
Conformational and Spectroscopic Behaviors of 2,4-xylyl isothiocyanate
8
Conformational and spectroscopic behaviors of 2,4-xylyl isothiocyanate Mehmet Cinar a , Mehmet Karabacak b,⇑ , Satish Chand c , Vikas K. Shukla c , Leena Sinha c , Onkar Prasad c , Manoj Pratap Singh d , Abdullah M. Asiri e,f a Department of Science Education, Bayburt University, 69000 Bayburt, Turkey b Department of Mechatronics Engineering, H.F.T. Technology Faculty, Celal Bayar University, Turgutlu, Manisa, Turkey c Department of Physics, University of Lucknow, 226 007 Lucknow, India d Advanced Instrumentation Research Facility, JNU, New Delhi, India e Department of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia f Center of Excellence for Advanced Materials Research, King Abdulaziz University, Jeddah, Saudi Arabia highlights The compound 2,4-xylyl isothiocyanate was characterized by FT-IR and FT-Raman spectroscopy. Vibrational assignments performed on the basis of the experimental data and total energy distribution. AN@C@S group containing non- bonded electrons act as a auxochrome. NBO analysis done to explore charge transfer or conjugative interaction. graphical abstract article info Article history: Received 21 August 2014 Received in revised form 25 December 2014 Accepted 29 December 2014 Available online 21 January 2015 Keywords: 2,4-xylyl isothiocyanate FT-IR and FT-Raman HOMO–LUMO DFT NLO and NBO analysis abstract This study aims to identify the conformational and spectroscopic characteristics of 2,4-xylyl isothiocya- nate (C 9 H 9 NS) compound via experimental and computational methods. To accomplish this, density func- tional theory (DFT), with the B3LYP functional was used to determine ground state conformation, vibrational wavenumbers and also isotropic chemical shifts of the title molecule. Experimentally, vibra- tional features of the compound were evaluated by FT-IR and FT-Raman spectroscopic analysis in the solid phase. On the basis of these studies, the conformational and spectroscopic behaviors of 2,4-xylyl isothiocyanate were interpreted. The fundamental vibrational wavenumbers as well as their intensities were computed, and a good correlation between experimental and scaled calculated wavenumbers was observed. The polarizability, first hyperpolarizability and dipole moment values of 2,4-xylyl isothi- ocyanate were calculated at the same level of theory and basis set. The results show that 2,4-xylyl isothi- ocyanate molecule possesses nonlinear optical (NLO) behavior with non-zero values. Stability of the molecule arising from hyper-conjugative interactions and charge delocalization was analyzed using nat- ural bond orbital (NBO) analysis. Ó 2015 Elsevier B.V. All rights reserved. Introduction Isothiocyanates occur naturally as glucosinolate conjugates in cruciferous vegetables, like broccoli, cauliflower, cabbage, Brussel http://dx.doi.org/10.1016/j.molstruc.2014.12.079 0022-2860/Ó 2015 Elsevier B.V. All rights reserved. ⇑ Corresponding author. Tel.: +90 236 314 10 10; fax: +90 236 314 20 20. E-mail address: [email protected] (M. Karabacak). Journal of Molecular Structure 1087 (2015) 113–120 Contents lists available at ScienceDirect Journal of Molecular Structure journal homepage: www.elsevier.com/locate/molstruc
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Conformational and Spectroscopic Behaviors of 2,4-xylyl isothiocyanate

Journal of Molecular Structure 1087 (2015) 113–120
Contents lists available at ScienceDirect
Journal of Molecular Structure
journal homepage: www.elsevier .com/ locate /molst ruc
Conformational and spectroscopic behaviors of 2,4-xylyl isothiocyanate
http://dx.doi.org/10.1016/j.molstruc.2014.12.0790022-2860/� 2015 Elsevier B.V. All rights reserved.
⇑ Corresponding author. Tel.: +90 236 314 10 10; fax: +90 236 314 20 20.E-mail address: [email protected] (M. Karabacak).
Mehmet Cinar a, Mehmet Karabacak b,⇑, Satish Chand c, Vikas K. Shukla c, Leena Sinha c, Onkar Prasad c,Manoj Pratap Singh d, Abdullah M. Asiri e,f
a Department of Science Education, Bayburt University, 69000 Bayburt, Turkeyb Department of Mechatronics Engineering, H.F.T. Technology Faculty, Celal Bayar University, Turgutlu, Manisa, Turkeyc Department of Physics, University of Lucknow, 226 007 Lucknow, Indiad Advanced Instrumentation Research Facility, JNU, New Delhi, Indiae Department of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabiaf Center of Excellence for Advanced Materials Research, King Abdulaziz University, Jeddah, Saudi Arabia
h i g h l i g h t s
� The compound 2,4-xylylisothiocyanate was characterized byFT-IR and FT-Raman spectroscopy.� Vibrational assignments performed
on the basis of the experimental dataand total energy distribution.� AN@C@S group containing non-
bonded electrons act as aauxochrome.� NBO analysis done to explore charge
transfer or conjugative interaction.
g r a p h i c a l a b s t r a c t
a r t i c l e i n f o
Article history:Received 21 August 2014Received in revised form 25 December 2014Accepted 29 December 2014Available online 21 January 2015
Keywords:2,4-xylyl isothiocyanateFT-IR and FT-RamanHOMO–LUMODFTNLO and NBO analysis
a b s t r a c t
This study aims to identify the conformational and spectroscopic characteristics of 2,4-xylyl isothiocya-nate (C9H9NS) compound via experimental and computational methods. To accomplish this, density func-tional theory (DFT), with the B3LYP functional was used to determine ground state conformation,vibrational wavenumbers and also isotropic chemical shifts of the title molecule. Experimentally, vibra-tional features of the compound were evaluated by FT-IR and FT-Raman spectroscopic analysis in thesolid phase. On the basis of these studies, the conformational and spectroscopic behaviors of 2,4-xylylisothiocyanate were interpreted. The fundamental vibrational wavenumbers as well as their intensitieswere computed, and a good correlation between experimental and scaled calculated wavenumberswas observed. The polarizability, first hyperpolarizability and dipole moment values of 2,4-xylyl isothi-ocyanate were calculated at the same level of theory and basis set. The results show that 2,4-xylyl isothi-ocyanate molecule possesses nonlinear optical (NLO) behavior with non-zero values. Stability of themolecule arising from hyper-conjugative interactions and charge delocalization was analyzed using nat-ural bond orbital (NBO) analysis.
