
Computed tomography studies on patients with mucopolysaccharidoses
15
Neuroradiology(1981) 21:9-23 Neuroradiologg © Springer-Verlag 1981 Computed Tomography Studies on Patients with Mucopolysaccharidoses R. W. E. Watts 1, Elizabeth Spellacy 1, B. E. Kendall2, G. du Boulay2 and Dorothy A. Gibbs 1 IDivision of InheritedMetabolicDiseases, MedicalResearch Council,Clinica!Research Centre, Harrow, Middlesex, and 2Lysholm Radiological Department,The NationalHospital for NervousDiseases, Queen Square, London,England Summary. The appearances on computed tomogra- phy (CT) in eight patients with mucopolysac- charidosis Type I [five with classical Type IH (Hurler disease)], two with mucopolysaccharidosis Type II (Hunter disease) and two with mucopolysac- charidosis Type IIIB (Sanfilippo B disease) are pre- sented. Reference is also made to two further cases [mucopolysaccharidosis VI and mucopolysac- charidosis IIIB] in which the CT showed special fea- tures. Follow-up scans were obtained to assess the evolution of the changes. The interplay of neuronal damage, cerebral atrophy and obstruction to the cerebrospinal fluid (CSF) circulation in the produc- tion of the cerebral manifestations, and the extent to which non-genetic factors influence the expression of the underlying biochemical lesion, are discussed. This series of patients illustrates the problem of classifying those with a-iduronidase deficiency, who do not have the classical Hurler disease phenotype (mucopolysac- charidosis IS and IH/S; Scheie disease and Hurler/ Scheie disease), on the basis of currently available criteria. The place of CT in the diagnosis of complica- tions due to thecal involvement is examined. The density of grey matter on the CT scans was similar to that of normal brain. Inhaled xenon did not produce any special enhancement which could be helpful in assessing the degree of the mucopolysaccharide deposition within cerebral ceils. Symmetrical low attenuation in the white matter was a very common finding. It was not specifically associated with hy- drocephalus and its relationship to some of the known neuropathological aspects of mucopolysac- charidoses is discussed. This work has shown that the stage in the evolution of the mucopolysaccharidoses at which hydrocephalus develops as a complication is highly variable and CSF diversion procedures are sometimes indicated to improve the quality of the patients' lives. These indications are briefly discus- sed. We consider that CT is essential for the adequate appraisal of these patients and to identify some treat- able complications. Key words: Mucopolysaccharidoses - Computed tomography - Hurler disease - Hunter disease - San- filippo disease - Morquio disease - Scheie disease - Hurler/Scheie disease Introduction The mucopolysaccharidoses are inborn errors of metabolism in which deficiency of a specific exogly- cosidase prevents the normal lysosomal degradation of the mucopolysaccharides (glycosaminoglycans). The mucopolysaccharide deposits in the lysosomes interfere with the degradation of other ma- cromolecules and this accounts for the intralysosomal accumulation of glycolipids as well as mucopolysac- charides. Cortical neurones have been described as developing large processes ("meganeurites") inter- posed between the axons and perikarya and contain- ing the loaded lysosomes or storage bodies [1, 2]. Inclusions have also been found in astrocytes and in capillary pericytes. The laminated appearance of those in the neurones and astrocytes suggests that they contain glycolipids as well as mucopolysac- charides, whereas the inclusions in the pericytes resemble those in hepatocytes which contain only, or almost entirely, mucopolysaccharides [3-5]. Table 1 summarises the main biochemical, genet- ic and clinical aspects of the mucopolysaccharidoses. The clinical manifestations of the individual mucopolysaccharidoses vary. This variation is partly inherited, so that mild and severe genetic variants exist, and partly non-genetic. Mechanical factors 0028-3940/81/0021/0009/$03.00
-
Upload
independent -
Category
Documents
-
view
0 -
download
0
Transcript of Computed tomography studies on patients with mucopolysaccharidoses

Neuroradiology (1981) 21:9-23 Neuroradiologg © Springer-Verlag 1981
Computed Tomography Studies on Patients with Mucopolysaccharidoses
R. W. E. Watts 1, Elizabeth Spellacy 1, B. E. Kendall 2, G. du Boulay 2 and Dorothy A. Gibbs 1
IDivision of Inherited Metabolic Diseases, Medical Research Council, Clinica! Research Centre, Harrow, Middlesex, and 2Lysholm Radiological Department, The National Hospital for Nervous Diseases, Queen Square, London, England
Summary. The appearances on computed tomogra- phy (CT) in eight patients with mucopolysac- charidosis Type I [five with classical Type IH (Hurler disease)], two with mucopolysaccharidosis Type II (Hunter disease) and two with mucopolysac- charidosis Type IIIB (Sanfilippo B disease) are pre- sented. Reference is also made to two further cases [mucopolysaccharidosis VI and mucopolysac- charidosis IIIB] in which the CT showed special fea- tures. Follow-up scans were obtained to assess the evolution of the changes. The interplay of neuronal damage, cerebral atrophy and obstruction to the cerebrospinal fluid (CSF) circulation in the produc- tion of the cerebral manifestations, and the extent to which non-genetic factors influence the expression of the underlying biochemical lesion, are discussed. This series of patients illustrates the problem of classifying those with a-iduronidase deficiency, who do not have the classical Hurler disease phenotype (mucopolysac- charidosis IS and IH/S; Scheie disease and Hurler/ Scheie disease), on the basis of currently available criteria. The place of CT in the diagnosis of complica- tions due to thecal involvement is examined. The density of grey matter on the CT scans was similar to that of normal brain. Inhaled xenon did not produce any special enhancement which could be helpful in assessing the degree of the mucopolysaccharide deposition within cerebral ceils. Symmetrical low attenuation in the white matter was a very common finding. It was not specifically associated with hy- drocephalus and its relationship to some of the known neuropathological aspects of mucopolysac- charidoses is discussed. This work has shown that the stage in the evolution of the mucopolysaccharidoses at which hydrocephalus develops as a complication is highly variable and CSF diversion procedures are sometimes indicated to improve the quality of the patients' lives. These indications are briefly discus-
sed. We consider that CT is essential for the adequate appraisal of these patients and to identify some treat- able complications.
Key words: Mucopolysaccharidoses - Computed tomography - Hurler disease - Hunter disease - San- filippo disease - Morquio disease - Scheie disease - Hurler/Scheie disease
Introduction
The mucopolysaccharidoses are inborn errors of metabolism in which deficiency of a specific exogly- cosidase prevents the normal lysosomal degradation of the mucopolysaccharides (glycosaminoglycans). The mucopolysaccharide deposits in the lysosomes interfere with the degradation of other ma- cromolecules and this accounts for the intralysosomal accumulation of glycolipids as well as mucopolysac- charides. Cortical neurones have been described as developing large processes ("meganeurites") i n t e r - posed between the axons and perikarya and contain- ing the loaded lysosomes or storage bodies [1, 2]. Inclusions have also been found in astrocytes and in capillary pericytes. The laminated appearance of those in the neurones and astrocytes suggests that they contain glycolipids as well as mucopolysac- charides, whereas the inclusions in the pericytes resemble those in hepatocytes which contain only, or almost entirely, mucopolysaccharides [3-5].
