
A subset of bacterial inner membrane proteins integrated by the twin-arginine translocase
14
Molecular Microbiology (2003) 49(5), 1377–1390 doi:10.1046/j.1365-2958.2003.03642.x © 2003 Blackwell Publishing Ltd Blackwell Science, LtdOxford, UKMMIMolecular Microbiology 1365-2958Blackwell Publishing Ltd, 200349 513771390Original ArticleTat-dependent integral membrane proteinsK. Hatzixanthis, T. Palmer and F. Sargent Accepted 27 May, 2003. *For correspondence. E-mail [email protected]; Tel. (+44) 1603 592 889; Fax (+44) 1603 592 250. A subset of bacterial inner membrane proteins integrated by the twin-arginine translocase Kostas Hatzixanthis 1 , Tracy Palmer 2 and Frank Sargent 1 * 1 Centre for Metalloprotein Spectroscopy and Biology, School of Biological Sciences, University of East Anglia, Norwich, NR4 7TJ, UK 2 Department of Molecular Microbiology, John Innes Centre, Norwich Research Park, Norwich, NR4 7UH, UK. Summary A group of bacterial exported proteins are synthe- sized with N-terminal signal peptides containing a SRRxFLK ‘twin-arginine’ amino acid motif. Proteins bearing twin-arginine signal peptides are targeted post-translationally to the twin-arginine translocation (Tat) system which transports folded substrates across the inner membrane. In Escherichia coli, most integral inner membrane proteins are assembled by a co-translational process directed by SRP/FtsY, the SecYEG translocase, and YidC. In this work we define a novel class of integral membrane proteins assem- bled by a Tat-dependent mechanism. We show that at least five E. coli Tat substrate proteins contain hydro- phobic C-terminal transmembrane helices (or ‘C- tails’). Fusions between the identified transmembrane C-tails and the exclusively Tat-dependent reporter proteins TorA and SufI render the resultant chimeras membrane-bound. Export-linked signal peptide pro- cessing and membrane integration of the chimeras is shown to be both Tat-dependent and YidC-indepen- dent. It is proposed that the mechanism of membrane integration of proteins by the Tat system is fundamen- tally distinct from that employed for other bacterial inner membrane proteins. Introduction Biosynthesis of biological membranes requires the con- trolled targeting and integration of proteins into lipid bilay- ers. Studies of the model prokaryote Escherichia coli have established in some detail the molecular mechanisms involved in insertion of integral membrane proteins into the energy-transducing cytoplasmic (inner) membrane. Most integral inner membrane proteins are assembled by a co-translational process coordinated by the general secretory (Sec) system (de Gier and Luirink, 2001) which is also the predominant route of protein export. In general, integral membrane proteins do not have cleavable N- terminal signal peptides but contain long hydrophobic transmembrane a -helices (TMs). As the first hydrophobic TMs emerge de novo from the ribosome they are imme- diately recognized by the bacterial signal recognition par- ticle (SRP). The nascent chain-ribosome-SRP complex is then targeted to the SecYEG translocase via the SRP receptor FtsY and translocation of the proteins proceeds by a co-translational ‘threading mechanism’ (de Gier and Luirink, 2001). The key process of clearing new TMs from the SecYEG channel into the lipid environment is facili- tated by the YidC membrane protein (Samuelson et al ., 2000). YidC is the E. coli homologue of the mitochondrial Oxa1p and thylakoidal Alb3 proteins required for mem- brane protein assembly in these organelles (Luirink et al ., 2001) and, although a proportion of cellular YidC has been shown to associate with the SecYEG translocase (Scotti et al ., 2000) and the SecDFYajC auxiliary complex (Nou- wen and Driessen, 2002), some integral membrane pro- teins are targeted directly to YidC and thus integrated by a completely Sec-independent mechanism (Samuelson et al ., 2000). Indeed, YidC is reportedly required for the integration of all inner membrane proteins so far tested (Chen et al ., 2002). In parallel to the Sec system E. coli utilizes a second general pathway for protein targeting. A subset of ex- ported proteins is synthesized with distinctive N-terminal signal peptides containing a SRRxFLK ‘twin-arginine’ amino acid motif. Proteins bearing twin-arginine signal peptides are directed post-translationally to the mem- brane-embedded twin-arginine translocation (Tat) system which transports fully folded proteins across the inner membrane using energy provided by the transmembrane D p (Berks et al ., 2000). In E. coli the TatA, TatB, TatC and TatE membrane proteins have been shown to be compo- nents of the Tat translocase (Bogsch et al ., 1998; Sargent et al ., 1998a, 1999; Weiner et al ., 1998; Berks et al ., 2000). The core Tat transporter is a large oligomeric com- plex of the TatA, TatB and TatC proteins (Bolhuis et al ., 2001; de Leeuw et al ., 2002). Within the fully assembled complex the TatA protein probably fulfils the role of the protein-conducting channel (Sargent et al ., 2001; Porcelli
Transcript of A subset of bacterial inner membrane proteins integrated by the twin-arginine translocase

Molecular Microbiology (2003)
49
(5), 1377–1390 doi:10.1046/j.1365-2958.2003.03642.x
© 2003 Blackwell Publishing Ltd
Blackwell Science, LtdOxford, UKMMIMolecular Microbiology 1365-2958Blackwell Publishing Ltd, 200349
513771390
Original Article
Tat-dependent integral membrane proteinsK. Hatzixanthis, T. Palmer and F. Sargent
Accepted 27 May, 2003. *For correspondence. [email protected]; Tel. (
+
44) 1603 592 889; Fax (
+
44) 1603 592250.
A subset of bacterial inner membrane proteins integrated by the twin-arginine translocase
Kostas Hatzixanthis
1
, Tracy Palmer
2
andFrank Sargent
1
*
1
Centre for Metalloprotein Spectroscopy and Biology, School of Biological Sciences, University of East Anglia, Norwich, NR4 7TJ, UK
2
Department of Molecular Microbiology, John Innes Centre, Norwich Research Park, Norwich, NR4 7UH, UK.
Summary
A group of bacterial exported proteins are synthe-sized with N-terminal signal peptides containing aSRRxFLK ‘twin-arginine’ amino acid motif. Proteinsbearing twin-arginine signal peptides are targetedpost-translationally to the twin-arginine translocation(Tat) system which transports folded substratesacross the inner membrane. In
Escherichia coli,
mostintegral inner membrane proteins are assembled by aco-translational process directed by SRP/FtsY, theSecYEG translocase, and YidC. In this work we definea novel class of integral membrane proteins assem-bled by a Tat-dependent mechanism. We show that atleast five
E. coli
Tat substrate proteins contain hydro-phobic C-terminal transmembrane helices (or ‘C-tails’). Fusions between the identified transmembraneC-tails and the exclusively Tat-dependent reporterproteins TorA and SufI render the resultant chimerasmembrane-bound. Export-linked signal peptide pro-cessing and membrane integration of the chimeras isshown to be both Tat-dependent and YidC-indepen-dent. It is proposed that the mechanism of membraneintegration of proteins by the Tat system is fundamen-tally distinct from that employed for other bacterialinner membrane proteins.
Introduction
Biosynthesis of biological membranes requires the con-trolled targeting and integration of proteins into lipid bilay-ers. Studies of the model prokaryote
Escherichia coli
haveestablished in some detail the molecular mechanismsinvolved in insertion of integral membrane proteins intothe energy-transducing cytoplasmic (inner) membrane.
Most integral inner membrane proteins are assembled bya co-translational process coordinated by the generalsecretory (Sec) system (de Gier and Luirink, 2001) whichis also the predominant route of protein export. In general,integral membrane proteins do not have cleavable N-terminal signal peptides but contain long hydrophobictransmembrane
a
-helices (TMs). As the first hydrophobicTMs emerge
de novo
from the ribosome they are imme-diately recognized by the bacterial signal recognition par-ticle (SRP). The nascent chain-ribosome-SRP complex isthen targeted to the SecYEG translocase via the SRPreceptor FtsY and translocation of the proteins proceedsby a co-translational ‘threading mechanism’ (de Gier andLuirink, 2001). The key process of clearing new TMs fromthe SecYEG channel into the lipid environment is facili-tated by the YidC membrane protein (Samuelson
et al
.,2000). YidC is the
E. coli
homologue of the mitochondrialOxa1p and thylakoidal Alb3 proteins required for mem-brane protein assembly in these organelles (Luirink
et al
.,2001) and, although a proportion of cellular YidC has beenshown to associate with the SecYEG translocase (Scotti
et al
., 2000) and the SecDFYajC auxiliary complex (Nou-wen and Driessen, 2002), some integral membrane pro-teins are targeted directly to YidC and thus integrated bya completely Sec-independent mechanism (Samuelson
et al
., 2000). Indeed, YidC is reportedly required for theintegration of all inner membrane proteins so far tested(Chen
et al
., 2002).In parallel to the Sec system
E. coli
utilizes a secondgeneral pathway for protein targeting. A subset of ex-ported proteins is synthesized with distinctive N-terminalsignal peptides containing a SRRxFLK ‘twin-arginine’amino acid motif. Proteins bearing twin-arginine signalpeptides are directed post-translationally to the mem-brane-embedded twin-arginine translocation (Tat) systemwhich transports fully folded proteins across the innermembrane using energy provided by the transmembrane
D
p
(Berks
et al
., 2000). In
E. coli
the TatA, TatB, TatC andTatE membrane proteins have been shown to be compo-nents of the Tat translocase (Bogsch
et al
., 1998; Sargent
et al
., 1998a, 1999; Weiner
et al
., 1998; Berks
et al
.,2000). The core Tat transporter is a large oligomeric com-plex of the TatA, TatB and TatC proteins (Bolhuis
et al
.,2001; de Leeuw
et al
., 2002). Within the fully assembledcomplex the TatA protein probably fulfils the role of theprotein-conducting channel (Sargent
et al
., 2001; Porcelli

1378
K. Hatzixanthis, T. Palmer and F. Sargent
© 2003 Blackwell Publishing Ltd,
Molecular Microbiology
,
49
, 1377–1390
et al
., 2002) while TatB and TatC are involved in initialsignal peptide recognition (de Leeuw
et al
., 2002).The physiological role of the Tat pathway is to transport
folded proteins, many of which contain redox active cofac-tors which must be incorporated in the cytoplasm prior toexport (Berks, 1996; Berks
et al
., 2000). In the currentwork, we demonstrate that of the approximately 26 Tatsubstrates in
E. coli
at least five are genuine integral innermembrane proteins. The
E. coli
Tat substrates in questionare shown to possess single transmembrane
a
-helices atthe extreme C-terminus with N
periplasm
-C
cytoplasm
topologies(herein termed ‘C-tails’). Using the exclusively Tat-dependent water-soluble reporter proteins TorA and SufIwe demonstrate that the
E. coli
Tat system can mediateintegration of transmembrane
a
-helices into the innermembrane. Remarkably, the integration of hydrophobicTMs by the Tat translocase is shown to have no require-ment for the YidC protein.