� 2015 Elsevier B.V. All rights reserved.
Introduction
Isothiocyanates occur naturally as glucosinolate conjugates incruciferous vegetables, like broccoli, cauliflower, cabbage, Brussel

114 M. Cinar et al. / Journal of Molecular Structure 1087 (2015) 113–120
sprouts, Kale, and are important families of natural chemo protec-tive agents [1,2]. They have been shown to inhibit cancer devel-opment, and are especially effective in fighting lung andesophageal cancers [3–5]. The risks of other cancers of the gastro-intestinal tract and the respiratory tract can also be reduced byconsuming isothiocyanate-rich vegetables. The diets high in thesevegetables have been shown to protect against a number ofmalignancies including non-Hodgkin’s lymphoma and cancers ofthe liver, prostate, ovary, lung, colon, and gastrointestinal tract[6]. In addition, organic isothiocyanates (R–NCS, R being an alkyl,phenoxy, sulfoxyl group for example) exhibit interesting anti-fungal properties. Organic isothiocyanates are also well-knownuseful reagents for hetero cycle formation by dipolar cyclo addi-tion or electrophilic addition [7] as well as important intermedi-ate and end products in chemical industry. Thus, theconformational and spectroscopic characterization of isothiocya-nates is gaining interest day by day. The vibrational spectrumof phenyl isocyanate was investigated by Stephenson et al. [8]and Chantry et al. [9] assuming C2v and Cs symmetry, respectively.Low resolution spectra of phenyl isothiocyanate and phenyl isocy-anate have been studied in microwave region by Higgins et al.[10]. Low resolution microwave, infrared and Raman spectra ofisopropylisothiocyanate [11] and, infrared and Raman spectra ofmethylisothiocyanate have been reported by Durig et al. [12].Clark and Williams analyzed infrared spectra of metal–isothiocyanate complexes in the region 3000–200 cm�1 togetherwith the vibrational assignments [13]. Torgrimsen and Klaboe[14] have reported the infrared spectra of allylisocyanate andallylisothiocyanate in vapor, liquid and amorphous and crystallinesolid state. The conformational behaviors of methoxycarbonyl-sulfenyl cyanide and methoxycarbonylsulfenyl thiocyanate wereaddressed by Torrico-Vallejos et al. [15] by using experimentalspectroscopic techniques such as 1H NMR, CG–MS, FT-IR andFT-Raman, and quantum chemical calculations at DFT and MP2methods. The spectroscopic characterization of isolated chlorocar-bonylthio- and isothiocyanate by IR (Ar matrix), Raman, 13C NMRand UV–visible spectroscopies was reported by Ramos et al. [16],and also the constitutional and rotational isomerism of the mole-cules were determined in this study by quantum chemical stud-ies. The electron transmission and the C 1s, N 1s, and S 2p coreexcitation spectra of CH3X (X = CN, NC, SCN and NCS) wererecorded and analyzed by Hitchcock et al. [17]. Tokue et al. [18]determined the electronic features of a number methylpseudoha-lides. Photoabsorption cross sections and fluorescence excitationspectra of CH3X (X = NCO, NCS and SCN) vapor measurements inthe vacuum ultraviolet using synchrotron radiation were reported.The He I and He II photoelectron spectra of methyl pseudohalideswere reported by Pasinszki et al. [19]. Cortes et al. investigated thedissociative photoionization of gas CH3SCN at the S 2p core levelusing time-of-flight mass spectrometry and synchrotron radiation[20]. The electronic structure and the dissociative ionization of theCH2ClSCN molecule in the valence region was investigated by Pira-ni et al. [21] using photoelectron spectroscopy and synchrotronbased photoelectron photoion coincidence spectroscopy. Recently,the conformational and vibration analyzes of 2-methoxyphenylisocyanate and 2-methoxyphenyl isothiocyanate were performedby Yenagi et al. for liquid samples [22].
In the present study, the vibrational interpretation of 2,4-xylylisothiocyanate based on infrared and Raman spectral analysisand quantum chemical computations have been carried out. Thesimulated UV spectrum was predicted in solutions with theoreticalTD-DFT results. Conformational and magnetic features of the titlecompound were also determined theoretically. NBO analysis hasbeen applied to study stability of the compound arising fromcharge delocalization.
Experimental studies
2,4-xylyl isothiocyanate was purchased from M/SSigma–Aldrich Chemical Co., USA and was used as received with-out further purification for spectroscopic measurements. The Per-kin Elmer spectrometer (version 10.03.06) was used to record theFourier transform Infrared (FT-IR) spectrum of 2,4-xylyl isothiocy-anate at room temperature using KBr pellet method. FT-IR spec-trum was recorded in the range of 4000–400 cm�1. The Varian7000 series spectrometer at AIRF, was used to record FT-Ramanspectrum of the title compound in the region 4000–100 cm�1.The 1064 nm laser line of Nd:YAG laser was used as the excitingwavelength.
Quantum chemical calculations
The ground state molecular geometry optimization, vibrationaland electronic transitions along with the magnetic properties of2,4-xylyl isothiocyanate were calculated by using B3LYP functional[23,24] with 6-311++G(d,p) basis set. Since the experimental crys-tallographic data of 2,4-xylyl isothiocyanate is not available, wecompared the predicted results with the data of 4-bromophenylis-othiocyanate [25]. The spectroscopic properties were calculated forthe optimized conformation. The harmonic vibrational wavenum-bers were computed and to correct overestimations scaled with0.983 and 0.958 for up to 1700 cm�1 and greater than1700 cm�1, respectively [26,27]. The assignments of the funda-mental vibrational modes were examined on the basis of the mea-sured data and total energy distribution (TED) of the vibrationalmodes, calculated with scaled quantum mechanical (SQM) method[28] using PQS program [29]. The NBO calculations [30] were per-formed in order to understand various second order interactionsbetween the filled orbitals of one subsystem and vacant orbitalsof another subsystem, which quantify the intermolecular delocal-ization or hyper conjugation. 1H and 13C NMR chemical shifts werecalculated with Gauge-Including Atomic Orbital (GIAO) approach[31]. Electronic absorption spectrum and electronic energy stateswere determined by time dependent-DFT (TD-DFT) method forvacuum and solvated phases. All calculations were accomplishedwith the Gaussian 09 program package [32].