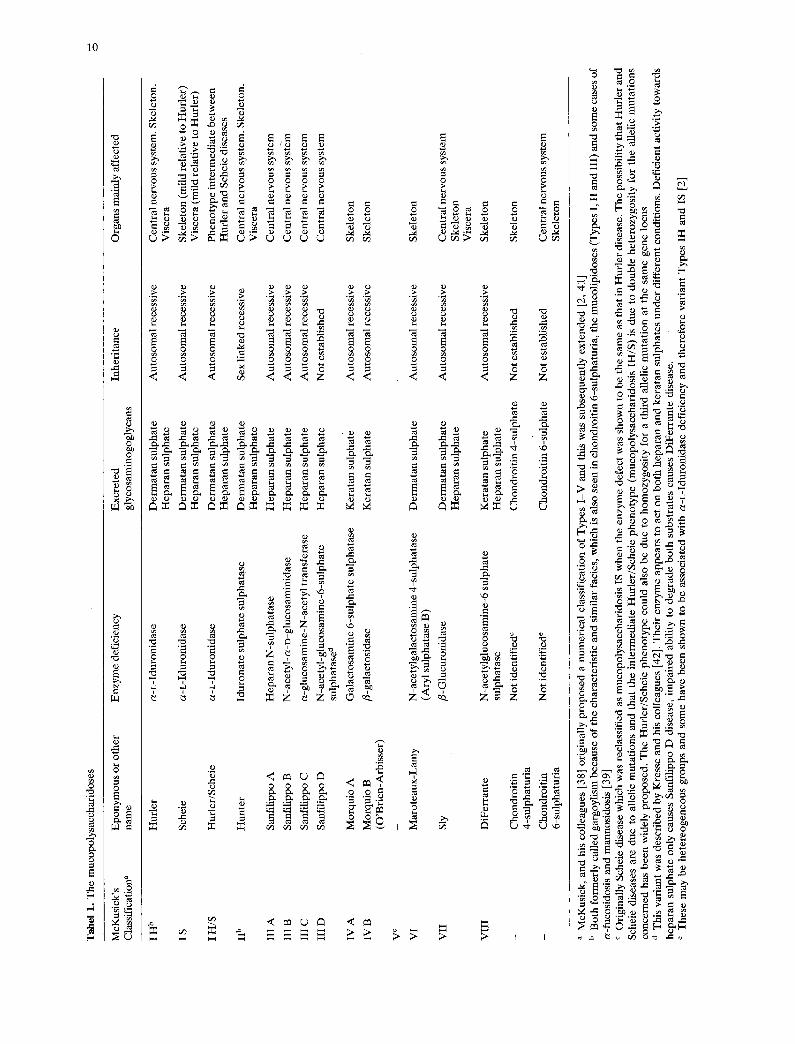
Table 1 summarises the main biochemical, genet- ic and clinical aspects of the mucopolysaccharidoses. The clinical manifestations of the individual mucopolysaccharidoses vary. This variation is partly inherited, so that mild and severe genetic variants exist, and partly non-genetic. Mechanical factors
0028-3940/81/0021/0009/$03.00
Tab
el 1
. T
he m
ucop
olys
acch
arid
oses
McK
usic
k's
Epo
nym
ous
or o
ther
E
nzy
me
defi
cien
cy
Exc
rete
d In
heri
tanc
e O
rgan
s m
ainl
y af
fect
ed
Cla
ssif
icat
ion a
na
me
glyc
osam
inog
ogly
cans
I H
b
Hur
ler
a-L
-Idu
roni
dase
D
erm
atan
sul
phat
e A
utos
omal
rec
essi
ve
Cen
tral
ner
vous
sys
tem
. S
kele
ton.
H
epar
an s
ulph
ate
Vis
cera
I S
Sch
eie
a-L
-Idu
roni
dase
D
erm
atan
sul
phat
e A
utos
omal
rec
essi
ve
Ske
leto
n (m
ild
rela
tive
to
Hur
ler)
H
epar
an s
ulph
ate
Vis
cera
(m
ild
rela
tive
to
Hur
ler)
I H
/S
Hur
ler/
Sch
eie
a-L
-Idu
roni
dase
D
erm
atan
sul
phat
e A
utos
omal
rec
essi
ve
Phe
noty
pe i
nter
med
iate
bet
wee
n H
epar
an s
ulph
ate
Hur
ler
and
Sch
eie
dise
ases
IIb
Hu
nte
r Id
uron
ate
sulp
hate
sul
phat
ase
Der
mat
an s
ulph
ate
Sex
lin
ked
rece
ssiv
e C
entr
al n
ervo
us s
yste
m.
Ske
leto
n.
Hep
aran
sul
phat
e V
isce
ra
III
A
San
fili
ppo
A
Hep
aran
N-s
ulph
atas
e H
epar
an s
ulph
ate
Aut
osom
al r
eces
sive
C
entr
al n
ervo
us s
yste
m
III
B
San
fili
ppo
B
N-a
cety
l-a-
o-gl
ucos
amin
idas
e H
epar
an s
ulph
ate
Aut
osom
al r
eces
sive
C
entr
al n
ervo
us s
yste
m
III
C
San
fili
ppo
C
a-gl
ucos
amin
e-N
-ace
tyl
tran
sfer
ase
Hep
aran
sul
phat
e A
utos
omal
rec
essi
ve
Cen
tral
ner
vous
sys
tem
III
D
San
fili
ppo
D
N-a
cety
l-gl
ucos
amin
e-6-
sulp
hate
H
epar
an s
ulph
ate
Not
est
abli
shed
C
entr
al n
ervo
us s
yste
m
sulp
hata
se a
IV A
M
orqu
io A
G
alac
tosa
min
e 6-
sulp
hate
sul
phat
ase
Ker
atan
sul
phat
e A
utos
omal
rec
essi
ve
Ske
leto
n
IV B
M
orqu
io B
fl
-gal
acto
sida
se
Ker
atan
sul
phat
e A
utos
omal
rec
essi
ve
Ske
leto
n (O
'Bri
en-A
rbis
ser)
V
c .
..
..
VI
Mar
otea
ux-L
amy
N-a
cety
lgal
acto
sam
ine
4-su
lpha
tase
D
erm
atan
sul
phat
e A
utos
omal
rec
essi
ve
Ske
leto
n (A
ryl
sulp
hata
se B
)
VII
Sl
y fi
-Glu
curo
nida
se
Der
mat
an s
ulph
ate
Aut
osom
al r
eces
sive
C
entr
al n
ervo
us s
yste
m
Hep
aran
sul
phat
e S
kele
ton
Vis
cera
VII
I D
iFer
rant
e N
-ace
tylg
luco
sam
ine-
6 su
lpha
te
Ker
atan
sul
phat
e A
utos
omal
rec
essi
ve
Ske
leto
n su
lpha
tase
H
epar
an s
ulph
ate
- C
hond
roit
in
Not
ide
ntif
ied e
C
hond
roit
in 4
-sul
phat
e N
ot e
stab
lish
ed
Ske
leto
n 4-
sulp
hatu
ria
- C
hond
roit
in
Not
ide
ntif
ied e
C
hond
roit
in 6
-sul
phat
e N
ot e
stab
lish
ed
Cen
tral
ner
vous
sys
tem
6-
sulp
hatu
ria
Ske
leto
n
a M
cKus
ick,
and
his
col
leag
ues
[38]
ori
gina
lly
prop
osed
a n
umer
ical
cla
ssif
icat
ion
of T
ypes
I-V
and
thi
s w
as s
ubse
quen
tly
exte
nded
[2,
41]
b
Bot
h fo
rmer
ly c
alle
d ga
rgoy
lism
bec
ause
of
the
char
acte
rist
ic a
nd s
imil
ar f
acie
s, w
hich
is
also
see
n in
cho
ndro
itin
6-s
ulph
atur
ia,
the
muc
olip
idos
es (
Typ
es I
, II
and
III)
and
som
e ca
ses
of
a-fu
cosi
dosi
s an
d m
anno
sido
sis
[39]
c
Ori
gina
lly
Sch
eie
dise
ase
whi
ch w
as r
ecla
ssif
ied
as m
ucop
olys
acch
arid
osis
IS
whe
n th
e en
zym
e de
fect
was
sho
wn
to b
e th
e sa
me
as t
hat
in H
urle
r di
seas
e. T
he p
ossi
bili
ty th
at H
urle
r an
d S
chei
e di
seas
es a
re d
ue t
o al
leli
c m
utat
ions
and
tha
t th
e in
term
edia
te H
urle
r/S
chei
e ph
enot
ype
(muc
opol
ysac
char
idos
is I
H/S
) is
due
to
doub
le h
eter
ozyg
osit
y fo
r th
e al
leli
c m
utat
ions
co
ncer
ned
has
been
wid
ely
prop
osed
. T
he H
urle
r/S
chei
e ph
enot
ype
coul
d al
so b
e du
e to
hom
ozyg
osit
y fo
r a
thir
d al
leli
c m
utat
ion
at t
he s
ame
gene
loc
us
d T
his
vari
ant
was
des
crib
ed b
y K
ress
e an
d hi
s co
llea
gues
[42
]. T
heir
enz
yme
appe
ars
to a
ct o
n bo
th h
epar
an a
nd k
erat
an s
ulph
ates
und
er d
iffe
rent
con
diti
ons.