Results and discussion
Transmembrane helices on
E. coli
Tat substrates
Sequence analysis suggests that at least five of theapproximately 26 probable endogenous
E. coli
Tat sub-
strates contain plausible hydrophobic TMs and are thusC-tail anchored integral membrane proteins (Fig. 1A; Sar-gent
et al
., 2002). The five gene products identified areHybO, HyaA, HybA, FdnH and FdoH and each are com-ponents of larger multisubunit [NiFe] hydrogenase or for-mate dehydrogenase respiratory complexes. In general,periplasmically oriented membrane-bound [NiFe] hydro-genases and formate dehydrogenases share a heterotri-meric ‘
abg
’ structure (Fig. 1B). The catalytic
a
-subunitforms a peripheral heterodimer with its
b
-subunit partnerand the
ab
complex is targeted as a preformed unit by asingle twin-arginine signal peptide located on only one ofthe subunits (e.g. Rodrigue
et al
., 1999). The
g
-subunit isan SRP-dependent integral membrane protein containingat least four transmembrane helices (e.g. Jormakka
et al
.,2002). Early sequence analysis had previously predictedthe existence of hydrophobic C-tail anchors on compo-nents of the peripheral heterodimers from membrane-bound [NiFe] hydrogenases and formate dehydrogenases(Berg
et al
., 1991; Wu and Mandrand, 1993; Berks
et al
.,1995). However, attempts to obtain experimental evidencefor their function in membrane association yielded conflict-ing results. For example, the peripheral subunits of theTat-dependent [NiFe] hydrogenases from
Wolinella succi-
Fig. 1.
Tat-dependent integral membrane proteins.A. Hydrophobic transmembrane C-tails on
Escherichia coli
Tat substrates. Possible transmembrane helices are shaded based on the crystal structure of formate dehydrogenase-N (Jormakka
et al
., 2002; Sargent
et al
., 2002). Positively charged side-chains located on the cytoplasmic boundary of the TMs are shown in italics. The averaged hydrophobic index (AHI) for each shaded sequence is given. Values were calculated using individual hydrophobic indices for each side-chain.B. Typical structure of membrane-bound [NiFe] hydrogenases and formate dehydrogenases. The Tat-dependent
a
- and
b
-subunits are shadedgrey.C. Western blot analysis of the core [NiFe] hydrogenase-2 subunits (HybOC). Strains MC4100 (parent strain), FTD674 (
hybO
D
C-tail) and BØD674 (
hybO
D
C-tail,
D
tatB
) were cultured anaerobically overnight in CR medium containing 0.5% (v/v) glycerol and 0.5% (w/v) fumarate. Cells were fractionated into periplasm (P), total membranes (M), and cytoplasm (C) and proteins separated by SDS–PAGE (10% w/v acrylamide), blotted and challenged with an anti-HybOC serum. The asterisk denotes a non-specific immunoreactive band.

Tat-dependent integral membrane proteins
1379
© 2003 Blackwell Publishing Ltd,
Molecular Microbiology
,
49
, 1377–1390
nogenes
,
Rhodobacter capsulatus
and
E. coli
are stillstrongly membrane associated in strains lacking the inte-gral membrane partner subunits (Gross
et al
., 1998; Mag-nani
et al
., 2000; Dubini
et al
., 2002) providing goodevidence for the presence of at least one TM associatedwith the peripheral subunits. Yet analogous experimentswith the closely related
Ralstonia eutropha
membrane-bound [NiFe] hydrogenase (Bernhard
et al
., 1997) and
E.coli
formate dehydrogenase-N (Stanley
et al
., 2002) led tothe accumulation of soluble periplasmic proteins with noapparent membrane affinity. In the current work we there-fore set out to address directly the intrinsic membraneaffinity and attachment properties of hydrophobic C-tailsfrom Tat-dependent proteins and assess the role of theTat translocase in the integration of Tat-dependent TMsinto the bilayer.
In the initial portion of this study we studied a nativeTat-dependent respiratory enzyme. The
E. coli
[NiFe]hydrogenase-2 isoenzyme is comprised of an
a
-subunitthat contains the Ni-Fe-CO-2CN cofactor (HybC) and a
b
-subunit which binds three Fe-S clusters (HybO). HybOCform the peripheral
ab
dimer and transport of the complexis dependent on a single Tat signal peptide located at theN-terminus of HybO (Sargent
et al
., 1998a; Rodrigue
et al
., 1999). Experimental evidence points to the pres-ence of a TM at the C-terminus of HybO. First, in the holo-complex the HybOC dimer associates with an integralmembrane protein (HybB) and a Tat-dependent ferredoxin(HybA); however, the HybOC dimer remains stronglymembrane-bound in the absence of either HybA or HybB(Dubini
et al
., 2002). Second, the HybOC dimer can bereleased from sphaeroplasts as a stable active fragmentby delicate protease treatment which specifically removes5 kDa from the C-terminus of HybO and leaves the HybCsubunit untouched (Sargent
et al
., 1998b). To obtain directevidence for the presence of a transmembrane C-tail atthe C-terminus of HybO we constructed a strain (FTD674,
hybO
D
C-tail) with a chromosomal in frame deletionbetween
hybO
codons 328 (encoding glutamate) and 355(encoding valine) thus removing a hydrophobic regionpredicted to form a TM (Fig. 1A). As expected the HybOCcomplex was found in the membrane fraction of the ‘wild-type’ parent strain (Fig. 1C). However, the mutant HybOCcomplex lacking the HybO TM no longer co-sedimentedwith the membrane vesicles and was instead located inthe periplasm (Fig. 1C). The soluble periplasmic HybOC(
D
C-tail) complex retained enzymatic activity with BenzylViologen (BV) as an artificial electron acceptor (data notshown) and transport remained completely dependent onthe Tat translocase (Fig. 1C). Taken together, we concludethat HybO contains a C-terminal TM and that the C-tail isneither required for cofactor insertion nor plays an obligaterole in targeting. Instead, the HybO C-tail is dedicated tothe membrane attachment of the hydrogenase.
Corroborating evidence for the existence of other Tat-dependent membrane proteins comes from the structureof
E. coli
formate dehydrogenase-N (Jormakka
et al
.,2002). The high-resolution three-dimensional crystalstructure suggests that the FdnH subunit of the Tat-dependent
ab
dimer contains a hydrophobic (probablytransmembrane) C-tail (Jormakka
et al
., 2002; Sargent
et al
., 2002).
Addition of the HybO C-tail renders TorAmembrane-bound
Detailed studies of the individual processes involved inassembly of native Tat-dependent hydrogenases and for-mate dehydrogenases are complicated. Careful coordina-tion of cofactor biosynthesis, cofactor insertion, subunitrecruitment and protein targeting is crucial to successfulbiosynthesis of such multisubunit enzymes. Therefore, inorder to study the process of Tat-dependent C-tail integra-tion in isolation it was desirable to develop a simplifiedassay based on Tat-dependent reporter proteins. Genefusions have been extensively exploited as tools to probeSec-dependent membrane protein assembly (e.g. Traxler
et al
., 1993; Drew
et al
., 2002). To adopt a similarapproach for Tat-dependent C-tail studies we initiallychose trimethylamine
N
-oxide (TMAO) reductase (TorA)as a reporter. TorA is a periplasmic, water-soluble, singlesubunit enzyme, and transport is absolutely dependent onan intact twin-arginine signal peptide and an active Tattranslocase (Sargent
et al
., 1998a; Buchanan
et al
.,2001). Importantly, cytoplasmic loading of a single molyb-dopterin cofactor into the monomeric apoenzyme is anessential prerequisite to TorA transport (Santini
et al
.,1998). Thus only fully assembled enzyme is presented forexport and no defaulting to the Sec pathway is observed.
A chromosomal fusion was constructed between the
torA
gene and the final 60 codons of the
hybO
gene(Fig. 2A). Such a gene fusion would result in the synthesisof a chimeric protein with the HybO C-tail located at theextreme C-terminus of TorA. We felt it was important toconstruct the
f
torA
::
hybO
gene fusion at the native
tor
locus to preserve native expression levels and correctstoichiometries with biosynthetic factors. Inspection of thecrystal structures of TorA-family proteins suggests that theC-termini of the native enzymes are surface exposed andthat any additions to this region should not affect assem-bly (e.g. Czjzek
et al
., 1998).The
E. coli
parent strain (DSS401;
tor
+
,
D
dmsABC
), inwhich all TMAO reductase activity is attributable to TorA,and the strain expressing the
f
TorA::HybO chimera(FTF20; as DSS401,
f
torA
::
hybO) were grown anaerobi-cally in the presence of TMAO to induce expression of thetor regulon. As expected, native TorA was located almostexclusively in the periplasm by Western analysis (Fig. 2B).

