 55.
(73) Gschwendtner, W.; Schneider, H.-J. J. Org. Chem. 1980, 45, 3507.
(74) Schneider, H.-J.; Buchheit, U.; Becker, N.; Schmidt, G.; Siehl, U. J. Am. Chem.
Soc. 1985, 107, 7027.
(75) Lambert, J. B.; Netzel, D. A.; Sun, H.-n.; Lilianstrom, K. K. J. Am. Chem. Soc.
1976, 98, 3778.
(76) Lambert, J. B.; Vagenas, A. R. Org. Magn. Reson. 1981, 77, 265.
(77) Lambert, J. B.; Vagenas, A. R. Org. Magn. Reson. 1981, 77, 270.
(78) Sachse, H. Chem. Ber. 1890, 23, 1363.
(79) Hueckel, W. Liebigs. Ann. Chem. 1925, 7, 441.
(80) Barton, D. H. R. Experientia 1950, 6, 316.
(81) Anet, F. A. L; Bourn, A. J. R. J. Am. Chem. Soc. 1967, 89, 760.
(82) Buys, H. R.; Geise, H. J. Tetrahedron Lett. 1970, 2991.
(83) Bovey, F. A.; Hood III, F. P.; Anderson, E. W.; Kornegay, R. L J. Chem. Phys.
1964, 41, 2041.
(84) Lambert, J. B.; Mixan, C. E.; Johnson, D. H. J. Am. Chem. Soc. 1973, 95, 4634.
(85) Lambert, J. B.; Goldstein, J. E. J. Am. Chem. Soc. 1977, 99, 5689.
(86) Ruzicka, L. Helv. Chim. Acta 1926, 9, 715.
(87) Ruzicka, L. Helv. Chim. Acta 1926, 9, 1008.
(88) Ruzicka, L; Stoll, M.; Huyser, H. W.; Boekenoogen, H. A. Helv. Chim. Acta
1930,73,1152.
(89) Ruzicka, L; Plattner, P. A.; Wild, H. Helv. Chim. Acta 1946, 29, 1611.

340
(90) Prelog, V. J. Chem. Soc. 1950, 420.
(91) Brockmann, H.; Henkel, W. Naturwissenschaften 1950, 37, 138.
(92) Woodward, R. B. Angew. Chem. 1957, 69, 50.
(93) Huber, E.; Dunitz, J. D.; Venkatesan, K. Proc. Chem. Soc. 1961, 463.
(94) Dale, J. J. Chem. Soc. 1963, 93.
(95) Dale, J. Angew. Chem. Int. Ed. Engl. 1966, 5, 1000.
(96) Dale, J. Acta Chem. Scand. 1973, 27, 1115.
(97) Anet, F. A. L; Cheng, K. J. Am. Chem. Soc. 1975, 97, 2420.
(98) Anet, F. A. L; Rawdah, T. N. J. Am. Chem. Soc. 1978, 100, 7810.
(99) Dale, J. Stereochemistry and Conformational Analysis; Verlag Chemie, New
York, 1978; p215.
(100) Saunders, M. Tetrahedron 1967, 23, 2105.
(101) Borgen, G.; Dale, J.; Teien, G. Acta Chem. Scand. B 1979, 33, 15.
(102) Neeland, E. G. Ph. D. Thesis, University of British Columbia, 1987.
(103) Ogura, H.; Furuhata, K.; Kuwano, H.; Harada, N. J. Am. Chem. Soc. 1975, 97,
1930.
(104) Furusaki, A.; Matsumoto, T.; Furuhata, K.; Ogura, H. Bull. Chem. Soc. Jpn.
1982, 55, 59.
(105) Dale, J. Acta Chem. Scand. 1973, 27, 1149.
(106) Groth, P. Acta Chem. Scand. A 1976, 30, 155.
(107) Anet, F. A. L; Cheng, A. K.; Wagner, J. J. J. Am. Chem. Soc. 1972, 94, 9250.
(108) Drotloff, H.; Rotter, H.; Emeis, D.; Moeller, M. J. Am. Chem. Soc. 1987, 109,
7797.
(109) Shannon, D. L; Strauss, H. L ; Snyder, R. G.; Elliger, C. A.; Mattice, W. L. J.
Am. Chem. Soc. 1989, 89, 1947.
(110) Bassi, I. W.; Scordamaglia, R.; Fiore, L. J. C. S. Perkin Trans. I11972, 1726.
(111) Groth, P. Acta Chem. Scand. A 1975, 29, 374.
(112) Groth, P. Acta Chem. Scand. A 1979, 33, 503.
(113) Dale, J. Top. Stereochem. 1976, 9, 199.
(114) Flippen, J. L. Acta Cryst. 1972, B28, 3618.
(115) Sim, G. A. Acta Cryst. 1987, C43, 778.

341
(116) Rubin, B. H.; Williamson, M.; Takeshita, M.; Menger, F. M.; Anet, F. A. L ;
Bacon, B.; Allinger, N. L. J. Am. Chem. Soc. 1984, 706, 2088.
(117) Anet, F. A. L; Anet, R. I. Dynamic Nuclear Magnetic Resonance Spectroscopy;
Academic Press, New York, 1975; Chapter 14.
(118) Dale, J. Acta Chem. Scand. 1973, 27, 1130.
(119) Jeffs, P. W.; Molina, G.; Cass, M. W.; Cortese, N. A. J. Org. Chem. 1982, 47,
3871.
(120) Farcasiu, D.; Jaehme, J.; Ruechardt, C. J. Am. Chem. Soc. 1985, 707, 5717.
(121) Lu, C.-S.; Hughes, E. W.; Giguere, P. A. J. Am. Chem. Soc. 1941, 63, 1507.
(122) Cooper, M. S.; Heaney, H.; Newbold, A. J.; Sanderson, W. R. Syn/eff 1990, 533.
(123) Heaney, H. Aldrichimica Acta 1993, 26, 35.
(124) Wiberg, K. B.; Boyd, R. H. J. Am. Chem. Soc. 1972, 94, 8426.
(125) Taguchi, H.; Yamamoto, H.; Nozaki, H. Bull. Chem. Soc. Jpn. 1977, 50, 1588.
(126) Lermer, L. Ph. D. Thesis, University of British Columbia, 1998.
(127) Taguchi, H.; Yamamoto, H.; Nozaki, H. Bull. Chem. Soc. Jpn. 1977, 50, 1592.
(128) Hoeger, C. A.; Johnston, A. D.; Okamura, W. H. J. Am. Chem. Soc. 1987, 709,
4690.
(129) Corey, E. J.; Lee, J.; Liu, D. R. Tetrahedron Lett. 1994, 35, 9149.
(130) Friedrich, E.; Kalinowski, H.-O.; Lutz, W. Tetrahedron 1980, 36, 1051.
(131) Nonoshita, K.; Banno, H.; Maruoka, K.; Yamamoto, H. J. Am. Chem. Soc. 1990,
772, 316.
(132) Maruoka, K.; Ooi, T.; Nagahara, S. Tetrahedron 1991, 47, 6983.
(133) Maruoka, K.; Sata, J.; Yamamoto, H. J. Am. Chem. Soc. 1992, 774", 4422.
(134) Giese, B.; Kopping, B. Tetrahedron Lett. 1989, 30, 681.
(135) Cannizzo, L. F.; Grubbs, R. H. J. Org. Chem. 1985, 50, 2386.
(136) Weast, R. C; Astle, M. J.; Beyer, W. H. CRC Handbook of Chemistry; 66th ed.;
CRC Press Inc., Boca Raton, 1985; p F-167.
(137) Schumann, H.; Wasserman, B. C; Hahn, F. E. Organometallics 1992, 77, 2803.
(138) Suginome, H.; Yamada, S. J. Org. Chem. 1985, 50, 2489.
(139) Larock, R. C. Comprehensive Organic Transformations; VCH Publishers, Inc.,
New York, 1989; p 843.

342
(140) Sharadendu, A. Ph. D. Thesis, University of British Columbia, 1996.
(141) Maurice, J. R. L. Ph.D. Thesis, University of British Columbia, 1991.
(142) Still, I. W. J.; Hasan, S. K.; Turnbull, K. Can. J. Chem. 1977, 56, 1423.
(143) Baechler, R. D.; Daley, S. K. Tetrahedron Lett. 1978, 101.
(144) Nicolaou, K. C; Hwang, C.-K.; Duggan, M. E.; Nugiel, D. A.; Abe, Y.; Bal Reddy,
K.; DeFrees, S. A. J. Am. Chem. Soc. 1995, 117, 10227.
(145) Kang, S.-K.; Kim, W.-S.; Moon, B.-H. Synthesis 1985, 1161.
(146) Rama Rao, A. V.; Deshmukh, M. N.; Sharma, G. V. M. Tetrahedron 1987, 43,
779.
(147) Miyashita, N.; Yoshikoshi, A.; Grieco, P. A. J. Org. Chem. 1977, 42, 3722.
(148) Corey, E. J.; Erickson, B. W. J. Org. Chem. 1971, 36, 3553.
(149) Cain, E. N.; Welling, L. L. Tetrahedron Lett. 1975, 1353.
(150) Bernardi, R.; Ghiringhelli, D. J. Org. Chem. 1987, 52, 5021.
(151) Robinson, R. A.; Clark, J. S.; Holmes, A. B. J. Am. Chem. Soc. 1993, 115,
10400.
(152) Pine, S. H.; Zahler, R.; Evans, D. A.; Grubbs, R. H. J. Am. Chem. Soc. 1980,
102, 1980.
(153) Cannizzo, L. F.; Grubbs, R. H. J. Org. Chem. 1985, 50, 2386.
(154) Meyers, A. I.; Reider, P. J.; Campbell, A. L. J. Am. Chem. Soc. 1980, 102, 6597.
(155) Mori, K.; Tomioka, H. LiebigsAnn. Chem. 1992, 1011.
(156) Denmark, S. E.; Edwards, J. P. J. Org. Chem. 1991, 56, 6974.
(157) Denmark, S. E.; Edwards, J. P.; Wilson, S. R. J. Am. Chem. Soc. 1992, 114,
2592.
(158) Piers, E.; Coish, P. D. Synthesis 1995, 47.
(159) Simmons, H. E.; Smith, R. D. J. Am. Chem. Soc. 1959, 81, 4256.
(160) Simmons, H. E.; Cairns, T. L; Vladuchick, S. A.; Hoiness, C. M. Organic
Reactions 1973, 20, 1.
(161) Buss, V.; Gleiter, R.; v. R. Schleyer, P. J. Am. Chem. Soc. 1970, 93, 3927.
(162) Rylander, P. N. Hydrogenation Methods; Academic Press, London, 1985; p 174.
(163) Omura, K.; Swern, D. Tetrahedron 1978, 34, 1651.
(164) Mancuso, A. J.; Swern, D. Synthesis 1981, 165.

343
(165) Bal, B. S.; Childers Jr., W. E.; Pinnick, H. W. Tetrahedron 1981, 37, 2091.
(166) Schwab, P.; Grubbs, R. H.; Ziller, J. W. J. Am. Chem. Soc. 1996, 118, 100.
(167) Shon, Y.-S.; Lee, T. R. Tetrahedron Lett. 1997, 38, 1283.
(168) Silverstein, R. M.; Bassler, G. C; Morrill, T. C. Spectrometric Identification of
Organic Compounds; 5th ed.; John Wiley & Sons, Inc., New York, 1991, p 221.
(169) Bernstein, H. J.; Powling, J. J. Am. Chem. Soc. 1951, 73, 1843.
(170) Kuivila, H. G.; Sommer, R. J. Am. Chem. Soc. 1967, 89, 5616.
(171) Chatgilialoglu, C.; Ballestri, M.; Ferreri, C.; Vecchi, D. J. Org. Chem. 1995, 60,
3826.
(172) Ferreri, C.; Ballestri, M.; Chatgilialoglu, C. Tetrahedron Lett. 1993, 34, 5147.
(173) Sonnet, P. E. Tetrahedron 1980, 36, 557.
(174) Sonnet, P.E.J. Org. Chem. 1974, 39, 3793.
(175) Moussebois, C; Dale, J. J. Chem. Soc. C 1966, 260.
(176) Dale, J.; Moussebois, C. J. Chem. Soc. C1966, 264.
(177) Murov, S. L.; Carmichael, I.; Hug, G. L. Handbook of Photochemistry; 2nd ed.;
Dekker, New York, 1993, p 317.
(178) Bowers, A.; Halsall, T. G.; Jones, E. R. H.; Lemin, A. J. J. Chem. Soc. 1953,
2548.
(179) Eisenbraun, E. J. Organic Synthesis 1965, 45, 28.
(180) Rubottom, G. R.; Kim, C.-W. J. Org. Chem. 1983, 48, 1550.
(181) Fuerstner, A.; Langemann, K. J. Org. Chem. 1996, 61, 3942.
(182) Still, W. C; Galynker, I. Tetrahedron 1981, 37, 3981.
(183) Aldrich Catalogue Handbook of Fine Chemicals, 1996, p 414.
(184) Aldrich Catalogue Handbook of Fine Chemicals, 1996, p 430.
(185) Perrin, D. D.; Armarego, W. L. F. Purification of Laboratory Chemicals; 3rd. ed.;
Pergamon Press, New York, 1988; .
(186) Winkle, M. R.; Lansinger, J. M.; Ronald, R. C. J.C.S. Chem. Comm. 1980, 87.
(187) Lu, C.-S.; Hughes, E. W.; Giguere, P. A. J. Am. Chem. Soc. 1941, 63, 1507.
(188) Kuivila, H. G.; Beumel, O. F. J. Am. Chem. Soc. 1961, 83, 1246.
(189) Shank, R.; Shechter, H. J. Org. Chem. 1959, 24, 1825.
(190) Still, W. C; Kahn, M.; Mirta, A. J. Org. Chem. 1978, 43, 2923.

(191) Mohamadi, F.; Richards, N. G. J.; Guida, W. C.; Liskamp, R.; Lipton, M.;
Caufield, C.; Chang, G.; Hendrickson, T.; Still, W. C. J. Comput. Chem. 1990,
11, 440.
(192) Allinger, N. L. J. Am. Chem. Soc. 1977, 99, 8127.
(193) Allinger, N. L; Yuh, Y. H.; Lii, J.-H. J. Am. Chem. Soc. 1989, 111, 8551.

SPECTRAL APPENDIX
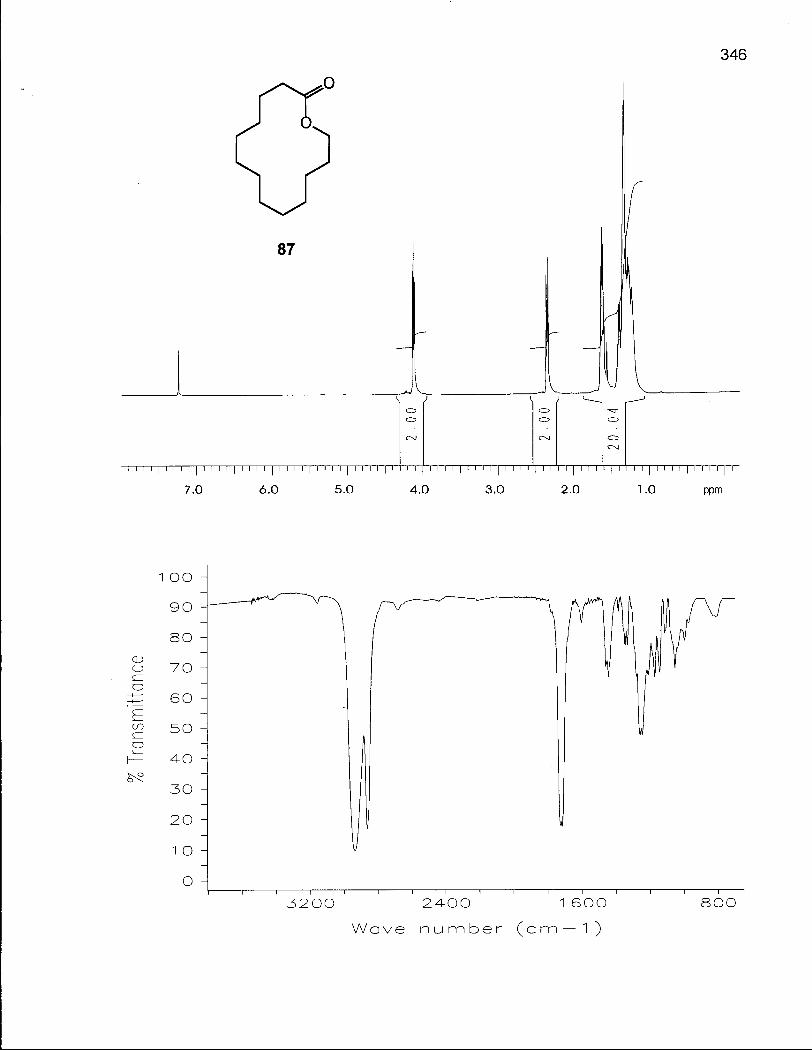
346
1 O O H
o -1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
3 2 0 0 2 4 0 0 1 6 0 0 S O O
W a v e n u m b e r ( c m — 1 )
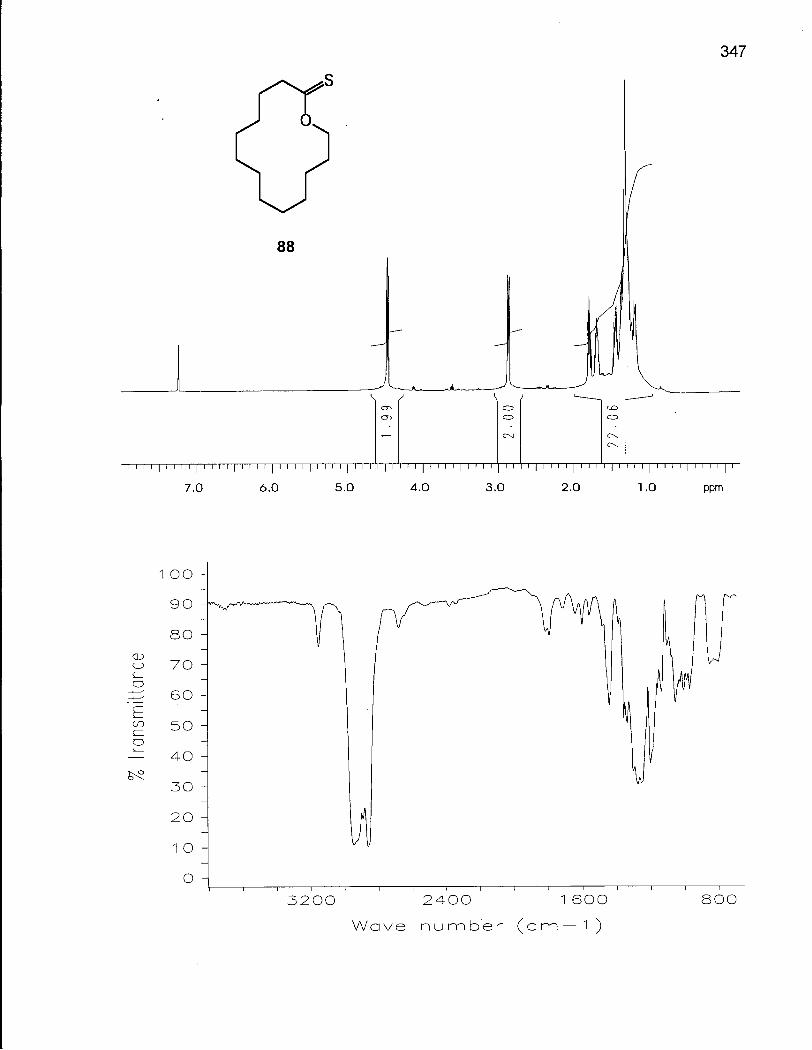
1 o o H
° i \ — i — i — i — i — i — i — i — i — i — i — i — i i i i
3 2 0 0 2 4 0 0 1 6 0 0 8 0 0 W a v e n u m b e r ( c m — 1 )
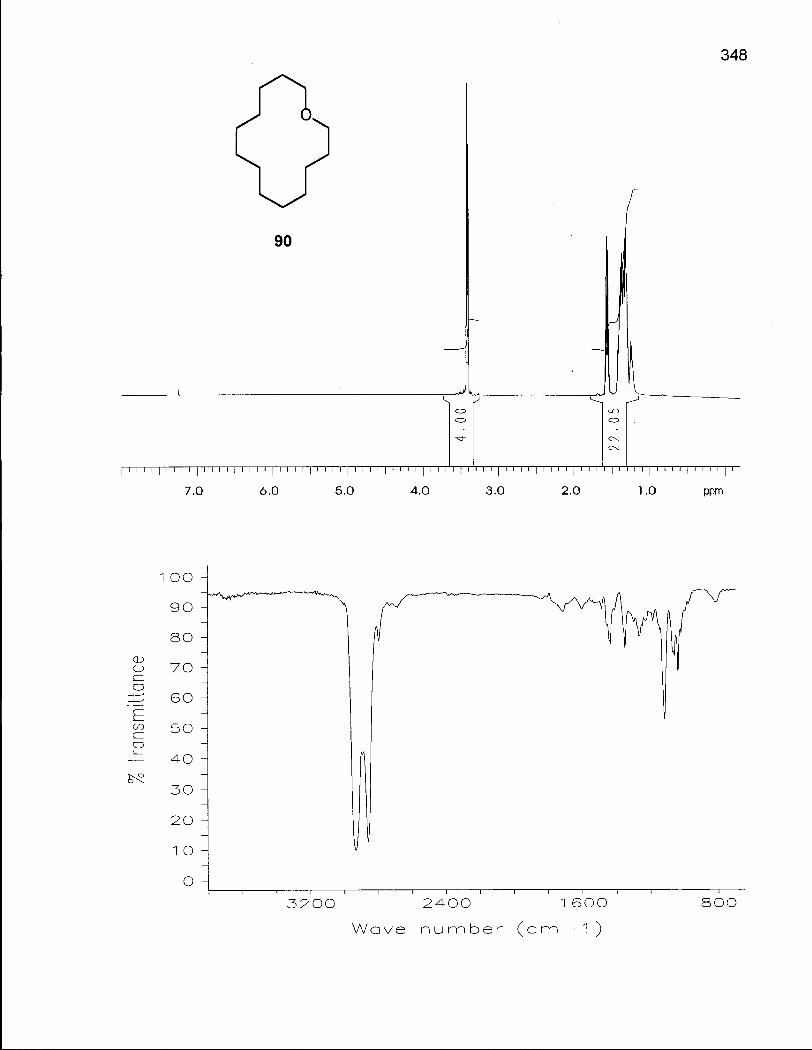
1 OO H
o -1 1 1 1 1 1 1 1 1 I I I I I I
3 2 0 0 2 4 0 0 1 6 0 0 SOO W a v e n u m b e r ( c m — 1 )
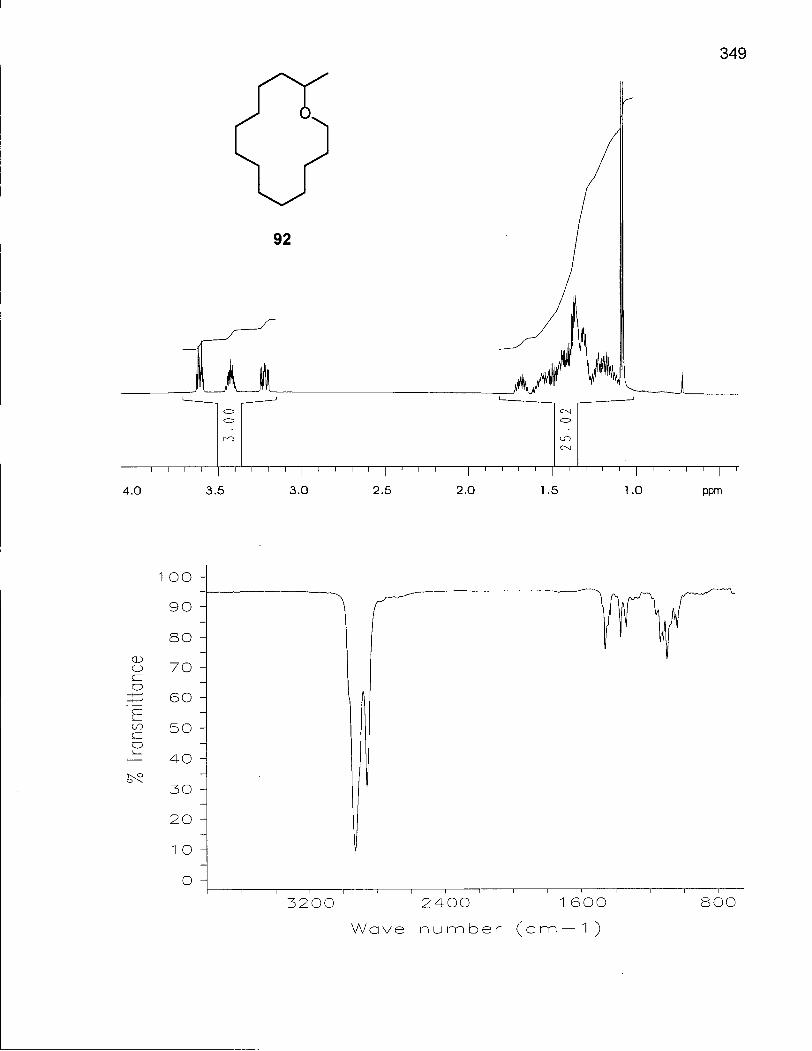
349