Results and discussions
Molecular geometry
Basing on the u(C3C4AN10C19) dihedral angle, the investi-gated molecule may has two conformations; u = 180� (conforma-tion 1, C1) or u = 0� (conformation 2, C2). The potential energycurve which was computed by using semi-empirical (Austin Model1, AM1) method is shown in Fig. 1 for internal rotation around theCAX (X = S and N) bond obtained by structure optimization atdihedral angle u(C3C4AN10C19) from 0� to 360� in steps of 10�.The C1 form was predicted to be lower in energy (DE) by0.41 kcal mol�1. The optimized most stable conformation of 2,4-xylyl isothiocyanate is presented in Fig. 2 with atom numbers.The lone pair of electrons at N atom of isothiocyanate resonate asACANþBCAS� $ AC@Nþ@C@S and consequently favoring theisothiocyanate group in plane of the phenyl ring. The predictedstructural parameters comparison with the X-ray data of 4-brom-ophenylisothiocyanate [25] are given in Table 1. As a consequenceof resonance, alteration in C4AN10 and N10AC19 bond lengthsfrom the standard bond lengths has been observed. In thesix-membered ring all the CAC and CAH bond distances are inthe range of 1.389–1.407 Å and 1.083–1.086 Å, respectively. One
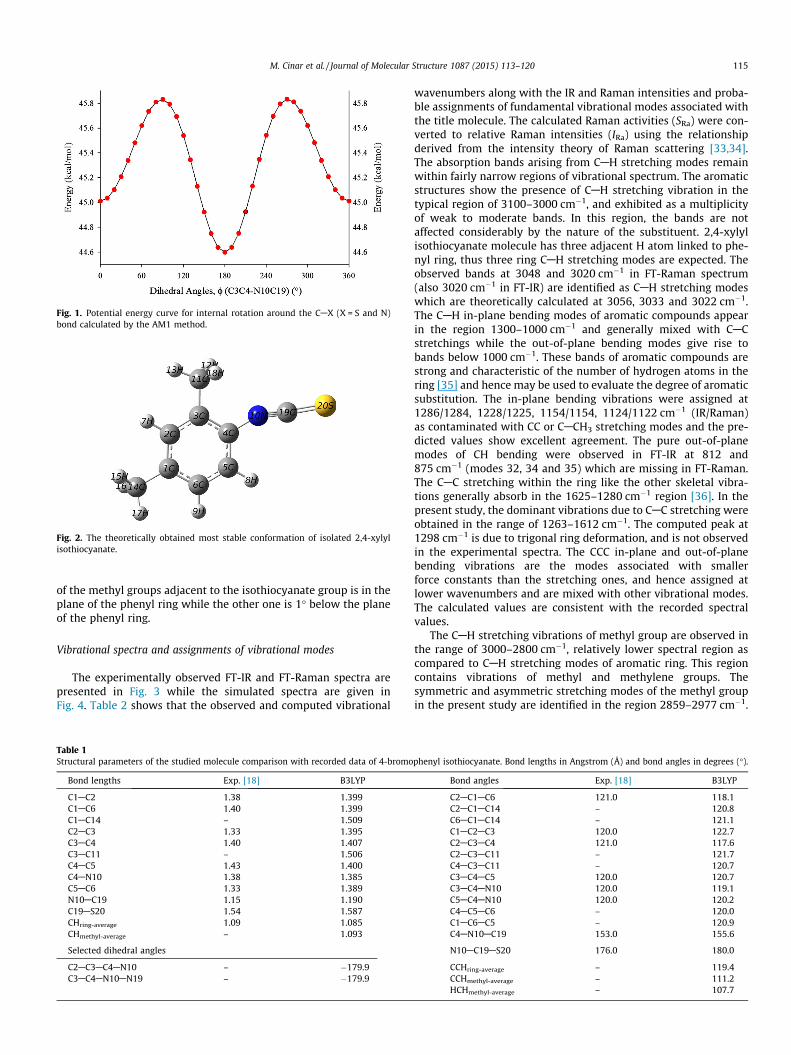
Fig. 1. Potential energy curve for internal rotation around the CAX (X = S and N)bond calculated by the AM1 method.
Fig. 2. The theoretically obtained most stable conformation of isolated 2,4-xylylisothiocyanate.
M. Cinar et al. / Journal of Molecular Structure 1087 (2015) 113–120 115
of the methyl groups adjacent to the isothiocyanate group is in theplane of the phenyl ring while the other one is 1� below the planeof the phenyl ring.
Vibrational spectra and assignments of vibrational modes
The experimentally observed FT-IR and FT-Raman spectra arepresented in Fig. 3 while the simulated spectra are given inFig. 4. Table 2 shows that the observed and computed vibrational
Table 1Structural parameters of the studied molecule comparison with recorded data of 4-bromo
Bond lengths Exp. [18] B3LYP
C1AC2 1.38 1.399C1AC6 1.40 1.399C1AC14 – 1.509C2AC3 1.33 1.395C3AC4 1.40 1.407C3AC11 – 1.506C4AC5 1.43 1.400C4AN10 1.38 1.385C5AC6 1.33 1.389N10AC19 1.15 1.190C19AS20 1.54 1.587CHring-average 1.09 1.085CHmethyl-average – 1.093
Selected dihedral angles
C2AC3AC4AN10 – �179.9C3AC4AN10AN19 – �179.9
wavenumbers along with the IR and Raman intensities and proba-ble assignments of fundamental vibrational modes associated withthe title molecule. The calculated Raman activities (SRa) were con-verted to relative Raman intensities (IRa) using the relationshipderived from the intensity theory of Raman scattering [33,34].The absorption bands arising from CAH stretching modes remainwithin fairly narrow regions of vibrational spectrum. The aromaticstructures show the presence of CAH stretching vibration in thetypical region of 3100–3000 cm�1, and exhibited as a multiplicityof weak to moderate bands. In this region, the bands are notaffected considerably by the nature of the substituent. 2,4-xylylisothiocyanate molecule has three adjacent H atom linked to phe-nyl ring, thus three ring CAH stretching modes are expected. Theobserved bands at 3048 and 3020 cm�1 in FT-Raman spectrum(also 3020 cm�1 in FT-IR) are identified as CAH stretching modeswhich are theoretically calculated at 3056, 3033 and 3022 cm�1.The CAH in-plane bending modes of aromatic compounds appearin the region 1300–1000 cm�1 and generally mixed with CACstretchings while the out-of-plane bending modes give rise tobands below 1000 cm�1. These bands of aromatic compounds arestrong and characteristic of the number of hydrogen atoms in thering [35] and hence may be used to evaluate the degree of aromaticsubstitution. The in-plane bending vibrations were assigned at1286/1284, 1228/1225, 1154/1154, 1124/1122 cm�1 (IR/Raman)as contaminated with CC or CACH3 stretching modes and the pre-dicted values show excellent agreement. The pure out-of-planemodes of CH bending were observed in FT-IR at 812 and875 cm�1 (modes 32, 34 and 35) which are missing in FT-Raman.The CAC stretching within the ring like the other skeletal vibra-tions generally absorb in the 1625–1280 cm�1 region [36]. In thepresent study, the dominant vibrations due to CAC stretching wereobtained in the range of 1263–1612 cm�1. The computed peak at1298 cm�1 is due to trigonal ring deformation, and is not observedin the experimental spectra. The CCC in-plane and out-of-planebending vibrations are the modes associated with smallerforce constants than the stretching ones, and hence assigned atlower wavenumbers and are mixed with other vibrational modes.The calculated values are consistent with the recorded spectralvalues.