1380 K. Hatzixanthis, T. Palmer and F. Sargent
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 49, 1377–1390
However, addition of the HybO C-tail to TorA rendered theprotein membrane-bound (Fig. 2B) and enzymatic assaysconfirmed that the majority of correctly assembled activeTorA co-sedimented with the membrane fraction (Fig. 2B).The fTorA::HybO chimera was found to be resistant toextraction by salt, urea or alkali indicating that the fusionprotein was an integral membrane protein (Fig. 2C). Inter-estingly, growth of the fTorA::HybO-expressing strain onglycerol and TMAO as sole carbon and energy sourceswas not significantly affected (data not shown). Finally, totest whether the TorA portion of the fusion protein hadbeen correctly translocated to the periplasm and thusconfirm an Nperi-Ccyto topology for the HybO C-tail weemployed a protease accessibility assay. Native HybO canbe released from the surface of sphaeroplasts by limitedtrypsinolysis (Sargent et al., 1998b) and our fusion con-struct contains the trypsin hyper-sensitive sites. Trypsinol-ysis released TorA activity from the surface of intactsphaeroplasts (Fig. 2D) confirming that the fTorA::HybO
fusion was correctly transported to the periplasm andanchored at the periplasmic face of the cytoplasmicmembrane.
The complete Tat-dependence on export and mem-brane attachment of the native HybOC holo-complex(Bogsch et al., 1998; Sargent et al., 1998a; 1999; Fig. 1C)and the fact that the entire TorA portion of thefTorA::HybO membrane-bound chimera is transportedacross the membrane (Fig. 2) suggests strongly that theHybO TM is inserted at some stage into the Tat channel.Thus the Tat transporter is clearly implicated in the inte-gration of a subset of membrane proteins into the lipidbilayer.
Fusions to the SufI protein identify five Tat-dependent membrane proteins in E. coli
While the chromosomal torA fusion approach (Fig. 2)demonstrated the ability of the E. coli Tat system to handlea hydrophobic transmembrane C-tail fused to a normallywater-soluble reporter protein, clearly a more tractableassay was required for possible high-throughput screen-ing of putative Tat-dependent membrane proteins and forkinetic and mechanistic studies of Tat-dependent C-tailintegration. We therefore turned our attention to the nativeE. coli Tat substrate SufI. SufI is a 50 kDa monomericperiplasmic protein and completely dependent upon theTat system for export (Stanley et al., 2000). SufI shares31% overall amino acid identity and 47% overall similarity
Fig. 2. TorA as a Tat-exclusive reporter protein.A. Cartoon representation of the torCAD locus in strain FTF20 (DdmsABC, ftorA::hybO). The relative locations of the twin-arginine signal peptide encoded by torA (RR-SS) and the position of the DNA encoding the HybO hydrophobic C-tail (HybO C-tail) are highlighted.B. A compound diagram showing (top panel) Western analysis of TorA in strain DSS401 (as MC4100, DdmsABC; parent strain) and FTF20 (as DSS401, ftorA::hybO). Cells were cultured anaerobically in CR medium containing 0.5% (v/v) glycerol, 0.5% (w/v) fumarate and 0.4% (w/v) TMAO and fractionated into periplasm (P), total membranes (M) and cytoplasm (C). Proteins were separated by SDS–PAGE (7.5%w/v acrylamide), blotted and challenged with anti-TorA. Total specific TMAO-dependent BV oxidoreductase activities for each fractionare shown (middle panel) with units as mmol BV oxidized min-1 gcells-1, together with percentage of total cellular activity (bottom panel).C. Total membrane vesicles prepared by sonication of strain FTF20 (DdmsABC, ftorA::hybO) were extracted with 50 mM NaCl, 1 M NaCl, 200 mM Na2CO3 and 4 M urea. Samples were separated into (P) pellet (membrane-bound) and (S) soluble fractions by ultracentrifuga-tion and analysed by immunoblotting using anti-TorA.D. Sphaeroplasts of strain FTF20 (DdmsABC, ftorA::hybO) were pre-pared and incubated with various amounts of trypsin. Reactions were stopped at time points by the addition of excess soybean trypsin inhibitor, sphaeroplasts removed by ultracentifugation and TMAO-dependent BV oxidoreductase activities measured for the remaining supernatants. TMAO reductase activity released after incubation with 0.2% (w/v) trypsin (�), 0.0002% (w/v) trypsin (�) or no protease added (�). Enzyme activities are expressed as relative to total enzyme activity determined after disruption of non-trypsin-treated sphaeroplasts.

Tat-dependent integral membrane proteins 1381
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 49, 1377–1390
with the E. coli copper protein CueO (Roberts et al., 2002)but does not bind metal and is thus an excellent substratefor in vivo and in vitro Tat transport studies (Stanley et al.,2000; Yahr and Wickner, 2001; Alami et al., 2002).
The sufI gene (minus stop codon) was placed down-stream of a T7 polymerase promoter on plasmid pT7.5(Fig. 3A). DNA encoding the HybO C-tail was cloneddownstream of, and in frame with, the plasmid-borne sufIgene. Again, the crystal structure of CueO suggests theC-terminus of this type of protein is accessible (Robertset al., 2002). Western analysis revealed that the additionof the HybO C-tail to SufI rendered the resultantfSufI::HybO chimera membrane-bound (Fig. 3B). More-over, expression of the fsufI::hybO chimera in the tatmutant strain demonstrated that membrane attachmentwas strictly Tat-dependent (Fig. 3C).
The establishment of a plasmid-based system allowedus to explore the structure and function of other putativeC-tails from E. coli Tat substrates. First, we tested theability of the formate dehydrogenase-N FdnH subunitC-tail to anchor SufI to the membrane. Formatedehydrogenase-N has a typical abg structure comprisinga molybdopterin-binding a-subunit (FdnG) complexed witha Fe-S cluster-binding b-subunit partner (FdnH). The abdimer associates with an integral membrane cytochromeb g-subunit (FdnI) on the periplasmic side of the membrane(Jormakka et al., 2002; Sargent et al., 2002). In contrastto the Tat-dependent [NiFe] hydrogenases the twin-argin-ine signal peptide is located on the a-subunit of formatedehydrogenase-N i.e. not on the same polypeptide as thepredicted TM. The final 60 codons of the fdnH gene wereincorporated in frame at the 3¢ end of the sufI gene. Theresultant fSufI::FdnH protein was found to be associatedwith the cell membranes (Fig. 3B) and membrane attach-ment of the fSufI::FdnH fusion protein was strictly Tat-dependent (Fig. 3C) and resistant to extraction by salt,urea and alkali (Fig. 3D). This was a very important resultin establishing the fidelity of our reporter protein assay inidentifying Tat-dependent transmembrane segments asprevious analysis of the native formate dehydrogenasecomplexes had resulted in some ambiguity (Berg et al.,1991; Berks et al., 1995; Benoit et al., 1998; Jormakkaet al., 2002; Sargent et al., 2002; Stanley et al., 2002).
Next, fusions of the last 60 amino acids of the HybOhomologue HyaA, and the FdnH homologue FdoH, weremade to SufI. Expression of the fSufI::C-tail constructsin Tat+ and Tat– strains demonstrated that these two pro-teins also contain Tat-dependent transmembrane C-tails(Fig. 3E). Finally, we tested the ability of the final 60 aminoacids of the HybA protein to anchor our Tat-dependentreporter SufI to the membrane. HybA is part of thehydrogenase-2 complex and is a member of the ‘16Fe’-ferredoxin family but is not closely related to the core[NiFe] hydrogenase or formate dehydrogenase subunits
Fig. 3. SufI as a Tat-exclusive reporter protein.A. Cartoon representation of the sufI gene (minus stop codon) down-stream of the T7 promoter on plasmid pT7.5 (pSuf-NOSTOP). The relative locations of the twin-arginine signal peptide encoded by sufI (RR-SS) and the DNA-encoding hydrophobic C-tail anchors (C-tail) are shown.B. Western analysis of SufI in strain NRS-3 (as MC4100, DsufI; transformed with the helper plasmid (pGP1-2) encoding T7 poly-merase and pNR14 (encoding native sufI) or pSuf-NOSTOP incorpo-rating DNA encoding the C-tails of HybO (fSufI::HybO) or FdnH (fSufI::FdnH). Cells were grown anaerobically in CR medium con-taining 0.4% (w/v) glucose and fractionated into periplasm (P), total membranes (M) and cytoplasm (C). Proteins were separated initially by SDS–PAGE (10% w/v acrylamide), blotted and challenged with an anti-SufI serum.C. Western analysis of total membranes isolated from NRS-3 (DsufI) and NRS-3C (DsufI, DtatC) strains expressing fSufI::HybO and fSufI::FdnH probed with anti-SufI serum.D. Total membrane vesicles prepared by sonication of strain NRS-3 (DsufI) expressing fSufI::FdnH were extracted with 50 mM NaCl, 1 M NaCl, 200 mM Na2CO3 and 4 M urea. Samples were separated into (P) pellet (membrane-bound) and (S) soluble fractions by ultracentrif-ugation and analysed by immunoblotting using anti-SufI.E. Western analysis of total membranes isolated from NRS-3 (DsufI) and NRS-3C (DsufI, DtatC) strains expressing fSufI::FdoH, fSufI::HyaA and fSufI::HybA probed with anti-SufI serum.