Def
icie
nt a
ctiv
ity
tow
ards
he
para
n su
lpha
te o
nly
caus
es S
anfi
lipp
o D
dis
ease
, im
pair
ed a
bili
ty t
o de
grad
e bo
th s
ubst
rate
s ca
uses
DiF
erra
nte
dise
ase.
e
The
se m
ay b
e he
tere
ogen
eous
gro
ups
and
som
e ha
ve b
een
show
n to
be
asso
ciat
ed w
ith
a-L
-Idu
roni
dase
def
icie
ncy
and
ther
efor
e va
rian
t T
ypes
IH
and
IS
[2]

11
associated with the anatomical location of mucopoly- saccharide deposits, and the tissue reactions to them, contribute to the non-genetic component. The men- tal handicap, which is a prominent feature of mucopolysaccharidoses I, II, III and VII, is primarily due to direct neuronal damage by the intralysosomal mucopolysaccharides, but the secondary anatomical abnormalities contribute to this and are mainly responsible for the other neurological manifestations.
Skeletal involvement dominates the clinical pic- ture in mucopolysaccharidoses IV and VI, and there is associated neurological damage due to vertebral subluxation especially at the atlantoaxial joint. A wider range of neurological complications also occurs in mucopolysaccharidosis VI; these include hy- drocephalus, arachnoid cysts, pachymeningitis and nerve entrapment.
Recognition of genetic variation within the mucopolysaccharidoses, with the existence of mild variants, emphasises the importance of measures to ameliorate the effects of the non-genetic contribu- tions to the clinical phenotypes. Until recently, further elucidation of such potentially treatable com- plications as arachnoid cysts and hydrocephalus required pneumoencephalography, and this was potentially hazardous because of the cardiorespira- tory complications associated with systemic mucopolysaccharide deposition. CT gives similar anatomical information and also allows examination of the cerebral substance.
This paper reports our experience with cranial CT in the assessment of a series of patients with mucopolysaccharidoses who were referred to the Clinical Research Centre for assessment because of the clinical trial of repeated fibroblast transplants as a means of enzyme replacement therapy, which is in progress there [6, 7]. Biochemical observations after single transplantations in mucopolysaccharidosis II were reported by others [8]. The clinical assessment of changes in the neurological manifestations during treatment is rendered more difficuk because of their complex aetiology. Direct examination of the central nervous system by CT is therefore essential in order to appraise the progress of the patient and to explain any changes which occur.
Methods
Twelve patients were examined by CT using the EMI 1010 scanner or a 5005 general purpose scanner. Nine of the patients needed general anaesthesia because of inability to cooperate, and one was man- aged with sedation alone. Plain CT and reexamina- tion after intravenous iodide enhancement was per-
formed on all patients. Seven were also examined with xenon enhancement. There were no adverse effects from any of the scanning procedures. Skull radiographs were available for review.
Biochemical Diagnosis
The diagnoses were established biochemically by the following criteria: (i) demonstration of the specific enzyme deficiency (leukocyte and fibroblast a- iduronidase [9] and iduronate sulphate sulphatase [8] in mucopolysaccharidoses IH, IS and IH/S and in mucopolysaccharidosis II respectively, and serum N- acetyl-a-D-glucosaminidase [10] in mucopolysac- charidosis IIIB); (ii) cross correction studies in which it was shown that the abnormal pattern of 35504 uptake and release which is typical of fibroblasts from patients with mucopolysaccharidoses IH, IS or IH/S was corrected by growing the cells in tissue cul- ture medium in which mucopolysaccharidosis II (iduronate Sulphate sulphatase deficient) cells had been grown and vice versa; (iii) demonstration of an abnormal excretion of heparan and dermatan sul- phates in mucopolysaccharidoses IH, IS, IH/S, II and of an abnormal excretion of heparan sulphate only in mucopolysaccharidosis IIIB. The urines were initially examined by precipitation of the total mucopolysac- charides with cetylpyridinium chloride [11]. Heparan and dermatan sulphates were later differentiated by thin layer chromatography [12].
Patients
Patient 1 (mucopolysaccharidosis IH) was referred to the Clinical Research Centre at age 5 years 6 months. Although her comprehension was appropriate to her age, her expressive abilities were several years behind her chronological age and her level of visuo- spatial integration corresponded to that of a 3-year- old. She was 86 cm tall (< 3rd centile), weighed 14.2 kg (< 3rd centile) and had the typical physiog- nomy of the Hurler phenotype (coarse features, depressed widened nasal bridge, excessive hair growth over forehead). Her head was scaphocephalic and large relative to her height with a skull circum- ference of 51 cm (50th centile) [13]. The other physi- cal components of the Hurler phenotype (corneal clouding, deafness, dysostosis multiplex, hepato- splenomegaly, thickened skin, hypertrichosis, stiff mitral valve on echocardiography and umbilical her- nia) were all present to a marked degree. She became blind due to a combination of glaucoma and an acute vascular occlusion affecting the retina and/or optic

12
Fig. 1 a-g. Patient 1. Mucopolysaccharidosis I/H. a and b CT sec- tions at age 6 years. There is moderate dilatation of lateral ventri- cles (Evans' index 0.36) and third ventricle. Interhemispheric fis- sure is slightly dilated. There is ill-defined low density in deep frontal and minimally in deep parietal white matter, e---e at age 7 years 10 months. The hydrocephalus has increased (Evans' index 0.48). Dilated third ventricle extends into sella turcica; fourth ven- tricle is dilated. More extensive low density in frontal and parietal white matter, f Lateral skull at same age. There is scaphocephaly associated with early fusion of sagittal suture; lambdoid suture is also fused. Vault is thin due to hydrocephalus and sella is large with wide outlet consistent with pressure from dilated third ventri- cle. Cervical vertebrae show changes of dysostosis multiplex and atlas is closely applied to foramen magnum, g CT sections at age 8 years, 18 days after ventriculoatrial shunt. Tip of the shunt tube is in right lateral ventricle and hydrocephalus has been relieved. Abnormal low density of frontal and parietal white matter persists. Interhemispheric fissure is wider and there is small low attenuation subdural effusion on left.
nerve when she was 6 years 5 mon ths old [14]. She made a good ad ju s tmen t at a school for b l ind chil- dren. Al lowing for her mul t ip le disabilit ies her m e n - tal deve lopmen t has paral le led bu t r ema ined abou t 2 years beh ind her chronological age unt i l she was last tes ted at age 9 years 0 months . She developed bi la tera l carpal t unne l m e d i a n nerve compress ion which was successfully decompressed surgically.
The CT scan t aken when she w a s . 6 years 0 mon ths old was repea ted at age 7 years t 0 mon ths and showed a m a r k e d increase in the degree of hy- drocephalus (Fig. 1). She began to compla in of headaches at abou t the t ime of t h e second examina- t ion and the skull X-ray (Fig. 1) indica ted raised in t racrania l pressure, bu t there had b e e n no change in her level of consciousness, vomit ing, fits or focal

13
Fig. 2a-c. Patient 2. Mucopolysaccharidosis, I/H. a and b are CT sections at age 23 months. There is enlargement of lateral ventri- cles, interhemispheric fissure and cortical sulci, and small areas of abnormal low density in white matter adjacent to lateral margin of occipital horus, more evident on left side. c lateral skull. Vault is large with thin bones and prominent lambdoid suture. Arias is closely applied to skull base. Appearances on CT and skull X-rays suggest communicating hydrocephalus
n e u r o l o g i c a l s igns, w h i c h m i g h t h a v e s u g g e s t e d t h e d e v e l o p m e n t o f h y d r o c e p h a l u s o n p u r e l y c l in ica l
s tudy . A v e n t r i c u l o a t r i a l s h u n t was i n s e r t e d a n d a
t h i rd C T scan 18 days p o s t o p e r a t i v e l y s h o w e d tha t t h e h y d r o c e p h a l u s h a d b e e n r e l i e v e d (Fig. 1). T h i s
a n a t o m i c a l i m p r o v e m e n t was m a i n t a i n e d at t h e t i m e of t h e las t e x a m i n a t i o n 14 m o n t h s l a t e r ( age 9 y e a r s
3 m o n t h s ) .
