
Electrical Characterization of Germanium Nanowires Using a ...
Upload
independentCategory
view
1download
0
JOURNAL OF CHEMICAL PHYSICS VOLUME 117, NUMBER 22 8 DECEMBER 2002
An L-shaped equilibrium geometry for germanium dicarbide „GeC2…?Interesting effects of zero-point vibration, scalar relativity,and core–valence correlation
Levent SariCenter for Computational Quantum Chemistry, University of Georgia, Athens, Georgia 30602-2525
Kirk A. PetersonDepartment of Chemistry, Washington State University, Richland, Washington 99352
Yukio Yamaguchi and Henry F. Schaefer IIIa)
Center for Computational Quantum Chemistry, University of Georgia, Athens, Georgia 30602-2525
~Received 11 July 2002; accepted 12 September 2002!
The ground state potential energy surface of the GeC2 molecule has been investigated at highlycorrelated coupled cluster levels of theory. Large basis sets including diffuse functions and functionsto describe core correlation effects were employed in order to predict the true equilibrium geometryfor GeC2. Like the much-studied valence isoelectronic SiC2, the linear (1(1), L-shaped (1A8), andT-shaped structures (1A1) must be investigated. The L-shaped Cs geometry is found to have realharmonic vibrational frequencies along every internal coordinate, and the linear stationary point hasan imaginary vibrational frequency along the bending mode at every level of theory employed. TheT-shaped geometry is found to have an imaginary vibrational frequency along the asymmetricstretching mode. At the coupled cluster with single and double excitations and perturbative tripleexcitations@CCSD~T!#/correlation consistent polarized valence quadrupole-z ~cc-pVQZ! level, thenonrelativistic classical relative energies of the T-shaped and linear structures with respect to theL-shaped minimum are 0.1 and 2.8 kcal/mol, respectively. Including zero-point vibrational energy,scalar relativistic, and core-valence corrections, the T-L energy separation is shifted to 0.4 kcal/moland the relative energy between the L-shaped and linear structures is still 2.8 kcal/mol. Allnonrelativistic and relativistic computations predict that the L-shaped (1A8) structure is mostfavored for the ground state. The linear structure is predicted to be a transition state, as the case ofSiC2. © 2002 American Institute of Physics.@DOI: 10.1063/1.1518966#
Sigen
ctoesrehiri
ffem
rg
nain
en-
o-t lin-
edind
ta-rnal
tate
I. INTRODUCTION
The group IV diatomics and triatomics such as CCSi2, SiC2, GeC, Ge2C, GeC2, and SnC have held a growininterest for both experimental and theoretical researchmainly due to their extraordinary electronic structures apotential usages in optoelectronic and semiconduapplications.1–6 The inconsistency in the electronic structurof these periodically related species gave rise to incorconclusions about the ground state structures, some of whad been held for many years. One reason for encountethese surprising results is that C, Si, Ge, and Sn have dient core sizes, which play a significant role in the minimuenergy geometries and in the extent of relativistic enecorrections. Also, the absence of occupiedd electrons for Cand Si causes an unbalanced competition for the valeelectrons compared to Ge or Sn. There have been many creported in the literature showing this inconsistency. Forstance, the ground state of C2
7 is 1(g1 , that of CSi5 is 3) ,
and for Ge28 it is 3(g
2 . Further, C3 has a linear structure9 in
a!Electronic mail: [email protected]
10000021-9606/2002/117(22)/10008/11/$19.00
Downloaded 20 Nov 2002 to 134.121.44.19. Redistribution subject to A
,
rs,dr
ctchngr-
y
ceses-
its ground state (1(g1), Si3 has a triangular structure10 (1A1),
while GeSi2 is predicted4 to have a triplet (3B2) rather thansinglet ground state.
The most discussed molecule of this kind is SiC2. Onceevidence was found for SiC2 in carbon-rich stars11 in 1926, itbecame a focus of interest. However, until 1982, experimtal and theoretical studies seemed to show that SiC2 has alinear structure in its ground state (1(1). In 1982,Bondybey12 carried out a time-resolved laser-induced flurescence spectroscopy experiment, and he concluded thaear ground state structure is nota priori obvious. The firstabinitio study of SiC2 was published by Green13 in 1983. Theyexcluded the possibility of the CSiC isomer, and concludthat the linear1(1 state is the ground state. However,1984, Grev and Schaefer14 carried out CISD/DZP studies anreported that the T-shaped1A1 state is, in fact, lower in en-ergy than the linear structure by 0.4 kcal/mol. Simulneously, Smalley and co-workers15 concluded that the linea1(1 structure is not consistent with the observed rotatiospectra. In 1988, Shepherd and Graham16 performed Fouriertransform infrared spectroscopy~FTIR! experiments andconfirmed the cyclic T-shaped geometry for the ground s(1A1).
8 © 2002 American Institute of Physics
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

yyio
geo
nl
m
nd
th
noyio
tthcen
ome,
oed
ll
do
a-
e-
he
to
Ge
nfor
nsta-i-de
d
-
10009J. Chem. Phys., Vol. 117, No. 22, 8 December 2002 Germanium dicarbide
The analogous GeC2 molecule was detected bSchmude, Gingerich, and Kingcade6 in a mass spectrometrexperiment in 1995. They reported enthalpies of formatfor GeC2 as well as Ge2C, Ge2C2, and Ge3C. However, theyassumed the structure of GeC2 to be T-shaped, considerinthe Si analog. Of course, the mass spectroscopic experimdid not provide any information concerning the geometrythe global minimum and the electronic structure. The otheoretical study to date was reported by Liet al.4 in 2001.They performed DFT calculations on AmBn ~A,B5Si,Ge,Candm1n,10). For GeC2, they excluded the possibility of alinear structure, stating that AB2 binary clusters of group IVelements have extremely low stabilities for linear geoetries. Li and co-workers reported that GeC2, Ge2C, SiC2,Si2C, and SiGe2 all have T-shaped geometries in their groustates.
Because there is no experimental data pertinent tostructure and energetics of the GeC2 system, as well as nohigh level theoretical study, we aimed to study the groustate electronic structure of this elusive molecule by empling highly correlated coupled-cluster theories in conjunctwith substantial basis sets. Especially, we wanted to seeeffects of relativistic and core–valence correlations onstructure and energetics of the system due to the existenthe Ge atom. The primary motivation here is that the groustate potential energy surface is extremely flat, like SiC2, andeven 2–3 kcal/mol energy is sufficient to invert the Ge ataround the molecule. Second, the main differences betwthe Si and Ge atoms are~a! the relatively large core of Gewhich makes relativity more important; and~b! the 3d elec-trons of Ge which gives rise to additional core–valence crelations with the 4s and 4p electrons. Therefore, the Gatom sometimes behaves differently from the Si atom, anturn, the ground state potential energy surface of GeC2 mightbe different from that of SiC2, due to the fact that very smaenergy differences determine the shape of the surface.
II. ELECTRONIC STRUCTURE CONSIDERATIONS
The three different geometries that have been studiethis work are given in Fig. 1. The ground electronic stateGeC2 has accordingly the following electronic configurtions: LinearC`v symmetry,
@core#~10s!2~11s!2~12s!2~13s!2~4p!4⇒1(1; ~1!
cyclic C2v symmetry~T-shaped geometry!,
@core#~9a1!2~10a1!2~5b2!2
3~4b1!2~11a1!2~12a1!2⇒1A1 ; ~2!
bentCs symmetry~L-shaped geometry!,
@core#~13a8!2~14a8!2~15a8!2
3~5a9!2~16a8!2~17a8!2⇒1A8. ~3!
Downloaded 20 Nov 2002 to 134.121.44.19. Redistribution subject to A
n
ntsfy
-
e
d-
nheeof
d
en
r-
in
inf
In the above equations,@core# denotes the 16 core~Ge:1s-, 2s-, 2p-, 3s-, 3p-, and 3d-like and C: 1s-like! orbit-als. One of the 4p molecular orbitals~MOs! of linear GeC2
becomes the 4b1 MO for the T-shaped structure and reprsents thep bonding. The 5a9 MO of the L-shaped structurecorresponds to thep molecular orbital. The in-planep MOin the L-shaped structure is mixed with other MOs of tsame symmetry. The 10s and 11s MOs of the linear struc-ture, 9a1 , 10a1 , and 5b2 MOs of the T-shaped, and 13a8and 14a8 MOs of the L-shaped arrangement correspondthe s~C–C!1s~C–Ge! and s~C–C!2s~C–Ge! bonds. The12s and 13s orbitals of the linear structure, 11a1 and 12a1
orbitals for T-shaped, and 16a8 and 17a8 orbitals forL-shaped are associated with the lone pairs of the C andatoms, respectively.