350
OTMS
95
i i i i i | i i i 11 i i i i | i i i 1 1 1 i i i | i i 11 11 11 i | i i i i 11 i i i | 1 1 1 1 | 1 1 i i | 11 1 1 | i i i 1 1
7.0 6.0 5.0 4.0 3.0 2.0 1.0 ppm
1 OO
90
80
8 70 H
60
£2 50 o
H = 40 H
30
20 -
1 O -
O i 1 1 1 1 1 1 1 1 1 1 1 i i
3 2 0 0 2 4 0 0 1 6 0 0 SOO W a v e n u m b e r ( c m — 1 )
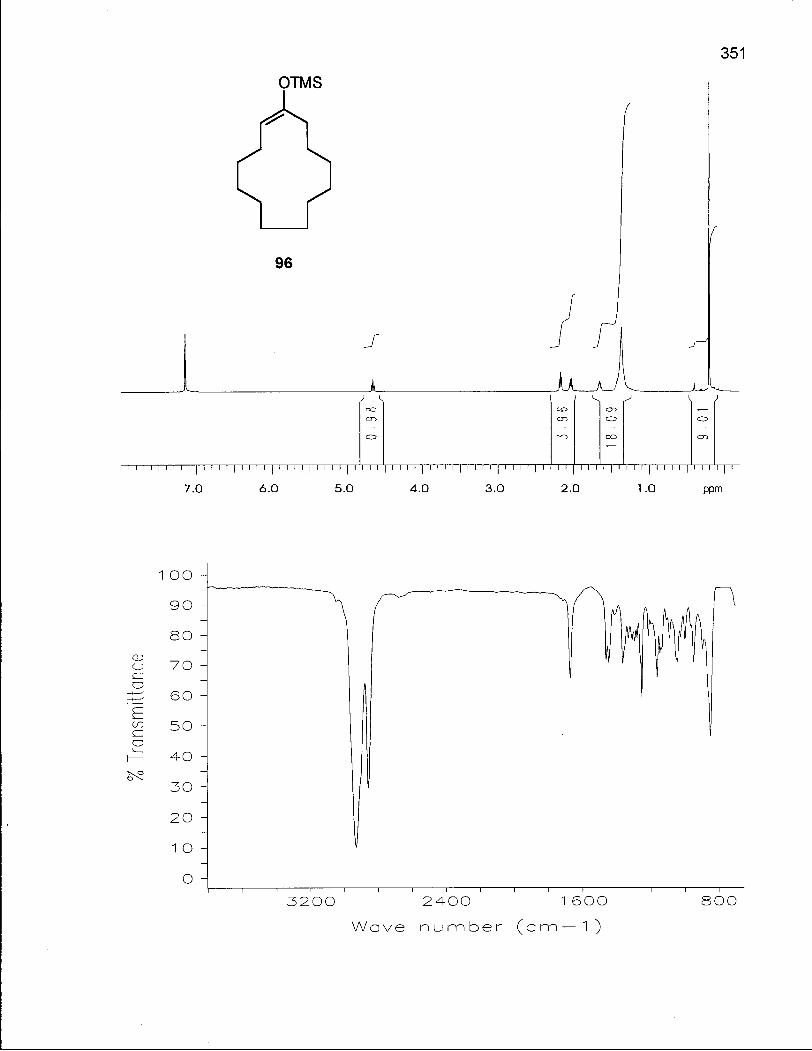
351
OTMS
7.0 6 .0 5 .0 4 .0 3 .0 2 .0 1.0 ppm
1 O O -\
o -1 1 1 1 1 1 i 1 1 1 1 1 1 1 1 1
3 2 0 0 2 4 0 0 1 6 0 0 8 0 0 W a v e n u m b e r ( c m — 1 )
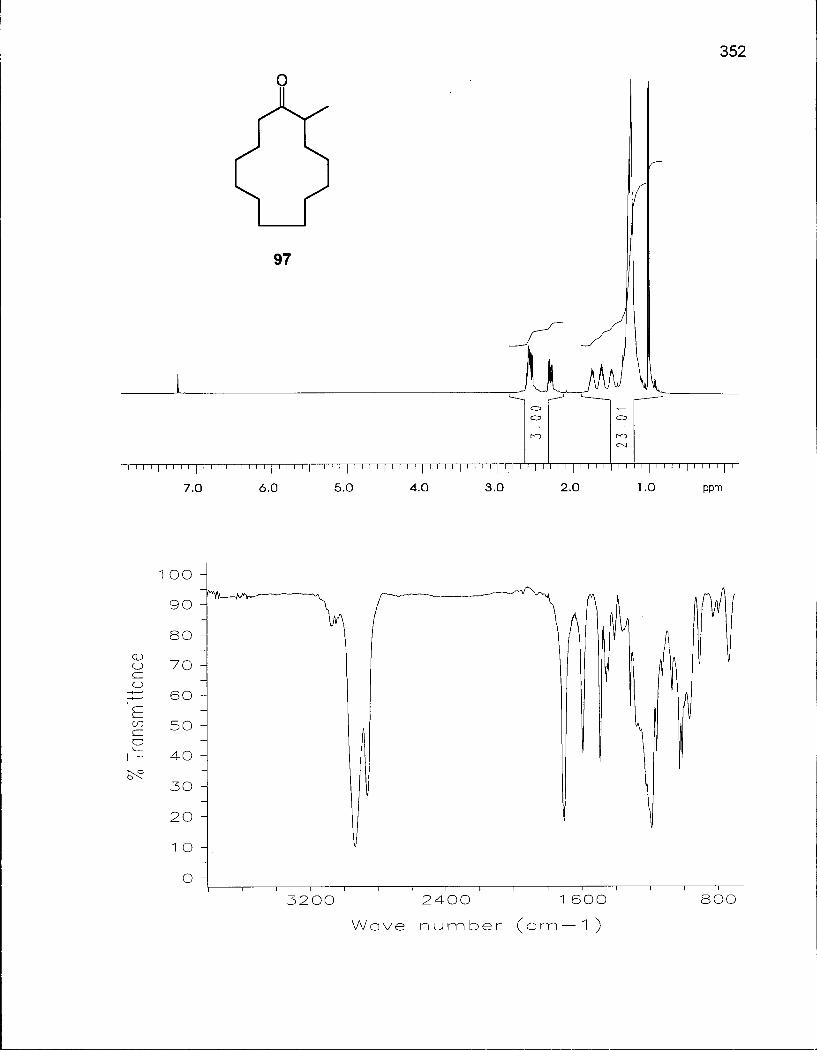
352