Fig. 3a--e. Patient 3, sister of Patient 4. Mucopolysaccharidosis I/H. a and b CT sections at 5 years 2 months. There is dilatation of Sylvian and interhemispheric fissures and of cortical sulci, and abnormal low density in deep white matter, most prominent in frontal lobes, c and d Similar sections at 6 years 4 months. Hy- drocephalus is more marked (Evans' index 0.48) and white matter low density has increased in extent, e lateral skull X-ray at 5 years 11 months: sagittal and lambdoid sutures are fused, coronal suture widened and sellar outlet open

14
Patient 2 (mucopolysaccharidosis IH) was referred to the Clinical Research Centre at age 13 months when she was judged to be functioning at a level corre- sponding to not more than 2 months behind her chronological age, but the physical stigmata of Hurler disease were already detectable. At age 23 months she was 75.8 cm long (< 3rd centile) weighed 10.8 kg (10-25th centile) and her skull circumference was 53 cm (> 97th centile). She died from cerebral ve- nous thrombosis complicating an acute dysenteric ill- ness when she was 2 years 8 months old. The post- mortem findings confirmed the diagnosis of Hurler disease.
The CT scan and skull X-ray (Fig. 2) were made when she was 2 years 4 months old and functioning at the level of a normal 20-month-old child except that her communication and social interactions were at a more retarded level.
Patient 3 (mucopolysaccharidosis IH, sib of Pa- tient 4) was referred to the Clinical Research Centre when she was 4 years 11 months old. Her lying length was 85.9 cm (< 3rd centile), weight 16.0 kg (10-25th centile) skull circumference 56 cm (> 90th centile) and she showed all the classical stigmata of mucopolysaccharidosis IH.
Her overall functional level was assessed as cor- responding to that of a normal 18-24-month-old child except that her communicative and social interactive skills corresponded to a lower age than this and could not be assessed. She had begun to retrogress before she was referred and this has con- tinued until, by the age of 7 years 2 months, she was mute, apathetic and lacking in all purposive activity.
The two CT scans (Fig. 3) were made when she was 5 years 2 months and 6 years 4 months old. Hydrocephalic changes and white matter low density had increased during the intervening period. A ven- triculoatrial shunt was not recommended in this case because of her low level of attainment initially and the severe degree of retrogression that had occurred.
Patient 4 (mucopolysaccharidosis IH, sib of Pa- tient 3) was referred to the Clinical Research Centre when she was 2 years 2 months old. At age 2 years 5 months her lying length was 78.9 cm (3rd centile), her weight was 12.3 kg (25th centile) and her head circumference was 51.5cm (> 97th centile). She showed all of the classical features of mucopolysac- charidosis IH although to a lesser degree than her sister. Her overall level of function was assessed as being at about the 2-year-old level except for com- munication and social interactions which were assess- ed at being no more than appropriate to a normal 1- year-old. She made some developmental progress so
that at chronological age 4 years 10 months her over- all level of function was at about the 21/2-3 year level with verbal comprehension and expressive language scores at the 2-year level having been static for 6 months. CT and skull X-rays (Fig. 4) were made when she was 2 years 6 months, 3 years 10 months, 4 years 1 month, and 5 years old. The last two of these being after the insertion of a ventriculoatrial shunt because hydrocephalus had developed between the first and second scans.
Patient 5 (mucopolysaccharidosis IH) was referred to the Clinical Research Centre when she was 9 months old and her overall functional level was assessed as 7 months. At that time the degree of apparent retarda- tion could have been contributed to by a somewhat deprived social background. She was 67 cm long (10th centile) and weighed 7.05 kg (<10th centile). Her physiognomy suggested, but was not diagnostic of, Hurler disease and her head circumference was 45 cm (50th centile). She had hepatosplenomegaly (liver edge 3 cm below costal margin, spleen tip just palpable), and skeletal changes of dysostosis multi- plex were demonstrable radiologically. CT and skull X-rays (Fig. 5) were made when she was 11, 16 and 25 months old. The circumference of her head had increased to 49 cm (75th centile) by age 28 months. Her clinical condition and developmental level did not change appreciably between ages 9 and 11 months, but by 16 months the clinical manifesta- tions of the Hurler disease phenotype were obvious and she had made little progress developmentally. Her most recent assessments, when she was 2 years old, showed that she was then functioning overall at a 9 month to 1 year level with communication skills at only the 3-4-month level.
Patient 6 (mucopolysaccharidosis II) was first seen for a genetic consultation at age i year 8 months well before the clinical trial was being planned and when the physical findings were compatible with a diag- nosis of Hunter disease [height 82.5 cm (10-25th centile); weight 13.9 kg (75-90th centile); head cir- cumference 52.3 cm (> 97th centile)]; but he was' not thought to be grossly retarded. His mother had an affected child by her previous marriage. The patient was seen again at age 4 years 8 months when the clinical and radiological features of Hunter dis- ease were more marked than on the previous occa- sion, growth had slowed [height 102.6 cm (10-25th centile), weight 21.7kg (90-97th centile)] and macrocrania was increasing with a head circumfer- ence of 55 cm (> 97th centile). His overall function was assessed at the 2 year level with verbal corn-

15
Fig. 4a--d. Patient 4, sister of Patient 3. Mucopolysaccharidosis I /H. CT scan at 2 years 6 months was normal. a lateral skull X-ray at 3 years 3 months shows vault enlarged and widening of coronal and lambdoid sutures, b CT at 3 years 10 months shows dilatation of lateral ventricles; cerebral substance appears normal. c and d CT sections at age 5 years, 1 year after insertion of ventricnlo- atrial shunt into right lateral ventri- cle. Hydrocephalus has been reliev- ed. There is slight widening of interhemispheric fissure. The density of parietal white mat ter is prominent and probably abnormal
prehension and expressive language scores at the 2 year 2 month, and 2 year 3 month levels. CT scans (Fig. 6) were made when he was 4 years 10 months and 5 years 11 months old, the overall functional level remaining unchanged at about 2 years. Com- prehension and expressive language were also asses- sed as being at about the 2-year level when he was 5 years 11 months old.
Patient 7 (mucopolysaccharidosis II) was referred to the Clinical Research Centre when he was 3 years 6 months old. He had the classical physical and radiological features of Hunter disease, was 101.5 cm tall (75th centile), weighed 20.1 kg (75-90th centile) and had a head circumference of 52 cm (97th cen- tile). His mother's brother had died when he was 24 years old after a prolonged period of progressively increasing mental handicap and with physical fea- tures compatible with a diagnosis of Hunter disease. CT scans (Fig. 7) were made at ages 3 years
7 months, and 4 years 7 months. At the time of the first scan the patient's overall function was assessed at "only a little below his chronological age but with marked deficiency in the area of communication". Although his mother reported developmental pro- gress one year later when the second scan was made, there was no objective evidence of this, his overall functional level being assessed at the 21/2-3 year old level with expressive speech at the 13-month-old level. The physical findings were essentially unchanged except for some overall growth [height 105.4 cm (50th centile), weight 23.3 kg (> 97th cen- tile)] and an increase in head circumference to 53 cm (> 97th centile).