The CAH stretching vibrations of methyl group are observed inthe range of 3000–2800 cm�1, relatively lower spectral region ascompared to CAH stretching modes of aromatic ring. This regioncontains vibrations of methyl and methylene groups. Thesymmetric and asymmetric stretching modes of the methyl groupin the present study are identified in the region 2859–2977 cm�1.
phenyl isothiocyanate. Bond lengths in Angstrom (Å) and bond angles in degrees (�).
Bond angles Exp. [18] B3LYP
C2AC1AC6 121.0 118.1C2AC1AC14 – 120.8C6AC1AC14 – 121.1C1AC2AC3 120.0 122.7C2AC3AC4 121.0 117.6C2AC3AC11 – 121.7C4AC3AC11 – 120.7C3AC4AC5 120.0 120.7C3AC4AN10 120.0 119.1C5AC4AN10 120.0 120.2C4AC5AC6 – 120.0C1AC6AC5 – 120.9C4AN10AC19 153.0 155.6
N10AC19AS20 176.0 180.0
CCHring-average – 119.4CCHmethyl-average – 111.2HCHmethyl-average – 107.7

Fig. 3. Experimental FT-Raman (top) and FT-IR (bottom) spectra of 2,4-xylylisothiocyanate.
Fig. 4. Simulated (with the scale factor) Raman (top) and infrared (bottom) spectraof isolated 2,4-xylyl isothiocyanate.
116 M. Cinar et al. / Journal of Molecular Structure 1087 (2015) 113–120
The difficult task of identification of in-plane and out-of-planedeformation modes of the methyl group due to presence of severalmodes becomes simpler with TED analysis, the modes 14–17which correspond to the 1457–1474 cm�1 region are assigneddue to the in-plane and out-of-plane deformation modes of themethyl groups. The CH3 symmetric deformation (or umbrella)bending modes were recorded at 1400 cm�1 and 1375 cm�1 inFT-Raman (1378 cm�1 in FT-IR) and computed at 1394 and1391 cm�1 for two CH3 groups. The in-phase rockings (qCH3) wereassigned from 987 to 1043 cm�1, and out-of-phase rockings (cCH3)at 147 and 197 cm�1. The torsion modes of methyl groups (sCH3)are not present in the experimental spectra and theoretical valuesare assigned at 36 and 107 cm�1. The methyl group assignmentsproposed in this study is also in agreement with the literature val-ues [36–39].
The stretching modes of isothiocyanates are observed in the2000–2280 cm�1 region [36]. The characteristic asymmetric dou-blet around 2100 cm�1 is due to the isothiocyanate group [22].For the title compound, a doublet around 2084 and 2094 cm�1 inFT-IR and FT-Raman is due to asymmetric NCS stretching mode.DFT calculations produce the correlated doublet around2076 cm�1. The TED of this vibration is 100%. Like many isothiocy-anate systems, this doublet is interpreted to be caused by theFermi resonance [22,40,8]. The C@S stretching mode was calcu-lated at 892 cm�1 and recorded at 900 cm�1 in FT-IR. This bandwas observed at 927 cm�1 for the phenyl isothiocyanate [8] and931 cm�1 for 2-methoxyphenylisothiocyanate [22]. The out-of-plane bending modes of isothiocyanate group were obtained in434–483 cm�1.
Polarizability and hyperpolarizability
NLO properties are at the forefront of current research becauseof its importance in providing the key functions of wavenumbersshifting, optical modulation, optical switching, optical logic, andoptical memory for emerging technologies in the areas such astelecommunications, signal processing, and optical interconnec-tions [41–45]. Urea is one of the prototypical molecules used inthe study of the NLO properties of molecular systems. Therefore,it is used often as a threshold value for comparative purposes.The calculated values of hai and bTOTAL for the title compound werefound to be 21.89 � 10�24 e.s.u. and 0.86 � 10�30 e.s.u. respectivelyat B3LYP/6-311++G(d,p) basis set (Table S1). The 2,4-xylyl isothio-cyanate has large bTOTAL value than urea (almost 4.4 times greaterthan urea), that indicates, 2,4-xylyl isothiocyanate is a good candi-date of NLO material. Also, the calculated value of dipole moment(l) of 2,4-xylyl isothiocyanate is found to be 4.07 Debye at B3LYP/6-311++G(d,p) basis set.
The theoretical calculations are more useful in the determina-tion of particular components of b tensor than in establishing thereal values of b. Dominance of particular components shows sub-stantial delocalization of charges in those directions. It is noticedthat in bxyy direction, the biggest values of hyperpolarizability arenoticed and subsequently delocalization of electron cloud is morein that direction.
Ultraviolet (UV) studies and electronic properties
The simulated UV spectrum of 2,4-xylyl isothiocyanate is pre-sented in Fig. 5. The low-lying excited states of the title compound

Table 2The observed (FT-IR and FT-Raman) and calculated vibrational frequencies along with their relative intensities, probable assignments and total energy distribution (TED) ofvibrational modes.