III. THEORETICAL APPROACH
SCF ~restricted open shell! wave functions have beeused for the zeroth-order descriptions of the ground stateeach geometry~T-shaped, linear, and L-shaped!. The con-figuration interaction with single and double excitatio~CISD!, the coupled cluster with single and double excitions ~CCSD!,17 and the CCSD with perturbative triple exctations@CCSD~T!#18 methods have been employed to inclucorrelation effects. The 11 lowest-lying MOs~Ge: 1s-, 2s-,2p-, 3s- 3p-like and C: 1s-like! were frozen and the twohighest-lying virtual MOs were deleted with all correlatelevels with the TZ2P1diff and TZ3P~2f! basis sets. Only the11 lowest-lying MOs were frozen for the correlation
FIG. 1. The three different geometries of GeC2 studied in this work.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
10010 J. Chem. Phys., Vol. 117, No. 22, 8 December 2002 Sari et al.
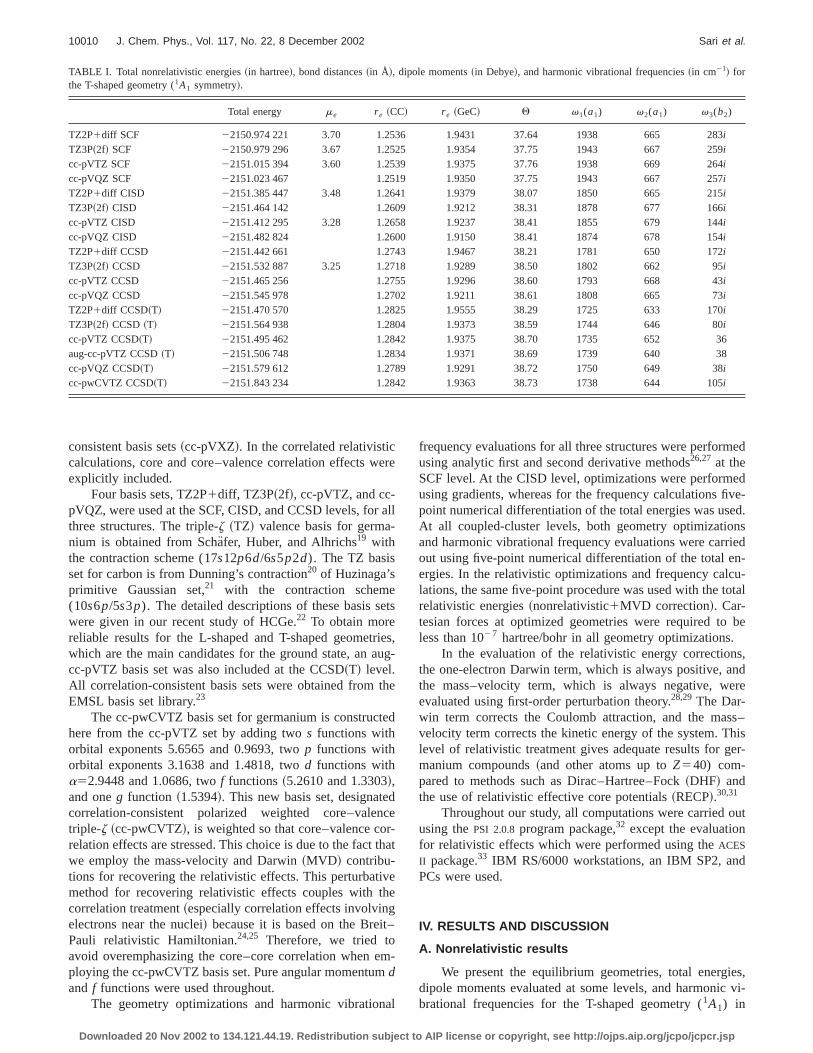
TABLE I. Total nonrelativistic energies~in hartree!, bond distances~in Å!, dipole moments~in Debye!, and harmonic vibrational frequencies~in cm21! forthe T-shaped geometry (1A1 symmetry!.
Total energy me r e ~CC! r e ~GeC! Q v1(a1) v2(a1) v3(b2)
TZ2P1diff SCF 22150.974 221 3.70 1.2536 1.9431 37.64 1938 665 283i
TZ3P~2f! SCF 22150.979 296 3.67 1.2525 1.9354 37.75 1943 667 259i
cc-pVTZ SCF 22151.015 394 3.60 1.2539 1.9375 37.76 1938 669 264i
cc-pVQZ SCF 22151.023 467 1.2519 1.9350 37.75 1943 667 257i
TZ2P1diff CISD 22151.385 447 3.48 1.2641 1.9379 38.07 1850 665 215i
TZ3P~2f! CISD 22151.464 142 1.2609 1.9212 38.31 1878 677 166i
cc-pVTZ CISD 22151.412 295 3.28 1.2658 1.9237 38.41 1855 679 144i
cc-pVQZ CISD 22151.482 824 1.2600 1.9150 38.41 1874 678 154i
TZ2P1diff CCSD 22151.442 661 1.2743 1.9467 38.21 1781 650 172i
TZ3P~2f! CCSD 22151.532 887 3.25 1.2718 1.9289 38.50 1802 662 95i
cc-pVTZ CCSD 22151.465 256 1.2755 1.9296 38.60 1793 668 43i
cc-pVQZ CCSD 22151.545 978 1.2702 1.9211 38.61 1808 665 73i
TZ2P1diff CCSD~T! 22151.470 570 1.2825 1.9555 38.29 1725 633 170i
TZ3P~2f! CCSD ~T! 22151.564 938 1.2804 1.9373 38.59 1744 646 80i
cc-pVTZ CCSD~T! 22151.495 462 1.2842 1.9375 38.70 1735 652 36aug-cc-pVTZ CCSD~T! 22151.506 748 1.2834 1.9371 38.69 1739 640 38cc-pVQZ CCSD~T! 22151.579 612 1.2789 1.9291 38.72 1750 649 38i
cc-pwCVTZ CCSD~T! 22151.843 234 1.2842 1.9363 38.73 1738 644 105i
e
a-
ee
rieu
th
te
dncr-th
veheg–
emm
a
ed
edve-ed.nsiedn-u-
otal
be
s,nd
ere
s–hiser-
ut
d
ies,vi-
consistent basis sets~cc-pVXZ!. In the correlated relativisticcalculations, core and core–valence correlation effects wexplicitly included.
Four basis sets, TZ2P1diff, TZ3P~2f!, cc-pVTZ, and cc-pVQZ, were used at the SCF, CISD, and CCSD levels, forthree structures. The triple-z ~TZ! valence basis for germanium is obtained from Scha¨fer, Huber, and Alhrichs19 withthe contraction scheme (17s12p6d/6s5p2d). The TZ basisset for carbon is from Dunning’s contraction20 of Huzinaga’sprimitive Gaussian set,21 with the contraction schem(10s6p/5s3p). The detailed descriptions of these basis swere given in our recent study of HCGe.22 To obtain morereliable results for the L-shaped and T-shaped geometwhich are the main candidates for the ground state, an acc-pVTZ basis set was also included at the CCSD~T! level.All correlation-consistent basis sets were obtained fromEMSL basis set library.23
The cc-pwCVTZ basis set for germanium is construchere from the cc-pVTZ set by adding twos functions withorbital exponents 5.6565 and 0.9693, twop functions withorbital exponents 3.1638 and 1.4818, twod functions witha52.9448 and 1.0686, twof functions~5.2610 and 1.3303!,and oneg function ~1.5394!. This new basis set, designatecorrelation-consistent polarized weighted core–valetriple-z ~cc-pwCVTZ!, is weighted so that core–valence corelation effects are stressed. This choice is due to the factwe employ the mass-velocity and Darwin~MVD ! contribu-tions for recovering the relativistic effects. This perturbatimethod for recovering relativistic effects couples with tcorrelation treatment~especially correlation effects involvinelectrons near the nuclei! because it is based on the BreitPauli relativistic Hamiltonian.24,25 Therefore, we tried toavoid overemphasizing the core–core correlation whenploying the cc-pwCVTZ basis set. Pure angular momentudand f functions were used throughout.