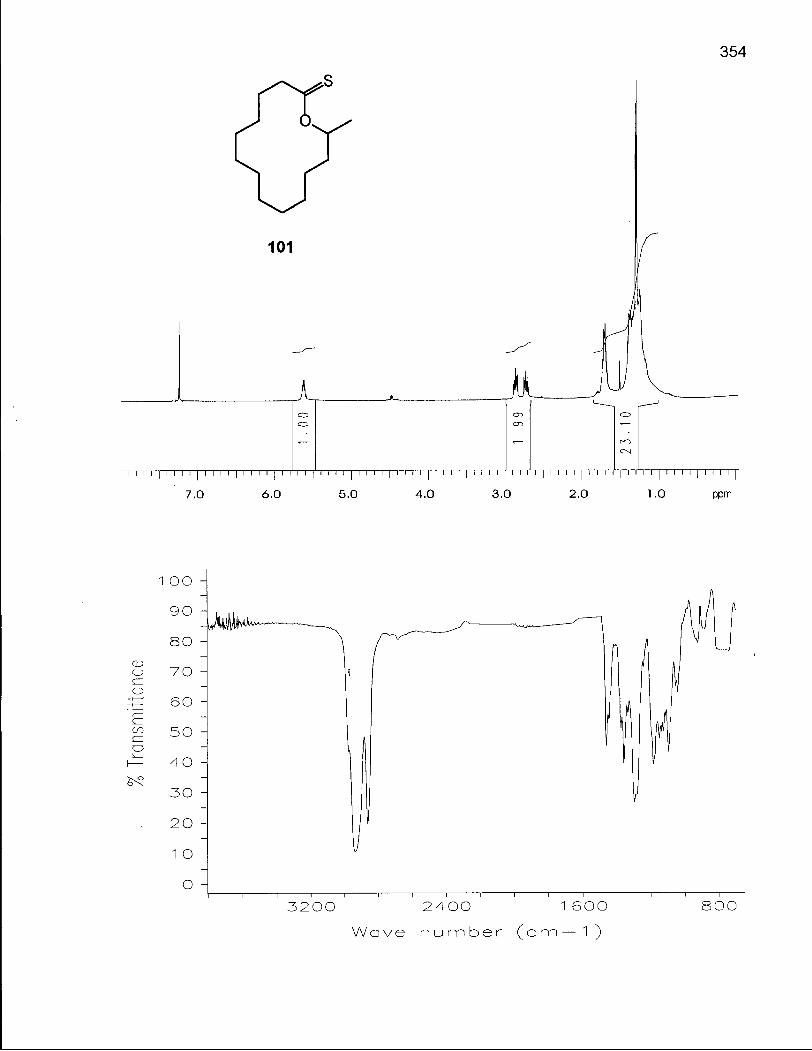
354
1 o o -{
— i 1 1 1 1 1 1 1 1 1 1 i i
3 2 0 0 2 4 0 0 1 6 0 0 S O O W a v e n u m b e r ( c m — 1 )
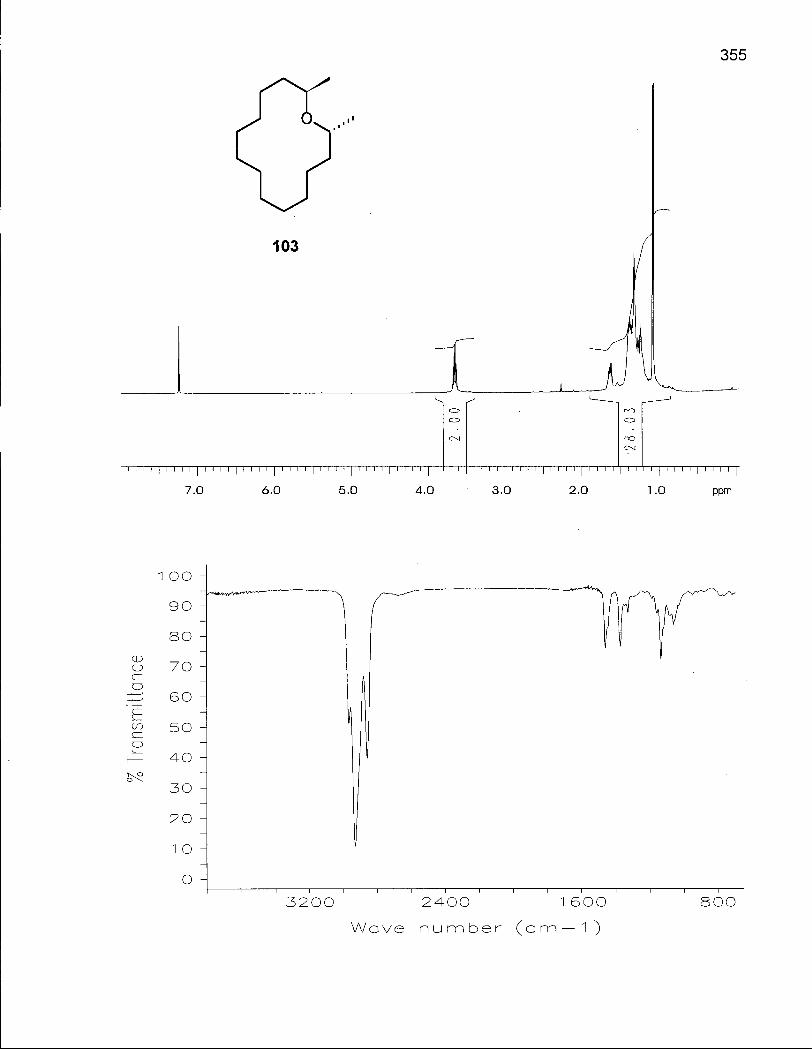
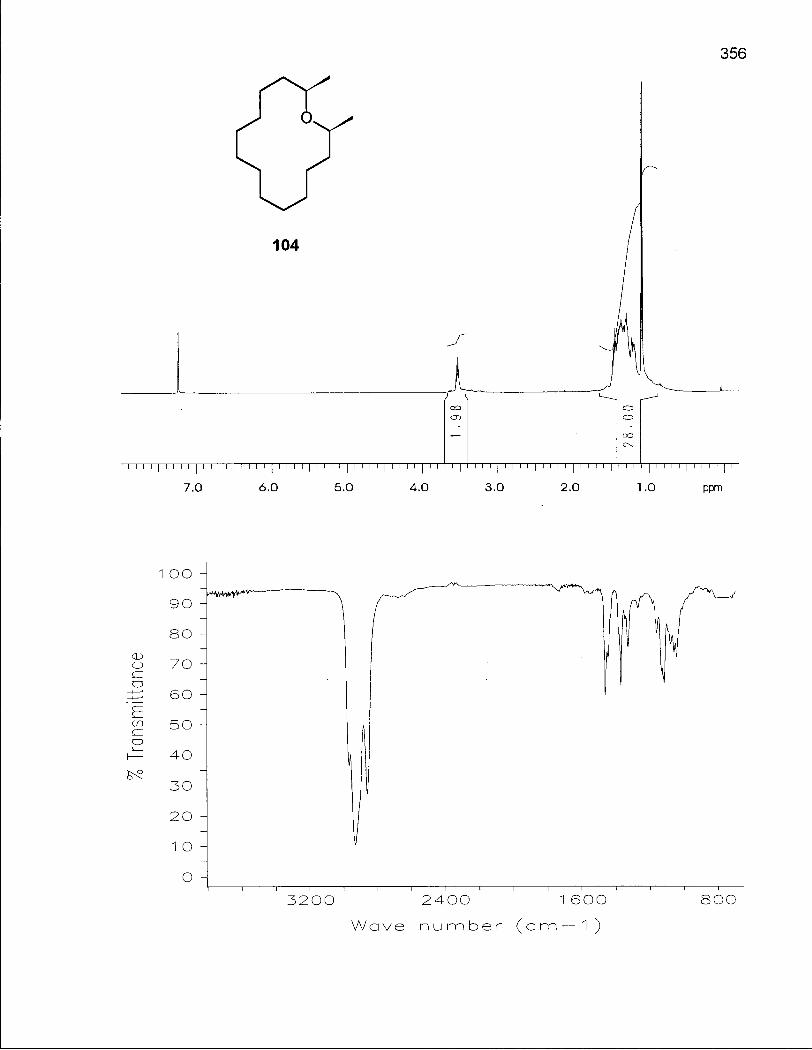
356
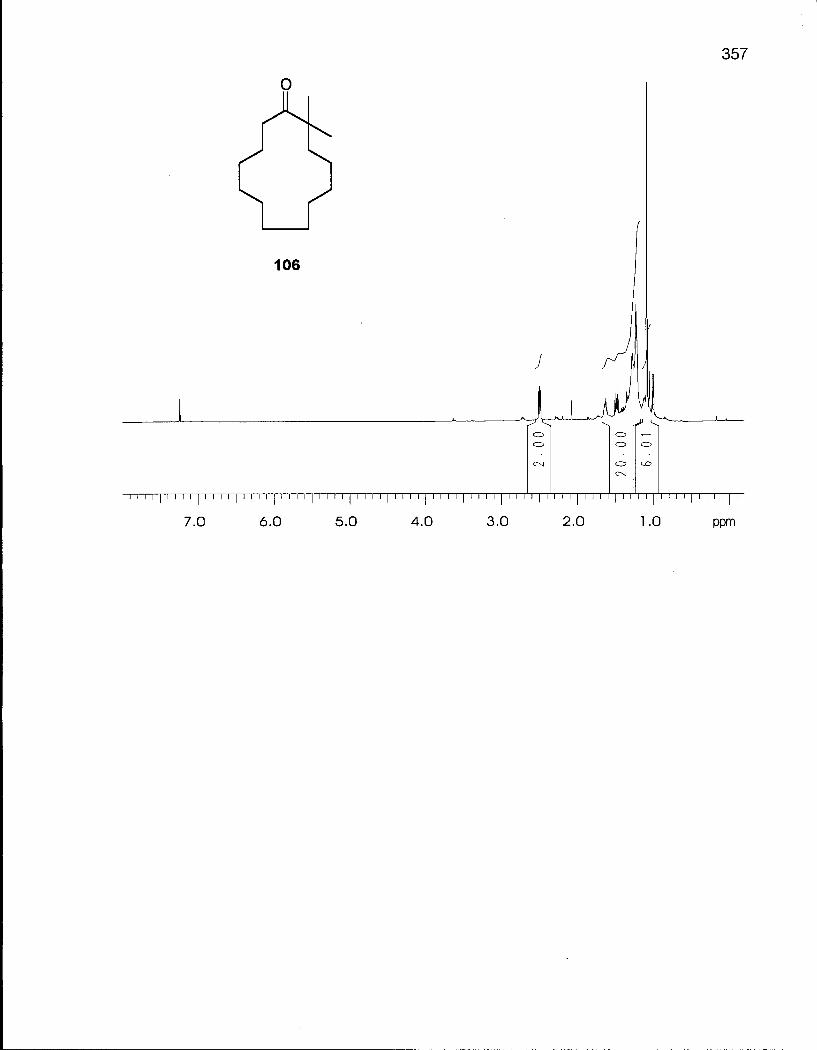
357
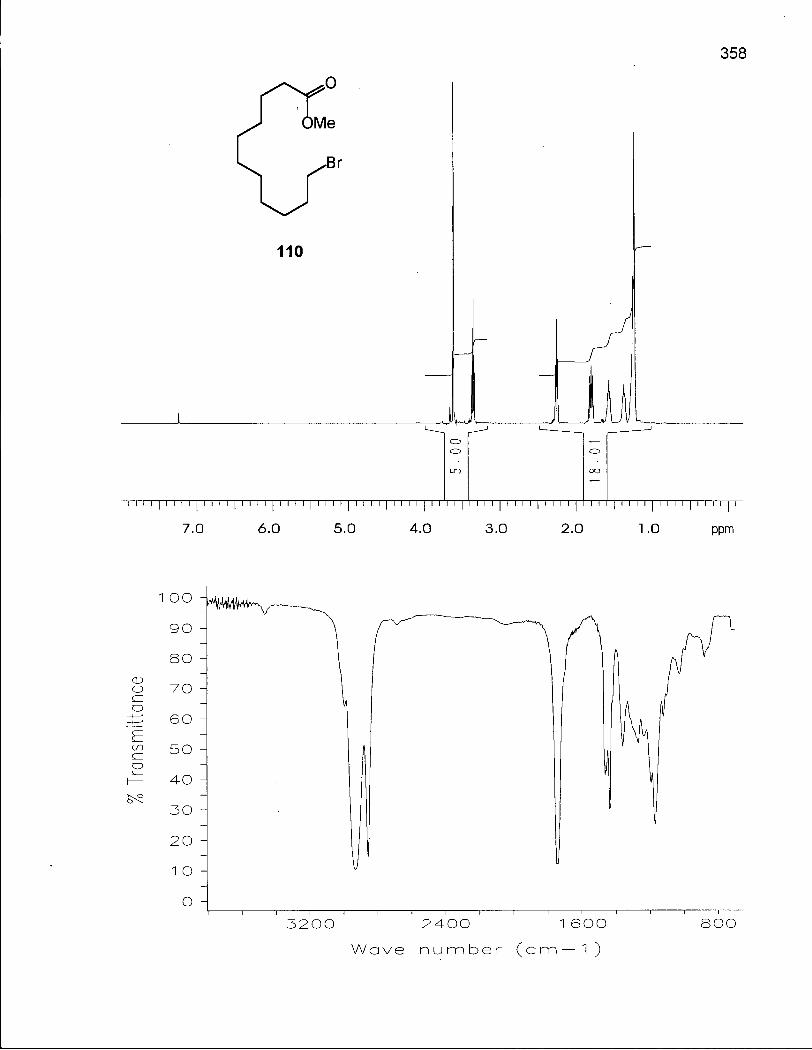
359
1 o - •
o -1 1 1 1 1 1 1 1 I I I I I I I
3 2 0 0 2 4 0 0 1 6 0 0 8 0 0 W a v e n u m b e r ( c m — 1 )
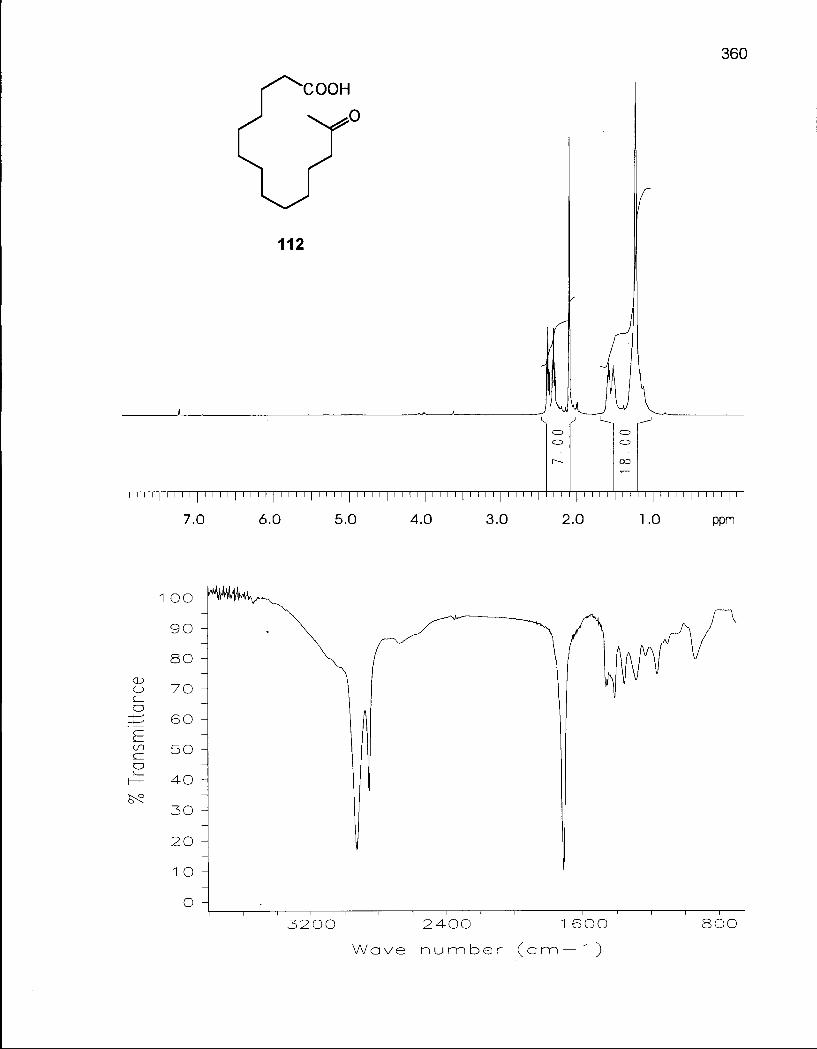
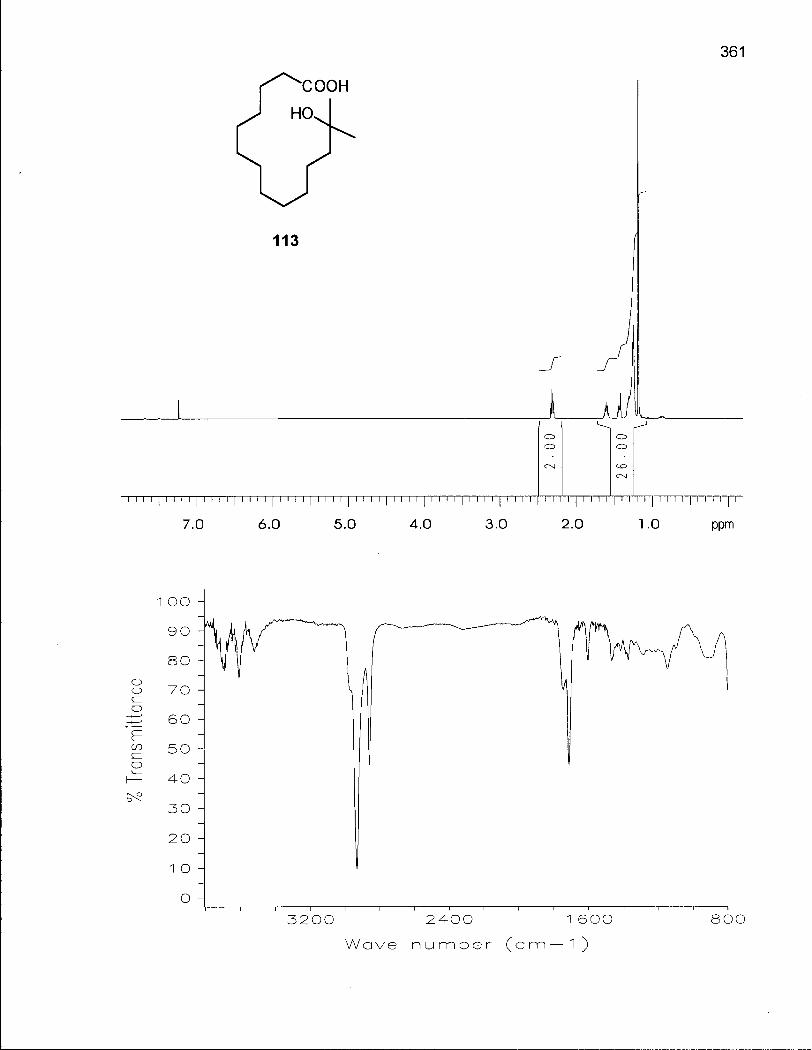
361
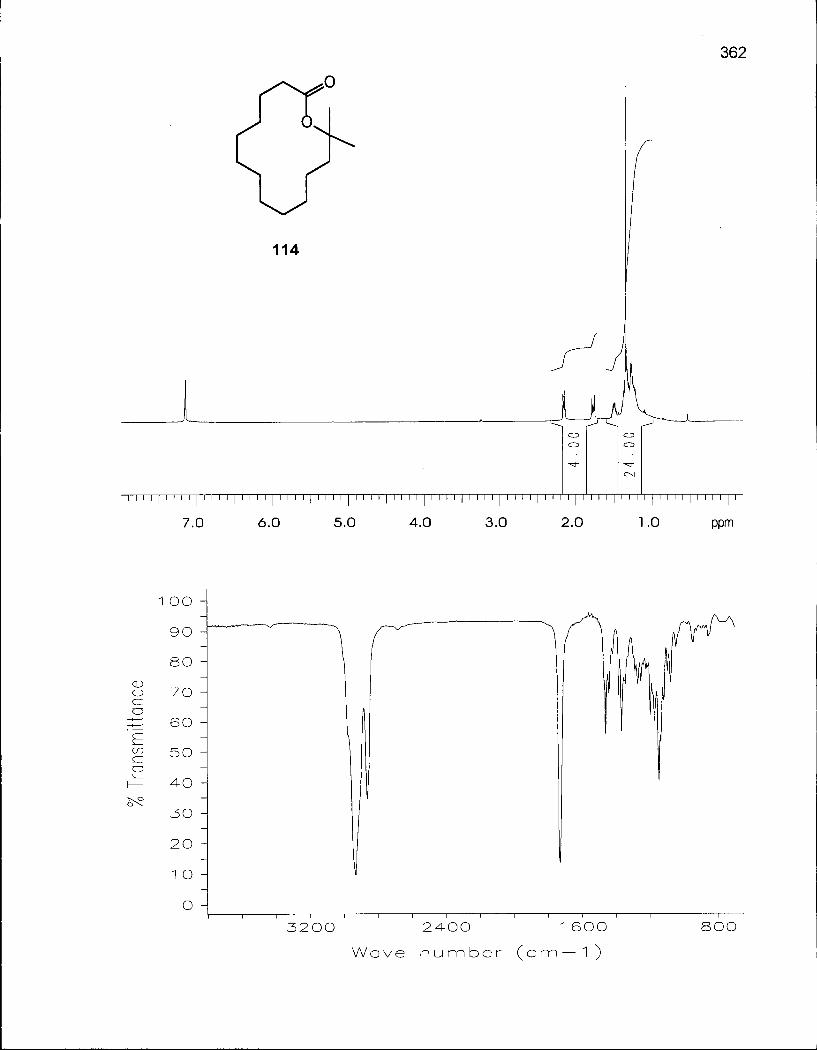
362
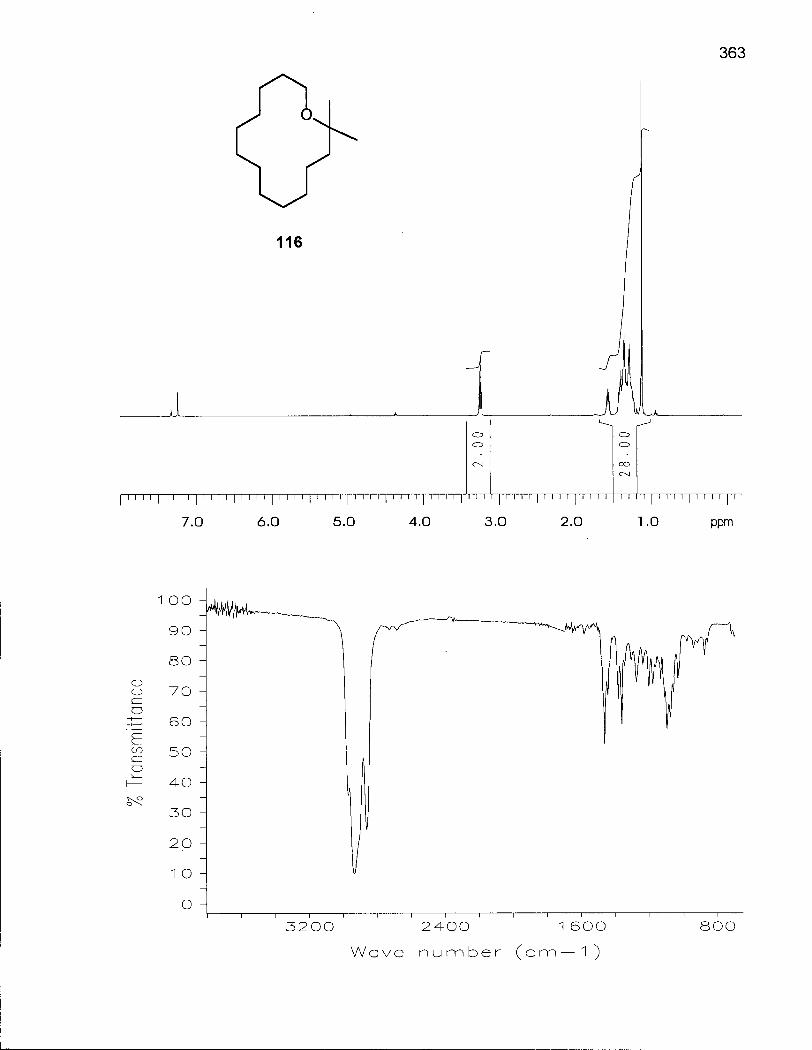
363
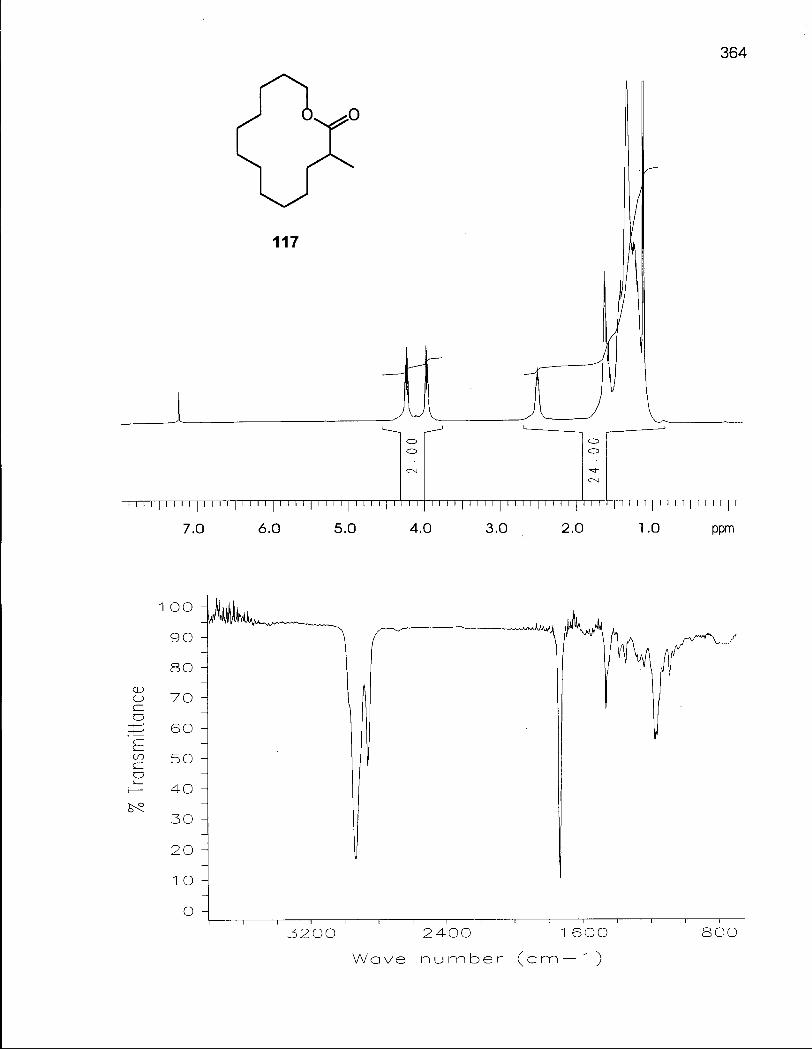
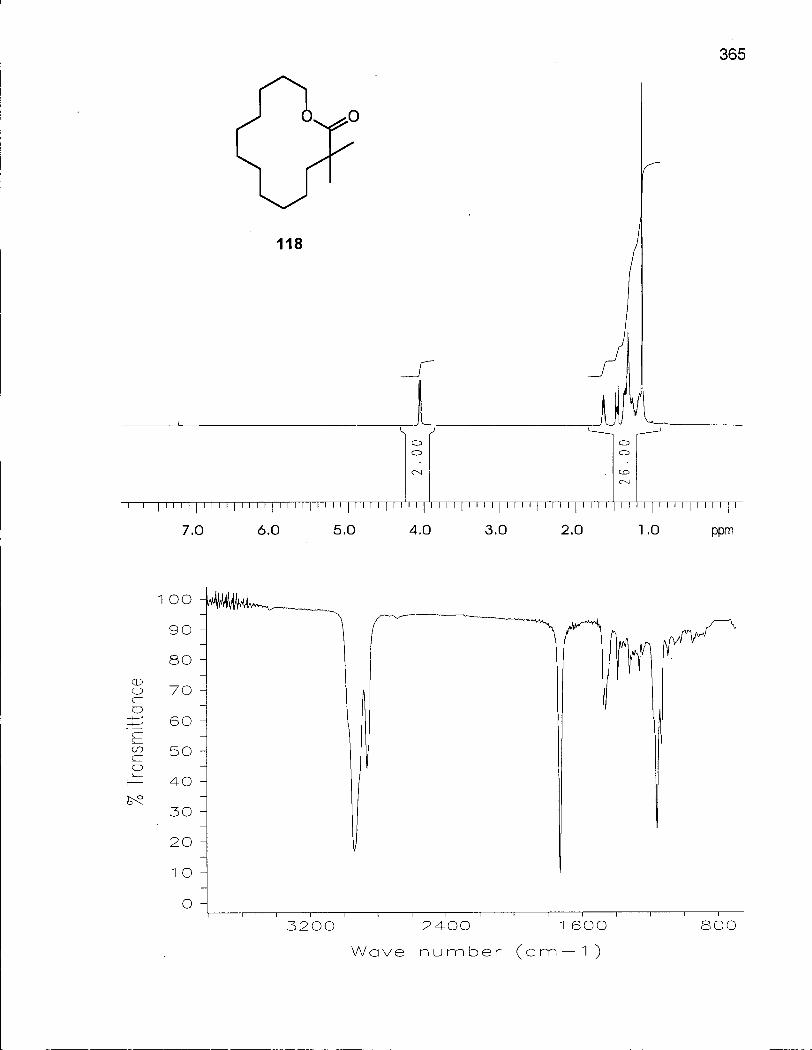
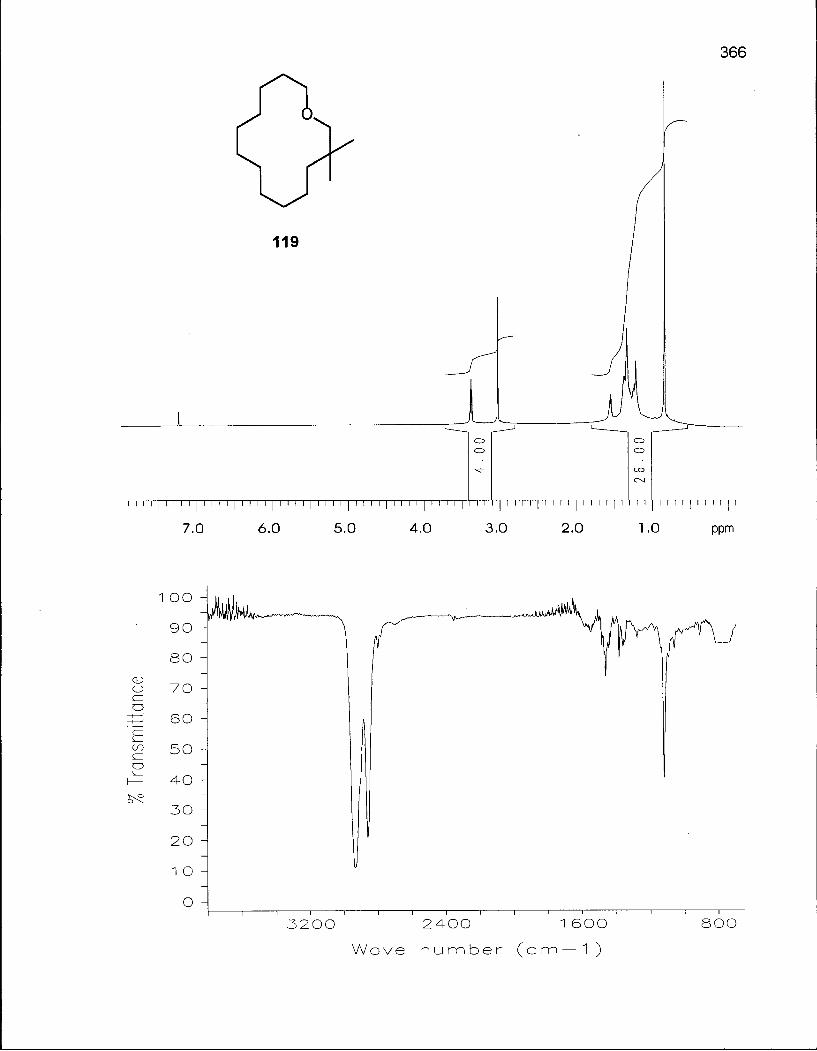
366
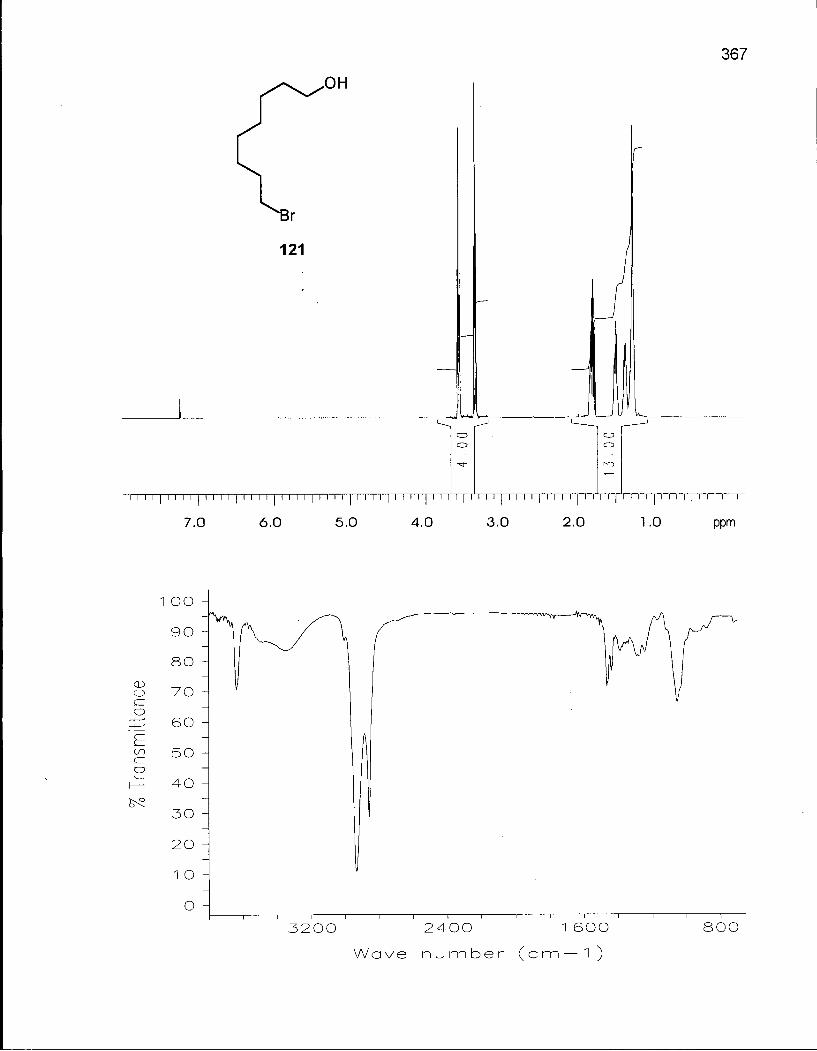
367
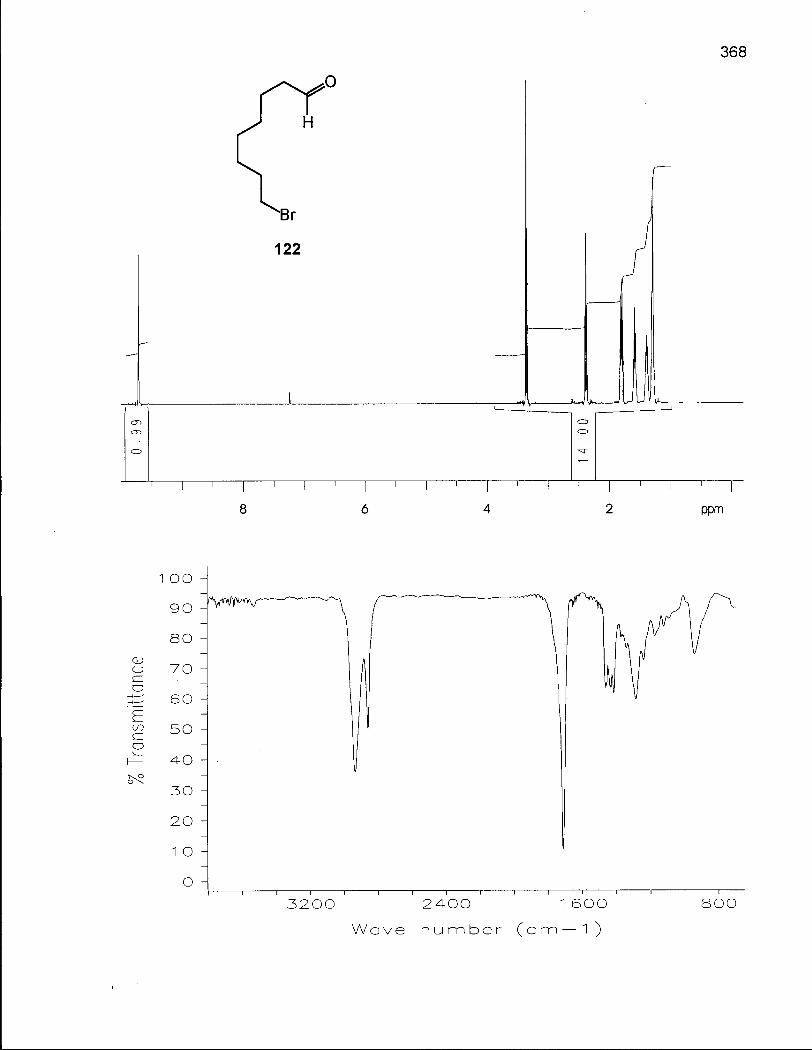
368
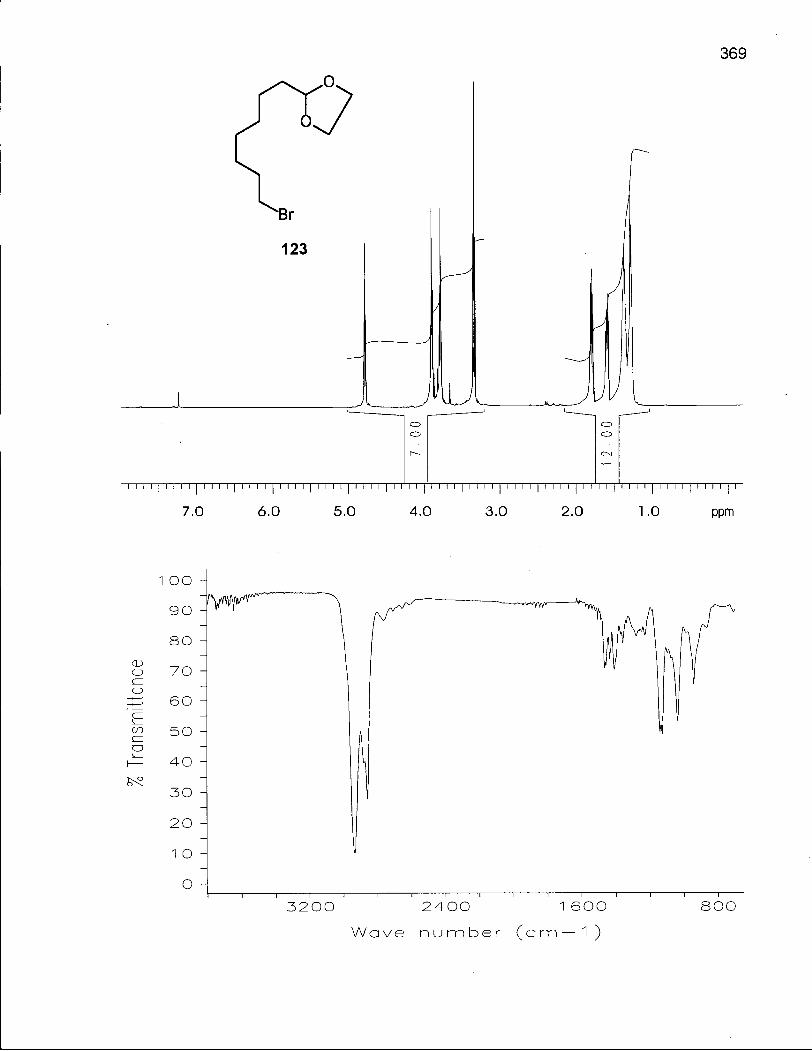
369
W
C 5 O
C N
1 1 1 1 j I 1 1 1 1 1 1 1 1
7 . 0
1 i i i i j i i i i
6 . 0
1 i i I I 1 i i i i | i i
5 . 0
M | I I I I
4 . 0
1 i i i i 1 i i i i
3 . 0
1 i i i i | i i
2 . 0
1 1 1 1 1 1 1 | 1 1 1 1
1 .0
I 1
ppm
o
Ul
p
OO
9 0
S O
7 0
6 0 H
5 0
4 0
3 0 H
2 0
1 o H
o n 1 r
3 2 0 0 2 4 0 0 1 6 0 0
W a v e n u m b e r ( c m — 1 )
S O O
371
1 oo H
o -I I 1 1 1 1 1 1 1 1 I I I I I I
3 2 0 0 2 4 0 0 1 6 0 0 S O O W a v e n u m b e r ( c m — 1 )