Patient 8 (mucopolysaccharidosis IH/S). This girl was referred to the Clinical Research Centre at age 5 years 3 months because of stiffness of all the joints of the upper and lower limbs. She was 93.4 cm tall (< 3rd centile), weighed 15.4 kg (10th centile) and

16
Fig. 5a-f. Patient 5. Mucopolysaccharidosis I/H. a and b CT sec- tions at age 16 months show slight widening of left trigone, anterior part of interhemispheric fissure prominent, and cerebral substance normal. Skull X-ray e AP and d lateral projections at 2 years of age show fusion of lambdoid and sagittal sutures and scaphocephaly. The coronal suture is markedly widened and sellar outlet is wide. Appearances indicate raised intracranial pressure and suggest that hydrocephalus has developed, e and f CT sections at levels similar to a and b at age 2 years 1 month. There has been minor increase in size of lateral ventricles and slightly diminished attenuation in deep parietal white matter
he r h e a d c i r cumfe rence was 53 cm ( > 97 th cent i le) . H e r in te l l ec tua l a t t a i nmen t s were a p p r o p r i a t e for he r age, as were he r func t iona l abi l i t ies excep t whe re these were l imi ted by the a r th ropa thy . T h e type of jo in t de fo rmi ty r e s e m b l e d tha t seen in H u r l e r dis- ease. She had a mi ld d e g r e e of m ic rogna th i a and her p h y s i o g n o m y did no t suggest a d iagnos is of H u r l e r d isease , a l though she had o t h e r typica l f ea tu res of the H u r l e r d i sease p h e n o t y p e . This c o m b i n a t i o n of well p r e s e r v e d in te l lec t wi th m a r k e d jo in t and viscera l
i n v o l v e m e n t at an ear ly age is c o m p a t i b l e with a d iag- nosis of H u r l e r / S c h e i e d i sease (mucopo lysac - char idos is I H / S ) . The a p p a r e n t b iochemica l a b n o r - mal i t ies a re the same in all t h r e e of the inhe r i t ed a - i du ron idase def ic iency s y n d r o m e s (Tab le 1) and were p re sen t in this pa t ien t . C T scans (Fig. 8) were m a d e at age 5 yea r s 2 months , 6 years 2 months , and 6 years 6 months . T h e pa t i en t ' s men ta l d e v e l o p m e n t had con t inued n o r m a l l y b e t w e e n the e xa mina t i ons and she had grown [height 102 cm ( < 3rd cent i le ) ,

17
Fig. 6 a and b. Patient 6. Mucopolysaccharidosis II. a CT at age 4 years 10 months. There is dilatation of lateral ventricles (Evans' index 0.33), widening of interhemispheric fissure and, to a minor degree, of cortical sulci, b CT scan at 5 years 11 months. Hy- drocephalus has increased (Evans' index 0.40) and interhemis- pheric fissure and cortical sulci have widened further
Fig. 8a and b. Patient 8. Mucopolysaccharidosis I H/S. a and b CT at age 6 years 2 months. There is abnormal low density in parietal white matter; ventricular system and intracranial subarachnoid spaces are normal
Fig. 7a and b. Patient 7. Mucopolysaccharidosis II. a Normal CT scan at 3 years 7 months of age. b Similar section at age 4 years 7 months. Density of deep parietal white matter is abnormally low
Fig, 9a and b. Patient 9. Mucopolysaccharidosis I H/S. a and b CT at age 3 years 9 months. There is abnormal low density in parietal and deep frontal white matter; ventricles are normal but Sylvian fissures are slightly prominent
weight 16 kg ( < 3 rd cen t i l e ) ] bu t t he re had been no fu r the r e n l a r g e m e n t of the head . H e r ma in dis- abi l i t ies were still a s soc ia t ed with stiffness of the jo in t s and deafness ; the l a t t e r had i m p r o v e d af te r a m y r i n g o t o m y and inse r t ion of g rommets . She had recen t ly c o m p l a i n e d of pa in r ad ia t ing f rom the cervi- cal sp ine to the shou lde r r eg ion on bo th s ides and a l though t h e r e we re no ob jec t ive a b n o r m a l sensory signs and the t e n d o n ref lexes in the a rms were no t pa tho log i ca l l y br isk , she had d e v e l o p e d a f inger j e r k and the k n e e and ank le j e rks were e x a g g e r a t e d a l though the p l an t a r r e sponses were f lexor . T h e s e changes suggest the beg inn ing of cervical co rd com- p re s s ion assoc ia ted with e x t r a d u r a l and du ra l m u c o p o l y s a c c h a r i d e depos i t s as occu r r ed in Pa- t ien t 10.
P a t i e n t 9 (mucopo lysaccha r idos i s / H / S ) . This b o y was r e f e r r ed at age 3 years 9 months . T h e ma in com- p la in ts were stiffness and de fo rmi ty of his f ingers and d e v e l o p m e n t a l delay. H e had heavy bu t no t typica l ly H u r l e r - l i k e facial fea tures . The o t h e r p h e n o t y p i c man i fe s t a t ions were s imi lar to those e n c o u n t e r e d in H u r l e r d i sease bu t to a r e la t ive ly mi ld degree . His he igh t was 9 2 . 2 c m (3rd cent i le) weight 1 5 . 2 k g ( 2 5 t h - 5 0 t h cent i le) and h e a d c i r cumfe rence 54.5 cm (97th cent i le) . T h e b iochemica l f indings were those which are c o m m o n to all of the a - i d u r o n i d a s e def i - c iency mucopo lysaccha r idose s . CT scan was o b t a i n e d at age 3 years 9 months , when his overa l l func t iona l level was assessed at the 2 yea r 6 m o n t h o ld level , with c o m p r e h e n s i v e and express ive l anguage bo th at the 2 yea r 1 m o n t h o ld level , This pa t i en t had also

18
Fig. 10a and b. Patient 10. Mucopolysaccharidosis IS. a and b CT at age 20 years. There is slight dilatation of lateral ventricles; third and fourth ventricles are normal. There is marked widening of subarachnoid space anterior to left temporal lobe suggesting small arachnoid cyst and less marked widening in similar position on right side. Suprasellar cisterns apparently extend into upper part of sella turcica, which was abnormally deep on plain X-rays
Fig. l l a and b° Patient 10. Myelogram a lateral, b oblique projections. Metal tracheostomy tube obscured contrast medium in AP position. Cervical subarachnoid space is narrowed and there is "increase in distance between it and adjacent bony canal due to thickening of meninges. Mid and upper cervical nerve root sheaths are occluded
had congenital posterior urethral valves which were treated by diathermy when he was 3 years old. Although the obstruction to the outflow of urine had been relieved he was left with bilateral hydro- nephrosis.
Patient 10 (mucopolysaccharidosis Type IS). This man (age 20 years 10 months) was referred as a case of Scheie disease; the diagnosis had been made on clinical and enzymological grounds elsewhere and we confirmed the latter. This patient presented particu- lar problems of clinical classification which are dis- cussed later. H e complained of stiffness in all his joints and gross difficulty in walking. He had also had attacks of severe sleep apnoea necessitating a tracheostomy. The presenting symptoms had been stiffness in the hands and an abnormal gait when he was about 4 years old and his disability had increased so that when he was referred to us he had bilateral claw hands and could walk only about 20 yards. H e was 143 cm tail, weighed 35.5 kg with a skull circum- ference of 57.5 cm (97th centile) and skeletal abnor- malities which were not entirely characteristic of dysostosis multiplex. H e had heavy but not specifi- cally Hurler-l ike features, corneal clouding, micro- gnathia, cardiac systolic and diastolic murmurs (the echoeardiogram suggested mild aortic stenosis) and hepatosplenomegaly. The results of pulmonary func-
tion tests indicated a severe restrictive defect. The physical signs of spinal cord compression at the C5-C 6 level, and of bilateral median nerve compression in the carpal tunnel, were present. His intellect was nor- mal. The CTs and myelograms are shown in Fig- ures 10 and 11. The patient had an older brother with the disease who had died following an aortic and mit- ral valve replacement operation.
Patient 11 (mucopolysaccharidosis I I IB) was referred at age 10 days. The diagnosis had been made in utero
at the 16th week of pregnancy, and the mother had subsequently changed her mind about having the pregnancy terminated. Clinically the patient was a normal baby, but the absence of N-acetyl -a-D- glucosaminidase confirmed the prenatal diagnosis. He r length, weight and head circumferance were: 51cm, 3 .22kg, and 3 6 c m (all 50-90th centile) respectively. CT scans (Fig. 12) were made at age 6 weeks, and 6 months. She was clinically normal when the second scan was made except for a large head with prominent frontal bossing and open anterior fontanelle (circumference 46.8 cm; > 95th centile) and a palpable liver edge 2 cm below the right costal margin.