No. Experimental B3LYP/6-311++G(d,p)
FT-IR FT-Raman Unscaled Scaled IIR SRa IRa Assignments and TED results (given in parenthesis, P8%)
1 – 3048 3190 3056 1825.91 917.58 0.42 m CHring (99)2 – – 3166 3033 73.07 178.86 0.23 m CHring (100)3 3020 3020 3154 3022 54.66 0.65 0.00 m CHring (99)4 2977 2979 3111 2981 35.81 2.64 0.05 masym CHmethyl (100)5 – – 3103 2973 35.03 0.37 0.00 masym CHmethyl (99)6 2947 – 3078 2948 33.39 426.10 0.06 masym CHmethyl (100)7 – – 3076 2947 28.33 6.95 0.03 masym CHmethyl (100)8 2919 2916 3029 2902 19.07 120.48 0.01 msym CHmethyl (100)9 2859 2862 3021 2895 18.08 216.07 0.03 msym CHmethyl (100)
– 2131 – – – – – 1560 + 564 = 212410 2084 2094 2167 2076 17.74 111.34 0.01 masymNCS (100)11 1612 1608 1644 1616 17.24 58.49 0.01 m CC (79)12 1556 1572 1605 1578 15.41 19.77 0.03 m CC (73)13 1493 1488 1527 1501 14.66 74.81 0.01 m CC (66)14 – – 1499 1474 12.63 8.11 0.11 b CHmethyl (56), m CC (8), c CHmethyl (8)15 – – 1490 1465 12.02 60.54 0.01 b CHmethyl (62), c CHmethyl (27)16 – – 1487 1462 10.97 99.79 0.01 b CHmethyl (59), c CHmethyl (24)17 1453 1443 1482 1457 10.05 1.14 0.00 b CHmethyl (70), c CHmethyl (20)18 – – 1434 1410 9.02 5.13 0.01 m CC (36), b CHring (19), b CHmethyl (11)19 – 1400 1419 1394 8.70 8.50 0.01 CH3 umbrella (92)20 1378 1375 1415 1391 7.10 38.14 0.04 CH3 umbrella (91)21 – – 1320 1298 6.62 12.57 0.02 Ring deformation (79)22 1286 1284 1310 1288 5.95 2.81 0.00 b CHring (60), m CC (24)23 1263 1252 1280 1258 5.93 44.72 0.08 m CN (38), m CC (27), b CHring (12), m SC (11)24 1228 1225 1252 1231 5.16 494.22 1.00 b CHring (27), m CACH3 (25), m CC (21)25 1154 1154 1178 1158 4.78 0.49 0.00 b CHring (47), m CACH3 (20), m CC (19)26 1124 1122 1152 1132 4.52 0.20 0.00 b CHring (35), m CC (24), m CACH3 (17), b CCCring (11)27 – – 1061 1043 4.48 478.91 0.49 q CH3 (83)28 1036 – 1059 1041 4.33 3.10 0.00 q CH3 (86)29 1013 – 1035 1017 4.18 92.18 0.01 q CH3 (68), m CC (10)30 – – 1004 987 3.77 0.81 0.01 q CH3 (56), m CC (12)31 947 – 963 947 3.70 2.79 0.03 m CC (40), m C@S (16), m CACH3 (13)32 – – 959 943 3.20 3.89 0.01 c CHring (88)33 900 – 908 892 3.16 32.73 0.26 m C@S (29), m CACH3 (22), m CC (25)34 875 – 893 877 3.13 1.40 0.00 c CHring (77)35 812 – 826 812 2.59 1.00 0.02 c CHring (87)36 724 726 735 722 2.48 0.24 0.01 b CCC (80)37 705 – 725 713 2.28 1.68 0.00 c CCCC (78)38 – – 639 628 2.07 0.42 0.01 b CAN„C (31), b CCC (23), m CACH3 (16), m C@S (14)39 566 564 574 564 1.96 7.75 0.06 b CCC (42), m CACH3 (19), b CCN (16)40 548 – 562 552 1.87 12.16 0.02 c CCCC (71)41 498 486 491 483 1.87 28.95 0.08 b (CN„C + N„C@S) (57), b CCACH3 (29)42 – 452 464 457 1.60 2.34 0.01 b (N„C@S) (38), b CCACH3 (37)43 – – 452 445 0.86 2.13 0.08 c CCCC (51), c SCNC (30)44 439 438 441 434 0.63 4.98 0.21 c SCNC (75)45 – 344 368 362 0.62 23.45 0.03 b (CN„C + N„C@S) (49), b CCC (33)46 – – 342 336 0.53 0.25 0.00 Ring twisting (81)47 – 273 296 291 0.50 2.87 0.02 b CACH3 (58), b CCN (19)48 – – 254 249 0.47 16.92 0.03 b CACH3 (53), b CCN (14)49 – 215 200 197 0.36 1.86 0.01 c CH3 (75), c CCCC (10)50 – 163 150 147 0.35 0.28 0.00 c CH3 (38), c SCNC (29), c CCCC (20)51 – – 109 107 0.27 50.28 0.08 s CH3 (96)52 – – 47 46 0.04 11.06 0.02 Butterfly (88)53 – – 37 36 0.01 1.79 0.00 s CH3 (100)54 – – 33 32 0.01 0.86 0.01 b CAN„C (93)
m; stretching, b; in-plane bending, c; out-of-plane bending, q; scissoring, s; torsion, [wavenumbers (cm�1), IR intensities; IIR (K mmol�1), Raman scattering activities; SRaman
(Å4 amu�1)].
M. Cinar et al. / Journal of Molecular Structure 1087 (2015) 113–120 117
were determined on the basis of TD-DFT/6311++G(d,p) method.The main absorption peak was observed at about 280 nm, and asecondary peak was recorded at about 225 nm. The calculatedresults involving the vertical excitation energies, oscillatorstrengths (f) and transition wavelengths for vacuum and in ethanolare given in Table 3. The predicted absorption spectra show devia-tion from the experimental spectrum which is due to the presenceAN@C@S group containing non-bonded electrons acting as auxo-chrome. Presence of an auxochrome modifies both the intensityand wavelength of absorption. Auxochrome in direct conjugation
with the p-electron system may increase the wavelength of lightabsorbed and also increase the absorption. Theoretical electronictransition is obtained around 280 nm with highest oscillatorstrength for all three solutions while for the gas phase it is calcu-lated ca. 2 nm below. It is clear from Fig. 6 showing the patternsof the molecular orbitals involved in the electronic transitions, thatthis corresponds to transition between HOMO and LUMO (p ? p⁄
transition). HOMO is a delocalized p orbital located over the entiremolecule while LUMO having spread over entire molecule havingmore anti-bonding character. The secondary transition is also

Fig. 5. The UV spectra of 2,4-xyly lisothiocyanate.