The geometry optimizations and harmonic vibration
Downloaded 20 Nov 2002 to 134.121.44.19. Redistribution subject to A
re
ll
ts
s,g-
e
d
e
at
-
l
frequency evaluations for all three structures were performusing analytic first and second derivative methods26,27 at theSCF level. At the CISD level, optimizations were performusing gradients, whereas for the frequency calculations fipoint numerical differentiation of the total energies was usAt all coupled-cluster levels, both geometry optimizatioand harmonic vibrational frequency evaluations were carrout using five-point numerical differentiation of the total eergies. In the relativistic optimizations and frequency calclations, the same five-point procedure was used with the trelativistic energies~nonrelativistic1MVD correction!. Car-tesian forces at optimized geometries were required toless than 1027 hartree/bohr in all geometry optimizations.
In the evaluation of the relativistic energy correctionthe one-electron Darwin term, which is always positive, athe mass–velocity term, which is always negative, wevaluated using first-order perturbation theory.28,29 The Dar-win term corrects the Coulomb attraction, and the masvelocity term corrects the kinetic energy of the system. Tlevel of relativistic treatment gives adequate results for gmanium compounds~and other atoms up toZ540) com-pared to methods such as Dirac–Hartree–Fock~DHF! andthe use of relativistic effective core potentials~RECP!.30,31
Throughout our study, all computations were carried ousing thePSI 2.0.8program package,32 except the evaluationfor relativistic effects which were performed using theACES
II package.33 IBM RS/6000 workstations, an IBM SP2, anPCs were used.
IV. RESULTS AND DISCUSSION
A. Nonrelativistic results
We present the equilibrium geometries, total energdipole moments evaluated at some levels, and harmonicbrational frequencies for the T-shaped geometry (1A1) in
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
10011J. Chem. Phys., Vol. 117, No. 22, 8 December 2002 Germanium dicarbide
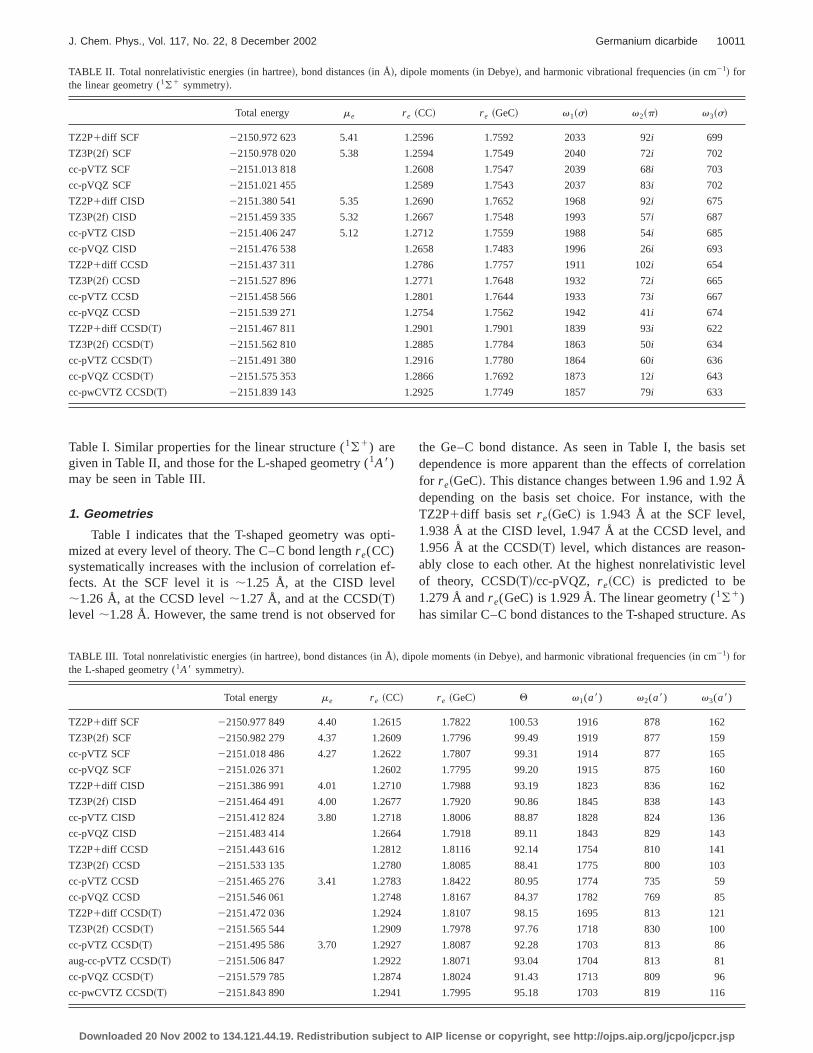
TABLE II. Total nonrelativistic energies~in hartree!, bond distances~in Å!, dipole moments~in Debye!, and harmonic vibrational frequencies~in cm21! forthe linear geometry (1(1 symmetry!.
Total energy me r e ~CC! r e ~GeC! v1~s! v2~p! v3~s!
TZ2P1diff SCF 22150.972 623 5.41 1.2596 1.7592 2033 92i 699
TZ3P~2f! SCF 22150.978 020 5.38 1.2594 1.7549 2040 72i 702
cc-pVTZ SCF 22151.013 818 1.2608 1.7547 2039 68i 703
cc-pVQZ SCF 22151.021 455 1.2589 1.7543 2037 83i 702
TZ2P1diff CISD 22151.380 541 5.35 1.2690 1.7652 1968 92i 675
TZ3P~2f! CISD 22151.459 335 5.32 1.2667 1.7548 1993 57i 687
cc-pVTZ CISD 22151.406 247 5.12 1.2712 1.7559 1988 54i 685
cc-pVQZ CISD 22151.476 538 1.2658 1.7483 1996 26i 693
TZ2P1diff CCSD 22151.437 311 1.2786 1.7757 1911 102i 654
TZ3P~2f! CCSD 22151.527 896 1.2771 1.7648 1932 72i 665
cc-pVTZ CCSD 22151.458 566 1.2801 1.7644 1933 73i 667
cc-pVQZ CCSD 22151.539 271 1.2754 1.7562 1942 41i 674
TZ2P1diff CCSD~T! 22151.467 811 1.2901 1.7901 1839 93i 622
TZ3P~2f! CCSD~T! 22151.562 810 1.2885 1.7784 1863 50i 634
cc-pVTZ CCSD~T! 22151.491 380 1.2916 1.7780 1864 60i 636
cc-pVQZ CCSD~T! 22151.575 353 1.2866 1.7692 1873 12i 643
cc-pwCVTZ CCSD~T! 22151.839 143 1.2925 1.7749 1857 79i 633
pt
ef
fo
settion
2 Åthe
,ndn-vel
. As
Table I. Similar properties for the linear structure (1(1) aregiven in Table II, and those for the L-shaped geometry (1A8)may be seen in Table III.
1. Geometries
Table I indicates that the T-shaped geometry was omized at every level of theory. The C–C bond lengthr e(CC)systematically increases with the inclusion of correlationfects. At the SCF level it is;1.25 Å, at the CISD level;1.26 Å, at the CCSD level;1.27 Å, and at the CCSD~T!level ;1.28 Å. However, the same trend is not observed
Downloaded 20 Nov 2002 to 134.121.44.19. Redistribution subject to A
i-
-
r
the Ge–C bond distance. As seen in Table I, the basisdependence is more apparent than the effects of correlafor r e~GeC!. This distance changes between 1.96 and 1.9depending on the basis set choice. For instance, withTZ2P1diff basis setr e~GeC! is 1.943 Å at the SCF level1.938 Å at the CISD level, 1.947 Å at the CCSD level, a1.956 Å at the CCSD~T! level, which distances are reasoably close to each other. At the highest nonrelativistic leof theory, CCSD~T!/cc-pVQZ, r e~CC! is predicted to be1.279 Å andr e(GeC) is 1.929 Å. The linear geometry (1(1)has similar C–C bond distances to the T-shaped structure
TABLE III. Total nonrelativistic energies~in hartree!, bond distances~in Å!, dipole moments~in Debye!, and harmonic vibrational frequencies~in cm21! forthe L-shaped geometry (1A8 symmetry!.