1382 K. Hatzixanthis, T. Palmer and F. Sargent
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 49, 1377–1390
(Sargent et al., 1998b). The fSufI::HybA chimera was alsoefficiently integrated into the membrane in a completelyTat-dependent manner (Fig. 3E). Note that these experi-ments were performed under conditions where synthesisof native Tat-dependent hydrogenases and formate dehy-drogenases is repressed (Gennis and Stewart, 1996).Note also that neither fSufI::C-tail construct hindered Tat-dependent anaerobic respiration on TMAO (data notshown) suggesting the chimeras are rapidly and efficientlyincorporated into the bilayer and do not block the Tattransporter.
Taken together, these data suggest that the E. coli Tatsystem is capable of integrating at least five differenthydrophobic C-tails of varying amino acid composition intothe membrane. Interestingly, the average hydrophobicityindices (AHI) for E. coli Tat-dependent C-tails (Fig. 1A)show no significant differences from those of SRP/Sec-targeted proteins. For example, the E. coli TatC proteincontains at least four TMs (Gouffi et al., 2002) and the AHIfor helices I to IV are 2.9, 1.9, 2.1 and 1.5 respectively.Thus beyond TatC Helix I, which would be expected to berecognized directly by SRP and hence demonstrate a‘higher’ average hydrophobicity, the Sec-dependent TMsshow no obvious differences from Tat-dependent TMs. Tat-dependent C-tail-anchored proteins of the [NiFe] hydroge-nase and formate dehydrogenase families are very con-served in other bacterial systems (Sargent et al., 2002).Moreover, there are also indications that both bacterialand plant Rieske Fe-S proteins are targeted to the mem-brane by bona fide twin-arginine signal peptides thatremain uncleaved and act as N-terminal signal anchorsafter transport (Berks, 1996; Molik et al., 2001). Thus han-dling of transmembrane segments is likely to be a univer-sal capability of the Tat translocase.
Membrane targeting and attachment properties of the FdnH transmembrane C-tail
The demonstration that hydrophobic Tat-dependent C-tails direct the cofactor-less reporter protein SufI to themembrane (Fig. 3) facilitates studies of Tat-dependent C-tail targeting and integration mechanisms uncomplicatedby cofactor loading into associated periplasmic domains.We therefore next established an in vivo transport assaybased on our construct expressing the sufI gene fused toDNA encoding the crystallographically defined FdnH C-tail. The fsufI::fdnH fusion was expressed together with ahelper plasmid encoding an inducible T7 RNA polymeraseand this system allowed the specific radio-labelling of thefSufI::FdnH chimera. Labelling of the fSufI::FdnH fusionwith [35S]-methionine followed by SDS–PAGE and autora-diography identified two protein species corresponding toprecursor (containing Tat signal) and mature (signal-processed) forms (Fig. 4A). Processing of the Tat signal
peptide on the fSufI::FdnH chimera was found to be com-pletely dependent upon an active Tat translocase(Fig. 4A). Furthermore, the full-length precursor form offSufI::FdnH visible in the tat mutant is stable in a pulse-chase experiment: after a 5 minute labelling reactionexcess non-radioactive methionine was added andsamples taken at various time-points. The full-lengthfSufI::FdnH precursor was present throughout the time-scale of the experiment (Fig. 4C and E) thus we concludethat processing of the fSufI::FdnH chimera is, like thenative SufI precursor (Fig. 4A and E; Stanley et al., 2000),completely dependent upon translocation to the periplasmby the Tat system and subsequent exposure to signalpeptidase in that compartment. This demonstrated stabil-ity of the non-exported fSufI::FdnH chimera enabled usto confidently use signal peptide processing as a goodindication of successful protein transport.
In a Tat+ strain the fSufI::FdnH precursor was found tobe processed to the mature form over time with no obvi-ous degradation of the fusion protein during the time-course of this experiment (Fig. 4B). Interestingly, the rateof export-linked precursor processing was found to bevery similar to that observed for native SufI under identicalconditions (Fig. 4E). Thus addition of a hydrophobic C-tailto a normally water-soluble Tat substrate does not changetransport kinetics of the protein and clearly does nothinder the normal operation of the Tat system.
The FdnH C-tail comprises a 21 residue hydrophobica-helix stretching from L-260 to G-280 inclusive (Fig. 5A).At the C-terminal side of the hydrophobic helix is a small,apparently unstructured, acidic cytoplasmic domain of 14amino acid residues. Positively charged residues are iden-tifiable in the cytoplasmic domain close to the hydrophobicC-tail possibly indicative of an adherence to the ‘positive-inside’ rule for membrane protein topology (von Heijne,1992). In order to test the role of the charged cytoplasmicdomain in FdnH C-tail integration we undertook a system-atic deletion analysis. The C-tail was truncated by oneamino acid at a time from E-294 to F-265 resultingin a total of 30 fSufI::FdnH mutants [fSufI::FdnH(-1) tofSufI::FdnH(-30)]. The ability of each mutant C-tail toanchor SufI to the membrane was then assessed by West-ern immunoblotting. Complete deletion of the entire cyto-plasmic domain (chimera fSufI::FdnH(-14)) did not hampermembrane attachment of the fusion protein (Fig. 5B). Thisindicates that the cytoplasmic domain is not itself actingas a trailing ‘plug’ preventing complete transport of thehydrophobic C-tail through the channel rather the TM itselfis recognized and integrated by the transport system.Continued truncation analysis of the FdnH C-tail showsthat membrane attachment becomes erratic betweenfSufI::FdnH(-18) and fSufI::FdnH(-21) (Fig. 5B). The short-est fSufI::FdnH fusion which remains membrane-boundis fSufI::FdnH(-21) which would contain a hydrophobic C-

Tat-dependent integral membrane proteins 1383
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 49, 1377–1390
tail of 14 amino acids (Fig. 5A) and the fSufI::FdnH(-21)
chimera remains resistant to chemical extraction and sois genuinely integral (Fig. 5D). Moreover, the fusion pro-teins containing the longest hydrophobic C-tails that donot integrate into the membrane [fSufI::FdnH(-18),fSufI::FdnH(-20) and fSufI::FdnH(-22)] are demonstrated tobe not hindered in their passage through the Tat translo-con and accumulate as soluble proteins in the periplasm(Fig. 5C). Thus the lack of membrane integration shownby these mutants is clearly not as a result of unsuccessfulprotein targeting.
Early sequence analysis compared Tat-dependent C-tails with some types of signal peptides (Wu and Man-drand, 1993). In order to test directly the intrinsic targetingactivity of the FdnH hydrophobic C-tail we modified ourreporter system. A fusion protein in which the SufI twin-
arginine signal peptide had been inactivated by the sub-stitution of the arginine pair by lysines was constructedand the fKKSufI::FdnH chimera subjected to pulse-chaseanalysis. No processing of the protein was evident(Fig. 6A) strongly suggesting protein transport had beencompletely blocked. Indeed, subsequent Western immu-noblotting showed that none of the fKKSufI::FdnH antigencould be located either in the membrane or periplasmicfractions (Fig. 6B). Therefore in our system the N-terminaltwin-arginine signal peptide is key to targeting and inte-gration and the C-terminal hydrophobic C-tail plays noobligate targeting role.
Interestingly, targeting of a plant thylakoidal integralmembrane protein (Pftf) was recently found to be Tat-dependent (Summer et al., 2000). The Pftf protein has atopology completely analogous to that of bacterial Tat-dependent membrane proteins (Fig. 1B) i.e. a lumenal N-terminal domain anchored to the thylakoid membrane bya single TM with the C-terminus in the stroma (Summeret al., 2000). However, mutagenesis of the Pftf Tat signalpeptide did not severely impair Tat-targeting or membraneintegration of the Pftf protein in vitro thus implicating thetransmembrane segment as a novel targeting element. Incontrast, we have found no evidence for targeting activityassociated with bacterial Tat-dependent C-tails. This maybe as a consequence of our experimental design; how-ever, note that Tat transport of the native C-tail anchoredW. succinogenes [NiFe] hydrogenase was completelyblocked when the conserved arginine pair in the Tat signalpeptide were replaced by glutamines pointing to no role
Fig. 4. Export-linked processing kinetics of fSufI::FdnH.A. PreSufI and fpreSufI::FdnH were expressed in strains NRS-3 (as MC4100, DsufI) ‘Tat +’ and NRS-3C (as NRS-3, DtatC) ‘Tat –’ with pGP1-2. Cultures were pulse-labelled for 5 min by the addition of [35S]-methionine before precursor synthesis and processing were assessed by SDS–PAGE of whole cells and autoradiography.B. fpreSufI::FdnH was pulse-labelled with [35S]-methionine and then chased from time 0 with non-radioactive methionine. Precursor syn-thesis and processing were assessed by SDS–PAGE and autorad-iography. Control lanes are pulse-labelled fSufI::FdnH from a tat mutant (C1), to align full-length precursor and pulse-labelled preSufI from a Tat+ strain (C2) to help locate any tail-less degradation products.C. Pulse-chase analysis of fSufI::FdnH in a tat mutant (NRS-3C; DsufI, DtatC). fpreSufI::FdnH was pulse-labelled with [35S]-methion-ine and then chased with non-radioactive methionine. Control lane is pulse-labelled fSufI::FdnH from a Tat+ strain (C).D. Pulse-chase of preSufI in a Tat+ strain (NRS-3; DsufI). PreSufI was pulse-labelled and then chased with unlabelled methionine. The con-trol lane is pulse-labelled preSufI from a tat mutant (C).E. Pulse-chase analysis of preSufI and fpreSufI::FdnH in Tat+ and Tat– backgrounds was performed. Precursor synthesis and process-ing were assessed by SDS–PAGE followed by analysis on a phos-phorimager. Following electronic quantification of labelled proteins, the mean percentage of total precursor remaining in whole cells at each indicated time-point was plotted (n = 3). The bars indicate the standard error of the mean. PreSufI processing in a Tat+ (�) and Tat– (▲) background. fpreSufI::FdnH processing in a Tat+ (�) and Tat– (¥) background.