372

373
C N
I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I I I I I I I I M I | I I I I | I
7.0 6.0 5.0 4.0 3.0 2.0 1.0 ppm
1 o o H
o -\ 1 1 1 1 1 1 1 1 1 1 1 1 1 i i
3 2 0 0 2 4 0 0 1 6 0 0 8 0 0 W a v e n u m b e r ( c m — 1 )
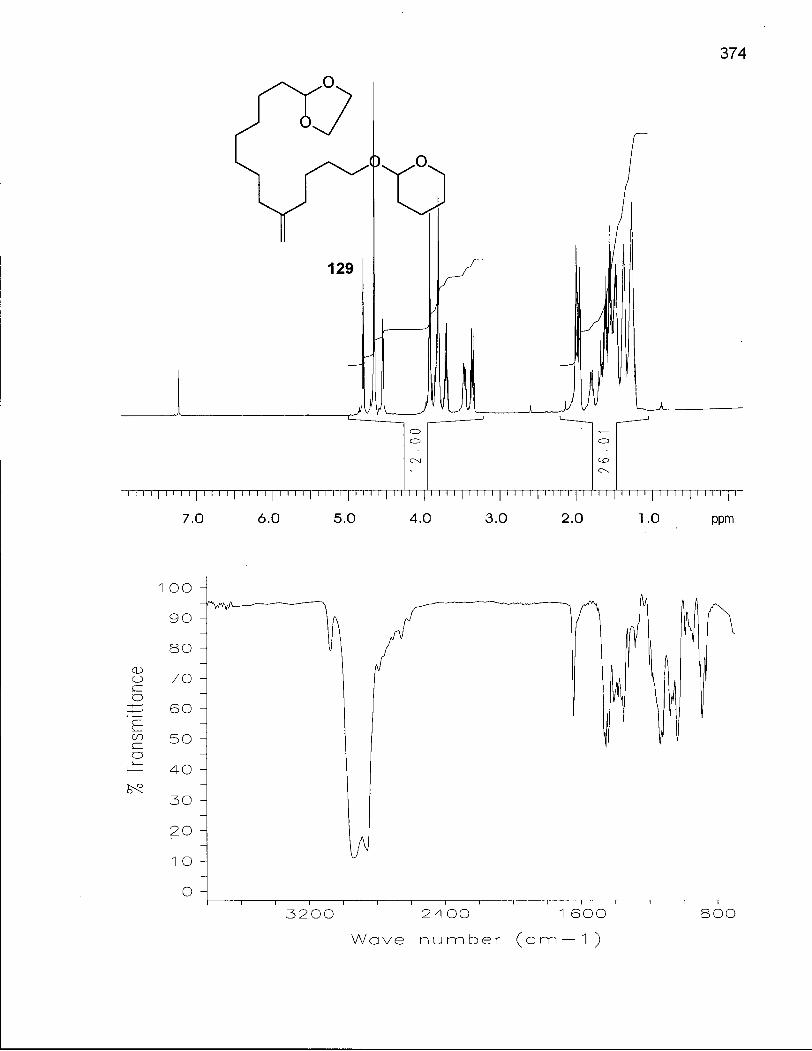
374
W a v e n u m b e r ( c m — 1 )
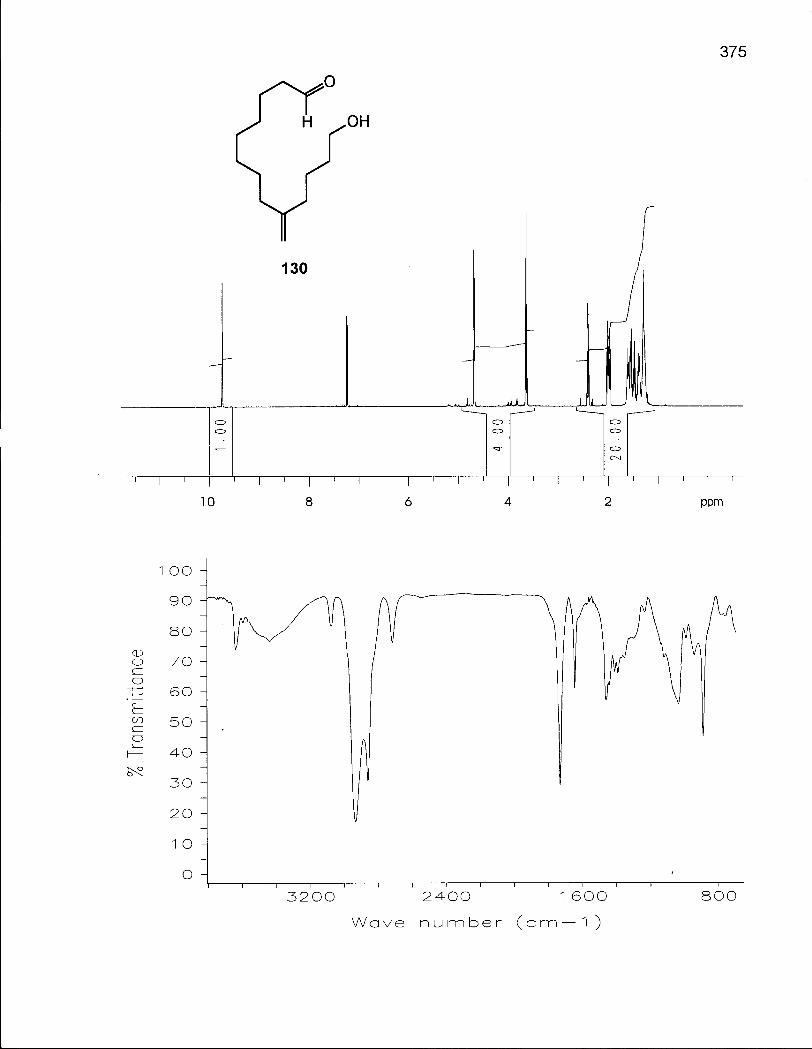
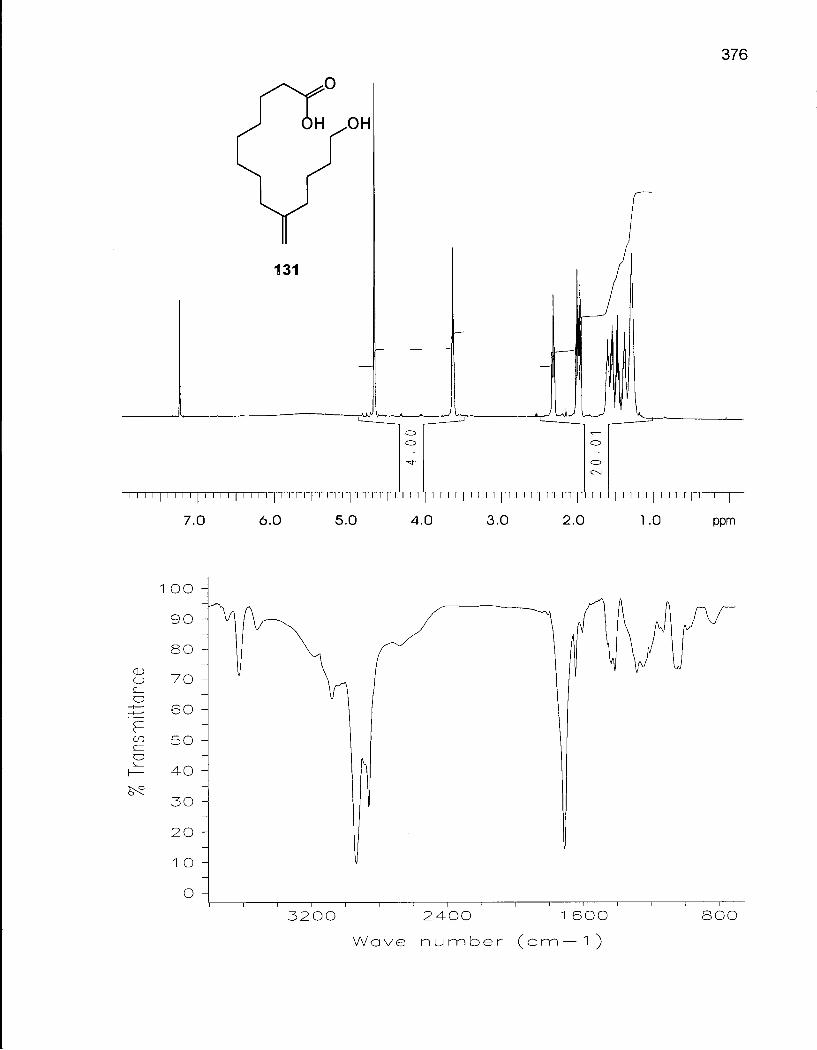
376