Total energy me r e ~CC! r e ~GeC! Q v1(a8) v2(a8) v3(a8)
TZ2P1diff SCF 22150.977 849 4.40 1.2615 1.7822 100.53 1916 878 162
TZ3P~2f! SCF 22150.982 279 4.37 1.2609 1.7796 99.49 1919 877 159
cc-pVTZ SCF 22151.018 486 4.27 1.2622 1.7807 99.31 1914 877 165
cc-pVQZ SCF 22151.026 371 1.2602 1.7795 99.20 1915 875 160
TZ2P1diff CISD 22151.386 991 4.01 1.2710 1.7988 93.19 1823 836 162
TZ3P~2f! CISD 22151.464 491 4.00 1.2677 1.7920 90.86 1845 838 143
cc-pVTZ CISD 22151.412 824 3.80 1.2718 1.8006 88.87 1828 824 136
cc-pVQZ CISD 22151.483 414 1.2664 1.7918 89.11 1843 829 143
TZ2P1diff CCSD 22151.443 616 1.2812 1.8116 92.14 1754 810 141
TZ3P~2f! CCSD 22151.533 135 1.2780 1.8085 88.41 1775 800 103
cc-pVTZ CCSD 22151.465 276 3.41 1.2783 1.8422 80.95 1774 735 59
cc-pVQZ CCSD 22151.546 061 1.2748 1.8167 84.37 1782 769 85
TZ2P1diff CCSD~T! 22151.472 036 1.2924 1.8107 98.15 1695 813 121
TZ3P~2f! CCSD~T! 22151.565 544 1.2909 1.7978 97.76 1718 830 100
cc-pVTZ CCSD~T! 22151.495 586 3.70 1.2927 1.8087 92.28 1703 813 86
aug-cc-pVTZ CCSD~T! 22151.506 847 1.2922 1.8071 93.04 1704 813 81
cc-pVQZ CCSD~T! 22151.579 785 1.2874 1.8024 91.43 1713 809 96
cc-pwCVTZ CCSD~T! 22151.843 890 1.2941 1.7995 95.18 1703 819 116
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

m5eorry
ytrupeaththent obveo
refag
tru
tankeaell
eth
un
gm-io
c3Z
ur
Zthat-ent-ch-
Al-weect,inar-
ereat-ing
seton-
ra-
re-ry.cylsped
rn
cary
fthisbe-
ansi-
ped
atallin
ithx-
g
rel-,
aryy to
qui-of
10012 J. Chem. Phys., Vol. 117, No. 22, 8 December 2002 Sari et al.
presented in Table II, the trend inr e(CC) with the inclusionof correlation effects parallels that for the T-shaped geoetry. However, the linear Ge–C bond length is about 0.10.20 Å shorter than that of the T-shaped structure at evlevel of theory. Our most reliable nonrelativistic values fthe r e(CC) and r e(GeC) distances at the linear stationapoint geometry are 1.287 and 1.769 Å, respectively.
The L-shaped (1A8) equilibrium geometry has a slightllonger C–C bond distance than the T-shaped and linear stures, by about 0.01 Å. On the other hand, the L-shaGe–C bond distance falls between that of the T-shapedlinear geometries. The effects of electron correlation onL-shaped geometry are similar to those found forT-shaped structure. The primary difference is an increasthe C–C bond distance. An important point is that the boangle in the L-shaped geometry is very much dependenboth the theoretical model and basis set. As shown in TaIII, larger basis sets produce smaller bond angles. Howeas we include electron correlation, no regular trend wasserved. Although the CISD and CCSD Ge–C–Cangles aresmaller than the SCF values, the CCSD~T! angles are largethan both CISD and CCSD. This is not very surprising bcause, as will be discussed later, the potential energy suris extremely flat, and even 2–3 kcal/mol of energy is enouto invert the Ge atom with respect to the C–C bond.
Nielsen et al.34 published a significant theoretical~abinitio! paper on SiC2 in 1997. Their predictions for the C–Cbond distances in the T-shaped, linear, and L-shaped stures are very similar to our predictions for GeC2. The Ge–Cbond distance is about 0.10, 0.06, and 0.10 Å longer thananalogous Si–C distances in the T-shaped, linear,L-shaped geometries, respectively. Nielsen and co-worreported that they could not locate an L-shaped stationpoint at several levels of theory. Here we find an L-shapGeC2 stationary point at every level of theory. As is now weknown, the true ground state geometry of SiC2 is T-shaped,and the experimental structural parameters35 are r 0(CC)51.269 Å,r 0(SiC)51.832 Å, while the C–Si–C bond anglis 40.4°. The predicted geometrical parameters forT-shaped GeC2 are r e(CC)51.279 Å, r e(GeC)51.929 Å,and Qe(CGeC)538.7° at the CCSD~T!/cc-pVQZ level oftheory. However, as we will discuss in the next section,like SiC2 the T-shaped geometry of GeC2 is found to be atransition state rather than the global minimum.
2. Harmonic vibrational frequencies
The T-shaped geometry (1A1), which is the true globalminimum for SiC2, is found to be a stationary point havinan imaginary harmonic vibrational frequency, for the asymetric stretching modev3(b2). As presented in Table I, although the imaginary value is getting smaller as correlateffects are included, our CCSD~T! GeC2 v3(b2) predictionsare still imaginary, except with the cc-pVTZ and aug-cpVTZ basis sets, for which real frequencies of 36 andcm21 were obtained, respectively. However, the cc-pVQCCSD~T! method yields an imaginaryv3(b2) of 38i cm21,and the core–valence correlation-consistent basis set~cc-pwCVTZ! produced an imaginary frequency of 105i cm21.Interestingly, as discussed later, relativistic corrections t
Downloaded 20 Nov 2002 to 134.121.44.19. Redistribution subject to A
-–ry
c-dndeeindn
ler,
b-
-ceh
c-
hedrsryd
e
-
-
n
-8
n
the realv3(b2) values for the cc-pVTZ and aug-cc-pVTbasis sets into imaginary values. Therefore, consideringalmost all nonrelativistic levels~except the two just mentioned!, the core–valence correlated correlation-consistbasis set at the CCSD~T! level, and all relativistic computations, produce imaginary values for the asymmetric streting v3(b2) mode, the T-shaped GeC2 is predicted to be atransition state between the two L-shaped geometries.though our theoretical results support this conclusion,realize that new experiments are necessary in this respbecause the system is elusive and the surface is extraordily flat.
The C–Cv1(a1) stretching vibrational frequency of thT-shaped structure decreases with more sophisticated tments of correlation effects, whereas the Ge–C stretchv2(a1) frequency is mainly dependent on the basischoice, similar to the Ge–C bond length. At the highest nrelativistic level of theory, CCSD~T!/cc-pVQZ, T-shaped fre-quencies of 1750, 649, and 38i cm21 are predicted for thev1(a1), v2(a1), andv3(b2) modes, respectively.
The linear structure has an imaginary harmonic vibtional frequency for the degenerate bending modev2(p).This imaginary frequency, as well as the C–C stretching fquencyv1(s), decreases as we improve the level of theoThe linear Ge–C stretching harmonic vibrational frequenv3(s) is slightly higher at the SCF, CISD, and CCSD levethan the values for the corresponding mode of the T-shav2(a1), whereas at the CCSD~T! level v3(s) is slightlylower than v2(a1). Although someab initio and DFTmethods34,36 give a spurious real vibrational frequency fothe bending mode of SiC2 in the linear structure, it has beeconcluded both from experiment and theory14–16,34 that thelinear geometry of SiC2 is a transition state to the cycliT-shaped structure. In contrast, we have obtained imaginfrequencies for the bendingv2(p) mode of GeC2 at all lev-els of theory~Table II!. Therefore, the linear geometry oGeC2 is a transition state. However, as mentioned above,transition state cannot connect two T-shaped structurescause the T-shaped geometry is also predicted to be a trtion state. Consequently, the linear geometry of GeC2 shouldbe a transition state between the two equivalent L-shageometries.
