A Machine Learning Approach for the Identification of Protein Secondary Structure Elements from...
11
Dong Si, Shuiwang Ji, Kamal Al Nasr, Jing He Department of Computer Science, Old Dominion University, Norfolk, VA 23529 Received 2 November 2011; revised 12 March 2012; accepted 20 March 2012 Published online 27 March 2012 in Wiley Online Library (wileyonlinelibrary.com). DOI 10.1002/bip.22063 This article was originally published online as an accepted preprint. The ‘‘Published Online’’ date corresponds to the preprint version. You can request a copy of the preprint by emailing the Biopolymers editorial office at biopolymers@wiley. com INTRODUCTION E lectron cryo-microscopy (cryoEM) has become a major experimental technique to study the structures of large protein complexes. 1–10 It is a structure deter- mination technique complementary to the X-ray crys- tallography and nuclear magnetic resonance. 11,12 A number of such complexes have been resolved to near atomic resolutions (2–5 A ˚ ) 7,8,13–15 and many more have reached me- dium resolutions (5–10 A ˚ ). 16–23 At the medium resolutions, secondary structure elements (SSEs) such as a-helices and b- sheets can be visually and computationally identified using image processing techniques. It was first demonstrated using Helixhunter that a-helices can be computationally detected from a density map at subnano resolution. 24 After that, a number of approaches have been developed to detect the a-helices from the medium resolution electron density maps, 25–30 and a few approaches have been developed to detect the b-sheets. 26,28,29,31 Most of the computational approaches use automatic detection, while a few of them are semi-automatic guided by user interpretation. 29 Although a number of methods have been developed to identify the SSEs, it is still challenging to identify them auto- matically and accurately. In general, the long a-helices and A Machine Learning Approach for the Identification of Protein Secondary Structure Elements from Electron Cryo-Microscopy Density Maps Additional Supporting Information may be found in the online version of this article. Correspondence to: Jing He; e-mail: [email protected] ABSTRACT: The accuracy of the secondary structure element (SSE) identification from volumetric protein density maps is critical for de-novo backbone structure derivation in electron cryo-microscopy (cryoEM). It is still challenging to detect the SSE automatically and accurately from the density maps at medium resolutions (5–10 A ˚ ). We present a machine learning approach, SSELearner , to automatically identify helices and b-sheets by using the knowledge from existing volumetric maps in the Electron Microscopy Data Bank. We tested our approach using 10 simulated density maps. The averaged specificity and sensitivity for the helix detection are 94.9% and 95.8%, respectively, and those for the b-sheet detection are 86.7% and 96.4%, respectively. We have developed a secondary structure annotator, SSID, to predict the helices and b-strands from the backbone Ca trace. With the help of SSID, we tested our SSELearner using 13 experimentally derived cryo-EM density maps. The machine learning approach shows the specificity and sensitivity of 91.8% and 74.5%, respectively, for the helix detection and 85.2% and 86.5% respectively for the b-sheet detection in cryoEM maps of Electron Microscopy Data Bank. The reduced detection accuracy reveals the challenges in SSE detection when the cryoEM maps are used instead of the simulated maps. Our results suggest that it is effective to use one cryoEM map for learning to detect the SSE in another cryoEM map of similar quality. # 2012 Wiley Periodicals, Inc. Biopolymers 97: 698–708, 2012. Keywords: machine learning; protein; electron cryo- microscopy; secondary structure; helix; beta sheet; density map; local structure tensor V V C 2012 Wiley Periodicals, Inc. 698 Biopolymers Volume 97 / Number 9
Transcript of A Machine Learning Approach for the Identification of Protein Secondary Structure Elements from...

A Machine Learning Approach for the Identification of Protein SecondaryStructure Elements from Electron Cryo-Microscopy Density Maps
Dong Si, Shuiwang Ji, Kamal Al Nasr, Jing HeDepartment of Computer Science, Old Dominion University, Norfolk, VA 23529
Received 2 November 2011; revised 12 March 2012; accepted 20 March 2012
Published online 27 March 2012 in Wiley Online Library (wileyonlinelibrary.com). DOI 10.1002/bip.22063
This article was originally published online as an accepted
preprint. The ‘‘Published Online’’ date corresponds to the
preprint version. You can request a copy of the preprint by
emailing the Biopolymers editorial office at biopolymers@wiley.
com
INTRODUCTION
Electron cryo-microscopy (cryoEM) has become a
major experimental technique to study the structures
of large protein complexes.1–10 It is a structure deter-
mination technique complementary to the X-ray crys-
tallography and nuclear magnetic resonance.11,12 A
number of such complexes have been resolved to near atomic
resolutions (2–5 A)7,8,13–15 and many more have reached me-
dium resolutions (5–10 A).16–23 At the medium resolutions,
secondary structure elements (SSEs) such as a-helices and b-sheets can be visually and computationally identified using
image processing techniques. It was first demonstrated using
Helixhunter that a-helices can be computationally detected
from a density map at subnano resolution.24 After that, a
number of approaches have been developed to detect the
a-helices from the medium resolution electron density
maps,25–30 and a few approaches have been developed to
detect the b-sheets.26,28,29,31 Most of the computational
approaches use automatic detection, while a few of them are
semi-automatic guided by user interpretation.29
Although a number of methods have been developed to
identify the SSEs, it is still challenging to identify them auto-
matically and accurately. In general, the long a-helices and
A Machine Learning Approach for the Identification of Protein SecondaryStructure Elements from Electron Cryo-Microscopy Density Maps
Additional Supporting Information may be found in the online version of this
article.
Correspondence to: Jing He; e-mail: [email protected]
ABSTRACT:
The accuracy of the secondary structure element (SSE)
identification from volumetric protein density maps is
critical for de-novo backbone structure derivation in
electron cryo-microscopy (cryoEM). It is still challenging
to detect the SSE automatically and accurately from the
density maps at medium resolutions (�5–10 A). We
present a machine learning approach, SSELearner, to
automatically identify helices and b-sheets by using the
knowledge from existing volumetric maps in the Electron
Microscopy Data Bank. We tested our approach using 10
simulated density maps. The averaged specificity and
sensitivity for the helix detection are 94.9% and 95.8%,
respectively, and those for the b-sheet detection are 86.7%
and 96.4%, respectively. We have developed a secondary
structure annotator, SSID, to predict the helices and
b-strands from the backbone Ca trace. With the help of
SSID, we tested our SSELearner using 13 experimentally
derived cryo-EM density maps. The machine learning
approach shows the specificity and sensitivity of 91.8%
and 74.5%, respectively, for the helix detection and
85.2% and 86.5% respectively for the b-sheet detection in
cryoEM maps of Electron Microscopy Data Bank. The
reduced detection accuracy reveals the challenges in SSE
detection when the cryoEM maps are used instead of the
simulated maps. Our results suggest that it is effective to
use one cryoEM map for learning to detect the SSE in
another cryoEM map of similar quality. # 2012 Wiley
Periodicals, Inc. Biopolymers 97: 698–708, 2012.