Patient 12 (mucopolysaccharidosis I I IB, brother of Patient 11) had been a normal full term baby but developmental delay had been apparent to the

19
Fig. 12a and b. Patient 11. sister of Patient 12. Mucopolysac- charidosis IIIB. a, b CT at 6 months of age, following intravenous contrast medium. All deep hemispheric white matter is prominent but is only below normal range of variation in parietal lobes. Ven- tricular system and intracranial subarachnoid spaces are normal
Fig. 13a and b. Patient 12, brother of Patient 11. Mucopolysac- charidosis IIIB. a and b CT at age 7 years 2 months. Ventricular system and intracranial subarachnoid spaces are enlarged
Fig. 14a and b. Mucopolysaccharidosis VI. a and b CT shows abnormal low density throughout frontal and parietal white matter. Ventricular system is dilated due to communicating hydrocephalus. Right lateral ventricle is larger than left, especially right frontal horn. There is more extensive low density anteriorly in right frontal lobe; right side of frontal bone is expanded. These additional findings are due to surgery performed for a suprasellar arachnoid cyst which has recurred and is enlarging sella
Fig. 15. Mucopolysaccharidosis IIIB. There is well-defined low attenuation in left lentiform nucleus consistent with brain destruction following previous infarct. Left Sylvian fissure is relatively wide due to some adjacent atrophy
parents by age 2 years and retrogression from about 41/2 years. When seen by us at age 7 years 2 months just before the CT scan (Fig. 13), he was a very hyperkinetic, whining child with aggressive tenden- cies and short attention span, who showed virtually no interaction with other children or the staff although he recognised his mother. He was 112 cm tall (3rd-10th centile), weighed 23.4 kg (50th cen- tile), and his skull circumference was 57 cm (> 97th centile). The main physical findings were coarse but not specifically Hurler-l ike facial features, moderate generalised hypertrichosis, restricted abduction at the shoulder joints, limited supination of the fore- arms, hepatomegaly (liver edge palpable 3.5 cm
below the right costal margin), a systolic murmur at the cardiac apex and brisk deep tendon reflexes with flexor plantar responses.
Results
The results of the CT examinations and skull radio- graphs are summarised in Table 2 and in Fig- ures 1-10, 12 and 13. Figure 11 shows the cervical myelogram of Patient 10.
Figures 14 and 15 are CT scans of two patients who do not belong to the main series. These scans show certain additional features which have also been encountered in the mucopolysaccharidoses.

2O
Table 2. Results of computed tomography and skull X-rays [Zero (0) = negative finding. Dash (-) -- no observation made.]
Case Type Age Computed Tomogram Skull y m Hydrocephalus White matter Raised pressure Other Changes
(Evans' Low density Large Wide Cranio- index a) (40) skull suture b stenosis b Sella
1 IH
2 IH
3 IH
4 IH
5 IH
6 II
7 II
8 IH/S
9 IH/S
10 dis
11 IIIB
12 IIIB
6 0 + +(0.36) + +F&P c + 0 +(S) + 7 10 + + + ( 0 . 4 8 ) + + + + 0 +(S) + + 8 0 0(after shunt) + + . . . .
2 4 +(0.3) +P + + (L/C) 0 0
5 2 + +(0.36) + F&P + 0 +(S/L) + 6 4 + + + (0.48) + + F&P + + (C) + (S/L) +
2 6 0 0 . . . . 3 3 - - + + 0 + 3 10 +(0.33) 0 + + + 0 0 4 1 0(after shunt) 0 . . . . 5 0 0 +P + 0 0 0
0 11 0 0 . . . . 1 4 0 0 . . . . 2 0 - - + + + (C) + (S/L) + 2 1 +(0.30) + P . . . .
4 10 +(0.33) 0 . . . . 5 11 + + + ( 0 . 4 0 ) 0 0 0 +(S/L/C) 0
3 7 0 ? P 0 0 + (S) 0 4 7 0 + P . . . .
5 2 0 + P . . . . 6 2 0 + + P . . . . 6 6 0 + + P 0 0 +(S) +
3 9 0 + P&F 0 0 0 0
20 10 +(0.30) 0 0 0 +(S/L/C) + Empty
0 1'/2 0 + P . . . . 0 6 0 + P 0 0 + (S/L/C) +
7 2 + + (0.37) 0 0 0 0 0
a Maximum width of frontal horns/maximum width between inner tables b C=coronal; L=lambdoid; S=sagittal c F=frontal, P=parietal d See text for further discussion of the classification of this patient as either Type IS or IH/S. The present investigations showed a left temporal arachnoid cyst and an empty sella. Myelography showed thick spinal dura
D i s c u s s i o n
T h e i n t e r p l a y of t h e e f fec t s of n e u r o n a l d a m a g e by
l y s o s o m e s d i s t e n d e d w i t h m u c o p o l y s a c c h a r i d e s a n d
g lyco l ip ids , c e r e b r a l a t r o p h y a n d o b s t r u c t i o n to t h e
C S F c i r c u l a t i o n d e t e r m i n e s t h e c e r e b r a l m a n i f e s t a - t i ons o f t h e m u c o p o l y s a c c h a r i d o s e s .
T h o s e d i seases in wh ich t h e r e is a l ack of a - i d u r o n i d a s e ac t iv i ty ( m u c o p o l y s a c c h a r i d o s e s I H , IS
a n d I H / S ) can p r e s e n t m o r e d i f f icu l t p r o b l e m s in c l in ica l d i agnos i s t h a n t h e u sua l d e s c r i p t i o n s sugges t .
In t he i r fu l ly d e v e l o p e d f o r m s , H u r l e r [15] and
S c h e i e [16] d i seases a r e v e r y d i f f e r e n t f r o m o n e a n o t h e r . T h e y b r e e d t r u e w i th in f ami l i e s a n d a re p r e - s u m a b l y d u e to a l le l ic m u t a t i o n s . T h e s ta tus of
H u r l e r / S c h e i e d i s ea se is less c lear . I t has b e e n w i d e l y
p r o p o s e d as b e i n g d u e to d o u b l e h e t e r o z y g o s i t y fo r t h e m u t a n t g e n e s w h i c h c a u s e H u r l e r a n d S c h e i e dis-
eases , a l t h o u g h it c o u l d r e p r e s e n t h o m o z y g o s i t y fo r a t h i rd a l le l ic m u t a t i o n at t h e a - i d u r o n i d a s e g e n e locus
[17, 18]. T h e r e is, h o w e v e r , c o n s i d e r a b l e c l in ica l
h e t e r o g e n e i t y w i th in t h e c l in ica l spec t r a of all t h r e e
d i seases a n d t h e e x t e n t to w h i c h this is g e n e t i c a n d n o n - g e n e t i c is u n k n o w n .
C e r v i c a l p a c h y m e n i n g i t i s a n d m e d i a n n e r v e c o m -
p r e s s i o n in t h e c a r p a l t u n n e l w e r e m a j o r f ac to r s in
d e t e r m i n i n g t h e p h e n o t y p e in P a t i e n t 10, w h o was
o r ig ina l l y c lass i f ied as h a v i n g S c h e i e d i s ea se o n t h e basis of n o r m a l i n t e l l ec t a n d a b s e n t a - i d u r o n i d a s e ac t iv i ty . H o w e v e r , h e h a d m a j o r s y s t e m i c a n d v is -

21
ceral abnormalities attributable to his metabolic lesion and it could be argued that he would be more accurately classified as having Hurler/Scheie disease. The physical signs suggest that cervical cord com- pression may also be beginning in Patient 8, who is classified as having Hurler/Scheie disease on the grounds of the severe systemic involvement, although unlike Patient 9, another case of Hurler/Scheie dis- ease, she is not mentally handicapped. Consideration of these three cases and those reported in the litera- ture [19-25] shows that clinically Hurler/Scheie and Scheie diseases should be regarded as forming a con- tinuum, and unless biochemical differences can be found by more refined techniques than have been used heretofore, there seems little justification for separating them completely.
Spinal cord compression in the mucopolysac- charidoses can be due to several mechanisms. It has been caused by atlantoaxial subluxation in Morquio disease [26], and in Maroteaux-Lamy disease [27]. Such subluxation is generally associated with laxity of the transverse odontoid ligament but in four of the cases of Blaw and Langer [26] there was also either hypoplasia or absence of the odontoid process. Gib- bus formation, which is most marked in Morquio dis- ease, may cause thoracic cord compression [26]. Dural thickening by abnormal deposition of collagen, causing changes identical to those in Patient 10, have been described in Maroteaux-Lamy and Hurler/ Scheie diseases [28, 29]. The importance of recognis- ing these surgically correctable causes of cord com- pression is apparent, but marked constriction of the cervical theca may also influence the development of hydrocephalus in the mucopolysaccharidoses. The thecal changes may be visible on CT, especially using 4th generation scanners, and if such patients are sub- mitted to head scanning, inclusion of a section through the cervical cord is suggested.