377
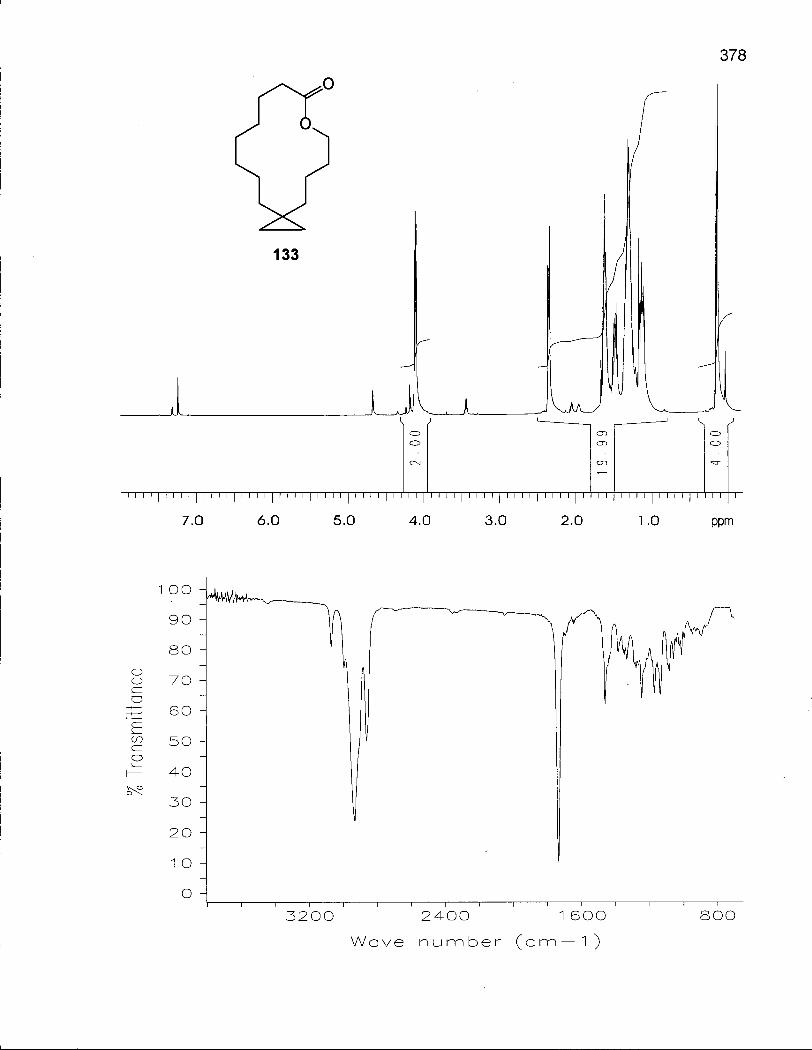
378
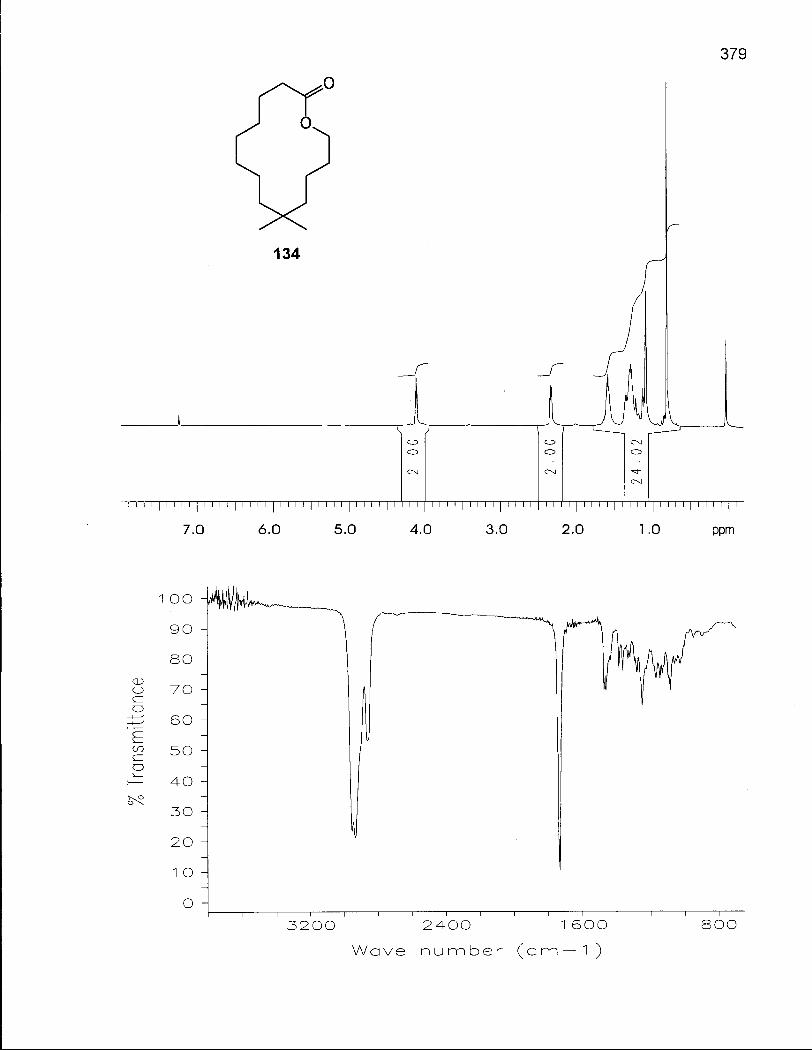
3 7 9
1 o -A
o -1 1 1 1 1 1 1 1 1 1 1 i i i i
3 2 0 0 2 4 0 0 1 6 0 0 8 0 0 W a v e n u m b e r ( c m — 1 )
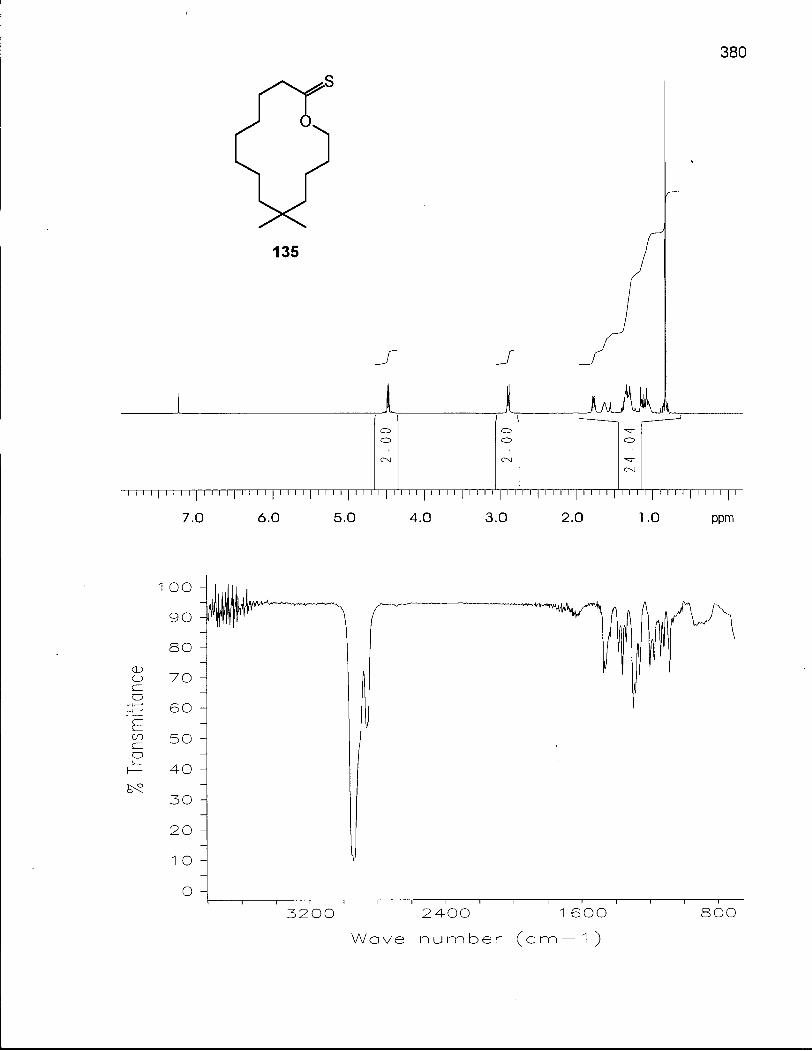
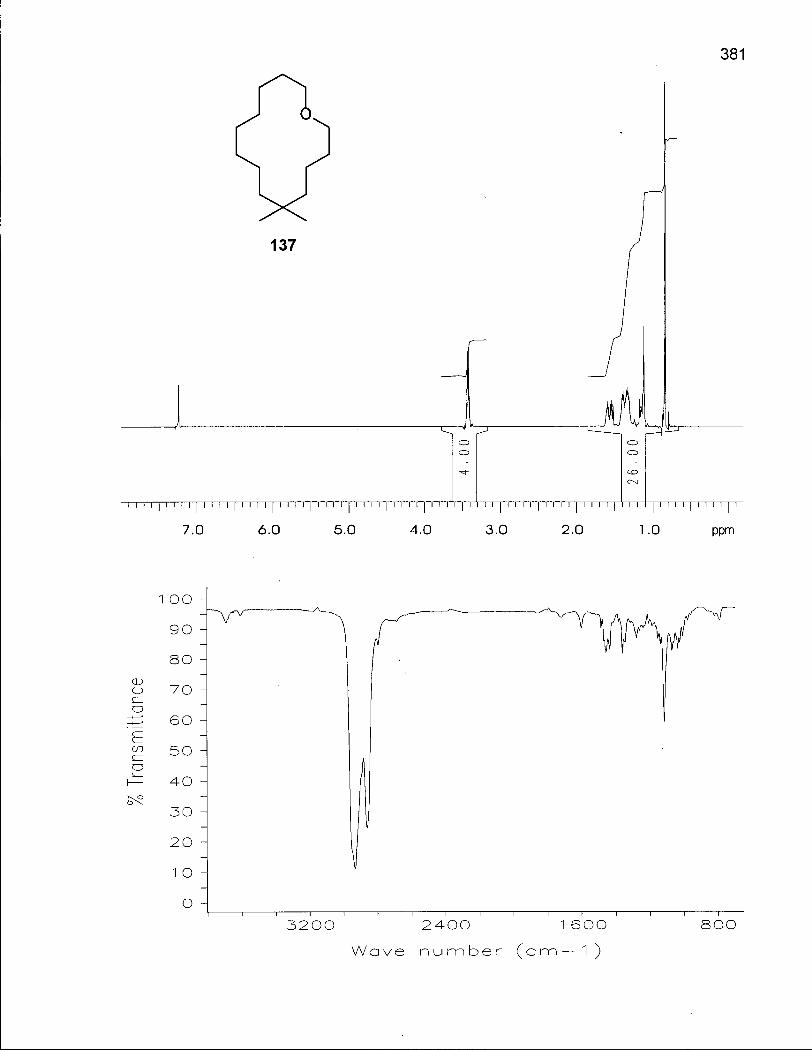
381
I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I I I I | I
7.0 6.0 5.0 4.0 3.0 2.0 1.0 ppm
1 OO H
1 1 1 1 1 1 1 1 1 1 1 1 1
3 2 0 0 2 4 0 0 1 6 0 0 S O O
W a v e n u m b e r ( c m — 1 )
382
1 o o H
20 -
1 0 - "
o -1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 —
3 2 0 0 2 4 0 0 1 6 0 0 SOO W a v e n u m b e r ( c m — 1 )
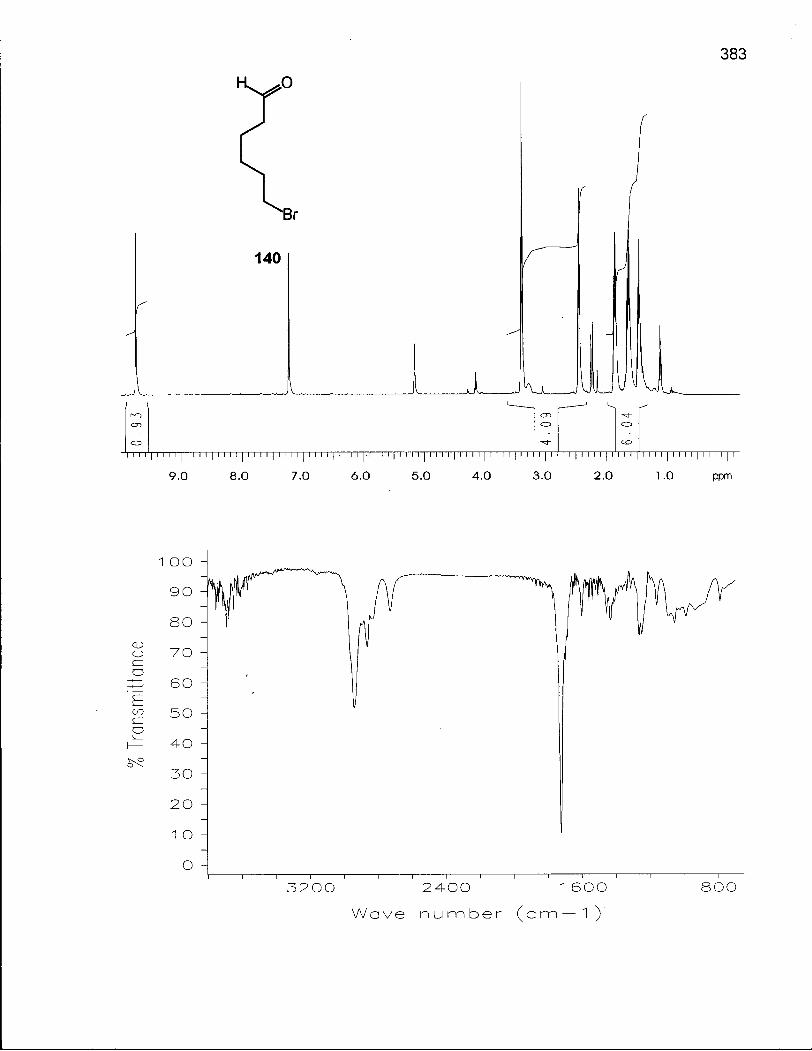
383
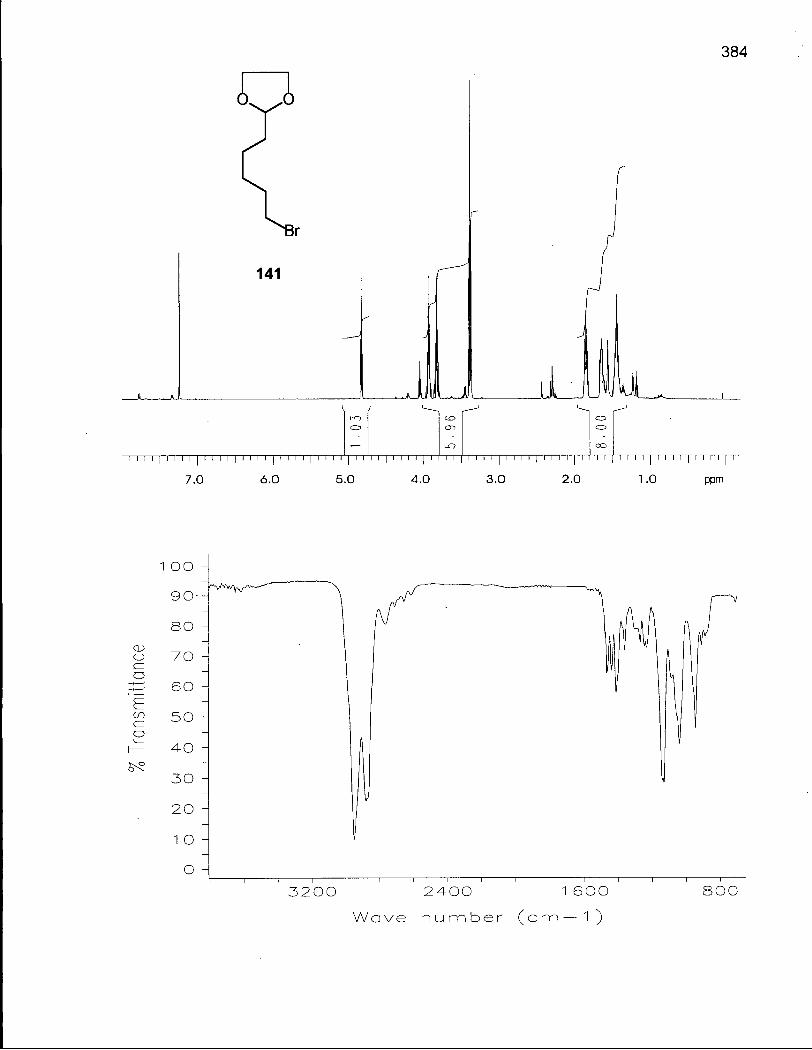
384
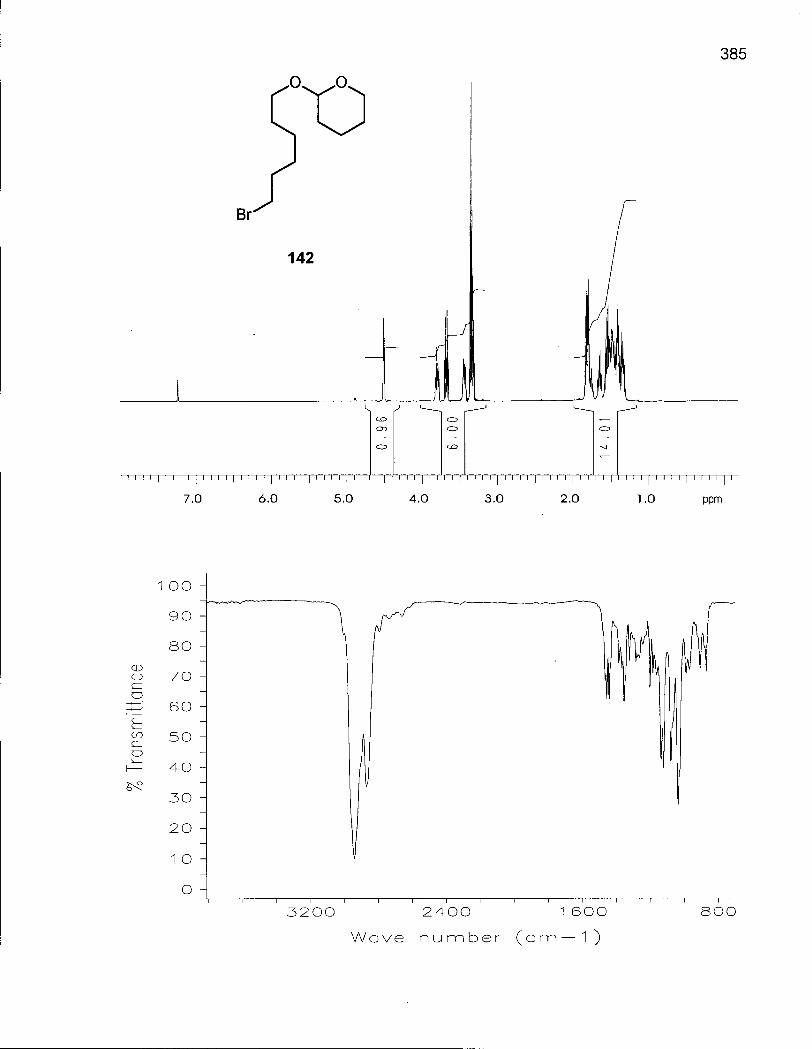
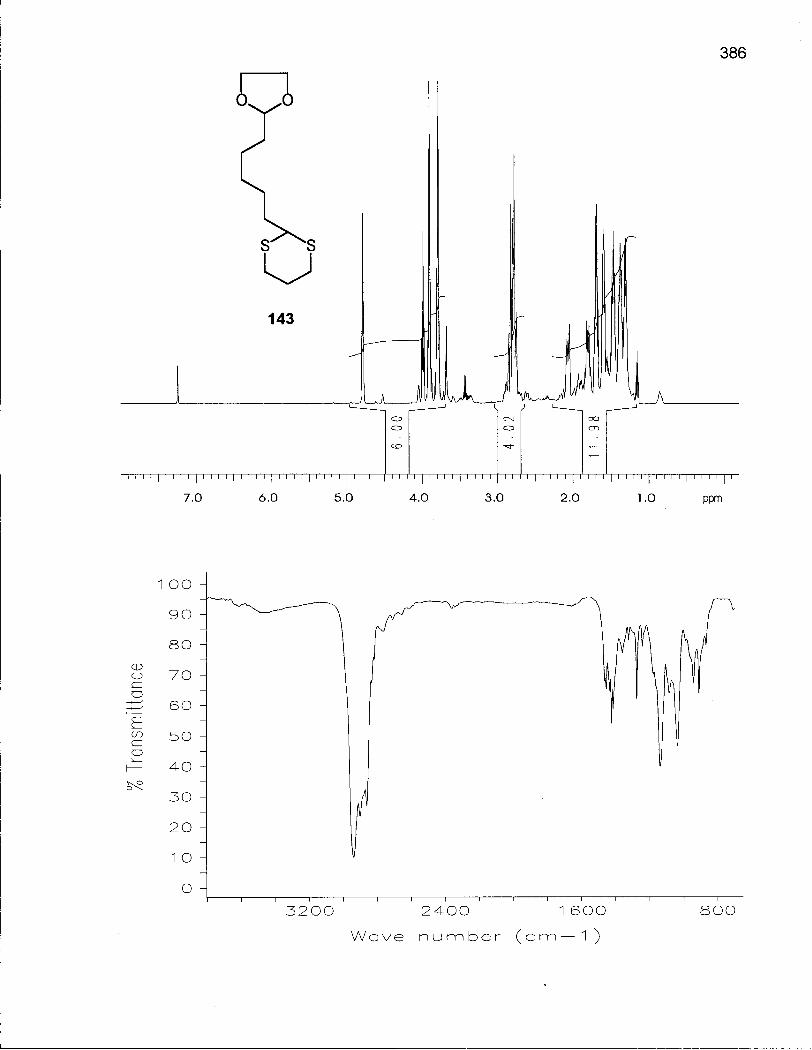
386
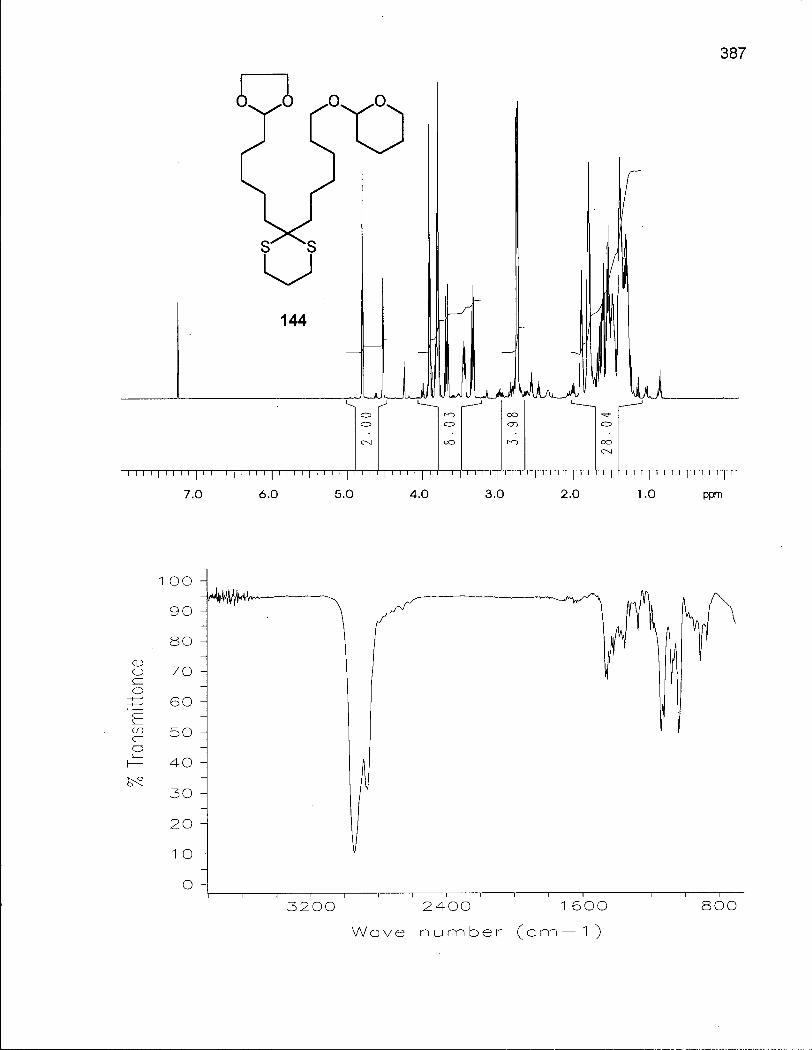
387
W a v e n u m b e r ( c m — 1 )
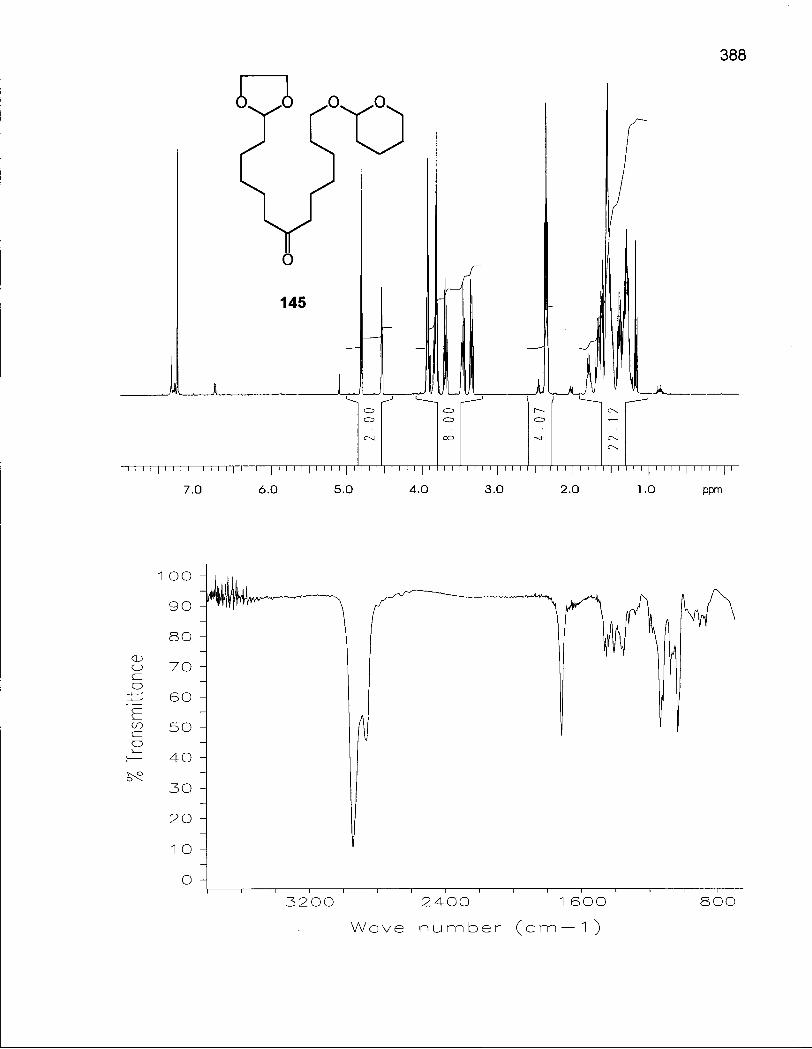
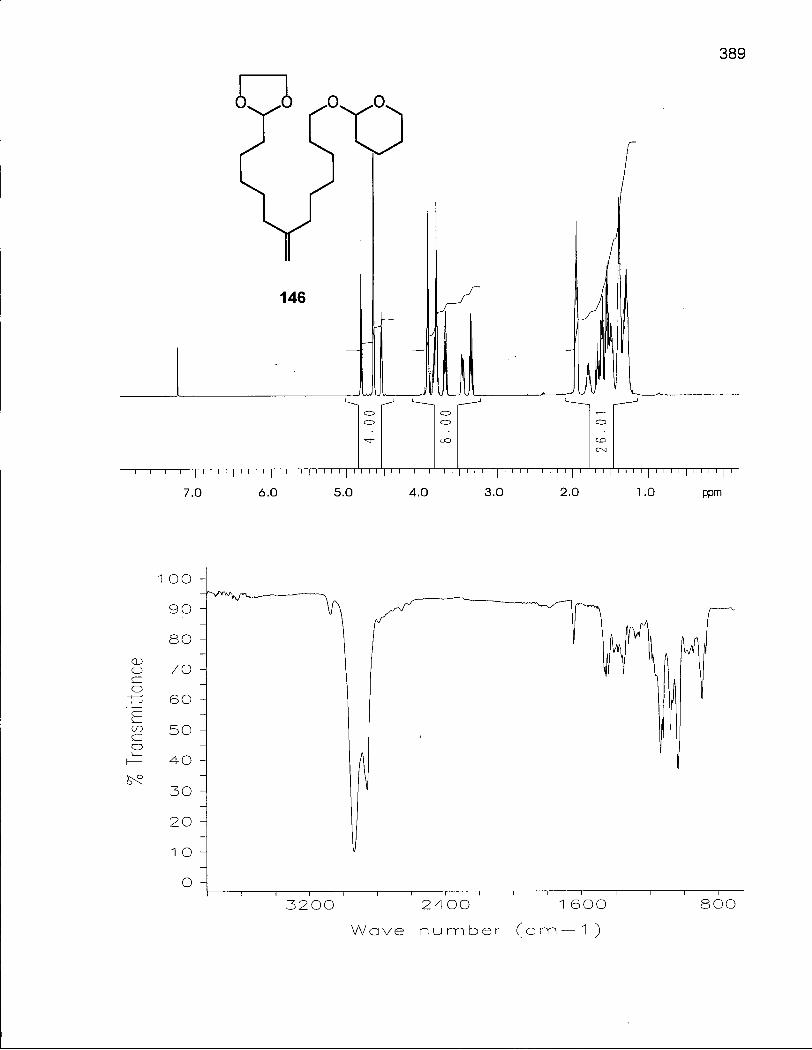
1 oo H
1 0 - v
o -1 1 1 1 1 1 < 1 1 \ 1 i i i i
3 2 0 0 2 4 0 0 1 6 0 0 S O O W a v e n u m b e r ( c m — 1 )
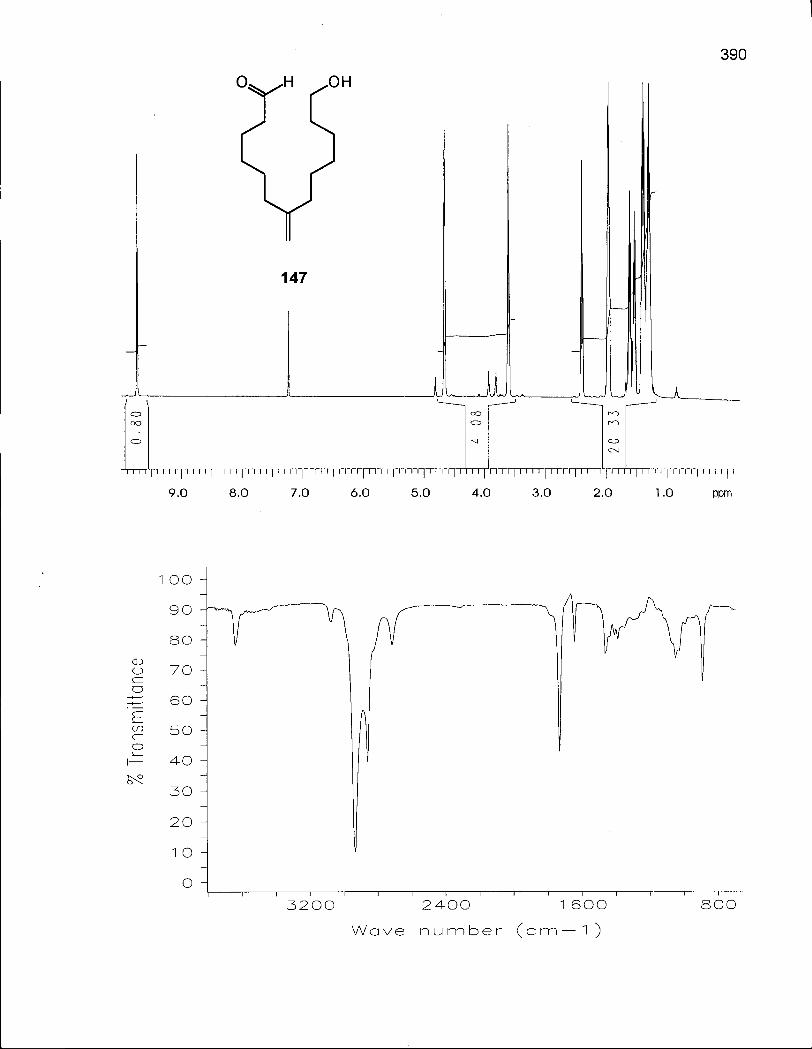
390