The L-shaped geometry was successfully optimizedevery level of theory, and real values are obtained forharmonic vibrational frequencies, as may be observedTable III. All three vibrational frequencies decrease whigher level treatments of electron correlation, with the eception that the CCSD~T! values for the Ge–C stretchinmode (v2) and Ge–C–Cbending mode (v3) are slightlyhigher than those predicted by CCSD. At the highest nonativistic level, CCSD~T!/cc-pVQZ, frequencies of 1713, 809and 96 cm21 are predicted for the C–C stretching (v1),Ge–C stretching (v2), and Ge–C–C bending (v3) modes,respectively. Because we did not encounter any imaginvibrational frequencies, and found this L-shaped geometrlie energetically lowest ~see Sec. IV A 4 below!, theL-shaped geometry is predicted to be the ground state elibrium geometry. As will be discussed later, the inclusion
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
10013J. Chem. Phys., Vol. 117, No. 22, 8 December 2002 Germanium dicarbide
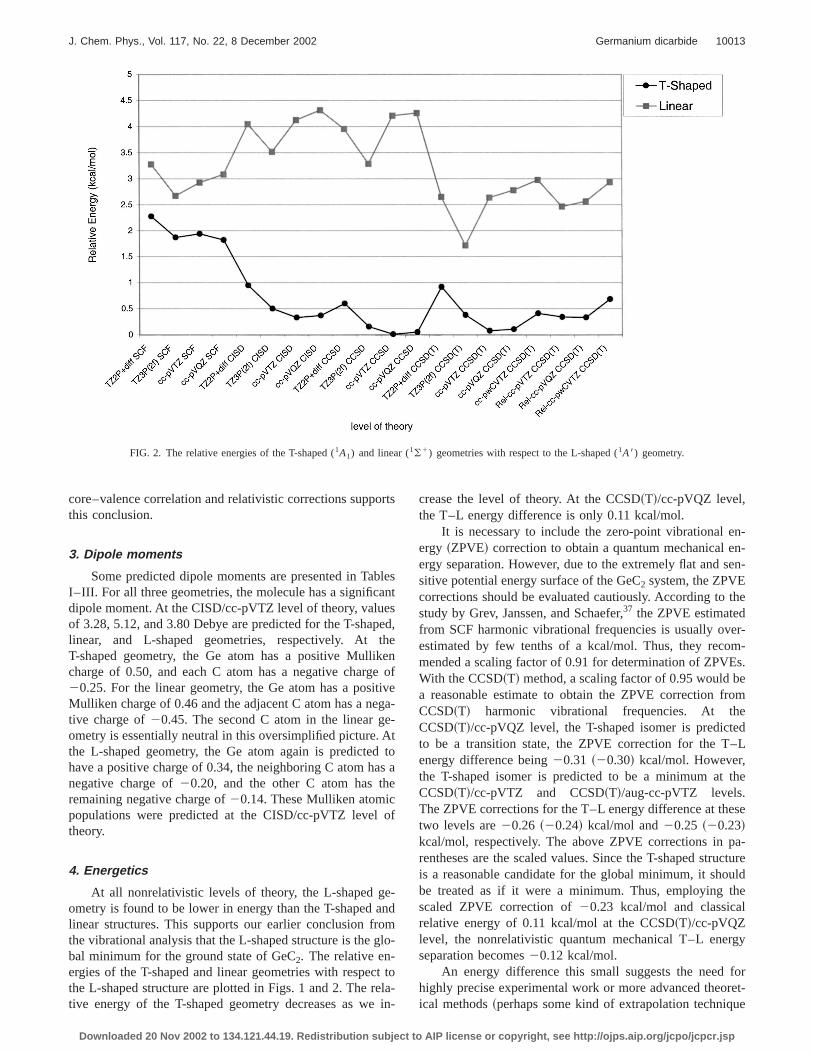
FIG. 2. The relative energies of the T-shaped (1A1) and linear (1(1) geometries with respect to the L-shaped (1A8) geometry.
or
blanespetheetivge-Adase
o
e-anomgl
ctel
n-n-en-
the
er-m-s.
eomeed–L
the
se
a-ctureuldhel
gy
forret-ue
core–valence correlation and relativistic corrections suppthis conclusion.
3. Dipole moments
Some predicted dipole moments are presented in TaI–III. For all three geometries, the molecule has a significdipole moment. At the CISD/cc-pVTZ level of theory, valuof 3.28, 5.12, and 3.80 Debye are predicted for the T-shalinear, and L-shaped geometries, respectively. AtT-shaped geometry, the Ge atom has a positive Mullikcharge of 0.50, and each C atom has a negative charg20.25. For the linear geometry, the Ge atom has a posiMulliken charge of 0.46 and the adjacent C atom has a netive charge of20.45. The second C atom in the linear gometry is essentially neutral in this oversimplified picture.the L-shaped geometry, the Ge atom again is predictehave a positive charge of 0.34, the neighboring C atom hnegative charge of20.20, and the other C atom has thremaining negative charge of20.14. These Mulliken atomicpopulations were predicted at the CISD/cc-pVTZ leveltheory.
4. Energetics
At all nonrelativistic levels of theory, the L-shaped gometry is found to be lower in energy than the T-shapedlinear structures. This supports our earlier conclusion frthe vibrational analysis that the L-shaped structure is thebal minimum for the ground state of GeC2. The relative en-ergies of the T-shaped and linear geometries with respethe L-shaped structure are plotted in Figs. 1 and 2. The rtive energy of the T-shaped geometry decreases as we
Downloaded 20 Nov 2002 to 134.121.44.19. Redistribution subject to A
ts
est
d,enofea-
ttoa
f
d
o-
toa-in-
crease the level of theory. At the CCSD~T!/cc-pVQZ level,the T–L energy difference is only 0.11 kcal/mol.
It is necessary to include the zero-point vibrational eergy ~ZPVE! correction to obtain a quantum mechanical eergy separation. However, due to the extremely flat and ssitive potential energy surface of the GeC2 system, the ZPVEcorrections should be evaluated cautiously. According tostudy by Grev, Janssen, and Schaefer,37 the ZPVE estimatedfrom SCF harmonic vibrational frequencies is usually ovestimated by few tenths of a kcal/mol. Thus, they recomended a scaling factor of 0.91 for determination of ZPVEWith the CCSD~T! method, a scaling factor of 0.95 would ba reasonable estimate to obtain the ZPVE correction frCCSD~T! harmonic vibrational frequencies. At thCCSD~T!/cc-pVQZ level, the T-shaped isomer is predictto be a transition state, the ZPVE correction for the Tenergy difference being20.31 ~20.30! kcal/mol. However,the T-shaped isomer is predicted to be a minimum atCCSD~T!/cc-pVTZ and CCSD~T!/aug-cc-pVTZ levels.The ZPVE corrections for the T–L energy difference at thetwo levels are20.26 ~20.24! kcal/mol and20.25 ~20.23!kcal/mol, respectively. The above ZPVE corrections in prentheses are the scaled values. Since the T-shaped struis a reasonable candidate for the global minimum, it shobe treated as if it were a minimum. Thus, employing tscaled ZPVE correction of20.23 kcal/mol and classicarelative energy of 0.11 kcal/mol at the CCSD~T!/cc-pVQZlevel, the nonrelativistic quantum mechanical T–L enerseparation becomes20.12 kcal/mol.
An energy difference this small suggests the needhighly precise experimental work or more advanced theoical methods~perhaps some kind of extrapolation techniq
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

Deeiumcee–T
thsa
thoifpo
vi-Cn
o-tivasthn
th
u
u
bt
eltwrs
–tcd
ndn
enwi
aethlufr
eet
end-
arytt is
witheli-
er-esisve
thee fa-herpednd
hein-
larm-
e-caldfirstryre
theriespeddif-ofing
e-if-
onehighi-
e
d,d
hatort-
10014 J. Chem. Phys., Vol. 117, No. 22, 8 December 2002 Sari et al.
to the full CI limit!. The basis set convergence at the CISCCSD, and CCSD~T! levels does not absolutely guarantour assignment of the L-shaped geometry to the equilibrgeometry~see Fig. 2!. However, the basis set convergenappears satisfactory, and the convergence seems to bproached near the cc-pVQZ basis set. The cc-pVQZ Tenergy differences are very slightly greater than the cc-pVresults at the correlated levels@by 0.038 kcal/mol higher withCISD, 0.039 kcal/mol with CCSD, and 0.031 kcal/mol wiCCSD~T!#. This observation may suggest that larger basets will not significantly lower the relative energy. Asresult, although all of our nonrelativistic and relativistic~seebelow! calculations put the T-shaped geometry aboveL-shaped structure, we are concerned that a definitive cclusion about the energetics of these two geometries is dcult due to the extremely flat nature of the ground statetential energy surface.
The energy relative to the global minimum is more edent for the linear structure. As displayed in Fig. 2, the Smethod puts the linear geometry about 3 kcal/mol, CISD aCCSD about 4 kcal/mol, and CCSD~T! 2.5–3.0 kcal/molabove the L-shaped structure. At the CCSD~T! level, exceptfor one basis set, TZ3P~2f!, the other seven basis sets prduced values between 2.5 and 3.0 kcal/mol for the relaenergy of the linear geometry. The best nonrelativistic clsical energy separation is predicted to be 2.78 kcal/mol atCCSD~T!/cc-pVQZ level and the quantum mechanical eergy difference~with the scaled ZPVE correction! to be 2.64kcal/mol. Therefore, the linear structure is certainly nottrue global minimum.