Keywords: machine learning; protein; electron cryo-
microscopy; secondary structure; helix; beta sheet; density
map; local structure tensor
VVC 2012 Wiley Periodicals, Inc.
698 Biopolymers Volume 97 / Number 9

large b-sheets can be detected more accurately. Small helices
appear to be similar to turns in the density maps at the me-
dium resolution and they are hard to distinguish. A b-sheetwith two strands can be confused with a helix. Ideally, the
detection methods should be tested using a large number of
experimentally derived cryoEM density maps for which the
backbone structures are known. However, due to the lack of
such paired data, the current detection methods were pre-
dominantly tested using simulated volumetric density
maps. Without a test of a large number of cryoEM maps,
the effectiveness of the current methods is still not
clear when the experimentally derived cryoEM maps are
presented.
The Electron Microscopy Data Bank (EMDB)32 (http://
www.emdatabank.org/) currently archives over 1000 den-
sity map entries with resolution ranging from 2.8 to 97
A. These density maps were derived from cryoEM experi-
ments. Some of them have been resolved to near-atomic
resolution structures and therefore, their PDB structures
are linked in the EMDB. For some cryoEM maps of the
medium resolution, the backbone trace of their compo-
nent proteins have been resolved using fitting or compara-
tive modeling methods.20,33–35 When the PDB structure is
available for a component of protein, fitting methods,
such as SITUS and Flex-EM, can be used.33,34 Alterna-
tively, modeling can be done using a homologous protein
for which the backbone trace is available in the PDB.20,35
In these cases, the cryoEM maps of medium resolution
are linked to their corresponding PDB structures, and the
volumetric density maps are aligned with their corre-
sponding PDB structures.32 However, some of the PDB
structures of the cryoEM maps only have Ca trace avail-
able. Without full trace of backbone, the hydrogen bonds
are not annotated in the PDB structure. Therefore, there
is no annotation of secondary structures in such PDB
files. We developed a secondary structure annotator, SSID,
to predict the secondary structures from the Ca trace.
With the help of this tool, it is possible to test our SSE
detection method on those cryoEM maps with the Catraces but not full backbone traces.
The current secondary structure detection methods are
mostly based on image-processing techniques. These
methods search for cylinder-like regions for helices and
plane-like regions for b-sheets.24–29,31 Although such
methods can recognize most of the helices and b-sheets,they face difficulties in recognizing the border-line cases.
The drawback of these methods is that they do not have
the capability of using existing data to assist with the
detection. As more and more protein backbones are
derived for the cryoEM maps, learning from the existing
data is more and more important. It has been suggested
recently that machine learning improves the helix detec-
tion in RENNSH.30 RENNSH method uses the nested k
Nearest Neighbors classifiers in machine learning for the
detection of a-helices. It uses the training data and the
test data from different proteins of the same cryoEM
map. In this article, we will demonstrate that the training
process and the test process can use different cryoEM
maps in EMDB. Our SSELearner detects both helices and
b-sheets through the supervised learning from the cryoEM
density map that is estimated to have a similar nature to
that in the target cryoEM map.
MATERIALS AND METHODSThere are three major components in our method (Figure 1). The
first component develops the features using image processing con-
cepts. The second component performs the multi-task classification
using Support Vector Machine (SVM). The post-processing step
performs additional filtering and clustering based on the relation-
ships among the classified voxels.
Feature ExtractionThe feature extraction step characterizes each voxel based on its
local geometrical features. Local gradient is often used to charac-
terize the geometrical features in volumetric density maps.24,25,28
We applied the local structure tensor to describe the local
shape.28,36
FIGURE 1 The flowchart of SSELearner.
Machine Learning Approach for the Identification of Protein SSE 699
Biopolymers

Let I(x, y, z) denote the density at voxel (x, y, z). The local struc-
ture tensor is a symmetric positive semi-definite matrix given by:
Ka �I2x IxIy IxIzIxIy I2y Iy Iz
IxIz Iy Iz I2z
24
35
where Ix, Iy, and Iz are the derivatives (or gradient) along x, y, and z
direction, respectively. The symbol ‘‘*’’ stands for component wise
convolution, and Ka is a Gaussian convolution kernel, with standard
deviation a over which the local structure is averaged. The orthogo-
nal eigenvectors of the structure tensor v1, v2, v3 provide the pre-
ferred local orientations. The corresponding eigenvalues k1, k2, k3(k1 � k2 � k3) provide the average contrast along these directions.
The first eigenvector v1 represents the direction with the maximum
variance of the density, whereas v3 represents the direction with the
minimum variance. The three eigenvalues could therefore be used,
based on their relative eigenvectors, to describe the local density na-
ture in three classes: cylinder-like, plane-like or isotropic structure:
� Cylinder-like structures: k1 � k2 � k3� Plane-like structures: k1 � k2 � k3� Isotropic structures: k1 � k2 � k3
Instead of using the three eigenvalues of the structure tensor to
distinguish different local structures, Yu and Bajaj proposed a prac-
tical parameter—thickness.28 The thickness we applied is defined by
the width of the region above a pre-chosen threshold along the
eigenvector. Let t1, t2, t3 be the thicknesses along direction v1, v2, v3.
The typical thicknesses for different local structures have the follow-
ing criteria:
� Cylinder-like structures: t1 � t2 � t3� Plane-like structures: t1 � t2 � t3
� Isotropic structures: t1 � t2 � t3
Based on the above local structure measurements, we derived
five features for each voxel in the density map: two ratios of the
eigenvalues k1/k2 and k2/k3, two ratios of the thickness t1/t2 and t2/
t3, and the normalized density value of this voxel.
Multi-Class Classification of the Voxels Using SVMFirst introduced by Boser et al. in 1992,37 SVM is one of the most
commonly used supervised learning methods. It employs a maxi-
mum margin criterion and is a powerful tool for classification and
regression tasks. We applied the SVM to classify the voxels from
the test density map into three different classes: helix, sheet, and
background voxels (Figure 2). Given a training set of instance-label
pairs (xi, yi), i 5 1,. . ., I where xi [ Rn is an n-dimensional feature
vector and yi is the corresponding class label of that instance. SVM
finds the parameters of a decision function D(x) 5 wT/(x) 1 b
during a learning phase, where /(x) maps xi into a higher dimen-
sional space.37 The idea is to find a linear separating hyper plane
with maximal margin between the classes in this higher dimen-
sional space.37,38 All the parameters found during this learning
phase can be stored in a model for future prediction on the test
data.