391
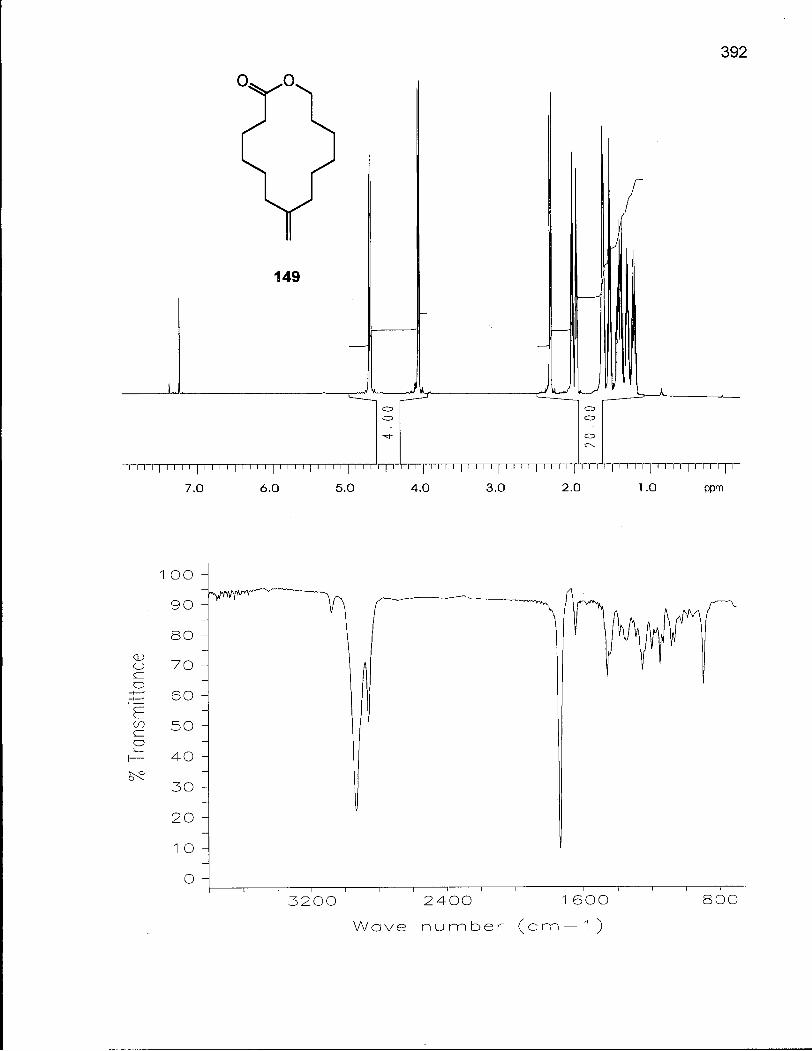
392
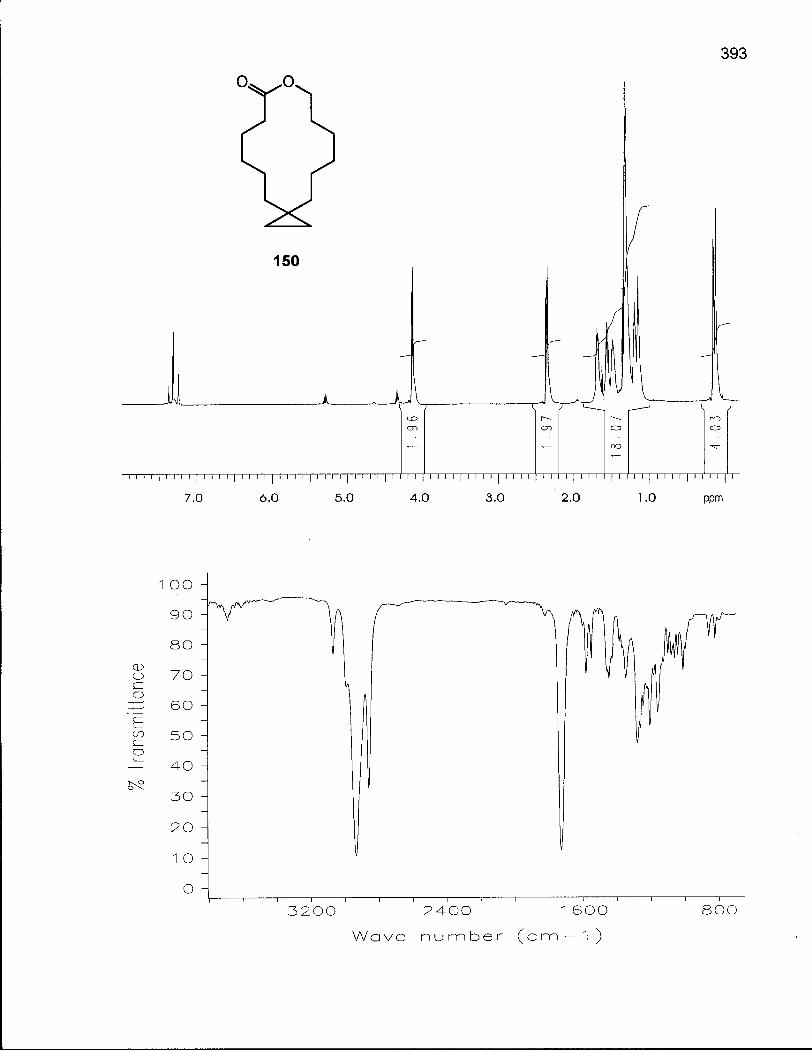
393
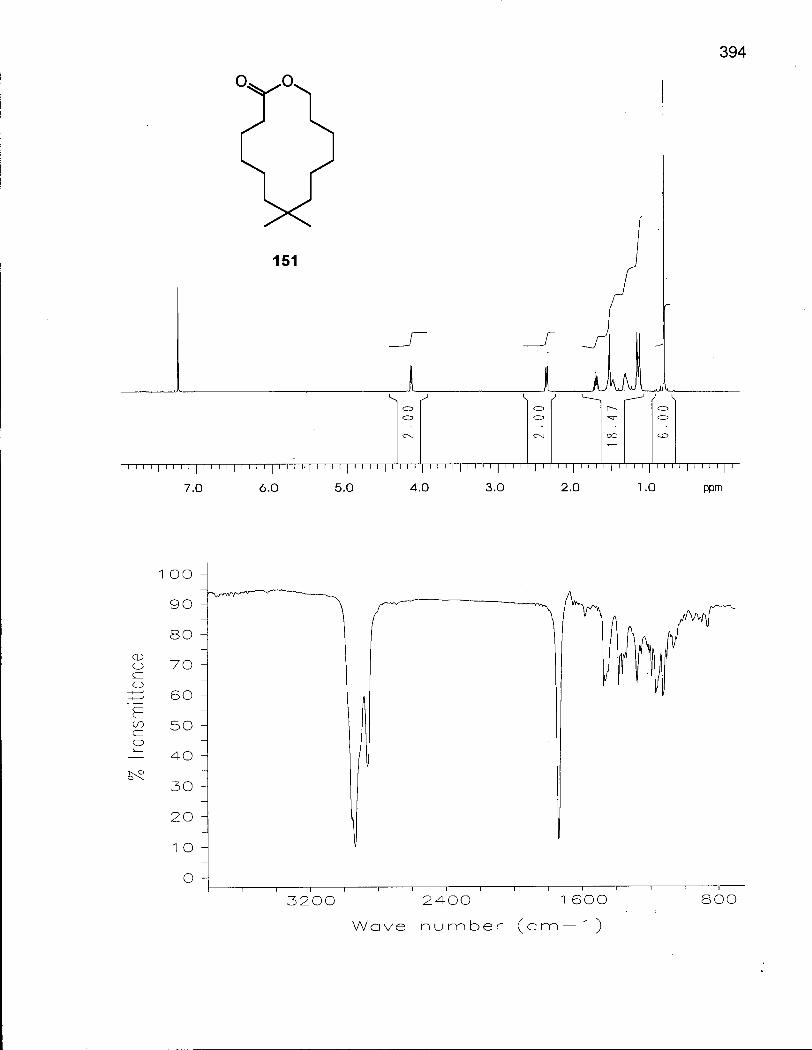
3 9 4
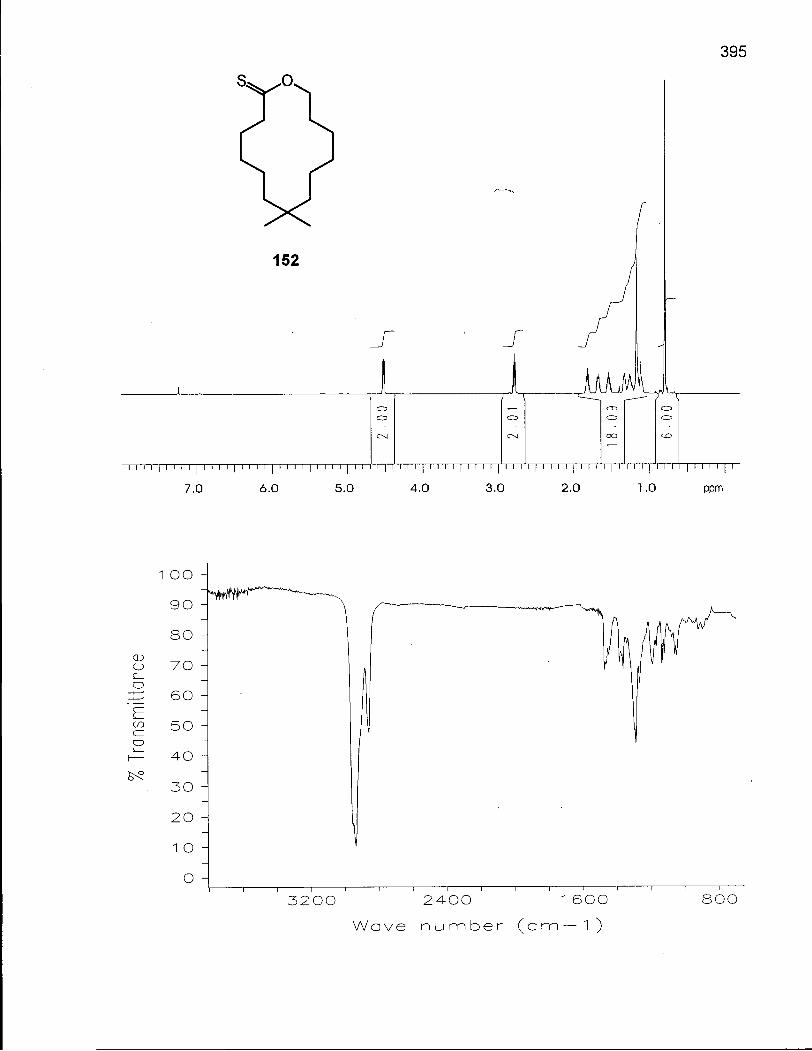
395
1 o o H
o -1 1 1 1 1 1 1 1 i i i i i i i
3 2 0 0 2 4 0 0 1 6 0 0 S O O W a v e n u m b e r ( c m — 1 )
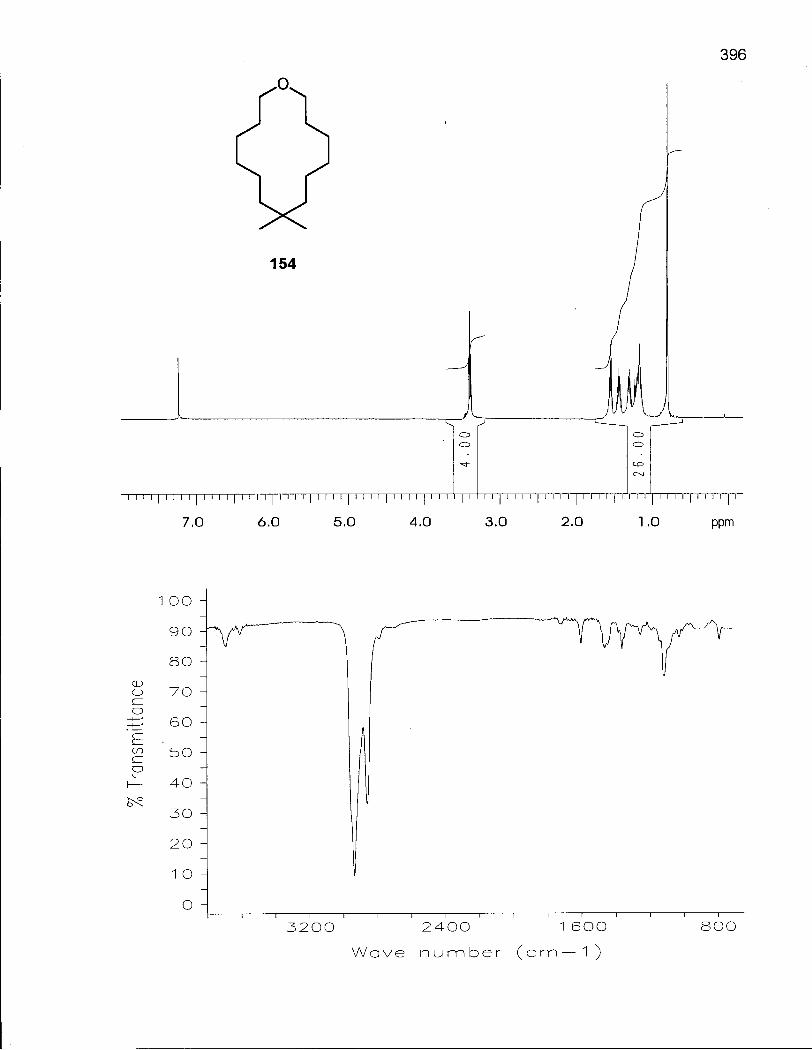
396
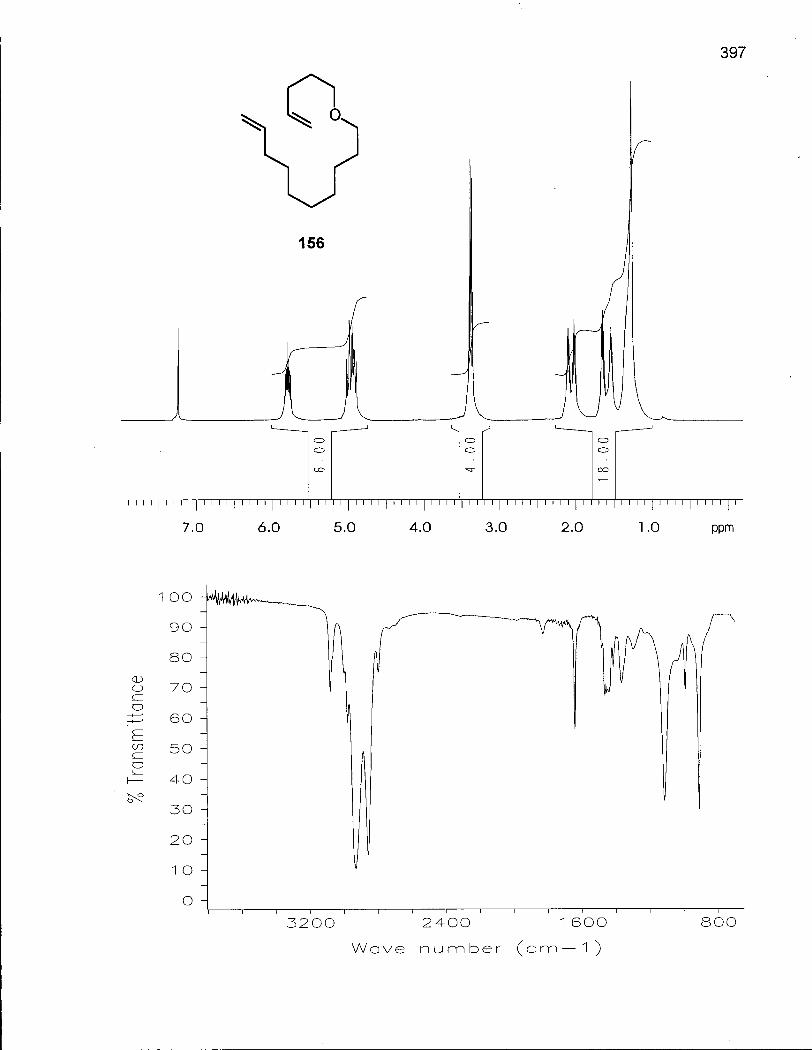
397
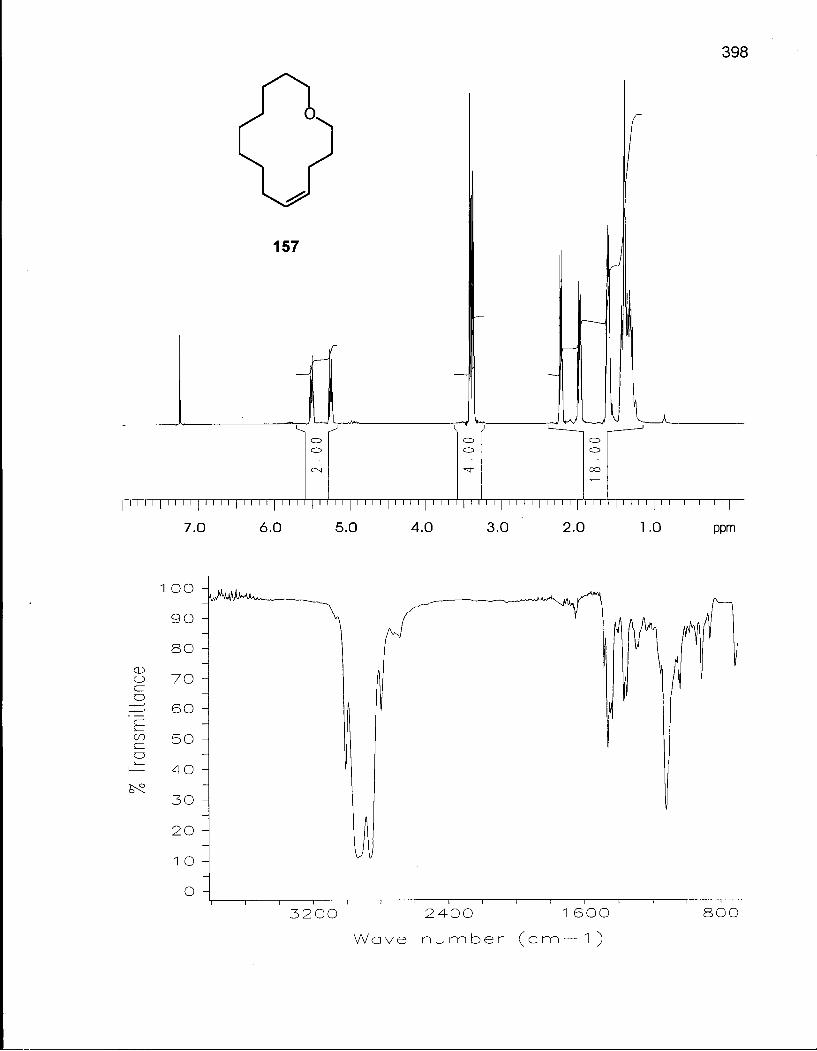
398
o -l ] ! ! ! ! ! ! ! f , , , , 1 | |
3 2 0 0 2 4 0 0 1 6 0 0 8 0 0 W a v e n u m b e r ( c m — 1 )
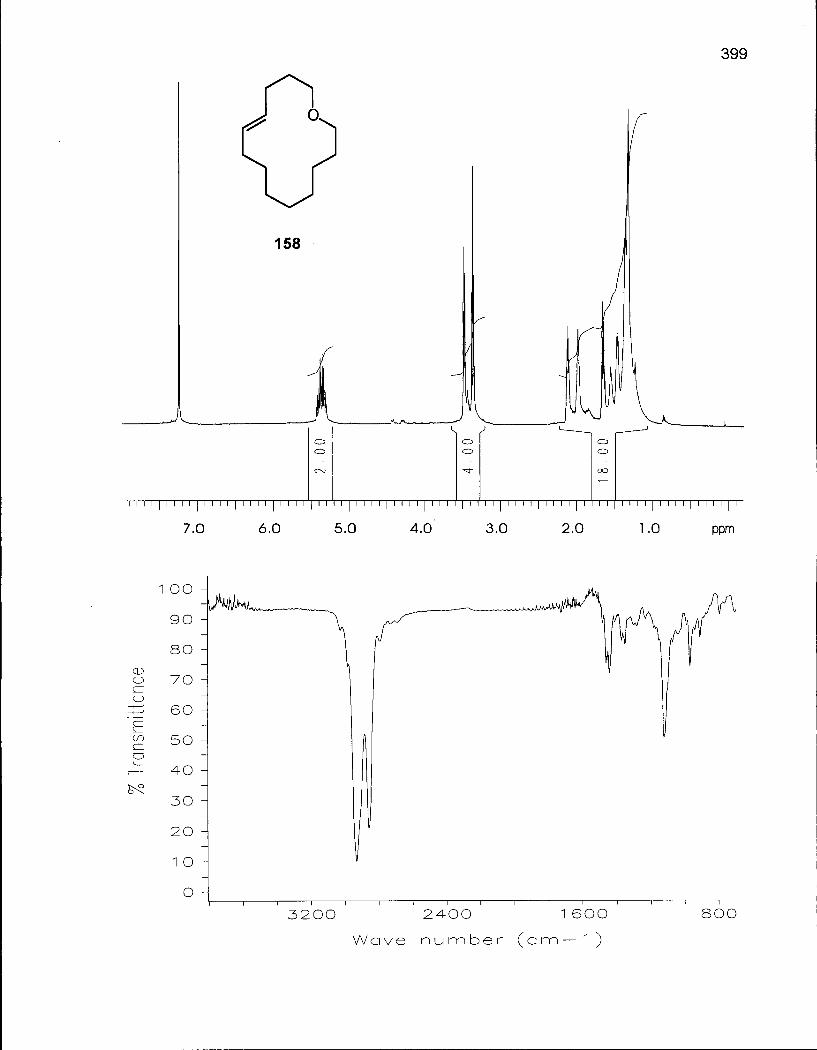
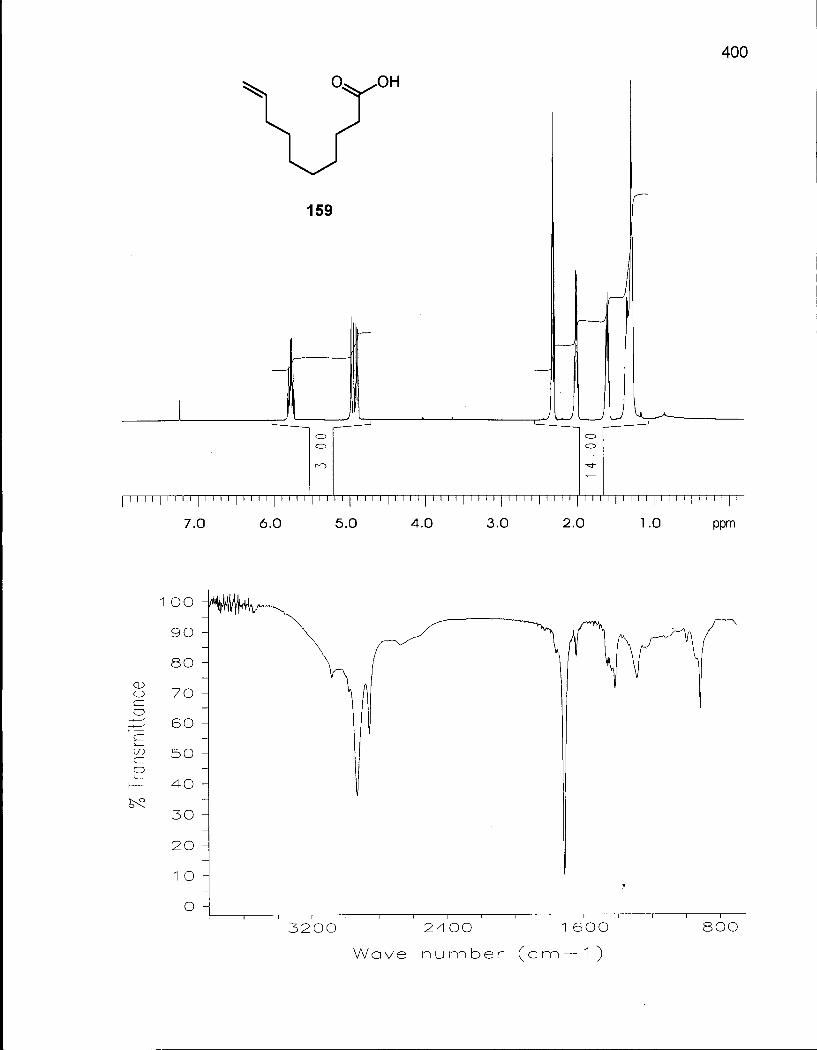
400
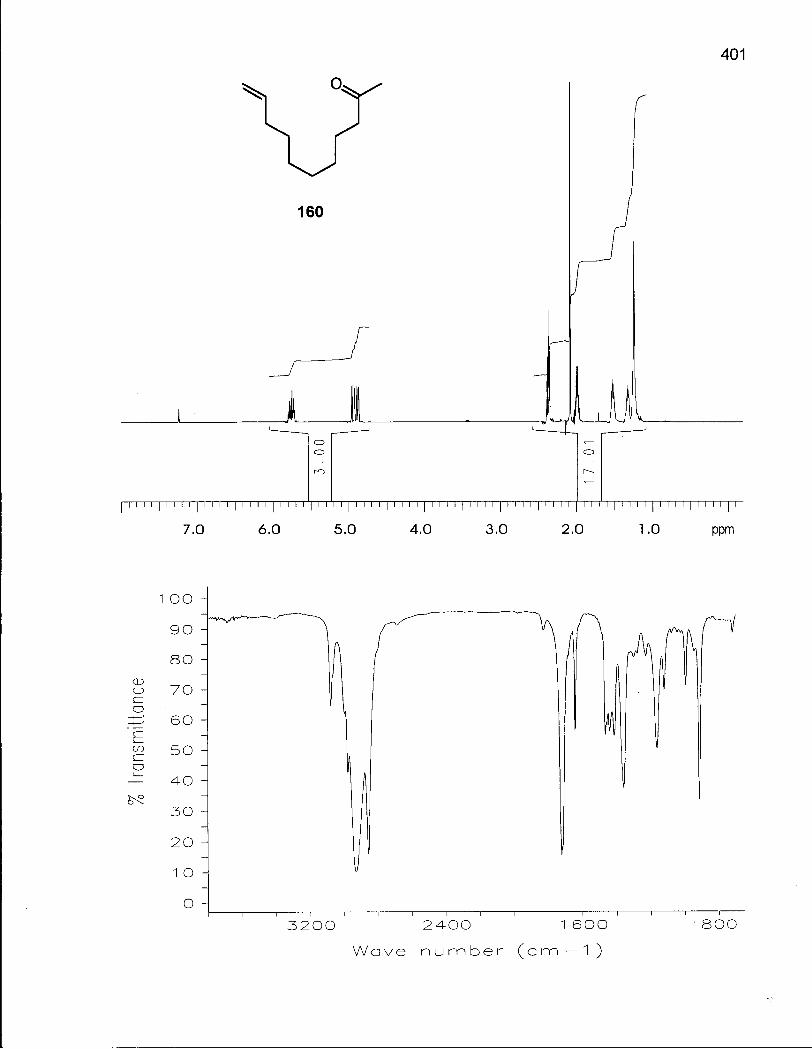