1384 K. Hatzixanthis, T. Palmer and F. Sargent
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 49, 1377–1390
for the hydrophobic C-tail in the targeting of this enzyme(Gross et al., 1999).
The role of YidC in Tat-dependent membraneprotein integration
A possible mechanism for the assembly of Tat-dependentmembrane proteins by the bacterial twin-arginine translo-case would involve the hydrophobic C-tails acting as ‘stop-transfer’ domains in which the hydrophobicity of the C-terminal a-helix would cause a stalling of the transportprocess. The TM would then escape laterally into the lipidbilayer and a stop-transfer model would thus require achannel-clearing mechanism. During TM integration bythe Sec system the YidC protein facilitates clearing of thetransport channel (Chen et al., 2002). It is noteworthy,however, that YidC can also operate completely indepen-dently of Sec and assemble some membrane proteinsdirectly into the membrane (Samuelson et al., 2000).Given that YidC is not a partisan Sec component it wasimportant to assess the role of YidC in targeting of Tat-dependent integral membrane proteins.
We constructed a strain in which the yidC gene wasunder the transcriptional control of the AraC protein. Ourstrain (FTL10) shared identical characteristics to the yidC
Fig. 5. Functional analysis of the FdnH C-tail.A. Amino acid sequence of the C-terminus of the E. coli FdnH protein. Residue numbers (from the extreme N-terminus) are given above the sequence and the hydrophobic helix is shaded. For truncation analysis reference points from the extreme C-terminus (-10, -20 and -30) are given below the sequence.B. Total membrane vesicles from strain NRS-3 (DsufI) expressing fusion constructs fSufI::FdnH(-1) to fSufI::FdnH(-30) were prepared and analysed by immunoblotting using an anti-SufI serum.C. Western analysis of fSufI::FdnH(-18), fSufI::FdnH(-20) and fSufI::FdnH(-22) in strain NRS-3 (as MC4100, DsufI). Cells were grown anaerobically in CR medium containing 0.4% (w/v) glucose and fractionated into periplasm (P), total membranes (M) and cytoplasm (C). Proteins were separated initially by SDS–PAGE (10% w/v acrylamide), electroblotted and challenged with an anti-SufI serum.D. Total membrane vesicles prepared by sonication of strain NRS-3 (DsufI) expressing fSufI::FdnH(-17) and fSufI::FdnH(-21) were extracted with 50 mM NaCl, 1 M NaCl, 200 mM Na2CO3 and 4 M urea. Samples were separated into (P) pellet (membrane-bound) and (S) soluble fractions by ultracentrifugation and analysed by immunoblotting using an anti-SufI serum.
Fig. 6. No targeting role for E. coli Tat-dependent C-tails.A. The mutant fKKSufI::FdnH fusion in which the essential arginine pair in the signal peptide had been substituted by lysines was pulse-labelled with [35S]-methionine for 5 min and then chased from time 0 with non-radioactive methionine. Precursor synthesis and processing were assessed by SDS–PAGE and autoradiography. The control lane is pulse-labelled fSufI::FdnH from a tat mutant (C).B. Western analysis of SufI in strain NRS-3 (DsufI) transformed with the helper plasmid (pGP1-2) encoding T7 polymerase and a plasmid encoding fKKSufI::FdnH. Cells were grown anaerobically in CR medium containing 0.4% (w/v) glucose and fractionated into peri-plasm (P), total membranes (M) and cytoplasm (C). Proteins were separated initially by SDS–PAGE (10% w/v acrylamide), electroblot-ted and challenged with an anti-SufI serum. Probable degraded fusion protein (fSufI¢) is indicated to the side of the blot.
Tat-dependent integral membrane proteins 1385
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 49, 1377–1390
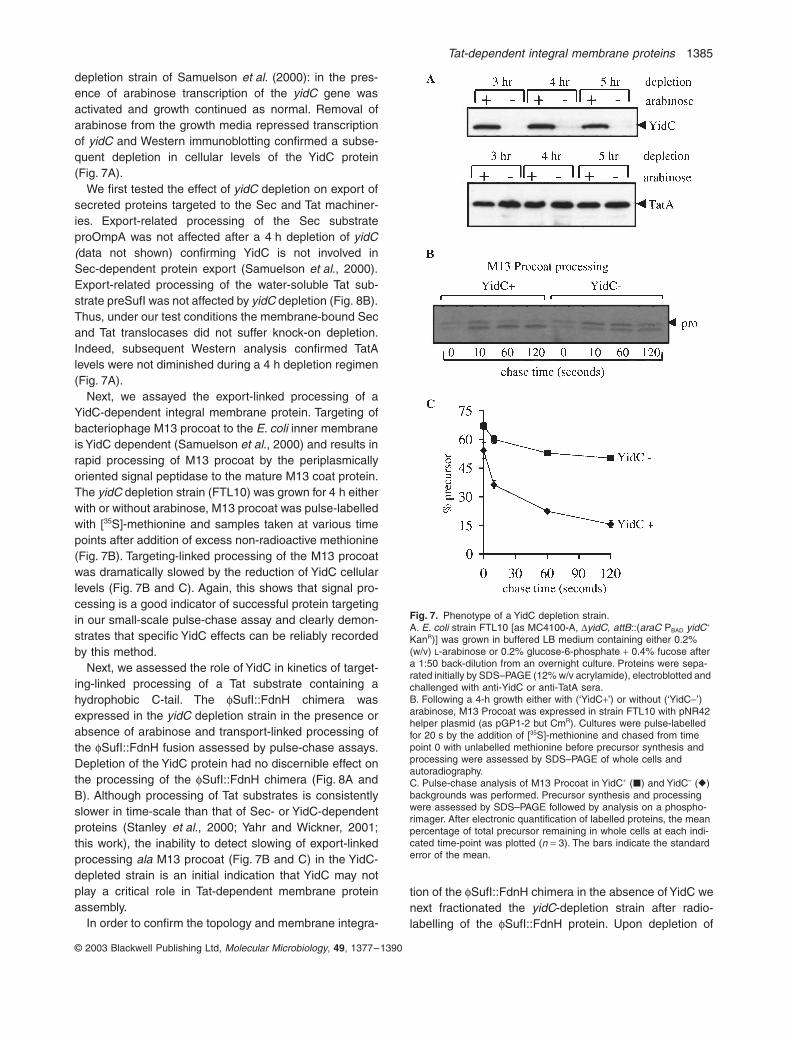
depletion strain of Samuelson et al. (2000): in the pres-ence of arabinose transcription of the yidC gene wasactivated and growth continued as normal. Removal ofarabinose from the growth media repressed transcriptionof yidC and Western immunoblotting confirmed a subse-quent depletion in cellular levels of the YidC protein(Fig. 7A).
We first tested the effect of yidC depletion on export ofsecreted proteins targeted to the Sec and Tat machiner-ies. Export-related processing of the Sec substrateproOmpA was not affected after a 4 h depletion of yidC(data not shown) confirming YidC is not involved inSec-dependent protein export (Samuelson et al., 2000).Export-related processing of the water-soluble Tat sub-strate preSufI was not affected by yidC depletion (Fig. 8B).Thus, under our test conditions the membrane-bound Secand Tat translocases did not suffer knock-on depletion.Indeed, subsequent Western analysis confirmed TatAlevels were not diminished during a 4 h depletion regimen(Fig. 7A).
Next, we assayed the export-linked processing of aYidC-dependent integral membrane protein. Targeting ofbacteriophage M13 procoat to the E. coli inner membraneis YidC dependent (Samuelson et al., 2000) and results inrapid processing of M13 procoat by the periplasmicallyoriented signal peptidase to the mature M13 coat protein.The yidC depletion strain (FTL10) was grown for 4 h eitherwith or without arabinose, M13 procoat was pulse-labelledwith [35S]-methionine and samples taken at various timepoints after addition of excess non-radioactive methionine(Fig. 7B). Targeting-linked processing of the M13 procoatwas dramatically slowed by the reduction of YidC cellularlevels (Fig. 7B and C). Again, this shows that signal pro-cessing is a good indicator of successful protein targetingin our small-scale pulse-chase assay and clearly demon-strates that specific YidC effects can be reliably recordedby this method.
Next, we assessed the role of YidC in kinetics of target-ing-linked processing of a Tat substrate containing ahydrophobic C-tail. The fSufI::FdnH chimera wasexpressed in the yidC depletion strain in the presence orabsence of arabinose and transport-linked processing ofthe fSufI::FdnH fusion assessed by pulse-chase assays.Depletion of the YidC protein had no discernible effect onthe processing of the fSufI::FdnH chimera (Fig. 8A andB). Although processing of Tat substrates is consistentlyslower in time-scale than that of Sec- or YidC-dependentproteins (Stanley et al., 2000; Yahr and Wickner, 2001;this work), the inability to detect slowing of export-linkedprocessing ala M13 procoat (Fig. 7B and C) in the YidC-depleted strain is an initial indication that YidC may notplay a critical role in Tat-dependent membrane proteinassembly.
In order to confirm the topology and membrane integra-
tion of the fSufI::FdnH chimera in the absence of YidC wenext fractionated the yidC-depletion strain after radio-labelling of the fSufI::FdnH protein. Upon depletion of
Fig. 7. Phenotype of a YidC depletion strain.A. E. coli strain FTL10 [as MC4100-A, DyidC, attB::(araC PBAD yidC+ KanR)] was grown in buffered LB medium containing either 0.2%(w/v) L-arabinose or 0.2% glucose-6-phosphate + 0.4% fucose after a 1:50 back-dilution from an overnight culture. Proteins were sepa-rated initially by SDS–PAGE (12% w/v acrylamide), electroblotted and challenged with anti-YidC or anti-TatA sera.B. Following a 4-h growth either with (‘YidC+’) or without (‘YidC–’) arabinose, M13 Procoat was expressed in strain FTL10 with pNR42 helper plasmid (as pGP1-2 but CmR). Cultures were pulse-labelled for 20 s by the addition of [35S]-methionine and chased from timepoint 0 with unlabelled methionine before precursor synthesis and processing were assessed by SDS–PAGE of whole cells andautoradiography.C. Pulse-chase analysis of M13 Procoat in YidC+ (�) and YidC– (�) backgrounds was performed. Precursor synthesis and processing were assessed by SDS–PAGE followed by analysis on a phospho-rimager. After electronic quantification of labelled proteins, the mean percentage of total precursor remaining in whole cells at each indi-cated time-point was plotted (n = 3). The bars indicate the standard error of the mean.