In the secondary structure identification problem, each voxel in
the training density maps is associated with five features and one
class label. The class label of each training voxel is determined based
on its estimated proximity to the secondary structures. The cut-off
values are empirical, by taking the consideration of the typical
thickness of a helix (�5 A in diameter), and the distance between
two adjacent b-stands (�4.5 A). In particular, the three classes were
defined as the following:
� 11, for a helix voxel, if it is within 3 A from the axis of a helix;
� 21, for a sheet voxel, if it is within 4.5 A from the Ca atoms
of a b-sheet;� 0, for a background voxel, if it is not a helix voxel or a sheet
voxel.
SVM is inherently two-class classifiers. Multiple two-class prob-
lems can be converted to a multi-class problem using the concept of
voting. We employed LIBSVM39 to solve our three-class prediction
problem. LIBSVM uses the ‘‘one-against-one’’ approach for multi-
class classification.40 If k is the total number of classes, this approach
trains k(k 2 1)/2 classifiers for all the possible combinations of the
class pairs. A voting strategy was applied in which each two-class
classification is considered as a vote.39 Each voxel from the test den-
sity map was then classified according to the class with the highest
number of ‘‘votes.’’
FIGURE 2 The training and prediction using the SVM.
700 Si et al.
Biopolymers

PostprocessingThe SVM classification determines the class label for each density
voxel in the target map. The postprocessing takes the class labels as
input and determines the exact position for the helices and b-sheets(Figure 3). A helix was represented as a set of voxels that are often
near the central axis of the helix. A b-sheet was represented as a set
of critical voxels on the sheet. The postprocessing includes two
steps: filtering and clustering.
The filtering step aims at identifying the voxels with high density
in a small neighborhood. We observed that such voxels are often more
reliable representatives for the SSEs. We applied a filter using the
local-peak-counter (LPC) proposed in sheettracer.41 For each voxel,
the average density was calculated within a sphere of 3 A in radius.
Those voxels in the sphere with density value greater than the average
have their LPC incremented. All the voxels were sorted according to
their LPC numbers after counting. A threshold parameter was used to
select the top ranked voxels. For example, the LPC filtering step
selected the top 50% of voxels as the candidate representatives for the
helices in the simulated density map. The top 75% of the voxels were
selected as the candidate representatives for the b-sheets (Figure 3D).The candidate voxels were further clustered to select the more
reliable clusters for the annotation of secondary structures. The
clusters were created based on the adjacency of voxels and then the
size of each cluster was measured. A cluster size parameter was used
to discard the small clusters that are often related to the turns. As an
example, we used the size of 3 and 8 A to discard the small clusters
for the helix and the sheet, respectively, in the simulated density
map. The two threshold parameters can be adjusted by the user
depending on the quality of the density maps.
Finally, a central axial line of helix voxel cluster was generated to
represent the helix. This was done by traveling along the locally highest
density voxels between the two ends of the helix voxel cluster. Because
the shape of b-sheets is different for different sheets, we used the sheet
voxels after postprocessing to represent the sheets (Figure 3E).
Annotation of the Secondary Structures from Ca
TraceOne of the current difficulties in testing the SSE identification meth-
ods is the lack of data pairs, each of which includes a cryoEM den-
sity map and its aligned backbone structure. In some cases, the
backbone structure is available in the Ca trace, but the secondary
structures were not annotated due to the lack of hydrogen-bond in-
formation. An earlier study has shown that the Ca trace of the back-
bone can be used to evaluate the overall quality of the structure
using certain geometrical measurement of the Ca atoms.42 In this
article, we defined two torsion angles to characterize a helix based
on the geometrical nature of the helix. T1 is the torsion angle
formed by Cai, Ci, Ci11, Cai14 and T2 is the torsion angle formed
by Ci, Ci11, Cai14, Cai15. We use Ci to denote the geometrical cen-
ter of the three consecutive Ca atoms Cai, Cai11, Cai12, i 5 1,. . ., n2 3. According to our survey of a dataset of PDB structures, the dis-
tribution of T1 and T2 shows characteristic angles for helices (data
not shown). There are two passes involved in the helix annotation.
In the first pass, the spiral nature of the helix was examined using
the torsion angle T1 and T2. The second pass of the helix annotation
refines the annotation and determines the ends of the helices. The
b-sheet annotation starts after the helix annotation finishes. The
first pass characterizes the parallelism of the strands. For each Cai, alist of its contacts (within 6.5 A) was built. For each amino acid j in
the contact list, Cai and Cai12 were used as a vector to compare
with the vectors formed by Caj and Caj12 and Caj and Caj22,
respectively. If the angle formed by the two vectors is less than 708and the distance between either ends of either vectors is less than
6.5 A, the vectors are considered parallel. The second pass in strand
annotation further refines the results from the first pass. In particu-
lar, it aims to distinguish the turns from the stands. This was done
by calculating the angle formed by three consecutive Ca atoms,
because this angle is usually restricted in a turn.
RESULTSWe tested the performance of SSELearner on 10 simulated
density maps and 13 experimental cryoEM density maps
from EMDB. The selected EMDB density maps are between
3.8 and 9 A resolution. Two types of evaluation were per-
formed. One measures the number of identified secondary
structures24–26 and the other measures the number of Caatoms24,31 that falls in the neighborhood of the secondary
structures. A helix is identified if its length is within one turn
difference from the length of the helix in the PDB structure.
A b-sheet is identified if the identified b-sheet voxels visually
FIGURE 3 Post-processing. (A) the structure of 2AW0 (PDB ID) with helices (red ribbon) and
b-sheet (blue ribbon); (B) the simulated density map at 8 A resolution; (C) the helix (red) and
sheet (blue) voxels labeled by SVM; (D) the helix (red) and sheet (blue) voxels after post-process-
ing; (E) the detected secondary structures superimposed on the PDB structure.
Machine Learning Approach for the Identification of Protein SSE 701
Biopolymers

overlay on the b-sheet of the PDB structure. To present a
more quantitative estimation about the size of the identified
helices and b-sheets, we estimated the number of Ca atoms
that are close to the identified helix voxels and b-sheet voxels.In particular, a Ca is considered as an identified helix Ca, if itis within 2.5 A distance from an identified helix voxel. A Cais considered as an identified sheet Ca, if it is within 3 A dis-
tance from an identified sheet voxel. The definition of the
secondary structures was based on the PDB file that is the
authors’ annotation of the protein structure. Note that the
authors’ annotation in the PDB file may be slightly different
from the annotation using DSSP.43 Although the definition
of a helix and a b-sheet is clear in almost all the PDB files in
our tests, it is necessary to visually decide the number and
length in rare cases. For example, there is an overlap in the
annotated helices with amino acid index 922107 and
1062111 of 1CV1 (PDB ID). Three strands with amino acid
index 37248, 3622375, 962110 of 2GSY were annotated in
two b-sheets.