118 M. Cinar et al. / Journal of Molecular Structure 1087 (2015) 113–120
predicted at ca. 223 nm for three solutions is due toHOMO�1(orbital 42) ? LUMO(orbital 44) transition for ethanoland THF. The energy gap between the HOMO–LUMO was calcu-lated around at 4.90 eV (Table 4).
Nuclear Magnetic Resonance (NMR) studies
The proton and carbon nuclear magnetic resonance (1H and 13CNMR) isotropic chemical shifts of 2,4-xlyl isothiocyanate werecomputed taking tetramethylsilane (TMS) as a reference, by usingoptimized molecular structure at DFT-B3LYP/6-311++G(d,p) leveland GIAO method which is an exponential term containing the vec-tor potential is included with each atomic orbital, for vacuum, inDMSO and chloroform solutions. The calculated results are pre-sented in Table 5. The chemical shifts of TMS obtained with thesame method and basis set are given in the bottom of the table.
The chemical shifts of aromatic protons of organic compoundsare usually observed in the range of 7.00–8.00 ppm. Three protonsattached to ring predicted in the range 6.98–7.37 ppm for solvatedphase, and in the lower range for vacuum. The methyl protonswere obtained at higher region, as expected. In general, aromaticcarbon resonances occur between 100 and 150 ppm [46,47]. How-ever, with the attachment of more than one electron withdrawingand electron releasing substituents, the full range of aromatic res-onances expand to 90–180 ppm. The ring hydrogen attached car-bon atoms (C2, C5 and C6) resonated at 129.0–142.1 ppm. Theresonance peaks of methyl groups attached C atoms were com-puted around 138.0 or 140.0 ppm which are shifted lower regionwhen compared to other ring carbons. The resonance peak of C4atom linked to isothiocyanate group was predicted at nearly132.0 ppm, and seemed to be not affected to much because ofthe ACAN„C@S character of this functional group which causethe reduction of electron density on nitrogen atom. The methyl
Table 3The calculated absorption wavelengths k (nm), excitation energies E (eV) and oscillator st
Gas Ethanol
E (eV) k (nm) f E (eV) k (nm) f E
4.4575 278.15 (43 ? 44) 0.3687 4.4251 280.19 (43 ? 44) 0.5478 44.4796 276.78 (43 ? 46) 0.0019 4.4395 279.28 (43 ? 45) 0.0034 44.6685 265.57 (42 ? 44) 0.0000 4.7383 261.66 (43 ? 46) 0.0261 44.7020 263.69 (43 ? 45) 0.0323 4.8046 258.05 (41 ? 44) 0.0000 45.3068 233.63 (43 ? 47) 0.0021 5.4161 228.92 (43 ? 47) 0.0083 55.5577 223.08 (41 ? 44) 0.2085 5.5392 223.83 (43 ? 46) 0.2923 5
HOMO is no. 43.
group carbons are attached to three hydrogen atoms and henceresonate at higher field, 16–18 ppm. On the other side, due to thetriple bond character of N„C, and electron withdrawing abilityof nitrogen group the chemical shift of C19 was computed at129.0 ppm for solvated media.
Natural bond orbital (NBO) analysis
NBO analysis is an essential tool for studying intra- and inter-molecular bonding and interaction among bonds, and also providesa convenient basis for investigating charge transfer or conjugativeinteraction in molecular systems. Some electron donor orbital,acceptor orbital and the interacting stabilization energy resultingfrom the second-order micro disturbance theory were reported[48,49]. The larger the E(2) value, the more intensive is the interac-tion between electron donors and electron acceptors, i.e. the moredonating tendency from electron donors to electron acceptors andthe greater the extent of conjugation of the whole system. Delocal-ization of electron density between occupied Lewis-type (bond orlone pair) NBO orbitals and formally unoccupied (anti-bonding orRydberg) non-Lewis NBO orbitals correspond to a stabilizingdonor–acceptor interaction. The direction of the line of centersbetween the two nuclei is compared with the hybrid direction todetermine the bending of the bond, expressed as the deviationangle between these two directions and are depicted in Table S2.
The hybrid directionality and bond bending analysis of Naturalhybrid orbitals (NHOs) offer indications of the substituent effectand steric effect. In rC3AC4 and rC4AN10, the C3 and C4 NHOsshow deviations of 1.3� and 1.2� with C3 and C4 atoms. InrC19AS20, NHOs also show a very large deviation of 47.8� and25.7� with C19 and S20 atoms. It is evident from deviations ofthese NHOs, a strong charge transfer path toward ANCS group.
Table S3 depicts the bonding concepts such as type of bondorbital, their occupancies, the natural atomic hybrids of whichthe NBO is composed, giving the percentage of the NBO on eachhybrid, the atom label, and a hybrid label showing the hybrid orbi-tal (spx) composition (the amount of s-character, p-character, etc.)of 2,4-xylyl isothiocyanate molecule determined by B3LYP/6-311++G(d,p) method with decent accuracy. The occupancies ofNBOs are reflecting their exquisite dependence on the chemicalenvironment. The bonding orbital for C4AN10 with 1.98312 elec-trons has 37.59% C4 character in a sp2.91 hybrid and has 62.41%N10 character in a sp1.07 hybrid orbital of 2–4-xylyl isothiocyanate.Similarly, for N10AC19, the bonding orbital with 1.77940 electronshas 61.95% N10 character in a sp1.01 hybrid and has 38.05% C19character in a sp1.21 hybrid orbital. A bonding orbital forC19AS20 with 1.83116 electrons has 49.6% C19 character in asp1.15 hybrid and has 50.4% S20 character in a sp5.04 orbital. TheCAC bonds of the aromatic ring possess more p character than scharacter. This is clearly indicates the delocalization of p electronsamong all the carbon atoms.
The intra-molecular interaction are formed by the orbital over-lap between r(CAC), r⁄(CAC), p(CAC), p⁄(CAC) bond orbital
rengths (f) of the studied molecule along with transition levels.
THF CCl4
(eV) k (nm) f E (eV) k (nm) f
.4144 280.86 (43 ? 44) 0.5605 4.3933 282.21 (43 ? 44) 0.5661
.4449 278.94 (43 ? 45) 0.0009 4.4606 277.95 (43 ? 46) 0.0002
.7341 261.89 (43 ? 46) 0.0255 4.7197 262.70 (42 ? 44) 0.0257
.7855 259.08 (41 ? 44) 0.0000 4.7310 262.07 (41 ? 44) 0.0000
.4066 229.32 (43 ? 47) 0.0075 5.3702 230.88 (43 ? 47) 0.0051
.5284 224.27 (43 ? 46) 0.2984 5.5080 225.10 (42 ? 44) 0.3025

Fig. 6. The frontier molecular orbitals scheme and energy values in ethanol solution of 2,4-xylyl isothiocyanate.