Mucopolysaccharides and glycolipids must have been present within the nerve cells of our cases, but on CT the density of grey matter was similar to that of normal brain. Inhaled xenon crosses the normal blood-brain barrier and is lipid soluble; because of the energy of its K-shell electron, it causes consider- able enhancement of the images of the brain sub- stance [30], but there is no detectable difference be- tween the enhancement of normal brain and of the abnormal tissue in the mucopolysaccharidoses; also, the blood-brain barrier behaves normally to intravenous contrast medium.
Symmetrical low attenuation in the white matter was seen in nine of our cases; such low density gener- ally indicates increased water content. Three of these patients, all with mucopolysaccharidoses IH, had marked communicating hydrocephalus and in two of
them the low attenuation, in being more marked around the dilated frontal horns, simulated that due to hydrocephalus [31]. It was, however, unusually extensive for this alone and in one of these cases in which a successful ventricular drainage system was inserted, it resolved only partly (Patient 1), and in another (Patient 4) it first developed in the presence of a functioning shunt (Table 2). It occurred in all types of mucopolysaccharidosis in this series except in Patient 10 and we have seen it in mucopolysac- charidosis VI also (Fig. 14). The low density, though often more pronounced adjacent to the ventricles, extended well laterally. The parietal white matter was involved in all cases, and the frontal region also in two. The latter was affected earlier in one of these. It would correlate well with the pitting and cavitation around blood vessels which is visible at autopsy on the cut surface of the white matter in Hurler disease [32] due to concentrations of foam cells within some of the Virchow-Robin spaces [25]. Dekaban and Constantopoulos [33], who differentiate clearly be- tween the parenchymal and leptomeningeal lesions in Hurler disease, also describe greatly dilated periad- ventitial spaces filled with viscous fluid, "gargoyl cells" and mesenchymal elements. This fluid would explain the similarity between the low attenuation of the white matter seen in some of our cases and cere- bral oedema. Patient 10 did not show changes i'n the white matter and this would correspond to the repor- tedly less severe leptomeningeal changes in Scheie disease than in Hurler disease [34]. These workers also report that the neuronal changes seen in Hurler disease are absent in Scheie disease. In Sanfilippo A disease also, the nerve damage by mucopolysac- charide and sphingolipid engorgement is accom- panied by dilatation of the perivascular spaces in the white matter by mucopolysaccharides [5, 35, 36].
There are several possible reasons for enlarge- ment of the head in the mucopolysaccharidoses. The bones of the vault may be thickened by mucopolysac- charide deposits. Such thickening is evident on skull radiography and on CT, but it was not a pronounced feature in any of our patients. Thickening of the vault may also occur due to deposition of bone on the inner table following relief of hydrocephalus, and this was seen in two of our cases. Remodelling of bone is abnormal in the skull as well as in other bones and may contribute to the abnormal shape of the head. Accumulation of mucopolysaccharides within the meninges and brain substance increases the volume of the intracranial contents. Thickening of the cere- bral meninges sufficient to cause enlargement of the head would be revealed by CT but it was not evident in this series. Leptomeningeal cysts are important determinants of the characteristic abnormalities in

22
the sphenoid, especially in the pituitary fossa region [37].
Distention of brain substance may enlarge the brain sufficiently to expand the skull, but growth of the head from this factor alone is slow and unlikely to separate the sutures. Should atrophy occur in such cases, an enlarged vault could be associated with widened intracranial CSF spaces. There was some widening of the Sylvian, and sometimes of the interhemispheric fissures or cortical sulci in ten of our patients. This was usually accompanied by other evi- dence of raised intracranial pressure.
The presence of moderately severe communicat- ing hydrocephalus, which was unsuspected before CT in three of the cases, and its marked progression over a period of two years in two of them, emphasises the importance of this investigation in the assessment of such patients. The slightly enlarged ventricles in two other cases with widened sutures on plain skull X- rays also strongly suggest communicating hy- drocephalus. In one case (Patient 4) aged 21/: years the scan was normal, but the cranial sutures were splayed on the plain skull films and the head was enlarged suggesting raised intracranial pressure. Her 4-year-old sister had manifest communicating hy- drocephalus on CT and the younger child subse- quently developed it (Figs. 3 and 4). Any difficulty in distinction between atrophy and communicating hy- drocephalus can usually be resolved by subsequent CT as in Patient 6. Examination of CSF dynamics using intrathecal indicators such as RHISA or metrizamide may be helpful if a more immediate decision regarding the shunt procedure is necessary.
Although hydrocephalus is a well known autopsy finding in the mucopolysaccharidoses, there have not been, to our knowledge, any studies which have indi- cated the stage in the evolution of the disease at which it begins. Our observations show this to be very variable, but it can occur early and therefore materially modify the overall prognosis. Ven- triculoatrial shunting operations are only a palliative in this group of diseases, but they are, in our opinion, justified in order to improve the prospect for the immediate quality of life. This is likely to be the case if the hydrocephalus is known to be of recent onset, if the child's overall level of function is relatively well preserved, and if the development of the hy- drocephalus has been associated with sudden deterioration or symptoms, such as severe headaches, specifically related to it.
The value of CT in the elucidation of acute cere- bral deterioration in general is well established, and it might have allowed antemortem diagnosis in the patient who died from an incidental sagittal sinus thrombosis though the clinical course would probably not have been modified by this knowledge.
There were no other abnormal findings in our series of patients, but we have seen the value of CT in showing other lesions in patients with mucopolysac- charidoses, not included in this series. Examples include a recurrent suprasellar arachnoid cyst associ- ated with communicating hydrocephalus in a patient with Maroteaux-Lamy disease (Fig. 14), and left hemiatrophy with a cavity low in the left putamen in another patient with Sanfilippo B disease and a pre- viously confusing clinical picture which included wasting of the right leg and grand mal convulsions (Fig. 15).
Acknowledgements. We are pleased to acknowledge the following who kindly referred patients to us: Dr. Barbara Ansell, (North- wick Park Hospital, Harrow) Dr. E. M. Carr-Saunders (Cumber- land Infirmary, Carlisle), Dr. P. F. Deasy (Our Lady's Hospital for Sick Children, Dublin), Dr. D. W. Fielding (Chester City Hospital, Chester), Dr. J. L, Greaves (Royal Hampshire County Hospital, Winchester), Dr. S. H. Green (Lea Castle Hospital, Wolverley), Dr. R. Lindop (Manor Hospital, Walsall), Dr. A.J. O'Donnell (The Health Centre, Steyning), Dr. K. Prowse (City General Hos- pital, Stoke-on-Trent), Dr. D. G. H. Stone (Royal Berkshire Hos- pital, Reading). We are greatly indebted to the sisters and nursing staff of Cavendish and Haldane Wards of Northwick Park Hospi- tal, and to the Occupational Therapists, Physiotherapists and Speech Therapists for their work in the management and assess- ment of these patients. We are most grateful to many colleagues who have advised us on the management of various complications which have arisen in these children in particular: Dr. B. Ansell, Mr. R. Auerbach, Mr. J. L. Kennerley Bankes, Mr. H. B. Eckstein, Mr. A. Kark, Mr. L. Klenerman, Dr. T.E. Stacey and Dr. H. B. Valman. Dr. P. Rudge generously allowed us to admit patients, who required temporary admission for CT scanning, to his beds at the National Hospital for Nervous Diseases. These investigations were approved by the Ethical Committees of North- wick Park Hospital and Clinical Research Centre and the National Hospital for Nervous Diseases.