B. Effects of core–valence correlation
As described in Sec. III, the cc-pwCVTZ basis set docmented here for Ge was used at the CCSD~T! level to con-sider core–valence electron correlation effects. The resfor this level of theory, CCSD~T!/cc-pwCVTZ, are includedin Tables I–III. The predictions with this basis set shouldcompared with those for the cc-pVTZ basis set in orderdeduce the differential effects of Ge core–valence corrtions. This is because the only difference between thebasis sets is the usage of the core–valence correlated veof the normal cc-pVTZ basis sets for the Ge atom~see theabove theoretical procedures for more information!.
The most obvious effect of the inclusion of Ge corevalence correlation was seen to be on the asymmetric streing modev3(b2) of the T-shaped geometry and on the bening harmonic vibrational frequencies of the linear aL-shaped geometries. This is expected because movemethe Ge atom in the molecular plane requires very littleergy, and all these harmonic vibrations are associatedthe motion of the Ge atom. As noted above, the CCSD~T!/cc-pVTZ level of theory predicts a real harmonic vibrationfrequency of 36 cm21 for the asymmetric stretching modv3(b2) of the T-shaped geometry, although most other meods predict imaginary values. As seen in Table I, the incsion of the core–valence correlation replaces the realquency with an imaginary value of 105i cm21. This is quiteimportant because this core–valence correlation effect vmuch supports our earlier finding that the T-shaped geom
Downloaded 20 Nov 2002 to 134.121.44.19. Redistribution subject to A
,
ap-LZ
is
en-fi--
Fd
e-e
-
e
-
lts
eoa-oion
h--
t of-th
l
--e-
ryry
of GeC2 is a transition state. The situation is similar in thlinear case. As presented in Table II, our result for the being harmonic vibrationv2(p) with the normal cc-pVTZ ba-sis set@at the CCSD~T! level# is 60i cm21 and the inclusionof the core–valence correlation produces a larger imaginvalue of 79i cm21. This confirms our earlier prediction thathe linear geometry is a transition state. The inverse effecseen for the L-shaped geometry~see Table III!. The Ge–C–Cbending harmonic vibrational frequency (v3) becomes morereal, 116 cm21 ~the cc-pVTZ prediction is 86 cm21!. In otherwords, the core–valence correlation effects associatedthe Ge atom in the L-shaped geometry decrease the likhood that the L-shaped structure is a transition state.
The effects of the core–valence correlation on the engetics of the GeC2 system are also important. The relativenergy of the T-shaped geometry with the cc-pwCVTZ baset is 0.41 kcal/mol, and it is larger by 0.33 kcal/mol relatito that ~0.078 kcal/mol! with the cc-pVTZ basis set. Thisstabilization of the L-shaped geometry with respect toT-shaped structure also suggests the L-shaped to be thvored choice for the ground state geometry. On the othand, the energy difference between the linear and L-shageometries is 2.98 kcal/mol with the cc-pwCTZ basis set a2.64 kcal/mol with the cc-pVTZ basis set. Therefore, trelative energy of the linear structure increases with theclusion of core–valence correction as well, by a simiamount of 0.34 kcal/mol with respect to the L-shaped geoetry.
C. Effects of relativistic corrections
The relativistic optimizations are computationally dmanding. They require nine single point and nine numerifirst derivative CCSD~T! calculations for the T-shaped anlinear geometries, and 13 single points and 13 numericalderivative CCSD~T! calculations for the L-shaped geometjust for oneoptimization cycle. Therefore, we focused moattention on the T-shaped geometry. This is also becausenonrelativistic results for the L-shaped and linear geometare definitive, whereas those for the L-shaped and T-shageometries are not. As discussed earlier, the T–L energyference is small, and two of our nonrelativistic levelstheory gave real frequencies for the asymmetric stretchvibration v3(b2) of the T-shaped geometry~see Table I!.Therefore, we carried out relativistic optimizations and frquency evaluations for the T-shaped geometry with four dferent basis sets at the CCSD~T! level. For the linear geom-etry we employed three different basis sets, whereas onlybasis set was used for the L-shaped geometry, due to thecomputational cost@a total of 52 single point and 52 numercal first derivative CCSD~T! calculations inCs symmetrywere performed for the relativistic optimization of thL-shaped geometry with one basis set#. We present the ef-fects of relativistic corrections in Table IV for the T-shapein Table V for the linear, and Table VI for the L-shapegeometries.
The effects of relativity on the geometries are somewdifferent among the three structures. The Ge–C bond shens by;0.003 Å in the T-shaped geometry, by;0.005 Å in
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
harmonic
i
10015J. Chem. Phys., Vol. 117, No. 22, 8 December 2002 Germanium dicarbide
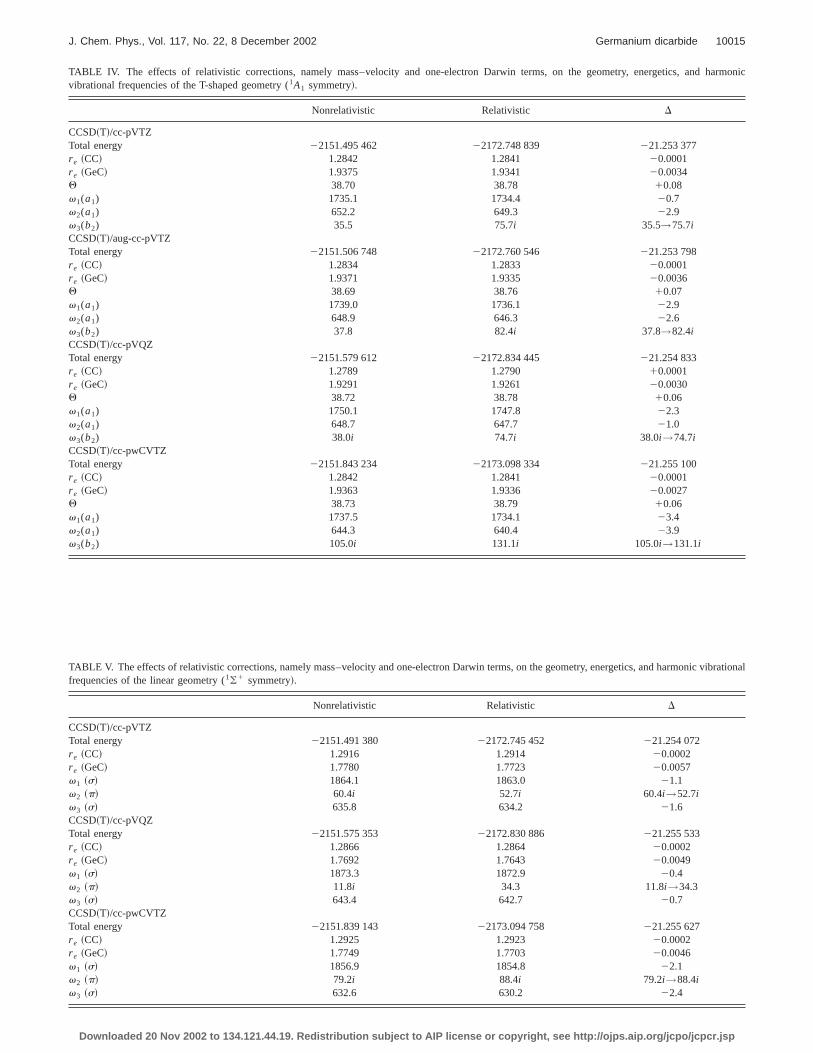
TABLE IV. The effects of relativistic corrections, namely mass–velocity and one-electron Darwin terms, on the geometry, energetics, andvibrational frequencies of the T-shaped geometry (1A1 symmetry!.
Nonrelativistic Relativistic D
CCSD~T!/cc-pVTZTotal energy 22151.495 462 22172.748 839 221.253 377r e ~CC! 1.2842 1.2841 20.0001r e ~GeC! 1.9375 1.9341 20.0034Q 38.70 38.78 10.08v1(a1) 1735.1 1734.4 20.7v2(a1) 652.2 649.3 22.9v3(b2) 35.5 75.7i 35.5→75.7iCCSD~T!/aug-cc-pVTZTotal energy 22151.506 748 22172.760 546 221.253 798r e ~CC! 1.2834 1.2833 20.0001r e ~GeC! 1.9371 1.9335 20.0036Q 38.69 38.76 10.07v1(a1) 1739.0 1736.1 22.9v2(a1) 648.9 646.3 22.6v3(b2) 37.8 82.4i 37.8→82.4iCCSD~T!/cc-pVQZTotal energy 22151.579 612 22172.834 445 221.254 833r e ~CC! 1.2789 1.2790 10.0001r e ~GeC! 1.9291 1.9261 20.0030Q 38.72 38.78 10.06v1(a1) 1750.1 1747.8 22.3v2(a1) 648.7 647.7 21.0v3(b2) 38.0i 74.7i 38.0i→74.7iCCSD~T!/cc-pwCVTZTotal energy 22151.843 234 22173.098 334 221.255 100r e ~CC! 1.2842 1.2841 20.0001r e ~GeC! 1.9363 1.9336 20.0027Q 38.73 38.79 10.06v1(a1) 1737.5 1734.1 23.4v2(a1) 644.3 640.4 23.9v3(b2) 105.0i 131.1i 105.0i→131.1i
TABLE V. The effects of relativistic corrections, namely mass–velocity and one-electron Darwin terms, on the geometry, energetics, and harmonic vbrationalfrequencies of the linear geometry (1(1 symmetry!.