Performance on the Simulated Maps
We tested our SSELearner using 10 simulated density maps
that were generated to 8 A resolution using the program
pdb2mrc of EMAN44 with a sampling size of 1 A/pixel. The
10 proteins were used for testing SSEhunter at the same reso-
lution.26 The training dataset contains four other proteins
(PDB ID: 1C3W, 1IRK, 1TIM, and 2BTV) previously used
for testing SSEhunter.26
Our method successfully identified all the 74 helices that
have more than four amino acids (Table I). Because we used
3 A as the minimum helix length in the post-process step,
only 4 out of the 14 extremely short helices were identified.
Most of the missed helices have three amino acids in length,
presumably of the three-helices. Our method detected all the
17 b-sheets, six of which have only two strands. Compared
with SSEhunter’s result (Table I), our SSELearner appears to
be able to detect more two-stranded b-sheets and is at least
comparable in helix identification. Note that we used the
same criteria to measure the number of the detected helices
and b-sheet as indicated in the SSEhunter paper.26
Although there were no false-positive helices and b-sheets, the detected secondary structures may not be accu-
rate in size. The most commonly over-determined region
for a helix is at the end of the helix (dotted region in Figure
4C). In this case, a turn at the end of the helix appears as
part of the helix. Similarly, the over determined area for a
b-sheet can be at the edge of the b-sheets or at the loop
region that appears to be a strand (Figure 4A and 4B).
Some of the false positive detections are at the arguable
regions of the structure where there is disagreement
between the author’s annotation and the DSSP annotation.
The DSSP annotation is available under the ‘‘sequence’’ tab
in the PDB website (http://www.rcsb.org). For example,
residue 136 and 137 of 2ITG (in the circle of Figure 4A)
were considered as a b-strand by the DSSP annotation, but
they were not annotated as a b-strand by the authors of
PDB file. Similar situation happens to residue 214 to 216 of
1AJZ (Figure 4B). In these two cases, our false positively
detected b-sheet Ca atoms would have not been false posi-
tive if we had used the DSSP annotation of the secondary
structures.
To quantify the size of the detected secondary structures,
particularly for b-sheets, we calculated the specificity and
Table I The Comparison of the Number of Detected Secondary Structures from the Simulated Maps
PDB ID
SSELearner SSEhuntera
Helix\ 5aa Helix 5–8aa Helix[ 8aa
Sheet 5 2
strands
Sheet[ 2
strands Helix\ 5aa Helix 5–8aa
Sheet 5 2
strands
1AJW 0/0 1/1 0/0 0/0 2/2 0/0 1/1 0/0
1AJZ 0/1 3/3 7/7 1/1 1/1 0/1 3/3 0/1
1AL7 0/3 4/4 10/10 2/2 1/1 1/3 4/4 0/2
1CV1 1/1 2/2 8/8 0/0 1/1 1/1 0/2 0/0
1DAI 1/2 2/2 5/5 2/2 1/1 2/2 2/2 0/2
1ENY 0/0 1/1 9/9 0/0 1/1 0/0 1/1 0/0
1WAB 1/3 0/0 6/6 0/0 1/1 1/3 0/0 0/0
2AW0 0/0 0/0 2/2 0/0 1/1 0/0 0/0 0/0
2ITG 0/0 1/1 5/5 0/0 1/1 0/0 1/1 0/0
3LCK 1/4 2/2 6/6 1/1 1/1 1/4 0/2 1/1
Total 4/14 16/16 58/58 6/6 11/11 6/14 12/16 1/6
a As a comparison, the columns for SSEhunter can be found in the supporting Table 1 of the SSEhunter paper.26
702 Si et al.
Biopolymers

sensitivity based on the detected helix and sheet Ca atoms
similar to the estimation used in sheetminer paper.31 Table II
shows the number of identified Ca atoms for the dataset in
Table I. A Ca is considered as an identified helix Ca, if it iswithin 2.5 A distance from an identified helix voxel. A Ca is
considered as an identified sheet Ca, if it is within 3 A dis-
tance from an identified sheet voxel. The sensitivity and the
specificity of helix identification is 95.8% and 94.9%, respec-
tively. The sensitivity and specificity for b-sheet identificationis 96.4% and 86.7%, respectively.
Table II The Accuracy of Identified Ca Atoms from the Simulated Maps
PDB ID Totala Hlxb tp Hlxc m Hlxd fp Hlxe Shtf tp Shtg m Shth fp ShtiSpec. Hlxj
(%)
Sen. Hlxk
(%)
Spec. Shtl
(%)
Sens. Shtm
(%)
1AJW 145 5 5 0 1 63 50 13 10 99.3 100.0 87.8 79.4
1AJZ 282 124 120 4 6 37 37 0 31 96.2 96.8 87.4 100.0
1AL7 350 159 145 14 7 46 46 0 26 96.3 91.2 91.5 100.0
1CV1 162 123 114 9 3 14 14 0 11 92.3 92.7 92.6 100.0
1DAI 219 84 81 3 7 47 47 0 32 94.8 96.4 81.4 100.0
1ENY 268 126 121 5 4 66 56 10 25 97.2 96.0 87.6 84.9
1WAB 212 96 90 6 6 24 24 0 21 94.8 93.8 88.8 100.0
2AW0 72 22 22 0 3 25 25 0 11 94.0 100.0 76.6 100.0
2ITG 160 66 66 0 10 21 21 0 18 89.4 100.0 87.1 100.0
3LCK 270 107 98 9 9 30 30 0 33 94.5 91.6 86.3 100.0
Average 94.9 95.8 86.7 96.4
a The total number of Ca atoms in the protein.b The total number of Ca atoms in the helices.c The number of true positive Ca atoms of helices.d The number of missed Ca atoms that are on helices but not detected.e The number of false positive Ca atoms for helices.f The total number of Ca atoms in the b-sheets.g The number of true positive Ca atoms for b-sheets.h The number of missed Ca atoms that are on b-sheets but were not detected.i The number of false positive Ca atoms for b-sheets.j The specificity of helix detection, calculated by the formula: 1 2 (e/(a2b)).k The sensitivity of helix detection, calculated by the formula: c/b.l The specificity of sheet detection, calculated by the formula: 12 (i/(a2f)).m The sensitivity of sheet detection, calculated by the formula: g/f.
FIGURE 4 Examples of identified false-positive regions. The detected helices (red lines) and b-sheets (blue voxels) were superimposed on the proteins with PDB ID: 2ITG (A), 1AJZ (B), and
2ITG (C). The dotted line highlights the false positively identified regions.