Table 4Significant molecular orbitals and corresponding energies for 2,4-xlyl isothiocyanate.
Gas Ethanol THF CCl4
ELUMO+3 �0.32 �0.22 �0.22 �0.25ELUMO+2 �0.63 �0.67 �0.68 �0.69ELUMO+1 �0.76 �0.78 �0.75 �0.70ELUMO �1.42 �1.46 �1.45 �1.43EHOMO �6.28 �6.36 �6.34 �6.31EHOMO�1 �7.13 �7.19 �7.20 �7.21EHOMO�2 �7.26 �7.36 �7.32 �7.23
DELUMO�HOMO 4.86 4.90 4.89 4.88DELUMO+1�HOMO 5.52 5.58 5.59 5.60DELUMO+2�HOMO 5.65 5.68 5.66 5.62DELUMO+3�HOMO 5.96 6.14 6.12 6.06DELUMO�HOMO�1 5.71 5.73 5.75 5.79DELUMO�HOMO�2 5.84 5.90 5.88 5.81
Table 5The theoretical 1H and 13C NMR chemical shifts (with respect to TMS) of 2,4-xlylisothiocyanate compound (atom positions were numbered as in Fig. 2).
Atom Gas DMSO CDCl3 Atom Gas DMSO CDCl3
H7 7.11 7.37 7.23 C1 141.4 142.0 141.8H8 6.94 7.24 7.08 C2 138.8 142.1 141.2H9 6.85 7.13 6.98 C3 136.9 138.7 138.2H12 2.36 2.49 2.39 C4 131.3 132.3 132.0H13 2.21 2.31 2.22 C5 130.7 132.0 131.6H15 2.21 2.30 2.22 C6 129.9 129.0 129.2H16 1.92 2.11 1.99 C19 128.1 129.0 128.7H17 1.86 2.06 1.94 C11 17.9 18.2 18.1H18 1.55 1.84 1.69 C14 16.1 16.3 16.3
Computed chemical shift values of TMS using B3LYP/6-311++G(d,p) method,1H NMR: 31.6702/31.7615/31.6706 ppm (Gas/DMSO/CDCl3) and13C NMR: 178.5432/179.1808/178.9865 ppm (Gas/DMSO/CDCl3).
M. Cinar et al. / Journal of Molecular Structure 1087 (2015) 113–120 119

120 M. Cinar et al. / Journal of Molecular Structure 1087 (2015) 113–120
which results intra-molecular charge transfer (ICT) causing stabil-ization of the system. These interactions are observed as increasein electron density (ED) in CAC anti-bonding orbital that weakensthe respective bonds. These intra-molecular charge transfer(r ? r⁄, p ? p⁄) can induce large nonlinearity of the molecule.Table S4 shows the strong intra molecular hyper conjugative inter-action of the r electron of (C1AC6) with r⁄(C1AC2), (C5AC6),(C2AH7), (C5AH8) of the ring. On the other hand the p(C1AC6)in the ring conjugate to the anti-bonding orbital of p⁄(C2AC3)and p⁄(C4AC5) which leads to strong delocalization of 18.57,23.27 kcal/mol. The intra-molecular interaction is formed by theorbital overlap between p(C2AC3) and p⁄(C1AC6) with the energyof 21.91 kcal/mol. The same p(C2AC3) bond also interact withp⁄(C4AC5) with the energy of 19.41 kcal/mol. This strong stabiliza-tion denotes the larger delocalization. The interesting interactionin 2–4 xylyl isothiocyanate is lone pair of S20 with that of anti-bonding r⁄ N10AC19. This interaction results in the stabilizationof the system with energy 19.34 kcal/mol. The strongest intramolecular hyper conjugative interaction of the p⁄ electron of(C4AC5) distribute to p⁄(C1AC6) and p⁄(C2AC3) of the ring witha very high stabilization energy of 217.99 and 212.51 kcal/mol,respectively.
Conclusions
From the analysis of the recorded FT-IR and FT-Raman spectraand quantum chemical calculations with application of DFT, thevibrational transitions of 2,4-xlyl isothiocyanate were interpretedand assignments of each vibrational mode were carried out. Thecharacteristic vibrational mode of isothiocyanate systems thatobserved as doublet band and invoking as Fermi resonance wasrecorded at 2084/2094 cm�1. Similarly, the asymmetric NSCstretching or C@S double bond stretching of isothiocyanate groupwere obtained in expected region from experimental and also com-putational studies, and these results show a fairly good agreementwith literature data. The ground state equilibrium structure wasoptimized and compared with the X-ray crystallographic structureof a similar molecule that reported earlier. The magnetic featuresof the studied molecule were investigated by the help of the com-putational NMR chemical shift analysis for the isolated molecule invacuum and solvated phase. The simulated UV spectra of the inves-tigated compound were predicted by using TD-DFT method for thegas phase and solutions. Additionally, the NBO analysis was donefor exploring charge transfer or conjugative interaction in molecu-lar systems, also for studying intra- and intermolecular bondingand interaction among bonds.
Appendix A. Supplementary material
Supplementary data associated with this article can be found, inthe online version, at http://dx.doi.org/10.1016/j.molstruc.2014.12.079.
References
[1] Y.S. Keum, W.S. Jeong, A.N. Kong, Mutat. Res. 555 (2004) 191–202.[2] S. Adsule, S. Banerjee, F. Ahmed, S. Padhye, F.H. Sarkar, Bioorg. Med. Chem. Lett.
20 (2010) 1247–1251.[3] P.M. Cinciripini, S.S. Hecht, J.E. Henningfield, M.W. Manley, B.S. Kramer, J. Natl
Cancer Inst. 89 (1997) 1852–1867.[4] S.J. London, J.-M. Yuan, F.-L. Chung, Y.-T. Gao, G.A. Coetzee, R.K. Ross, M.C. Yu,
Lancet 356 (2000) 724–729.