Nonrelativistic Relativistic D
CCSD~T!/cc-pVTZTotal energy 22151.491 380 22172.745 452 221.254 072r e ~CC! 1.2916 1.2914 20.0002r e ~GeC! 1.7780 1.7723 20.0057v1 ~s! 1864.1 1863.0 21.1v2 ~p! 60.4i 52.7i 60.4i→52.7iv3 ~s! 635.8 634.2 21.6CCSD~T!/cc-pVQZTotal energy 22151.575 353 22172.830 886 221.255 533r e ~CC! 1.2866 1.2864 20.0002r e ~GeC! 1.7692 1.7643 20.0049v1 ~s! 1873.3 1872.9 20.4v2 ~p! 11.8i 34.3 11.8i→34.3v3 ~s! 643.4 642.7 20.7CCSD~T!/cc-pwCVTZTotal energy 22151.839 143 22173.094 758 221.255 627r e ~CC! 1.2925 1.2923 20.0002r e ~GeC! 1.7749 1.7703 20.0046v1 ~s! 1856.9 1854.8 22.1v2 ~p! 79.2i 88.4i 79.2i→88.4iv3 ~s! 632.6 630.2 22.4
Downloaded 20 Nov 2002 to 134.121.44.19. Redistribution subject to AIP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
y.th
cto
in
3
esfseles
hiscttheuevis-
recy
theueary
sticr-
, aseC
f
emetry
tser-
ta-21,
ityo
10016 J. Chem. Phys., Vol. 117, No. 22, 8 December 2002 Sari et al.
the linear case, and by;0.013 Å in the L-shaped geometrThe C–C bond distance decreases very slightly forT-shaped and linear geometries, whereas it elongates;0.001 Å for the L-shaped geometry. The relativistic effeon the L-shaped geometry are considerably larger than thfor the other two structures.
For the T-shaped geometry, the asymmetric stretchvibrational frequencyv3(b2) becomesmore imaginarywhenrelativity is considered. The two real frequencies, 36 andcm21 obtained at the CCSD~T!/cc-pVTZ and CCSD~T!/aug-cc-pVTZ levels, shifted to imaginary values of 76i and 82icm21, respectively, as presented in Table IV. At the highlevel of theory, CCSD~T!/cc-pVQZ, the imaginary value o38i cm21 becomes 75i cm21 for the same mode. This shift iin the same direction as the effects of core–valence corrtion ~see the previous section!. Therefore, depending on threlativistic and core–valence correlated results, we canthat both of these corrections tend to move the Ge atom
TABLE VI. The effects of relativistic corrections, namely mass–velocand one-electron Darwin terms, on the geometry, energetics, and harmvibrational frequencies of the L-shaped geometry (1A8 symmetry!.
Nonrelativistic Relativistic D
CCSD~T!/cc-pVTZTotal energy 22151.495 586 22172.749 386 221.253 800r e ~CC! 1.2927 1.2937 10.0010r e ~GeC! 1.8087 1.7960 20.0127Q 92.28 96.38 14.1v1(a8) 1703.0 1705.9 12.9v2(a8) 812.7 827.4 114.7v3(a8) 86.1 106.7 120.6
Downloaded 20 Nov 2002 to 134.121.44.19. Redistribution subject to A
ebysse
g
8
t
a-
ayin
the T-shaped geometry toward the L-shaped geometry. Tinteresting effect of relativity may be attributed to the fathat the valence electrons, which mainly contribute tochemical bonding, also display direct relativistic effects dto their core-penetrating natures as well as indirect relatitic effects.
The main relativistic effects for the linear structure aobserved for the bending harmonic vibrational frequenv2(p), as shown in Table V. The CCSD~T!/cc-pVTZ basisset produces a lower imaginary frequency, whereasCCSD~T!/cc-pwCVTZ generates a higher imaginary valcompared to the corresponding nonrelativistic imaginbendingv2(p) frequencies. The CCSD~T!/cc-pVQZ basisset shifts the small imaginary value of 12i cm21 to a realvalue of 34 cm21 when relativity is included. This shiftshould not be taken too seriously because the relativiCCSD~T!/cc-pwCVTZ level, which has a core–valence corelation, presents a larger imaginary bending frequencymentioned above. For the L-shaped structure, the Gstretching (v2) and Ge–C–Cbending (v3) frequencies in-crease by 15 and 21 cm21, respectively, upon the inclusion othe relativity.
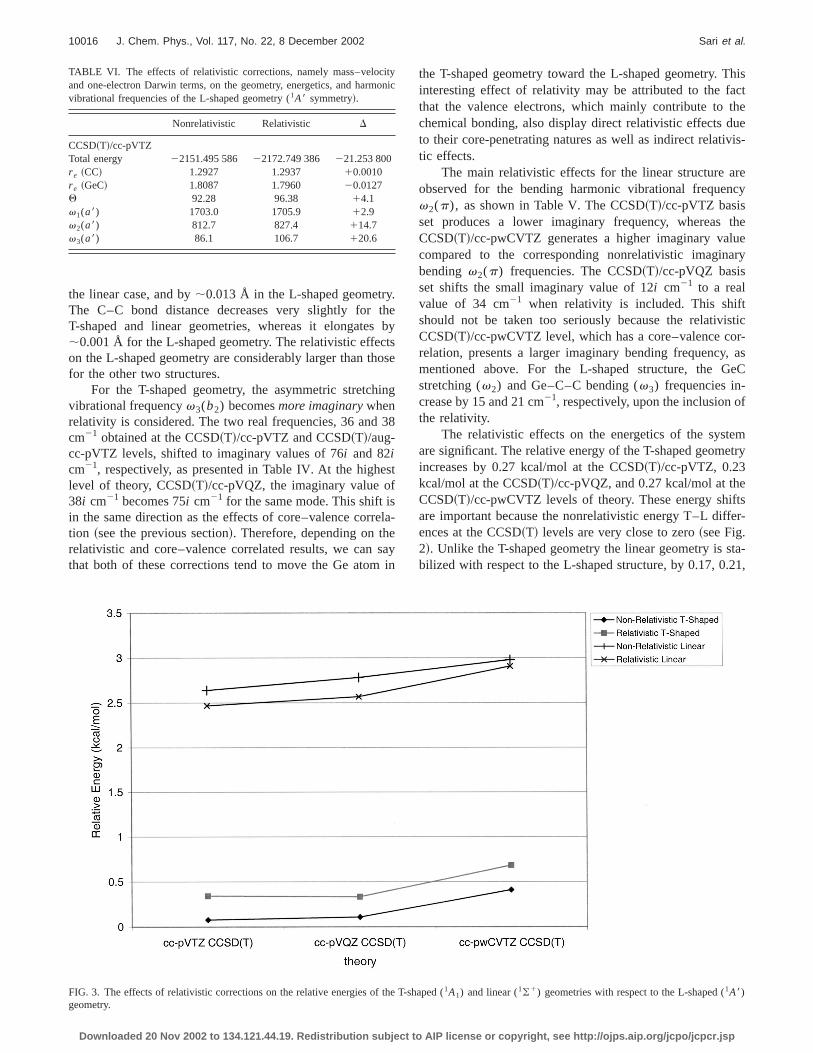
The relativistic effects on the energetics of the systare significant. The relative energy of the T-shaped geomincreases by 0.27 kcal/mol at the CCSD~T!/cc-pVTZ, 0.23kcal/mol at the CCSD~T!/cc-pVQZ, and 0.27 kcal/mol at theCCSD~T!/cc-pwCVTZ levels of theory. These energy shifare important because the nonrelativistic energy T–L diffences at the CCSD~T! levels are very close to zero~see Fig.2!. Unlike the T-shaped geometry the linear geometry is sbilized with respect to the L-shaped structure, by 0.17, 0.
nic
FIG. 3. The effects of relativistic corrections on the relative energies of the T-shaped (1A1) and linear (1(1) geometries with respect to the L-shaped (1A8)geometry.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

y
–
10017J. Chem. Phys., Vol. 117, No. 22, 8 December 2002 Germanium dicarbide
FIG. 4. The proposed potential energsurface for the ground state of GeC2 atthe CCSD~T!/cc-pVQZ level with theinclusion of zero-point vibrationalenergy, scalar relativistic, and corevalence corrections.