402
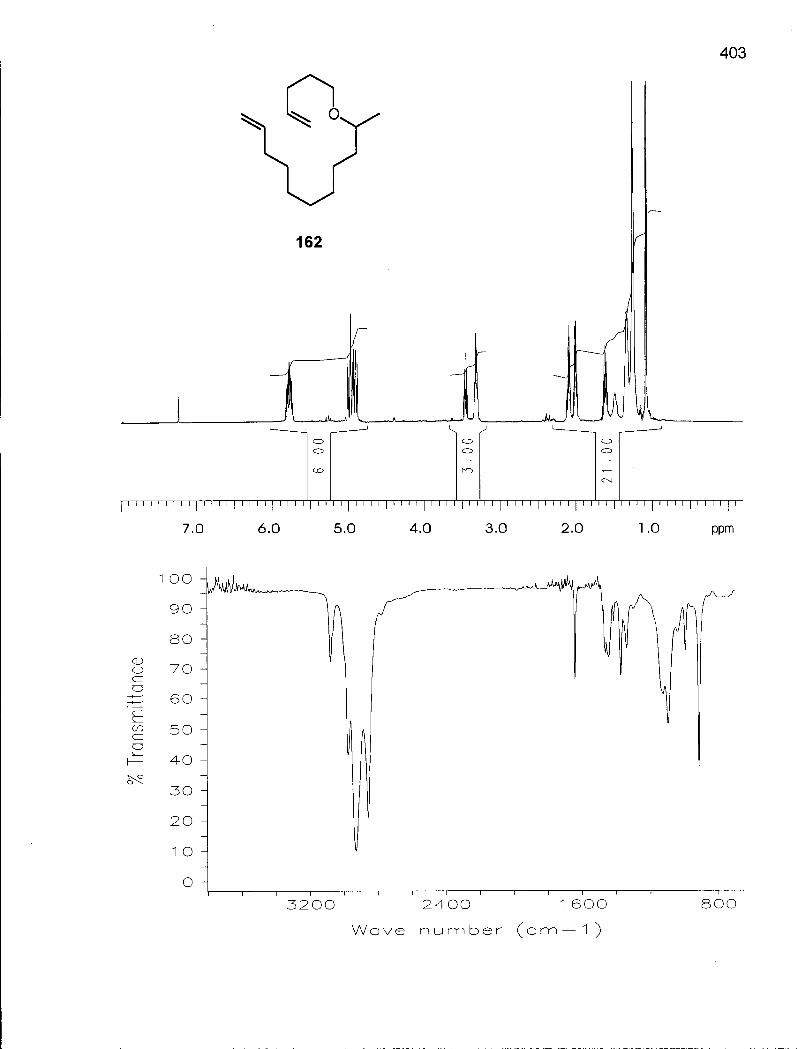
403
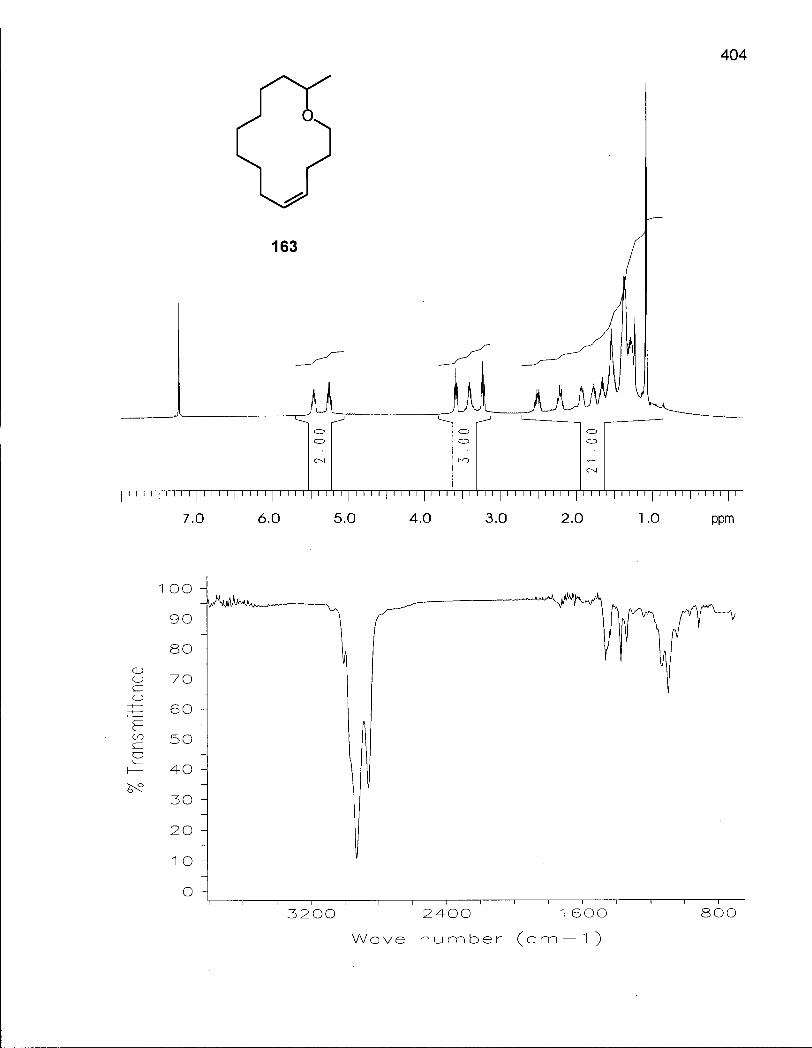
404
W a v e n u m b e r ( c m —
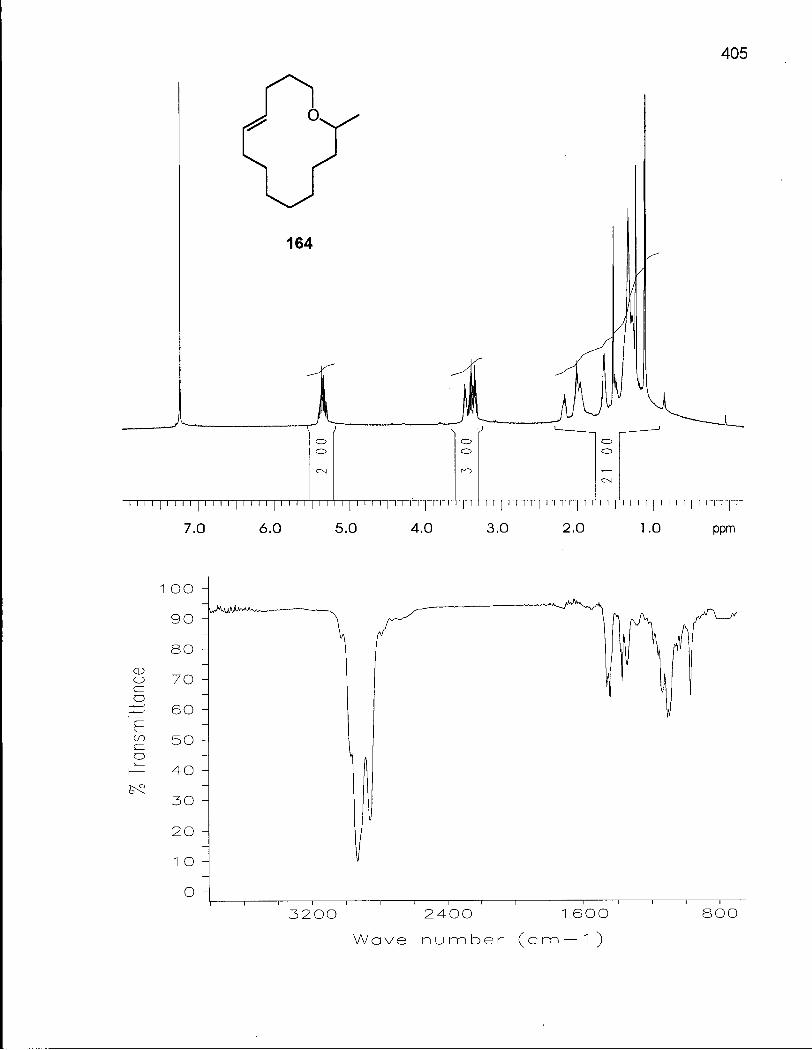
405

406

407
1 o o -\
o -1 1 1 1 1 1 1 1 1 1 1 i i i i
3 2 0 0 2 4 0 0 1 6 0 0 8 0 0 W a v e n u m b e r ( c m — 1 )