References
1. Purpura DP, Suzuki K (1976) Distortion of neuronal geometry and formation of aberrant synapses in neuronal storage dis- ease. Brain Res 116:1-21
2. McKusick VA, Neufeld EF, Kelly TE (1978) The mucopoly- saccharide storage dieseases. In: Stanbury JB, Wyngaarden JB, Fredrickson DS (eds) The metabolic basis of inherited diseases. 4th ed. McGraw-Hill, New York
3. Dorfman A, Matalon R (1972) The mucopolysaccharidoses. In: Stanbury JB, Wyngaarden JB, Fredrickson DS (eds). The metabolic basis of inherited disease. 3rd ed. McGraw-Hill, New York
4. Doshi R, Sandry SA, Churchill AW, Brownell B (1974) The cerebellum in mucopolysaecharidosis. A histological, histo- chemical and ultra-structural study. J Neurol Neurosurg Psychiatry 37:1133-1138
5. Kriel RL, Hauser WA, Sung JH, Posalaky Z (1978) Neuroanatomical and electroencephalographic correlations in Sanfilippo Syndrome, Type A. Arch Neurol 35:838-843
6. Gibbs DA, Spellacy E, Roberts AE, Watts RWE (1977) Pro- gress report on the treatment of Hurler's disease by enzyme replacement therapy. Glycoconjugate Res 2 :885-888
7. Gibbs DA, Spellacy E, Roberts AE, Watts RWE (1980) The treatment of lysosomal storage diseases by fibroblast trans- plantation: some preliminary observations. Birth Defects 16: 457-474

8. Dean MF, Stevens RL, Muir H, Benson PF, Button LR, Anderson RL, Boylston A, Mowbray J (1979) Enzyme replacement therapy by fibroblast transplantation. J Clin Invest 63:138-145
9. Hall CW, Neufeld EF (1973) a-L-Iduronidase activity in cul- tured skin fibroblasts and amniotic fluid cells. Arch Biochem Biophys 158:817-821
10. Hall CW, Liebaers I, Di Natale P, Neufeld EF (1978) Enzy- matic diagnosis of the genetic mucopoly saccharide storage disorders. Methods Enzymol 50:439-456
11. Pennock CA (1976) A review and selection of simple labora- tory methods used for the study of glycosaminoglycan excre- tion and the diagnosis of mucopolysaccharidoses. J Clin Pathol 29:111-123
12. Humel R, Chamoles NA (1972) Sequential thin layer chromatography of urinary acid glycosaminoglycans. Clin Chim Acta 40:290-293
13. Westrupp CK, Barber CR (1956) Growth of skull in young children. I. Standards of head circumference. J Neurol Neurosurg Psychiatry 19:52-56
14. Spellacy E, Kennerley, Bankes JL, Crow J, Dourmashkin R, Shah D, Watts RWE (1980) Glaucoma in a case of Hurler disease. Br J Ophthalmol 64:773-778
15. Hurler G (1919) ~ber einen Typ multipler Abartungen, vor- wiegend am Skelettsystem. Z Kinderheilkd 24:220-234
16. Scheie HG, Hambrick GW Jr, Barness LA (1962) A newly reeognised forme fruste of Hurler's disease (gargoylism). Am J Ophthalmol 53:753-769
17. Fortuin JJH, Kleijer WJ (1980) Hybridization studies of fi- broblasts from Hurler, Scheie and Hurler/Scheie compound patients. Support for the hypothesis of allelic mutants. Hum Genet 53:155-159
18. Kaibara N, Eguchi M, Shibata K, Takagishi K (1979) Hurler- Scheie phenotype: a report of two pairs of inbred sibs. Hum Genet 53:37-41
19. Danes BS (1977) Variant of iduronidase deficient mucopoly- saccharidoses. Further evidence for genetic heterogeneity. J Med Genet 14:246--351
20. Elliot DE, Dorst JP (1974) Mucopolysaceharidoses: possible Hurler/Scheie genetic coumpound (MPS 1-H/S). Birth Defects 10:453-457
21. Kajii T, Matsuda I, Ohsawa T, Katsunum H, Ichida T, Arashima S (1974) Hurler/Scheie genetic compound (mucopolysaccharidosis IH/S) in Japanese brothers. Clin Genet 6:394-400
22. Leisti J, Rimoin DL, Kaback M, Shapiro LJ, Matalon R (1976) Allelic mutations in the mucopolysaccharidoses. Birth Defects 12:81-91
23. Thompson JN, Finley SC, Lorinez AE, Finley WH (1975) Absence of a-L-iduronidase activity in various tissues from two sibs affected with presumbly the Hurler-Scheie compound syndrome. Birth Defects 11:341-346
24. Tondeur M, Vamos-Hurwitz E, Cantz M, Cremer N, Libert J, Pardou A (1976) Clinical, ultrastructural and tissue culture studies in a possible compound Hurler-Scheie case. Aeta Paediatr Belg 29:109-115
25. Winters PR, Harrod MJ, Molenich-Heetred SA, Kirkpatrick J, Rosenberg RG (1976) a-L-iduronidase deficiency and pos- sible Hurler-Scheie genetic compound. Neurology 26: 1003-1007
23
26. Blaw ME, Langer LO (1969) Spinal cord compression in Mor- quio-Brailsford's disease. J Paediatr 74:592-700
27, McKusick VA (1972) Heritable disorders of connective tissue. 4th ed. Mosby, St. Louis, pp 524-686
28. Sostrin RD, Hasso AN, Peterson DI, Thompson JR (1977) Myelographic features of mucopolysaccharidosis: A new sign. Radiology 125:421-424
29. Kennedy P, Swash M, Dean MF (1972) Cervical cord com- pression in mucopolysaccharidoses. Dev Med Child Neurol 15:194-199
30. Kendall BE, Radii EW, Zilkha E (1978) Xenon enhancement in computed tomography. Xtraet 4 :7 -12
31. Moseley IF, Radii EW (1979) Factors influencing the develop- ment of periventricular lucencies in patients with raised intra- cranial pressure. Neuroradiology 17:65-70
32. Crome L, Stern J (1972) Pathology of mental retardation. 4thed. Churchill Livingstone, Edinburgh London, pp 320-324
33. Dekaban AS, Constantopoulos G (1977) Mucopolysacchari- doses types I, II, IIIA and IV. Pathological and biochemical abnormalities in the neural and mesenchymal elements of the brain. Acta Neuropathol (Berl) 39 :1-7
34. Dekaban AS, Constantopoulos G, Herman M, Steussing JK (1976) Mucopolysaccharidosis Type V (Scheie syndrome). A postmortem study by multidisciplinary techniques with emphasis on the brain. Arch Pathol Lab Med 100:237-245
35. Witting C, Miiller KM, Kresse H, von Figura K, Marx H (1975) Morphological and biochemical findings in a case of mucopolysaccharidosis type IIIA. (Sanfilippo disease type A). Beitr Pathol 154:324-338
36. Cain H, Egner E, Kresse H (1977) Mucopolysaccharidosis IIIA (Sanfilippo Disease Type A). Histochemical, electron microscopical and biochemical findings. Beitr Pathol 160: 58-72
37. Neuhauser EBD, Griscom NT, Gilles FH, Cricker AC (1967) Arachnoid cysts in the Hurler-Hunter syndrome. Ann Radiol 11:434-469
38. McKusick VA, Kaplan D, Wise D, Hanley WD, Suddarth SD, Sevick ME, Maumenee AW (1965) The genetic mucopolysac- charidoses. Medicine 44:445-483
39. Van Hoof F (1974) Mucopolysaccharidoses and mucolipi- doses. J Clin Pathol [Suppl] 8 :1-11
40. Evans WA (1942) An encephalographic ratio for estimating ventricular enlargement and cerebral atrophy. Arch Neurol Psychiatry 47:931-937
41. McKusick VA (1980) Genetic heterogeneity and allelic varia- tion in the mucopolysaccharidoses. Johns Hopkins Medical Journal 146:71-79
42. Kresse H Paschke G, Figura K, Gilberg W, Fuchs W (1980) Sanfilippo disease type D: Deficiency of N-acetylglucosamine -6-sulphate sulphatase required for heparan sulphate degra- dation Proc Natl Acad Sci USA 77:6822-6826
Received: 28 October 1980
Dr. R. W. E. Watts Division of Inherited Metabolic Diseases Medical Research Council Clinical Research Centre Watford Road, Harrow Middlesex, HA1 3UJ England
Note Added in Proof
"An indium-DTPA intrathecal scan was performed on Patient 6 at age 6 years 8 months. The isotope had entered the dilated lateral ventricles and basal cisterns half an hour after it had been injected into the lumbar theea, and even after 24 hours there was very little isotope over the vertex although isotope was present in the lateral ventricles. These changes show that communicating hydrocephalus was the major abnormality and presumably reflect involvement of the Pachionian granulations in the abnormal storage process.




