1386 K. Hatzixanthis, T. Palmer and F. Sargent
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 49, 1377–1390
YidC, no soluble periplasmic form of the fSufI::FdnH pro-tein was evident (Fig. 8C). When compared with mem-branes from a YidC+ strain, protease treatment of isolatedmembranes from the YidC– strain indicated that thefSufI::FdnH chimera was correctly transported (Fig. 8D).Moreover, a chemical wash of isolated membrane vesiclescould not extract the radio-labelled fSufI::FdnH fusionprotein indicating that the protein was correctly localizedin the lipid bilayer in the absence of YidC (Fig. 8D). Takentogether, we conclude from these experiments that YidCprobably plays no role in the integration of Tat-dependentC-tails.
Concluding remarks
From the data presented here it is now apparent that theTat system does not just transport water-soluble proteinsbut is also involved in the post-translational biogenesis ofcertain integral membrane proteins. The identification ofTat-dependent TMs is an exciting new development thatraises fundamental questions about membrane protein
assembly. For example, it is difficult to imagine a mecha-nism of Tat-dependent membrane protein assembly inwhich the Tat-targeted TM does not enter the Tat transportchannel at some point. Thus, given the key role of the Sec-translocase in orienting TMs with respect to the positive-inside rule (Prinz et al., 1998), how does the Tat translo-case correctly orient TMs before membrane integration?Moreover, identification of a subset of E. coli inner mem-brane proteins assembled independently of the YidCprotein is unprecedented. If Tat-dependent C-tails areassembled by a ‘stop-transfer’ mechanism then our datamust point to the existence of a method of channelclearing and TM integration fundamentally differentfrom currently characterized systems. However, the Tattranslocase may not in fact operate a ‘stop-transfer’ sys-tem and it should be alternatively considered that peri-plasmic intermediates may play a role in Tat-dependentC-tail assembly. In such a ‘periplasmic re-entry’ model theentire protein, including TM, would be exported to theperiplasm and then re-associate with the membrane fromthe periplasmic side. Such a mechanism is used by someSec-dependent peripheral membrane proteins (e.g. Jack-son and Pratt, 1987). However a truly transmembrane
Fig. 8. Targeting of Tat-dependent C-tails is YidC independent.A. Following a 4-h growth either with (‘YidC+’) or without (‘YidC–’) arabinose, fSufI::FdnH was expressed in strain FTL10 with pNR42 helper plasmid (encoding T7 polymerase). Cultures were pulse-labelled for 20 s by the addition of [35S]-methionine and chased from time point 0 with unlabelled methionine. Precursor synthesis and processing were assessed by SDS–PAGE of whole cells andautoradiography.B. Pulse-chase analysis of PreSufI and fSufI::FdnH in YidC+ and YidC– backgrounds was performed. Precursor synthesis and process-ing were assessed by SDS–PAGE followed by analysis on a phos-phorimager. After electronic quantification of labelled proteins, the mean percentage of total precursor remaining in whole cells at each indicated time-point was plotted (n = 3). The bars indicate the stan-dard error of the mean. PreSufI expressed in a YidC+ (�) and a YidC– (�) background. fSufI::FdnH expressed in a YidC+ (▲) and a YidC– (¥) background.C. PreSufI and fpreSufI::FdnH were expressed in strain FTL10 with pNR42 helper plasmid (encoding T7 polymerase) either with (‘YidC+’) or without (‘YidC–’) arabinose. Cultures were pulse-labelled for 5 min by the addition of [35S]-methionine (no chase step) before cells were harvested, fractionated and assessed by SDS–PAGE and autorad-iography. Samples analysed were whole cells (WC), periplasmic frac-tion (P) and sphaeroplasts (SP). The position of mature fSufI::FdnH is indicated.D. fpreSufI::FdnH was expressed in strain FTL10 with pNR42 helper plasmid (encoding T7 polymerase) either with (‘YidC+’) or without (‘YidC–’) arabinose. Cultures were pulse-labelled for 5 min by the addition of [35S]-methionine before being chased with excess unla-belled methionine for 45 min to ensure complete processing of the Tat substrate. Cells were then harvested, fractionated and assessed by SDS–PAGE and autoradiography. Samples analysed were whole cells (WC), right-side out membrane vesicles (M), membrane vesicles treated with 0.1 mg ml-1 Proteinase K (PK) and membrane pellet after a wash in 200 mM sodium carbonate (W). The position of mature fSufI::FdnH is indicated and the control lane (C) is whole cells con-taining both radio-labelled fpreSufI::FdnH precursor and mature fSufI::FdnH.

Tat-dependent integral membrane proteins 1387
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 49, 1377–1390
inner membrane protein has never been shown to beintegrated in such a way and note that we have no indi-cation from our current experiments for the existence ofperiplasmic intermediates during Tat-dependent C-tailintegration. This in vivo study of Tat-dependent TMs pavesthe way for detailed analysis of the novel integration mech-anism at the molecular level.
Experimental procedures
Bacterial strains
E. coli strain MC4100 (F-, DlacU169, araD139, rpsL150,relA1, ptsF, rbs, flbB5301) was the parental strain used forall experiments. Strains carrying deletions in hybO wereFTD674 (as MC4100, hybO DC-tail) and BØD674 (asFTD674, DtatB). PCR-based methods were used to assem-ble an in frame deletion allele covering codons 328–355 ofhybO on plasmid pMAK705 (Hamilton et al., 1989). The dele-tion allele was transferred to the chromosome of strainsMC4100 and BØD (as MC4100, DtatB; Sargent et al., 1999)by homologous recombination as described (Hamilton et al.,1989).
The chromosomal torA fusion was constructed as follows:an ~500 bp DNA fragment covering the 3¢ end of the torAgene (minus stop codon) was amplified by PCR and clonedinto plasmid pFAT210 (Dubini et al., 2002) to give plasmidpFRF8. Next, an ~500 bp DNA fragment covering the 5¢region of torD (including Shine–Dalgarno possible se-quences) was cloned into pFRF8 to give pFRF9 which con-tains a torA-his allele plus upstream and downstream flankingsequences. The histag sequence was replaced by a DNAfragment covering the final 60 codons of hybO to give plasmidpFRF11. The resultant ftorA::hybO allele on pFRF11 wastransferred to pMAK705 and then to the chromosome ofDSS401 (as MC4100, DdmsABC, KanR); Sambasivarao andWeiner, 1991) as described (Hamilton et al., 1989) to yieldstrain FTF20 (as DSS401, ftorA::hybO).
Pulse-chase and steady-state experiments with plasmidsencoding fSufI::C-tail chimeras were carried out with NRS-3(as MC4100, DsufI; Stanley et al., 2001) and NRS-3C (asNRS-3, DtatC; Buchanan et al., 2002).
Our yidC depletion strain [FTL10; as MC4100-A, DyidC,attB::(araC+, PBAD, yidC+) KanR] which contains a single copyof the yidC gene under control of the arabinose promoter wasconstructed in an arabinose-resistant isolate of MC4100termed MC4100-A (Ize et al., 2002). First, the entire yidCgene (including upstream Shine–Dalgarno elements anddownstream transcriptional termination motifs) was amplifiedby PCR and cloned downstream of the arabinose-regulatedpromoter on plasmid pBAD18 (Guzman et al., 1995). Next,the yidC gene, arabinose promoter region and araC gene onpBAD18 were amplified by PCR and cloned into the vectorpRS552 (Simons et al., 1987). The pRS552-based constructwas then used to transfer the entire arabinose-regulated yidCallele to the lamba attachment site (attB) on the chromosomeof E. coli host strain MC1061 (Simons et al., 1987). TheattB::(araC+, PBAD, yidC+) allele (which was now 100% linkedto a kanamycin resistance marker) was then moved by P1transduction to the chromosome of MC4100-A. Finally, a
DyidC allele containing the first two and final two codons ofyidC was constructed by PCR-based methods on plasmidpMAK705. In media containing 0.2% (w/v) L-arabinose and0.5% (v/v) glycerol to ensure expression of yidC from thearabinose-regulated promoter at the attB site, the native yidCgene was then deleted according to the method of Hamiltonet al. (1989) to yield strain FTL10.
Plasmids
For experiments with fSufI::C-tail chimeras, the sufI gene(minus stop codon) was amplified by PCR and cloned asan EcoRI/BamHI fragment into pT7.5 (Tabor and Richard-son, 1985) to give plasmid pSuf-NOSTOP. DNA encodingthe final 60 codons of hybO, fdnH, hybA, hyaA and fdoHwere amplified by PCR and cloned in frame as BamHI/XbaIfragments downstream of sufI on pSuf-NOSTOP. The KK-sufI gene (minus stop codon), in which codons for the con-served arginine pair in the signal peptide had been substi-tuted by lysine codons, was amplified by PCR using pNR68(Stanley et al., 2000) as template and cloned as an EcoRI/BamHI fragment into pT7.5. The final 60 codons of fdnHwere amplified by PCR and cloned in frame downstreamof the mutant sufI gene to give a plasmid encodingfKKSufI::FdnH.