[5] S.S. Hecht, Drug Metab. Rev. 32 (2000) 395–411.[6] G. Murillo, R.G. Mehta, Nutr. Cancer 41 (2001) 17–28.[7] Y-J Kim, J-T Han, S. Kang, W.S. Han, S.W. Lee, Dalton Trans. 17 (2003) 3357–
3364.[8] C.V. Stephenson, W.C. Coburn Jr., W.S. Wilcox, Spectrochim. Acta 17 (1961)
933–946.[9] G.W. Chantry, E.A. Nicol, D.J. Harrison, A. Bouchy, G. Roussy, Spectrochim. Acta
A 30 (1974) 1717–1722.[10] R.J. Higgins, L.L. Combs, T.B. Malloy Jr., R.L. Cook, J. Mol. Struct. 28 (1975) 121–
127.[11] J.R. Durig, J.F. Sullivan, T.S. Little, S. Cradock, J. Mol. Struct. 118 (1984) 103–
117.[12] J.R. Durig, J.F. Sullivan, H.L. Heusel, J. Mol. Struct. 100 (1983) 241–257.[13] R.J.H. Clark, C.S. Williams, Spectrochim. Acta 22 (1966) 1081–1090.[14] T. Torgrimsen, P. Klaboe, J. Mol. Struct. 20 (1974) 213–227.[15] S. Torrico-Vallejos, M.F. Erben, M-Fa Ge, H. Willner, C.O. Della Vedova, J. Phys.
Chem. A 114 (2010) 3703–3712.[16] L.A. Ramos, S.E. Ulic, R.M. Romano, et al., J. Phys. Chem. A 117 (2013) 2383–
2399.[17] A.P. Hitchcock, M. Tronc, A. Modelli, J. Phys. Chem. 93 (1989) 3068–3077.[18] I. Tokue, A. Hiraya, K. Shobatake, Chem. Phys. 117 (1987) 315–324.[19] T. Pasinszki, T. Veszpremi, M. Feher, B. Kovac, L. Klasinc, S.P. Mcglynn, Int. J.
Quantum Chem. 26 (1992) 443–453.[20] E. Cortes, M.F. Erben, M. Gerones, R.M. Romano, C.O. Della Vedova, J. Phys.
Chem. A 113 (2009) 564–572.[21] L.S.R. Pirani, M. Gerones, C.O. Della Vedova, R.M. Romano, A. Fantoni, R.
Cavasso-Filho, C. Ma, M. Ge, M.F. Erben, J. Phys. Chem. A 116 (2012) 231–241.[22] J. Yenagi, A.R. Nandurkar, J. Tonannavar, Spectrochim. Acta A 91 (2012) 261–
268.[23] W. Kohn, L.J. Sham, Phys. Rev. A 140 (1965) 1133–1138.[24] A.D. Becke, J. Chem. Phys. 98 (1993) 5648–5652.[25] L. Ulicky, J. Soldanova, Chem. Pap. 39 (1985) 473–480.[26] N. Sundaraganesan, S. Ilakiamani, H. Saleem, P.M. Wojciechowski, D.
Michalska, Spectrochim. Acta A 61 (2005) 2995–3001.[27] M. Karabacak, L. Sinha, O. Prasad, Z. Cinar, M. Cinar, Spectrochim. Acta A 93
(2012) 33–46.[28] J. Baker, A.A. Jarzecki, P. Pulay, J. Phys. Chem. A 102 (1998) 1412–1424.[29] SQM version 1.0, Scaled Quantum Mechanical Force Field, 2013 Green Acres
Road, Fayetteville, Arkansas 72703.[30] E.D. Glendening, C.R. Landis, F. Weinhold, WIREs Comput. Mol. Sci. 2 (2011) 1–
42.[31] K. Wolinski, R. Haacke, J.F. Hinton, P. Pulay, J. Compos. Chem. 18 (1997) 816–
825.[32] M.J. Frisch et al., Gaussian 09, Revision A.1, Gaussian, Inc., Wallingford CT,
2009.[33] G. Kereztury, S. Holly, J. Varga, G. Besenyei, A.Y. Wang, J.R. Durig, Spectrochim.
Acta 49A (2007) 1993–2019.[34] G. Kereztury, in: J.M. Chalmers, P.R. Griffith (Eds.), Raman Spectroscopy:
Theory, Handbook of Vibrational Spectroscopy, vol. 1, John Wiley & Sons Ltd,New York, 2002.
[35] B. Stuart, Infrared Spectroscopy: Fundamentals and Applications, Wiley IndiaEd., 2010.
[36] R.M. Silverstein, F.X. Webster, D.J. Kiemle, Spectrometric Identification ofOrganic Compounds, seventh ed., John Wiley & Sons, 2005.
[37] M. Govindarajan, K. Ganasan, S. Periandy, M. Karabacak, Spectrochim. Acta,Part A 79 (2011) 646–653.
[38] G. Varsanyi, Vibrational Spectra of Benzene Derivatives, Academic Press, NewYork, 1969.
[39] M. Karabacak, M. Cinar, M. Kurt, J. Mol. Struct. 968 (2010) 108–114.[40] E. Lieber, C.N.R. Rao, J. Ramachandran, Spectrochim. Acta 13 (1959) 296–299.[41] Y.X. Sun, Q.L. Hao, W.X. Wei, Z.X. Yu, L.D. Lu, X. Wang, Y.S. Wang, J. Mol. Struct.:
Theochem 904 (2009) 74–82.[42] C. Andraud, T. Brotin, C. Garcia, F. Pelle, P. Goldner, B. Bigot, A. Collet, J. Am.
Chem. Soc. 116 (1994) 2094–2102.[43] V.M. Geskin, C. Lambert, J.L. Bredas, J. Am. Chem. Soc. 125 (2003) 15651–
15658.[44] M. Nakano, H. Fujita, M. Takahata, K. Yamaguchi, J. Am. Chem. Soc. 124 (2002)
9648–9655.[45] D. Sajan, N. Vijayan, K. Safakath, Reji Philip, M. Karabacak, Spectrochim. Acta A
108 (2013) 186–196.[46] H.O. Kalinowski, S. Berger, S. Braun, Carbon-13 NMR Spectroscopy, John Wiley
& Sons, Chichester, 1988.[47] K. Pihlaja, E. Kleinpeter (Eds.), Carbon-13 Chemical Shifts in Structural and
Stereochemical Analysis, VCH Publishers, Deerfield Beach, 1994.[48] C. James, A.A. Raj, R. Reghunathan, I.H. Joe, V.S.J. Kumar, J. Raman Spectrosc. 37
(2006) 1381–1392.[49] L.J. Na, C.Z. Rang, Y.S. Fang, J. Zhejiang Univ. Sci. 6B (2005) 584–589.




