Machine Learning Approach for the Identification of Protein SSE 703
Biopolymers
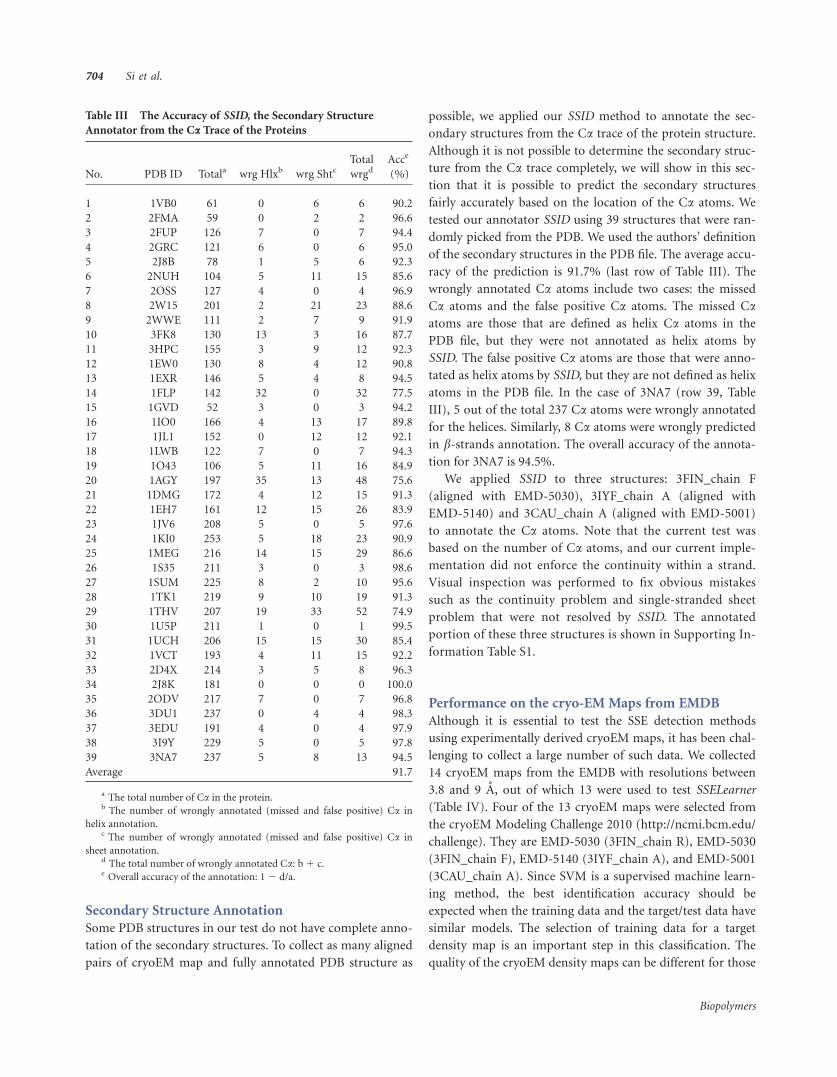
Secondary Structure Annotation
Some PDB structures in our test do not have complete anno-
tation of the secondary structures. To collect as many aligned
pairs of cryoEM map and fully annotated PDB structure as
possible, we applied our SSID method to annotate the sec-
ondary structures from the Ca trace of the protein structure.
Although it is not possible to determine the secondary struc-
ture from the Ca trace completely, we will show in this sec-
tion that it is possible to predict the secondary structures
fairly accurately based on the location of the Ca atoms. We
tested our annotator SSID using 39 structures that were ran-
domly picked from the PDB. We used the authors’ definition
of the secondary structures in the PDB file. The average accu-
racy of the prediction is 91.7% (last row of Table III). The
wrongly annotated Ca atoms include two cases: the missed
Ca atoms and the false positive Ca atoms. The missed Caatoms are those that are defined as helix Ca atoms in the
PDB file, but they were not annotated as helix atoms by
SSID. The false positive Ca atoms are those that were anno-
tated as helix atoms by SSID, but they are not defined as helix
atoms in the PDB file. In the case of 3NA7 (row 39, Table
III), 5 out of the total 237 Ca atoms were wrongly annotated
for the helices. Similarly, 8 Ca atoms were wrongly predicted
in b-strands annotation. The overall accuracy of the annota-tion for 3NA7 is 94.5%.
We applied SSID to three structures: 3FIN_chain F
(aligned with EMD-5030), 3IYF_chain A (aligned with
EMD-5140) and 3CAU_chain A (aligned with EMD-5001)
to annotate the Ca atoms. Note that the current test was
based on the number of Ca atoms, and our current imple-
mentation did not enforce the continuity within a strand.
Visual inspection was performed to fix obvious mistakes
such as the continuity problem and single-stranded sheet
problem that were not resolved by SSID. The annotated
portion of these three structures is shown in Supporting In-
formation Table S1.
Performance on the cryo-EM Maps from EMDB
Although it is essential to test the SSE detection methods
using experimentally derived cryoEM maps, it has been chal-
lenging to collect a large number of such data. We collected
14 cryoEM maps from the EMDB with resolutions between
3.8 and 9 A, out of which 13 were used to test SSELearner
(Table IV). Four of the 13 cryoEM maps were selected from
the cryoEM Modeling Challenge 2010 (http://ncmi.bcm.edu/
challenge). They are EMD-5030 (3FIN_chain R), EMD-5030
(3FIN_chain F), EMD-5140 (3IYF_chain A), and EMD-5001
(3CAU_chain A). Since SVM is a supervised machine learn-
ing method, the best identification accuracy should be
expected when the training data and the target/test data have
similar models. The selection of training data for a target
density map is an important step in this classification. The
quality of the cryoEM density maps can be different for those
Table III The Accuracy of SSID, the Secondary Structure
Annotator from the Ca Trace of the Proteins
No. PDB ID Totala wrg Hlxb wrg ShtcTotal
wrgdAcce
(%)
1 1VB0 61 0 6 6 90.2
2 2FMA 59 0 2 2 96.6
3 2FUP 126 7 0 7 94.4
4 2GRC 121 6 0 6 95.0
5 2J8B 78 1 5 6 92.3
6 2NUH 104 5 11 15 85.6
7 2OSS 127 4 0 4 96.9
8 2W15 201 2 21 23 88.6
9 2WWE 111 2 7 9 91.9
10 3FK8 130 13 3 16 87.7
11 3HPC 155 3 9 12 92.3
12 1EW0 130 8 4 12 90.8
13 1EXR 146 5 4 8 94.5
14 1FLP 142 32 0 32 77.5
15 1GVD 52 3 0 3 94.2
16 1IO0 166 4 13 17 89.8
17 1JL1 152 0 12 12 92.1
18 1LWB 122 7 0 7 94.3
19 1O43 106 5 11 16 84.9
20 1AGY 197 35 13 48 75.6
21 1DMG 172 4 12 15 91.3
22 1EH7 161 12 15 26 83.9
23 1JV6 208 5 0 5 97.6
24 1KI0 253 5 18 23 90.9
25 1MEG 216 14 15 29 86.6
26 1S35 211 3 0 3 98.6
27 1SUM 225 8 2 10 95.6
28 1TK1 219 9 10 19 91.3
29 1THV 207 19 33 52 74.9
30 1U5P 211 1 0 1 99.5
31 1UCH 206 15 15 30 85.4
32 1VCT 193 4 11 15 92.2
33 2D4X 214 3 5 8 96.3
34 2J8K 181 0 0 0 100.0
35 2ODV 217 7 0 7 96.8
36 3DU1 237 0 4 4 98.3
37 3EDU 191 4 0 4 97.9
38 3I9Y 229 5 0 5 97.8
39 3NA7 237 5 8 13 94.5
Average 91.7
a The total number of Ca in the protein.b The number of wrongly annotated (missed and false positive) Ca in
helix annotation.c The number of wrongly annotated (missed and false positive) Ca in
sheet annotation.d The total number of wrongly annotated Ca: b1 c.e Overall accuracy of the annotation: 12 d/a.