408
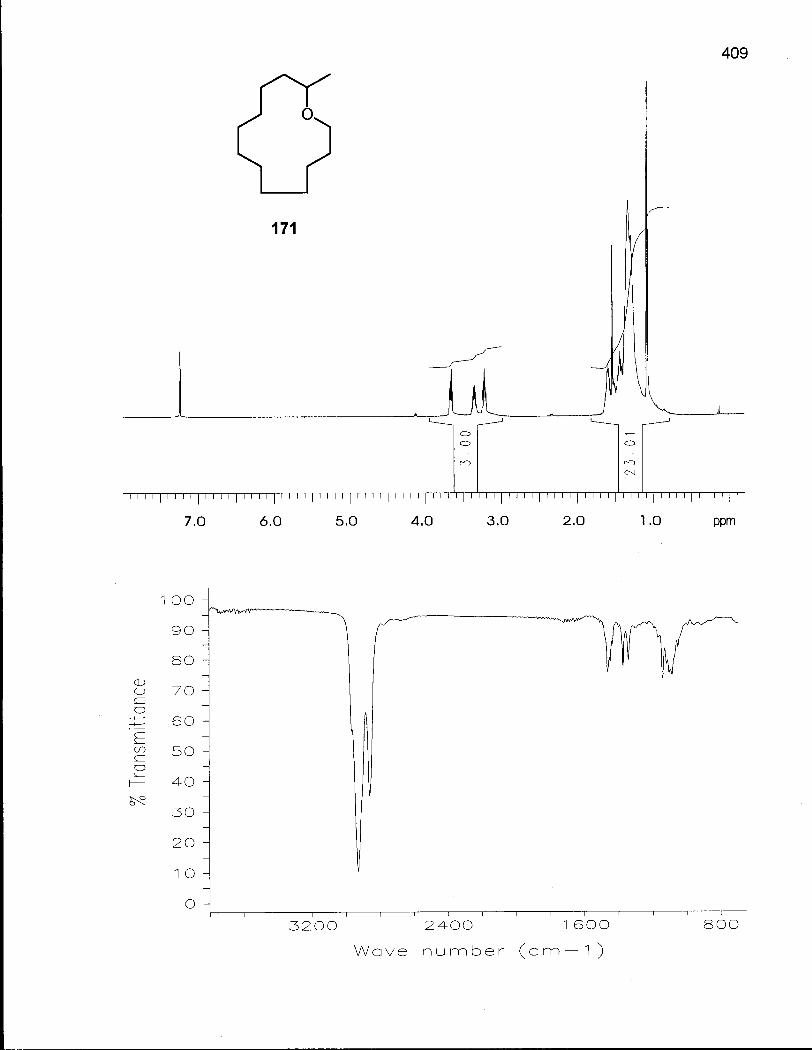
4 1 0

411
412

413
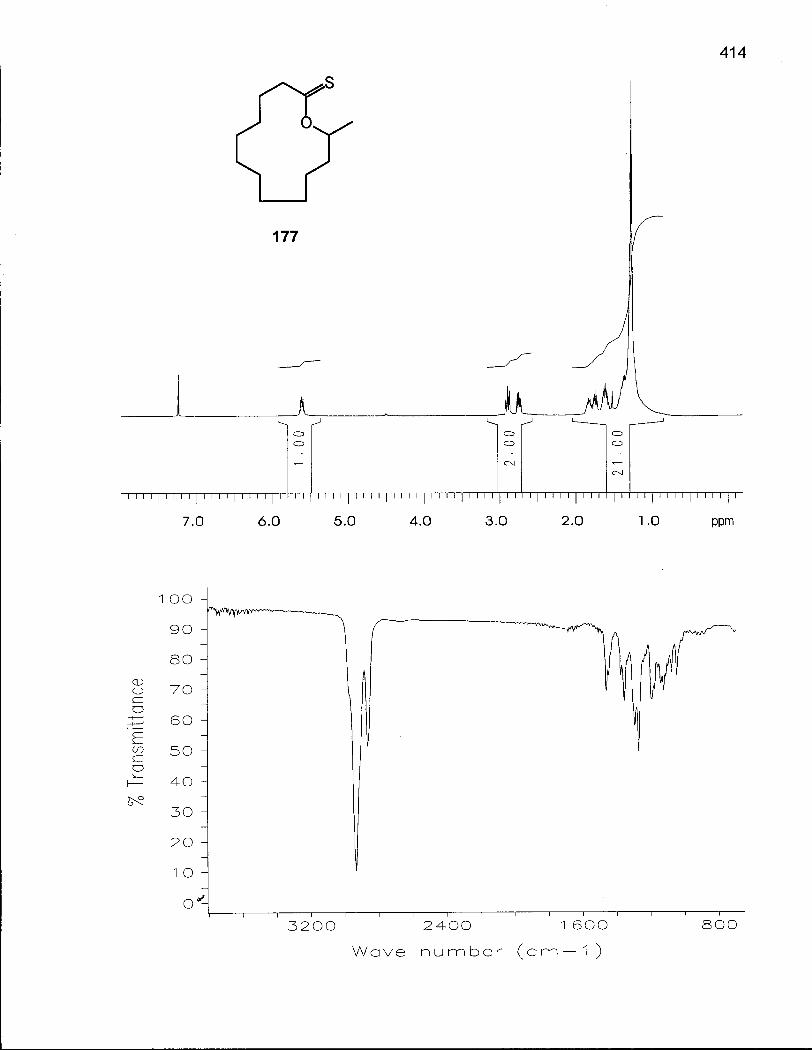
414

415
1 o o -\
90
s o
70
60 H
50
40
30
20
1 O
O H
3 2 0 0 2 4 0 0 1 6 0 0
W a v e n u m b e r ( c m — 1 )
8 0 0
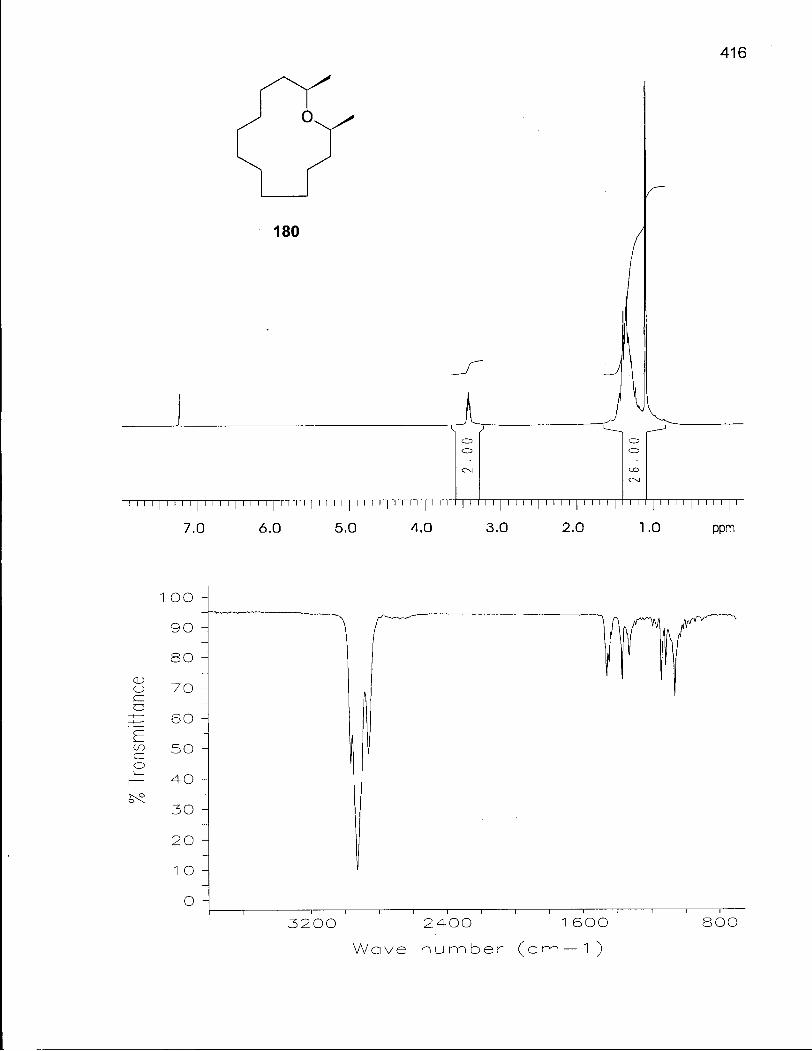
416
1 oo H
£2 50 -o
H= 40 -
30 -20 H
O -1 1 1 1 1 1 1 1 1 i i i i i i
3 2 0 0 2 4 0 0 1 6 0 0 8 0 0 W a v e n u m b e r ( c m — 1 )
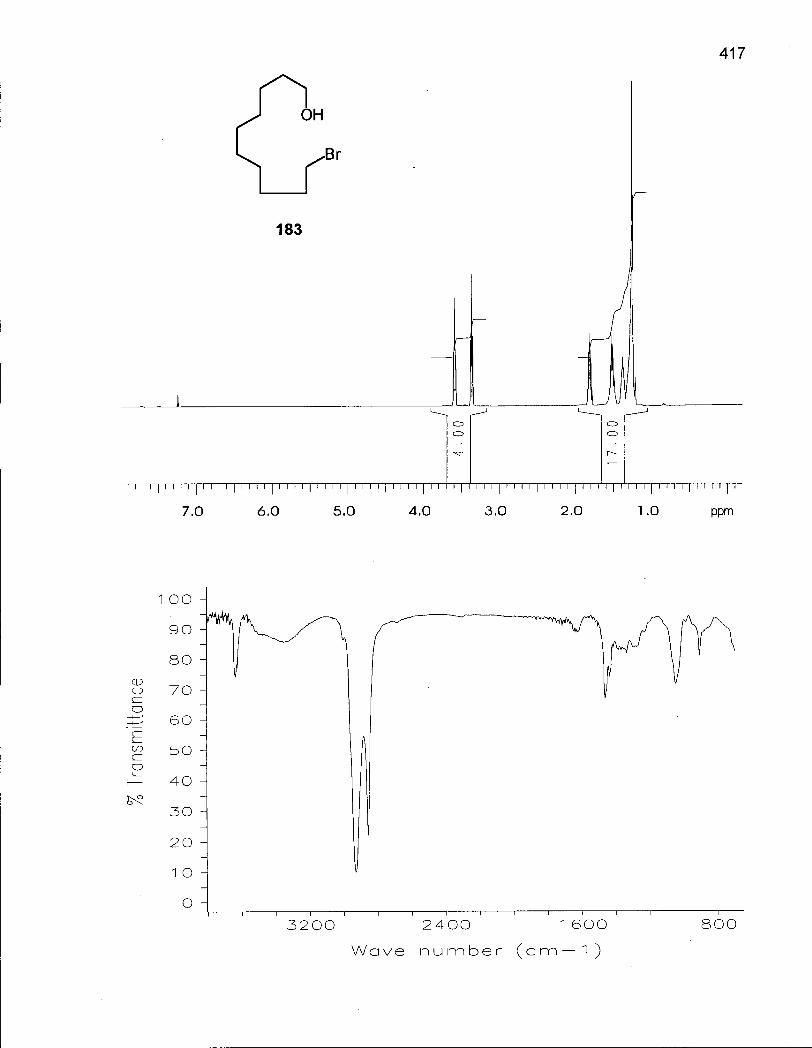
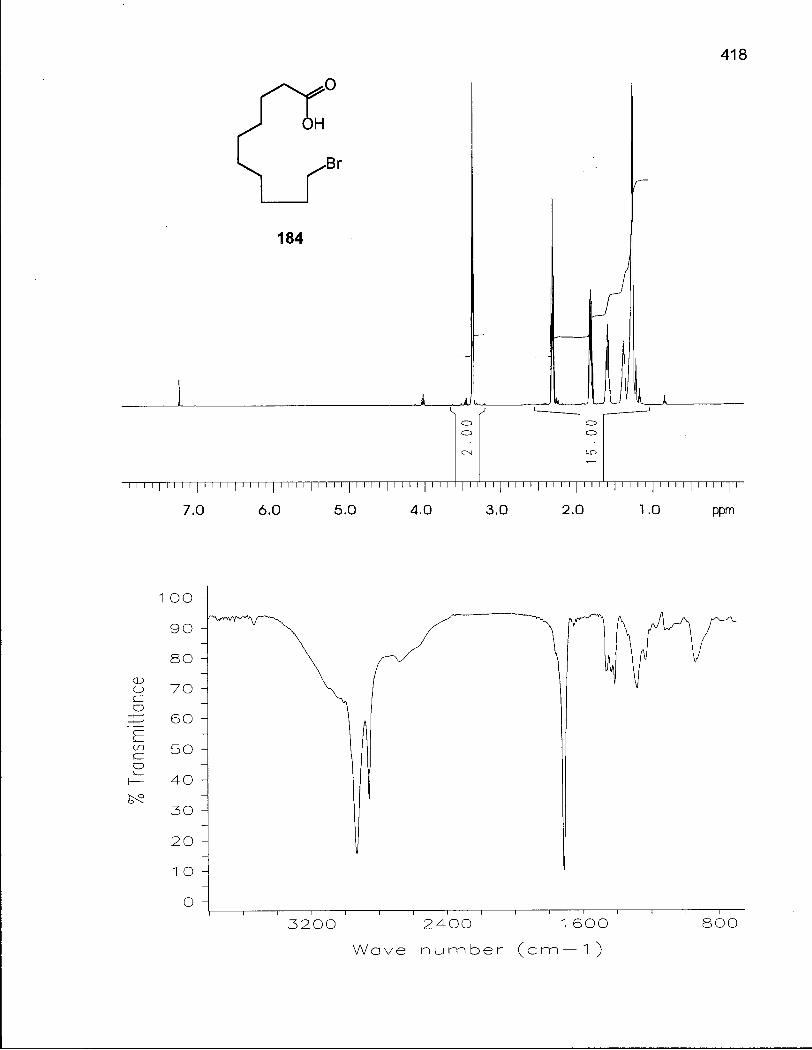
418

419
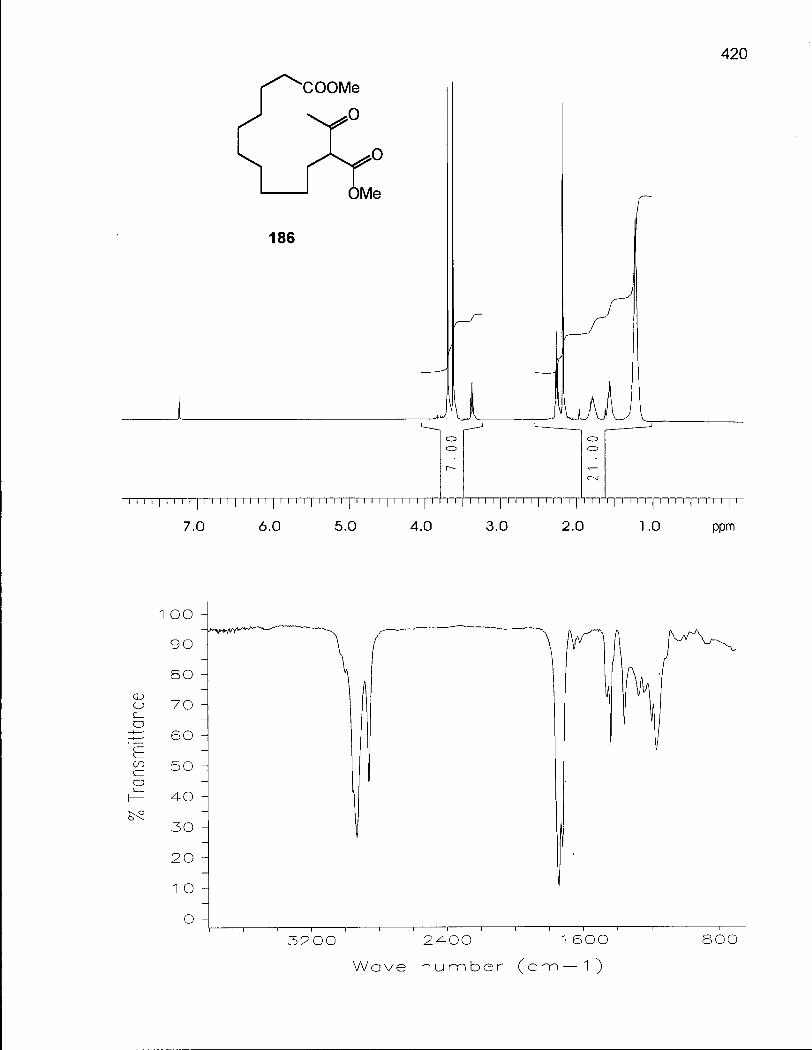
420
421

422
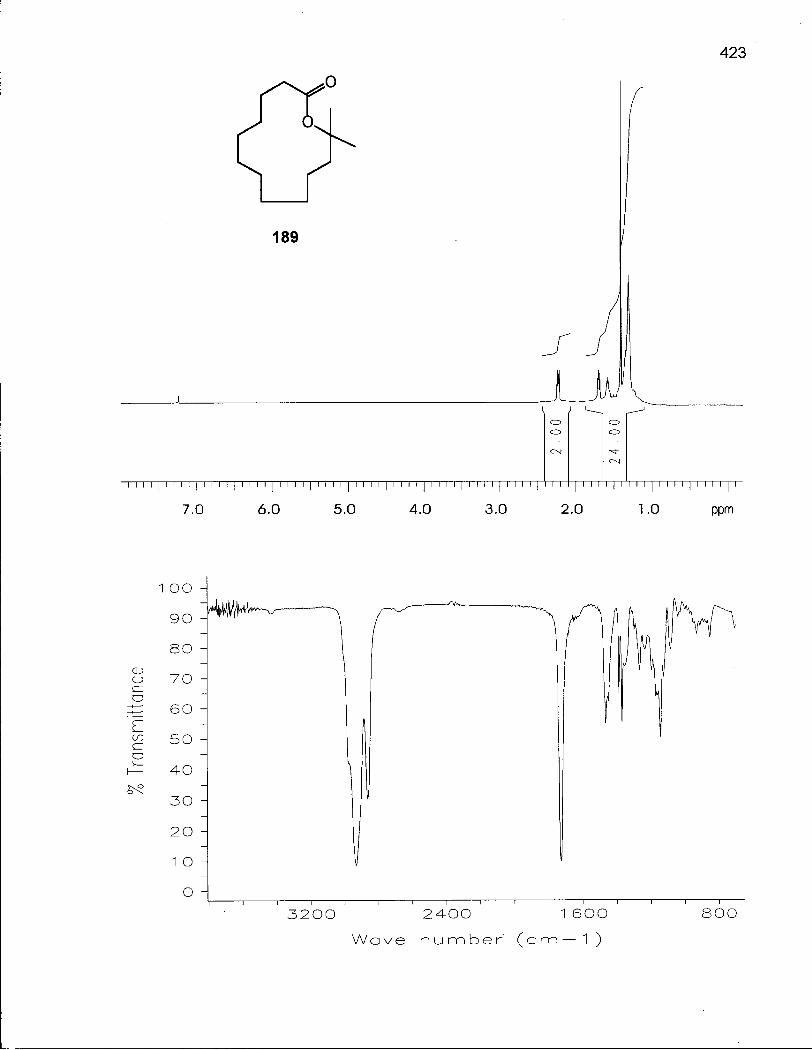
423
189
i i i i | i i i i 11 i i i | i i i i | i i i i | 11 i i | i i i i | i i i 11 i i i i | i i M | i i i 11 i i i i | i i i i | i i i i | i i M | i i i i | i
7.0 6 .0 5 .0 4 .0 3 .0 2 .0 1.0 ppm
CD O <ZZ
o
*E CO cz p
00 H
90
SO
70
60
50 H
40
30
20 -\
1 O
O 3 2 0 0 2 4 0 0 1 6 0 0
W a v e n u m b e r ( c m — 1)
SOO
424
1 o - »
o -\ 1 1 1 1 1 1 1 1 1 1 1 1 1 I I
3 2 0 0 2 4 0 0 1 6 0 0 SOO W a v e n u m b e r ( c m — 1 )
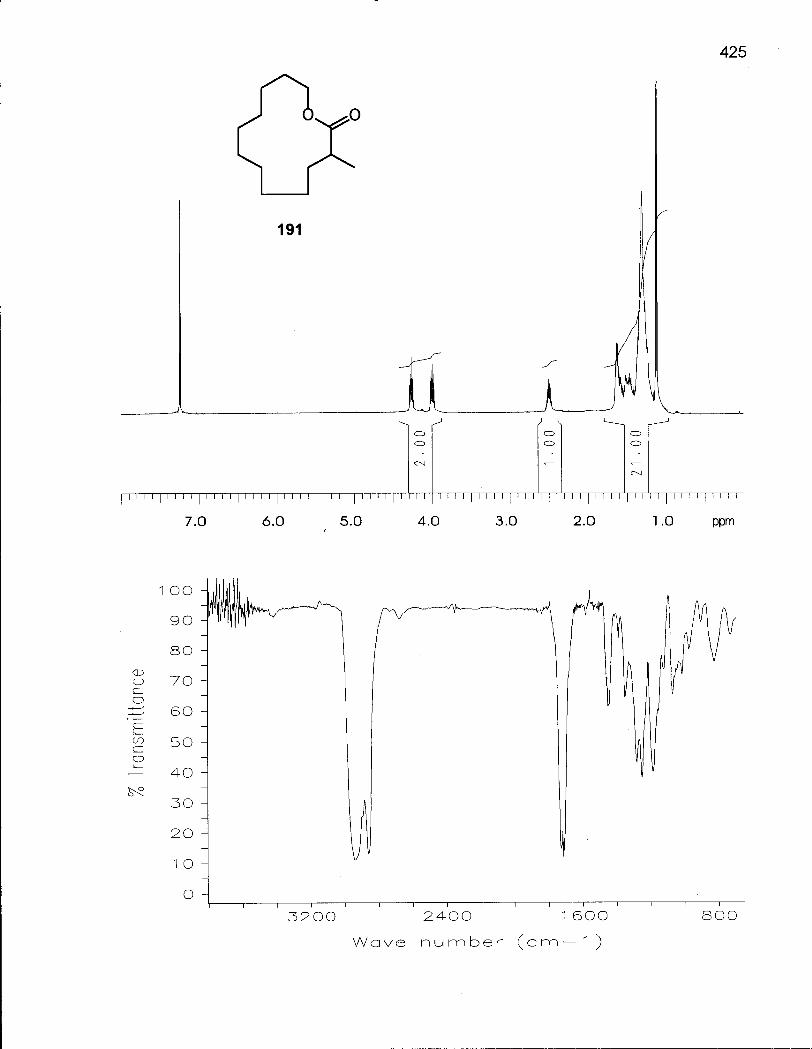

426
C N
I I I I I I I I I I I I I I I I I I I I I I I I I II i I I I I I I I I ( I I I I I I I I I I I I I I I I I I I I I I I I I I I I II I I I I I I I I i I I I I I
7 . 0 6 . 0 5 . 0 4 . 0 3 . 0 2 . 0 1 .0 ppm
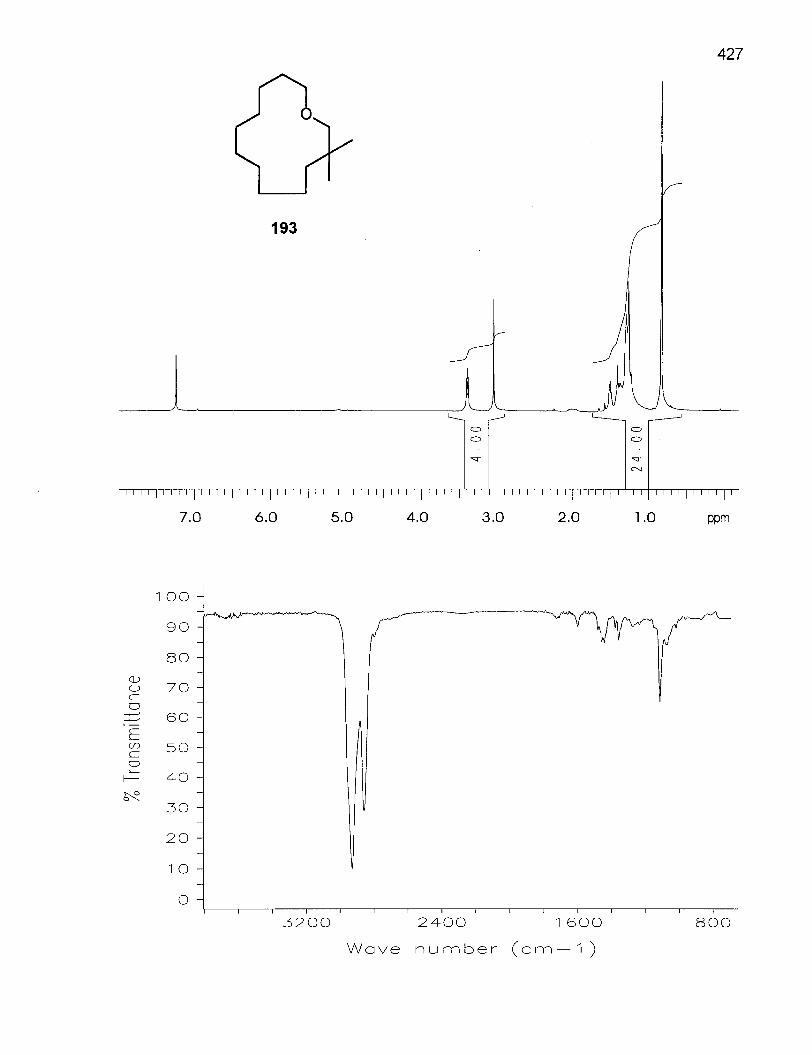
1 o o H
£2 50 -o
H= 40 -
30 - U
20 -
1 0 - '
o -1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
3 2 0 0 2 4 0 0 1 6 0 0 8 0 0 W a v e n u m b e r ( c m — 1 )
Copyright © 2022 FDOKUMEN




