Control experiments involved pT7-derivatives expressingM13 procoat and proOmpA (kindly provided by J.-W. de Gier,Stockholm). The T7 RNA polymerase was coexpressed intrans from plasmids pGP1-2 (KanR) and pNR42 (CmR) (Stan-ley et al., 2000).
Bacterial growth and processing
During genetic manipulations strains were grown aerobicallyin Luria–Bertani (LB) medium (Russell and Sambrook,2001). For biochemical characterizations strains were cul-tured anaerobically or aerobically in Cohen–Rickenberg(CR) medium containing combinations of 0.4% (w/v) glu-cose, 0.5% (v/v) glycerol, and 0.4% (w/v) fumarate and0.4% (w/v) TMAO where indicated (Sargent et al., 1998a).Subcellular fractionation of large unlabelled cultures wasperformed by the lysozyme/EDTA protocol with ultimate celllysis by French Press to generate inside-out membrane ves-icles or sonication to form right-side-out membrane vesicles(Sambasivarao et al., 1990; Stanley et al., 2000). For small-scale fractionation of radio-labelled cultures cells were har-vested by centrifugation and washed twice in 1 ml ice-cold20 mM Tris-HCl (pH 7.6). For sphaeroplast formation cellpellets were resuspended in 350 ml 20% (w/v) sucrose and20 mM Tris-HCl (pH 7.6). EDTA was added to 5 mM (finalconcentration) together with lysozyme (0.6 mg ml-1 final con-centration) and the mixture incubated at 30∞C for 20 min.Sphaeroplasts were harvested by centrifugation and thesupernatant retained as the periplasmic fraction. For gener-ation of right-side-out membrane vesicles by sonication (e.g.Sambasivarao et al., 1990) cell pellets were resuspended in20 mM Tris-HCl (pH 7.6), 5 mM EDTA, 0.6 mg ml-1 lyso-zyme and sonicated on ice. Unbroken cells were removedby centrifugation and membrane vesicles harvested byultracentrifugation.

1388 K. Hatzixanthis, T. Palmer and F. Sargent
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 49, 1377–1390
SDS–PAGE and immunoblotting analyses were carried outaccording to the methods of Lämmli (1970) and Towbin et al.(1979). Immuno-reactive bands were detected by ECL (APBiotech). Hydrogen:benzyl viologen and TMAO:benzyl violo-gen oxidoreductase activities were measured as previouslypublished and sphaeroplast formation/tryptic digest protocolsfor unlabelled cells have also been described elsewhere (Sil-vestro et al., 1988; Sargent et al., 1998a, b). Protein concen-trations were estimated according to the method of Lowryet al. (1951).
Pulse-chase experiments
Pulse-chase experiments were carried out in aerobicallygrown whole cells essentially as described by Stanley et al.(2000). Quantification of bands was carried out with a FujiBAS-1000 phosphorimager and the software packageMacBAS version 2.0.
YidC-depletion strain FTL10 was treated as follows: anovernight culture of FTL10 transformed with appropriate plas-mids was back-diluted 1:50 and grown for 4 h in phosphate-buffered LB medium (pH 7.5) at 30∞C either in the presenceof 0.2% (w/v) L-arabinose or 0.2% (w/v) glucose-6-phosphate+0.4% (w/v) fucose (Guzman et al., 1995). Whole cell sam-ples for SDS–PAGE were collected by centrifugation and re-suspended directly in Lämmli disaggregation buffer. For pulsechase analysis cultures were washed and resuspended inM9 medium containing the appropriate carbon sources andamino acids, treated with rifampicin for 5 min, pulse-labelledfor 20 s by the addition of [35S]-methionine, and chased with0.2% (w/v) unlabelled methionine before samples withdrawnand snap-frozen in liquid nitrogen.
Labelling of small-scale cultures for subsequent fraction-ation was carried out as follows: an overnight culture wasback-diluted 1:50 in 5 ml of LB medium plus any appropriateadditives. After 3 h aerobic growth at 30∞C the entire culturewas then pelleted by centrifugation, washed twice in 10 ml ofice-cold M9 medium, and resuspended in 5 ml of M9 mediumcontaining all appropriate additives plus all amino acids(minus methionine and cysteine) each at 0.01% (w/v) finalconcentration. Cultures were returned to 30∞C for 30 minbefore heat-shock at 42∞C for 5 min to induce synthesis ofT7 polymerase. Rifampicin was added (200 mg ml-1 final con-centration) to block E. coli RNA polymerase and cultureswere maintained at 42∞C for a further 10 min before beingreturned to 30∞C for 20 min. T7-dependent gene productswere then labelled by the addition of [35S]-methionine for5 min. If required, unlabelled methionine was added to a finalconcentration of 0.075% (w/v).
Acknowledgements
We thank Dr Ben C. Berks (Oxford, UK) for critical readingof the manuscript, Professor Matthias Müller (Freiburg, Ger-many) for anti-YidC serum, Dr Jan-Willem de Gier (Stock-holm, Sweden) for plasmids encoding M13 procoat andproOmpA, and Mr Phil A. Lee (Norwich, UK) for technicaladvice. This work was funded by BBSRC research grant 83/P15018 (to F.S.). F.S. and T.P. are Royal Society UniversityResearch Fellows.
References
Alami, M., Trescher, D., Wu, L.-F., and Müller, M. (2002)Separate analysis of Tat-specific membrane- binding andtranslocation in Escherichia coli. J Biol Chem 277: 20499–20503.
Benoit, S., Abaibou, H., and Mandrand-Berthelot, M.-A.(1998) Topological analysis of the aerobic membrane-bound formate dehydrogenase of Escherichia coli. J Bac-teriol 180: 6625–6634.
Berg, B.L., Li, J., Heider, J., and Stewart, V. (1991) Nitrate-inducible formate dehydrogenase in Escherichia coli K-12.I. Nucleotide sequence of the fdnGHI operon and evidencethat opal (UGA) endcodes selenocysteine. J Biol Chem266: 22380–22385.
Berks, B.C. (1996) A common export pathway for proteinsbinding complex redox cofactors? Mol Microbiol 22: 393–404.
Berks, B.C., Page, M.D., Reilly, A., Richardson, D.J., Outen,F., and Ferguson, S.J. (1995) Sequence analysis of sub-units of the membrane-bound nitrate reductase from adenitrifying bacterium: the integral membrane subunit pro-vides a prototype for the dihaem electron carrying arm ofa redox loop. Mol Microbiol 15: 319–331.
Berks, B.C., Sargent, F., and Palmer, T. (2000) The Tatprotein export pathway. Mol Microbiol 35: 260–274.
Bernhard, M., Benelli, B., Hochkoeppler, A., Zannoni, D., andFriedrich, B. (1997) Functional and structural role of thecytochrome b subunit of the membrane-bound hydroge-nase complex of Alcaligenes eutrophus H16. Eur J Bio-chem 248: 179–186.
Bogsch, E.G., Sargent, F., Stanley, N.R., Berks, B.C., Rob-inson, C., and Palmer, T. (1998) An essential componentof a novel bacterial protein export system with homologuesin plastids and mitochondria. J Biol Chem 273: 18003–18006.
Bolhuis, A., Mathers, J.E., Thomas, J.D., Barrett, C.M., andRobinson, C. (2001) TatB and TatC form a functional andstructural unit of the twin-arginine translocase from Escher-ichia coli. J Biol Chem 276: 20213–20219.
Buchanan, G., Sargent, F., Berks, B.C., and Palmer, T.(2001) A genetic screen for suppressors of Escherichia coliTat signal peptide mutations establishes a critical role forthe second arginine within the twin-arginine motif. ArchMicrobiol 177: 107–112.
Buchanan, G., de Leeuw, E., Stanley, N.R., Wexler, M.,Berks, B.C., Sargent, F., and Palmer, T. (2002) Functionalcomplexity of the twin-arginine translocase TatC compo-nent revealed by site-directed mutagenesis. Mol Microbiol43: 1457–1470.
Chen, M., Xie, K., Jiang, F., Yi, L., and Dalbey, R.E. (2002)YidC, a newly defined evolutionarily conserved protein,mediates membrane protein assembly in bacteria. BiolChem 383: 1565–1572.
Czjzek, M., Dos Santos, J.P., Pommier, J., Giordano, G.,Mejean, V., and Haser, R. (1998) Crystal structure ofoxidized trimethylamine N-oxide reductase fromShewanella massilia at 2.5Å resolution. J Mol Biol 284:435–447.
Drew, D., Sjöstrand, D., Nilsson, J., Urbig, T., Chin, C.N., deGier, J.W., and von Heijne, G. (2002) Rapid topology map-

Tat-dependent integral membrane proteins 1389
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 49, 1377–1390
ping of Escherichia coli inner-membrane proteins by pre-diction and PhoA/GFP fusion analysis. Proc Natl Acad SciUSA 99: 2690–2695.
Dubini, A., Pye, R.L., Jack, R.L., Palmer, T., and Sargent, F.(2002) How bacteria get energy from hydrogen: a geneticanalysis of periplasmic hydrogen oxidation in Escherichiacoli. Int J Hydrogen Energy 27: 1413–1420.
Gennis, R.B., and Stewart, V. (1996) Respiration. In Escher-ichia coli and Salmonella: Cellular and Molecular Biology,2nd edn. Neidhardt, F.C., Curtiss, R., Ingraham, J.L., Lin,E.C.C., Low, K.B., Magasanik, B., et al. (eds). WashingtonDC: American Society for Microbiology Press, pp. 217–261.
de Gier, J.W., and Luirink, J. (2001) Biogenesis of innermembrane proteins in Escherichia coli. Mol Microbiol 40:314–322.
Gouffi, K., Santini, C.-L., and Wu, L.-F. (2002) Topologydetermination and functional analysis of the Escherichiacoli TatC protein. FEBS Lett 525: 65–70.