.a
-
ndof
ren-arth8ofrgphest
erf
thpelar-b
Allicttheforte,-pedntialtome
ci-
ev.
s.
B
ys.
and 0.07 kcal/mol, at the CCSD~T!/cc-pVTZ, CCSD~T!/cc-pVQZ, and CCSD~T!/cc-pwCVTZ levels, respectivelyThese relativistic effects on the energetics of the systemplotted in Fig. 3.
In Sec. IV A 4, the nonrelativistic classical and ZPVEcorrected T–L energy separations at the CCSD~T!/cc-pCVQZ level of theory were determined to be 0.11 a20.12 kcal/mol, respectively. Assuming the additivitycore–valence correlation effects of10.33 kcal/mol in Sec.IV B and the relativistic effects of10.23 kcal/mol, the rela-tivistic quantum mechanical T–L energy difference is pdicted to be10.44 kcal/mol. On the other hand, the norelativistic classical and quantum mechanical energy septions between the linear and L-shaped structures atCCSD~T!/cc-pCVQZ level of theory were found to be 2.7and 2.64 kcal/mol, respectively. Including core–valence crelation effects of10.34 kcal/mol and relativistic effects o20.21 kcal/mol, the relativistic quantum mechanical enedifference between the linear and L-shaped structures isdicted to be12.77 kcal/mol. The final predicted shape of tground state potential energy surface at the relativiCCSD~T!/cc-pVQZ level of theory is depicted in Fig. 4.
V. CONCLUDING REMARKS
The ground state potential energy surface of the expmentally observed GeC2 molecule reflects the same kind oelusive structure as that of the much discussed SiC2. Al-though the T- and L-shaped geometries are very closeenergy, and a conclusive result is difficult to reach due tovery flat nature of the potential energy surface, the L-shastructure is predicted to be the global minimum. The scarelativistic corrections~mass–velocity and one-electron Dawin terms! and core–valence corrections are found to sta
Downloaded 20 Nov 2002 to 134.121.44.19. Redistribution subject to A
re
-
a-e
r-
yre-
ic
i-
inedr
i-
lize the L-shaped geometry over the T-shaped structure.of our nonrelativistic and relativistic computations predthe L-shaped structure to be lower in energy thanT-shaped geometry, which is the equilibrium geometrySiC2. The linear structure is predicted to be a transition staas is the case with SiC2. However, this transition state connects the two L-shaped geometries, not the two T-shastructures. The predicted shape of the ground state poteenergy surface is given in Fig. 4. Finally, it is importantnote that new experimental work will be necessary to coto definitive conclusions.
ACKNOWLEDGMENT
This research was supported by the U.S. National Sence Foundation, Grant No. CHE-0136186.
1P. Venezuela, G. M. Dalpian, A. J. R. da Silva, and A. Fazzio, Phys. RB 64, 193202~2001!.
2A. Benzair, B. Bouhafs, B. Khelifa, C. Mathieu, and H. Aourag, PhyLett. A 282, 299 ~2001!.
3C. Jo and K. Lee, J. Chem. Phys.113, 7268~2000!.4S.-D. Li, Z.-G. Zhao, X.-F. Zhao, H.-S. Wu, and Z.-H. Jin, Phys. Rev.64, 195312~2001!.
5C. R. Brazier, L. C. O’Brien, and P. F. Bernath, J. Chem. Phys.91, 7384~1989!.
6R. W. Schmude, K. A. Gingerich, and J. E. Kingcade, J. Phys. Chem.99,15294~1995!.
7P. J. Bruna, S. D. Peyerimhoff, and R. J. Buenker, J. Chem. Phys.72, 5437~1980!.
8K. Balasubramanian, J. Mol. Spectrosc.123, 228 ~1987!.9M. Izuha and K. Yamanouchi, J. Chem. Phys.109, 1810~1998!.
10C. Xu, T. R. Taylor, G. R. Burton, and D. M. Neumark, J. Chem. Ph108, 1395~1998!.
11P. W. Merrill, Publ. Astron. Soc. Pac.38, 175 ~1926!.12V. E. Bondybey, J. Phys. Chem.86, 3396~1982!.13S. Green, Astrophys. J.266, 895 ~1983!.14R. S. Grev and H. F. Schaefer, J. Chem. Phys.80, 3552~1984!.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp

E
em
asn
oryndl
Ab
ity
d P.
.
.
10018 J. Chem. Phys., Vol. 117, No. 22, 8 December 2002 Sari et al.
15D. L. Michalopoulos, M. E. Geusic, P. R. R. Langridge-Smith, and R.Smalley, J. Chem. Phys.80, 3556~1984!.
16R. A. Shepherd and W. R. M. Graham, J. Chem. Phys.88, 3399~1988!.17G. D. Purvis and R. J. Bartlett, J. Chem. Phys.76, 1910~1982!.18K. Raghavachari, G. W. Trucks, J. A. Pople, and M. Head-Gordon, Ch
Phys. Lett.157, 479 ~1989!.19A. Schafer, C. Huber, and R. Ahlrichs, J. Chem. Phys.100, 5829~1994!.20T. H. Dunning, J. Chem. Phys.55, 716 ~1971!.21S. Huzinaga, J. Chem. Phys.42, 1293~1965!.22L. Sari, Y. Yamaguchi, and H. F. Schaefer, J. Chem. Phys.115, 5932
~2001!.23Extensible Computational Chemistry Environment Basis Set Datab
Version 1/29/01, as developed and distributed by the Molecular ScieComputing Facility, Environmental and Molecular Sciences Laboratwhich is part of the Pacific Northwest Laboratory, P.O. Box 999, RichlaWashington 99352; http://www.emsl.pnl.gov:2080/forms/basisform.htm
24K. Balasubramanian,Relativistic Effects in Chemistry Part A~Wiley, NewYork, 1997!.
25E. M. Rose,Relativistic Electron Theory~Wiley, New York, 1961!.26P. Pulay, inModern Theoretical Chemistry, edited by H. F. Schaefer~Ple-
num, New York, 1977!, Vol. 4, pp. 153–185.27Y. Yamaguchi, Y. Osamura, J. D. Goddard, and H. F. Schaefer,A New
Downloaded 20 Nov 2002 to 134.121.44.19. Redistribution subject to A
.
.
e,ce,,
Dimension to Quantum Chemistry: Analytic Derivative Methods inInitio Molecular Electronic Structure Theory~Oxford University Press,New York, 1994!.
28R. D. Cowan and D. C. Griffin, J. Opt. Soc. Am.66, 1010~1976!.29R. L. Martin, J. Phys. Chem.87, 730 ~1983!.30E. Ottschofski and W. Kutzelnigg, J. Chem. Phys.102, 1752~1995!.31K. G. Dyall, J. Chem. Phys.96, 1210~1992!.32PSI 2.0.8; C. L. Janssen, E. T. Seidl, G. E. Scuseriaet al., PSITECH, Inc.,
Watkinsville, GA 30677.33ACES II is a program product of the Quantum Theory Product, Univers
of Florida. Authors: J. F. Stanton, J. Gauss, J. D. Wattset al., Integralpackages included areVMOL ~J. Almlof and P. R. Taylor!; VPROPS~P. Tay-lor!, ABACUS ~T. Helgaker, H. J. Aa. Jensen, P. Jørgensen, J. Olsen, anR. Taylor!.
34I. M. B. Nielsen, W. D. Allen, A. G. Csa´szar, and H. F. Schaefer, J. ChemPhys.107, 1195~1997!.
35J. Cernicharo, M. Gue´lin, C. Kahane, M. Bogey, C. Demuynck, and J. LDestombes, Astron. Astrophys.246, 213 ~1991!.
36Y. Zhang, C.-Y. Zhao, W.-H. Fang, and Z.-H. Lu, J. Mol. Struct.454, 31~1998!.
37R. S. Grev, C. L. Janssen, and H. F. Schaefer, J. Chem. Phys.95, 5128~1991!.
IP license or copyright, see http://ojps.aip.org/jcpo/jcpcr.jsp
Copyright © 2022 FDOKUMEN




