704 Si et al.
Biopolymers

at the similar resolutions. We carefully selected a training
density map for each target cryoEM map that is to be tested
(Table IV). A number of factors were taken into considera-
tion in the selection of training data. These factors include
the resolution, the estimated range of helix density, the esti-
mated range of sheet density and the estimated noise level in
the density map.
An example of the detected secondary structures is shown
in Figure 5. In this case, the cryoEM density map EMD-5030
was aligned with 3FIN_chain R. SSELearner detected all the
four helices and the b-sheet (row 1, Table V). The specificity
and the sensitivity for the detected helix Ca atoms are 93.1%
and 96.6%, and those for the sheet detection are 89.3% and
100%, respectively (row 1, Table VI).
The test of 13 cryoEM maps suggests that the helices longer
than eight amino acids and sheets with more than two strands
can be mostly detected. SSELearner detected 89 of 107 such
helices and all 26 such b-sheets (Table V). Note that SSE-
Learner detected 100% such helices and sheets in the simulated
data (Table I). The contrast shows the challenges to the SSE
detection method for the experimentally derived density maps.
This is only visible when a large number of the experimental
cryoEM density maps are used for testing. Our test also sug-
gests that SSELearner detects the b-sheets fairly well in the cry-
oEM maps. It detected all the 26 b-sheets that have more than
two strands, and 9 of 16 b-sheets with two strands (Table V).
For helices with no more than eight amino acids, SSELearner
was only able to detect 30 of 61 such helices (Table V).
We analyzed the performance of our SSE detection method
using the number of identified Ca atoms to reveal the size ac-
curacy of the SSE (Table VI). The overall specificity and sensi-
tivity are 91.8% and 74.5%, respectively, in helix detection.
The main reason for the reduced sensitivity between the simu-
lated maps verses the EMDB maps is in the short helix detec-
tion. For example, 8 out of 11 helices in EMD-1237 and 13
out of 26 helices in EMD-5100 are no more than eight amino
acids. Another reason is the reduced quality of the experimen-
tal cryoEM maps compared with that of the simulated density
maps. The experimental density maps often have incomplete
density data, particularly for the short helices. The overall
specificity and sensitivity in sheet identification are 85.2% and
86.5%, respectively. Our test using 13 experimentally derived
cryoEM maps shows the challenges in the SSE detection from
the real cryoEM maps. It is not possible to detect them as
accurately as in the simulated maps at this point.
DISCUSSIONWe have developed an approach to detect the secondary
structures from the medium resolution density maps using a
combination of image processing and supervised machine
learning techniques. The supervised machine learning allows
the input of user knowledge, and that was reflected in the
Table IV The Target CryoEM Density Maps (EMDB ID, PDB
ID, and Resolution) and Their Corresponding Training Data
Test Data Training Data
5030 (3FIN_R), 6.4 A 5168 (3MFP_A), 6.6 A
5030 (3FIN_F), 6.4 A 5168 (3MFP_A), 6.6 A
5140 (3IYF_A), 8 A 5030 (3FIN_F), 6.4 A
1733 (3C91_H), 6.8 A 1780 (3IZ6_K), 5.5 A
5168 (3MFP_A), 6.6 A 5030 (3FIN_R), 6.4 A
1237 (2GSY_A), 7.2 A 1780 (3IZ6_K), 5.5 A
5100 (3IXV_A), 6.8 A 5030 (3FIN_R), 6.4 A
5199 (3N09_C), 3.8 A 1780 (3IZ6_K), 5.5 A
1780 (3IZ6_K), 5.5 A 1740 (3C92_A), 6.8 A
5223 (3IZ0_A), 8.6 A 1340 (2P4N_A), 9 A
1340 (2P4N_A), 9 A 5223 (3IZ0_A), 8.6 A
1780 (3IZ6_T), 5.5 A 5030 (3FIN_F), 6.4 A
5001 (3CAU_A), 4.2 A 1740 (3C92_A), 6.8 A
FIGURE 5 Secondary structures detected using SSELearner. (A) Part of EMDB entry EMD-5030
at resolution 6.4 A with fitted secondary structure of protein 3FIN_chain R; (B) identified helix and
sheet locations.
Machine Learning Approach for the Identification of Protein SSE 705
Biopolymers

selection of the training density map for each target density
map. We found that this step is important for achieving good
accuracy, and it requires certain knowledge about the nature
of the density map. We visually estimated the density ranges
for helices and sheets then used such estimated parameters in
searching for the density maps for training. RENNSH demon-
strated that selecting training data from the density map
within which the test data are located is a viable approach for
the detection of a-helices.30 We extended their finding by
showing that it is possible to use different cryoEM maps for
training and test and to obtain fairly good detection accuracy.