Gross, R., Simon, J., Theis, F., and Kröger, A. (1998) Twomembrane anchors of Wolinella succinogenes hydroge-nase and their function in fumarate and polysulfide respi-ration. Arch Microbiol 170: 50–58.
Gross, R., Simon, J., and Kröger, A. (1999) The role of thetwin-arginine motif in the signal peptide encoded by thehydA gene of the hydrogenase from Wolinella succino-genes. Arch Microbiol 172: 227–232.
Guzman, L.M., Belin, D., Carson, M.J., and Beckwith, J.(1995) Tight regulation, modulation, and high-level expres-sion by vectors containing the arabinose PBAD promoter. JBacteriol 177: 4121–4130.
Hamilton, C.M., Aldea, M., Washburn, B.K., Babitzke, P., andKushner, S.R. (1989) New method for generating deletionsand gene replacements in Escherichia coli. J Bacteriol171: 4617–4622.
von Heijne, G. (1992) Membrane protein structure prediction.Hydrophobicity analysis and the positive-inside rule. J MolBiol 225: 487–494.
Ize, B., Gérard, F., Zhang, M., Chanal, A., Voulhoux, R.,Palmer, T., et al. (2002) In vivo dissection of the Tat trans-location pathway in Escherichia coli. J Mol Biol 317: 327–335.
Jackson, M.E., and Pratt, J.M. (1987) An 18 amino acidamphiphilic helix forms the membrane-anchoring domainof the Escherichia coli penicillin-binding protein 5. MolMicrobiol 1: 23–28.
Jormakka, M., Törnroth, S., Byrne, B., and Iwata, S. (2002)Molecular basis of proton motive force generation: struc-ture of formate dehydrogenase-N. Science 295: 1863–1868.
Lämmli, U.K. (1970) Cleavage of structural proteins duringthe assembly of the head of bacteriophage T4. Nature 277:680–685.
de Leeuw, E., Granjon, T., Porcelli, I., Alami, M., Carr, S.B.,Müller, M., et al. (2002) Oligomeric properties and signalpeptide binding by Escherichia coli Tat protein transportcomplexes. J Mol Biol 322: 1135–1146.
Lowry, O.H., Rosebrough, N.J., Farr, A.L., and Randall, R.J.(1951) Protein measurements with the Folin phenolreagent. J Biol Chem 193: 265–275.
Luirink, J., Samuelsson, T., and de Gier, J.-W. (2001) YidC/
Oxa1p/Alb3: evolutionarily conserved mediators of mem-brane protein assembly. FEBS Lett 501: 1–5.
Magnani, P., Doussiere, J., and Lissolo, T. (2000) Diphe-nylene iodonium as an inhibitor for the hydrogenase com-plex of Rhodobacter capsulatus: evidence for two distinctelectron donor sites. Biochim Biophys Acta 1459: 169–178.
Molik, S., Karnauchov, I., Weidlich, C., Herrmann, R.G., andKlösgen, R.B. (2001) The Rieske Fe/S protein of the cyto-chrome b6f complex in chloroplasts: missing link in theevolution of protein transport pathways in chloroplasts? JBiol Chem 276: 42761–42766.
Nouwen, N., and Driessen, A.J.M. (2002) SecDFYajC formsa heterotetrameric complex with YidC. Mol Microbiol 44:1397–1405.
Porcelli, I., de Leeuw, E., Wallis, R., van den Brink-van derLaan, E., de Kruijff, B., Wallace, B.A., et al. (2002) Char-acterisation and membrane assembly of the TatA compo-nent of the Escherichia coli twin-arginine protein transportsystem. Biochemistry 41: 13690–13697.
Prinz, W.A., Boyd, D.H., Ehrmann, M., and Beckwith, J.(1998) The protein translocation apparatus contributes todetermining the topology of an integral membrane proteinin Escherichia coli. J Biol Chem 273: 8419–8424.
Roberts, S.A., Weichsel, A., Grass, G., Thakali, K., Hazzard,J.T., Tollin, G., et al. (2002) Crystal structure and electrontransfer kinetics of CueO, a multicopper oxidase requiredfor copper homeostasis in Escherichia coli. Proc Natl AcadSci USA 99: 2766–2771.
Rodrigue, A., Chanal, A., Beck, K., Müller, M., and Wu, L.-F.(1999) Co-translocation of a periplasmic enzyme complexby a hitchhiker mechanism through the bacterial Tat path-way. J Biol Chem 274: 13223–13228.
Russell, D.W., and Sambrook, J. (2001) Molecular Cloning:A Laboratory Manual, 3rd edn. Cold Spring Harbor, NY:Cold Spring Laboratory Press.
Sambasivarao, D., and Weiner, J.H. (1991) Dimethyl sulfox-ide reductase of Escherichia coli: an investigation of func-tion and assembly by use of in vivo complementation. JBacteriol 173: 5935–5943.
Sambasivarao, D., Scraba, D.G., Treiber, C., and Weiner,J.H. (1990) Organisation of dimethyl sulfoxide reductase inthe plasma membrane of Escherichia coli. J Bacteriol 172:5938–5948.
Samuelson, J.C., Chen, M., Jiang, F., Möller, I., Wiedmann,M., Kuhn, A., et al. (2000) YidC mediates membrane pro-tein insertion in bacteria. Nature 406: 637–641.
Santini, C.-L., Ize, B., Chanal, A., Müller, M., Giordano, G.,and Wu, L.-F. (1998) A novel Sec-independent periplasmicprotein translocation pathway in Escherichia coli. EMBO J17: 101–112.
Sargent, F., Bogsch, E., Stanley, N.R., Wexler, M., Robinson,C., Berks, B.C., and Palmer, T. (1998a) Overlapping func-tions of components of a bacterial Sec-independent proteinexport pathway. EMBO J 17: 3640–3650.
Sargent, F., Ballantine, S.P., Rugman, P.A., Palmer, T., andBoxer, D.H. (1998b) Reassignment of the gene encodingthe Escherichia coli hydrogenase 2 small subunit: identifi-cation of a soluble precursor of the small subunit in a hypBmutant. Eur J Biochem 255: 746–754.
Sargent, F., Stanley, N.R., Berks, B.C., and Palmer, T. (1999)Sec-independent protein translocation in Escherichia coli.

1390 K. Hatzixanthis, T. Palmer and F. Sargent
© 2003 Blackwell Publishing Ltd, Molecular Microbiology, 49, 1377–1390
a distinct and pivotal role for the TatB protein. J Biol Chem274: 36073–36082.
Sargent, F., Gohlke, U., de Leeuw, E., Stanley, N.R., Palmer,T., Saibil, H.R., and Berks, B.C. (2001) Purified compo-nents of the Escherichia coli Tat protein transport systemform a double-layered ring structure. Eur J Biochem 268:3361–3367.
Sargent, F., Berks, B.C., and Palmer, T. (2002) Assembly ofmembrane-bound respiratory complexes by the Tat protein-transport system. Arch Microbiol 178: 77–84.
Scotti, P.A., Urbanus, M.L., Brunner, J., de Gier, J.W., vonHeijne, G., van der Does, C., et al. (2000) YidC, theEscherichia coli homologue of mitochondrial Oxa1p, is acomponent of the Sec translocase. EMBO J 19: 542–549.
Silvestro, A., Pommier, J., and Giordano, G. (1988) Theinducible trimethylamine N-oxide reductase of Escherichiacoli K-12: biochemical and immunological studies. BiochimBiophys Acta 954: 1–13.
Simons, R.W., Houman, F., and Kleckner, N. (1987)Improved single and multicopy lac-based cloning vectorsfor protein and operon fusions. Gene 53: 85–96.
Stanley, N.R., Palmer, T., and Berks, B.C. (2000) The twin-arginine consensus motif of Tat signal peptides is involvedin Sec-independent protein targeting in Escherichia coli. JBiol Chem 275: 11591–11596.
Stanley, N.R., Findlay, K., Berks, B.C., and Palmer, T. (2001)Escherichia coli strains blocked in Tat-dependent proteinexport exhibit pleiotropic defects in the cell envelope. JBacteriol 183: 139–144.
Stanley, N.R., Sargent, F., Buchanan, G., Shi, J., Stewart,V., Palmer, T., and Berks, B.C. (2002) Behaviour of topo-logical marker proteins targeted to the Tat protein transportpathway. Mol Microbiol 43: 1005–1021.
Summer, E.J., Hori, H., Settles, M.A., and Cline, K. (2000)The thylakoid DpH-dependent pathway machinery facili-tates RR-independent N-tail protein integration. J BiolChem 275: 23483–23490.
Tabor, S., and Richardson, C.C. (1985) A bacteriophage T7RNA polymerase/promoter system for controlled exclusiveexpression of specific genes. Proc Natl Acad Sci USA 82:1074–1078.
Towbin, H., Staehelin, T., and Gordon, J. (1979) Electro-phoretic transfer of proteins from polyacrylamide gels tonitrocellulose sheets: procedure and some applications.Proc Natl Acad Sci USA 76: 4350–4354.
Traxler, B., Boyd, D., and Beckwith, J. (1993) The topologicalanalysis of integral cytoplasmic membrane proteins. JMembr Biol 132: 1–11.
Weiner, J.H., Bilous, P.T., Shaw, G.M., Lubitz, S.P., Frost,L., Thomas, G.H., et al. (1998) A novel and ubiquitoussystem for membrane targeting and secretion of cofactor-containing proteins. Cell 93: 93–101.
Wu, L.-F., and Mandrand, M.-A. (1993) Microbial hydrogena-ses: Primary structure, classification, signatures and phy-logeny. FEMS Microbiol Rev 104: 143–270.
Yahr, T.L., and Wickner, W.T. (2001) Functional reconstitu-tion of bacterial Tat translocation in vitro. EMBO J 20:2472–2479.




