We have demonstrated that it is possible to find a training
Table V The Identified Secondary Structures from the Experimental CryoEM Density Maps
EMDB (PDB) ID Helix\ 5aa Helix 5–8aa Helix[ 8aa Sheet 5 2 strands Sheet[ 2 strands
5030 (3FIN_R) 0/0 0/0 4/4 0/0 1/1
5030 (3FIN_F) 1/1 0/0 6/6 2/2 1/1
5140 (3IYF_A) 0/0 4/8 9/16 1/3 4/4
1733 (3C91_H) 0/0 0/0 5/5 1/1 2/2
5168 (3MFP_A) 0/0 6/9 8/10 0/1 2/2
1237 (2GSY_A) 0/3 2/5 3/3 1/1 4/4
5100 (3IXV_A) 0/2 5/11 12/13 0/1 2/2
5199 (3N09_C) 0/2 3/5 6/6 1/3 2/2
1780 (3IZ6_K) 0/0 0/1 2/2 0/0 1/1
5223 (3IZ0_A) 3/3 1/2 8/11 0/0 2/2
1340 (2P4N_A) 0/1 2/4 9/11 0/0 2/2
1780 (3IZ6_T) 0/0 0/0 4/4 1/1 0/0
5001 (3CAU_A) 0/0 3/4 13/16 2/3 3/3
Total 4/12 26/49 89/107 9/16 26/26
Table VI The Identified Ca Atoms from the Experimental cryoEM Maps
EMDB (PDB) ID Totala Hlxb tp Hlxc m Hlxd fp Hlxe Shtf tp Shtg m Shth fp ShtiSpec. Hlxj
(%)
Sen. Hlxk
(%)
Spec. Shtl
(%)
Sens. Shtm
(%)
5030 (3FIN_R) 117 59 57 2 4 14 14 0 11 93.1 96.6 89.3 100.0
5030 (3FIN_F) 208 80 71 9 4 37 37 0 15 96.9 88.8 91.2 100.0
5140 (3IYF_A) 491 263 136 127 42 74 47 27 69 81.6 51.7 83.5 63.5
1733 (3C91_H) 203 86 69 17 0 62 54 8 25 100.0 80.2 82.3 87.1
5168 (3MFP_A) 374 186 138 48 17 60 41 19 56 91.0 74.2 82.2 68.3
1237 (2GSY_A) 428 69 46 23 5 187 180 7 71 98.6 66.7 70.5 96.3
5100 (3IXV_A) 626 277 195 82 35 99 86 13 51 90.0 70.4 90.3 86.9
5199 (3N09_C) 397 144 118 26 24 128 114 14 41 90.5 81.9 84.8 89.1
1780 (3IZ6_K) 119 37 25 12 1 29 29 0 11 98.8 67.6 87.8 100.0
5223 (3IZ0_A) 412 186 116 70 34 55 42 13 24 85.0 62.4 93.3 76.4
1340 (2P4N_A) 412 202 124 78 21 55 39 16 75 90.0 61.4 79.0 70.9
1780 (3IZ6_T) 82 54 45 9 0 7 7 0 8 100.0 83.3 89.3 100.0
5001 (3CAU_A) 526 255 214 41 61 82 71 11 72 77.5 83.9 83.8 86.6
Average 91.8 74.5 85.2 86.5
a The total number of Ca atoms in the protein.b The total number of Ca atoms in the helices.c The number of true positive Ca atoms of helices.d The number of missed Ca atoms that are on helices but not detected.e The number of false positive Ca atoms for helices.f The total number of Ca atoms in the b-sheets.g The number of true positive Ca atoms for b-sheets.h The number of missed Ca atoms that are on b-sheets but were not detected.i The number of false positive Ca atoms for b-sheets.j The specificity of helix detection, calculated by the formula: 12 (e/(a2b)).k The sensitivity of helix detection, calculated by the formula: c/b.l The specificity of sheet detection, calculated by the formula: 12 (i/(a2f)).m The sensitivity of sheet detection, calculated by the formula: g/f.
706 Si et al.
Biopolymers

density map that shares similar density nature with that of the
target map in the current EMDB. We have tested using thir-
teen cryoEM density maps from the EMDB to demonstrate
that this approach is feasible. As more and more cryoEM
maps are available in the EMDB database, it is promising that
the combined image processing and machine learning techni-
ques will result in improved accuracy in SSE detection.
In principle, the resolution of the density map can affect
the accuracy of the SSE detection. We generated the density
map at 6 A (Figure 6A), 8 A (Figure 3B), and 10 A (Figure 6B)
resolution using EMAN.44 The detection of the helices appears
to be less affected by the resolution at the resolution range of
6210 A. In fact both helices were detected to similar lengths
at the three resolutions sampled (Figures 3E, 6A, and 6B). The
b-sheet was all detected at the three resolution ranges. How-
ever, the size of the detected b-sheet appears to be affected
slightly by the resolution. It appears that more true-positive
sheet voxels were detected in the 6 A density map (Figure 6A
cyan) than in the 10 A map (Figure 6B cyan).
We have developed the SSELearner for the detection of heli-
ces and b-sheets from cryoEM density maps at the medium
resolution range of 5210 A. The approach has been tested
using 10 simulated density maps as well as 13 cryoEM maps
from the EMDB. Our results show that although the detection
can be fairly accurate in the simulated density maps, the accu-
racy decreases significantly for the short helices from the
experimentally derived density maps. The specificity and the
sensitivity for the helix detection are 94.9% and 95.8%, respec-
tively, in the simulated density maps and 91.8% and 74.5% in
the experimentally derived cryoEM maps. The specificity and
the sensitivity for the b-sheet detection are 86.7% and 96.4%,
respectively, in the simulated maps and 85.2% and 86.5% in
the experimentally derived maps. The overall detection accu-
racy demonstrated that it is feasible to select a specific density
map from the current EMDB as training data to detect the
SSE of a target cryoEM map.
Secondary structure detection from the cryoEM density
map at the medium resolution is still a challenging problem
despite the multiple proposed methods. To speed-up the
research in this direction, coordinated effort is needed to
promote the public sharing of the developed software and
the development of the benchmark data that is available to
the public. However, the comparison of the software is still
challenging. Some of the methods are automatic and others
are semi-automatic.29 The continuing maintenance of the
software has also been inadequate in this area.
Old Dominion University MDS research fund and NSF DBI-1147134.
REFERENCES1. Bottcher, B.; Wynne, S. A.; Crowther, R. A. Nature 1997, 386,
88–91.
2. Mancini, E. J.; Clarke, M.; Gowen, B. E.; Rutten, T.; Fuller, S. D.
Mol Cell 2000, 5, 255–266.
3. Zhou, Z. H.; Dougherty, M.; Jakana, J.; He, J.; Rixon, F. J.; Chiu,
W. Science 2000, 288, 877–880.
4. van Heel, M.; Gowen, B.; Matadeen, R.; Orlova, E. V.; Finn, R.;
Pape, T.; Cohen, D.; Stark, H.; Schmidt, R.; Schatz, M.; Patward-
han, A. Quarterly Rev Biophys 2000, 33, 307–369.
5. Zhou, Z. H.. Curr Opin Structural Biol 2008, 18, 218–228.
6. Hryc, C. F.; Chen, D. H.; Chiu, W. Curr Opin Virol 2011, 1,
110–117.
7. Zhang, R.; Hryc, C. F.; Cong, Y.; Liu, X. G.; Jakana, J.; Gorcha-
kov, R.; Baker, M. L.; Weaver, S. C.; Chiu, W. Embo J 2011, 30,
3854–3863.
8. Settembre, E. C.; Chen, J. Z.; Dormitzer, P. R.; Grigorieff, N.;
Harrison, S. C. Embo J 2011, 30, 408–416.
9. Daban, J. R. Electron microscopy and atomic force microscopy
studies of chromatin and metaphase chromosome structure.
Micron 2011, 42, 733–750.
FIGURE 6 SSE identification at different resolutions. Protein 2AW0 (PDB ID) was used to gener-
ate the simulated density map at 6 A resolution (A) and 10 A resolution (B). The detected helix
(red line) and b-sheet (blue voxels) were superimposed on the PDB structure.
Machine Learning Approach for the Identification of Protein SSE 707
Biopolymers

10. Grigorieff, N.; Harrison, S. C. Curr Opin Struct Biol 2011, 21,
265–273.
11. Chiu, W.; Baker, M. L.; Jiang, W.; Dougherty, M.; Schmid, M. F.
Structure 2005, 13, 363–372.
12. Zhou, Z. H. Adv Protein Chem Struct Biol 2011, 82, 1–35.
13. Ludtke, S. J.; Baker, M. L.; Chen, D. H.; Song, J. L.; Chuang, D.
T.; Chiu, W. Structure 2008, 16, 441–448.
14. Jiang, W.; Baker, M. L.; Jakana, J.; Weigele, P. R.; King, J.; Chiu,
W. Nature 2008, 451, 1130–1134.
15. Yu, X.; Jin, L.; Zhou, Z. H. Nature 2008, 453, 415–419.
16. Schuette, J.-C.; Murphy, F. V.; Kelley, A. C.; Weir, J. R.; Giese-
brecht, J.; Connell, S. R.; Loerke, J.; Mielke, T.; Zhang, W.; Penc-
zek, P. A.; Ramakrishnan, V.; Spahn, C. M. T. Embo J 2009, 28,
755–765.
17. Luque, D.; Saugar, I.; Rodriguez, J. F.; Verdaguer, N.; Garriga,
D.; Martin, C. S.; Velazquez-Muriel, J. A.; Trus, B. L.; Carra-
scosa, J. L.; Caston, J. R. J Virol 2007, 81, 6869–6878.
18. Rabl, J.; Smith, D. M.; Yu, Y.; Chang, S.-C.; Goldberg, A. L.;
Cheng, Y. Molecular Cell 2008, 30, 360–368.
19. Fujii, T.; Iwane, A. H.; Yanagida, T.; Namba, K. Direct visualiza-
tion of secondary structures of F-actin by electron cryomicro-
scopy. Nature 2010, 467, 724–728.
20. Zhang, J.; Baker, M. L.; Schroder, G. F.; Douglas, N. R.; Reiss-
mann, S.; Jakana, J.; Dougherty, M.; Fu, C. J.; Levitt, M.; Ludtke,
S. J.; Frydman, J.; Chiu, W. Nature 2010, 463, 379–383.
21. Cong, Y.; Zhang, Q.; Woolford, D.; Schweikardt, T.; Khant, H.;
Dougherty, M.; Ludtke, S. J.; Chiu, W.; Decker, H. Structure
2009, 17, 749–758.
22. Sindelar, C. V.; Downing, K. H. J Cell Biol 2007, 177, 377–385.
23. Alushin, G. M.; Ramey, V. H.; Pasqualato, S.; Ball, D. A.; Grigor-
ieff, N.; Musacchio, A.; Nogales, E. Nature 2010, 467, 805–810.
24. Jiang, W.; Baker, M. L.; Ludtke, S. J.; Chiu, W. J Mol Biol 2001,
308, 1033–1044.
25. Del Palu, A.; He, J.; Pontelli, E.; Lu, Y.Proceeding of Computa-
tional Systems Bioinformatics Conference(CSB), 2006, pp 89–
98.
26. Baker, M. L.; Ju, T.; Chiu, W. Structure 2007, 15, 7–19.
27. Lasker, K.; Dror, O.; Shatsky, M.; Nussinov, R.; Wolfson, H. J.
IEEE/ACM Trans Comput Biol Bioinform 2007, 4, 28–39.
28. Zeyun, Y.; Bajaj, C. IEEE/ACM Trans Comput 2008, 5, 568–582.
29. Baker, M. L.; Abeysinghe, S. S.; Schuh, S.; Coleman, R. A.;
Abrams, A.; Marsh, M. P.; Hryc, C. F.; Ruths, T.; Chiu, W.; Ju, T.
J Structural Biol 2011, 174, 360–373.
30. Ma, L.; Reisert, M.; Burkhardt, H. Comput Biol Bioinformatics,
IEEE/ACM Trans 2011, ( 99), 1.
31. Kong, Y.; Ma, J. J Mol Biol 2003, 332, 399–413.
32. Lawson, C. L.; Baker, M. L.; Best, C.; Bi, C.; Dougherty, M.;
Feng, P.; van Ginkel, G.; Devkota, B.; Lagerstedt, I.; Ludtke, S. J.;
Newman, R. H.; Oldfield, T. J.; Rees, I.; Sahni, G.; Sala, R.;
Velankar, S.; Warren, J.; Westbrook, J. D.; Henrick, K.; Kleywegt,
G. J.; Berman, H. M.; Chiu, W. Nucleic Acids Res 2011, 39
(Database issue), D456–D464.
33. Wriggers, W.; Milligan, R. A.; McCammon, J. A. J Struct Biol
1999, 125, 185–195.
34. Topf, M.; Lasker, K.; Webb, B.; Wolfson, H.; Chiu, W.; Sali, A.
Structure 2008, 16, 295–307.
35. Schroder, G. F.; Brunger, A. T.; Levitt, M. Structure 2007, 15,
1630.
36. Fernandez, J.-J.; Li, S. J Structural Biol 2003, 144, 152–161.
37. Boser, B. E.; Guyon, I. M.; Vapnik, V. N. In Proceedings of the
Fifth Annual Workshop on Computational Learning Theory;
ACM: Pittsburgh, PA, 1992; pp 144–152.
38. Cortes, C.; Vapnik, V. Mach. Learn. 1995, 20, 273–297.
39. Chang, C.-C.; Lin, C.-J. ACM Trans Intell Syst Technol 2011, 2,
1–27.
40. Knerr, S.; Personnaz, L.; Dreyfus, G. In Neurocomputing: Algo-
rithms, Architectures and Applications; Fogelman-Soulie, F.;
Herault, J., Eds. Springer-Verlag, LLC: New York, 1990; Vol.
F68, pp 41–50.
41. Kong, Y.; Zhang, X.; Baker, T. S.; Ma, J. J Mol Biol 2004, 339,
117–130.
42. Kleywegt, G. J. J Mol Biol 1997, 273, 371–376.
43. Kabsch, W.; Sander, C. Biopolymers 1983, 22, 2577–2637.
44. Ludtke, S. J.; Baldwin, P. R.; Chiu, W. J Struct Biol 1999, 128,
82–97.
Reviewing Editor: Steven J. Ludtke
708 Si et al.
Biopolymers